 Alvaro A. Elorza
Alvaro A. Elorza Juan Pablo Soffia
Juan Pablo Soffia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 22 February 2021
Sec. Cellular Biochemistry
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.625020
This article is part of the Research TopicMitochondrial Remodeling and Dynamic Inter-Organellar Contacts in Cardiovascular PhysiopathologyView all 12 articles
The most common aging-associated diseases are cardiovascular diseases which affect 40% of elderly people. Elderly people are prone to suffer aging-associated diseases which are not only related to health and medical cost but also to labor, household productivity and mortality cost. Aging is becoming a world problem and it is estimated that 21.8% of global population will be older than 65 years old in 2050; and for the first time in human history, there will be more elderly people than children. It is well accepted that the origin of aging-associated cardiovascular diseases is mitochondrial dysfunction. Mitochondria have their own genome (mtDNA) that is circular, double-stranded, and 16,569 bp long in humans. There are between 500 to 6000 mtDNA copies per cell which are tissue-specific. As a by-product of ATP production, reactive oxygen species (ROS) are generated which damage proteins, lipids, and mtDNA. ROS-mutated mtDNA co-existing with wild type mtDNA is called mtDNA heteroplasmy. The progressive increase in mtDNA heteroplasmy causes progressive mitochondrial dysfunction leading to a loss in their bioenergetic capacity, disruption in the balance of mitochondrial fusion and fission events (mitochondrial dynamics, MtDy) and decreased mitophagy. This failure in mitochondrial physiology leads to the accumulation of depolarized and ROS-generating mitochondria. Thus, besides attenuated ATP production, dysfunctional mitochondria interfere with proper cellular metabolism and signaling pathways in cardiac cells, contributing to the development of aging-associated cardiovascular diseases. In this context, there is a growing interest to enhance mitochondrial function by decreasing mtDNA heteroplasmy. Reduction in mtDNA heteroplasmy is associated with increased mitophagy, proper MtDy balance and mitochondrial biogenesis; and those processes can delay the onset or progression of cardiovascular diseases. This has led to the development of mitochondrial therapies based on the application of nutritional, pharmacological and genetic treatments. Those seeking to have a positive impact on mtDNA integrity, mitochondrial biogenesis, dynamics and mitophagy in old and sick hearts. This review covers the current knowledge of mitochondrial physiopathology in aging, how disruption of OXPHOS or mitochondrial life cycle alter mtDNA and cardiac cell function; and novel mitochondrial therapies to protect and rescue our heart from cardiovascular diseases.
The world is aging at a very high speed and with it, the aging-associated diseases are going up. According to Lutz et al. (2008), the proportion of the global population of 60 + years was 10% in 2000 but is projected to be 21.8% in 2050 and then to 32.2% in 2100. Current projections on aging show that in 2050 there will be more elderly people (65+) than children (0-14 years old) for the first time in human history. This is enormous pressure for the health care system and for the economy of any country because older adults are affected by several and diverse chronic diseases - aging-associated diseases- that contribute to disability, diminish the quality of life and increase health- and long term- care costs. The cost of aging-associated diseases is not only related to medical care but also to labor and household productivity losses and mortality costs. Furthermore, old people in many cases are affected by a combination of diseases which increases the cost even more. Older people are more susceptible to get ill when they work or live under environmental risk factors such as air pollution, tobacco smoke, particulate matter and ozone and/or under low quality of life risk factors such as sedentarism and bad food quality, leading to obesity. This negative environmental exposure may cause or exacerbate respiratory and cardiovascular diseases (Van Houtven et al., 2008). Cardiovascular diseases are the leading cause of death among older people reaching up to 40% of deaths in people over 65 years old (Van Houtven et al., 2008; Ginneken, 2017). Therefore, new heart failure treatments are of high relevance to improve the life quality and lifespan of older people and also to decrease the pressure and costs on the public health system. Working on new methodologies and technologies will make a contribution to expediting and advancing drug discovery and genetic therapies in this area.
Our heart is a wonderful organ built at the beginning of our existence, during embryonic development, and never ever stops working until we die. It is a perfect machine that pumps, day and night, blood into all tissues, resting only between beats. Furthermore, if we exercise or have an increase in oxygen demand, our heart beats even harder to satisfy tissue demands. It has been estimated that our heart beats 36 million times per year and to support such a high load of work, our heart must be able to produce an enormous amount of ATP that has been estimated in about 30-40 Kg per day (Ferrari et al., 2003) which is about one-third of total ATP produced by our body (a normal individual produces 90-100 Kg of ATP daily). Cardiac cells are contractile striated cells with an average render cell volume of 30.5 pL (picoLiters) and 140 μm × 32 μm × 13 μm (LxWxD) average dimension in mammals. The cardiac cell capacitance, which is proportional to the amount of membrane and membrane invaginations, varies among different mammal species between 138 and 300 pF (picoFaraday). The amount of membrane invaginations is explained by the presence of T-tubules (50% membrane), endoplasmic reticulum and mitochondria which are all involved in cardiac cell contraction (Satoh et al., 1996). The ultrastructural analysis of cardiac cells also reveals that the volume density of mitochondria is over 25.3%, the myofibrils 52.3% and the cytoplasm 22.3%. One-third of the cardiac volume is filled with mitochondria (Barth et al., 1992); although Huang et al. (2013) estimate mitochondrial volume to be up to 40%.
Mitochondria are cellular organelles that are well known as the powerhouse of the cell in charge of satisfying ATP demands. However, today, mitochondria are also acknowledged as a metabolic hub that integrates intracellular signaling to elaborate and execute a cellular response to adapt cell metabolism to external or internal environmental changes, taking primary action in cell fate determination. Fit and healthy mitochondria will be able to satisfy the heart’s energy demands, but dysfunctional mitochondria won’t, therefore a cardiac failure is expected.
In general, for all of our cells (excluding erythrocytes that do not have mitochondria) two convoluted mitochondrial systems work collaboratively to maintain a healthy mitochondrial population. The Oxidative Phosphorylation (OXPHOS) system and the Mitochondrial Quality Control System also known as the Mitochondrial Life Cycle (MLC). Together they complement each other to keep mitochondrial function i.e., energy production (ATP), reactive oxygen species (ROS) detoxification, proper metabolism and retrograde signaling (Figure 1).
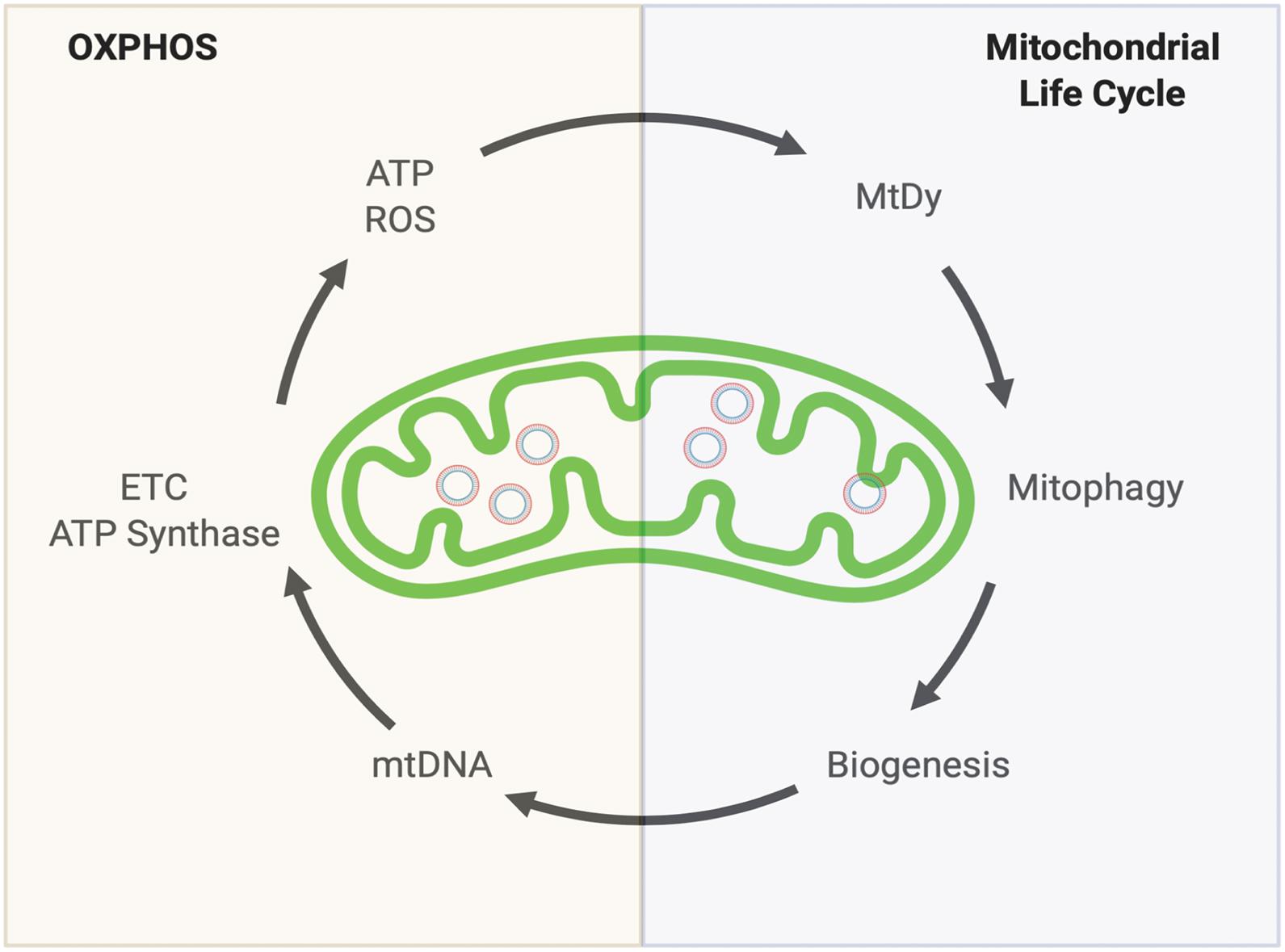
Figure 1. Keeping a healthy mitochondrial population. Mitochondria’s health depends on two complementary systems. The first system is the OXPHOS. This will provide energy (ATP) and reactive oxygen species (ROS) which are both necessary to drive the second system: The Mitochondrial Life cycle. Through mitochondrial fusion and fission events (Mitochondrial dynamics, MtDy), mitochondria are segregated to be eliminated by mitophagy. The induction of mitophagy is coupled with mitochondrial biogenesis which involves mitochondrial DNA (mtDNA) replication. mtDNA encodes for 11 respiratory complex subunits (7 subunits for complex I, 1 for complex III, 3 for complex IV) plus 2 subunits of the ATP synthase that will allow OXPHOS assembly to keep going the virtuous circle. Any disruption or delay in this running circle will end up in the loss of energy, mutations in mtDNA, decrease in mtDNA copy number and increased ROS leading to mitochondrial dysfunction, cell death, aging and aging-associated diseases.
OXPHOS is made of the Electron Transport Chain (ETC) to generate the proton motive force (mitochondrial membrane potential) and the ATP synthase that utilizes the proton motive force to generate ATP. The ETC or respiratory chain is formed by 4 respiratory complexes plus the ubiquinone and the cytochrome c, located in the inner mitochondrial membrane that forms cristae. OXPHOS is fed by NADH at the level of complex I and FADH2 at the level of complex II. Both reductant equivalents are mostly generated by the Krebs (TCA) cycle which in turn is dependent on calcium and respiratory substrates such as carbohydrates and lipids. As a byproduct of the respiratory chain, the superoxide radical (a ROS molecule) is generated, which has been involved in aging and aging-associated diseases. In this way, a very simplistic OXPHOS equation is:
The OXPHOS system is encoded by two independent genetic material: nuclear DNA and mitochondrial DNA (mtDNA). The mtDNA is a plasmid DNA of 16 kb (circular, closed), doubled stranded and codes for 37 genes (13 OXPHOS protein-encoding mRNA, 2 rRNA and 22 tRNA). It is composed of a heavy strand and a light strand based on the proportion of heavier nucleotides (adenine and guanine). It also has a large non-coding region (around 1 kb) that contains the regulatory elements for the initiation and termination of the transcription of both strands. The D-Loop (Displacement Loop) or control region, is within this non-coding zone and contains the origin of replication of the heavy strand and high-variability zones called HVRI, HVR II and HVR III where most of the polymorphisms associated with mtDNA occur. Each molecule of mtDNA is packaged in structures called nucleoids made of proteins involved in their replication and transcription, such as TFAM. Nucleoids are located in the mitochondrial matrix associated with the internal membrane and the oxidative phosphorylation system (Brown et al., 2011).
