
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 19 February 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.627295
This article is part of the Research Topic Genetic Mutations Associated with Ocular Diseases View all 19 articles
 Xiao-Fang Wang1†
Xiao-Fang Wang1† Hui Chen1†Peng-Juan Huang1Zhuo-Kun Feng1
Hui Chen1†Peng-Juan Huang1Zhuo-Kun Feng1 Zi-Qi Hua1Xiang Feng1Fang Han2Xiao-Tao Xu1
Zi-Qi Hua1Xiang Feng1Fang Han2Xiao-Tao Xu1 Ren-Juan Shen2
Ren-Juan Shen2 Yang Li2
Yang Li2 Zi-Bing Jin1,2*Huan-Yun Yu1*
Zi-Bing Jin1,2*Huan-Yun Yu1*Purpose: Congenital nystagmus (CN) is a genetically and clinically heterogeneous ocular disorder that manifests as involuntary, periodic oscillations of the eyes. To date, only FRMD7 and GPR143 have been reported to be responsible for causing CN. Here, we aimed to identify the disease-causing mutations and describe the clinical features in the affected members in our study.
Methods: All the subjects underwent a detailed ophthalmic examination. Direct sequencing of all coding exons and splice site regions in FRMD7 and GPR143 and a mutation assessment were performed in each patient.
Results: We found 14 mutations in 14/37 (37.8%) probands, including nine mutations in the FRMD7 gene and five mutations in the GPR143 gene, seven of which are novel, including c.284G>A(R95K), c.964C>T(P322S), c.284+10T>G, c.901T>C (Y301H), and c.2014_2023delTCACCCATGG(S672Pfs*12) in FRMD7, and c.250+1G>C, and c.485G>A (W162*) in GPR143. The mutation detection rate was 87.5% (7/8) of familial vs. 24.1% (7/29) of sporadic cases. Ten mutations in 24 (41.7%) non-syndromic subjects and 4 mutations in 13(30.8%) syndromic subjects were detected. A total of 77.8% (7/9) of mutations in FRMD7 were concentrated within the FERM and FA domains, while all mutations in GPR143 were located in exons 1, 2, 4 and 6. We observed that visual acuity tended to be worse in the GPR143 group than in the FRMD7 group, and no obvious difference in other clinical manifestations was found through comparisons in different groups of patients.
Conclusions: This study identified 14 mutations (seven novel and seven known) in eight familial and 29 sporadic patients with congenital nystagmus, expanding the mutational spectrum and validating FRMD7 and GPR143 as mutation hotspots. These findings also revealed a significant difference in the screening rate between different groups of participants, providing new insights for the strategy of genetic screening and early clinical diagnosis of CN.
Nystagmus is an involuntary, periodic oscillation of unilateral or bilateral eyes. It can be classified into congenital nystagmus (CN) and acquired nystagmus according to the age at onset. The prevalence of nystagmus in the general population has been estimated to be 24/10,000 of the population, and CN is the most common type of all forms of nystagmus (Sarvananthan et al., 2009; Watkins et al., 2012). CN usually appears at birth or in early childhood, and is predominantly characterized by horizontal pendular or jerk nystagmus with various degrees of visual impairment. Abnormal head position (AHP) that is often linked to an eccentric horizontal null position can be observed in patients with CN (Watkins et al., 2012; Brodsky and Dell'Osso, 2014; Papageorgiou et al., 2014; Richards and Wong, 2015). Previous studies have demonstrated that CN may occur as an isolated trait or may be accompanied by other ocular abnormalities such as ocular albinism, strabismus, aniridia, achromatopsia, congenital cataract, Leber congenital amaurosis, retinitis pigmentosa, cone-rod dystrophy and optic nerve hypoplasia (Sarvananthan et al., 2009; Brodsky and Dell'Osso, 2014; Richards and Wong, 2015). At present, although there have been a large number of studies focusing on CN over the past years, the mechanisms of pathogenesis remain unclear. A review in 2015 concluded two main hypotheses to explain this phenotype: dysfunction in the ocular motor control pathways and developmental abnormalities in the anterior visual pathway (Richards and Wong, 2015). Currently, no cure is available for CN, but many treatments, including non-surgical options (prisms, contact lenses, and afferent stimulation) and surgical extraocular muscle surgery, have been reported to improve visual acuity by directly or indirectly reducing but not eliminating nystagmus (Dell'Osso, 2002; Hertle et al., 2010).
Multiple modes of inheritance of CN have been reported in the literature, and X-linked CN is the most common form of hereditary nystagmus with significant clinical and genetic heterogeneity (Forssman, 1971). To date, only FRMD7 and GPR143 are considered the major disease-causing genes for CN. However, a recent study found that mutations within the C-terminal region of CASK disrupt the interaction, which is crucial for correct development of oculomotor control between FRMD7 and CASK, leading to nystagmus (Schnur et al., 1998; Watkins et al., 2013). Mutations were first identified in the FRMD7 (Xq26-27) gene in both X-linked and sporadic CN cases in 2006 (Tarpey et al., 2006). Since then, more than 90 mutations in the FRMD7 gene have been reported to date. G-protein coupled receptor 143 (GPR143), also known as OA1, was primarily described to cause ocular albinism with nystagmus as a prominent concomitant symptom. Pigmentation loss in the iris and retina is usually not obvious in Asian individuals, therefore, nystagmus may be the main manifestation in patients with GPR143 mutations (Zhou et al., 2008). Currently, over 100 mutations in GPR143 have been collected in the HGMD (Human Gene Mutation Database) (http://www.hgmd.cf.ac.uk). The mutation detection rate was found to be 20–57% and 62.5–95% in FRMD7 and GPR143, respectively, in X-linked cases, and much higher than the rates in sporadic cases. The genetic causes of sporadic cases are still poorly understood (Richards and Wong, 2015; Jia et al., 2017a).
In our study, FRMD7 and GPR143 mutation analysis and detailed clinical characteristics evaluation were performed in a group of Chinese patients with CN. A total of 9 mutations in the FRMD7 gene and five mutations in the GPR143 gene were identified in this study, including 7 novel and 7 previously reported mutations.
This study was in compliance with the Declaration of Helsinki and was approved by the Institutional Review Board. Informed written consent was obtained from all participants. All participants were enrolled from The Affiliated Eye Hospital of Wenzhou Medical University in this study. The average age was 12 ± 10 years, ranging between 1 year and 46 years old. After a detailed ophthalmic examination including visual acuity, intraocular pressure measurement, a slit-lamp examination, fundus photography, OCT and other specialist review. All the patients were diagnosed with congenital nystagmus by a specialist. Among these participants, 24 were non-syndromic CN cases, 13 were syndromic CN cases (11 cases with strabismus, four cases with ocular albinism, and two patients suffered from both strabismus and ocular albinism).
Dna was extracted from each participant's peripheral blood using a DNA extraction kit (TIANGEN, Beijing, China) following the manufacturer's instructions. Nanodrop 2000 (Thermal Fisher Scientific, Delaware, USA) was used to determine the concentration and purity of the extracted DNA.
The primers of all coding exons and splice site regions in FRMD7 and GPR143 were obtained from previous literature (Zhang Q. et al., 2007; Hu et al., 2011). After PCR amplification and direct sequencing of the coding regions and splice site junctions in FRMD7 and GPR143, we analyzed the sequencing of results. The reference genomic sequence versions of FRMD7 and GPR143 used were NM_194277.3 and NM_000273.3 from the GenBank database. The potential pathogenicity of the detected mutations in this study was evaluated by the following bioinformatics tools: SIFT (http://sift.jcvi.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (http://mutationtaster.org/), and PROVEAN (http://provean.jcvi.org/index.php). Allele frequency was assessed by the ExAC database (http://exac.broadinstitute.org/), and 1,000 Genomes Project (ftp://1000genomes.ebi.ac.uk/vol1/ftp). Co-segregation analysis was performed for mutations detected in our study when members in families were available.
