 Emmanuelle Canet-Soulas
Emmanuelle Canet-Soulas Laurence Bessueille
Laurence Bessueille Laura Mechtouff
Laura Mechtouff David Magne
David Magne- 1CarMeN Laboratory, INSERM, INRA, INSA Lyon, University of Lyon, Université Claude Bernard Lyon 1, Lyon, France
- 2ICBMS, CNRS, INSA Lyon, CPE, University of Lyon, Université Claude Bernard Lyon 1, Lyon, France
- 3Stroke Department, Hospices Civils de Lyon, Lyon, France
It has been known for decades or even centuries that arteries calcify as they age. Vascular calcification probably affects all adults, since virtually all have atherosclerotic plaques: an accumulation of lipids, inflammatory cells, necrotic debris, and calcium phosphate crystals. A high vascular calcium score is associated with a high cardiovascular mortality risk, and relatively recent data suggest that even microcalcifications that form in early plaques may destabilize plaques and trigger a cardiovascular event. If the cellular and molecular mechanisms of plaque calcification have been relatively well characterized in mice, human plaques appear to calcify through different mechanisms that remain obscure. In this context, we will first review articles reporting the location and features of early calcifications in human plaques and then review the articles that explored the mechanisms though which human and mouse plaques calcify.
Atherosclerotic Plaque Calcifications: The Smaller, the Scarier
Different Types of Human Plaque Calcification
Cardiovascular diseases are the leading cause of death worldwide (Roth et al., 2017). Atherosclerotic plaque rupture is the primary mechanism responsible for myocardial infarction and accounts for about 20% of cases of ischemic stroke (Ornello et al., 2018). Since coronary artery calcium scores correlate with cardiovascular mortality in asymptomatic individuals (Greenland et al., 2004), it was long believed that plaque calcification had a detrimental impact on plaque stability. In the last two decades, however, clinical and preclinical studies suggested that plaque calcification may have either beneficial or detrimental effects depending on the amount of calcium and type of calcification. Human plaques, in particular, can present very different types of calcification (Figure 1; Herisson et al., 2011; Jinnouchi et al., 2020). Microcalcifications, defined by size <15 or 50 μm depending on the author, can be observed in early type I lesions (Roijers et al., 2011; Chatrou et al., 2015). They sometimes coalesce to generate punctate calcifications with a size between 15 μm (or 50 μm) and 1 mm (Jinnouchi et al., 2020). Bigger calcifications comprise fragment calcifications, which measure more than 1 mm, and sheet calcifications, defined by size more than 3 mm (Jinnouchi et al., 2020). Nodular calcifications result from the fracture of calcified sheets under mechanical stress, such as that associated with coronary hinge motion (Lee et al., 2017). Finally, plaque ossification, with trabecular-like structures and bone marrow, is also sometimes observed although predominantly in peripheral arteries (Herisson et al., 2011). If macrocalcifications were historically considered to be harmful, a new paradigm has recently emerged, suggesting that heavily calcified plaques are in fact more stable. This paradigm relies in particular on the assumption that, with progressive calcification, plaque inflammation becomes pacified, and the necrotic core walled off from the blood (Dweck et al., 2016). On the other hand, biomechanical studies still indicate that macrocalcifications likely generate a significant mechanical stress that may negatively affect plaque stability [reviewed in Barrett et al. (2019)]. It is not our aim in this article to discuss the clinical impact of macrocalcifications but to describe the molecular mechanisms through which calcification is initiated in atherosclerotic plaques.
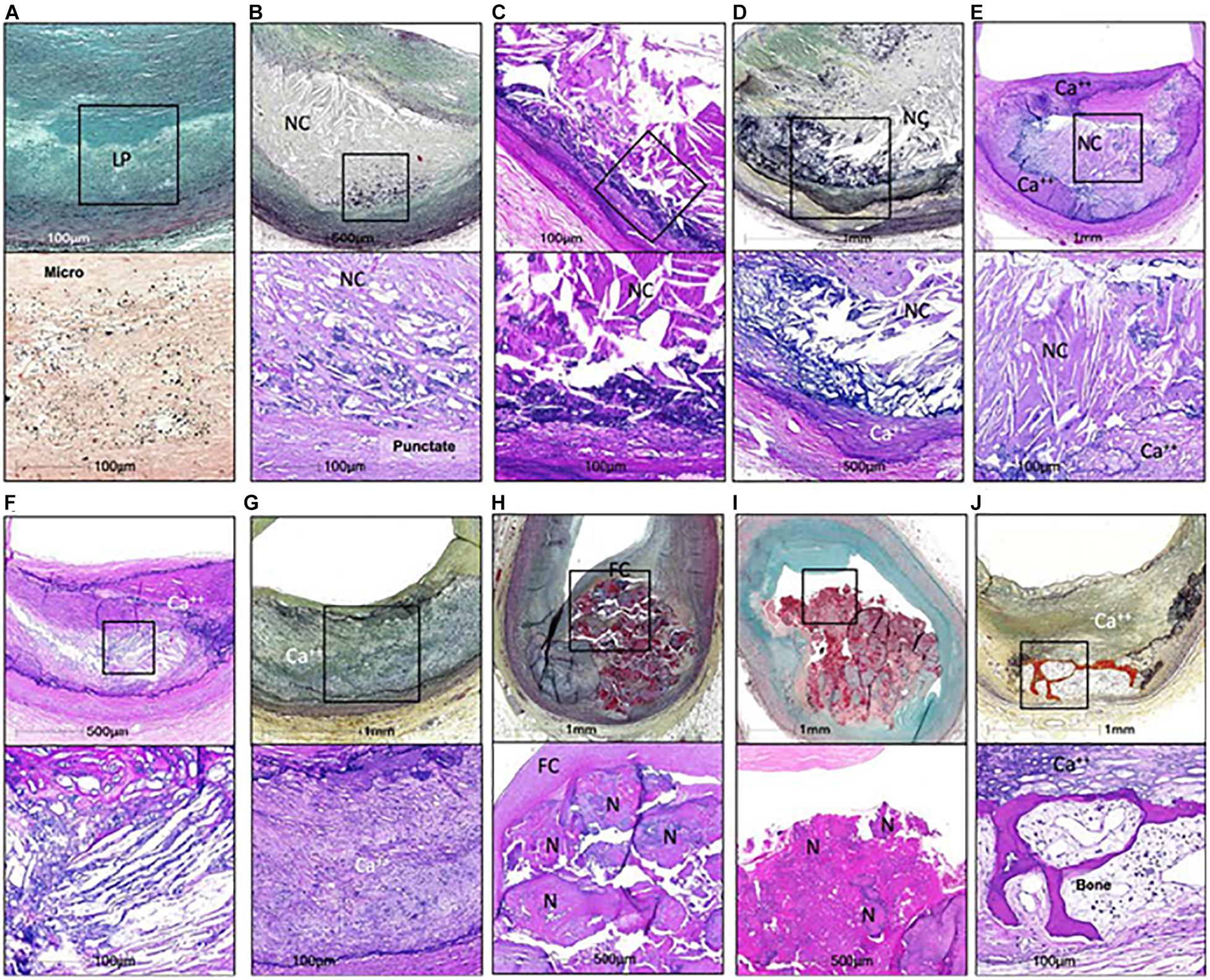
Figure 1. Calcifications in human coronaries. (A,B,D,G,H,I,J) Images in the top row are Movat pentachrome stained, and (C,E,F) Images are H&E stained; lower row shows high power image; corresponding to (A) Is Von Kossa staining and all others are H&E. Non-decalcified arterial segments (A) And all others are decalcified segments (B–J). (A) Pathological intimal thickening characterized by a lipid pool (IP) that lacks VSMCs. Corresponding high power image (Von Kossa staining) of the boxed area shows microcalcification <15 μm in diameter within the LP. (B) Fibroatheroma showing an early necrotic core (NC) infiltrated by macrophages which are calcified, seen as punctate (≥15 μm) areas of calcification. (C) Fibroatheroma with a late NC and fragmented calcification seen toward the medial wall. (D) Late fibroatheroma with larger area of calcification occupying an area greater than 1 mm that shows calcification of the NC. (E) Fibrocalcific plaque with sheet calcification and calcifying NC, which is incompletely calcified. (F) Fibrocalcific plaque showing sheet calcium with both fibrous tissue and NC completely calcified. (G) Fibrocalcific plaque with sheet calcium without a NC. (H) Nodular calcification showing fragment of calcium separated by fibrin and lumina I coverage by fibrous cap. (I) Calcified nodule showing nodules of calcium within the lumen and an overlying thrombus. (J) Fibrocalcific plaque with an area of ossification at the edge of sheet calcification. Ca++, calcification; FC, fibrous cap; H&E, hematoxylin and eosin; N, nodule. From Jinnouchi et al. (2020), with permission from Elsevier.
