 Zihao Li
Zihao Li Ziyu Huang2
Ziyu Huang2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 11 February 2021
Sec. Cell Death and Survival
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.628330
This article is part of the Research Topic Programmed Cell Death 2.0: The Quality of Cell Death View all 11 articles
Osteoarthritis (OA) is the most common joint disease. With the increasing aging population, the associated socio-economic costs are also increasing. Analgesia and surgery are the primary treatment options in late-stage OA, with drug treatment only possible in early prevention to improve patients’ quality of life. The most important structural component of the joint is cartilage, consisting solely of chondrocytes. Instability in chondrocyte balance results in phenotypic changes and cell death. Therefore, cartilage degradation is a direct consequence of chondrocyte imbalance, resulting in the degradation of the extracellular matrix and the release of pro-inflammatory factors. These factors affect the occurrence and development of OA. The P2X7 receptor (P2X7R) belongs to the purinergic receptor family and is a non-selective cation channel gated by adenosine triphosphate. It mediates Na+, Ca2+ influx, and K+ efflux, participates in several inflammatory reactions, and plays an important role in the different mechanisms of cell death. However, the relationship between P2X7R-mediated cell death and the progression of OA requires investigation. In this review, we correlate potential links between P2X7R, cartilage degradation, and inflammatory factor release in OA. We specifically focus on inflammation, apoptosis, pyroptosis, and autophagy. Lastly, we discuss the therapeutic potential of P2X7R as a potential drug target for OA.
Osteoarthritis (OA), as an age-related degenerative joint disease, presents with physical pain and disability in patients. Finding effective therapies for OA remains a pertinent health problem (Carr et al., 2012). The occurrence and development of OA can be attributed to multifaceted factors, such as age, obesity, exercise, and trauma (Uthman et al., 2013). From a clinical perspective, its features include articular cartilage loss, synovitis, subchondral bone sclerosis, and osteophyte formation. From a cellular perspective, it is attributed to morphological, biochemical, and biomechanical changes affecting the extracellular matrix (ECM).
The most important structural part of the joint is the cartilage, and its condition directly affects the occurrence and development of OA. Cartilage consists of structural proteins, such as collagen (primarily type II collagen), non-collagen proteins, proteoglycan, elastin, and aminoglycan, which form a stable network structure providing elasticity and compression resistance to joints. In OA tissues, this network structure loses its integrity with a resulting loss of tensile strength (Speziali et al., 2015). The composition and integrity of the cartilage matrix can be maintained by chondrocytes—which account for a small proportion of the total cartilage volume—to provide mechanical support and joint lubrication (Archer and Francis-West, 2003). In response to certain chemical and mechanical factors, chondrocytes produce and release inflammatory factors (IL-1β, IL-6, and TNF-α) and secrete matrix-degrading enzymes [metalloproteinases (MMPs)] and proteoglycan-degrading enzymes (ADAMTS), to regulate the shape and structure of cartilage. Unfortunately, these inflammatory factors and enzymes are the primary contributors to cartilage degradation. Therefore, the occurrence and development of OA can be ascribed to the state and the catabolism balance of chondrocytes and cartilage. In OA, the changes in the chondrocytes can be categorized into three groups, namely: (i) cell proliferation (early stage)/cell death (late stage); (ii) anabolic and catabolic balance disorder (production of matrix-degrading enzymes); and (iii) phenotypic change (Sandell and Aigner, 2001). The influence of the cell phenotypic changes in the development of OA is particularly critical. In this article, we focus on chondrocyte inflammation, apoptosis, pyroptosis, and autophagy, and analyze the correlation between these phenotypes and cartilage degradation in OA.
The P2X7 purinergic receptor (P2X7R) is a trimeric adenosine triphosphate (ATP)-gated cation channel, which is expressed in several eukaryotic cells, such as immune cells and bone cells. As a key inflammatory switch, the activation of P2X7R mediates several downstream reactions, including the release of inflammatory factors, cell proliferation, death, and phenotypic changes (North, 2002). Owing to the important role of P2X7R in various immune, inflammatory, musculoskeletal, and nervous system diseases, it could be a potential drug treatment target for OA. In this article, we review the association between P2X7R and OA. We explain the structure and function and the inhibitory effect of P2X7R and emphasize the correlation and intersection between P2X7R, inflammation, and OA. From the perspective of apoptosis, pyroptosis, and autophagy, we discuss the possible association between P2X7R, cartilage degradation, and inflammatory factor release in OA. Further, we summarize the possible treatment methods, including the use of P2X7R as a drug target, and highlight the potential future mechanistic research.
Most studies on OA focus on the synovium-mediated development of inflammation. Synoviocytes identify the foreign fragments that fall into the joint cavity as byproducts of cartilage degradation. This results in synovial angiogenesis, an increase in the release of auto-inflammatory factors, and the stimulation of chondrocytes to synthesize and secrete MMPs. Through this mechanism, synovitis can promote cartilage degradation and aggravate OA (van Lent et al., 2004). Synovial fluid factors are increased in the damaged cartilage of OA patients (Kim et al., 2006), including toll-like receptor (TLR)-2 and TLR-4 ligands, such as alarm proteins [S100 protein and high mobility group protein B1 (HMGB1)], low molecular weight hyaluronic acid, tenascin C, and fibronectin (Scanzello et al., 2008; García-Arnandis et al., 2010; van Lent et al., 2012). These mediators, together with low-grade inflammation present in the compromised joints, induce inflammation of the synoviocytes and affect the catabolism balance of the chondrocytes (Wang et al., 2011). Therefore, the innate immunity may be a key factor driving the development of OA. As for mild systemic inflammation, plasma and peripheral white blood cells can reflect the level of inflammation in the joint tissues. Adipokines secreted from visceral fat, such as leptin, resistin, and adiponectin also play an important role (Gomez et al., 2011), having both pro- or anti-inflammatory effects on OA development (Distel et al., 2009). The release of inflammatory factors into the blood can cause diseases in other parts as well, such as the inflammation of the nervous system and Alzheimer’s disease (Kyrkanides et al., 2011).
Although our understanding of the mechanism underlining OA has improved, limited progress has been made with respect to its treatment. Currently, analgesia and joint replacement are primarily used to treat end-stage OA (Bijlsma et al., 2011; Carr et al., 2012; Pivec et al., 2012) which neglects the problem of early disease incidence. Fortunately, the continuous advancement in our understanding of OA pathogenesis and the improvement in detection methods have shifted the focus toward the prevention and treatment of early OA. Lifestyle adjustments, such as weight loss and exercise for obese individuals, can enhance muscle strength and joint stability, improve cardiovascular function, and reduce the risk of OA (Felson et al., 1992; Gudbergsen et al., 2012; Uthman et al., 2013).
Drug treatment often goes hand-in-hand with prevention, with commonly used drugs in clinical practice, such as paracetamol (acetaminophen) and non-steroidal anti-inflammatory drugs (NSAIDs) also being able to effectively relieve pain symptoms (Palmer et al., 2013). Furthermore, the anti-inflammatory and anti-catabolism properties of chondroitin and glucosamine have been proven in clinical trials to alleviate the occurrence and development of OA (Henrotin and Lambert, 2013). Hyaluronic acid, a glycosaminoglycan, can also act as a lubricant in the synovial fluid. In patients with OA, the concentration of hyaluronic acid in the joint cavity is low, and the friction experienced by the joint surfaces during limb movement increases pain. It must be noted, however, the clinical efficacy and safety of injecting hyaluronic acid into the joint cavity remains controversial (Rutjes et al., 2012). Another lubricating element, lubricin, has been shown to work synergistically with hyaluronic acid with limited effects (Schmidt et al., 2007). Several targeted drugs have also been developed to treat OA, such as MMP inhibitors (doxycycline) (da Costa et al., 2012), osteoclast inhibitors (bisphosphonates and strontium ranelate) (Henrotin et al., 2001), IL-1R inhibitors (anakinra) (Chevalier et al., 2009), immunoglobulins (AMG 108) (Cohen et al., 2011), TNF-α inhibitors (adalimumab) (Verbruggen et al., 2012), cartilage repair factors [recombinant osteogenic protein-1 (Nishida et al., 2004) and kartogenin (Johnson et al., 2012)]. The application of these drugs still needs improvement as there is still a gap between the desired therapeutic effect and the clinical outcomes. This implies that more effective and targeted drugs are required in the prevention and treatment of OA. Therefore, an in-depth exploration of the particular etiology and pathogenesis of OA is critical.
Purine receptors can be divided into two categories: adenosine (ADO) activated P1 receptors and purine and pyrimidine nucleotides (ATP and ADP) activated P2 receptors. P2 receptors can further be divided into P2X ion channel receptors and P2Y metabotropic receptors. There are seven different members of the P2X family (P2X1-7), of which the P2X7 receptor (P2X7R, encoded by the P2RX7 gene) is most closely related to inflammation and immunity, and belongs to the trimeric ligand-gated cation channel. Compared with other P2X receptors, P2X7R requires a higher concentration of ATP for activation and has a higher affinity for the selective agonist BzATP, with 10–30 times more potency than ATP (North, 2002). In addition, natural splice variants (P2X7A-J) and (P2X7a, P2X7k, P2X713b, and P2X713c) were found in human and rodent tissues, respectively, with P2X7R sharing 77–85% sequence identity. Therefore, several experiments have used rodent models to study the function of P2X7R.
Structurally, P2X7R contains relatively short intracellular amino- (N) and long carboxyl- (C) termini, and two hydrophobic transmembrane fragments separated by glycosylated extracellular ATP binding domains (transmembrane domains); its topology is similar to that of other ionic P2X receptors (Khakh and North, 2006; Karasawa and Kawate, 2016). The functional channel toward the plasma membrane is composed of stable trimers (Nicke, 2008; Jiang et al., 2013). Moderate activation occurs when the receptor is bound to ATP. The gated state of P2X7R is opened, mediating non-inactivated Na+ and Ca2+ influx and K+ efflux, resulting in rapid depolarization (Surprenant et al., 1996). When the activation time is prolonged, P2X7R can induce the formation of membrane pores, allowing molecules up to hundreds of Da to pass (Pelegrin and Surprenant, 2006).
The transcription and expression of P2X7R can be regulated by microRNAs [e.g., miR-373 (Zhang et al., 2018)], long-coding RNAs (e.g., lncRNA NONRATT021972), and transcription factors [e.g., specific protein 1 (Sp1)], and P2X7R will also undergo post-translational modifications, including N-linked glycosylation, palmitoylation, and ADP-ribosylation (Sluyter, 2017). The most important mediator for the activation of receptors is ATP and its role in inflammation and immunity has been proven. Cell death in inflammatory tissues releases large amounts of ATP, increasing the extracellular ATP concentration to hundreds of μM, which is enough to activate P2X7R. In contrast, the extracellular ATP concentration in healthy tissues is very low (Pellegatti et al., 2008; Wilhelm et al., 2010; Barbera-Cremades et al., 2012). In addition to passive release, ATP can also cross incomplete cell membranes through specific membrane protein channels, or can be stored in cytoplasmic vesicles and secreted outside the cell (Burnstock, 2006). Other mechanisms, such as the one mediated by the gap junction protein pannexin-1, control the release of ATP from living or apoptotic cells. Under certain conditions, such as hypoxia, increased extracellular K+ concentration, and mechanical stress stimulation, pannexin-1 has high ATP permeability (Wang and Dahl, 2018). In undamaged tissues, a small amount of ATP in the extracellular environment will be rapidly degraded by enzymes (Plesner, 1995). Therefore, to activate P2X7R, the ATP release channel and the receptor should be in close proximity, which explains the closely related protein functions and structures of pannexin-1 and P2X7R (Bao et al., 2004; Pelegrin and Surprenant, 2006; Locovei et al., 2007).
The P2X7 receptor activation mediates numerous signaling pathways and cellular responses, such as the activation of the NLRP3 inflammasome via K+ efflux, which induces IL-1β release; the formation of mitochondrial reactive oxygen species (mtROS) via ATP signaling, which can also regulate the activity of P2X7 ion channels; the regulation of caspase, cathepsin, and MMP release; and the regulation of the pro-inflammatory mediator prostaglandin E2 (PGE-2). PGE-2 is an important downstream inflammatory pathway of P2X7R, which may make P2X7R a potential anti-inflammatory target to replace cyclo-oxygenase that regulates the activation of transcription factors, such as NF-κB p65, HIF-1α, and PI3K-AKT; glutamate efflux; endocytosis; cell proliferation; and cell death. Briefly, P2X7R regulates ion flow, protease activation, and various secretory responses, which constitute the most common signaling pathways in inflammation (Bartlett et al., 2014). Therefore, P2X7R is also known as an inflammatory switch.
The importance of P2X7R in inflammation has recently attracted attention in the field of bone-related diseases. It has critical influence on OA, synovitis, and rheumatoid arthritis. This has opened a way for P2X7R inhibitors (as a specific target-directed approach) to serve as potential new therapeutics for these diseases.
When OA occurs, the most significant cellular response is inflammation, which can be triggered by external and internal mechanisms. External factors include mechanical stress (compression, stretching, hydrostatic pressure, and shear stress), while the mechanoreceptors (ion channels and integrins) present on the surface of joint cells convert abnormal mechanical stress into activated intracellular signals (Guilak, 2011). Accordingly, the activation of the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways is commonly observed (Signaling, 2004).
In daily life, physical exercise causes mechanical stress on joints that can be beneficial. Studies have shown that regular exercise can alleviate low-intensity inflammatory conditions, as are observed in OA, cancer, and cardiovascular disease, and reduce the risks associated with a high-fat diet. P2X7R is one of the factors that determine the level of IL-1β in the plasma after exercise. Furthermore, P2X7R, NF-κB, NLRP3, and caspase-1 levels have been shown to progressively increase between sedentary individuals, trained athletes, and endurance athletes (Comassi et al., 2017). Post-exercise, NLRP3 and caspase-1 levels increased in sedentary individuals (pro-inflammatory), while they decreased in endurance athletes (anti-inflammatory). Regardless of the degree of fitness, acute exercise increased P2X7R expression and function. In another study, exercise reduced the expression levels of P2X7R, NLRP3, caspase-1, and IL-1β in plasma caused by a high-fat diet in Sprague Dawley rats, inhibiting inflammation and apoptosis, enhancing autophagy, and reducing myocardial damage (Chen et al., 2019). It is thus evident that P2X7R plays an important role during exercise to relieve inflammation and other risk factors.