Protein sequences were obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/), and multiple sequence alignments were performed using Clustalx1.83 software. Sequence logos were made with WebLogo3 (http://weblogo.threeplusone.com/). Schematic of protein domain structures were created using DOG 2.0 (Ren et al., 2009). The crystal structures of several wild-type and mutant proteins were predicted by Swiss Model (https://swissmodel.expasy.org/), and the predicted PDB files were visualized by PyMol software (version 2.1.1).
We used Mann–Whitney U-tests to compare the VA of the two groups. Onset age in two groups was analyzed using a t-test. Fisher's exact test was used to evaluate the significance of proportions (strabismus, stereopsis, and AHP) between groups. Non-parametric Kruskal-Wallis tests and Pearson chi-square tests were used to compare multiple groups.
A total of 37 patients with CN (29 male, eight female) were recruited for this study, and the mean age of the participants was 12 ± 10 years. Pedigrees of the 8 (21.6%) families followed an X-linked pattern of inheritance, and the remaining 29 (78.4%) sporadic patients had no positive family history. Different degrees of reduced visual acuity, stereopsis, and AHP were observed among the patients: eleven of them had associated strabismus (29.7%), four of them had associated ocular albinism (10.8%), and two patients had both conditions. A total of 93.8% (30/32) of these patients presented horizontal nystagmus, indicating that horizontal nystagmus was the most common form, which is consistent with previous reports (Zhao et al., 2016).
Sanger sequencing of FRMD7 and GPR143 in this Chinese cohort revealed 14 different mutations (nine in GPR143, five in FRMD7) in seven unrelated families and five sporadic cases. Seven mutations are novel: c.284G>A(R95K), c.964C>T(P322S), c.901T>C(Y301H), c.2014_2023delTCACCCATGG(S672Pfs*12), and c.284+10T>G in FRMD7 and c.250+1G>C, and c.485G>A(W162*) in GPR143. The remaining seven mutations have been reported before. The molecular and clinical results of the 12 participants with identified mutation are shown in Table 1. Mutations identified in our study were assessed for pathogenicity with four different bioinformatics tools, as shown in Table 2. Figure 1 illustrates the pedigrees and the sequencing data. Figure 2 shows the sequence conservation of the FRMD7 protein of three novel missense mutations and the structural modeling of the novel missense mutation c.284G>A.
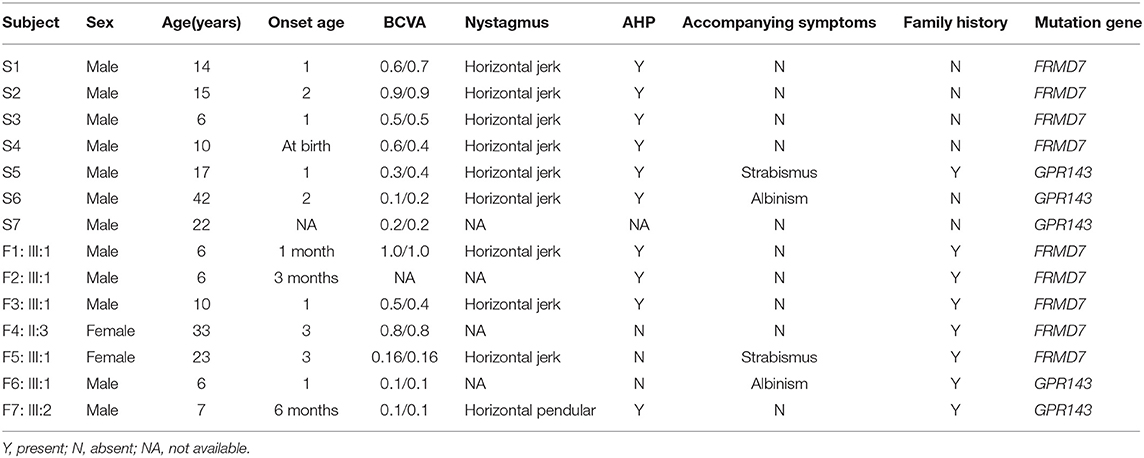
Table 1. Clinical features of subjects.
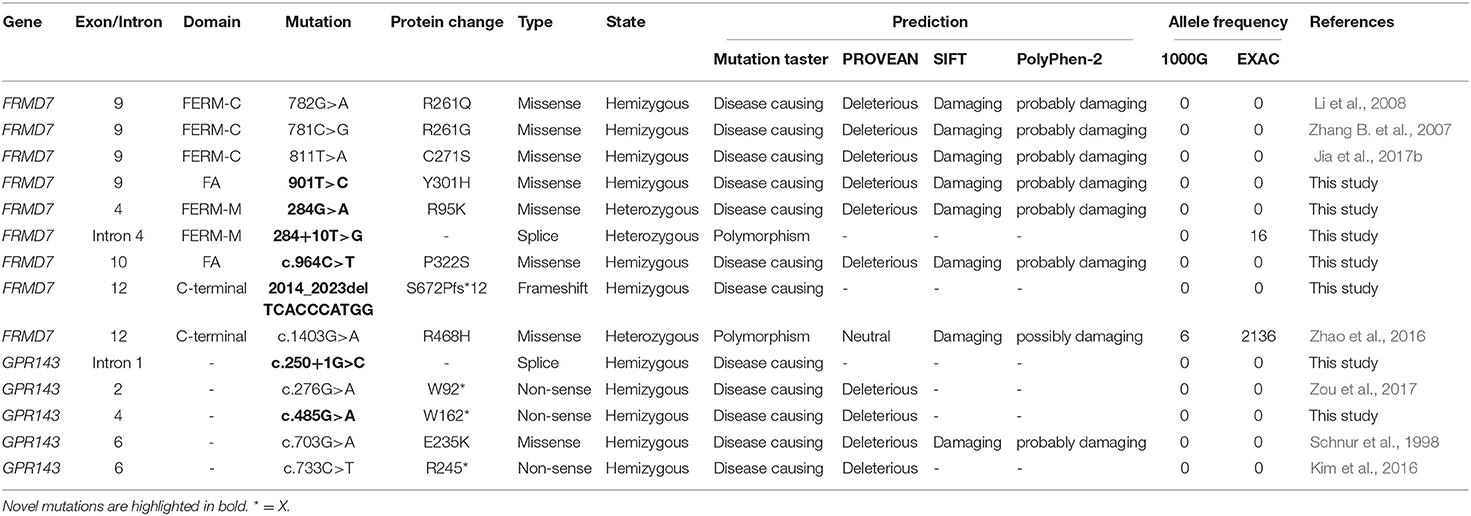
Table 2. Overview of the mutations found in FRMD7 and GPR143 and their assessment.