Microcalcifications: New Suspects for Plaque Rupture
The discovery of microcalcifications, as opposed to large calcifications, in soft plaques undergoing pathology analysis (Vengrenyuk et al., 2006; Maldonado et al., 2012; Kelly-Arnold et al., 2013) suggested a role in acute events involving stress-induced plaque rupture, a mechanism long suspected in coronary arteries (Richardson et al., 1989). Microcalcifications are probably dangerous when they form in the fibrous cap (Vengrenyuk et al., 2006; Maldonado et al., 2012; Kelly-Arnold et al., 2013). Fibrous cap thickness is known to correlate with plaque vulnerability (Burke et al., 1997), and the presence of >5 μm microcalcifications might be particularly harmful, generating mechanical stress (Kelly-Arnold et al., 2013). In addition, microcalcifications exacerbate plaque inflammation, stimulating macrophages to release more tumor necrosis factor alpha (TNF-α) (Nadra et al., 2005, 2008), which also likely has detrimental effects on plaque progression.
While there is still no experimental proof that microcalcifications are indeed the most likely to trigger plaque rupture, an increasing number of studies using positron emission tomography (PET) with sodium fluoride 18 radiotracer (18F-NaF) have spotlighted the risk. Fluoride ions replace hydroxyl ions preferentially in newly formed, immature apatite crystals and are therefore a very interesting tool for detecting microcalcification (Hawkins et al., 1992). 18F-NaF PET has long been used to detect abnormal bone formation (Hawkins et al., 1992), but it was not until 2010 that a hybrid PET-CT camera enabled detection of 18F-NaF fixation in atherosclerosis plaque (Derlin et al., 2010, 2011). Seminal studies showed that 18F-NaF uptake was associated with features of instability and was more pronounced in clinically adjudicated culprit plaques in patients with myocardial infarction or ischemic stroke (Joshi et al., 2014; Vesey et al., 2017). However, it is now established that not only microcalcifications but also bigger ones that are actively growing are 18F-NaF positive, complicating the clinical interpretation of 18F-NaF PET-CT (Irkle et al., 2015; Høilund-Carlsen et al., 2020). Nevertheless, whereas the classical CT calcium score for macrocalcification is used to assess atherosclerotic load in coronary arteries, whole-body 18F-NaF PET-CT may be used as a marker of atherosclerotic plaque burden characterizing the patient’s global risk rather than focusing exclusively on the culprit plaque (Arbab-Zadeh and Fuster, 2015). In the clinical setting, PET-MRI will certainly help to better characterize the microcalcification process in plaque composition and will be more suitable for longitudinal studies. In a recent review on PET-MRI, Evans et al. (2020) pointed out the importance of combining 18F-NaF molecular calcification imaging and high-resolution MRI plaque characterization. An initial study by our group found that 18F-NaF-positive lesions were not associated with known MRI criteria of vulnerability (Mechtouff et al., 2020), but further longitudinal studies could helpfully use contrast-enhanced MRI to check whether 18F-NaF uptake precedes the vulnerable state. As longitudinal clinical studies are still lacking and are also rare in preclinical models of microcalcification (Hsu et al., 2020), the mechanisms underlying 18F-NaF uptake and the link with specific inflammatory processes are not fully elucidated. We are currently performing a preclinical study in mice with 18F-NaF PET to determine the impact of microcalcifications on plaque development.
If, in the near future, the harmful impact of microcalcifications is experimentally proven, it will be crucial to better understand how they form. Two models can be drawn from the literature. First, vascular smooth muscle cells (VSMCs) may undergo phenotypic changes to transform into osteochondrocyte-like cells. Many factors have indeed been shown to induce this phenotypic transformation. An alternative hypothesis is that calcification initiates independently of osteochondrocyte markers; this is supported by histopathological findings. In the next two chapters, we review the arguments in favor of each hypothesis.
Arguments in Favor of the Phenotypic Change Hypothesis
Plaques Are Calcified by Endochondral Ossification in Mice
Since it is obviously extremely difficult to analyze the longitudinal process of plaque calcification in humans, atherosclerotic mice have been widely used to investigate how plaques calcify. Two mouse models have been explored in depth: mice deficient in apolipoprotein E (ApoE) were used in most studies and mice deficient in low-density lipoprotein receptor (Ldlr) in some others. When ApoE-deficient mice are given a high-fat diet from 10 weeks of age, calcification can be detected in the aorta from the age of 20 weeks (Aikawa et al., 2007). Histological examination of animals aged between 45 and 75 weeks revealed the presence of chondrocyte-like cells, expressing type II collagen in calcified regions, suggesting that plaque calcification develops through a process mimicking endochondral ossification (Qiao et al., 1995; Rattazzi et al., 2005). Importantly, in these studies, the authors observed chondrocytes before calcifications and concluded that plaque calcification results from cartilage metaplasia (Rattazzi et al., 2005).