The P2X7 receptor regulates the intensity and duration of several inflammatory reactions (Ferrari et al., 2006; Khakh and North, 2006; Dubyak, 2012). In macrophages, monocytes, and microglia, P2X7R mediates Ca2+ influx and K+ efflux inducing the activation and release of cytokines (Kahlenberg and Dubyak, 2004; Ferrari et al., 2006). This initiates inflammasome assembly, while caspase-1 pro-IL-1β cleavage releases a large amount of mature IL-1β (MacKenzie et al., 2001; Pelegrin et al., 2008; Dubyak, 2012). IL-1β can induce reactive oxygen species (ROS) and the expression of protein-degrading enzymes, leading to the loss of type II collagen and proteoglycans, thereby destroying cartilage structure and affecting joint function and stability (Sitia and Rubartelli, 2020). IL-1β is an atypical cytokine lacking secreted fragments which does not follow the standard endoplasmic reticulum-Golgi pathway for extracellular release (Dinarello, 2002). Instead, it functions via an ATP-dependent P2X7R-inflammasome pathway by TLR stimulation-induced accumulation of pro-IL-1β in the cytoplasm followed by its subsequent release (Perregaux et al., 2000; Ferrari et al., 2006). Other mechanisms of cytokine release occur through passive release after cell death, and secretion by modified lysosomes, exosomes, or plasma membrane-derived microvesicles (MacKenzie et al., 2001; Lopez-Castejon and Brough, 2011; Piccioli and Rubartelli, 2013). In all these instances, P2X7R is the main driver (Bianco et al., 2005; Pizzirani et al., 2007; Qu et al., 2007), further emphasizing its key role in the release of biologically active IL-1β.
Among the internal factors contributing to the development of OA, ROS play a pivotal role. ROS comprise molecules containing free radicals, including oxygen free radicals (OH–), hypochlorite ions (OCl–), superoxide anions (O2–), nitric oxide (NO), and hydrogen peroxide (H2O2). ROS is mainly produced through the following pathways: mitochondrial (through oxidative phosphorylation) and non-mitochondrial membrane-bound nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and xanthine oxidase (XO) pathways (Turrens, 2003). When the catabolic cell balance is disturbed, the accumulation of ROS leads to an increase in the production of inflammatory mediators, and an ineffective elimination of the oxidized proteins. This ultimately triggers oxidative damage that exacerbates inflammation (Licastro et al., 2005). Oxidative stress can promote cell senescence, especially affecting chondrocytes (Loeser, 2011). Chondrocytes have a low ability to divide and proliferate with high ability to synthesize and secrete, a circumstance called the senescence-associated secretory phenotype (SASP) (Coppe et al., 2010). ROS normally occurs at low levels in chondrocytes but can still regulate gene expression, affect ECM synthesis and catabolism balance, drive the production of cytokines, such as IL-1β (Forsyth et al., 2005), and induce cell apoptosis. The exacerbated oxidative stress in chondrocytes inhibits the PI3K/Akt pathway; activates the NF-κB pathway to promote the transcription of MMPs; activates the extracellular signal-regulated kinase (ERK)/MAPK pathway to reduce the expression of type II collagen, proteoglycans, and Sox-9; reduces ECM synthesis (Yin et al., 2009; Yu and Kim, 2015); and acts as a signal transduction intermediate for IL-1β and TNF-α in the c-Jun N-terminal kinase (JNK) pathway activation (Lo et al., 1996). Therefore, ROS plays an important role in the intracellular signal transduction mechanisms and is closely related to cartilage homeostasis (Rathakrishnan et al., 1992; Henrotin et al., 1993; Henrotin et al., 2003).
Summarily, the ion flow mediated by P2X7R activation is closely related to the homeostasis of the intracellular environment. Mitochondrial dysfunction caused by Ca2+ overload can induce the production of ROS. Activation of inflammasomes triggered by K+ efflux can induce the production of IL-1β and the activation of inflammation-related pathways. This indicates the important role of P2X7R in inflammation and its potential influence on the occurrence and development of OA. In terms of preventive treatment and cytokine treatments were not found to significantly improve the symptoms of OA or relieve the deterioration of bone structure. The results of pilot and controlled studies using anti-IL-1 and anti-TNF molecules lack credibility (Chevalier et al., 2009; Verbruggen et al., 2012). In this context, P2X7R-targeted therapy could present as a new direction for the prevention and treatment of OA.
When a cell undergoes apoptosis, the chromatin condenses around the nucleus, the cell membrane shrinks and bubbles, and apoptotic bodies with intact membranes form around organelles (Kerr et al., 1972). Upon recognition by the immune system, inflammation is induced. This can be prevented, however, by the phagocytic engulfment of these vesicles (Kurosaka et al., 2003). Apoptosis is a programmed cell death, which differs from passive necrosis (or cell membrane disintegration) caused by pyroptosis. At the molecular level, initiation, execution, degradation, and clearance are the established sequential steps of apoptosis.
Apoptosis can be induced via extrinsic mediation by death receptors and via intrinsic mediation by mitochondria (Elmore, 2007). The extrinsic pathways include damage or pathogen-related molecular patterns (DAMPs or PAMPs) and cytokines that activate the TNF superfamily (e.g., death receptors). The death receptor Fas and its ligand FasL combine to drive the assembly of the death-inducing signaling complex (DISC). Thereafter, the recruitment and activation of caspase-8 are mediated by Fas-related proteins and death domain (FADD) adaptor molecules, which leads to caspase-3 activation, and finally apoptosis (Fuentes-Prior and Salvesen, 2004). P2X7R is closely related to apoptosis. IL-1β, mediated by P2X7R, can further induce the production of TNF-α, both having pro-apoptotic effects (Lee et al., 2016). The membrane pores formed by P2X7R (Donnelly-Roberts et al., 2004), together with its mediated K+ efflux (Delarasse et al., 2009; Aguirre et al., 2013), can induce the activation of caspase-8 and subsequently cleave caspase-3, the key executor of apoptosis.
Intrinsic pathways include those mediated by DNA damage, cytoplasmic Ca2+ overload, oxidative or endoplasmic reticulum stress (ERS), decreased cytokine levels, and the response to intracellular damage (Vanden Berghe et al., 2015). Bcl-2 family members respond by changing the permeability of the mitochondrial outer membrane (Kroemer and Reed, 2000). The activation of its priming members (e.g., Bid) not only inhibit survival supervisory members (e.g., Bcl-2), but also oligomerize pro-apoptotic members (e.g., Bax), thereby destroying the mitochondrial outer membrane. A large number of proteins are released into the cytoplasm, such as cytochrome c (Cyt c) (Qiu et al., 2000), activating a key component of apoptotic bodies, namely the apoptotic protease activator factor-1 (APAF-1) (Green, 2003). Cyt c combines with oligomeric APAF-1 to form a multimeric structure, recruiting and activating caspase-9 in the mitochondrial pathway (Bao and Shi, 2007). Caspase-9 directly activates downstream caspase-3, 6, and 7, ultimately leading to substrate cleavage and apoptosis. In addition to the inflammasome assembly caused by the intracellular K+ consumption, K+ efflux can also induce apoptosis by promoting APAF-1 (Bortner et al., 1997; Karki et al., 2007). Furthermore, Ca2+ influx can lead to mitochondrial dysfunction and caspase-3 activation. As P2X7R plays a crucial role ion transport, P2X7R inhibitors have shown promise in preventing cell death dependent on these mechanisms (Nishida et al., 2012).
Reactive oxygen species (e.g., H2O2) damages mitochondria as part of the apoptotic intrinsic pathway, destroying its DNA integrity and repair ability (Grishko et al., 2009). This can induce chondrocyte apoptosis through PI3K/Akt, p38 MAPK, and JNK signaling pathways (Yu and Kim, 2014; Li and Dong, 2016; Rao et al., 2017). As mentioned earlier, P2X7R can induce the generation of ROS, and the activation of P2X7 will increase the phosphorylation of tyrosine protein, activating MAPK (Panenka et al., 2001; Papp et al., 2007), including ERK, JNK, and p38 MAPK. ERK is essential for cell survival, while JNK and p38 MAPK can be activated under stress stimulation (Wada and Penninger, 2004; Yang et al., 2012). JNK phosphorylates Bcl-2 to reduce its anti-apoptotic activity (Cui et al., 2007), and p38 MAPK reduces the expression of Bcl-2 and induces the production of caspase-3, leading to apoptosis (Nishida et al., 2012). The NF-κB signaling pathway closely relates to P2X7R involvement in chondrocyte apoptosis, since it can affect the expression of apoptosis-related proteins (Bcl-2, Bax, Cyt c, and caspase-3) (Pan et al., 2018).
Unregulated apoptosis can lead to pathological changes, such as tumors and inflammation. Apoptosis is both the initiator (Hashimoto et al., 1998) and feedback in the development of OA (Blanco et al., 1998; Heraud et al., 2000). Specifically, chondrocyte apoptosis is considered a prerequisite for the development of OA. Compared with the normal cartilage, the number of apoptotic chondrocytes is increased in the cartilage of OA patients (Hashimoto et al., 1998), while that of normal chondrocytes is decreased (Aigner et al., 2006), and a dysregulation of apoptosis-related genes occurs (Bobinac et al., 2003). In addition, animal studies showed that low levels of mechanical stress stimulation can induce chondrocyte apoptosis, with high levels of mechanical stress stimulation leading to the degradation of the cartilage matrix (Loening et al., 2000). This indicates that chondrocyte apoptosis occurs first, followed by tissue damage, reflecting early features of OA. Therefore, chondrocyte apoptosis also represents the feedback of the outcome of OA (Zamli et al., 2013). In transgenic mice with type II collagen gene knockout, chondrocytes undergo increased apoptosis, have lower survival, and no longer interact with the surrounding matrix, and there is presentation of severe cartilage degradation, compared to the wild-type mice (Zemmyo et al., 2003). Several studies have confirmed the positive relationship between the severity of cartilage degradation and the rate of chondrocyte apoptosis (Blanco et al., 1998; Hashimoto et al., 1998; Thomas et al., 2007). Some studies used collagenase to destroy the matrix, increasing cell permeability to expose the chondrocytes to apoptosis inducers (NO and cytokines) secreted by either synoviocytes or other chondrocytes, ultimately leading to cell apoptosis (Zamli and Sharif, 2011). Taken together, although the sequence of cartilage degradation and chondrocyte death is still controversial, they are undeniably closely related in OA (Zamli and Sharif, 2011).
Apoptosis does not always occur in isolation and often accompanies other phenotypes. In the early stage of OA, autophagy plays a protective role to prevent cell death on the surface and middle layers of cartilage (Almonte-Becerril et al., 2010; Carames et al., 2010); while in the late stage of OA, autophagy induces apoptosis and accelerates cell death (Almonte-Becerril et al., 2010). In the cartilage of patients with OA, the apoptotic bodies that have not been engulfed in time, together with increasing cartilage calcification, aggravate OA through secondary pyroptosis (Roach et al., 2004). The cell death caused by apoptosis and pyroptosis have also been confirmed in a model of OA induced by mechanical stress (Chen et al., 2001; Stolberg-Stolberg et al., 2013). In vivo and in vitro experiments showed that the application of mechanical stress to the chondrocytes of human cartilage explants (D’Lima et al., 2001), end plates (Kong et al., 2013), or growth plates (Sun et al., 2017) can lead to the loss of type II collagen, proteoglycans, and glycosaminoglycans, accompanied by cell death. The protective role of antioxidants must briefly be mentioned as they can reduce cell death caused by shear stress (Martin and Buckwalter, 2006) and abnormal cyclic loading (Beecher et al., 2007).
In summary, P2X7R-mediated ion flow, oxidative stress, and related signaling pathways are closely related to apoptosis. Apoptosis is complemented by OA under the action of autophagy and pyroptosis. Thus, P2X7R plays an important role in the interaction between apoptosis and OA. It participates in cartilage degradation and inflammatory factor release, aiding the progression to OA.
Apoptosis and pyroptosis can occur independently, sequentially, and simultaneously (Zeiss, 2003). Both have common triggers and biochemical networks. The intensity and duration of different stimuli, the amount of ATP that can be consumed in the cell, and the potency of caspases can convert the ongoing process of apoptosis into pyroptosis (Leist et al., 1997; Denecker et al., 2001; Zeiss, 2003). The process of accidental and passive cell death caused by harmful stimuli (ultraviolet radiation, heat, hypoxia, and cytotoxic drugs), accompanied by cytoplasmic granulation, mitochondrial swelling, cell swelling, rupture of the plasma membrane, and uncontrolled release of intracellular substances (including pro-inflammatory cytokines, DAMPs, and lysosomal enzymes), causes an inflammatory response called pyroptosis (Festjens et al., 2006; Kaczmarek et al., 2013).
NLRP3, as one of the components of the inflammasome (the core component of pyroptosis) exists in the cytoplasm and can recognize PAMPs (such as pore-forming toxins and microbial cell wall components) or DAMPs (such as ATP derived from endogenous stress and uric acid crystals) (McGilligan et al., 2013; Guo et al., 2015; Sharma and Kanneganti, 2016). NLRP3 activates caspase-1, promoting the secretion of IL-1β and IL-18 to exacerbate inflammation (Shao et al., 2015), resulting in rapid cell death (Latz et al., 2013). There are two main ways to activate inflammasomes. The first is through the recognition of PAMPs or DAMPs by TLRs to activate the NF-κB signaling pathway, increasing the synthesis of NLRP3, pro-IL-1β, and pro-IL-18; the second is by initiating oligomerization, leading to the assembly of inflammasomes (Shao et al., 2015). Both kinase activity and autophagy can regulate the activity of the NLRP3 inflammasome (Yang et al., 2017). Autophagy relieves pyroptosis by degrading pro-IL-1β, inflammasome components, and damaged mitochondria (Zhou et al., 2011; Shi et al., 2012).
As a key activator of inflammasomes, P2X7R participates in both the classical pathway (e.g., NLRP3) and non-classical pathway (e.g., caspase-11). The latter induces cell death similar to pyroptosis (Vigano and Mortellaro, 2013; de Gassart and Martinon, 2015). The C-terminal part of pannexin-1 can be cleaved by caspase-11, leading to ATP release and K+ efflux (Yang et al., 2015). Hypoxia-induced caspase-11 expression and activation (Kim et al., 2003) are involved in ERS-dependent cell death (Fradejas et al., 2010). P2X7R and NLRP3 can also interact physically. Immunoprecipitation and confocal microscopy studies have found that changes in the local ionic microenvironment in cells caused by P2X7R or by a membrane pore opening can result in inflammasome assembly (Franceschini et al., 2015).