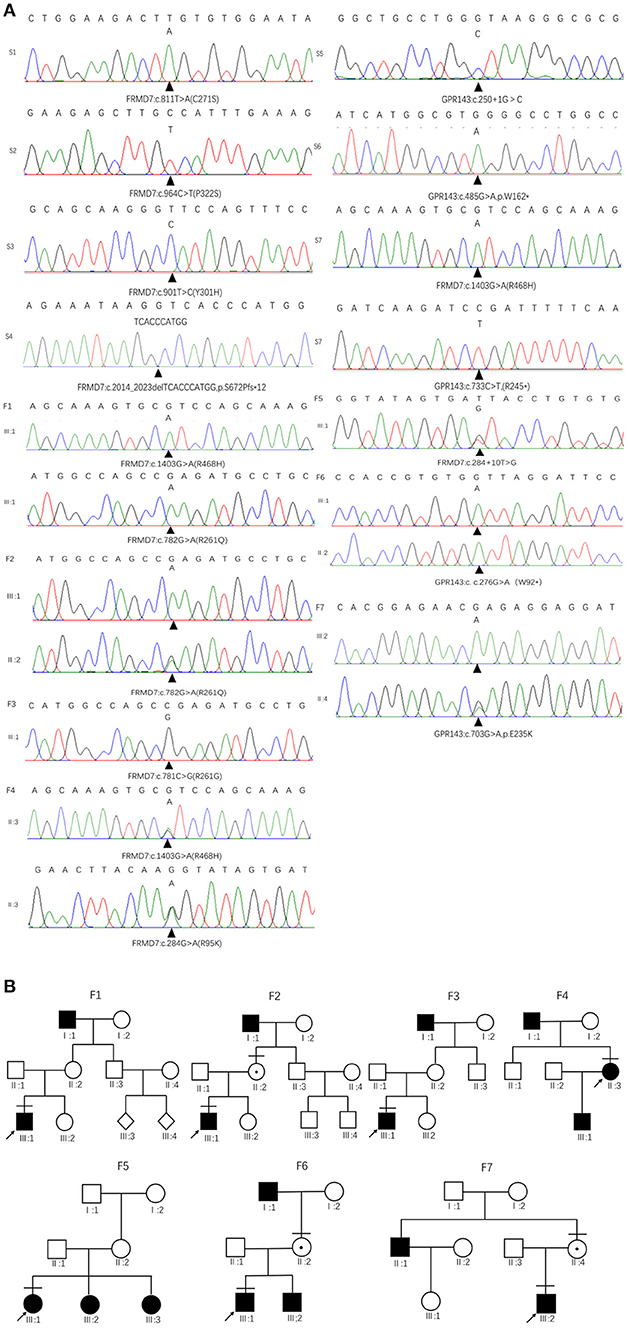
Figure 1. Family pedigrees and Sanger sequencing results. (A) The chromatograms of sequencing results: F 1-F 7 represent family 1 to family 7, S 1-S 7 represent sporadic 1 to sporadic 7. (B) Pedigrees of FRMD7 and GPR143 mutation–positive families: F 1 Family 1; F 2, family 2; F 3, family 3; F 4, family 4; F5, family 5; F 6, family 6; F 7, family 7. Filled symbols indicate affected individuals, unfilled symbols indicate unaffected individuals, and a dotted circle indicates a heterozygous carrier. Bars over the symbols indicate subjects enrolled in this study. Arrows indicate the probands.
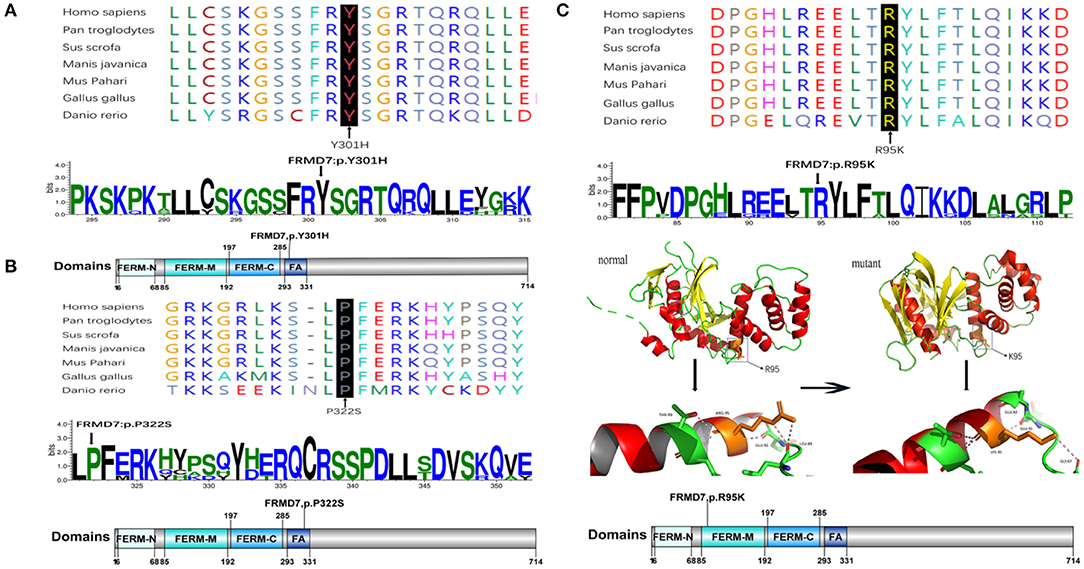
Figure 2. Missense mutations identified in this study. One-dimensional and two-dimensional multiple sequence alignment of the FRMD7 protein in different species and the predicted three-dimensional structure of the FRMD7 protein show the high evolutionary conservation of amino acid residues. Black arrows show the location of the novel missense mutations identified in this study in the FRMD7 protein. (A) The alignment of amino acids around residue p.301. (B) The alignment of amino acids around residue p.322. (C) The alignment of amino acids around residue p.95.The position of the missense mutation c.284G>A around residue p.R95K of FRMD7 is highlighted in the 3D structure model. Hydrogen bonds between amino acids are shown as pink dashed lines. The normal protein is on the left, and the mutant protein is on the right.
We detected a reported missense c.782G>A mutation in exon 9 in family 1 (III1) and family 2 (III1 and II2), which causes the substitution of arginine to glutamine at position 261 (p.R261Q). Li et al. (2008) already identified R261Q in two Chinese families with CN in 2008, and the pathogenicity of this mutation is well-established. In family 2, the unaffected mother of the proband carried c.782G>A, and the results demonstrated the co-segregation of the c.782G>A mutation with CN in family 2. The known missense mutation c.781C>G in exon 9 was found in family 3 (III1), which results in the substitution of an amino acid at a highly conserved position, p.R261G, and has been reported three times in the Chinese population, indicating the high possibility that it is a Chinese-specific variant (Zhang B. et al., 2007; Song et al., 2013; Zhao et al., 2016). The novel missense mutation c.284G>A, leading to a replacement of arginine by lysine at position 95, was detected in a female proband in family 4. A different substitution of amino acid arginine to methionine at 95 has ever been reported by Bai et al. (2017). The substitution occurred in the highly conserved domain FERM-M, and the predicted three-dimensional structure showed a change in the mutant protein. The Arg-95 residue was predicted to locate in the alpha helix of the FERM domain, and the side chain of Arg-95 projected outwards from the alpha helix forming hydrogen bonds with the side chains of Leu-89. In the mutant protein, two hydrogen bonds disappeared between Lys-95 and Leu-89, and Lys-95 side chains formed another one hydrogen bond with the amino acid residues Glu-92 and Gly-87, respectively (Figure 2C). Furthermore, all four software tools predicted the p.R95K mutation to be pathogenic (Table 2). In family 5, the heterozygous donor splice site mutation c.284+10T>G in intron4 was identified in the female proband (III1). This variant was considered to be possibly pathogenic in this family, because the frequency was < 0.01. The other two family members (III2, III3) who were unable to give a peripheral blood sample in this study also showed similar clinical signs of CN and esotropia. In sporadic cases, four mutations, c.811T>A, c.901T>C, c.964C>T, and c.2014_2023delTCACCCATGG, were found in S1, S2, S3, and S4, respectively; the latter three are first reported here. The missense mutation c.811T>A in exon 9 which caused a protein change at p.C271S, has been described before in a previous Chinese study (Jia et al., 2017b). The two novel missense variants resulted in amino acid substitutions at p.Y301H and p.P322S, and both of them occurred in the highly conserved residue of the FERM-adjacent (FA) domain. Multiple sequence alignment suggested that these sites were evolutionarily conserved from Danio to humans (Figures 2A,B). The 10-bp deletion mutation (c.2014_2023delTCACCCATGG) in exon 12 caused a frameshift in the ORF, leading to pre-mature translation termination of the FRMD7 protein at position 683 (p. S672Pfs*12) (Figure 1A). Apart from the mutations above, the c.1403G>A mutation that changed arginine to histidine at position 468 (p.R468H) was identified in three unrelated probands [III1 in family 1, III1 in family 4 and sporadic 2 (S2)] in our study, which was predicted to be non-pathogenic by PROVEAN and Mutation Taster and damaging by SIFT and PolyPhen-2. Based on the fact that its minor allele frequency (MAF)>0.01 and all three probands harbored another disease-causing mutation, in addition to published report (Zhao et al., 2016), the c.1403G>A mutation did not appear to be the main causative mutation in the three probands.