Mitochondria have their own genome is maternally inherited and this configures each individual with their own maternal surname - called a haplogroup. All mitochondrial haplogroups derive from a common ancestor, from a black African woman, called “The Mitochondrial Eve” from 200,000 years ago (Cann et al., 1987). At present, a total of 20 haplogroups are recognized, each with its respective subdivisions, which were established by the acquisition of mutations and natural selection, according to the patterns of migration and human settlement, and fixed in each of the different ethnic groups. This is how the haplogroup J, D4a, D4b2b, D5 have been associated with longevity; the H5 and United Kingdom, at risk of suffering from Alzheimer’s disease; the U, with a predisposition to psychosis in bipolar disorders; and others associated with resistance to altitude, with athletic performance, predisposition to diabetes, cancer or cardiovascular diseases (Hiona, 2008; Lakatos et al., 2010; Nishigaki et al., 2010; Picard et al., 2016; Chocron et al., 2019). Knowing that the haplogroup confers adaptive advantages and/or disease propensity and, therefore, their identification can also become a tool of preventive diagnosis that can be applied since the birth of a new being.
mtDNA is prone to oxidative or replicative damage. Each mitochondrion has 3 to 10 mtDNA copies which undergo mutations by either the reactive oxygen species (ROS) generated as a byproduct of electron transfer chain due to electron leak mainly from respiratory complex I and III (Brand et al., 2013) or alterations in the mtDNA replication or repair. A higher mutation load in mtDNA means lower OXPHOS functionality giving rise to more ROS and more mtDNA mutations in a vicious cycle. The coexistence of wild type mtDNA and mutated mtDNA in the same mitochondrion is known as heteroplasmy. As the percentage of mutant mtDNA rises above certain thresholds (60-70%), cellular homeostatic mechanisms are disrupted, leading to mitochondrial dysfunction, cellular atrophy, and death, contributing to age-related diseases. Heteroplasmy also leads to broad changes in gene expression that can shift abruptly as the percentage of mutant mtDNA increases (Lakshmanan et al., 2018). To date, more than 400 mtDNA mutations have been associated with human diseases (Li et al., 2010).
The mitochondrial life cycle involves three processes: mitochondrial dynamics, mitochondrial selective autophagy (mitophagy) and mitochondrial biogenesis. Through MtDy, mitochondria share or dilute components and also segregate dysfunctional mitochondrial units for degradation by mitophagy (Legros et al., 2002; Liesa et al., 2009). While mitochondria are being degraded, mitochondrial biogenesis is turned on to maintain mitochondrial population and homeostasis (Palikaras et al., 2015a; Sin et al., 2016). In addition, mitochondrial biogenesis responds to energy demands and environmental factors such as cold and hypoxia (Tohme et al., 2017; Gureev, 2019).
Mitochondrial Dynamics (MtDy) refers to the ability of two mitochondria to fuse each other (fusion) or one mitochondrion to divide into two daughter mitochondria (fission). Mitochondrial morphology and size are dependent on mitochondrial fusion and fission events. Importantly, changes in MtDy regulate bioenergetics outputs including respiratory rate, energy expenditure and ATP synthesis as well as apoptosis and the segregation and elimination of dysfunctional mitochondrial units by mitophagy (Bénard et al., 2007; Sheridan and Martin, 2010; Westermann, 2012). Mitochondrial fission is dependent on the protein DRP1 that has been recognized as a mitochondrial fission promoter (Smirnova et al., 2001). It is located in the cytosol, but translocate to mitochondria through its binding to the mitochondrial receptor proteins FIS1, MFF, MiD49, and MiD51 (Losón et al., 2013), which are all located in the mitochondrial outer membrane (MOM). Mitochondrial fusion is dependent on MFN1 and MFN2, located in the MOM, and OPA1, which is located in the mitochondrial inner membrane (Liesa et al., 2009). A fusion event is often followed by a fission event generating one hyperpolarized and one depolarized daughter mitochondrion. The depolarized unit loses OPA1, MFN1, and MFN2 and thus its ability to fuse, becoming a target for degradation by mitophagy; while the hyperpolarized unit remains in the cytosol and can fuse again. In this way, cells keep a more active, healthier and functional mitochondrial web (Twig et al., 2008).
Mitochondrial Selective Autophagy or Mitophagy is a type of macroautophagy that delivers mitochondria to lysosomes for degradation. It is essential for the recycling and protective process of mitochondria to maintain cellular homeostasis. Mitophagy initiates with the formation of a double-membrane phagophore which elongates and closes to generate a mature, double-membrane autophagosome that engulfs mitochondria. Then, autophagosomes are fused to lysosomes for content degradation (Youle and Narendra, 2011). Mitophagy allows the removal of dysfunctional, depolarized and/or damaged mitochondria for mitochondrial turnover, ROS management, and programmed mitochondrial clearance, as seen in erythropoiesis (Schweers et al., 2007), lens differentiation (Bassnett, 2002), and mature T-lymphocytes (Pua et al., 2009). Mitophagy is also essential for embryonic development by removal of paternal sperm mitochondria from the fertilized eggs, leaving only the maternal ones (Rojansky et al., 2016; Pickles et al., 2018). In this regard, cells display basal mitophagy, stress-induced mitophagy and programmed mitophagy, being the cardiac muscle one of the tissues with enhanced basal mitophagy (Palikaras et al., 2018).
The process of mitophagy requires two systems: The autophagic core machinery, which is common to all types of autophagy; and the mitochondrial receptors and adaptors, a specific set of mitochondrial and/or cytosolic proteins needed for the assembly of mitochondria with the autophagic core machinery. The Autophagic Core Machinery is composed of ATG proteins that are found from yeast to mammals, indicating that autophagy is an evolutionarily conserved process. Phagophore initiation and formation is dependent on the ATG16L1 complex, made of ATG16L1, ATG5, and ATG12 which localize to the isolation membrane and dissociates from it upon the completion of autophagosome formation; and ATG8/LC3, which upon the formation of autophagosome, remains in on both sides—inside and outside of the double-membrane structure. While in yeast there is a single ATG8 protein, in mammals there are seven orthologs of ATG8 named LC3A, LC3B, LC3C, GABARAP, GABARAPL1, GABARAPL2, GABARAPL3 (thereafter they will be called LC3) suggesting a complex diversification of their function. LC3/ATG8 can be found in two isoforms: LC3-I and LC3-II. LC3-I is the inactive form that is constitutively expressed and found in the cytosol. Upon induction of autophagy, LC3-I is converted to LC3-II by site-specific proteolysis and lipidation near to the C-terminus to generate the autophagosome. LC3 is considered a reliable marker for on-going autophagy (Ashrafi and Schwarz, 2013; Lee and Lee, 2016).
The Mitochondrial Receptors interact directly or indirectly through adaptors, with LC3 via the LC3-interacting Region (LIR), a tetrapeptide sequence W/YXXL/I. Independent and non-redundant mitophagy pathways have been defined according to the mitochondrial receptors and adaptors that are involved in it. Those are (i) Ubiquitin-Dependent Mitophagy, with the PINK1-PARKIN protein axis. PINK1 (PTEN-induced putative protein kinase 1) is a mitochondrial protein whose import is dependent on mitochondrial membrane potential. Once imported, PINK1 is degraded by mitochondrial proteases. However, if mitochondria become depolarized, PINK1 is no longer imported and accumulates on the mitochondrial surface to phosphorylate ubiquitin and Mfn2, allowing the recruitment of the E3 ubiquitin ligase PARKIN from the cytosol. PARKIN mediates a hyper-ubiquitination of the mitochondrial outer membrane proteins, which are recognized by ubiquitin-binding adaptors such as P62/SQSTM1, OPTN, TAX1BP1, NDP52, NBR1, TOLLIP and HDAC6. These adaptor proteins interact and bind LC3 by means of their LIR domain. Of note, P62/SQSTM1 has been also used as a reliable indicator of the mitophagic flux, because under mitophagic defects it accumulates in response to stress stimuli (Xu et al., 2015; Yamaguchi et al., 2016). This PINK/PARKIN mitophagy is the canonical pathway for mitochondrial turnover and mitochondrial quality control. (ii) Ubiquitin-Independent Mitophagy is dependent on mitochondrial receptors interacting directly with LC3 through the LIR domain. Several and diverse mitochondrial LC3 receptors have been discovered, which also account for the complexity of mitophagy. NIX, BNIP3 (Zhang and Ney, 2009), FUNDC (Liu et al., 2012), FKBP8 (Bhujabal et al., 2017; Yoo et al., 2020), NLRX1 (Zhang et al., 2019) are located in the MOM; Prohibitins (Wei et al., 2017), in the MIM and, MsrB2 in the mitochondrial matrix (Lee et al., 2019). Not only proteins may bind LC3, but also the membrane phospholipid cardiolipin (Chu et al., 2013) mainly found in the MIM. NIX protein is highly expressed in erythroid precursors and is needed for erythropoiesis (Novak et al., 2009). Also, NIX, BNIP3 and FUNDC trigger mitophagy in response to hypoxia (Sowter et al., 2001; Bellot et al., 2009; Liu et al., 2012), Prohibitins, MsrB2 and cardiolipin could be accessible to cytosolic proteins if MOM or MIM are disrupted.
Mitochondrial dynamics and the mitophagy pathways with their mitochondrial receptors, adaptors and regulator proteins that localize to mitochondria, seem to have not redundant functions and might act cooperatively, providing multiple mechanisms to clear out damaged mitochondria under different conditions. In fact, the mitophagy protein FUNDC1 has been shown to interact with the MtDy proteins DRP1 or OPA1 to coordinate mitochondrial fission or fusion and mitophagy. Upon mitochondrial stress, FUNDC1-OPA1 interaction is dismissed, promoting DRP1 translocation to mitochondria for fission and a new association FUNDC1-DRP1 for mitophagy. This reveals the complexity and importance that MtDy and Mitophagy play for cellular physiology.
Mitochondrial Biogenesis: When mitochondria are eliminated by mitophagy or when more mitochondria are needed to meet energy demands, the mitochondrial biogenesis program is started. Interestingly, mitochondrial biogenesis involves four independent processes: mtDNA replication, mtDNA transcription, protein translation and mitochondrial membrane biogenesis, which are not necessarily synchronized adding more complexity and regulation capabilities to mitochondria. Unfortunately, the mechanisms controlling these three processes are not fully understood or discovered. In fish acclimated to cold, it was shown that mitochondria are able to remodel differentially according to ATP demands, oxygen demands, or both. Under oxygen demands, membrane biogenesis is upregulated increasing the mitochondrial volume/mass but not OXPHOS protein densities. Having an extra membrane improves oxygen diffusion since oxygen diffuses easily and faster in lipidic than in an aqueous environment. Under ATP demand, mitochondria increase the OXPHOS densities without increasing their volume. In the case of ATP and oxygen demands, both protein density and membrane are increased (O’Brien, 2010). On the other hand, it has been reported that mtDNA replication is controlled by oxidative damage in the D-loop region which favors TFAM (a mitochondrial transcription factor) binding to mtDNA for replication. This oxidative damage may be caused by hypoxia, linking the lack of oxygen to mtDNA replication (Pastukh et al., 2016).
The aforementioned mitochondrial features for OXPHOS and mitochondrial life cycle are general for all mitochondria. But what is distinctive for heart mitochondria? (Table 1). Heart striated cells are special and different from other cell types. They have a particular shape and contract throughout our lives from early development until we die. Heart cells demand an enormous amount of energy and then mitochondria must do the work. Mitochondria in cardiac cells have been subdivided into interfibrillar, subsarcolemmal and perinuclear mitochondria, according to their distribution in the cell; and are tightly packed between myofibrils with almost no chance of free or flexible movement. However, Hendgen-Cotta et al. (2018) claimed that all cardiac mitochondria in mice are interconnected and, therefore, they are not different populations. Cardiac mitochondria seen under transmission electron microscopy look well organized to each other and actually mitochondrial cristae seem to be aligned between two adjacent mitochondria. Three-dimensional reconstruction of murine heart mitochondria has shown that they have an oval but irregular shape. Changes in mitochondrial morphology have been associated with metabolic and bioenergetic reprogramming. The heart is not the exception since most of the cardiopathies are associated with alteration in mitochondrial morphology. Fragmented, swollen, reduced mitochondrial number, reduced cristae densities are the main morphological features of human heart failure which are nicely tabulated in Daghistani et al. (2018). Interestingly, different cardiac pathologies have shown distinct morphological mitochondria and cristae patterns. Heart mitochondrial cristae are very long, mostly going all throughout the mitochondrial matrix. In comparison, cristae length in non-cardiac mitochondria is about 1/3-1/4 of mitochondrial width. Cristae hold the OXPHOS system and considering the length of cristae and the electrodense nature of mitochondrial matrix seen by TEM (in accordance with condense mitochondrial state and opposed to orthodox one), it is possible to suggest that cardiac mitochondria are fully packed with OXPHOS having a highly oxidative metabolism. In fact, nice work from Williams et al. (2018) analyzed the mitochondrial proteome from 5 different mouse tissues and showed that in heart tissues half of the total proteome (55%) correspond to mitochondrial proteins, which was higher than brain, liver, skeletal muscle and brown adipose tissue. When looking at OXPHOS protein expression, cardiac mitochondria displayed the highest OXPHOS protein density for both all ETC respiratory complexes and the ATP synthase as compared with other tissues.