Under normal circumstances, inflammasomes activate the innate immune system, producing IL-1β, IL-18, and other pro-inflammatory cytokines to protect cells from infection and reduce damage (Man and Kanneganti, 2015). However, excessive activation of inflammasomes overproduce cytokines, causing inflammation and metabolic disorders (Strowig et al., 2012). NLRP3 is an established potential target for diseases, such as atherosclerosis, rheumatoid arthritis, and gout. NLRP3 is also activated in the synovial tissue contributing to cartilage degradation (Bougault et al., 2012). In patients with OA, high NLRP3 expression in tissue has been linked to high XO levels (an enzyme producing ROS and uric acid), further confirming the association between OA and NLRP3 inflammasomes (Clavijo-Cornejo et al., 2016).
As stated before, cartilage degradation in OA can result from disrupted anabolic and catabolic chondrocyte balance (Bijlsma et al., 2011). Once initiated, the primary cartilage-degrading enzymes, IL-1β and TNF-α, are produced by the NLRP3 inflammasome (Mueller and Tuan, 2011; Haseeb and Haqqi, 2013). IL-1β both induces cell apoptosis (Haseeb and Haqqi, 2013) and stimulates the secretion of other cartilage-degrading enzymes, such as MMP3/13 and ADAMTS-4/5 (Mueller and Tuan, 2011), leading to the degradation of the type II collagen and the proteoglycans of the ECM (Haseeb and Haqqi, 2013). The released collagen and proteoglycan particles stimulate the production of IL-18 (Olee et al., 1999; Man and Mologhianu, 2014), which inhibits proteoglycan synthesis and chondrocyte proliferation, promotes prostaglandin production, and further induces apoptosis (Jin et al., 2011).
Targeting the components of inflammasomes and upstream and downstream pathways, such as K+ efflux, ROS, mitochondrial-, and lysosome dysfunction, is an important approach in the treatment of OA (Di Virgilio, 2013; He et al., 2016). The small molecule inhibitor MCC950 inhibits the oligomerization of inflammasomes and IL-1β release (Coll et al., 2015). Drugs that target IL-1R, such as rilonacept, anakinra, and canakinumab, can also reduce cartilage destruction (Dinarello et al., 2012). As an important driving force of the inflammasome pathway, P2X7R has potential as a drug target treatment to alleviate cartilage damage caused by pyroptosis.
Cells degrade excess or damaged lipids, proteins, and organelles, to maintain cell viability through autophagy. Autophagy also plays a role in maintaining mitochondrial function (Lo Verso et al., 2014), as the downregulation of autophagy will induce the accumulation of damaged mitochondria, increase the levels of ROS, and lead to tissue degeneration (Mizushima and Komatsu, 2011). Autophagic flux can be divided into five stages: (i) induction, (ii) nucleation, (iii) extension, (iv) maturation, and (v) lysis. A complex is formed in each of the first three stages. In the induction phase, the Atg1 complex (including Atg1/Ulk1, Atg13, and Atg17/FIP200) and the activity of mammalian target of rapamycin complex 1 (mTORC1) is inhibited, Atg13 phosphorylation level is reduced, and the complex is formed (Ganley et al., 2009). The Vps34 (PI3K)-Atg6 (Beclin-1) complex acts on the nucleation of the membrane vesicles and mediates the formation of the pre-autophagosome structure. The extension of autophagosomes mainly depends on two ubiquitin-like systems: the binding process of Atg12-Atg5 and the modification process of LC3. LC3-II is a multi-signal transduction regulatory protein that is located on the autophagy vesicle membrane and is often used as a marker for autophagy formation.
Cartilage lacks blood vessels to supply oxygen, thus cells exist in a somewhat hypoxic environment. Hypoxia promotes autophagy (e.g., through an increase in Ulk1 and Atg5 mRNA levels), maintains the chondrocyte phenotype (e.g., increased Sox9 and type II collagen mRNA levels, decreased MMP13 and ADAMTS-5 mRNA levels), and further induces hypoxia factors (HIFs) (Coimbra et al., 2004), which play important roles as key regulators of autophagy in chondrocytes. HIF-1 is a heterodimer composed of two different subunits, HIF-1α and HIF-1β. Under hypoxic conditions, HIF-1α degradation is prevented and transferred to the nucleus, where it combines with β subunits to form an active HIF-1 transcription factor. HIF-1α is essential for cell survival, and its knockout induces mass chondrocyte death in the cartilage growth plate (Schipani et al., 2001). Bcl-2 plays a role in HIF-1α-mediated autophagy (Zhang F. et al., 2015), while HIF-1α modulates the Beclin-1/Bcl-2 complex (Bohensky et al., 2007), activates AMPK, inhibits mTOR (Bohensky et al., 2010), induces chondrocyte autophagy, and prevents apoptosis. P2X7R is also closely related to HIF-1α and autophagy (Mascanfroni et al., 2015). The ATP-P2X7R signal axis driven by oxidative metabolism participates in the differentiation of bone marrow-derived macrophages into their M2 type (Barbera-Cremades et al., 2016). HIF-1α also plays a key role in this process. For example, HIF-1α promotes lactic acid-dependent M2 polarization in the tumor microenvironment (Colegio et al., 2014). Interestingly, P2X7R is a strong stimulator of aerobic glycolysis and lactic acid production (Amoroso et al., 2012). P2X7R receives ATP signals to stimulate the alkalinization of lysosomes (Guha et al., 2013), which leads to an increase in the lysosomal pH. The accumulated autophagosomes cannot be fused with lysosomes for degradation and release outside the cell, thereby reducing autophagy flux (Takenouchi et al., 2009a, b). P2X7R activation downregulates the expression of glutamate transporter and promotes neuro-autophagy, which increases excitatory amino acids (Zhang et al., 2008; Działo et al., 2013; Kulbe et al., 2014) caused by an abnormal stimulation of glutamate receptors, ultimately resulting in cognitive impairment (Sun et al., 2015). P2X7R activates PI3K/Akt/GSK3β/β-catenin and/or mTOR/HIF1α/VEGF pathways to promote the proliferation and metastasis of osteosarcoma and increase bone destruction (Zhang et al., 2019).
Whether P2X7R promotes or inhibits autophagy is still controversial (Sluyter, 2017). For instance, one study showed ATP-activated P2X7R to inhibit the PI3K/AKT pathway, activate the AMPK-PRAS40-mTOR pathway, promote autophagy, inhibit cell proliferation, and exert anti-tumor effects (Bian et al., 2013). In another study, LL-37 activated AMPK and PI3K through P2X7R-mediated Ca2+ influx, promoted autophagy, and aided in inducing resistence against Mycobacterium tuberculosis in macrophages (Rekha et al., 2015). In another case, P2X7R-mediated Ca2+ influx activated mTOR and inhibited Treg cell conversion (Bohensky et al., 2010). However, to the contrary, in memory CD8+ T cells, P2X7R receives extracellular ATP signals to activate AMPK through Ca2+ influx and increase the AMP/ATP ratio, inhibit mTOR, and enhance mitochondrial function (Borges da Silva et al., 2018).
The level of autophagy is also related to the activation state of P2X7R. In the early stage of activation, P2X7R-mediated K+ efflux and Ca2+ influx activate mtROS, and Ca2+ activates CaMK, which in turn activates the AMPK pathway, inhibits mTOR, and promotes mitophagy and lysosome biogenesis. In the late stage of activation, lysosome stability decreases and cell death occurs (Sekar et al., 2018). Energy receptor AMPK (Garcia and Shaw, 2017) is also regulated by exercise (Garber, 2012). High-intensity exercise activates AMPKα to increase autophagic flux (Schwalm et al., 2015). In skeletal muscle, exercise induces mitophagy to degrade damaged mitochondria through the AMPK-Ulk1 signaling pathway (Laker et al., 2017). When energy is severely lacking, exercise inhibits AMPK/Ulk1/Beclin-1 phosphorylation, and the accumulated p62/SQATM1 inhibits autophagy to reduce muscle loss (Martin-Rincon et al., 2019). As an important downstream signaling molecule of P2X7R, AMPK is involved in the regulation of several physiological and pathological functions (Jeon, 2016), including autophagy in OA.
Compared with microautophagy and chaperone-mediated autophagy, macroautophagy is most studied and understood (Parzych and Klionsky, 2014). However, the relationship between cartilage damage, the degree of autophagy, and cell death remains unclear (Caramés et al., 2015).
The consensus is that autophagy exerts a protective effect on chondrocytes in OA. In mice, as cartilage damage increases, and resulting cell death ensues, the expression levels of autophagy-related genes decrease (Caramés et al., 2015). In cell lines and primary chondrocytes, decreased autophagy leads to cartilage degradation (Ribeiro et al., 2016a). Rapamycin (an autophagy inducer) promotes the degradation and clearance of damaged mitochondria, reduces IL-1β-induced ROS generation, and reduces the OA-like phenotype of chondrocytes (Sasaki et al., 2012). Decreased levels of autophagy are often accompanied by increased levels of apoptosis [e.g., presentation of activated PARP (the caspase-3 substrate)], further exacerbating OA characteristics (Caramés et al., 2015). Moreover, Beclin-1 silencing which reduces autophagy exacerbates cell death (Bohensky et al., 2007). mTOR overexpression also inhibits chondrocyte autophagy and promotes apoptosis, leading to increased cartilage degeneration (Zhang Y. et al., 2015). Autophagy promoted through oligomycin stimulation can effectively eliminate dysfunctional mitochondria, thereby protecting cells from apoptosis (Lopez de Figueroa et al., 2015).
Exercise promotes autophagy which relieves inflammation (Deretic et al., 2013). During exercise, the innate immune molecule TLR-9 interacts with Beclin-1 to strengthen and regulate AMPK activation in muscles (Liu Y. et al., 2020). AMPK interacts with sestrins to participate in exercise-induced autophagy to maintain skeletal muscle glucose metabolism (Liu et al., 2015), and relieve aging-related muscle atrophy (Fan et al., 2017). Exercise-induced AMPK activation can also inhibit mTOR, thereby alleviating other diseases as well by promoting autophagy, reducing the transformation of fatty liver to hepatitis and tumors (Guarino et al., 2020), and alleviating heart damage caused by exhaustive exercise (Liu and Pan, 2019).
Excessive autophagy can be a double-edged sword (Carames et al., 2010; Lopez de Figueroa et al., 2015; Ribeiro et al., 2016a, b), with no protective effect on cells (Chang et al., 2013), resulting in cell death (Shapiro et al., 2014) through synergistic participation in the process of cell apoptosis (e.g., ATP-dependent apoptosis). Related genes, such as Ulk1, Beclin-1, and LC3, are highly expressed in the early stage of OA, reflecting the protective effect of autophagy on chondrocytes; while in the late stage of OA, they are weakly expressed, as a consequence of autophagy-induced apoptotic cell death (Almonte-Becerril et al., 2010; Carames et al., 2010; Sasaki et al., 2012; Caramés et al., 2015). The opening of membrane pores induced by the ATP-P2X7R axis in muscle injury mediates autophagic cell death (Young et al., 2015). P2X7R receives ATP signals induced by ivermectin to promote autophagy, leading to tumor cell death. Although the role of P2X7R in infection and inflammation has been confirmed (Di Virgilio et al., 2017), studies have shown that inflammation leads to the downregulation of P2X7R expression, which in turn inhibits the PI3K-AKT-mTOR pathway, and promotes the osteogenesis of periodontal ligament stem cells (Xu et al., 2019).
Contrary to the uncertain relationship between autophagy and apoptosis, autophagy usually alleviates pyroptosis (Gudipaty et al., 2018). This can be verified through various mechanisms in many studies. First, miR-103 targets BNIP3 (Bcl2/Adenovirus EIB 19 kDa Interacting Protein 3) to mediate late autophagy and relieve H2O2-induced oxidative stress and pyroptosis (Wang et al., 2020). Second, electrical stimulation affects THP-1 macrophages, activates Sirt3, promotes autophagy, and relieves ROS-induced pyroptosis (Cong et al., 2020). Third, adrenomedullin promotes autophagy and inhibits pyroptosis in testicular stromal cells through the ROS-AMPK-mTOR pathway (Li et al., 2019). Fourth, resveratrol reduces mitochondrial damage and increases autophagy, thereby inhibiting NLRP3 activation and reducing inflammation (Chang et al., 2015). Fifth, baicalein, a Chinese herbal ingredient, promotes autophagy degradation, and reduces unfolded protein accumulation and mitochondrial dysfunction caused by spinal cord ischemia-reperfusion injury, thereby alleviating pyroptosis (Wu et al., 2020). Sixth, metformin activates AMPK, inhibits mTOR, relieves pyroptosis, and treats diabetic heart disease in obese mice (Yang et al., 2019). And seventh, SP1 transcription increases the expression of lnc ZFS1, which downregulates miR-590-3p, inhibits AMPK, activates mTOR, inhibits autophagy, increases pyroptosis, and aggravates sepsis-induced cardiac dysfunction (Liu Y. et al., 2020).
In addition to removing harmful components in the cell, autophagy can also prevent pyroptosis by degrading inflammasome components. The NLRP3 inhibitor CP-456773 and the NF-κB inhibitor celastrol work together to induce autophagy through the AMPK-mTOR pathway and inhibit HSP-90, thereby increasing the autophagic degradation of NLRP3 to inhibit pyroptosis (Saber et al., 2020). Furthermore, bone marrow-derived mesenchymal stem cell (BMSC)-derived exosomes activate AMPK in hypoglycemic/reoxygenation-induced pheochromocytoma cells, inhibiting mTOR, promoting autophagic flux, while LC3 results in NLRP3 degradation to inhibit pyroptosis (Zeng et al., 2020). Also, immunity-related GTPase M (IRGM) interacts with NLRP3 and ASC to inhibit inflammasome oligomerization, thereby inhibiting its assembly and activation, and selectively degrades NLRP3 and ASC by autophagy, alleviating inflammatory cell death (Mehto et al., 2019).