We detected the known non-sense mutation c.276G>A in exon 2 in the proband (III1) and his unaffected mother (II2) in family 6, which introduced a pre-mature stop codon into the ORF and generated a truncated protein. In family 7, III2 and his asymptomatic mother had the missense mutation c.703G>A (from glutamate to lysine), which was first described in a North American family (Schnur et al., 1998). All the pedigrees in which mothers' blood samples were available are consistent with X-linked transmission. In sporadic patients, two new mutations, a splice mutation c.250+1G>C on intron1 and a non-sense mutation c.485G>A truncating the translated protein at position 162, which were not present in the 1,000 Genomes Project or ExAC database and assessed to be pathogenic by prediction tools, were identified in S5 and S6. The known non-sense mutation c.733C>T created an early termination in the GPR143 protein was present in S7.
In our cohort of 37 participants with CN, 9 patients with FRMD7 mutations and five patients with GPR143 mutations were identified here. To further investigate the relationship of phenotype-to-genotype, we compared whether there was a statistical difference in terms of clinical characteristics between different groups of patients.
We found no significant difference (p>0.05) in onset age was observed between the FRMD7 group and the GPR143 group (t-test, P = 0.2846) or between the FRMD7 group and the non-FRMD7 group (t-test, P = 0.4009 > 0.05). The mean age of onset in our study was later than that in other studies.
We tested eight of nine patients (accurate visual acuity was not available for one child) in the FRMD7 group and five patients in the GPR143 group. The median logMAR visual acuity was −0.22 and −1.00 in these two groups of patients, respectively. The group with GPR143 mutations had worse vision than in the group with FRMD7 mutations (Mann–Whitney U-test, P = 0.0062 < 0.05). However, we did not see an obvious difference in patients' acuity between the FRMD7 group and non-FRMD7 group (Mann–Whitney U-test, P = 0.5279 > 0.05).
Only 1 case (11.11%) had esotropia in the FRMD7 group. By comparison, in the GPR143 group, one patient (20%) had exotropia, and two patients (40%) had ocular albinism (OA). No observable difference was found in the incidence of strabismus between the two groups (Fisher's exact test, P > 0.99). In the non-FRMD7 group, strabismus was identified in nine of 23 (39.13%) patients (five with esotropia, four with exotropia), and no significant difference was observed between the FRMD7 group and the non-FRMD7 group (Fisher's exact test, P = 0.21 > 0.05).
Seven of nine subjects were tested for stereopsis in the FRMD7 group, and one subject (14.29%) with by esotropia demonstrated no stereopsis by the TNO and Titmus tests. In the GPR143 group, two patients (100%) (test data were not available in the other three patients) with OA and exotropia were found to have no stereopsis in either TNO or Titmus test. In the non-FRMD7 group, 59.09% (13/22) and 54.55% (12/22) did not have stereopsis by TNO test and Titmus test, respectively, and 61.54% (8/13) and 58.33% (7/12) of which had manifested strabismus in the TNO and Titmus tests, respectively. Fisher's exact test was performed between the FRMD7 group and non-FRMD7 group (p = 0.08 > 0.05), and statistical comparison between the GPR143 group and FRMD7 group was not considered due to the meager data.
AHP was recorded in 7 (77.78%) patients in the FRMD7 group compared with 3 (75%) patients in the GPR143 group (Fisher's exact test, P > 0.99). Likewise, a similar proportion of AHP was achieved between the FRMD7 group (7/9, 77.78%) and the non-FRMD7 group (13/23, 56.52%) (Fisher's exact test, p = 0.42 > 0.05).
In addition to the subjects in our study, we also reviewed all samples as well as mutations in FRMD7 reported in literature and conducted correlation analysis on patients with CN.
A total of 110 patients were recorded in the literature. We conducted two sets of analysis depending on the mutation locations and types in the patients in terms of VA, onset age, and the proportion of AHP. One-way analysis of variance (ANOVA) was performed and there were no significant differences among the FERM domain group, FA domain group, and other domain group (Kruskal-Wallis test, p > 0.05; Pearson chi-square test, p > 0.05). Similarly, we did not observe any differences among the missense group, non-sense group, deletion/insertion group, and splice group (Kruskal-Wallis test, p > 0.05; Pearson chi-square test, p > 0.05).
In this study, a total of fourteen mutations (nine mutations in the FRMD7 gene and five mutations in the GPR143 gene) in 37.8% (14/37) of the probands were identified. Seven of them were new mutations.
The mutation detection rate differs significantly between the familial cases (87.5%, 7/8) and the sporadic cases (24.1%, 7/29), and is higher than published studies. These data further support that FRMD7 and GPR143 are two major causative genes for CN. Tarpey et al. (2006) first detected mutations in 84.6% (22/26) of the familial cases and 7% (3/42) of the sporadic cases. Zhang Q. et al. (2007) detected mutations in 4 of 14 (28.5%) Chinese families with X-linked nystagmus. Self et al. (2007) identified mutations in 20% (2 of 10) of apparent X-linked families and 3.6% (1/28) singleton cases. A total of 33.3% (7/21) familial and 14.3% (4/28) of sporadic cases had mutations. In 2015, 40% (2/5) of X-linked IN families were found to have mutations, while no mutations were identified in 15 sporadic cases (AlMoallem et al., 2015). The screening rate was 38.89% (7/18) in another Chinese cohort published in 2017 (Jia et al., 2017b). Unexpectedly, 41.7% (10/24) of simple cases had mutations, comparable to 30.8% (4/13) of syndromic cases in this study. This illustrates that mutation screening is equally necessary in both X-linked cases and sporadic cases.
The FRMD7 gene contains 12 exons and encodes a 714 amino acid protein that is composed of an N-terminal FERM domain, FERM-adjacent domain (FA), and C-terminal domain. The conserved FERM domain comprises three tightly folded clover-leaf structures (F1, F2, F3). Despite several studies, the specific functions of FRMD7 protein are still uncertain. However, the two closest homologous proteins FARP1 and FARP2 have been found to be involved in the regulation of neural development, indicating that FRMD7 may play a role in this process. A later study discovered that knockdown of FRMD7 can cause abnormalities in neurite development, partially confirming the above conjecture, thus, mutations in FRMD7 can influence neuronal functions, consequently leading to nystagmus (Betts-Henderson et al., 2010).