Table 1. Summary of main characteristics found in mitochondria from non-cardiac cells, cardiomyocytes and aged cardiomyocytes.
The super oxidative metabolism of cardiac cells is explained by the use of triacylglycerols and fatty acids as the main oxidative fuels, followed by glucose and other carbohydrates, which enter to the TCA cycle-OXPHOS to produce 80-90% of total ATP. The relative contribution of fat and carbohydrate to energy provision for the heart is 70% and 30%, respectively (Stoll et al., 2018) (Taegtmeyer et al., 2016). Furthermore, OXPHOS in cardiac mitochondria is equipped with specific subunit isoforms for the cytochrome c oxidase (complex IV) to make the ETC more efficient (Sinkler et al., 2017). The assembly of respiratory complexes in supercomplexes or respirosomes, reported for bovine, ovine and porcine hearts (Letts et al., 2016; Sousa et al., 2016) plus post-translational modifications in respiratory complexes such as O-GlcNAcylation, the ability to increase calcium uptake, and de-sensitize the opening of the mPTP can also improve OXPHOS efficiency (Ma et al., 2015; Stoll et al., 2018). OXPHOS efficiency is not exclusively dependent on OXPHOS proteins but also on other proteins involved in the bioenergetic capacity of cardiac cells. This is the case of the Adenine Nucleotide Transporter (ANT) protein, which is nuclear-encoded and allows the exchange of ATP by ADP through the inner mitochondrial membrane. ANT KO mouse display impaired activity of complex I, increase in ROS and oxidative damage. They also display a reduction in OPA1 leading to a fragmented mitochondrial web and sensitization to the opening of mPTP which has been associated with cardiopathies (Strauss et al., 2013).
Heart mitochondria have an active mitochondrial life cycle. Albeit to be highly packed with almost no freedom for movement, mitochondrial dynamics, mitophagy and biogenesis are critical for heart function (Eisner et al., 2017; Wu et al., 2019). Heart and skeletal muscle mitochondria feature specialized structures called nanotunnels that allow component mixing between mitochondria which resemble a fusion event (Huang et al., 2013). Disruption of MtDy has drastic consequences for cell physiology which are mainly associated with the accumulation of dysfunctional and ROS-generating mitochondria and heart failure. In humans, mutations in MFN2 and OPA1 cause Charcot–Marie–Tooth disease type 2A and dominant optic atrophy respectively. Also, complete loss of mitochondrial fusion results in a dramatic decrease in mtDNA content, loss of membrane potential and reduced respiratory chain function in both cultured cells and tissues. After conditionally knocking out Mfn1 and Mfn2 genes in adult rats, the mitochondrial fission in cardiomyocytes increased leading to abnormal cellular respiration, eventually leading to progressive dilated cardiomyopathy. Excessive mitochondrial fission has been associated with decreased mitochondrial function and increased ROS (Jheng et al., 2011; Haileselassie et al., 2019). FIS1 up-regulation decreased cellular ATP levels in anoxic cardiomyocytes (Wang et al., 2012). On the other hand, an excessive fusion is also deleterious for cardiac cells. MFF mutant mice died at 13 weeks due to heart failure caused by severe dilated cardiomyopathy. Mutant tissues showed decreased mitochondrial density and respiratory chain activity, and increased mitochondria size (García-Palmer, 2008; Zhou et al., 2017; Zhou and Tian, 2018). A thoughtful review on mitochondrial dynamics and cardiac biology is found in Vasquez-Trincado et al. (2016).
Although the role and the importance of the proper balance of MtDy for cell physiology are well accepted, there is no clarity about the signaling mechanisms that might be associated with heart failure. In our laboratory studying the role of MtDy in erythropoiesis, it was discovered a link between the mPTP and MtDy to control cell differentiation in erythropoiesis (Gonzalez-Ibanez et al., 2020). Interestingly, the role of the mPTP in cellular differentiation has been reported in the past for early embryonic cardiomyocytes, where a frequent opening of the mPTP is needed to maintain an immature mitochondrial morphology with low oxidative capacity. On the other hand, the closure of the pore was required for proper differentiation into cardiac muscle cells (Hom et al., 2011; Folmes et al., 2012). One of the triggers of mPTP opening is an elevated Ca++ concentration in the mitochondrial matrix (Bernardi and Di Lisa, 2015) and the up-regulation of FIS-1 by means of its interaction with the ER protein BAP31 might favor calcium entrance. In fact, mitochondrial calcium overload is a key determinant in heart failure (Iwasawa et al., 2011; Santulli et al., 2015).
Mitophagy is the cellular process to dismiss dysfunctional mitochondria and secure ATP demand for heart cells. As it was discussed before, many pathways and proteins are implicated in mitophagy which responds to different stimuli like ROS, hypoxia and mitochondrial depolarization. The great variability and diversity of proteins and pathways controlling mitochondria elimination reflect the importance and complexity of mitophagy for cell function that we are still far to fully understand. In cardiac cells, it is clear that mitophagy plays a major role in keeping healthy mitochondria and many or all known mitophagy pathways have been shown to play a critical role. In fact, it was recently published a novel and astonishing mechanism in cardiac cells to get rid of dysfunctional mitochondria, especially under cardiac stress. Dysfunctional mitochondria are expelled out from cardiomyocytes in special vesicles called exophers which are made by means of the cardiac mitophagic machinery. These expelled mitochondria are engulfed by macrophages which are resident in the cardiac tissue. Blocking the mitophagy machinery, reduction of macrophages or ablation of the specific exopher receptor caused and accumulation of damaged mitochondria and dysfunction in the heart (Nicolás-Ávila et al., 2020). In general, disruption of mitophagy is associated with heart failure and cardiopathies Deep details on mitophagy and cardiovascular diseases can be found in Morciano et al. (2020). On the other hand, induction of mitophagy, by any means (exercise, food, specific drugs or hypothermia), has shown a protective effect on many cardiovascular diseases.
Much of the cardiac literature describes a relationship between different cardiopathies and mitochondrial dysfunction. Daghistani et al. (2018), Chaanine (2019) showed that mitochondria changed their morphology from elongated to fragmented in heart failures. Lin and Kerkelä (2020), that there is a reduction in ETC and bioenergetic capacity in cardiomyopathies; and Vasquez-Trincado et al. (2016), Bravo-San Pedro et al. (2017) that mitochondrial dynamics and mitophagy are altered or reduced in cardiovascular diseases. In the end, most of those pathologies are related to an exacerbated amount of ROS, dysregulation of calcium homeostasis and the aperture of the mPTP. Mitochondria are constantly adapting bioenergetics, metabolism and signaling to allow cell function and survival. Thus, aged mitochondria (Table 1), with altered ROS and calcium homeostasis and dysregulation of the mPTP leading to ATP crisis, are one of the main causes of heart failure (Brand et al., 2013). Recently, the ANT protein was shown to increase mitochondrial proton leak - mitoflashes - in aged cardiomyocytes. This pathological proton leak is associated with inefficient ATP synthesis and increased electron flux and oxygen consumption, increasing ROS production and mPTP sensitivity. Interestingly, the treatment with the drug SS-31, a tetrapeptide that binds to cardiolipin-containing membranes and improves membrane stability and cristae curvatures (Birk et al., 2014), arrested proton leak, restored mitochondrial functionality, reduced ROS production and more surprisingly rejuvenated old cardiomyocytes (Zhang et al., 2020). Cardiac aging is characterized by a decrease in the number of mitochondria in the heart and a reduction in the area of the inner mitochondrial membrane implicating OXPHOS at the center of cardiac mitochondrial metabolism deficiency during aging. Respiratory complex III and IV deficiency in aging caused a decreased efficiency of OXPHOS, affecting also the number and mass of interfibrillar myofibrils (Stoll et al., 2018; Lin and Kerkelä, 2020).
The main driver of mitochondrial dysfunction in aging is the mtDNA, which progressively accumulates mutations and reduces its copy number as humans are getting old. In fact, mtDNA mutations cause aging, heart failure and many other aging-associated diseases. In 2004, it was published in Nature journal the “mutator mouse” whose mitochondrial DNA polymerase (POLG) had a dysfunctional proofreading domain which introduced random point mutations in the mtDNA, increasing mtDNA heteroplasmy (Trifunovic et al., 2004). The “mutator mouse” at the early age of 6 months, presented all the symptoms of old age such as blindness, deafness, alopecia, muscular atrophy, anemia, hump development and heart hypertrophy. The creation of this mutator mouse is an irrefutable proof that mtDNA mutations are a cause of aging and cardiovascular diseases. mtDNA mutations lead to OXPHOS inefficiency and then energy shortages especially in the tissues with the highest energy demands such as the heart, muscle and brain, which are the most vulnerable. By 40 weeks of age, mutator mice presented an enlarged volume of the left ventricle along with an increase in weight in relation to body weight. Histochemical analysis of the mutator mouse’s heart revealed a mosaic pattern with cytochrome c oxidase deficiency in some cardiomyocytes, which also occurs in the aged human hearts. TEM analysis of mitochondria ultrastructure confirmed the accumulation of fragmented, abnormal mitochondria. In addition, a decreased ATP production rate was noted (Trifunovic et al., 2004).
Another form of mtDNA damage during aging in humans is large mtDNA deletions. Mitochondrial diseases are usually associated with 5kb deletions of mtDNA which have been shown to accumulate in the human’s brain, muscle and heart (Chocron et al., 2019). Mutant mtDNA co-exists with wild type mtDNA giving rise to mtDNA heteroplasmy affecting the proper function of OXPHOS in a vicious cycle. Some pathogenic mtDNA mutations and a reduction in their copy number in cardiac cells have been specifically associated with impaired cardiac function and cardiac diseases and then, mtDNA mutations and copy number can be used as potential biomarkers of heart diseases (Ashar et al., 2017; Bray and Ballinger, 2017; Elosua, 2018; Galera-Monge et al., 2019). In fact, Elosua (2018) is emphatic when claiming that “mtDNA has been largely forgotten in cardiovascular research.”
The increase in mtDNA heteroplasmy not only affects mitochondrial function but also affects and influences nuclear genome mutations and the severity of cardiovascular diseases. This is also proof of mitochondria acting as a powerful signaling platform regulating both transcriptional and epigenetic nuclear gene expression. This mitochondria-nucleus retrograde communication acts in a synergistic way to produce cardiomyopathies or increase their severity. McManus et al. (2019) showed that ablation of ANT (the mitochondrial Adenine Nucleotide Transporter) in nuclear DNA is associated with cardiopathies where OXPHOS, MtDy and mitochondrial morphology are affected. However, in the presence of mtDNA mutations, cardiopathies are much more severe. mtDNA-related mitochondrial genetic diseases are associated with the development of cardiomyopathies in 40% approximately (Bates et al., 2012). Along with mtDNA mutations, the mtDNA haplogroups are also predictors of both lifespan and risk of several age diseases. Dr. Wallace’s group showed the type of mitochondrial haplogroups influences the severity of cardiovascular diseases with haplogroups U and H being the most affected (Strauss et al., 2013). On the other hand, the haplogroups J and T are associated with a reduced risk of cardiovascular disease (Veronese et al., 2019).
Reduction in heteroplasmy and restoration of mtDNA copy number is achieved by the coupling of OXPHOS with the Mitochondrial Life Cycle. It has been reported damaged DNA invokes mitophagy (Dan et al., 2020) and that adult post-mitotic tissue can eliminate mitochondria carrying damaged mtDNA by mitophagy which help to preserve cellular and mitochondrial function, suggesting that removal of the mutant mtDNA is protective for cells (Kandul et al., 2016). Mitochondrial turnover decreases with age, so there is growing interest in enhancing the pathways of mitophagy by discovering new pharmacological targets or cell mechanisms to counteract mitochondrial heteroplasmy (Diot et al., 2016). The aggravation of cardiac aging together with the accumulation of damaged mitochondria in cells can be caused by impaired and deficient mitophagy that results in increased heteroplasmy and altered mitochondrial metabolism (Quan et al., 2020). In fact, many of the pathways that improve health and extend longevity in various organisms all converge on mitophagy which is dependent on mitochondrial biogenesis and dynamics. A decrease in mitochondrial fission, functional mitochondria and NIX and FUNDC-1 receptor-mediated mitophagy may result from mtDNA mutations associated with aging (Lampert et al., 2019; Wu et al., 2019). Furthermore, Woodall et al. (2019) examined the role and expression levels of Parkin on accelerated cardiac failure in the mtDNA POLG mutator mice. It was observed that Parkin decreases in the mutator mouse’s heart with age. However, the restoration of Parkin level by means of its overexpression did not rescue the cardiac hypertrophy in the POLG mouse. Besides, deletion of parkin did not worsen heart disease. The authors claimed that mitochondria were not altered in their bioenergetics and that their function did not decline with age. This author’s interpretation is controversial since they observed a clear and significant decrease in complex II and IV protein levels, and TEM images showed altered cristae morphology in mutant mice with age, which might be indicative of mitochondrial dysfunction. An interesting discovery in the POLG mutant mouse’s heart is the upregulation of the receptor-mediated mitophagy genes and the appearance of larger mitochondria, which is in agreement with the upregulation of NIX protein myocardial hypertrophy (Yussman et al., 2002). This larger mitochondrial phenotype might be due, among other causes, to a decreased production and expulsion of exospheres that are dependent on the autophagic machinery (Nicolás-Ávila et al., 2020). However, the reduction of parkin in aging may be compensated by the upregulation of MUL1, a ubiquitin E3 ligase that is involved in regulating mitochondrial dynamics by promoting MFN2 degradation and leading to mitochondrial fragmentation and Parkin independent-mitophagy (Yun et al., 2014; Cai and Jeong, 2020). In cardiac cells, upregulation of MUL1 has been shown to fragment mitochondria and alter cristae structure, reduce mitochondrial membrane potential, promote cytochrome c release and sensitize mPTP, making it a potential mechanism of cell death and heart failure in aging (Yang et al., 2016; Wang et al., 2020).