Sometimes, autophagy is also the trigger point of pyroptosis. Autophagy mediates the release of IL-1β (Claude-Taupin et al., 2018). In macrophages, IL-1β is released outside the cell through a hole in the plasma membrane of N-GSDMD, while in neutrophils, N-GSDMD is not localized on the plasma membrane. IL-1β is released through the LC3+ autophagosome pathway (Karmakar et al., 2020). Arsenic can also promote autophagy, with lysosome degradation releasing cathepsin, resulting in NLRP3 activation (Qiu et al., 2018). Autophagy is often inhibited when pyroptosis occurs. During the resting state of macrophages, NLRC4 and Beclin-1/Atg6 form a complex to inhibit autophagy. Under a low degree of infection, NAIP5 recruits NLRC4 and pro-caspase-1 to form a complex to relieve autophagy inhibition, and induce cell protection. When autophagy cannot eliminate an intracellular infection, caspase-1 is activated to initiate pyroptosis (Byrne et al., 2013). NLRP3 inflammasomes in hepatocellular carcinoma cells inhibit autophagy through the 17β-estradiol (E2)/ ERβ/AMPK/mTOR pathway (Wei et al., 2019).
When the two are activated together, autophagy negatively regulates pyroptosis. Inflammasome activation can induce autophagy, but the recruitment of LC3 and p62 induces the co-localization of inflammasomes with autophagosomes, thereby degrading them (Shi et al., 2012). Furthermore, when acrolein induces NLRP3 activation and autophagy through ROS, autophagy inhibits pyrolysis and mitochondrial damage (Jiang et al., 2018). Moreover, H2O2 induces nucleus pulposus cells to produce ROS, inducing autophagy, pyroptosis, and the upregulation of nuclear factor erythroid 2 like 2 (NFE2L2, Nrf2). Both Nrf2 and autophagy can alleviate pyroptosis and intervertebral disc degeneration (Bai et al., 2020). Tumor necrosis factor receptor-associated factor 3 (TRAF3) mediates the ubiquitination and degradation of Ulk1 and induces ROS and pyroptosis, while Ulk1 inhibits ROS and apoptosis inducing factor (AIF) translocation into the nucleus (Shen et al., 2020). In another mechanism, zearalenone inhibits the Akt/mTOR pathway to promote autophagy, while also promoting pyroptosis through NF-κB, but the upregulation of autophagy inhibits pyroptosis (Wang et al., 2019). In macrophages, NF-κB mediates the delayed accumulation of p62, forming the NF-κB-p62-mitophagy regulatory loop eliminating damaged mitochondria caused by pyroptosis, and limiting the intracellular pro-inflammatory activity (Zhong et al., 2016).
Autophagolysosomes formed by the fusion of autophagosomes and lysosomes play an important role in pyroptosis. Lysosomes act as AMPK-mTOR signaling hubs and an instability may lead to apoptosis and pyroptosis (Zhu et al., 2020). An impaired autophagy-lysosomal pathway in macular corneal dystrophic cornea can also lead to pyroptosis (Zheng et al., 2020). Moreover, BpV(phen) increases the ubiquitination of p62, affects the binding of p62 and HDAC6, activates the deacetylation of α-tubulin, and affects the stability of acetylated microtubules, resulting in hindered autophagosome and lysosome fusion, while autophagy inhibition leads to apoptosis and pyroptosis (Chen et al., 2015). Lastly, hypoxia-induced autophagy/lysosomal dysfunction leads to ERS, leading to unfolded protein accumulation and impaired autophagy, which in turn activates NLRP3 inflammasomes and induces pyroptosis (Cheng et al., 2019).
The interrelationships among phenotypes, such as autophagy, apoptosis, and pyroptosis, are also reflected in P2X7R-mediated cell metabolism and nutrition (Orioli et al., 2017). P2X4/P2X7/pannexin-1 mediates ROS, Ca2+/CaMK II, mitochondrial membrane potential, and caspase-1 activation caused by NADPH oxidase to promote cell pyroptosis and necrosis (Draganov et al., 2015). In ischemic stroke disease, P2X7R-mediated Ca2+ and K+ flow leads to mitochondrial dysfunction, caspase-8 and MAPK activation, and induces apoptosis. Ca2+ influx induces lysosomal dysfunction, which leads to decreased autophagic flow and apoptosis. K+ efflux leads to the activation of inflammasomes and induces pyroptosis (Zhao et al., 2018). Ca2+ overload, caused by P2X7R-mediated Ca2+ influx under the stimulation by high ATP concentrations in macrophages, leads to mitochondrial dysfunction, which in turn causes cell pyroptosis. However, when P2X7R is stimulated by low ATP concentrations or is positively allosterically regulated by compound K, the accumulation of mtROS and the activation of caspase-1 and 3 alter cell death mechanism from pyroptosis to apoptosis (Bidula et al., 2019).
In summary, autophagy primarily plays the role of a protector, resisting stimuli that cause damage to cells through key signaling pathways, such as AMPK-mTOR and HIF-1α, and supporting homeostasis. When autophagy decreases, P2X7R-mediated ion flux and damage to organelles, such as mitochondria and lysosomes, prevent autophagy flux, and the balance shifts. At this time, autophagy may induce or convert into apoptosis and pyroptosis. On several occasions, autophagy is the initiator of apoptosis, pyroptosis, and inflammation. Therefore, autophagy is a double-edged sword, and the right balance, together with proper activation of P2X7R, is the key to its power.
The P2X7 receptor inhibition can occur via synthetic reagents, ions, natural molecular compounds, and Chinese herbal medicines. However, due to P2X7R’s central role in inflammation, P2X7R inhibitors are receiving more attention for the development of targeted receptor therapies (Khakh and North, 2006; Arulkumaran et al., 2011; North and Jarvis, 2013; Bartlett et al., 2014; Di Virgilio et al., 2017). Various inhibitors display differences in chemical structure, species selectivity, competitive or non-competitive antagonistic methods, and specificity. The first-generation inhibitors, such as Reactive Blue 2, KN-62, PPADS, brilliant blue G (BBG), and oxidized ATP (oATP), are not highly selective, inhibiting other purinergic receptors as well. Second-generation inhibitors have improved target specificity for P2X7R, such as A438079, A740003, A839977, AZ10606120, AZ11645373, GSK314181, and JNJ-47965567. To date, most in vivo experiments have been conducted with first-generation inhibitors, but a select few studies have been carried out with specific inhibitors, like A438079, showing good efficacy in vitro and in vivo. Pretreatment of a DC/CD4+ T cell co-culture system with A438079, and the ensuing inhibition of P2X7R, can reduce the levels of pro-inflammatory factors IL-1β, IL-6, IL-23p19, and TGF-β1, derived from Th17 cells. In the arthritis mice model, induced by the activation of related collagen, A438079 relieved the swelling of the hind paw and the pathological changes in the ankle joint (Fan et al., 2016).
In view of the positive therapeutic effects of P2X7R inhibitors in rodent studies, drugs targeting human P2X7R are being used in clinical settings for the treatment of several diseases, such as pain, arthritis, and multiple sclerosis (Bartlett et al., 2014). The therapeutic effects of inhibitors, such as oATP, BBG, KN-62, and A438079, have been confirmed in preclinical models of inflammatory diseases, such as contact allergy, inflammatory pain, endotoxin-induced fever, and antibody-induced nephritis (Matute et al., 2007; Taylor et al., 2009; Weber et al., 2010; Barbera-Cremades et al., 2012). Two clinical trials of patients with rheumatoid arthritis receiving P2X7R-specific inhibitors, AZD9056 and CE-224, reported on its safety and clinical efficacy. The dosages were well tolerated, but efficacy was not improved in patients already resistant to methotrexate or sulfasalazine (used in the treatment of joint swelling and pain) (Keystone et al., 2012; Stock et al., 2012). This would point to a need to use P2X7R inhibitors in conjunction with other targeted therapies capable of resensitizing mechanistic pathways involved in drug-resistance.
Although rodent models and receptor inhibitors are regularly encountered in P2X7R research, the selectivity and specificity of inhibitors still require attention. Recent development of therapeutic antibodies, such as nanobodies or single-domain antibodies, can also have the potential to specifically inhibit membrane proteins. Therefore, we expect the development of more effective drugs and treatments targeting P2X7R.
This article systematically analyzed and elucidated the association between P2X7R and OA. The most typical manifestation of OA is cartilage degradation. As the only cellular component of cartilage, chondrocytes are in a stable state and balance is crucial. The ECM degradation of chondrocytes and the release of inflammatory factors are important events leading to cartilage degradation. Inflammation is a direct manifestation of OA. To explore whether P2X7, as a key switch of inflammation, is involved in the occurrence and development of OA, we considered the network (Figure 1) of interaction from the perspectives of inflammation, apoptosis, pyroptosis, and autophagy, and presented new OA prevention and treatment strategies.
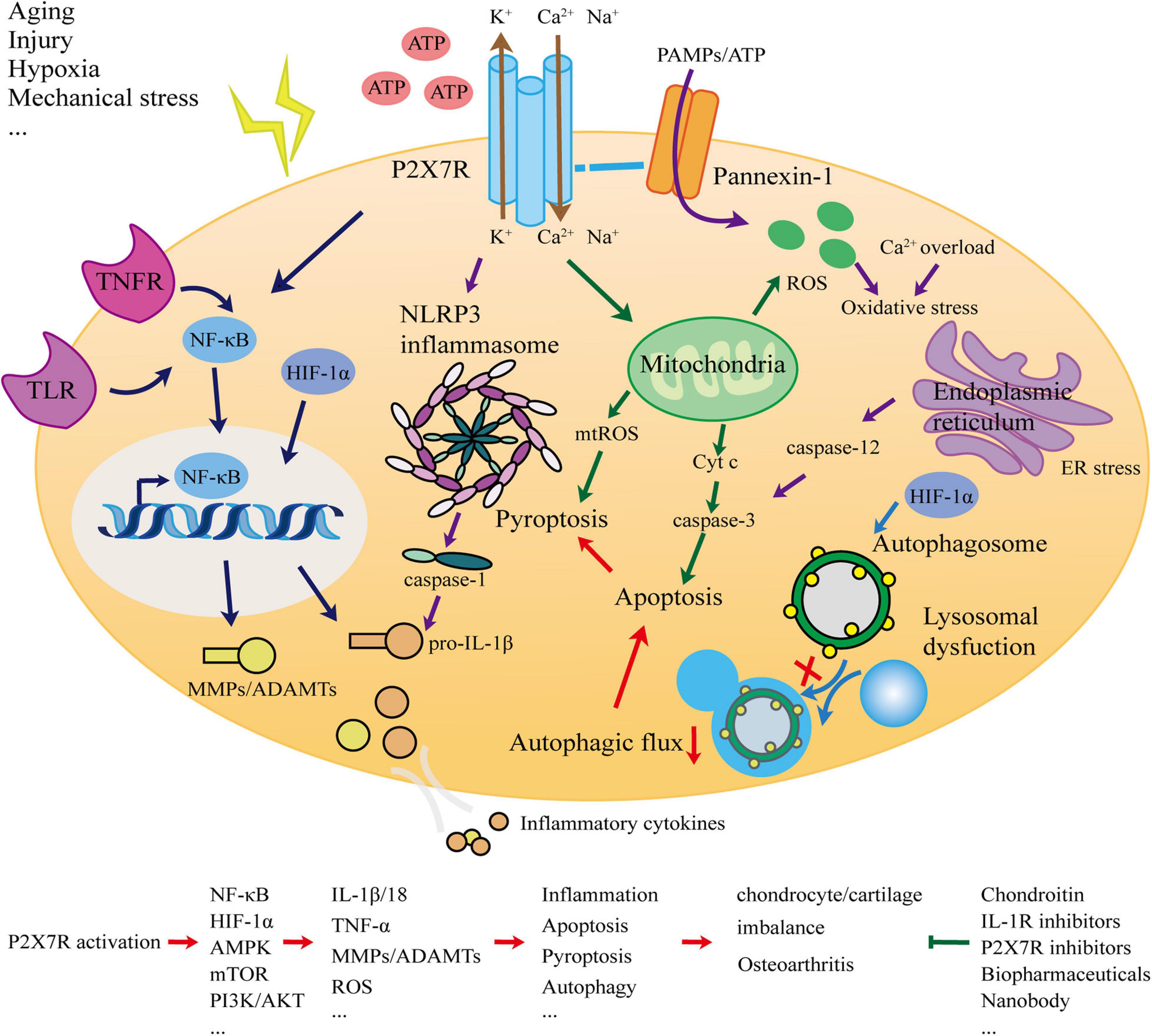
Figure 1. Schematic for the role of the P2X7 receptor in osteoarthritis.
Currently, there is no treatment method that can completely prevent the occurrence and development of OA. Delaying and reducing the death of chondrocytes to prevent the degradation of the cartilage matrix could be a potential therapeutic focus. As various factors affect chondrocyte cell death progression, including the degree of cartilage degradation, pro-inflammatory cytokines, and ATP availability, we propose the therapeutic focus to begin with the recognition of ATP as the first step in activating inflammation to narrow our approach to anti-inflammatory designs. Caspase-knockout studies have found that cells require both ATP energy and caspases to transform pyroptosis into apoptosis, thereby moving toward programmed death, and away from necrosis (Zeiss, 2003). zVAD-fmk is often used as a caspase inhibitor in OA research (Hwang and Kim, 2015), which can significantly reduce chondrocyte apoptosis and cartilage degradation (D’Lima et al., 2006). Moreover, when these factors are managed, mitochondrial integrity remains intact, reducing oxidative stress levels. Retaining a positive antioxidant balance exerts anti-inflammatory and anti-apoptotic effects. Oxidative stress can further be balanced by promoting autophagy, thus promoting mitochondrial housekeeping. However, autophagy can be a double-edged sword, and needs careful consideration when promoted. Excessive autophagy can trigger cell apoptosis, and the rapid loss of chondrocytes may worsen OA (Shapiro et al., 2014). Nevertheless, the mainstream view remains that autophagy stimulation in the early stages of OA can protect chondrocytes.
Treatment strategies should also focus on the role of P2X7R. Soon after it was cloned, the P2X7R protein received widespread attention as a key switch for inflammation with great therapeutic potential. For some chronic inflammatory diseases, small molecule (drug-like) inhibitors targeting P2X7R have been used in phase I and II clinical studies (Gunosewoyo and Kassiou, 2010; Sluyter and Stokes, 2011; Park and Kim, 2017). To date, more than 30 clinical studies in this regard have been conducted. Although the safety of inhibitors has been satisfactory, the clinical efficacy has been disappointing. In knee OA, rheumatoid arthritis, and chronic obstructive pulmonary disease, the efficacy is poor, while in Crohn’s disease fairly positive (Arulkumaran et al., 2011; Keystone et al., 2012; Stock et al., 2012; Eser et al., 2015). Biopharmaceuticals may present a new way to replace small drug-like compounds. Antibodies against non-functional variants of human P2X7R have been used to treat cancer. The highly purified goat IgG can effectively reduce pathological changes in basal cell carcinoma size (Gilbert et al., 2017). In addition, nanobodies (e.g., 13A7 nanobody, single-domain antibody fragments elicited in camelids) that can bind to human or mouse P2X7R with high affinity, after inoculation in mice, effectively relieve the symptoms of experimental allergic contact dermatitis and glomerulonephritis. Dano1 nano antibody is selective for human P2X7R and can effectively reduce the level of IL-1β in the blood after endotoxin treatment (Danquah et al., 2016). Therefore, in the case of poor efficacy of anti-P2X7 receptor active drugs, biologics targeting P2X7R indicate a new direction for future research.