If plaque calcification is indeed an active process relying on chondrocyte differentiation, and if VSMCs are involved, then deletion of RUNX2 in VSMCs should prevent it. RUNX2 is the master transcription factor governing the differentiation and maturation of mineralizing cells, i.e., hypertrophic chondrocytes and osteoblasts (Komori et al., 1997). Among other transcriptional targets, RUNX2 stimulates the expression of tissue-non-specific alkaline phosphatase (TNAP), a promineralizing enzyme (Murshed et al., 2005). TNAP allows mineralization to occur by hydrolyzing inorganic pyrophosphate (PPi), a constitutive mineralization inhibitor (Hessle et al., 2002; Murshed et al., 2005). Mineralization is physiologically restricted to growth-plate cartilage and bone despite TNAP being relatively ubiquitous because mineralization requires a fibrillar collagen as a template for crystal deposition and because TNAP and fibrillar collagens are only coexpressed in growth-plate cartilage and bone (Murshed et al., 2005). Two independent groups produced and analyzed atherosclerotic mice deficient in RUNX2 specifically in VSMCs. In ApoE–/– mice, RUNX2 deletion almost completely prevented TNAP expression and calcification (Sun et al., 2012). VSMC-specific deletion of RUNX2 in Ldlr–/– mice reduced plaque calcification, with a decrease in several RUNX2 transcriptional targets such as TNAP and type X collagen (Lin et al., 2016). These two articles clearly suggest that most calcium deposition in mouse plaque relies on VSMC phenotypic change into hypertrophic chondrocytes. Many factors have been shown to stimulate VSMCs to transdifferentiate into mineralizing cells, but the vast majority are associated with inflammation and oxidative stress, which often go hand in hand (Demer and Tintut, 2011).
Plaque Ossification Appears to Be Stimulated by Inflammation in Mice
Interestingly, early calcification was associated with inflammation in the aorta of ApoE–/– mice (Aikawa et al., 2007). A wide range of inflammatory molecules relevant to atherosclerosis have been shown in vitro to trigger the phenotypic change of VSMCs into RUNX2-expressing osteoblast-like cells and/or chondrocyte-like cells. Inflammatory phenotypic change cytokines such as TNF-α, interleukin (IL)-1β, and IL-6 are particularly potent calcifying factors in human and murine VSMCs [reviewed in Bessueille and Magne (2015)]. These promineralizing effects of inflammatory cytokines on VSMCs may not only be in vitro findings, since treatment of Ldlr–/– mice with an anti-IL-1β (Awan et al., 2015) or anti-TNF-α (Al-Aly et al., 2007) antibody reduced plaque calcification. In addition, toll-like receptor (TLR)-2 and TLR-4 agonists such as lipopolysaccharide also stimulated VSMC change into chondrocytes in vitro, and ApoE–/– mice also deficient in TLR-2 developed reduced plaque calcification with reduced cartilage metaplasia (Lee et al., 2019). This effect of LPS might be relevant to plaque calcification, since endotoxemia occurs after virtually all fatty meals (Herieka and Erridge, 2014), and is associated with atherosclerosis (Wiedermann et al., 1999).
This stimulatory effect of inflammation on VSMC phenotypic change into RUNX2-expressing chondrocytes is in contrast to the known inhibitory effect of inflammation on chondrocyte differentiation (Lencel et al., 2011). One possible explanation is that inflammation stimulates expression of bone morphogenetic protein 2 (BMP2), a strong bone anabolic factor, in VSMCs (Ikeda et al., 2012), which may induce VSMC differentiation into chondrocytes when inflammation begins to resolve (Li et al., 2008; Liberman et al., 2011). Interestingly, reduced plaque calcification in Ldlr–/– mice with anti-TNF-α treatment was associated with reduced BMP2 levels (Al-Aly et al., 2007). In addition, VSMC-targeted overexpression of BMP2 in ApoE–/– mice resulted in increased plaque calcification (Nakagawa et al., 2010). More importantly, inhibition of BMP2 with a small chemical inhibitor or with a recombinant BMP antagonist decreased plaque calcification, lipid deposition, and inflammation in Ldlr–/– mice (Derwall et al., 2012). Furthermore, ApoE–/– mice also deficient in BMP endothelial cell precursor-derived regulator (Bmper) exhibited increased BMP activity in endothelial cells and developed larger and more calcified atherosclerotic lesions (Pi et al., 2012). Finally, inhibition of plaque calcification by overexpression of matrix Gla protein (MGP), which is suspected to act as an inhibitor of BMP2 signaling (Zebboudj et al., 2002; Malhotra et al., 2015), decreased plaque calcification and lesion development in ApoE–/– mice, in association with reduced BMP activity, strongly reduced macrophage infiltration and inflammation (Yao et al., 2010). Taken together, these results suggest that plaque calcification is dependent on BMP2 and therefore on chondrocyte differentiation. They also suggest that the development of calcified cartilage adversely impacts plaque development. This hypothesis, however, has to be considered with caution because chemical BMP inhibition reduced cholesterol biosynthesis and attenuated liver steatosis, suggesting that indirect effects might account for BMP2’s impact on plaques (Derwall et al., 2012). Moreover, MGP may inhibit vascular calcification independently of BMP2-stimulated VSMC change into chondrocytes (Leroux-Berger et al., 2011; Khavandgar et al., 2014), and MGP deficiency, which strongly increases vascular calcification, also protected against lesion development and inflammation in ApoE–/– mice (Yao et al., 2010).
Similarly to pathogen-associated molecular patterns (PAMPs), such as LPS, and inflammatory cytokines, oxidized lipids may represent danger-associated molecular patterns (DAMPs), activating receptors in the toll-like receptor superfamily (Miller et al., 2011), and stimulate calcification in VSMCs culture (Demer and Tintut, 2011). Oxidative stress generated by H2O2 induced RUNX2 expression through AKT in mouse VSMCs (Mody et al., 2001; Byon et al., 2008). Acetylated low-density lipoproteins (LDLs) induced greater calcification than native LDL in human VSMCs (Proudfoot et al., 2002). In bovine VSMCs, oxidized LDL increased TNAP activity and calcification (Parhami et al., 2002). Oxidized LDL stimulated calcification in human VSMCs through TLR-4 and expression of osteochondrogenic factors (Song et al., 2017). Chondrogenic differentiation and calcification in response to oxidized LDL in human VSMCs involve activation of transforming growth factor (TGF)-β (Yan et al., 2011). These in vitro findings are, nevertheless, in contradiction with the recent report that VSMC-specific ablation of TGF-β signaling in ApoE–/– mice leads to aortic aneurysms, with extensive lipid and calcium accumulation throughout the aorta (Chen et al., 2020); in this study, deletion of TGF-β signaling in VSMCs led to their dedifferentiation toward mesenchymal stem cells, enabling commitment toward chondrocytes and adipocytes (Chen et al., 2020). Finally, while inflammatory and oxidized molecules promote VSMC phenotypic change and calcification in vitro, how they act in vivo and whether they activate BMP and TGF-β growth factors still needs to be better understood. Moreover, there are solid arguments supporting the hypothesis that, in vivo, plaque calcification begins independently of VSMC phenotypic change.