Stressed mitochondria have been shown to release mtDNA into cytosol which is recognized as a Damage-Associated Mitochondrial Patterns (DAMPs), that trigger an inflammatory response. This pro-inflammatory environment may activate fibroblast proliferation and excessive production of extracellular matrix proteins which correlates with cardiomyopathies (West et al., 2011; Lin and Kerkelä, 2020). These alterations at molecular levels are associated with cellular changes such as hypertrophy, fibrosis, accumulation of misfolded proteins, loss of cardiac cells, extracellular matrix remodeling and amyloid deposition and structural changes in the myocardium like left ventricle hypertrophy and left auricle hypertrophy, which causes diastolic dysfunction (Steenman and Lande, 2017; Liang and Gustafsson, 2020). It has also been reported that mtDNA and whole mitochondria are released into bloodstream which offers a new biomarker of mitochondrial function and stress. mtDNA may travel naked (circulating cell-free mtDNA) which is recognized by immune cells to initiate the immune response, or they can go inside extracellular vesicles (EVs). Circulating EVs have been described as a novel mechanism of intercellular communication. In aging, Lazo et al. (2020) showed that mtDNA in EVs is diminished as compared with young people and even more, EVs containing old and damaged mtDNA affects mitochondrial bioenergetic in other cells and tissues. Picca et al. (2019) pointed out that EVs allow the elimination of dysfunctional or damage mitochondrial components; and Freeman et al. (2018) proved that EV secretion is dependent on autophagy. All these antecedents support the hypothesis that a decline in mtDNA containing EVs is due to a defect in mitophagy with aging.
The master regulator of mitochondrial biogenesis is the protein PGC-1α which is a transcriptional co-activator that induces nuclear- and mitochondrial- encoded gene transcription coordinating both genomes. PGC-1α can be stimulated/induced by physical exercise, cold and hypoxia. The signaling pathways to activate PGC-1α are diverse and well described (see Gureev, 2019; Oka et al., 2020). In mammals, an increase in the AMP/ATP ratio given by an overwhelming ATP demand or reduction in ATP synthesis activates AMPK (AMP protein kinase) which in turn will activate PGC-1α and inhibit mTOR (mechanistic target of rapamycin kinase). In this way, AMPK activation stimulates mitochondrial biogenesis and mitophagy (Kupr and Handschin, 2015). As a transcriptional co-activator, PGC-1α does not bind directly to DNA promoters but interacts and activates the transcription factors Nuclear Respiratory Factor 1 and 2 (NRF1 and NRF2), Estrogen-Related Receptor (ERR) and Peroxisome Proliferator Activated Receptor (PPAR). In general, NRF2 induces nuclear-encoded OXPHOS protein expression and NRF1 induces the expression of mitochondrial transcription factors TFAM and TFB to initiate mtDNA replication and transcription. ERR and PPAR are involved in mitochondrial biogenesis and dynamics, Krebs’ cycle and mitochondrial fatty acid oxidation expression genes. Furthermore, the increase in NAD + /NADH ratio as a result of the electron transport chain induces SIRT1 that also stimulates PGC-1α.
An essential component in mitochondrial signaling is ROS. In addition, ROS regulates mitochondrial dynamics by promoting mitochondrial fission and disrupting mitochondrial fusion, decreasing the OPA1 active isoform and degrading MFN1/2; and mitophagy. In cardiac myocytes, increased ROS induces a non-canonical function of the human Telomerase Reverse Transcriptase (hTERT), which reversibly translocate from the nucleus to the mitochondria to bind and protect mtDNA, reduce ROS production and increase OXPHOS efficiency (Haendeler et al., 2009; Ait-Aissa et al., 2019). Besides, the up-regulation of TERT is dependent on PGC-1α (Zhang et al., 2018) and positively regulates PINK1 function and stabilizes its mitochondrial localization to promote mitophagy (Shin and Chung, 2020). PINK1 other than phosphorylating ubiquitin and Parkin to initiate mitophagy, phosphorylates PARIS and leads to its Parkin ubiquitination and elimination. Therefore, when the PINK/Parkin axis is inhibited, PARIS accumulates and represses PGC-1α (Lee et al., 2017); and the knockdown of PARIS with CRISPR/Cas9 regains mitochondrial biogenesis (Kumar et al., 2020).
There is another parallel and independent pathway in controlling mitochondrial biogenesis given by the transcription factor Nuclear Erythroid-Related Factor 2 (NFE2L2. ∗It is also named NRF2, but in this work will be called NFE2L2 to avoid confusion). It has been described that NFE2L2 can translocate to the nucleus under an increase in ROS, specifically H2O2 and binds to the antioxidant response elements (ARE) in the promotor of NRF1 to induce TFAM. In nematodes, it was found that the orthologous protein of human NIX/BNIP3, named DCT-1 that works together with PINK and PRD (Parkin orthologous) is controlled by SKN-1, which is also linked to mtDNA replication. SKN-1 deficient nematodes display reduced mitophagy and reduced mitochondrial biogenesis. Even stimulation of mitophagy does not work in SKN-1 KO cells (Palikaras et al., 2015b). SKN-1 is a sensor of oxidative stress generated by defective mitochondria and is required for the expression of several genes related to mitochondrial biogenesis. SKN-1 is the orthologous of human NFE2L2 (Palikaras et al., 2015b). In mammals, Ivankovic et al. (2016) established the relationship among mitochondrial damage, NFE2L2 induction, P62, mitochondrial biogenesis (mtDNA replication) and protection against oxidative stress, in addition to lysosomal biogenesis to support the formation and degradation of autolysosomes. Increased autophagy involves an interaction between the autophagy adaptor p62/SQSTM1 and KEAP1, the cytosolic inhibitor of NFE2L2 allowing the accumulation of NFE2L2 in the nucleus, and then increased expression of nuclear-encoded mitochondrial genes needed for mitochondrial biogenesis and antioxidant response (Ichimura et al., 2013; Katsuragi et al., 2016).
In aging, it has been widely reported a decreased mitophagy (Wu et al., 2019; Bakula and Scheibye-Knudsen, 2020) commanded by a reduction of Parkin protein expression and the concomitant upregulation of the Nuclear Receptor Corepressor 1 (NCoR1) and its homologous protein SMRT, which counteracts the transcriptional control commanded by the SIRT1-AMPK/PGC1-alpha axis. In addition, NCoR1 will downregulate lipogenic and antioxidant genes favoring the metabolic switch from lipid oxidation to glucose oxidation and diminishing the antioxidant capacity of cells. NCoR1 KO animal models have greater mitochondrial biogenesis, mitophagy and lifespan (Perez-Schindler et al., 2012; Fan et al., 2013; Liu et al., 2013; Ou-Yang et al., 2018). The stabilization of NCoR1 in aging is because this corepressor is normally bound to LC3 family protein and degraded by mitophagy. Arrested mitophagy will cause its accumulation and downstream effects (Saito et al., 2019). Despite these antecedents, the role of NCoR1 in cardiac aging is still controversial and needs more research. Li et al. (2019a) suggested that NCoR1 has a protective cardiac effect by suppressing cardiac hypertrophy and Wang et al. (2018) have shown that NCoR1 is downregulated in old mouse hearts. In this regard, NCoR1 is not the only transcriptional co-repressor involved in aging but also there is SMRT and RIP140 that might play a relevant role (Fan et al., 2013; Liu et al., 2013).
The core of our model and the main driver of aging is mtDNA heteroplasmy (Figure 2). In young people with a healthy heart and cardiomyocytes, OXPHOS efficiency is high and the mtDNA mutation level, is low (mutated mtDNA is represented with one or more “X” on the double-stranded circular DNA). Under this scenario operates the mitochondrial life cycle to dilute mutated copies of mtDNA by means of MtDy and mitophagy. This is what it is called in our model The Virtuous Cycle. One of the main controllers of mitophagy is the PINK/PARKIN/P62 axis. When active, two repressors of mitochondrial biogenesis, PARIS and KEAP1 are inhibited and degraded, unleashing the transcriptional coactivator PGC-1α and the transcription factor NFE2L2 which are also activated by AMPK -via increased AMP/ATP ratio- and ROS respectively. AMPK will also inhibit the mTOR pathway to induce mitophagy. In addition, increased NAD + /NADH activates SIRT1 which also induces PGC-1α for mitochondrial biogenesis. Once the autophagosomes are formed, they are eliminated via their fusion with lysosomes or expelled out of the cell as exospheres, to be engulfed by macrophages. The latter is a new mechanism described only for cardiomyocytes.
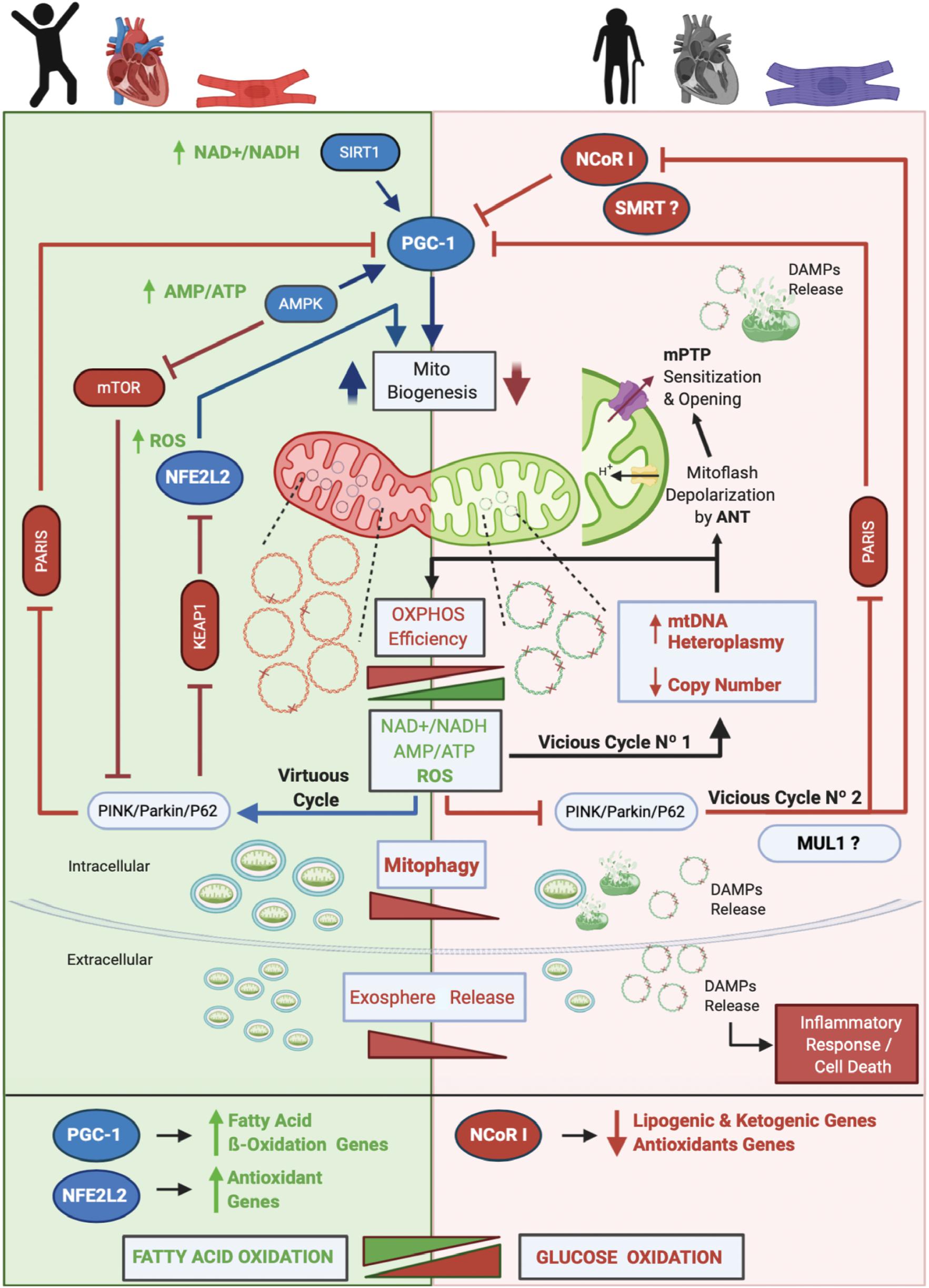
Figure 2. Proposed Model of Cardiac Aging. The core of our model and the main driver of aging is mtDNA heteroplasmy. In aging, two vicious cycles were defined. Vicious cycle No1 will progressively increase the level of mtDNA heteroplasmy and decrease OXPHOS efficiency at the point to affect mitophagy and set the onset of Vicious cycle No2 that will progressively inhibit mitochondrial biogenesis and antioxidant response due to inhibition of PGC-1α and stimulation and upregulation of NCoR1. As a result, dysfunctional mitochondria will accumulate, increasing the number of mitoflashes and pathological ROS to finally trigger the mPTP opening and the release of DAMPs to initiate an inflammatory cell response and an apoptotic/necrotic stimulus. Furthermore, a metabolic switch from fatty acid oxidation to glucose oxidation will occur. Altogether, these events lead to cardiovascular diseases and cardiac failure. On the other hand, in young people, a low level of mtDNA heteroplasmy will keep the efficiency of the OXPHOS system which will maintain low NAD + /NADH and AMP/ATP ratios and a physiological amount of ROS. These conditions allow the Virtuous Cycle where MtDy will segregate dysfunctional mitochondrial units for degradation by mitophagy and will activate PGC-1α and NFE2L2 for mitochondrial biogenesis. Of note, cardiac cells are able to expel out mitochondria in vesicles called exospheres that are degraded by surrounding macrophages. In this model, we hypothesized that exosphere release, which is dependent on the autophagic machinery, is decreased in aging. Also, it is not clear yet whether in cardiac aging the protein MUL1 is involved in mitophagy, but it has been shown to be upregulated under cardiac damage. Along with NCoR1, another transcriptional corepressor like SMRT has been shown to be involved in aging.