There are 98 mutations reported to date (Supplementary Excel 1 in Supplementary Material). Over half (51.02%) of the mutations are missense and are predicted to change the protein conformation, resulting in affecting the function of FRMD7. Non-sense, deletion/insertion, and splice mutations accounted for 6.12, 20.41, and 21.43%, respectively (Figure 3B). A significant number of mutations (80.6%) are primarily concentrated in the highly conserved FERM and FA domains (Figure 3C). In our study, 77.8% (7/9) were missense mutations, and all of them were located in the FERM and FA domains. Notably, the predicted molecular structural modeling of the c.284G>A mutation was similar with the normal protein. Lysine and arginine are both alkaline amino acids with similar molecular properties, but previous study has ever reported mutation of this locus causing nystagmus, and amino acids are well-conserved in the FERM domain, we therefore speculated that this variation of chemical bonds may affect the overall stability and functions of the protein (Bai et al., 2017). Of these 12 exons, exon 9 has been regarded as the mutational hot spot region, containing 21.4% of mutations (Figure 3A). Here, we found a higher proportion, with 4 (44.4%) mutations in exon 9. This indicates that alterations in this region severely affect the function of FRMD7. Remarkably, a new 10-bp deletion (c.2014_2023delTCACCCATGG) in exon 12 of C-terminal of FRMD7 was detected in S4 in our study. As a whole, ten mutations have been identified in exon 12, all of which are predicted to result in severe loss of function of FRMD7 due to the pre-mature termination of the protein. Only gross structural defects in this region may be responsible for the nystagmus phenotype. Therefore, we speculate that the specific biological function of this region may not be as important as the FERM and FA domains. In addition, we also noticed that c.781C>G and c.782G>A were most frequently reported in the Chinese population, and have not been found in other populations to date. c.782G>A were observed twice in 2 male probands of two unrelated families in our patients. The above findings to some extent suggest that these mutations are unique to Chinese people.
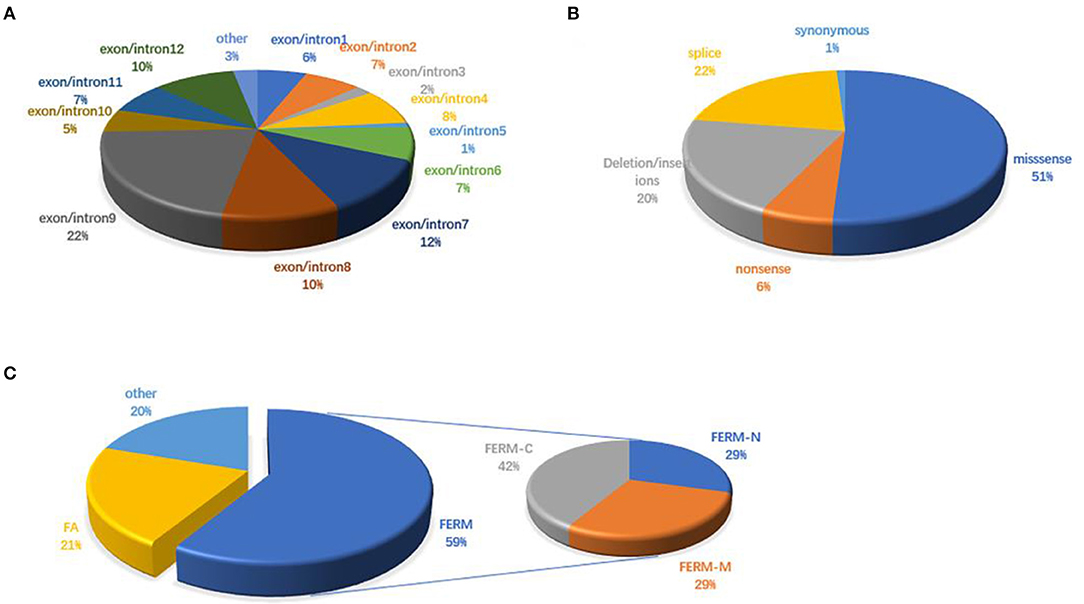
Figure 3. FRMD7 mutations published in the previous literature. (A) Distribution of FRMD7 mutations identified before on exon/introns. (B) Proportions of different types of FRMD7 mutations. (C) Distribution of FRMD7 mutations identified before on domains in the FRMD7 protein.
The GPR143 gene consists of 9 exons and encodes the G protein-coupled receptor 143 that is present on the membrane of melanosomes in pigment cells. Recent research has proven that GPR143 expression can be detected during the earliest stage of melanosome formation in the RPE. Mutations in GPR143 may cause isolated albinism in the eye or a series of other abnormalities, such as reduced visual acuity, nystagmus, and strabismus (Surace et al., 2000). The underlying pathogenic mechanisms of ocular abnormities caused by the GPR143 mutation have not been fully studied. It is well-known that melanin synthesis is disrupted in albinism, when this pathogenic condition occurs in the RPE, leading to severe defects in development and maintenance of vision eyes, including foveal hypoplasia, decreased numbers of photoreceptors and ganglion cells and misrouting of the optic tracts at the chiasm. However, mutations in GPR143, one of the genes associated with albinism, result in the impairment of visual pathway, but with intact melanin synthesis machinery (Schiaffino, 2010). This suggests that it is not the melanin synthesis and accumulation but the GPR143 signaling activity responsible for the developmental defects in retina. Researchers proposed that GPR143 signaling was the downstream of pigmentation, and all forms of albinism were dependent upon GPR143 signaling pathway to affect retinal development. Later studies found GPR143 signaling in RPE regulated the secretion of pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) and exosome release. All of these signaling activities were involved in the protection from retinal diseases. However, it remains unclear that how the exosome release influences the retina (Locke et al., 2014; McKay, 2019; Figueroa and McKay, 2020). Mutations in GPR143 contributing to the aberrant protein function might disrupt the GPR143 signaling pathway leading to the developmental visual disorder. It was concluded that mutations in GPR143 tend to cluster in exons 1-7; similarly, in this paper, all of these mutations in GPR143 were found in exons 1, 2, 4, and 6. These results suggested that exons 1-7 are the most frequently mutated regions in the GPR143 gene (Schnur et al., 1998; Fang et al., 2008).
However, several studies have reported that Chinese patients with GPR143 mutations manifested CN as the most dominant, consistent phenotype. Researchers have proposed that the typical symptoms of OA caused by GPR143 mutations are seldom observed in Asian populations, mainly because they have dark irises (Table 3) (Preising et al., 2001; Liu et al., 2007; Zhou et al., 2008; Peng et al., 2009; Xiao and Zhang, 2009; Hu et al., 2011; Gao et al., 2019). Three of five mutations (c.703G>A, c.733C>T, c.250+1G>C) in GPR143 were detected in three cases without ocular albinism in our study. Unlike the study here, c.703G>A was reported to cause typical ocular albinism, while patients with the c.733C>T mutation exhibited foveal hypoplasia in previous studies (Schnur et al., 1998; Kim et al., 2016). In addition, Janecke et al. (2012) reported a patient misdiagnosed with CN first the time who was examined to have slight hypopigmentation in the fundus and was finally identified as ocular albinism through genetic screening. Herein, we summarized all the mutations in GPR143 identified in Chinses populations and phenotypes of the probands. It is believed that nystagmus, foveal hypoplasia, and hypopigmentation in the fundus are all associated with GPR143 mutations (Table 3). We did not perform OCT to re-evaluate the three subjects more meticulously in our study due to the difficulty in children's examinations. Early and precise diagnosis is crucial to individual management and correct therapy. Several studies have shown that ultrahigh resolution OCT could detect minor differences in structural changes in the retina between albinism- and FRMD7- associated nystagmus (Thomas et al., 2014). Thus, comprehensive fundus examinations (OCT, fundus photograph) and molecular analysis are all required to distinguish between CN caused by FRMD7 mutations and atypical OA caused by GPR143 mutations more accurately. Targeted screening will be helpful to substantially increase the detection rate.