In conclusion, there remains a need for more specific OA therapies to be developed. P2X7R of the purinoceptor family, is closely related to inflammation and a promising drug target for OA. However, research into P2X7R and its role in the pathogenesis of OA requires further investigation. For the benefit of patients and scientific progress, we believe that these expectations will be realized in the near future.
ZL performed the literature review, drafted the manuscript, and prepared the figure. ZH and LB edited and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81772420, 81272050, and 31900847); China Postdoctoral Fund (2019M66169); and Liaoning Provincial Doctor Start-up Fund (2019JH3/10100299).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the lab affiliated to China Medical University for core support.
Aguirre, A., Shoji, K. F., Saez, J. C., Henriquez, M., and Quest, A. F. (2013). FasL-triggered death of Jurkat cells requires caspase 8-induced, ATP-dependent cross-talk between Fas and the purinergic receptor P2X(7). J. Cell. Physiol. 228, 485–493. doi: 10.1002/jcp.24159
Aigner, T., Fundel, K., Saas, J., Gebhard, P. M., Haag, J., Weiss, T., et al. (2006). Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 54, 3533–3544. doi: 10.1002/art.22174
Almonte-Becerril, M., Navarro-Garcia, F., Gonzalez-Robles, A., Vega-Lopez, M. A., Lavalle, C., and Kouri, J. B. (2010). Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis 15, 631–638. doi: 10.1007/s10495-010-0458-z
Amoroso, F., Falzoni, S., Adinolfi, E., Ferrari, D., and Di Virgilio, F. (2012). The P2X7 receptor is a key modulator of aerobic glycolysis. Cell Death Dis. 3:e370. doi: 10.1038/cddis.2012.105
Archer, C. W., and Francis-West, P. (2003). The chondrocyte. Int. J. Biochem. Cell Biol. 35, 401–404.
Arulkumaran, N., Unwin, R. J., and Tam, F. W. (2011). A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin. Investig. Drugs 20, 897–915. doi: 10.1517/13543784.2011.578068
Bai, Z., Liu, W., He, D., Wang, Y., Yi, W., Luo, C., et al. (2020). Protective effects of autophagy and NFE2L2 on reactive oxygen species-induced pyroptosis of human nucleus pulposus cells. Aging 12, 7534–7548. doi: 10.18632/aging.103109
Bao, L., Locovei, S., and Dahl, G. (2004). Pannexin membrane channels are mechanosensitive conduits for ATP. FEBS Lett. 572, 65–68. doi: 10.1016/j.febslet.2004.07.009
Bao, Q., and Shi, Y. (2007). Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 14, 56–65. doi: 10.1038/sj.cdd.4402028
Barbera-Cremades, M., Baroja-Mazo, A., Gomez, A. I., Machado, F., Di Virgilio, F., and Pelegrin, P. (2012). P2X7 receptor-stimulation causes fever via PGE2 and IL-1beta release. FASEB J. 26, 2951–2962. doi: 10.1096/fj.12-205765
Barbera-Cremades, M., Baroja-Mazo, A., and Pelegrin, P. (2016). Purinergic signaling during macrophage differentiation results in M2 alternative activated macrophages. J. Leukoc. Biol. 99, 289–299. doi: 10.1189/jlb.1a0514-267rr
Bartlett, R., Stokes, L., and Sluyter, R. (2014). The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol. Rev. 66, 638–675. doi: 10.1124/pr.113.008003
Beecher, B. R., Martin, J. A., Pedersen, D. R., Heiner, A. D., and Buckwalter, J. A. (2007). Antioxidants block cyclic loading induced chondrocyte death. Iowa Orthop. J. 27, 1–8.
Bian, S., Sun, X., Bai, A., Zhang, C., Li, L., Enjyoji, K., et al. (2013). P2X7 integrates PI3K/AKT and AMPK-PRAS40-mTOR signaling pathways to mediate tumor cell death. PLoS One 8:e60184. doi: 10.1371/journal.pone.0060184
Bianco, F., Pravettoni, E., Colombo, A., Schenk, U., Moller, T., Matteoli, M., et al. (2005). Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J. Immunol. 174, 7268–7277. doi: 10.4049/jimmunol.174.11.7268
Bidula, S., Dhuna, K., Helliwell, R., and Stokes, L. (2019). Positive allosteric modulation of P2X7 promotes apoptotic cell death over lytic cell death responses in macrophages. Cell Death Dis. 10:882.
Bijlsma, J. W. J., Berenbaum, F., and Lafeber, F. P. J. G. (2011). Osteoarthritis: an update with relevance for clinical practice. Lancet 377, 2115–2126. doi: 10.1016/s0140-6736(11)60243-2
Blanco, F. J., Guitian, R., Vazquez-Martul, E., de Toro, F. J., and Galdo, F. (1998). Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 41, 284–289. doi: 10.1002/1529-0131(199802)41:2<284::aid-art12>3.0.co;2-t
Bobinac, D., Spanjol, J., Zoricic, S., and Maric, I. (2003). Changes in articular cartilage and subchondral bone histomorphometry in osteoarthritic knee joints in humans. Bone 32, 284–290. doi: 10.1016/s8756-3282(02)00982-1
Bohensky, J., Leshinsky, S., Srinivas, V., and Shapiro, I. M. (2010). Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 25, 633–642. doi: 10.1007/s00467-009-1310-y
Bohensky, J., Shapiro, I. M., Leshinsky, S., Terkhorn, S. P., Adams, C. S., and Srinivas, V. (2007). HIF-1 regulation of chondrocyte apoptosis: induction of the autophagic pathway. Autophagy 3, 207–214. doi: 10.4161/auto.3708
Borges da Silva, H., Beura, L. K., Wang, H., Hanse, E. A., Gore, R., Scott, M. C., et al. (2018). The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 559, 264–268. doi: 10.1038/s41586-018-0282-0
Bortner, C. D., Hughes, F. M. Jr., and Cidlowski, J. A. (1997). A primary role for K+ and Na+ efflux in the activation of apoptosis. J. Biol. Chem. 272, 32436–32442. doi: 10.1074/jbc.272.51.32436
Bougault, C., Gosset, M., Houard, X., Salvat, C., Godmann, L., Pap, T., et al. (2012). Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum. 64, 3972–3981. doi: 10.1002/art.34678
Byrne, B. G., Dubuisson, J. F., Joshi, A. D., Persson, J. J., and Swanson, M. S. (2013). Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. mBio 4:e00620-12.
Caramés, B., Olmer, M., Kiosses, W. B., and Lotz, M. K. (2015). The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheum. 67, 1568–1576. doi: 10.1002/art.39073
Carames, B., Taniguchi, N., Otsuki, S., Blanco, F. J., and Lotz, M. (2010). Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 62, 791–801. doi: 10.1002/art.27305
Carr, A. J., Robertsson, O., Graves, S., Price, A. J., Arden, N. K., Judge, A., et al. (2012). Knee replacement. Lancet 379, 1331–1340.
Chang, J., Wang, W., Zhang, H., Hu, Y., Wang, M., and Yin, Z. (2013). The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 32, 1311–1318. doi: 10.3892/ijmm.2013.1520
Chang, Y. P., Ka, S. M., Hsu, W. H., Chen, A., Chao, L. K., Lin, C. C., et al. (2015). Resveratrol inhibits NLRP3 inflammasome activation by preserving mitochondrial integrity and augmenting autophagy. J. Cell. Physiol. 230, 1567–1579. doi: 10.1002/jcp.24903
Chen, C. T., Burton-Wurster, N., Borden, C., Hueffer, K., Bloom, S. E., and Lust, G. (2001). Chondrocyte necrosis and apoptosis in impact damaged articular cartilage. J. Orthop. Res. 19, 703–711. doi: 10.1016/s0736-0266(00)00066-8
Chen, Q., Yue, F., Li, W., Zou, J., Xu, T., Huang, C., et al. (2015). Potassium bisperoxo(1,10-phenanthroline)oxovanadate (bpV(phen)) induces apoptosis and pyroptosis and disrupts the P62-HDAC6 protein interaction to suppress the acetylated microtubule-dependent degradation of autophagosomes. J. Biol. Chem. 290, 26051–26058. doi: 10.1074/jbc.m115.653568
Chen, X., Li, H., Wang, K., Liang, X., Wang, W., Hu, X., et al. (2019). Aerobic exercise ameliorates myocardial inflammation, fibrosis and apoptosis in high-fat-diet rats by inhibiting P2X7 purinergic receptors. Front. Physiol. 10:1286. doi: 10.3389/fphys.2019.01286
Cheng, S.-B., Nakashima, A., Huber, W. J., Davis, S., Banerjee, S., Huang, Z., et al. (2019). Pyroptosis is a critical inflammatory pathway in the placenta from early onset preeclampsia and in human trophoblasts exposed to hypoxia and endoplasmic reticulum stressors. Cell Death Dis. 10:927.
Chevalier, X., Goupille, P., Beaulieu, A. D., Burch, F. X., Bensen, W. G., Conrozier, T., et al. (2009). Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 61, 344–352. doi: 10.1002/art.24096
Claude-Taupin, A., Bissa, B., Jia, J., Gu, Y., and Deretic, V. (2018). Role of autophagy in IL-1beta export and release from cells. Semin. Cell Dev. Biol. 83, 36–41. doi: 10.1016/j.semcdb.2018.03.012
Clavijo-Cornejo, D., Martinez-Flores, K., Silva-Luna, K., Martinez-Nava, G. A., Fernandez-Torres, J., Zamudio-Cuevas, Y., et al. (2016). The overexpression of NALP3 inflammasome in knee osteoarthritis is associated with synovial membrane prolidase and NADPH oxidase 2. Oxid. Med. Cell. Longev. 2016:1472567.
Cohen, S. B., Proudman, S., Kivitz, A. J., Burch, F. X., Donohue, J. P., Burstein, D., et al. (2011). A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res. Ther. 13:R125.
Coimbra, I. B., Jimenez, S. A., Hawkins, D. F., Piera-Velazquez, S., and Stokes, D. G. (2004). Hypoxia inducible factor-1 alpha expression in human normal and osteoarthritic chondrocytes. Osteoarthritis Cartilage 12, 336–345.
Colegio, O. R., Chu, N. Q., Szabo, A. L., Chu, T., Rhebergen, A. M., Jairam, V., et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. doi: 10.1038/nature13490
Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Munoz-Planillo, R., Inserra, M. C., et al. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–255.
Comassi, M., Santini, E., Rossi, C., Vitolo, E., Seghieri, M., Tocchini, L., et al. (2017). The level of physical training modulates cytokine levels through P2X7 receptor in healthy subjects. Eur. J. Clin. Invest. 48:e12880. doi: 10.1111/eci.12880
Cong, L., Gao, Z., Zheng, Y., Ye, T., Wang, Z., Wang, P., et al. (2020). Electrical stimulation inhibits Val-boroPro-induced pyroptosis in THP-1 macrophages via sirtuin3 activation to promote autophagy and inhibit ROS generation. Aging 12, 6415–6435. doi: 10.18632/aging.103038
Coppe, J. P., Desprez, P. Y., Krtolica, A., and Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118. doi: 10.1146/annurev-pathol-121808-102144
Cui, J., Zhang, M., Zhang, Y. Q., and Xu, Z. H. (2007). JNK pathway: diseases and therapeutic potential. Acta Pharmacol. Sin. 28, 601–608. doi: 10.1111/j.1745-7254.2007.00579.x
da Costa, B. R., Nuesch, E., Reichenbach, S., Juni, P., and Rutjes, A. W. (2012). Doxycycline for osteoarthritis of the knee or hip. Cochrane Database Syst. Rev. 11:CD007323.
Danquah, W., Meyer-Schwesinger, C., Rissiek, B., Pinto, C., Serracant-Prat, A., Amadi, M., et al. (2016). Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci. Transl. Med. 8:366ra162. doi: 10.1126/scitranslmed.aaf8463
de Gassart, A., and Martinon, F. (2015). Pyroptosis: caspase-11 unlocks the gates of death. Immunity 43, 835–837. doi: 10.1016/j.immuni.2015.10.024
Delarasse, C., Gonnord, P., Galante, M., Auger, R., Daniel, H., Motta, I., et al. (2009). Neural progenitor cell death is induced by extracellular ATP via ligation of P2X7 receptor. J. Neurochem. 109, 846–857. doi: 10.1111/j.1471-4159.2009.06008.x
Denecker, G., Vercammen, D., Declercq, W., and Vandenabeele, P. (2001). Apoptotic and necrotic cell death induced by death domain receptors. Cell. Mol. Life Sci. 58, 356–370. doi: 10.1007/pl00000863
Deretic, V., Saitoh, T., and Akira, S. (2013). Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737.
Di Virgilio, F. (2013). The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol. Rev. 65, 872–905. doi: 10.1124/pr.112.006171
Di Virgilio, F., Dal Ben, D., Sarti, A. C., Giuliani, A. L., and Falzoni, S. (2017). The P2X7 receptor in infection and inflammation. Immunity 47, 15–31. doi: 10.1016/j.immuni.2017.06.020
Dinarello, C. A. (2002). The IL-1 family and inflammatory diseases. Clin. Exp. Rheumatol. 20(5 Suppl. 27), S1–S13.