Arguments Against the Phenotypic Change Hypothesis
Not So Many Human Plaques Calcify Through an Ossification-Like Process
While plaque calcification incontestably develops by a process similar to endochondral ossification in mice (Rosenfeld et al., 2000; Rattazzi et al., 2005; Lin et al., 2016), studies in humans showing the presence of chondrocytes or cartilage are rare to say the least and remain controversial (Tyson et al., 2003; Aigner et al., 2008; Kuzan et al., 2017). In addition, bone-like structures, regardless of whether they formed through endochondral or intramembranous ossification, are not the most common type of calcification in human plaques (Jinnouchi et al., 2020): they are frequently observed in femoral arteries but not, for instance, in carotids (Herisson et al., 2011). Furthermore, the increasing number of studies of microcalcifications in early plaques reported that they formed independently of chondrocyte- or osteoblast-like cells or markers. In coronary arteries, for instance, microcalcifications were observed in preatheroma type I lesions before BMP2, or the RUNX2 transcriptional target osteocalcin could be detected (Roijers et al., 2011; Chatrou et al., 2015). It seems unlikely that, in these studies, chondrocytes or osteoblasts were present and but not be detected because, in most cases, microcalcifications that had grown and coalesced generated macrocalcifications still devoid of osteoblast or chondrocyte activity (Herisson et al., 2011; Jinnouchi et al., 2020).
How can we explain that mouse plaques calcify through endochondral ossification, whereas human plaques more frequently develop independently of chondrocytes or osteoblasts? One explanation could be that, in mice as in humans, calcification initially occurs independently of chondrocyte differentiation, which is induced later on. It is noteworthy that, in mice, VSMC-specific deletion of RUNX2 reduces but does not fully prevent plaque calcification (Sun et al., 2012; Lin et al., 2016). It is, however, difficult to know whether calcifications that formed despite the absence of RUNX2 in VSMCs did so independently of RUNX2 or rather under the control of RUNX2 in cells other than VSMCs (Lin et al., 2016). In the next section, we will review the main mechanisms that may lead to calcification independently of RUNX2 and of chondrocytes or osteoblasts.
Calcification May Begin on Cell Debris
One very plausible mechanism of early microcalcification formation involves cell debris (Kim, 1995). Whereas extracellular calcium phosphate precipitation is mainly prevented by the presence of mineralization inhibitors such as PPi, intracellular calcification is normally prevented by the physical separation of calcium ions and inorganic phosphate (Pi). Calcium is mainly stored in the endoplasmic reticulum and is present at very low concentrations in the cytoplasm, where Pi levels are higher than in the extracellular fluids (Romero-Garcia and Prado-Garcia, 2019). Apoptosis normally does not compromise this separation, or at least does not allow intracellular calcification to propagate extracellularly, because integrity of the cell membrane and of the membrane of apoptotic bodies (ABs) is preserved. However, when ABs are not phagocytosed rapidly enough by macrophages, they undergo necrosis, characterized by membrane rupture, allowing calcium, and Pi to precipitate. It is well known that apoptotic debris clearance is impaired in atherosclerotic plaque (Schrijvers et al., 2005), and calcification on apoptotic debris is frequent in early plaques. Histological examination of thousands of coronary arteries revealed that microcalcifications often form in proximity to apoptotic VSMCs, and that calcifying apoptotic macrophages are often seen in association with punctate calcifications resulting from microcalcifications (Jinnouchi et al., 2020). The role of apoptosis in initiating calcification is strengthened by the report that inhibition of apoptosis by a broad caspase inhibitor reduced calcification in cultured human VSMCs (Proudfoot et al., 2000). Impaired AB clearance rather than ABs themselves is likely involved in calcification, since induction of apoptosis specifically in VSMCs in vivo does not lead to arterial inflammation or calcification in wild-type mice, whereas it leads to reduced fibrous cap thickness and collagen content, together with increased inflammation and calcification in ApoE–/– mice (Clarke et al., 2006, 2008).
These data strongly suggest that necrosis secondary to VSMC apoptosis induces or increases plaque calcification. Alternatively, or in addition to secondary necrosis, programmed necrosis, also known as necroptosis, may participate in plaque calcification. Necroptosis was discovered relatively recently as a proinflammatory type of programmed cell death controlled by receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3 (Choi et al., 2019). Upon induction of necroptosis, RIPK3 phosphorylates the mixed-lineage kinase domain-like (MLKL) protein, which leads to MLKL oligomerization, membrane translocation, and formation of a pore allowing extracellular release of intracellular molecules (Kolbrink et al., 2020). Interestingly, Ldlr–/–;Ripk3–/– mice develop smaller necrotic cores than Ldlr–/– mice, indicating that necroptosis is a significant form of cell death in plaques, and exerts negative effects (Lin et al., 2013). This was confirmed in ApoE–/–;Ripk3–/– mice, which showed less plaque inflammation and later mortality than ApoE–/– mice (Meng et al., 2015). In addition, treatment of ApoE–/– mice with necrostatin-1, an inhibitor of RIPK1–RIPK3 interaction and subsequent necroptosis, reduces lesion size and necrotic core formation (Karunakaran et al., 2016). Taken together, these studies suggest that necroptosis, like necrosis induced by impaired apoptotic cell clearance, has a detrimental effect in plaque development that might include induction of calcification. However, this interpretation, according to which necroptosis induces calcification and has harmful effects only in atherosclerosis, might be oversimplistic. Indeed, specific Ripk3 deletion in macrophages or endothelial cells protects ApoE–/– mice from lipid accumulation, suggesting that necroptosis impacts plaque development differently depending on the cell lineage in which it takes place (Colijn et al., 2020). Moreover, a recent study surprisingly showed that, while inhibition of MLKL expression in ApoE–/– mice predictably impaired necroptosis, it increased lipid accumulation within the plaques (Rasheed et al., 2020). To our knowledge, whether plaque calcification can be prevented by inhibition of necroptosis has not yet been specifically investigated; since necrosis is known to induce calcification and since microcalcifications are often associated with dying macrophages and VSMCs (Jinnouchi et al., 2020), more studies are warranted.
In addition to necroptosis, another form of cell death, called pyroptosis, may participate in plaque calcification. Pyroptosis is the form of cell death associated with secretion of IL-1β relying on NOD-like receptor family pyrin-domain-containing 3 (NLRP3) activation (Kesavardhana et al., 2020). It has long been known that NLRP3 activation leads to caspase-1-mediated cleavage of pro-IL-1β into mature IL-1β; however, how this mature IL-1β is released extracellularly was discovered only recently. Activated caspase-1 not only cleaves the cytoplasmic protein gasdermin D, allowing the N-terminal fragment of gasdermin D to polymerize in the membrane and form pores through which IL-1β is released (Shi et al., 2015), but also leads to cell death (Kesavardhana et al., 2020). IL-1β is a very important cytokine in atherosclerosis, which modulates multiple aspects of plaque formation (Bhaskar et al., 2011) and development (Gomez et al., 2018) and has emerged as a promising target in patients with previous myocardial infarction (Ridker et al., 2017). However, probably because IL-1β has both beneficial and detrimental effects on atherosclerotic plaque development in ApoE–/– mice (Bhaskar et al., 2011; Gomez et al., 2018), manipulation of caspase-1 and NLRP3 levels in ApoE–/– mice provided contradictory results (Menu et al., 2011; Zheng et al., 2014; Yin et al., 2015; van der Heijden et al., 2017). To our knowledge, the possibility that pyroptosis participates in plaque calcification in vivo has not been specifically addressed, but in vitro inhibition of inflammasome activation reduced IL-1β secretion and inhibited VSMC calcification (Wen et al., 2013).