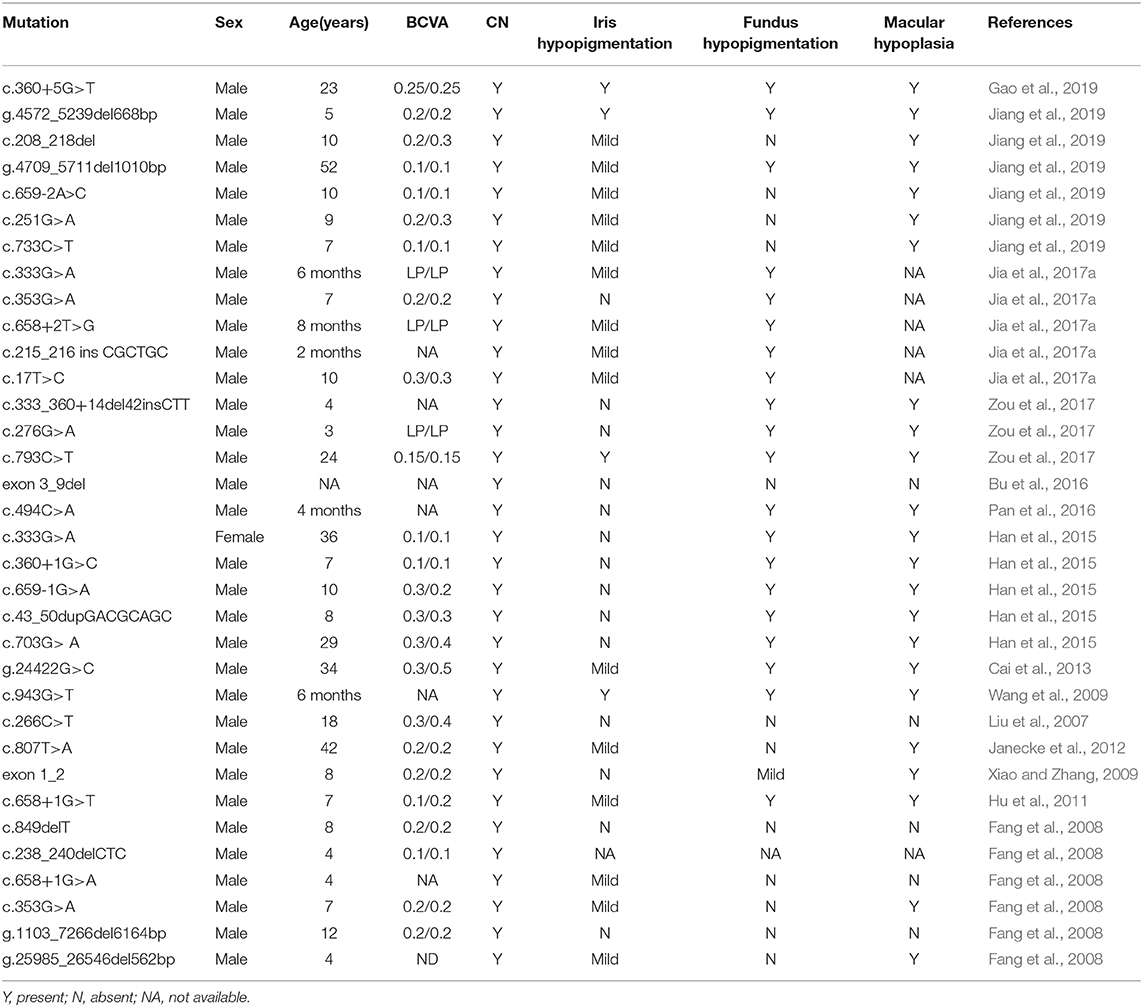
Table 3. Mutations identified in GPR143 and the clinical characteristics of the probands in the Chinese population.
We did witness a considerable difference in visual acuity between the FRMD7-group and the GPR143-group. This result was in keeping with previous observational studies in which patients with GPR143 mutations suffered more severe damage to vision, which may be explained by sensory defects in albinism-associated mechanisms, although this was not determined till now. GPR143 signaling might be essential for the process of RPE pigmentation protecting neurosensory retina. Although no typical albinism symptoms were observed, GPR143 mutation screening should be required for patients with poor visual function. In contrast to the earlier findings in 2011, comparisons of other clinical characteristics between the two groups are similar in our groups (Kumar et al., 2011).
Thomas et al. has described a lower proportion of AHP in the FRMD7 group than the non-FRMD7 group, which was not observed in our study. In addition, the remaining features of strabismus and stereopsis in the different groups here did not exhibit significant differences, which is compatible with previous studies (Thomas et al., 2008; Kumar et al., 2011). Noticeably, 100% of patients in FRMD7 group and 61.54% (8/13) by TNO test and 58.33% (7/12) by Titmus test in the non-FRMD7 group manifested strabismus in the group in which no stereopsis was recorded. This implies that strabismus has a strong adverse effect on binocular visual function. Excluding those with strabismus could be beneficial to improve the reliability of stereopsis phenotype analysis in different groups.
Unexpectedly, subjects with different mutation types exhibited no clear phenotypic-mutant link. In our study, we analyzed the clinical features of all the subjects reported before, but we did not conclude any differences among the non-sense, missense, deletion/insertion, and splice mutation groups. It seems that mutation types don not response to the severity of phenotype, and research by Thomas et al. (2008) also supported this finding. Other factors including the environment, modifier genes, and other unknown factors contributing to modulating the disease, may explain the phenotypic diversity.
In this research, we identified 14 mutations in two different genes, FRMD7 and GPR143, and seven of them are novel. However, we cannot exclude the possibility that mutations are present in non-coding regions of FRMD7 and GPR143. The combination of whole-genome and whole-exon sequencing is needed to identify novel candidate genes or variants in our unsolved cases.
However, several limitations in this study need to be acknowledged. First, it was difficult to obtain peripheral blood from some family members, and family segregation analysis was performed in only three families. Second, we did not re-evaluate patients without perfect clinical information, and the sample size may not be large enough, which might affect the credibility of the genotype–phenotype associations. Finally, the pathogenicity of new mutations was predicted by bioinformatic tools, and further experiments on cells and animals were not been performed in this study given the limited time and cost.