Dinarello, C. A., Simon, A., and van der Meer, J. W. M. (2012). Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652. doi: 10.1038/nrd3800
Distel, E., Cadoudal, T., Durant, S., Poignard, A., Chevalier, X., and Benelli, C. (2009). The infrapatellar fat pad in knee osteoarthritis: an important source of interleukin-6 and its soluble receptor. Arthritis Rheum. 60, 3374–3377. doi: 10.1002/art.24881
D’Lima, D., Hermida, J., Hashimoto, S., Colwell, C., and Lotz, M. (2006). Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 54, 1814–1821. doi: 10.1002/art.21874
D’Lima, D. D., Hashimoto, S., Chen, P. C., Colwell, C. W., and Lotz, M. K. (2001). Human chondrocyte apoptosis in response to mechanical injury. Osteoarthritis Cartilage 9, 712–719. doi: 10.1053/joca.2001.0468
Donnelly-Roberts, D. L., Namovic, M. T., Faltynek, C. R., and Jarvis, M. F. (2004). Mitogen-activated protein kinase and caspase signaling pathways are required for P2X7 receptor (P2X7R)-induced pore formation in human THP-1 cells. J. Pharmacol. Exp. Ther. 308, 1053–1061. doi: 10.1124/jpet.103.059600
Draganov, D., Gopalakrishna-Pillai, S., Chen, Y. R., Zuckerman, N., Moeller, S., Wang, C., et al. (2015). Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via Ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci. Rep. 5:16222.
Dubyak, G. R. (2012). P2X7 receptor regulation of non-classical secretion from immune effector cells. Cell. Microbiol. 14, 1697–1706. doi: 10.1111/cmi.12001
Działo, J., Tokarz-Deptuła, B., and Deptuła, W. (2013). Excitotoxicity and Wallerian degeneration as a processes related to cell death in nervous system. Arch. Ital. Biol. 151, 67–75.
Eser, A., Colombel, J. F., Rutgeerts, P., Vermeire, S., Vogelsang, H., Braddock, M., et al. (2015). Safety and efficacy of an oral inhibitor of the purinergic receptor P2X7 in adult patients with moderately to severely active crohn’s disease: a randomized placebo-controlled, double-blind, phase IIa study. Inflamm. Bowel Dis. 21, 2247–2253.
Fan, J., Yang, X., Li, J., Shu, Z., Dai, J., Liu, X., et al. (2017). Spermidine coupled with exercise rescues skeletal muscle atrophy from D-gal-induced aging rats through enhanced autophagy and reduced apoptosis via AMPK-FOXO3a signal pathway. Oncotarget 8, 17475–17490. doi: 10.18632/oncotarget.15728
Fan, Z. D., Zhang, Y. Y., Guo, Y. H., Huang, N., Ma, H. H., Huang, H., et al. (2016). Involvement of P2X7 receptor signaling on regulating the differentiation of Th17 cells and type II collagen-induced arthritis in mice. Sci. Rep. 6:35804.
Felson, D. T., Zhang, Y., Anthony, J. M., Naimark, A., and Anderson, J. J. (1992). Weight loss reduces the risk for symptomatic knee osteoarthritis in women. The Framingham Study. Ann. Intern. Med. 116, 535–539. doi: 10.7326/0003-4819-116-7-535
Ferrari, D., Pizzirani, C., Adinolfi, E., Lemoli, R. M., Curti, A., Idzko, M., et al. (2006). The P2X7 receptor: a key player in IL-1 processing and release. J. Immunol. 176, 3877–3883.
Festjens, N., Vanden Berghe, T., and Vandenabeele, P. (2006). Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta 1757, 1371–1387. doi: 10.1016/j.bbabio.2006.06.014
Forsyth, C. B., Cole, A., Murphy, G., Bienias, J. L., Im, H.-J., and Loeser, R. F. Jr. (2005). Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J. Gerontol. A Biol. Sci. Med. Sci. 60, 1118–1124. doi: 10.1093/gerona/60.9.1118
Fradejas, N., Pastor, M. D., Burgos, M., Beyaert, R., Tranque, P., and Calvo, S. (2010). Caspase-11 mediates ischemia-induced astrocyte death: involvement of endoplasmic reticulum stress and C/EBP homologous protein. J. Neurosci. Res. 88, 1094–1105.
Franceschini, A., Capece, M., Chiozzi, P., Falzoni, S., Sanz, J. M., Sarti, A. C., et al. (2015). The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 29, 2450–2461. doi: 10.1096/fj.14-268714
Fuentes-Prior, P., and Salvesen, G. (2004). The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J. 384(Pt 2), 201–232. doi: 10.1042/bj20041142
Ganley, I. G., Lam du, H., Wang, J., Ding, X., Chen, S., and Jiang, X. (2009). ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 284, 12297–12305. doi: 10.1074/jbc.m900573200
Garber, K. (2012). Autophagy. Explaining exercise. Science 335:281. doi: 10.1126/science.335.6066.281
Garcia, D., and Shaw, R. J. (2017). AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800. doi: 10.1016/j.molcel.2017.05.032
García-Arnandis, I., Guillén, M. I., Gomar, F., Pelletier, J.-P., Martel Pelletier, J., and Alcaraz, M. J. (2010). High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1 b in osteoarthritic synoviocytes. Arthritis Res. Ther. 12:R165.
Gilbert, S. M., Gidley Baird, A., Glazer, S., Barden, J. A., Glazer, A., Teh, L. C., et al. (2017). A phase I clinical trial demonstrates that nfP2X7 -targeted antibodies provide a novel, safe and tolerable topical therapy for basal cell carcinoma. Br. J. Dermatol. 177, 117–124. doi: 10.1111/bjd.15364
Gomez, R., Conde, J., Scotece, M., Gomez-Reino, J. J., Lago, F., and Gualillo, O. (2011). What’s new in our understanding of the role of adipokines in rheumatic diseases? Nat. Rev. Rheumatol. 7, 528–536. doi: 10.1038/nrrheum.2011.107
Green, D. R. (2003). Overview: apoptotic signaling pathways in the immune system. Immunol. Rev. 193, 5–9. doi: 10.1034/j.1600-065x.2003.00045.x
Grishko, V. I., Ho, R., Wilson, G. L., and Pearsall, A. W. (2009). Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthritis Cartilage 17, 107–113. doi: 10.1016/j.joca.2008.05.009
Guarino, M., Kumar, P., Felser, A., Terracciano, L. M., Guixe-Muntet, S., Humar, B., et al. (2020). Exercise attenuates the transition from fatty liver to steatohepatitis and reduces tumor formation in mice. Cancers (Basel) 12:1407. doi: 10.3390/cancers12061407
Gudbergsen, H., Boesen, M., Lohmander, L. S., Christensen, R., Henriksen, M., Bartels, E. M., et al. (2012). Weight loss is effective for symptomatic relief in obese subjects with knee osteoarthritis independently of joint damage severity assessed by high-field MRI and radiography. Osteoarthritis Cartilage 20, 495–502. doi: 10.1016/j.joca.2012.02.639
Gudipaty, S. A., Conner, C. M., Rosenblatt, J., and Montell, D. J. (2018). Unconventional ways to live and die: cell death and survival in development, homeostasis, and disease. Annu. Rev. Cell Dev. Biol. 34, 311–332. doi: 10.1146/annurev-cellbio-100616-060748
Guha, S., Baltazar, G. C., Coffey, E. E., Tu, L. A., Lim, J. C., Beckel, J. M., et al. (2013). Lysosomal alkalinization, lipid oxidation, and reduced phagosome clearance triggered by activation of the P2X7 receptor. FASEB J. 27, 4500–4509. doi: 10.1096/fj.13-236166
Guilak, F. (2011). Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 25, 815–823. doi: 10.1016/j.berh.2011.11.013
Gunosewoyo, H., and Kassiou, M. (2010). P2X purinergic receptor ligands: recently patented compounds. Expert Opin. Ther. Pat. 20, 625–646. doi: 10.1517/13543771003702424
Guo, H., Callaway, J. B., and Ting, J. P. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687. doi: 10.1038/nm.3893
Haseeb, A., and Haqqi, T. M. (2013). Immunopathogenesis of osteoarthritis. Clin. Immunol. 146, 185–196. doi: 10.1016/j.clim.2012.12.011
Hashimoto, S., Ochs, R. L., Komiya, S., and Lotz, M. (1998). Linkage of chondrocyte apoptosis and cartilage degradation in human osteoarthritis. Arthritis Rheum. 41, 1632–1638. doi: 10.1002/1529-0131(199809)41:9<1632::aid-art14>3.0.co;2-a
He, Y., Hara, H., and Nunez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021. doi: 10.1016/j.tibs.2016.09.002
Henrotin, Y., Deby-Dupont, G., Deby, C., De Bruyn, M., Lamy, M., and Franchimont, P. (1993). Production of active oxygen species by isolated human chondrocytes. Br. J. Rheumatol. 32, 562–567. doi: 10.1093/rheumatology/32.7.562
Henrotin, Y., Labasse, A., Zheng, S. X., Galais, P., Tsouderos, Y., Crielaard, J. M., et al. (2001). Strontium ranelate increases cartilage matrix formation. J. Bone Min. Res. 16, 299–308. doi: 10.1359/jbmr.2001.16.2.299
Henrotin, Y., and Lambert, C. (2013). Chondroitin and glucosamine in the management of osteoarthritis: an update. Curr. Rheumatol. Rep. 15:361.
Henrotin, Y. E., Bruckner, P., and Pujol, J. P. L. (2003). The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage 11, 747–755. doi: 10.1016/s1063-4584(03)00150-x
Heraud, F., Héraud, A., and Harmand, M. F. (2000). Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 59, 959–965. doi: 10.1136/ard.59.12.959
Hwang, H. S., and Kim, H. A. (2015). Chondrocyte apoptosis in the pathogenesis of osteoarthritis. Int. J. Mol. Sci. 16, 26035–26054. doi: 10.3390/ijms161125943
Jeon, S. M. (2016). Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 48:e245. doi: 10.1038/emm.2016.81
Jiang, C., Jiang, L., Li, Q., Liu, X., Zhang, T., Dong, L., et al. (2018). Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology 410, 26–40. doi: 10.1016/j.tox.2018.09.002
Jiang, L. H., Baldwin, J. M., Roger, S., and Baldwin, S. A. (2013). Insights into the molecular mechanisms underlying mammalian P2X7 receptor functions and contributions in diseases, revealed by structural modeling and single nucleotide polymorphisms. Front. Pharmacol. 4:55. doi: 10.3389/fphar.2013.00055
Jin, C., Frayssinet, P., Pelker, R., Cwirka, D., Hu, B., Vignery, A., et al. (2011). NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc. Natl. Acad. Sci. U.S.A. 108, 14867–14872. doi: 10.1073/pnas.1111101108
Johnson, K., Zhu, S., Tremblay, M. S., Payette, J. N., Wang, J., Bouchez, L. C., et al. (2012). A stem cell-based approach to cartilage repair. Science 336, 717–721.
Kaczmarek, A., Vandenabeele, P., and Krysko, D. V. (2013). Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223. doi: 10.1016/j.immuni.2013.02.003
Kahlenberg, J. M., and Dubyak, G. R. (2004). Mechanisms of caspase-1 activation by P2X 7 receptor-mediated K + release. Am. J. Physiol. Cell Physiol. 286, C1100–C1108.
Karasawa, A., and Kawate, T. (2016). Structural basis for subtype-specific inhibition of the P2X7 receptor. elife 5:e22153.
Karki, P., Seong, C., Kim, J. E., Hur, K., Shin, S. Y., Lee, J. S., et al. (2007). Intracellular K(+) inhibits apoptosis by suppressing the Apaf-1 apoptosome formation and subsequent downstream pathways but not cytochrome c release. Cell Death Differ. 14, 2068–2075. doi: 10.1038/sj.cdd.4402221
Karmakar, M., Minns, M., Greenberg, E. N., Diaz-Aponte, J., Pestonjamasp, K., Johnson, J. L., et al. (2020). N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 11:2212.
Kerr, J. F., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239–257. doi: 10.1038/bjc.1972.33
Keystone, E. C., Wang, M. M., Layton, M., Hollis, S., McInnes, I. B., and Team, D. C. S. (2012). Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 71, 1630–1635.
Khakh, B. S., and North, R. A. (2006). P2X receptors as cell-surface ATP sensors in health and disease. Nature 442, 527–532. doi: 10.1038/nature04886
Kim, H. A., Cho, M. L., Choi, H. Y., Yoon, C. S., Jhun, J. Y., Oh, H. J., et al. (2006). The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 54, 2152–2163. doi: 10.1002/art.21951
Kim, N.-G., Lee, H., Son, E., Kwon, O.-Y., Park, J.-Y., Park, J.-H., et al. (2003). Hypoxic induction of caspase-11/caspase-1/interleukin-1β in brain microglia. Mol. Brain Res. 114, 107–114. doi: 10.1016/s0169-328x(03)00135-9
Kong, D., Zheng, T., Zhang, M., Wang, D., Du, S., Li, X., et al. (2013). Static mechanical stress induces apoptosis in rat endplate chondrocytes through MAPK and mitochondria-dependent caspase activation signaling pathways. PLoS One 8:e69403. doi: 10.1371/journal.pone.0069403
Kulbe, J. R., Mulcahy Levy, J. M., Coultrap, S. J., Thorburn, A., and Bayer, K. U. (2014). Excitotoxic glutamate insults block autophagic flux in hippocampal neurons. Brain Res. 1542, 12–19. doi: 10.1016/j.brainres.2013.10.032
Kurosaka, K., Takahashi, M., Watanabe, N., and Kobayashi, Y. (2003). Silent cleanup of very early apoptotic cells by macrophages. J. Immunol. 171, 4672–4679. doi: 10.4049/jimmunol.171.9.4672
Kyrkanides, S., Tallents, R. H., Miller, J. N., Olschowka, M. E., Johnson, R., Yang, M., et al. (2011). Osteoarthritis accelerates and exacerbates Alzheimer’s disease pathology in mice. J. Neuroinflammation 8:112. doi: 10.1186/1742-2094-8-112
Laker, R. C., Drake, J. C., Wilson, R. J., Lira, V. A., Lewellen, B. M., Ryall, K. A., et al. (2017). Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8:548.
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Lee, M. S., Kwon, H., Lee, E. Y., Kim, D. J., Park, J. H., Tesh, V. L., et al. (2016). Shiga toxins activate the NLRP3 inflammasome pathway to promote both production of the proinflammatory cytokine interleukin-1beta and apoptotic cell death. Infect. Immun. 84, 172–186. doi: 10.1128/iai.01095-15
Leist, M., Single, B., Castoldi, A. F., Kuhnle, S., and Nicotera, P. (1997). Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185, 1481–1486. doi: 10.1084/jem.185.8.1481
Li, J., and Dong, S. (2016). The signaling pathways involved in chondrocyte differentiation and hypertrophic differentiation. Stem Cells Int. 2016:2470351.