Calcification May Result From the Release of Extracellular Vesicles and the Activation of TNAP
Finally, there are arguments suggesting that initiation of plaque calcification may be due to VSMCs that have acquired some functions of mineralizing cells, without truly differentiating into chondrocytes or osteoblasts. For instance, numerous in vitro studies and genetic models have shown that, often, the mere deficiency of a mineralization inhibitor or the mere upregulation of a promineralizing factor is sufficient to trigger calcification. In particular, several genetic diseases or mouse models indicate that a single enzyme, TNAP, is sufficient to induce arterial calcification. As described above, TNAP induces mineralization by hydrolyzing PPi (Hessle et al., 2002; Murshed et al., 2005), and constant physiological production of PPi is required to prevent vascular calcification. Deficient generation of PPi from extracellular ATP, due to mutations in the gene encoding ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1), leads to a disease known as generalized arterial calcification of infancy (GACI) (Rutsch et al., 2003). Logically, VSMC-specific overexpression of TNAP is sufficient to induce medial calcification in mice (Sheen et al., 2015). Interestingly, calcification in this model is associated with increased transcript levels of the osteochondrocyte markers Bmp2, Sox9, Acan, and Runx2 (Sheen et al., 2015), suggesting that TNAP not only stimulates calcification but also launches the whole phenotypic change process of VSMCs into osteochondrocyte-like cells. Molecular investigation of the mechanisms involved suggests that TNAP induces calcification in VSMCs, which in turn activates the bone anabolic factor BMP2 (Fakhry et al., 2017). Such a molecular sequence implies that TNAP is expressed before VSMC differentiation and independently of RUNX2. Another genetic disease, arterial calcification due to deficiency of CD73 (ACDC), offers a likely explanation (St Hilaire et al., 2011). CD73 is a relatively ubiquitous nucleotidase that dephosphorylates extracellular AMP into adenosine, to participate in the resolution of inflammation (Antonioli et al., 2013). Calcification in ACDC is due to upregulation of TNAP expression in the absence of adenosine, to compensate for decreased AMP dephosphorylation (Jin et al., 2016). TNAP has indeed recently been described as an anti-inflammatory nucleotidase, which explains its ubiquitous expression (Bessueille et al., 2020). Therefore, induction of TNAP expression in association with its inflammatory function may result in induction of plaque calcification, independently of RUNX2. The fact that TNF-α, IL-1β, or IL-6 stimulates TNAP expression in VSMCs supports this paradigm (Tintut et al., 2000; Shioi et al., 2002; Lee et al., 2010; Lencel et al., 2011; Zhao et al., 2012).
Finally, if TNAP emerges as a possible important contributor to microcalcification, it must be added that the role of TNAP in physiological mineralization is not to trigger crystal nucleation but to allow calcium phosphate crystals to grow (Fleisch et al., 1966; Millán and Whyte, 2016; Bottini et al., 2018). Crystal nucleation is thought to occur inside extracellular vesicles (EVs) released by hypertrophic chondrocytes and osteoblasts and generally named matrix vesicles (MVs) in the bone biology field (Bottini et al., 2018). Although still controversial, MVs are suspected to concentrate calcium and Pi through the channeling activity of annexins and Pit transporters, respectively (Yadav et al., 2016; Bottini et al., 2018). In addition, MVs may further concentrate Pi from phosphatidylcholine through the sequential activity of phospholipase A2, ectonucleotide pyrophosphatase/phosphodiesterase 6, and PHOSPHO1 (Roberts et al., 2007; Yadav et al., 2016; Stewart et al., 2018). Crystal formation inside MVs would then rely on phosphatidylserine-mediated nucleation (Wu et al., 1993; Cruz et al., 2020).
Increasing data suggest that VSMCs release EVs that may initiate vascular calcification similarly to the way MVs released by hypertrophic chondrocytes induce growth plate mineralization (Hutcheson et al., 2016). This suspected involvement of EVs in plaque calcification has been nicely reviewed recently (Kapustin and Shanahan, 2016; Bakhshian Nik et al., 2017; Blaser and Aikawa, 2018; Aikawa and Blaser, 2021). We will therefore briefly present what is known of their suspected contribution to plaque calcification. Electron microscopic exploration of human carotid plaques revealed that vulnerable plaques may contain more calcifying EVs than stable ones (Bobryshev et al., 2008). Interestingly, several distinct multilamellar vesicles were visible, suggesting that several types of vesicles may be involved in plaque calcification. In culture of VSMCs, calcification is reduced by inhibition of annexin A6 expression (Kapustin et al., 2011) or PHOSPHO1 activity (Kiffer-Moreira et al., 2013), two proteins thought to be important for MV-associated mineralization. However, there might be significant differences in the mechanisms governing EV and MV release and mineralization/calcification. It was particularly shown that, in VSMCs, sortilin regulates the load of TNAP into EVs and that sortilin deficiency reduces plaque calcification but not bone mineralization (Goettsch et al., 2016). Finally, not only MV-like EVs, which are membrane blebs but also exosomes, which have an intracellular origin, may be involved in VSMC-mediated calcification. Exosomes released from VSMCs were indeed shown to be enriched in factors involved in calcification, such as annexin A6 and phosphatidylserine (Kapustin et al., 2015; Kapustin and Shanahan, 2016). Finally and to add more complexity, macrophages have also been shown to release calcifying EVs (New et al., 2013; Aikawa and Blaser, 2021). Therefore, the origin and respective contribution EVs and exosomes to plaque calcification in vivo will be therefore a difficult task to assess, but which deserves intense efforts.
Conclusion
Arguments in favor of the phenotypic change hypothesis mainly come from mouse models of atherosclerosis, whereas human studies rather suggest that calcification begins independently of osteoblast or chondrocyte differentiation (Table 1). If microcalcification in mice, like in humans, originates independently of chondrocyte differentiation, then it will be interesting to understand why microcalcifications always lead to ossification in mice, but so infrequently in humans. Apatite crystals stimulate mouse VMSCs in vitro to express BMP2 (Sage et al., 2011), which triggers their chondrocyte differentiation (Fakhry et al., 2017). Human coronary plaques express BMP2 in association with calcifications (Boström et al., 1993; Dhore et al., 2001; Chatrou et al., 2015). Whether and why BMP2 is less potent in humans deserves investigation. Finally, it cannot be excluded that several different mechanisms initiate plaque calcification, since microcalcifications can be seen in the necrotic core of human plaques as floating debris or in the fibrous cap (Jinnouchi et al., 2020).

Table 1. Arguments in favor of, and arguments against the hypothesis that calcification is initiated by osteochondrocyte-like cells.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
We acknowledge financial support from the European Research Area Network on Cardiovascular Diseases (ERA-NET CVD, Microexploration project 2018-2021) to work on atherosclerotic plaque microcalcification.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aigner, T., Neureiter, D., Câmpean, V., Soder, S., and Amann, K. (2008). Expression of cartilage-specific markers in calcified and non-calcified atherosclerotic lesions. Atherosclerosis 196, 37–41. doi: 10.1016/j.atherosclerosis.2007.01.020
Aikawa, E., and Blaser, M. C. (2021). 2020 Jeffrey M. Hoeg Award Lecture: calcifying extracellular vesicles as building blocks of microcalcifications in cardiovascular disorders. Arterioscler. Thromb. Vasc. Biol. 41, 117–127.