In conclusion, despite the limitations of this study, our research certainly provides another large-scale cohort with CN since the first four cohorts (Tarpey et al., 2006; Thomas et al., 2008; AlMoallem et al., 2015; Choi et al., 2018). For the first the time, we compared the phenotypes between groups with FRMD7 and GPR143 mutations and reviewed all the patients reported to explore the relationship of mutations and clinical characteristics. Here, 14 mutations were detected in 37.8% (14/37) of the probands (seven familiar cases, seven sporadic cases), seven of which are novel, demonstrating that mutations in FRMD7 and GPR143 appear to be two major causative factors in both X-linked and sporadic cases. Worse visual acuity was observed in patients with GPR143 mutations than in patients with FRMD7 mutations. These results broaden the mutation spectrum of FRMD7 and GPR143, and add to our knowledge on Chinese patients with congenital nystagmus, providing new insights for the strategy of precise diagnosis and genetic counseling of CN.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethics Committee of Eye Hospital of Wenzhou Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Z-BJ conceived, supervised the study, and provided funding supports. H-YY and HC evaluated clinical characteristics for the enrolled patients. X-FW, P-JH, Z-KF, Z-QH, XF, X-TX, FH, and R-JS carried out the experiments. X-FW performed the data analysis and wrote the manuscript. Z-BJ and YL revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Key R&D Program of China (2017YFB0403700), National Natural Science Foundation of China (81970838), Beijing Natural Science Foundation (Z200014), and Institutional Grant (YNCX201503 to H-YY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the subjects for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.627295/full#supplementary-material
AlMoallem, B., Bauwens, M., Walraedt, S., Delbeke, P., De Zaeytijd, J., Kestelyn, P., et al. (2015). Novel FRMD7 mutations and genomic rearrangement expand the molecular pathogenesis of X-linked idiopathic infantile nystagmus. Invest. Ophthalmol. Vis. Sci. 56, 1701–1710. doi: 10.1167/iovs.14-15938
Bai, D., Shi, W., Qi, Z., Li, W., Wei, A., Cui, Y., et al. (2017). Clinical feature and waveform in infantile nystagmus syndrome in children with FRMD7 gene mutations. Sci. China Life Sci. 60, 707–713. doi: 10.1007/s11427-017-9089-5
Betts-Henderson, J., Bartesaghi, S., Crosier, M., Lindsay, S., Chen, H.-L., Salomoni, P., et al. (2010). The nystagmus-associated FRMD7 gene regulates neuronal outgrowth and development. Hum. Mol. Genet. 19, 342–351. doi: 10.1093/hmg/ddp500
Brodsky, M. C., and Dell'Osso, L. F. (2014). A unifying neurologic mechanism for infantile nystagmus. JAMA Ophthalmol. 132, 761–768. doi: 10.1001/jamaophthalmol.2013.5833
Bu, J., Liu, J., Jia, Y., and Wang, L. (2016). A previously unidentified deletion in G protein-coupled receptor 143 causing X-linked congenital nystagmus in a Chinese family. Indian J. Ophthalmol. 64, 813–817. doi: 10.4103/0301-4738.195593
Cai, C. Y., Zhu, H., Shi, W., Su, L., Shi, O., Cai, C. Q., et al. (2013). A novel splicing site mutation of the GPR143 gene in a Chinese X-linked ocular albinism pedigree. Genet. Mol. Res. 12, 5673–5679. doi: 10.4238/2013.November.18.16
Choi, J.-H., Jung, J.-H., Oh, E. H., Shin, J.-H., Kim, H.-S., Seo, J. H., et al. (2018). Genotype and phenotype spectrum ofFRMD7-associated infantile nystagmus syndrome. Invest. Opthalmol. Vis. Sci. 59, 3181–3188. doi: 10.1167/iovs.18-24207
Dell'Osso, L. F. (2002). Development of new treatments for congenital nystagmus. Ann. N. Y. Acad. Sci. 956, 361–379. doi: 10.1111/j.1749-6632.2002.tb02834.x
Fang, S., Guo, X., Jia, X., Xiao, X., Li, S., and Zhang, Q. (2008). Novel GPR143 mutations and clinical characteristics in six Chinese families with X-linked ocular albinism. Mol. Vis. 14, 1974–1982.
Figueroa, A. G., and McKay, B. S. (2020). A G-protein coupled receptor and macular degeneration. Cells 9:910. doi: 10.3390/cells9040910
Forssman, B. (1971). Hereditary studies of congenital nystagmus in a Swedish population. Ann. Hum. Genet. 35, 119–138. doi: 10.1111/j.1469-1809.1956.tb01385.x
Gao, X., Liu, T., Cheng, X., Dai, A., Liu, W., Li, R., et al. (2019). A novel GPR143 mutation in a Chinese family with X-linked ocular albinism type 1. Mol. Med. Rep. 21, 240–248. doi: 10.3892/mmr.2019.10813
Han, R., Wang, X., Wang, D., Wang, L., Yuan, Z., Ying, M., et al. (2015). GPR143 gene mutations in five Chinese families with X-linked congenital nystagmus. Sci. Rep. 5:12031. doi: 10.1038/srep12031
Hertle, R. W., Yang, D., Adams, K., and Caterino, R. (2010). Surgery for the treatment of vertical head posturing associated with infantile nystagmus syndrome: results in 24 patients. Clin. Exp. Ophthalmol. 39, 37–46. doi: 10.1111/j.1442-9071.2010.02380.x
Hu, J., Liang, D., Xue, J., Liu, J., and Wu, L. (2011). A novel GPR143 splicing mutation in a Chinese family with X-linked congenital nystagmus. Mol. Vis. 17, 715–722.
Janecke, A. R., Yan, N., Liao, X., Cai, S. P., Lan, C., Wang, Y., et al. (2012). A novel nonsense mutation of the GPR143 gene identified in a Chinese pedigree with ocular albinism. PLoS ONE 7:e43177. doi: 10.1371/journal.pone.0043177
Jia, X., Yuan, J., Jia, X., Ling, S., Li, S., and Guo, X. (2017a). GPR143 mutations in Chinese patients with ocular albinism type 1. Mol. Med. Rep. 15, 3069–3075. doi: 10.3892/mmr.2017.6366
Jia, X., Zhu, X., Li, Q., Jia, X., Li, S., and Guo, X. (2017b). Novel mutations of FRMD7 in Chinese patients with congenital motor nystagmus. Mol. Med. Rep. 16, 1753–1758. doi: 10.3892/mmr.2017.6824
Jiang, J., Yang, L., Li, H., Huang, L., and Li, N. (2019). Evaluation of the iris thickness changes for the Chinese families with GPR143 gene mutations. Exp. Eye Res. 189:107819. doi: 10.1016/j.exer.2019.107819
Kim, U. S., Cho, E., and Kim, H. J. (2016). A novel nonsense mutation of GPR143 gene in a Korean kindred with X-linked congenital nystagmus. Int. J. Ophthalmol. 9, 1367–1370. doi: 10.18240/ijo.2016.09.25
Kumar, A., Gottlob, I., McLean, R. J., Thomas, S., Thomas, M. G., and Proudlock, F. A. (2011). Clinical and oculomotor characteristics of albinism compared toFRMD7 associated infantile nystagmus. Invest. Opthalmol. Vis. Sci. 52, 2306–2313. doi: 10.1167/iovs.10-5685
Li, N., Wang, L., Cui, L., Zhang, L., Dai, S., Li, H., et al. (2008). Five novel mutations of the FRMD7 gene in Chinese families with X-linked infantile nystagmus. Mol. Vis. 14, 733–738.