Li, M. Y., Zhu, X. L., Zhao, B. X., Shi, L., Wang, W., Hu, W., et al. (2019). Adrenomedullin alleviates the pyroptosis of Leydig cells by promoting autophagy via the ROS-AMPK-mTOR axis. Cell Death Dis. 10:489.
Licastro, F., Candore, G., Lio, D., Porcellini, E., Colonna-Romano, G., Franceschi, C., et al. (2005). Innate immunity and inflammation in ageing: a key for understanding age-related diseases. Immun. Ageing. 2:8.
Liu, H. T., and Pan, S. S. (2019). Late exercise preconditioning promotes autophagy against exhaustive exercise-induced myocardial injury through the activation of the AMPK-mTOR-ULK1 pathway. Biomed. Res. Int. 2019:5697380.
Liu, J. J., Li, Y., Yang, M. S., Chen, R., and Cen, C. Q. (2020). SP1-induced ZFAS1 aggravates sepsis-induced cardiac dysfunction via miR-590-3p/NLRP3-mediated autophagy and pyroptosis. Arch. Biochem. Biophys. 695:108611. doi: 10.1016/j.abb.2020.108611
Liu, X., Niu, Y., Yuan, H., Huang, J., and Fu, L. (2015). AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy. Metabolism 64, 658–665. doi: 10.1016/j.metabol.2015.01.015
Liu, Y., Nguyen, P. T., Wang, X., Zhao, Y., Meacham, C. E., Zou, Z., et al. (2020). TLR9 and beclin 1 crosstalk regulates muscle AMPK activation in exercise. Nature 578, 605–609. doi: 10.1038/s41586-020-1992-7
Lo, Y. Y., Wong, J. M., and Cruz, T. F. (1996). Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J. Biol. Chem. 271, 15703–15707. doi: 10.1074/jbc.271.26.15703
Lo Verso, F., Carnio, S., Vainshtein, A., and Sandri, M. (2014). Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 10, 1883–1894. doi: 10.4161/auto.32154
Locovei, S., Scemes, E., Qiu, F., Spray, D. C., and Dahl, G. (2007). Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 581, 483–488. doi: 10.1016/j.febslet.2006.12.056
Loening, A. M., James, I. E., Levenston, M. E., Badger, A. M., Frank, E. H., Kurz, B., et al. (2000). Injurious mechanical compression of bovine articular cartilage induces chondrocyte apoptosis. Arch. Biochem. Biophys. 381, 205–212. doi: 10.1006/abbi.2000.1988
Lopez de Figueroa, P., Lotz, M. K., Blanco, F. J., and Carames, B. (2015). Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 67, 966–976. doi: 10.1002/art.39025
Lopez-Castejon, G., and Brough, D. (2011). Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev. 22, 189–195. doi: 10.1016/j.cytogfr.2011.10.001
MacKenzie, A., Wilson, H. L., Kiss-Toth, E., Dower, S. K., North, R. A., and Surprenant, A. (2001). Rapid secretion of interleukin-1β by microvesicle shedding. Immunity 15, 825–835. doi: 10.1016/s1074-7613(01)00229-1
Man, G., and Mologhianu, G. (2014). Osteoarthritis pathogenesis: a complex process that involves the entire joint. J. Med. Life 7, 37–41.
Man, S., and Kanneganti, T. D. (2015). Regulation of inflammasome activation. Immunol. Rev. 265, 6–21.
Martin, J. A., and Buckwalter, J. A. (2006). Post-traumatic osteoarthritis: the role of stress induced chondrocyte damage. Biorheology 43, 517–521.
Martin-Rincon, M., Perez-Lopez, A., Morales-Alamo, D., Perez-Suarez, I., de Pablos-Velasco, P., Perez-Valera, M., et al. (2019). Exercise mitigates the loss of muscle mass by attenuating the activation of autophagy during severe energy deficit. Nutrients 11:2824. doi: 10.3390/nu11112824
Mascanfroni, I. D., Takenaka, M. C., Yeste, A., Patel, B., Wu, Y., Kenison, J. E., et al. (2015). Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat. Med. 21, 638–646. doi: 10.1038/nm.3868
Matute, C., Torre, I., Perez-Cerda, F., Perez-Samartin, A., Alberdi, E., Etxebarria, E., et al. (2007). P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 27, 9525–9533. doi: 10.1523/jneurosci.0579-07.2007
McGilligan, V. E., Gregory-Ksander, M. S., Li, D., Moore, J. E., Hodges, R. R., Gilmore, M. S., et al. (2013). Staphylococcus aureus activates the NLRP3 inflammasome in human and rat conjunctival goblet cells. PLoS One 8:e74010. doi: 10.1371/journal.pone.0074010
Mehto, S., Jena, K. K., Nath, P., Chauhan, S., Kolapalli, S. P., Das, S. K., et al. (2019). The Crohn’s disease risk factor irgm limits nlrp3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol. Cell 73, 429–445.e7.
Mizushima, N., and Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. doi: 10.1016/j.cell.2011.10.026
Mueller, M. B., and Tuan, R. S. (2011). Anabolic/Catabolic balance in pathogenesis of osteoarthritis: identifying molecular targets. PM R 3(6 Suppl. 1), S3–S11.
Nicke, A. (2008). Homotrimeric complexes are the dominant assembly state of native P2X7 subunits. Biochem. Biophys. Res. Commun. 377, 803–808. doi: 10.1016/j.bbrc.2008.10.042
Nishida, K., Nakatani, T., Ohishi, A., Okuda, H., Higashi, Y., Matsuo, T., et al. (2012). Mitochondrial dysfunction is involved in P2X7 receptor-mediated neuronal cell death. J. Neurochem. 122, 1118–1128. doi: 10.1111/j.1471-4159.2012.07868.x
Nishida, Y., Knudson, C. B., and Knudson, W. (2004). Osteogenic Protein-1 inhibits matrix depletion in a hyaluronan hexasaccharide-induced model of osteoarthritis. Osteoarthritis Cartilage 12, 374–382. doi: 10.1016/j.joca.2004.01.008
North, R. A. (2002). Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1067. doi: 10.1152/physrev.00015.2002
North, R. A., and Jarvis, M. F. (2013). P2X receptors as drug targets. Mol. Pharmacol. 83, 759–769. doi: 10.1124/mol.112.083758
Olee, T., Hashimoto, S., Quach, J., and Lotz, M. (1999). IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J. Immunol. 162, 1096–1100.
Orioli, E., Marchi, E. D., Giuliani, A. L., and Adinolfi, E. (2017). P2X7 receptor orchestrates multiple signalling pathways triggering inflammation, autophagy and metabolic/trophic responses. Curr. Med. Chem. 24, 2261–2275.
Palmer, A. J., Thomas, G. E., Pollard, T. C., Rombach, I., Taylor, A., Arden, N., et al. (2013). The feasibility of performing a randomised controlled trial for femoroacetabular impingement surgery. Bone Joint Res. 2, 33–40. doi: 10.1302/2046-3758.22.2000137
Pan, T., Shi, X., Chen, H., Chen, R., Wu, D., Lin, Z., et al. (2018). Geniposide suppresses interleukin-1beta-induced inflammation and apoptosis in rat chondrocytes via the PI3K/Akt/NF-kappaB signaling pathway. Inflammation 41, 390–399. doi: 10.1007/s10753-017-0694-2
Panenka, W., Jijon, H., Herx, L. M., Armstrong, J. N., Feighan, D., Wei, T., et al. (2001). P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J. Neurosci. 21, 7135–7142. doi: 10.1523/jneurosci.21-18-07135.2001
Papp, L., Vizi, E. S., and Sperlagh, B. (2007). P2X7 receptor mediated phosphorylation of p38MAP kinase in the hippocampus. Biochem. Biophys. Res. Commun. 355, 568–574. doi: 10.1016/j.bbrc.2007.02.014
Park, J. H., and Kim, Y. C. (2017). P2X7 receptor antagonists: a patent review (2010-2015). Expert Opin. Ther. Pat. 27, 257–267. doi: 10.1080/13543776.2017.1246538
Parzych, K. R., and Klionsky, D. J. (2014). An overview of autophagy: morphology, mechanism, and regulation. Antioxid. Redox Signal. 20, 460–473. doi: 10.1089/ars.2013.5371
Pelegrin, P., Barroso-Gutierrez, C., and Surprenant, A. (2008). P2X7 receptor differentially couples to distinct release pathways for IL-1β in mouse macrophage. J. Immunol. 180, 7147–7157. doi: 10.4049/jimmunol.180.11.7147
Pelegrin, P., and Surprenant, A. (2006). Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 25, 5071–5082. doi: 10.1038/sj.emboj.7601378
Pellegatti, P., Raffaghello, L., Bianchi, G., Piccardi, F., Pistoia, V., and Di Virgilio, F. (2008). Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One 3:e2599. doi: 10.1371/journal.pone.0002599
Perregaux, D. G., McNiff, P., Laliberte, R., Conklyn, M., and Gabel, C. A. (2000). ATP acts as an agonist to promote stimulus-induced secretion of IL-1 beta and IL-18 in human blood. J. Immunol. 165, 4615–4623. doi: 10.4049/jimmunol.165.8.4615
Piccioli, P., and Rubartelli, A. (2013). The secretion of IL-1beta and options for release. Semin. Immunol. 25, 425–429. doi: 10.1016/j.smim.2013.10.007
Pivec, R., Johnson, A. J., Mears, S. C., and Mont, M. A. (2012). Hip arthroplasty. Lancet 380, 1768–1777.
Pizzirani, C., Ferrari, D., Chiozzi, P., Adinolfi, E., Sandona, D., Savaglio, E., et al. (2007). Stimulation of P2 receptors causes release of IL-1beta-loaded microvesicles from human dendritic cells. Blood 109, 3856–3864. doi: 10.1182/blood-2005-06-031377
Plesner, L. (1995). Ecto-ATPases: identities and functions. Int. Rev. Cytol. 158, 141–214. doi: 10.1016/s0074-7696(08)62487-0
Qiu, J. H., Asai, A., Chi, S., Saito, N., Hamada, H., and Kirino, T. (2000). Proteasome inhibitors induce cytochrome c-caspase-3-like protease-mediated apoptosis in cultured cortical neurons. J. Neurosci. 20, 259–265. doi: 10.1523/jneurosci.20-01-00259.2000
Qiu, T., Pei, P., Yao, X., Jiang, L., Wei, S., Wang, Z., et al. (2018). Taurine attenuates arsenic-induced pyroptosis and nonalcoholic steatohepatitis by inhibiting the autophagic-inflammasomal pathway. Cell Death Dis. 9:946.
Qu, Y., Franchi, L., Nunez, G., and Dubyak, G. R. (2007). Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 179, 1913–1925. doi: 10.4049/jimmunol.179.3.1913
Rao, Z., Wang, S., and Wang, J. (2017). Peroxiredoxin 4 inhibits IL-1beta-induced chondrocyte apoptosis via PI3K/AKT signaling. Biomed. Pharmacother. 90, 414–420. doi: 10.1016/j.biopha.2017.03.075
Rathakrishnan, C., Tiku, K., Raghavan, A., and Tiku, M. L. (1992). Release of oxygen radicals by articular chondrocytes: a study of luminol- dependent chemiluminescence and hydrogen peroxide secretion. J. Bone Min. Res. 7, 1139–1148. doi: 10.1002/jbmr.5650071005
Rekha, R. S., Rao Muvva, S. S., Wan, M., Raqib, R., Bergman, P., Brighenti, S., et al. (2015). Phenylbutyrate induces LL-37-dependent autophagy and intracellular killing of Mycobacterium tuberculosis in human macrophages. Autophagy 11, 1688–1699. doi: 10.1080/15548627.2015.1075110
Ribeiro, M., López de Figueroa, P., Blanco, F. J., Mendes, A. F., and Carames, B. (2016a). Insulin decreases autophagy and leads to cartilage degradation. Osteoarthritis Cartilage 24, 731–739. doi: 10.1016/j.joca.2015.10.017
Ribeiro, M., Lopez de Figueroa, P., Nogueira-Recalde, U., Centeno, A., Mendes, A. F., Blanco, F. J., et al. (2016b). Diabetes-accelerated experimental osteoarthritis is prevented by autophagy activation. Osteoarthritis Cartilage 24, 2116–2125. doi: 10.1016/j.joca.2016.06.019
Roach, H. I., Aigner, T., and Kouri, J. B. (2004). Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis 9, 265–277. doi: 10.1023/b:appt.0000025803.17498.26
Rutjes, A. W., Jüni, P., da Costa, B. R., Trelle, S., Nüesch, E., and Reichenbach, S. (2012). Viscosupplementation for osteoarthritis of the knee: a systematic review and meta-analysis. Ann. Intern. Med. 157, 180–191.
Saber, S., Abd El-Kader, E. M., Sharaf, H., El-Shamy, R., El-Saeed, B., Mostafa, A., et al. (2020). Celastrol augments sensitivity of NLRP3 to CP-456773 by modulating HSP-90 and inducing autophagy in dextran sodium sulphate-induced colitis in rats. Toxicol. Appl. Pharmacol. 400:115075. doi: 10.1016/j.taap.2020.115075
Sandell, L. J., and Aigner, T. (2001). Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 3, 107–113.
Sasaki, H., Takayama, K., Matsushita, T., Ishida, K., Kubo, S., Matsumoto, T., et al. (2012). Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 64, 1920–1928. doi: 10.1002/art.34323
Scanzello, C. R., Plaas, A., and Crow, M. K. (2008). Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr. Opin. Rheumatol. 20, 565–572. doi: 10.1097/bor.0b013e32830aba34
Schipani, E., Ryan, H. E., Didrickson, S., Kobayashi, T., Knight, M., and Johnson, R. S. (2001). Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 15, 2865–2876.
Schmidt, T. A., Gastelum, N. S., Nguyen, Q. T., Schumacher, B. L., and Sah, R. L. (2007). Boundary lubrication of articular cartilage: role of synovial fluid constituents. Arthritis Rheum. 56, 882–891. doi: 10.1002/art.22446
Schwalm, C., Jamart, C., Benoit, N., Naslain, D., Premont, C., Prevet, J., et al. (2015). Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 29, 3515–3526. doi: 10.1096/fj.14-267187
Sekar, P., Huang, D. Y., Hsieh, S. L., Chang, S. F., and Lin, W. W. (2018). AMPK-dependent and independent actions of P2X7 in regulation of mitochondrial and lysosomal functions in microglia. Cell Commun. Signal. 16:83.