As mtDNA heteroplasmy exceeds 60%, mtDNA replication and transcription will be affected, reducing mtDNA copy number and proteins needed for respiratory chain assembly, leading to mitochondrial dysfunction and disruption of mitophagy. As a result, dysfunctional mitochondria will accumulate within cells, and cellular bioenergetics needs will not be satisfied causing an ATP-crisis, cell atrophy or death, ultimately leading to aging and aging-associated diseases. In fact, aging-associated diseases such as cancer, diabetes, heart disease, muscle weakness or atrophy, Alzheimer’s disease and Parkinson’s disease among others, are mainly due to a loss of mtDNA integrity and a failure in the mitochondrial life cycle (biogenesis, MtDy, mitophagy). To date, more than 400 mtDNA mutations, as well as a reduction in mtDNA copy number and a decrease in mitophagy, have been associated with human diseases (Tuppen et al., 2010; Li et al., 2019b). mtDNA mutations and heteroplasmy will affect the performance of the OXPHOS system reducing its efficiency due to the progressive loss of respiratory complex assembly and coupling, generating an abnormal amount of ROS. This ROS will produce more mtDNA mutations, increasing progressively the level of mtDNA heteroplasmy. This process corresponds to the Vicious Cycle No1 in our model. ROS not only affect mtDNA but also proteins and lipids. Thus, the mtDNA replication machinery, as well as membrane proteins and membrane phospholipids, including cardiolipin will be damaged. In this regard, ANT protein has been associated with increased mitoflash generation (transient mitochondrial depolarization), which uncouples ATP synthesis from the electron transport chain and ROS generation. Under these conditions, the mPTP is prone to open, collapse mitochondrial function, release mtDNA and other DAMPs, and trigger an inflammatory response and cell death. In parallel, an excessive amount of ROS inhibits the PINK/PARKIN/P62 axis meaning that PARIS and KEAP1 will be active and then NFE2L2 and PGC-1α inactive. Then, mitochondrial biogenesis will be reduced. In addition, inhibition of the PINK/PARKIN/P62 axis will activate NCoR1 which in turn blocks PGC-1α and mitochondrial biogenesis. This is called the vicious cycle No2. As mitophagy gets decreased, it is expected that exosphere release is also affected, accumulating in the cytosol many dysfunctional mitochondria prone to release more DAMPs and to induce cell death. Another important feature, in aging, is the metabolic switch from fatty acid oxidation to glucose oxidation, given the inhibition of NFE2L2 and PGC-1α which upregulates the fatty acid oxidation and antioxidant genes; and the activation of NCoR1 which down-regulates the lipogenic and ketogenic genes, and also the antioxidant genes. All these metabolic changes occurring in aging find their onset in the heteroplasmic mtDNA; and the final consequence of this, is having dysfunctional and hypertrophic cardiomyocytes leading to heart failure.
Decreased heteroplasmy, increased mtDNA copy number, increased mitochondrial biogenesis and mitophagy have been shown to cause rejuvenation. Old mice that had a substantial loss of mtDNA copy number and reduced mtDNA gene expression showed improved memory performance after mtDNA replication and transcription were stimulated. The transgenic mouse called “mtDNA depleter” evidenced skin wrinkles and hair loss, associated with reduced copies of mtDNA. However, by restoring mtDNA copies, the same animal rejuvenated and returned to normal. In transgenic mice expressing PGC-1α under the MKC promoter, overexpression of PGC-1α promoted mitochondrial biogenesis in skeletal and cardiac muscle from fetal life onward, ameliorated aging phenotypes and prevented aging-associated cardiomyopathies in adults (Yuan et al., 2016). Regarding mitophagy, pharmaceutic compounds, calorie restriction or genetic manipulation have been proven to augment lifespan in different organisms via increased mitophagy. Upregulation of systemic ATG5 expression, a protein involved in autophagosome formation, in transgenic mice have increased mitophagy resulting in improved health, longer life spans and less weight gain with aging (Pyo et al., 2019). Besides, enhanced mitophagy and less fibrosis during aging were found in those mouse hearts. This was also established by the Levine group and others that constitutive activation of the autophagic gene Beclin1 enhanced autophagy resulting in remarkable health improvement and a longer lifespan in mice. Additionally, those mutant mice when aged, have less interstitial fibrosis and cardiac hypertrophy, meaning that maintenance of autophagy in the heart postpones or avoids cardiac aging and protects the heart during sepsis (Fernández et al., 2018; Sun et al., 2018). Even in injured cardiac tissue, mitophagy stimulation improves cellular function via a decrease in ROS and apoptosis, and mitochondrial function improvement (Torrealba et al., 2017). After ischemia/reperfusion injured heart, hypothermia has been shown to protect the cardiac tissues by increasing autophagy, autophagy flux and mitochondrial content in farm pigs (Marek-Iannucci et al., 2019). On the other hand, autophagy and autophagic flux impairment in the heart caused its accelerated aging, remodeling it toward heart hypertrophy and dysfunctional mitochondria, which are associated with heart failure and cardiomyocyte degeneration (Takemura et al., 2018; Liang and Gustafsson, 2020).
In the biomedical field, there is a growing interest in enhancing mitochondrial function to combat aging-associated diseases and improve lifespan. This has led to the development of nutraceutical therapies based on the application of antioxidants, vitamins or respiratory substrates and the discovery of new drugs, which have shown a positive antiaging effect. Some of the well-known compounds are resveratrol, an antioxidant found in red fruits and red wines; and rapamycin, a macrolide compound that is a blocker of the mTOR pathway, and then a powerful inducer of mitophagy, prolonging life in mice and other model organisms and it prevents age-associated symptoms in mammals including humans. The ketogenic diets (low glucose, high ketone bodies), albeit their mechanism of action is not well understood, have been shown to inhibit mTOR, stimulate mitophagy, mitochondrial biogenesis and mtDNA replication, and reduce oxidative stress (Santra et al., 2004; Wallace et al., 2010; Gano et al., 2014). The novel discoveries, drugs or technologies include the catechinic acid, a polyphenol widely present in tea and fruits, that is able to induce mitophagy, improve fitness and extend lifespan in aged nematodes (C. elegans) (Wu et al., 2020); the catalpol, an iridoid glucoside widely abundant in the root of Rehmannia glutinosa, that has a powerful antioxidant effect, decreases DNA damage and stimulates the PGC-1α TERT axis (Zhang et al., 2018). The Telomerase Activator 65 (TA-65), a compound extracted from Astragalus membranaceus has been also shown to extend lifespan by increasing TERT expression and reducing markers of cardiovascular disease and inflammation (Fernandez et al., 2018). The tetrapeptide SS-31 binds to cardiolipin from the inner mitochondrial membrane, stabilizing membranes and the integral membrane mitochondrial proteins, in particular, ANT protein to prevent and even revert aging-associated mitochondrial dysfunction in cardiomyocytes (Szeto, 2006; Birk et al., 2014; Zhang et al., 2020). Finally, the thiazolidinedione pioglitazone has been shown to be a potent inductor of the PGC-1α, mitochondrial biogenesis and antioxidant mitochondrial defense (Bogacka et al., 2005; Butterick et al., 2016).
The correlation between mtDNA integrity loss and aging has allowed the development of experimental strategies that seek to eliminate mutated mtDNA or increase wild type mtDNA to restore mitochondrial function and reverse the aging phenotype. The rationale of these strategies is that, if the ratio between the wild type mtDNA and the mutated mtDNA can be adjusted, the presence of wild type molecules will mitigate the effect of the mutants and therefore the defect will be corrected. Four different strategies have been developed (Taylor and Turnbull, 2005; Patananan et al., 2016; Aravintha Siva et al., 2019),” seems to be incomplete. Kindly check and advise. (i) Transfer of isolated xenogeneic mitochondria, which are deposited in the cell culture medium and enter the cells by macropinocytosis (Kitani et al., 2014). It has recently been shown that mitochondria therapy is capable of promoting the regeneration of damaged hippocampal neurons (Katrangi et al., 2007; Chien et al., 2018; Kim et al., 2018). Other mitochondria transfer technique is by centrifugation and also by the generation of mitocytoplasts (Yang and Koob, 2012). (ii) Transfer of exogenous wild type mtDNA, by direct microinjection of mtDNA into the host cell or by the use of “carriers” such as the mitochondrial transduction domain (MTD) conjugated to the mitochondrial transcription factor A (MTD-TFAM), MITO Porters, DQAsomes, and nanoagents (Niazi et al., 2013; Zhang and Zhang, 2016; Aravintha Siva et al., 2019). Exogenous mtDNA has also been transferred into muscle cells via hydrodynamic limb venous injection (Yasuzaki et al., 2013). These methodologies have shown promising and surprising results in the rescue of mitochondrial and cellular function. (iii) Elimination of heteroplasmy. These techniques are named the antigenomic mtDNA Therapy; the mitochondrial-targeted restriction endonucleases; zinc-finger nucleases type; and the effector nucleases of the transcription activator type (Transcription Activator-like effector Endonucleases), are all based on the delivery of endonucleases into mitochondria, which will specifically recognize and degrade the mutated mtDNA. An attempt has also been made to carry out the CRISPR/Cas9 technology in mitochondria, but the results have not been reproducible (Jo et al., 2015). (iv) Mixed mtDNA transfer system. This technology proposes direct electroporation of isolated exogenous mitochondria with wild type mtDNA. Next, mitochondria must then be transferred to the zygotes or cultured cells through microinjections (Collombet et al., 1997; Yoon et al., 2010). Recently, it was published in Nature journal a new technology to edit mitochondrial DNA which combines mitotalen proteins, Cas9 and the Uracyl glycosidase inhibitor with a bacterial cytidine deaminase toxin. This technology acts as a repair/edit system on mutant forms of mtDNA. It does not eliminate mtDNA copies, but rather, restore them (Mok et al., 2020). This revolutionary system was tested in HEK293T cells and it remains open to the question if it will also be efficient in other cellular models like cardiac cells.
In summary, heart mitochondria must satisfy an enormous amount of energy to keep the heart beating from conception to death. To do that, mitochondria are equipped with two complex systems that work collaboratively, the OXPHOS system and the Mitochondrial Life Cycle to keep safe and healthy the mtDNA and then mitochondria. Any disruption of these processes is associated with heart failure. On the other hand, the potentiation of these is beneficial to improve heart health. In aging, mtDNA is prone to undergo mutations leading to heteroplasmy which is associated with increased ROS affecting negatively both OXPHOS and the mitochondrial life cycle. Mitochondrial therapies have been developed to deal with mtDNA heteroplasmy. Many of them focus on Mitophagy stimulation which decreases mtDNA heteroplasmy and has a protective effect in cardiac cells. The development of new therapies aims to decrease mtDNA heteroplasmy in cells either by the transfer of mutation-free wild type mtDNA or by the direct elimination of mutant mtDNA. The results of these new technologies are promising in restoring mitochondrial and cellular function in vitro and open a new era in the field of mitochondrial medicine to combat aging-associated heart failure.