Aikawa, E., Nahrendorf, M., Figueiredo, J. L., Swirski, F. K., Shtatland, T., Kohler, R. H., et al. (2007). Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation 116:2841. doi: 10.1161/circulationaha.107.732867
Al-Aly, Z., Shao, J. S., Lai, C. F., Huang, E., Cai, J., Behrmann, A., et al. (2007). Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 27, 2589–2596. doi: 10.1161/atvbaha.107.153668
Antonioli, L., Pacher, P., Vizi, E. S., and Haskó, G. (2013). CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 19, 355–367. doi: 10.1016/j.molmed.2013.03.005
Arbab-Zadeh, A., and Fuster, V. (2015). The myth of the “vulnerable plaque:: transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J. Am. Coll. Cardiol. 65, 846–855.
Awan, Z., Denis, M., Roubtsova, A., Essalmani, R., Marcinkiewicz, J., Awan, A., et al. (2015). Reducing vascular calcification by anti-IL-1β monoclonal antibody in a mouse model of familial hypercholesterolemia. Angiology 67, 157–167. doi: 10.1177/0003319715583205
Bakhshian Nik, A., Hutcheson, J. D., and Aikawa, E. (2017). Extracellular vesicles as mediators of cardiovascular calcification. Front. Cardiovasc. Med. 4:78. doi: 10.3389/fcvm.2017.00078
Barrett, H. E., Van der Heiden, K., Farrell, E., Gijsen, F. J. H., and Akyildiz, A. C. (2019). Calcifications in atherosclerotic plaques and impact on plaque biomechanics. J. Biomech. 87, 1–12. doi: 10.1016/j.jbiomech.2019.03.005
Bessueille, L., Briolay, A., Como, J., Mebarek, S., Mansouri, C., Gleizes, M., et al. (2020). Tissue-nonspecific alkaline phosphatase is an anti-inflammatory nucleotidase. Bone 133:115262. doi: 10.1016/j.bone.2020.115262
Bessueille, L., and Magne, D. (2015). Inflammation: a culprit for vascular calcification in atherosclerosis and diabetes. Cell. Mol. Life Sci. 72, 2475–2489. doi: 10.1007/s00018-015-1876-4
Bhaskar, V., Yin, J., Mirza, A. M., Phan, D., Vanegas, S., Issafras, H., et al. (2011). Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in Apolipoprotein E-deficient mice. Atherosclerosis 216, 313–320. doi: 10.1016/j.atherosclerosis.2011.02.026
Blaser, M. C., and Aikawa, E. (2018). Roles and regulation of extracellular vesicles in cardiovascular mineral metabolism. Front. Cardiovasc. Med. 5:187. doi: 10.3389/fcvm.2018.00187
Bobryshev, Y. V., Killingsworth, M. C., Lord, R. S., and Grabs, A. J. (2008). Matrix vesicles in the fibrous cap of atherosclerotic plaque: possible contribution to plaque rupture. J. Cell. Mol. Med. 12, 2073–2082. doi: 10.1111/j.1582-4934.2008.00230.x
Boström, K., Watson, K. E., Horn, S., Wortham, C., Herman, I. M., and Demer, L. L. (1993). Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Invest. 91, 1800–1809. doi: 10.1172/jci116391
Bottini, M., Mebarek, S., Anderson, K. L., Strzelecka-Kiliszek, A., Bozycki, L., Simão, A. M. S., et al. (2018). Matrix vesicles from chondrocytes and osteoblasts: their biogenesis, properties, functions and biomimetic models. Biochim. Biophys. Acta Gen. Subj. 1862, 532–546. doi: 10.1016/j.bbagen.2017.11.005
Burke, A. P., Farb, A., Malcom, G. T., Liang, Y. H., Smialek, J., and Virmani, R. (1997). Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N. Engl. J. Med. 336, 1276–1282. doi: 10.1056/nejm199705013361802
Byon, C. H., Javed, A., Dai, Q., Kappes, J. C., Clemens, T. L., Darley-Usmar, V. M., et al. (2008). Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 283, 15319–15327. doi: 10.1074/jbc.m800021200
Chatrou, M. L., Cleutjens, J. P., van der Vusse, G. J., Roijers, R. B., Mutsaers, P. H., and Schurgers, L. J. (2015). Intra-section analysis of human coronary arteries reveals a potential role for micro-calcifications in macrophage recruitment in the early stage of atherosclerosis. PLoS One 10:e0142335. doi: 10.1371/journal.pone.0142335
Chen, P. Y., Qin, L., Li, G., Malagon-Lopez, J., Wang, Z., Bergaya, S., et al. (2020). Smooth muscle cell reprogramming in aortic aneurysms. Cell Stem Cell 26, 542–557.e11.
Choi, M. E., Price, D. R., Ryter, S. W., and Choi, A. M. K. (2019). Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight 4:e128834.
Clarke, M. C., Figg, N., Maguire, J. J., Davenport, A. P., Goddard, M., Littlewood, T. D., et al. (2006). Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med. 12, 1075–1080. doi: 10.1038/nm1459
Clarke, M. C., Littlewood, T. D., Figg, N., Maguire, J. J., Davenport, A. P., Goddard, M., et al. (2008). Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ. Res. 102, 1529–1538. doi: 10.1161/circresaha.108.175976
Colijn, S., Muthukumar, V., Xie, J., Gao, S., and Griffin, C. T. (2020). Cell-specific and athero-protective roles for RIPK3 in a murine model of atherosclerosis. Dis. Model. Mech. 13:dmm041962. doi: 10.1242/dmm.041962
Cruz, M. A. E., Ferreira, C. R., Tovani, C. B., de Oliveira, F. A., Bolean, M., Caseli, L., et al. (2020). Phosphatidylserine controls calcium phosphate nucleation and growth on lipid monolayers: a physicochemical understanding of matrix vesicles-driven biomineralization. J. Struct. Biol. 212:107607. doi: 10.1016/j.jsb.2020.107607
Demer, L., and Tintut, Y. (2011). The roles of lipid oxidation products and receptor activator of nuclear factor-κB signaling in atherosclerotic calcification. Circ. Res. 108, 1482–1493. doi: 10.1161/circresaha.110.234245
Derlin, T., Richter, U., Bannas, P., Begemann, P., Buchert, R., Mester, J., et al. (2010). Feasibility of 18F-sodium fluoride PET/CT for imaging of atherosclerotic plaque. J. Nucl. Med. 51, 862–865. doi: 10.2967/jnumed.110.076471