Liu, J. Y., Ren, X., Yang, X., Guo, T., Yao, Q., Li, L., et al. (2007). Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J. Hum. Genet. 52, 565–570. doi: 10.1007/s10038-007-0152-3
Locke, C. J., Congrove, N. R., Dismuke, W. M., Bowen, T. J., Stamer, W. D., and McKay, B. S. (2014). Controlled exosome release from the retinal pigment epithelium in situ. Exp. Eye Res. 129, 1–4. doi: 10.1016/j.exer.2014.10.010
McKay, B. S. (2019). Pigmentation and vision: is GPR143 in control? J. Neurosci. Res. 97, 77–87. doi: 10.1002/jnr.24246
Pan, Q., Yi, C., Xu, T., Liu, J., Jing, X., Hu, B., et al. (2016). A novel mutation, c.494C>A (p.Ala165Asp), in the GPR143 gene causes a mild phenotype in a Chinese X-linked ocular albinism patient. Acta Ophthalmol. 94, 417–418. doi: 10.1111/aos.12854
Papageorgiou, E., McLean, R. J., and Gottlob, I. (2014). Nystagmus in childhood. Pediatr. Neonatol. 55, 341–351. doi: 10.1016/j.pedneo.2014.02.007
Peng, Y., Meng, Y., Wang, Z., Qin, M., Li, X., Dian, Y., et al. (2009). A novel GPR143 duplication mutation in a Chinese family with X-linked congenital nystagmus. Mol. Vis. 15, 810–814.
Preising, M., Op de Laak, J. P., and Lorenz, B. (2001). Deletion in the OA1 gene in a family with congenital X linked nystagmus. Br. J. Ophthalmol. 85, 1098–1103. doi: 10.1136/bjo.85.9.1098
Ren, J., Wen, L., Gao, X., Jin, C., Xue, Y., and Yao, X. (2009). DOG 1.0: illustrator of protein domain structures. Cell Res. 19, 271–273. doi: 10.1038/cr.2009.6
Richards, M. D., and Wong, A. (2015). Infantile nystagmus syndrome: clinical characteristics, current theories of pathogenesis, diagnosis, and management. Can. J. Ophthalmol. 50, 400–408. doi: 10.1016/j.jcjo.2015.07.010
Sarvananthan, N., Surendran, M., Roberts, E. O., Jain, S., Thomas, S., Shah, N., et al. (2009). The prevalence of nystagmus: the leicestershire nystagmus survey. Invest. Opthalmol. Vis. Sci. 50, 5201–5206. doi: 10.1167/iovs.09-3486
Schiaffino, M. V. (2010). Signaling pathways in melanosome biogenesis and pathology. Int. J. Biochem. Cell Biol. 42, 1094–1104. doi: 10.1016/j.biocel.2010.03.023
Schnur, R. E., Gao, M., Wick, P. A., Keller, M., Benke, P. J., Edwards, M. J., et al. (1998). OA1 mutations and deletions in X-linked ocular albinism. Am. J. Hum. Genet. 62, 800–809. doi: 10.1086/301776
Self, J., Shawkat, F., Malpas, C., Thomas, N., Harris, C., Hodgkins, P., et al. (2007). Allelic variation of the FRMD7 gene in congenital idiopathic nystagmus. Arch. Ophthalmol. 125, 1255–1263. doi: 10.1001/archopht.125.9.1255
Song, F. W., Chen, B. B., Sun, Z. H., Wu, L. P., Zhao, S. J., Miao, Q., et al. (2013). Novel mutation c.980_983delATTA compound with c.986C>A mutation of the FRMD7 gene in a Chinese family with X-linked idiopathic congenital nystagmus. J. Zhejiang Univ. Sci. B 14, 479–486. doi: 10.1631/jzus.B1200259
Surace, E. M., Angeletti, B., Ballabio, A., and Marigo, V. (2000). Expression pattern of the ocular albinism type 1 (Oa1) gene in the murine retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 41, 4333–4337. doi: 10.1097/00004397-200040010-00021
Tarpey, P., Thomas, S., Sarvananthan, N., Mallya, U., Lisgo, S., Talbot, C. J., et al. (2006). Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat. Genet. 38, 1242–1244. doi: 10.1038/ng1893
Thomas, M. G., Crosier, M., Lindsay, S., Kumar, A., Araki, M., Leroy, B. P., et al. (2014). Abnormal retinal development associated with FRMD7 mutations. Hum. Mol. Genet. 23, 4086–4093. doi: 10.1093/hmg/ddu122
Thomas, S., Proudlock, F. A., Sarvananthan, N., Roberts, E. O., Awan, M., McLean, R., et al. (2008). Phenotypical characteristics of idiopathic infantile nystagmus with and without mutations in FRMD7. Brain 131, 1259–1267. doi: 10.1093/brain/awn046
Wang, Y., Guo, X., Wei, A., Zhu, W., Li, W., and Lian, S. (2009). Identification of a novel mutation in a Chinese family with X-linked ocular albinism. Eur. J. Ophthalmol. 19, 124–128. doi: 10.1177/112067210901900118
Watkins, R. J., Patil, R., Goult, B. T., Thomas, M. G., Gottlob, I., and Shackleton, S. (2013). A novel interaction between FRMD7 and CASK: evidence for a causal role in idiopathic infantile nystagmus. Hum. Mol. Genet. 22, 2105–2118. doi: 10.1093/hmg/ddt060
Watkins, R. J., Thomas, M. G., Talbot, C. J., Gottlob, I., and Shackleton, S. (2012). The role of FRMD7 in idiopathic infantile nystagmus. J. Ophthalmol. 2012, 1–7. doi: 10.1155/2012/460956
Xiao, X., and Zhang, Q. (2009). Iris hyperpigmentation in a Chinese family with ocular albinism and theGPR143mutation. Am. J. Med. Genet., Part A 149A, 1786–1788. doi: 10.1002/ajmg.a.32818
Zhang, B., Liu, Z., Zhao, G., Xie, X., Yin, X., Hu, Z., et al. (2007). Novel mutations of the FRMD7 gene in X-linked congenital motor nystagmus. Mol. Vis. 13, 1674–1679.
Zhang, Q., Xiao, X., Li, S., and Guo, X. (2007). FRMD7 mutations in Chinese families with X-linked congenital motor nystagmus. Mol. Vis. 13, 1375–1378.
Zhao, H., Huang, X.-F., Zheng, Z.-L., Deng, W.-L., Lei, X.-L., Xing, D.-J., et al. (2016). Molecular genetic analysis of patients with sporadic and X-linked infantile nystagmus. BMJ Open 6:e010649. doi: 10.1136/bmjopen-2015-010649
Zhou, P., Wang, Z., Zhang, J., Hu, L., and Kong, X. (2008). Identification of a novel GPR143 deletion in a Chinese family with X-linked congenital nystagmus. Mol. Vis. 14, 1015–1019.
Keywords: congenital nystagmus, FRMD7, GPR143, mutation, genotype-phenotype
Citation: Wang X-F, Chen H, Huang P-J, Feng Z-K, Hua Z-Q, Feng X, Han F, Xu X-T, Shen R-J, Li Y, Jin Z-B and Yu H-Y (2021) Genotype-Phenotype Analysis and Mutation Spectrum in a Cohort of Chinese Patients With Congenital Nystagmus. Front. Cell Dev. Biol. 9:627295. doi: 10.3389/fcell.2021.627295
Received: 09 November 2020; Accepted: 06 January 2021;
Published: 19 February 2021.
Edited by:
Minzhong Yu, Case Western Reserve University, United StatesReviewed by:
Mousumi Mutsuddi, Banaras Hindu University, IndiaCopyright © 2021 Wang, Chen, Huang, Feng, Hua, Feng, Han, Xu, Shen, Li, Jin and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huan-Yun Yu, aHVhbnl1bnl1QDEyNi5jb20=; Zi-Bing Jin, amluemliaW5nQGZveG1haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.