Shao, B. Z., Xu, Z. Q., Han, B. Z., Su, D. F., and Liu, C. (2015). NLRP3 inflammasome and its inhibitors: a review. Front. Pharmacol. 6:262. doi: 10.3389/fphar.2015.00262
Shapiro, I. M., Layfield, R., Lotz, M., Settembre, C., and Whitehouse, C. (2014). Boning up on autophagy: the role of autophagy in skeletal biology. Autophagy 10, 7–19. doi: 10.4161/auto.26679
Sharma, D., and Kanneganti, T. D. (2016). The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J. Cell Biol. 213, 617–629.
Shen, Y., Liu, W. W., Zhang, X., Shi, J. G., Jiang, S., Zheng, L., et al. (2020). TRAF3 promotes ROS production and pyroptosis by targeting ULK1 ubiquitination in macrophages. FASEB J. 34, 7144–7159. doi: 10.1096/fj.201903073r
Shi, C. S., Shenderov, K., Huang, N. N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. doi: 10.1038/ni.2215
Signaling, F. B. (2004). Transduction: target in osteoarthritis. Curr. Opin. Rheumatol. 16, 616–622. doi: 10.1097/01.bor.0000133663.37352.4a
Sitia, R., and Rubartelli, A. (2020). Evolution, role in inflammation, and redox control of leaderless secretory proteins. J. Biol. Chem. 295, 7799–7811. doi: 10.1074/jbc.rev119.008907
Sluyter, R., and Stokes, L. (2011). Significance ofP2X7receptor variants to human health and disease. Recent Pat. DNA Gene Seq. 5, 41–54. doi: 10.2174/187221511794839219
Speziali, A., Delcogliano, M., Tei, M., Placella, G., Chillemi, M., Tiribuzi, R., et al. (2015). Chondropenia: current concept review. Musculoskelet. Surg. 99, 189–200. doi: 10.1007/s12306-015-0377-9
Stock, T. C., Bloom, B., Wei, N., Ishaq, S., Park, W., Wang, X., et al. (2012). Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 39, 720–727. doi: 10.3899/jrheum.110874
Stolberg-Stolberg, J. A., Furman, B. D., Garrigues, N. W., Lee, J., Pisetsky, D. S., Stearns, N. A., et al. (2013). Effects of cartilage impact with and without fracture on chondrocyte viability and the release of inflammatory markers. J. Orthop. Res. 31, 1283–1292. doi: 10.1002/jor.22348
Strowig, T., Henao-Mejia, J., Elinav, E., and Flavell, R. (2012). Inflammasomes in health and disease. Nature 481, 278–286. doi: 10.1038/nature10759
Sun, K., Liu, F., Wang, J., Guo, Z., Ji, Z., and Yao, M. (2017). The effect of mechanical stretch stress on the differentiation and apoptosis of human growth plate chondrocytes. In Vitro Cell. Dev. Biol. Anim. 53, 141–148. doi: 10.1007/s11626-016-0090-5
Sun, L., Gao, J., Zhao, M., Cui, J., Li, Y., Yang, X., et al. (2015). A novel cognitive impairment mechanism that astrocytic p-connexin 43 promotes neuronic autophagy via activation of P2X7R and down-regulation of GLT-1 expression in the hippocampus following traumatic brain injury in rats. Behav. Brain Res. 291, 315–324. doi: 10.1016/j.bbr.2015.05.049
Surprenant, A., Rassendren, F., Kawashima, E., North, R. A., and Buell, G. (1996). The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272, 735–738. doi: 10.1126/science.272.5262.735
Takenouchi, T., Fujita, M., Sugama, S., Kitani, H., and Hashimoto, M. (2009a). The role of the P2X7 receptor signaling pathway for the release of autolysosomes in microglial cells. Autophagy 5, 723–724. doi: 10.4161/auto.5.5.8478
Takenouchi, T., Nakai, M., Iwamaru, Y., Sugama, S., Tsukimoto, M., Fujita, M., et al. (2009b). The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J. Immunol. 182, 2051–2062. doi: 10.4049/jimmunol.0802577
Taylor, S. R., Turner, C. M., Elliott, J. I., McDaid, J., Hewitt, R., Smith, J., et al. (2009). P2X7 deficiency attenuates renal injury in experimental glomerulonephritis. J. Am. Soc. Nephrol. 20, 1275–1281. doi: 10.1681/asn.2008060559
Thomas, C. M., Fuller, C. J., Whittles, C. E., and Sharif, M. (2007). Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthritis Cartilage 15, 27–34. doi: 10.1016/j.joca.2006.06.012
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552(Pt 2), 335–344. doi: 10.1113/jphysiol.2003.049478
Uthman, O. A., van der Windt, D. A., Jordan, J. L., Dziedzic, K. S., Healey, E. L., Peat, G. M., et al. (2013). Exercise for lower limb osteoarthritis: systematic review incorporating trial sequential analysis and network meta-analysis. BMJ 347:f5555. doi: 10.1136/bmj.f5555
van Lent, P. L., Blom, A. B., Schelbergen, R. F., Sloetjes, A., Lafeber, F. P., Lems, W. F., et al. (2012). Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 64, 1466–1476. doi: 10.1002/art.34315
van Lent, P. L., Blom, A. B., van der Kraan, P., Holthuysen, A. E., Vitters, E., van Rooijen, N., et al. (2004). Crucial role of synovial lining macrophages in the promotion of transforming growth factor beta-mediated osteophyte formation. Arthritis Rheum. 50, 103–111. doi: 10.1002/art.11422
Vanden Berghe, T., Kaiser, W. J., Bertrand, M. J., and Vandenabeele, P. (2015). Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2:e975093. doi: 10.4161/23723556.2014.975093
Verbruggen, G., Wittoek, R., Vander Cruyssen, B., and Elewaut, D. (2012). Tumour necrosis factor blockade for the treatment of erosive osteoarthritis of the interphalangeal finger joints: a double blind, randomised trial on structure modification. Ann. Rheum. Dis. 71, 891–898. doi: 10.1136/ard.2011.149849
Vigano, E., and Mortellaro, A. (2013). Caspase-11: the driving factor for noncanonical inflammasomes. Eur. J. Immunol. 43, 2240–2245. doi: 10.1002/eji.201343800
Wada, T., and Penninger, J. M. (2004). Mitogen-activated protein kinases in apoptosis regulation. Oncogene 23, 2838–2849. doi: 10.1038/sj.onc.1207556
Wang, J., and Dahl, G. (2018). Pannexin1: a multifunction and multiconductance and/or permeability membrane channel. Am. J. Physiol. Cell Physiol. 315, C290–C299.
Wang, Q., Rozelle, A. L., Lepus, C. M., Scanzello, C. R., Song, J. J., Larsen, D. M., et al. (2011). Identification of a central role for complement in osteoarthritis. Nat. Med. 17, 1674–1679.
Wang, X., Jiang, L., Shi, L., Yao, K., Sun, X., Yang, G., et al. (2019). Zearalenone induces NLRP3-dependent pyroptosis via activation of NF-kappaB modulated by autophagy in INS-1 cells. Toxicology 428:152304. doi: 10.1016/j.tox.2019.152304
Wang, Y., Song, X., Li, Z., Liu, N., Yan, Y., Li, T., et al. (2020). MicroRNA-103 protects coronary artery endothelial cells against H2O2-induced oxidative stress via BNIP3-mediated end-stage autophagy and antipyroptosis pathways. Oxid. Med. Cell Longev. 2020:8351342.
Weber, F. C., Esser, P. R., Muller, T., Ganesan, J., Pellegatti, P., Simon, M. M., et al. (2010). Lack of the purinergic receptor P2X(7) results in resistance to contact hypersensitivity. J. Exp. Med. 207, 2609–2619. doi: 10.1084/jem.20092489
Wei, Q., Zhu, R., Zhu, J., Zhao, R., and Li, M. (2019). E2-induced activation of the NLRP3 inflammasome triggers pyroptosis and inhibits autophagy in HCC cells. Oncol. Res. 27, 827–834. doi: 10.3727/096504018x15462920753012
Wilhelm, K., Ganesan, J., Muller, T., Durr, C., Grimm, M., Beilhack, A., et al. (2010). Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 16, 1434–1438. doi: 10.1038/nm.2242
Wu, C., Xu, H., Li, J., Hu, X., Wang, X., Huang, Y., et al. (2020). Baicalein attenuates pyroptosis and endoplasmic reticulum stress following spinal cord ischemia-reperfusion injury via autophagy enhancement. Front. Pharmacol. 11:1076. doi: 10.3389/fphar.2020.01076
Xu, X. Y., He, X. T., Wang, J., Li, X., Xia, Y., Tan, Y. Z., et al. (2019). Role of the P2X7 receptor in inflammation-mediated changes in the osteogenesis of periodontal ligament stem cells. Cell Death Dis. 10:20.
Yang, D., He, Y., Munoz-Planillo, R., Liu, Q., and Nunez, G. (2015). Caspase-11 requires the Pannexin-1 channel and the Purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43, 923–932. doi: 10.1016/j.immuni.2015.10.009
Yang, F., Qin, Y., Wang, Y., Meng, S., Xian, H., Che, H., et al. (2019). Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int. J. Biol. Sci. 15, 1010–1019. doi: 10.7150/ijbs.29680
Yang, J., Liu, Z., and Xiao, T. S. (2017). Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 14, 65–79. doi: 10.1038/cmi.2016.29
Yang, X., Zhou, G., Ren, T., Li, H., Zhang, Y., Yin, D., et al. (2012). Beta-Arrestin prevents cell apoptosis through pro-apoptotic ERK1/2 and p38 MAPKs and anti-apoptotic Akt pathways. Apoptosis 17, 1019–1026. doi: 10.1007/s10495-012-0741-2
Yin, W., Park, J. I., and Loeser, R. F. (2009). Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J. Biol. Chem. 284, 31972–31981. doi: 10.1074/jbc.m109.056838
Young, C. N., Sinadinos, A., Lefebvre, A., Chan, P., Arkle, S., Vaudry, D., et al. (2015). A novel mechanism of autophagic cell death in dystrophic muscle regulated by P2RX7 receptor large-pore formation and HSP90. Autophagy 11, 113–130. doi: 10.4161/15548627.2014.994402
Yu, S. M., and Kim, S. J. (2014). Withaferin A-caused production of intracellular reactive oxygen species modulates apoptosis via PI3K/Akt and JNKinase in rabbit articular chondrocytes. J. Korean Med. Sci. 29, 1042–1053. doi: 10.3346/jkms.2014.29.8.1042
Yu, S. M., and Kim, S. J. (2015). The thymoquinone-induced production of reactive oxygen species promotes dedifferentiation through the ERK pathway and inflammation through the p38 and PI3K pathways in rabbit articular chondrocytes. Int. J. Mol. Med. 35, 325–332. doi: 10.3892/ijmm.2014.2014
Zamli, Z., Adams, M. A., Tarlton, J. F., and Sharif, M. (2013). Increased chondrocyte apoptosis is associated with progression of osteoarthritis in spontaneous Guinea pig models of the disease. Int. J. Mol. Sci. 14, 17729–17743. doi: 10.3390/ijms140917729
Zamli, Z., and Sharif, M. (2011). Chondrocyte apoptosis: a cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 14, 159–166. doi: 10.1111/j.1756-185x.2011.01618.x
Zeiss, C. J. (2003). The apoptosis-necrosis continuum: insights from genetically altered mice. Vet. Pathol. 40, 481–495. doi: 10.1354/vp.40-5-481
Zemmyo, M., Meharra, E. J., Kuhn, K., Creighton-Achermann, L., and Lotz, M. (2003). Accelerated, aging-dependent development of osteoarthritis in alpha1 integrin-deficient mice. Arthritis Rheum. 48, 2873–2880.
Zeng, Q., Zhou, Y., Liang, D., He, H., Liu, X., Zhu, R., et al. (2020). Exosomes secreted from bone marrow mesenchymal stem cells attenuate oxygen-glucose deprivation/reoxygenation-induced pyroptosis in PC12 cells by promoting AMPK-dependent autophagic flux. Front. Cell. Neurosci. 14:182. doi: 10.3389/fncel.2020.00182
Zhang, F., Wang, J., Chu, J., Yang, C., Xiao, H., Zhao, C., et al. (2015). MicroRNA-146a induced by hypoxia promotes chondrocyte autophagy through Bcl-2. Cell. Physiol. Biochem. 37, 1442–1453.
Zhang, W., Zhong, B., Zhang, C., Luo, C., and Zhan, Y. (2018). miR-373 regulates inflammatory cytokine-mediated chondrocyte proliferation in osteoarthritis by targeting the P2X7 receptor. FEBS Open Bio. 8, 325–331.
Zhang, Y., Cheng, H., Li, W., Wu, H., and Yang, Y. (2019). Highly-expressed P2X7 receptor promotes growth and metastasis of human HOS/MNNG osteosarcoma cells via PI3K/Akt/GSK3beta/beta-catenin and mTOR/HIF1alpha/VEGF signaling. Int. J. Cancer 145, 1068–1082.
Zhang, Y., Vasheghani, F., Li, Y. H., Blati, M., Simeone, K., Fahmi, H., et al. (2015). Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 74, 1432–1440.
Zhang, Y. B., Li, S. X., Chen, X. P., Yang, L., Zhang, Y. G., Liu, R., et al. (2008). Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci. Bull. 24, 143–149.
Zhao, H., Chen, Y., and Feng, H. (2018). P2X7 receptor-associated programmed cell death in the pathophysiology of hemorrhagic stroke. Curr. Neuropharmacol. 16, 1282–1295.
Zheng, T., Zhao, C., Zhao, B., Liu, H., Wang, S., Wang, L., et al. (2020). Impairment of the autophagy-lysosomal pathway and activation of pyroptosis in macular corneal dystrophy. Cell Death Discov. 6:85.
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., Wong, J., et al. (2016). NF-kappaB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164, 896–910.
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225.
Keywords: osteoarthritis, P2X7 receptor, inflammation, apoptosis, pyroptosis, autophagy
Citation: Li Z, Huang Z and Bai L (2021) The P2X7 Receptor in Osteoarthritis. Front. Cell Dev. Biol. 9:628330. doi: 10.3389/fcell.2021.628330
Received: 11 November 2020; Accepted: 22 January 2021;
Published: 11 February 2021.
Edited by:
Yinan Gong, University of Pittsburgh, United StatesReviewed by:
Yansheng Feng, The University of Texas Health Science Center at San Antonio, United StatesCopyright © 2021 Li, Huang and Bai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lunhao Bai, bHVuaGFvYmFpX2FjZUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.