Derlin, T., Wisotzki, C., Richter, U., Apostolova, I., Bannas, P., Weber, C., et al. (2011). In vivo imaging of mineral deposition in carotid plaque using 18F-sodium fluoride PET/CT: correlation with atherogenic risk factors. J. Nucl. Med. 52, 362–368. doi: 10.2967/jnumed.110.081208
Derwall, M., Malhotra, R., Lai, C. S., Beppu, Y., Aikawa, E., Seehra, J. S., et al. (2012). Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 613–622. doi: 10.1161/atvbaha.111.242594
Dhore, C. R., Cleutjens, J. P., Lutgens, E., Cleutjens, K. B., Geusens, P. P., Kitslaar, P. J., et al. (2001). Differential expression of bone matrix regulatory proteins in human atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 21, 1998–2003. doi: 10.1161/hq1201.100229
Dweck, M. R., Aikawa, E., Newby, D. E., Tarkin, J. M., Rudd, J. H., Narula, J., et al. (2016). Noninvasive molecular imaging of disease activity in atherosclerosis. Circ. Res. 119, 330–340. doi: 10.1161/circresaha.116.307971
Evans, N. R., Tarkin, J. M., Le, E. P., Sriranjan, R. S., Corovic, A., Warburton, E. A., et al. (2020). Integrated cardiovascular assessment of atherosclerosis using PET/MRI. Br. J. Radiol. 93:20190921. doi: 10.1259/bjr.20190921
Fakhry, M., Roszkowska, M., Briolay, A., Bougault, C., Guignandon, A., Diaz-Hernandez, J. I., et al. (2017). TNAP stimulates vascular smooth muscle cell trans-differentiation into chondrocytes through calcium deposition and BMP-2 activation: Possible implication in atherosclerotic plaque stability. Biochim. Biophys. Acta 1863, 643–653. doi: 10.1016/j.bbadis.2016.12.003
Fleisch, H., Russell, R. G., and Straumann, F. (1966). Effect of pyrophosphate on hydroxyapatite and its implications in calcium homeostasis. Nature 212, 901–903. doi: 10.1038/212901a0
Goettsch, C., Hutcheson, J. D., Aikawa, M., Iwata, H., Pham, T., Nykjaer, A., et al. (2016). Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Invest. 126, 1323–1336. doi: 10.1172/jci80851
Gomez, D., Baylis, R. A., Durgin, B. G., Newman, A. A. C., Alencar, G. F., Mahan, S., et al. (2018). Interleukin-1β has atheroprotective effects in advanced atherosclerotic lesions of mice. Nat. Med. 24, 1418–1429. doi: 10.1038/s41591-018-0124-5
Greenland, P., LaBree, L., Azen, S. P., Doherty, T. M., and Detrano, R. C. (2004). Coronary artery calcium score combined with Framingham score for risk prediction in asymptomatic individuals. JAMA 291, 210–215. doi: 10.1001/jama.291.2.210
Hawkins, R. A., Choi, Y., Huang, S. C., Hoh, C. K., Dahlbom, M., Schiepers, C., et al. (1992). Evaluation of the skeletal kinetics of fluorine-18-fluoride ion with PET. J. Nucl. Med. 33, 633–642.
Herieka, M., and Erridge, C. (2014). High-fat meal induced postprandial inflammation. Mol. Nutr. Food Res. 58, 136–146. doi: 10.1002/mnfr.201300104
Herisson, F., Heymann, M. F., Chétiveaux, M., Charrier, C., Battaglia, S., Pilet, P., et al. (2011). Carotid and femoral atherosclerotic plaques show different morphology. Atherosclerosis 216, 348–354. doi: 10.1016/j.atherosclerosis.2011.02.004
Hessle, L., Johnson, K. A., Anderson, H. C., Narisawa, S., Sali, A., Goding, J. W., et al. (2002). Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc. Natl. Acad. Sci. U.S.A. 99, 9445–9449. doi: 10.1073/pnas.142063399
Høilund-Carlsen, P. F., Sturek, M., Alavi, A., and Gerke, O. (2020). Atherosclerosis imaging with 18F sodium fluoride PET: state of the art review. Eur. J. Nucl. Med. Mol. Imaging 47, 1538–1551. doi: 10.1007/s00259-019-04603-1
Hsu, J. J., Fong, F., Patel, R., Qiao, R., Lo, K., Soundia, A., et al. (2020). Changes in microarchitecture of atherosclerotic calcification assessed by 18F-NaF PET and CT after a progressive excercice regimen in hyperlipidemic mice. J. Nucl. Cardiol. doi: 10.1007/s12350-019-02004-3
Hutcheson, J. D., Goettsch, C., Bertazzo, S., Maldonado, N., Ruiz, J. L., Goh, W., et al. (2016). Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 15, 335–343. doi: 10.1038/nmat4519
Ikeda, K., Souma, Y., Akakabe, Y., Kitamura, Y., Matsuo, K., Shimoda, Y., et al. (2012). Macrophages play a unique role in the plaque calcification by enhancing the osteogenic signals exerted by vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 425, 39–44. doi: 10.1016/j.bbrc.2012.07.045
Irkle, A., Vesey, A. T., Lewis, D. Y., Skepper, J. N., Bird, J. L., Dweck, M. R., et al. (2015). Identifying active vascular microcalcification by (18)F-sodium fluoride positron emission tomography. Nat. Commun. 6:7495.
Jin, H., St Hilaire, C., Huang, Y., Yang, D., Dmitrieva, N. I., Negro, A., et al. (2016). Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci. Signal. 9:ra121. doi: 10.1126/scisignal.aaf9109
Jinnouchi, H., Sato, Y., Sakamoto, A., Cornelissen, A., Mori, M., Kawakami, R., et al. (2020). Calcium deposition within coronary atherosclerotic lesion: implications for plaque stability. Atherosclerosis 306, 85–95. doi: 10.1016/j.atherosclerosis.2020.05.017
Joshi, N. V., Vesey, A. T., Williams, M. C., Shah, A. S., Calvert, P. A., Craighead, F. H., et al. (2014). (18)F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: a prospective clinical trial. Lancet 383, 705–713. doi: 10.1016/s0140-6736(13)61754-7
Kapustin, A. N., Chatrou, M. L., Drozdov, I., Zheng, Y., Davidson, S. M., Soong, D., et al. (2015). Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 116, 1312–1323. doi: 10.1161/circresaha.116.305012
Kapustin, A. N., Davies, J. D., Reynolds, J. L., McNair, R., Jones, G. T., Sidibe, A., et al. (2011). Calcium regulates key components of vascular smooth muscle cell-derived matrix vesicles to enhance mineralization. Circ. Res. 109, e1–e12.