JS helped with the writing and discussion of the manuscript. AE conceived, wrote, and funded the manuscript. Both authors contributed to the article and approved the submitted version.
This work was funded by the grants: Fondecyt 1180983 (AE) and Millennium Institute on Immunology and Immunotherapy P09-016-F (AE).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Roberta Gottlieb from Cedars-Sinai Medical Center for her valuable comments and suggestions on the manuscript.
Ait-Aissa, K., Heisner, J. S., Norwood Toro, L. E., Bruemmer, D., Doyon, G., Harmann, L., et al. (2019). Telomerase Deficiency Predisposes to Heart Failure and Ischemia-Reperfusion Injury. Front. Cardiovasc. Med. 6:1–13. doi: 10.3389/fcvm.2019.00031
Aravintha Siva, M., Mahalakshmi, R., Bhakta-Guha, D., and Guha, G. (2019). Gene therapy for the mitochondrial genome: Purging mutations, pacifying ailments. Mitochondrion 46, 195–208. doi: 10.1016/j.mito.2018.06.002
Ashar, F. N., Zhang, Y., Longchamps, R. J., Lane, J., Moes, A., Grove, M. L., et al. (2017). Association of Mitochondrial DNA Copy Number With Cardiovascular Disease. JAMA Cardiol. 2, 1247–1249. doi: 10.1001/jamacardio.2017.3683
Ashrafi, G., and Schwarz, T. L. (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 20, 31–42. doi: 10.1038/cdd.2012.81
Bakula, D., and Scheibye-Knudsen, M. (2020). MitophAging: Mitophagy in Aging and Disease. Front. Cell Dev. Biol. 8:239. doi: 10.3389/fcell.2020.00239
Barth, E., Stämmler, G., Speiser, B., and Schaper, J. (1992). Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 24, 669–681. doi: 10.1016/0022-2828(92)93381-s
Bates, M. G. D., Bourke, J. P., Giordano, C., d’Amati, G., Turnbull, D. M., and Taylor, R. W. (2012). Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur. Heart J. 33, 3023–3033. doi: 10.1093/eurheartj/ehs275
Bellot, G., Garcia-Medina, R., Gounon, P., Chiche, J., Roux, D., Pouysségur, J., et al. (2009). Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 29, 2570–2581. doi: 10.1128/MCB.00166-09
Bénard, G., Bellance, N., James, D., Parrone, P., Fernandez, H., Letellier, T., et al. (2007). Mitochondrial bioenergetics and structural network organization. J. Cell. Sci. 120, 838–848. doi: 10.1242/jcs.03381
Bernardi, P., and Di Lisa, F. (2015). The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 78, 100–106. doi: 10.1016/j.yjmcc.2014.09.023
Bhujabal, Z., Birgisdottir, ÅB., Sjøttem, E., Brenne, H. B., Øvervatn, A., Habisov, S., et al. (2017). FKBP8 recruits LC3A to mediate Parkin−independent mitophagy. EMBO Rep. 18, 947–961. doi: 10.15252/embr.201643147
Birk, A. V., Chao, W. M., Bracken, C., Warren, J. D., and Szeto, H. H. (2014). Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 171, 2017–2028. doi: 10.1111/bph.12468
Bogacka, I., Xie, H., Bray, G. A., and Smith, S. R. (2005). Pioglitazone induces mitochondrial biogenesis in human subcutaneous adipose tissue in vivo. Diabetes 54, 1392–1399. doi: 10.2337/diabetes.54.5.1392
Brand, M. D., Orr, A. L., Perevoshchikova, I. V., and Quinlan, C. L. (2013). The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 169, 1–8. doi: 10.1111/bjd.12208
Bravo-San Pedro, J. M., Kroemer, G., and Galluzzi, L. (2017). Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 120, 1812–1824. doi: 10.1161/CIRCRESAHA.117.311082
Bray, A. W., and Ballinger, S. W. (2017). Mitochondrial DNA mutations and cardiovascular disease. Curr. Opin. Cardiol. 32, 267–274. doi: 10.1097/HCO.0000000000000383
Brown, T. A., Tkachuk, A. N., Shtengel, G., Kopek, B. G., Bogenhagen, D. F., Hess, H. F., et al. (2011). Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell. Biol. 31, 4994–5010. doi: 10.1128/MCB.05694-11
Butterick, T. A., Hocum Stone, L., Duffy, C., Holley, C., Cabrera, J. A., Crampton, M., et al. (2016). Pioglitazone increases PGC1-α signaling within chronically ischemic myocardium. Basic Res. Cardiol. 111, 37–39. doi: 10.1007/s00395-016-0555-4
Cai, Q., and Jeong, Y. Y. (2020). Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 9, 150–128. doi: 10.3390/cells9010150
Cann, R. L., Stoneking, M., and Wilson, A. C. (1987). Mitochondrial DNA and human evolution. Nature 325, 31–36. doi: 10.1038/325031a0
Chaanine, A. H. (2019). Morphological Stages of Mitochondrial Vacuolar Degeneration in Phenylephrine-Stressed Cardiac Myocytes and in Animal Models and Human Heart Failure. Medicina 55, 239–218. doi: 10.3390/medicina55060239
Chien, L., Liang, M.-Z., Chang, C.-Y., Wang, C., and Chen, L. (2018). Mitochondrial therapy promotes regeneration of injured hippocampal neurons. Biochimica et biophysica acta 1864, 3001–3012. doi: 10.1016/j.bbadis.2018.06.012
Chocron, E. S., Munkácsy, E., and Pickering, A. M. (2019). Cause or casualty: The role of mitochondrial DNA in aging and age-associated disease. Biochim Biophys Acta Mol. Basis Dis. 1865, 285–297. doi: 10.1016/j.bbadis.2018.09.035
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Publis. Group 15, 1197–1205. doi: 10.1038/ncb2837
Collombet, J. M., Wheeler, V. C., Vogel, F., and Coutelle, C. (1997). Introduction of plasmid DNA into isolated mitochondria by electroporation. A novel approach toward gene correction for mitochondrial disorders. J. Biol. Chem. 272, 5342–5347. doi: 10.1074/jbc.272.8.5342
Daghistani, H. M., Rajab, B. S., and Kitmitto, A. (2018). Three−dimensional electron microscopy techniques for unravelling mitochondrial dysfunction in heart failure and identification of new pharmacological targets. Br. J. Pharmacol. 176, 4340–4359. doi: 10.1111/bph.14499
Dan, X., Babbar, M., Moore, A., Wechter, N., Tian, J., Mohanty, J. G., et al. (2020). DNA damage invokes mitophagy through a pathway involving Spata18. Nucleic Acids Res. 48, 6611–6623. doi: 10.1093/nar/gkaa393
Diot, A., Morten, K., and Poulton, J. (2016). Mitophagy plays a central role in mitochondrial ageing. Mamm Genome 27, 381–395. doi: 10.1007/s00335-016-9651-x
Eisner, V., Cupo, R. R., Gao, E., Csordás, G., Slovinsky, W. S., Paillard, M., et al. (2017). Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc. Natl. Acad. Sci. U S A. 114, E859–E868. doi: 10.1073/pnas.1617288114
Elosua, R. (2018). Mitochondrial DNA and cardiovascular diseases: A long road ahead. Eur. J. Prev. Cardiol. 26, 973–975. doi: 10.1177/2047487319843388
Fan, W., Atkins, A. R., Yu, R. T., Downes, M., and Evans, R. M. (2013). Road to exercise mimetics: targeting nuclear receptors in skeletal muscle. J. Mol. Endocrinol. 51, T87–T100. doi: 10.1530/JME-13-0258
Fernández, ÁF., Sebti, S., Wei, Y., Zou, Z., Shi, M., McMillan, K. L., et al. (2018). Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140. doi: 10.1038/s41586-018-0162-7
Fernandez, M. L., Thomas, M. S., Lemos, B. S., DiMarco, D. M., Missimer, A., Melough, M., et al. (2018). TA-65, A Telomerase Activator improves Cardiovascular Markers in Patients with Metabolic Syndrome. CPD 24, 1905–1911. doi: 10.2174/1381612824666180316114832
Ferrari, R., Censi, S., and Journal, F. M. E. H. (2003). Prognostic benefits of heart rate reduction in cardiovascular disease. Eur. Heart J. Suppl. 5, G10–G14.
Folmes, C. D. L., Dzeja, P. P., Nelson, T. J., and Terzic, A. (2012). Mitochondria in control of cell fate. Circ. Res. 110, 526–529. doi: 10.1161/RES.0b013e31824ae5c1
Freeman, D. W., Noren Hooten, N., Eitan, E., Green, J., Mode, N. A., Bodogai, M., et al. (2018). Altered Extracellular Vesicle Concentration, Cargo, and Function in Diabetes. Diabetes 67, 2377–2388. doi: 10.2337/db17-1308
Galera-Monge, T., Zurita-Díaz, F., Garesse, R., and Gallardo, M. E. (2019). The mutation m.13513G>A impairs cardiac function, favoring a neuroectoderm commitment, in a mutant-load dependent way. J. Cell. Physiol. 234, 19511–19522. doi: 10.1002/jcp.28549
Gano, L. B., Patel, M., and Rho, J. M. (2014). Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 55, 2211–2228. doi: 10.1194/jlr.R048975
García-Palmer, F. J. (2008). Lack of functional assembly in mitochondrial supercomplexes: a new insight into impaired mitochondrial function? Cardiovasc. Res. 80, 3–4. doi: 10.1093/cvr/cvn213
Ginneken, V. V. (2017). Are there any Biomarkers of Aging? Biomarkers of the Brain. BJSTR 1, 1–16. doi: 10.26717/BJSTR.2017.01.000151
Gonzalez-Ibanez, A. M., Ruiz, L. M., Jensen, E., Echeverria, C. A., Romero, V., Stiles, L., et al. (2020). Erythroid Differentiation and Heme Biosynthesis Are Dependent on a Shift in the Balance of Mitochondrial Fusion and Fission Dynamics. Front. Cell Dev. Biol. 8:592035. doi: 10.3389/fcell.2020.592035
Gureev, A. P. (2019). Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 10:1–12. doi: 10.3389/fgene.2019.00435
Haendeler, J., Dröse, S., Büchner, N., Jakob, S., Altschmied, J., Goy, C., et al. (2009). Mitochondrial Telomerase Reverse Transcriptase Binds to and Protects Mitochondrial DNA and Function From Damage. ATVB 29, 929–935. doi: 10.1161/ATVBAHA.109.185546
Haileselassie, B., Mukherjee, R., Joshi, A. U., Napier, B. A., Massis, L. M., Ostberg, N. P., et al. (2019). Drp1/Fis1 interaction mediates mitochondrial dysfunction in septic cardiomyopathy. J. Mol. Cell. Cardiol. 130, 160–169. doi: 10.1016/j.yjmcc.2019.04.006
Hendgen-Cotta, U. B., Esfeld, S., Jastrow, H., Totzeck, M., Altschmied, J., Goy, C., et al. (2018). Mouse cardiac mitochondria do not separate in subsarcolemmal and interfibrillar subpopulations. Mitochondrion 38, 1–5. doi: 10.1016/j.mito.2017.07.005
Hiona, A. (2008). The role of mitochondrial DNA mutations in aging and sarcopenia: Implications for the mitochondrial vicious cycle theory of aging. Exp. Gerontol. 43, 24–33. doi: 10.1016/j.exger.2007.10.001
Hom, J. R., Quintanilla, R. A., Hoffman, D. L., de Mesy Bentley, K. L., Molkentin, J. D., Sheu, S.-S., et al. (2011). The Permeability Transition Pore Controls Cardiac Mitochondrial Maturation and Myocyte Differentiation. Dev. Cell 21, 469–478. doi: 10.1016/j.devcel.2011.08.008
Huang, X., Sun, L., Ji, S., Zhao, T., Zhang, W., Xu, J., et al. (2013). Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc. Natl. Acad. Sci. 110, 2846–2851. doi: 10.1073/pnas.1300741110
Ichimura, Y., Waguri, S., Sou, Y.-S., Kageyama, S., Hasegawa, J., Ishimura, R., et al. (2013). Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 51, 618–631. doi: 10.1016/j.molcel.2013.08.003
Ivankovic, D., Chau, K.-Y., Schapira, A. H. V., and Gegg, M. E. (2016). Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin-mediated mitophagy. J. Neurochem. 136, 388–402. doi: 10.1111/jnc.13412
Iwasawa, R., Mahul-Mellier, A.-L., Datler, C., Pazarentzos, E., and Grimm, S. (2011). Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 30, 556–568. doi: 10.1038/emboj.2010.346
Jheng, H. F., Tsai, P. J., Guo, S. M., Kuo, L. H., Chang, C. S., Su, I. J., et al. (2011). Mitochondrial Fission Contributes to Mitochondrial Dysfunction and Insulin Resistance in Skeletal Muscle. Mol. Cell. Biol. 32, 309–319. doi: 10.1128/MCB.05603-11
Jo, A., Ham, S., Lee, G. H., Lee, Y.-I., Kim, S., Lee, Y.-S., et al. (2015). Efficient Mitochondrial Genome Editing by CRISPR/Cas9. BioMed Res. Int. 2015, 1–10. doi: 10.1155/2015/305716
Kandul, N. P., Zhang, T., Hay, B. A., and Guo, M. (2016). Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat. Commun. 7, 1–11. doi: 10.1038/ncomms13100
Katrangi, E., D’Souza, G., Boddapati, S. V., Kulawiec, M., Singh, K. K., Bigger, B., et al. (2007). Xenogenic transfer of isolated murine mitochondria into human rho0 cells can improve respiratory function. Rejuvenation Res. 10, 561–570. doi: 10.1089/rej.2007.0575
Katsuragi, Y., Ichimura, Y., and Komatsu, M. (2016). Regulation of the Keap1-Nrf2 pathway by p62/SQSTM1. Curr. Opin. Toxicol. 1, 54–61. doi: 10.1016/j.cotox.2016.09.005
Kim, M. J., Hwang, J. W., Yun, C.-K., Lee, Y., and Choi, Y.-S. (2018). Delivery of exogenous mitochondria via centrifugation enhances cellular metabolic function. Sci. Rep. 8, 1–13. doi: 10.1038/s41598-018-21539-y
Kitani, T., Kami, D., Matoba, S., and Gojo, S. (2014). Internalization of isolated functional mitochondria: involvement of macropinocytosis. J. Cell. Mol. Med. 18, 1694–1703. doi: 10.1111/jcmm.12316
Kumar, M., Acevedo-Cintrón, J., Jhaldiyal, A., Wang, H., Andrabi, S. A., Eacker, S., et al. (2020). Defects in Mitochondrial Biogenesis Drive Mitochondrial Alterations in PARKIN-Deficient Human Dopamine Neurons. Stem Cell Rep. 15, 629–645. doi: 10.1016/j.stemcr.2020.07.013
Kupr, B., and Handschin, C. (2015). Complex Coordination of Cell Plasticity by a PGC-1α-controlled Transcriptional Network in Skeletal Muscle. Front. Physiol. 6:1008–1007. doi: 10.3389/fphys.2015.00325
Lakatos, A., Derbeneva, O., Younes, D., Keator, D., Bakken, T., Lvova, M., et al. (2010). Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol. Aging 31, 1355–1363. doi: 10.1016/j.neurobiolaging.2010.04.031
Lakshmanan, L. N., Yee, Z., Ng, L. F., Gunawan, R., Halliwell, B., and Gruber, J. (2018). Clonal expansion of mitochondrial DNA deletions is a private mechanism of aging in long-lived animals. Aging Cell 17:e12814. doi: 10.1111/acel.12814
Lampert, M. A., Orogo, A. M., Najor, R. H., Hammerling, B. C., Leon, L. J., Wang, B. J., et al. (2019). BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15, 1182–1198. doi: 10.1080/15548627.2019.1580095
Lazo, S., Noren Hooten, N., Green, J., Eitan, E., Mode, N. A., Liu, Q. R., et al. (2020). Mitochondrial DNA in extracellular vesicles declines with age. Aging Cell 20, 267–215. doi: 10.1111/acel.13283
Lee, S. H., Lee, S., Du, J., Jain, K., Ding, M., Kadado, A. J., et al. (2019). Mitochondrial MsrB2 serves as a switch and transducer for mitophagy. EMBO Mol. Med. 11, 1–16. doi: 10.15252/emmm.201910409
Lee, Y., Stevens, D. A., Kang, S.-U., Jiang, H., Lee, Y.-I., Ko, H. S., et al. (2017). PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. CellReports 18, 918–932. doi: 10.1016/j.celrep.2016.12.090
Lee, Y.-K., and Lee, J.-A. (2016). Role of the mammalian ATG8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 49, 424–430. doi: 10.5483/BMBRep.2016.49.8.081
Legros, F., Lombès, A., Frachon, P., and Rojo, M. (2002). Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell 13, 4343–4354. doi: 10.1091/mbc.e02-06-0330
Letts, J. A., Fiedorczuk, K., and Sazanov, L. A. (2016). The architecture of respiratory supercomplexes. Nature 537, 644–648. doi: 10.1038/nature19774
Li, C., Sun, X. N., Chen, B. Y., Zeng, M. R., Du, L. J., Liu, T., et al. (2019a). Nuclear receptor corepressor 1 represses cardiac hypertrophy. EMBO Mol. Med. 11, 1–14. doi: 10.15252/emmm.201809127
Li, H., Slone, J., Fei, L., and Huang, T. (2019b). Mitochondrial DNA Variants and Common Diseases: A Mathematical Model for the Diversity of Age-Related mtDNA Mutations. Cells 8:608. doi: 10.3390/cells8060608
Li, M., Schönberg, A., Schaefer, M., Schroeder, R., Nasidze, I., and Stoneking, M. (2010). Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 87, 237–249. doi: 10.1016/j.ajhg.2010.07.014
Liang, W. J., and Gustafsson, ÅB. (2020). The Aging Heart: Mitophagy at the Center of Rejuvenation. Front. Cardiovasc. Med. 7:1428–1427. doi: 10.3389/fcvm.2020.00018
Liesa, M., Palacin, M., and Zorzano, A. (2009). Mitochondrial Dynamics in Mammalian Health and Disease. Physiol. Rev. 89, 799–845. doi: 10.1152/physrev.00030.2008
Lin, R., and Kerkelä, R. (2020). Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging. IJMS 21, 1359–1318. doi: 10.3390/ijms21041359
Liu, L., Feng, Du, Chen, G., Chen, M., Zheng, Q., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, S., Reilly, S. M., and Lee, C.-H. (2013). Transcriptional Repression of Mitochondrial Function in Aging: A Novel Role for the Silencing Mediator of Retinoid and Thyroid Hormone Receptors Co-Repressor. Antioxidants Redox Signal. 19, 299–309. doi: 10.1089/ars.2011.4413
Losón, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lutz, W., Sanderson, W., and Scherbov, S. (2008). The coming acceleration of global population ageing. Nature 451, 716–719. doi: 10.1038/nature06516
Ma, J., Liu, T., Wei, A.-C., Banerjee, P., O’Rourke, B., and Hart, G. W. (2015). O-GlcNAcomic Profiling Identifies Widespread O-Linked Î2-N-Acetylglucosamine Modification (O-GlcNAcylation) in Oxidative Phosphorylation System Regulating Cardiac Mitochondrial Function∗âTM|. J. Biol. Chem. 290, 29141–29153. doi: 10.1074/jbc.M115.691741
Marek-Iannucci, S., Thomas, A., Hou, J., Crupi, A., Sin, J., Taylor, D. J., et al. (2019). Myocardial hypothermia increases autophagic flux, mitochondrial mass and myocardial function after ischemia-reperfusion injury. Sci. Rep. 9, 10001–10017. doi: 10.1038/s41598-019-46452-w
McManus, M. J., Picard, M., Chen, H.-W., De Haas, H. J., Potluri, P., Leipzig, J., et al. (2019). Mitochondrial DNA Variation Dictates Expressivity and Progression of Nuclear DNA Mutations Causing Cardiomyopathy. Cell Metabol. 29, 78.e–90.e. doi: 10.1016/j.cmet.2018.08.002
Mok, B. Y., de Moraes, M. H., Zeng, J., Bosch, D. E., Kotrys, A. V., Raguram, A., et al. (2020). A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 19, 1–27. doi: 10.1038/s41586-020-2477-4
Morciano, G., Patergnani, S., Bonora, M., Pedriali, G., Tarocco, A., Bouhamida, E., et al. (2020). Mitophagy in Cardiovascular Diseases. JCM 9, 892–826. doi: 10.3390/jcm9030892
Niazi, A. K., Mileshina, D., Cosset, A., Val, R., Weber-Lotfi, F., and Dietrich, A. (2013). Targeting nucleic acids into mitochondria: Progress and prospects. Mitochondrion 13, 548–558. doi: 10.1016/j.mito.2012.05.004
Nicolás-Ávila, J. A., Lechuga-Vieco, A. V., Esteban-Martínez, L., Sánchez-Díaz, M., Díaz-García, E., Santiago, D. J., et al. (2020). A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 183, 94.e–109.e. doi: 10.1016/j.cell.2020.08.031
Nishigaki, Y., Fuku, N., and Tanaka, M. (2010). Mitochondrial haplogroups associated with lifestyle-related diseases and longevity in the Japanese population. Geriatr. Gerontol. Int. 10, S221–S235. doi: 10.1111/j.1447-0594.2010.00599.x
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2009). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
O’Brien, K. M. (2010). Mitochondrial biogenesis in cold-bodied fishes. J. Exp. Biol. 214, 275–285. doi: 10.1242/jeb.046854
Oka, S.-I., Sabry, A. D., Cawley, K. M., and Warren, J. S. (2020). Multiple Levels of PGC-1α Dysregulation in Heart Failure. Front. Cardiovasc. Med. 7:829–817. doi: 10.3389/fcvm.2020.00002
Ou-Yang, Q., Lin, X.-M., Zhu, Y.-J., Zheng, B., Li, L., Yang, Y.-C., et al. (2018). Distinct role of nuclear receptor corepressor 1 regulated de novo fatty acids synthesis in liver regeneration and hepatocarcinogenesis in mice. Hepatology 67, 1071–1087. doi: 10.1002/hep.29562
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015a). Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 22, 1399–1401. doi: 10.1038/cdd.2015.86
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015b). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1–10. doi: 10.1038/s41556-018-0176-2
Pastukh, V. M., Gorodnya, O. M., Gillespie, M. N., and Ruchko, M. V. (2016). Regulation of mitochondrial genome replication by hypoxia: The role of DNA oxidation in D-loop region. Free Radic. Biol. Med. 96, 78–88. doi: 10.1016/j.freeradbiomed.2016.04.011
Patananan, A. N., Wu, T.-H., Chiou, P.-Y., and Teitell, M. A. (2016). Modifying the Mitochondrial Genome. Cell Metabol. 23, 785–796. doi: 10.1016/j.cmet.2016.04.004
Perez-Schindler, J., Summermatter, S., Salatino, S., Zorzato, F., Beer, M., Balwierz, P. J., et al. (2012). The Corepressor NCoR1 Antagonizes PGC-1 and Estrogen-Related Receptor in the Regulation of Skeletal Muscle Function and Oxidative Metabolism. Mol. Cell. Biol. 32, 4913–4924. doi: 10.1128/MCB.00877-12
Picard, M., Wallace, D. C., and Burelle, Y. (2016). The rise of mitochondria in medicine. Mitochondrion 30, 105–116. doi: 10.1016/j.mito.2016.07.003
Picca, A., Guerra, F., Calvani, R., Bucci, C., Monaco, Lo, M., et al. (2019). Mitochondrial Dysfunction and Aging: Insights from the Analysis of Extracellular Vesicles. IJMS 20, 805–812. doi: 10.3390/ijms20040805
Pickles, S., Vigié, P., and Youle, R. J. (2018). Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 28, R170–R185. doi: 10.1016/j.cub.2018.01.004
Pua, H. H., Guo, J., Komatsu, M., and He, Y.-W. (2009). Autophagy Is Essential for Mitochondrial Clearance in Mature T Lymphocytes. J. Immunol. 182, 4046–4055. doi: 10.4049/jimmunol.0801143
Pyo, J.-O., Yoo, S. M., Ahn, H.-H., Nah, J., Hong, S.-H., Kam, T.-I., et al. (2019). Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 4:3300. doi: 10.1038/ncomms3300
Quan, Y., Xin, Y., Tian, G., Zhou, J., and Liu, X. (2020). Mitochondrial ROS-Modulated mtDNA: A Potential Target for Cardiac Aging. Oxidat. Med. Cell. Longevity 2020:9423593. doi: 10.1155/2020/9423593
Rojansky, R., Cha, M. Y., and Elife, D. C. (2016). Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife Sci. 5:e17896. doi: 10.7554/eLife.17896.001
Saito, T., Kuma, A., Sugiura, Y., Ichimura, Y., Obata, M., Kitamura, H., et al. (2019). Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 10, 1–16. doi: 10.1038/s41467-019-08829-3
Santra, S., Gilkerson, R. W., Davidson, M., and Schon, E. A. (2004). Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann. Neurol. 56, 662–669. doi: 10.1002/ana.20240
Santulli, G., Xie, W., Reiken, S. R., and Marks, A. R. (2015). Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. U S A. 112, 11389–11394. doi: 10.1073/pnas.1513047112
Satoh, H., Delbridge, L. M., Blatter, L. A., and Bers, D. M. (1996). Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys. J. 70, 1494–1504. doi: 10.1016/S0006-3495(96)79711-4
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. 104, 19500–19505. doi: 10.1073/pnas.0708818104
Sheridan, C., and Martin, S. J. (2010). Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 10, 640–648. doi: 10.1016/j.mito.2010.08.005
Shin, W. H., and Chung, K. C. (2020). Human telomerase reverse transcriptase positively regulates mitophagy by inhibiting the processing and cytoplasmic release of mitochondrial PINK1. Cell Death Dis. 11:425. doi: 10.1038/s41419-020-2641-7
Sin, J., Andres, A. M., Taylor, D. J. R., Weston, T., Hiraumi, Y., Stotland, A., et al. (2016). Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 12, 369–380. doi: 10.1080/15548627.2015.1115172
Sinkler, C. A., Kalpage, H., Shay, J., Lee, I., Malek, M. H., Grossman, L. I., et al. (2017). Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome cOxidase Subunits: From Function to Human Disease. Oxidat. Med. Cell. Longevity 2017, 1–19. doi: 10.1155/2017/1534056
Smirnova, E., Griparic, L., Shurland, D. L., and van der Bliek, A. M. (2001). Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 12, 2245–2256. doi: 10.1091/mbc.12.8.2245
Sousa, J. S., Mills, D. J., Vonck, J., and Kühlbrandt, W. (2016). Functional asymmetry and electron flow in the bovine respirasome. eLife Sci. 5:805. doi: 10.7554/eLife.21290
Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H., and Harris, A. L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673.
Steenman, M., and Lande, G. (2017). Cardiac aging and heart disease in humans. Biophys. Rev. 9, 1–7. doi: 10.1007/s12551-017-0255-9
Stoll, S., Leimena, C., and Qiu, H. (2018). “Mitochondria and Heart Disease,” in Mitochondrial Diseases, ed. P. Lestienne (London: intechopen), 1–31.
Strauss, K. A., DuBiner, L., Simon, M., Zaragoza, M., Sengupta, P. P., Li, P., et al. (2013). Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc. Natl. Acad. Sci. 110, 3453–3458. doi: 10.1073/pnas.1300690110
Sun, Y., Yao, X., Zhang, Q.-J., Zhu, M., Liu, Z.-P., Ci, B., et al. (2018). Beclin-1-Dependent Autophagy Protects the Heart During Sepsis. Circulation 138, 2247–2262. doi: 10.1161/CIRCULATIONAHA.117.032821
Szeto, H. H. (2006). Mitochondria-targeted peptide antioxidants: Novel neuroprotective agents. AAPS J. 8, E521–E531. doi: 10.1208/aapsj080362
Taegtmeyer, H., Young, M. E., Lopaschuk, G. D., Abel, E. D., Brunengraber, H., Darley-Usmar, V., et al. (2016). Assessing Cardiac Metabolism: A Scientific Statement From the American Heart Association. Circ. Res. 118, 1659–1701. doi: 10.1161/RES.0000000000000097
Takemura, G., Kanamori, H., Okada, H., Miyazaki, N., Watanabe, T., Tsujimoto, A., et al. (2018). Anti-apoptosis in nonmyocytes and pro-autophagy in cardiomyocytes: two strategies against postinfarction heart failure through regulation of cell death/degeneration. Heart Fail Rev. 23, 759–772. doi: 10.1007/s10741-018-9708-x
Taylor, R. W., and Turnbull, D. M. (2005). Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402. doi: 10.1038/nrg1606
Tohme, S., Yazdani, H. O., Liu, Y., and Loughran, P. (2017). Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 66, 182–197. doi: 10.1002/hep.29184/suppinfo
Torrealba, N., Aranguiz, P., Alonso, C., Rothermel, B. A., and Lavandero, S. (2017). “Mitochondria in Structural and Functional Cardiac Remodeling,” in Mitochondrial Dynamics in Cardiovascular Medicine Advances in Experimental Medicine and Biology, ed. G. Santulli (Cham: Springer), 277–306. doi: 10.1007/978-3-319-55330-6_15
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Tuppen, H. A. L., Blakely, E. L., Turnbull, D. M., and Taylor, R. W. (2010). Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 1797, 113–128. doi: 10.1016/j.bbabio.2009.09.005
Twig, G., Elorza, A., Molina, A. J. A., Mohamed, H., Wikstrom, J. D., Walzer, G., et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. doi: 10.1038/sj.emboj.7601963
Van Houtven, G., Honeycutt, A., Gilman, B., and McCall, N. (2008). Costs of Illness Among Older Adults: An Analysis of Six Major Health Conditions with Significant Environmental Risk Factors. RTI Int. 2008:809. doi: 10.3768/rtipress.2008.rr.0002.0809
Vasquez-Trincado, C., Garcia-Carvajal, I., Pennanen, C., Parra, V., Hill, J. A., Rothermel, B. A., et al. (2016). Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. London 594, 509–525. doi: 10.1113/JP271301
Veronese, N., Stubbs, B., Koyanagi, A., Vaona, A., Demurtas, J., Schofield, P., et al. (2019). Mitochondrial genetic haplogroups and cardiovascular diseases: Data from the Osteoarthritis Initiative. PLoS One 14:213656–213610e. doi: 10.1371/journal.pone.0213656
Wallace, D. C., Fan, W., and Procaccio, V. (2010). Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 5, 297–348. doi: 10.1146/annurev.pathol.4.110807.092314
Wang, K., Long, B., Jiao, J.-Q., Wang, J.-X., Liu, J.-P., Li, Q., et al. (2012). miR-484 regulates mitochondrial network through targeting Fis1. Nat. Commun. 3:781. doi: 10.1038/ncomms1770
Wang, S., Ge, W., Harns, C., Meng, X., Zhang, Y., and Ren, J. (2018). Ablation of toll-like receptor 4 attenuates aging-induced myocardial remodeling and contractile dysfunction through NCoRI-HDAC1-mediated regulation of autophagy. J. Mol. Cell. Cardiol. 119, 40–50. doi: 10.1016/j.yjmcc.2018.04.009
Wang, S.-J., Chen, H., Tang, L.-J., Tu, H., Liu, B., Li, N.-S., et al. (2020). Upregulation of mitochondrial E3 ubiquitin ligase 1 in rat heart contributes to ischemia/reperfusion injury. Can. J. Physiol. Pharmacol. 98, 259–266. doi: 10.1139/cjpp-2019-0285
Wei, Y., Chiang, W.-C., Sumpter, R. Jr., Mishra, P., and Levine, B. (2017). Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168, 224.e–238.e. doi: 10.1016/j.cell.2016.11.042
West, A. P., Shadel, G. S., and Ghosh, S. (2011). Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402. doi: 10.1038/nri2975
Westermann, B. (2012). Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta Bioenerget. 1817, 1833–1838. doi: 10.1016/j.bbabio.2012.02.033
Williams, E. G., Wu, Y., Wolski, W., Kim, J. Y., Lan, J., Hasan, M., et al. (2018). Quantifying and Localizing the Mitochondrial Proteome Across Five Tissues in A Mouse Population. Mol. Cell Proteomics 17, 1766–1777. doi: 10.1074/mcp.RA118.000554
Woodall, B. P., Orogo, A. M., Najor, R. H., Cortez, M. Q., Moreno, E. R., Wang, H., et al. (2019). Parkin does not prevent accelerated cardiac aging in mitochondrial DNA mutator mice. JCI Insight 4, 2297–2219. doi: 10.1172/jci.insight.127713
Wu, N. N., Zhang, Y., and Ren, J. (2019). Mitophagy, Mitochondrial Dynamics, and Homeostasis in Cardiovascular Aging. Oxidat. Med. Cell. Longevity 2019, 1–15. doi: 10.1155/2019/9825061
Wu, X., Al-Amin, M., Zhao, C., An, F., Wang, Y., Huang, Q., et al. (2020). Catechinic acid, a natural polyphenol compound, extends the lifespan of Caenorhabditis elegans via mitophagy pathways. Food Funct. 11, 5621–5634. doi: 10.1039/d0fo00694g
Xu, Z., Yang, L., Xu, S., Zhang, Z., and Cao, Y. (2015). The receptor proteins: pivotal roles in selective autophagy. Acta Biochim. Biophys. Sin 47, 571–580. doi: 10.1093/abbs/gmv055
Yamaguchi, O., Murakawa, T., Nishida, K., and Otsu, K. (2016). Receptor-mediated mitophagy. J. Mol. Cell. Cardiol. 95, 50–56. doi: 10.1016/j.yjmcc.2016.03.010
Yang, Y., Noonan, M., Cilenti, L., Qiu, Y., Zervos, A., and Liao, R. (2016). Abstract 19341: The Role of Mul1, a Mitochondrial Localized E3 Ligase, in the Heart. Circulation 134:A19341. doi: 10.1161/circ.134.suppl_1.19341
Yang, Y.-W., and Koob, M. D. (2012). Transferring isolated mitochondria into tissue culture cells. Nucleic Acids Res. 40:639. doi: 10.1093/nar/gks639
Yasuzaki, Y., Yamada, Y., Kanefuji, T., and Harashima, H. (2013). Localization of exogenous DNA to mitochondria in skeletal muscle following hydrodynamic limb vein injection. J. Control. Release 172, 805–811. doi: 10.1016/j.jconrel.2013.09.029
Yoo, S. M., Yamashita, S.-I., Kim, H., Na, D., Lee, H., Kim, S. J., et al. (2020). FKBP8 LIRL−dependent mitochondrial fragmentation facilitates mitophagy under stress conditions. FASEB J. 34, 2944–2957. doi: 10.1096/fj.201901735R
Yoon, Y. G., Koob, M. D., and Yoo, Y. H. (2010). Re-engineering the mitochondrial genomes in mammalian cells. Anat. Cell Biol. 43, 97–13. doi: 10.5115/acb.2010.43.2.97
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Yuan, Y., Cruzat, V. F., Newsholme, P., Cheng, J., Chen, Y., and Lu, Y. (2016). Regulation of SIRT1 in aging: Roles in mitochondrial function and biogenesis. Mech. Ageing Dev. 155, 10–21. doi: 10.1016/j.mad.2016.02.003
Yun, J., Puri, R., Yang, H., Lizzio, M. A., Wu, C., Sheng, Z.-H., et al. (2014). MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife Sci. 3:e16038. doi: 10.7554/eLife.01958
Yussman, M. G., Toyokawa, T., Odley, A., Lynch, R. A., Wu, G., Colbert, M. C., et al. (2002). Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat. Med. 8, 725–730. doi: 10.1038/nm719
Zhang, H., Alder, N. N., Wang, W., Szeto, H., Marcinek, D. J., and Rabinovitch, P. S. (2020). Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. eLife Sci. 9, 1180–1118. doi: 10.7554/eLife.60827
Zhang, J., and Ney, P. A. (2009). Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 16, 939–946. doi: 10.1038/cdd.2009.16
Zhang, X.-Y., and Zhang, P.-Y. (2016). Mitochondria targeting nano agents in cancer therapeutics. Oncol. Lett. 12, 4887–4890. doi: 10.3892/ol.2016.5302
Zhang, Y., Wang, C., Jin, Y., Yang, Q., Meng, Q., Liu, Q., et al. (2018). Activating the PGC-1 α/TERT Pathway by Catalpol Ameliorates Atherosclerosis via Modulating ROS Production, DNA Damage, and Telomere Function: Implications on Mitochondria and Telomere Link. Oxidat. Med. Cell. Longevity 2018, 1–16. doi: 10.1155/2018/2876350
Zhang, Y., Yao, Y., Qiu, X., Wang, G., Hu, Z., Chen, S., et al. (2019). Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat. Immunol. 20, 1–19. doi: 10.1038/s41590-019-0324-2
Zhou, B., and Tian, R. (2018). Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investigat. 128, 3716–3726. doi: 10.1172/JCI120849
Zhou, H., Hu, S., Jin, Q., Shi, C., Zhang, Y., Zhu, P., et al. (2017). Mff-Dependent Mitochondrial Fission Contributes to the Pathogenesis of Cardiac Microvasculature Ischemia/Reperfusion Injury via Induction of mROS-Mediated Cardiolipin Oxidation and HK2/VDAC1 Disassociation-Involved mPTP Opening. J. Am. Heart Assoc. 6:H689. doi: 10.1161/JAHA.116.005328
Keywords: cardiac, OXPHOS, ROS, mtDNA heteroplasmy, aging, heart failure, mitophagy, biogenesis
Citation: Elorza AA and Soffia JP (2021) mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology. Front. Cell Dev. Biol. 9:625020. doi: 10.3389/fcell.2021.625020
Received: 02 November 2020; Accepted: 25 January 2021;
Published: 22 February 2021.
Edited by:
Giampaolo Morciano, University of Ferrara, ItalyReviewed by:
Vito De Pinto, University of Catania, ItalyCopyright © 2021 Elorza and Soffia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alvaro A. Elorza, YWx2YXJvLmVsb3J6YUB1bmFiLmNs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.