Kapustin, A. N., and Shanahan, C. M. (2016). Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J. Physiol. 594, 2905–2914. doi: 10.1113/jp271340
Karunakaran, D., Geoffrion, M., Wei, L., Gan, W., Richards, L., Shangari, P., et al. (2016). Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2:e1600224. doi: 10.1126/sciadv.1600224
Kelly-Arnold, A., Maldonado, N., Laudier, D., Aikawa, E., Cardoso, L., and Weinbaum, S. (2013). Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. U.S.A. 110, 10741–10746. doi: 10.1073/pnas.1308814110
Kesavardhana, S., Malireddi, R. K. S., and Kanneganti, T. D. (2020). Caspases in cell death, inflammation, and pyroptosis. Annu. Rev. Immunol. 38, 567–595. doi: 10.1146/annurev-immunol-073119-095439
Khavandgar, Z., Roman, H., Li, J., Lee, S., Vali, H., Brinckmann, J., et al. (2014). Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. 29, 327–337. doi: 10.1002/jbmr.2039
Kiffer-Moreira, T., Yadav, M. C., Zhu, D., Narisawa, S., Sheen, C., Stec, B., et al. (2013). Pharmacological inhibition of PHOSPHO1 suppresses vascular smooth muscle cell calcification. J. Bone Miner. Res. 28, 81–91. doi: 10.1002/jbmr.1733
Kolbrink, B., Riebeling, T., Kunzendorf, U., and Krautwald, S. (2020). Plasma membrane pores drive inflammatory cell death. Front. Cell Dev. Biol. 8:817. doi: 10.3389/fcell.2020.00817
Komori, T., Yagi, H., Nomura, S., Yamaguchi, A., Sasaki, K., Deguchi, K., et al. (1997). Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89, 755–764. doi: 10.1016/s0092-8674(00)80258-5
Kuzan, A., Chwiłkowska, A., Pezowicz, C., Witkiewicz, W., Gamian, A., Maksymowicz, K., et al. (2017). The content of collagen type II in human arteries is correlated with the stage of atherosclerosis and calcification foci. Cardiovasc. Pathol. 28, 21–27. doi: 10.1016/j.carpath.2017.02.003
Lee, G. L., Yeh, C. C., Wu, J. Y., Lin, H. C., Wang, Y. F., Kuo, Y. Y., et al. (2019). TLR2 promotes vascular smooth muscle cell chondrogenic differentiation and consequent calcification via the concerted actions of osteoprotegerin suppression and IL-6-mediated RANKL induction. Arterioscler. Thromb. Vasc. Biol. 39, 432–445. doi: 10.1161/atvbaha.118.311874
Lee, H. L., Woo, K. M., Ryoo, H. M., and Baek, J. H. (2010). Tumor necrosis factor-alpha increases alkaline phosphatase expression in vascular smooth muscle cells via MSX2 induction. Biochem. Biophys. Res. Commun. 391, 1087–1092. doi: 10.1016/j.bbrc.2009.12.027
Lee, T., Mintz, G. S., Matsumura, M., Zhang, W., Cao, Y., Usui, E., et al. (2017). Prevalence, predictors, and clinical presentation of a calcified nodule as assessed by optical coherence tomography. JACC Cardiovasc. Imaging 10, 883–891. doi: 10.1016/j.jcmg.2017.05.013
Lencel, P., Delplace, S., Pilet, P., Leterme, D., Miellot, F., Sourice, S., et al. (2011). Cell-specific effects of TNF-α and IL-1β on alkaline phosphatase: implication for syndesmophyte formation and vascular calcification. Lab. Invest. 91, 1434–1442. doi: 10.1038/labinvest.2011.83
Leroux-Berger, M., Queguiner, I., Maciel, T. T., Ho, A., Relaix, F., and Kempf, H. (2011). Pathologic calcification of adult vascular smooth muscle cells differs on their crest or mesodermal embryonic origin. J. Bone Miner. Res. 26, 1543–1553. doi: 10.1002/jbmr.382
Li, X., Yang, H. Y., and Giachelli, C. M. (2008). BMP-2 promotes phosphate uptake, phenotypic modulation, and calcification of human vascular smooth muscle cells. Atherosclerosis 199, 271–277. doi: 10.1016/j.atherosclerosis.2007.11.031
Liberman, M., Johnson, R. C., Handy, D. E., Loscalzo, J., and Leopold, J. A. (2011). Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 413, 436–441. doi: 10.1016/j.bbrc.2011.08.114
Lin, J., Li, H., Yang, M., Ren, J., Huang, Z., Han, F., et al. (2013). A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 3, 200–210. doi: 10.1016/j.celrep.2012.12.012
Lin, M. E., Chen, T. M., Wallingford, M. C., Nguyen, N. B., Yamada, S., Sawangmake, C., et al. (2016). Runx2 deletion in smooth muscle cells inhibits vascular osteochondrogenesis and calcification but not atherosclerotic lesion formation. Cardiovasc. Res. 112, 606–616. doi: 10.1093/cvr/cvw205
Maldonado, N., Kelly-Arnold, A., Vengrenyuk, Y., Laudier, D., Fallon, J. T., Virmani, R., et al. (2012). A mechanistic analysis of the role of microcalcifications in atherosclerotic plaque stability: potential implications for plaque rupture. Am. J. Physiol. Heart Circ. Physiol. 303, H619–H628.
Malhotra, R., Burke, M. F., Martyn, T., Shakartzi, H. R., Thayer, T. E., O’Rourke, C., et al. (2015). Inhibition of bone morphogenetic protein signal transduction prevents the medial vascular calcification associated with matrix Gla protein deficiency. PLoS One 10:e0117098. doi: 10.1371/journal.pone.0117098
Mechtouff, L., Sigovan, M., Douek, P., Costes, N., Le Bars, D., Mansuy, A., et al. (2020). Simultaneous assessment of microcalcifications and morphological criteria of vulnerability in carotid artery plaque using hybrid. J. Nucl. Cardiol. doi: 10.1007/s12350-020-02400-0
Meng, L., Jin, W., and Wang, X. (2015). RIP3-mediated necrotic cell death accelerates systematic inflammation and mortality. Proc. Natl. Acad. Sci. U.S.A. 112, 11007–11012. doi: 10.1073/pnas.1514730112
Menu, P., Pellegrin, M., Aubert, J. F., Bouzourene, K., Tardivel, A., Mazzolai, L., et al. (2011). Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2:e137. doi: 10.1038/cddis.2011.18
Millán, J. L., and Whyte, M. P. (2016). Alkaline phosphatase and hypophosphatasia. Calcif. Tissue Int. 98, 398–416.
Miller, Y. I., Choi, S. H., Wiesner, P., Fang, L., Harkewicz, R., Hartvigsen, K., et al. (2011). Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 108, 235–248. doi: 10.1161/circresaha.110.223875
Mody, N., Parhami, F., Sarafian, T. A., and Demer, L. L. (2001). Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med. 31, 509–519. doi: 10.1016/s0891-5849(01)00610-4
Murshed, M., Harmey, D., Millán, J. L., McKee, M. D., and Karsenty, G. (2005). Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 19, 1093–1104. doi: 10.1101/gad.1276205
Nadra, I., Boccaccini, A. R., Philippidis, P., Whelan, L. C., McCarthy, G. M., Haskard, D. O., et al. (2008). Effect of particle size on hydroxyapatite crystal-induced tumor necrosis factor alpha secretion by macrophages. Atherosclerosis 196, 98–105. doi: 10.1016/j.atherosclerosis.2007.02.005
Nadra, I., Mason, J. C., Philippidis, P., Florey, O., Smythe, C. D., McCarthy, G. M., et al. (2005). Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ. Res. 96, 1248–1256. doi: 10.1161/01.res.0000171451.88616.c2
Nakagawa, Y., Ikeda, K., Akakabe, Y., Koide, M., Uraoka, M., Yutaka, K. T., et al. (2010). Paracrine osteogenic signals via bone morphogenetic protein-2 accelerate the atherosclerotic intimal calcification in vivo. Arterioscler. Thromb. Vasc. Biol. 30, 1908–1915. doi: 10.1161/atvbaha.110.206185
New, S. E., Goettsch, C., Aikawa, M., Marchini, J. F., Shibasaki, M., Yabusaki, K., et al. (2013). Macrophage-derived matrix vesicles: an alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 113, 72–77. doi: 10.1161/circresaha.113.301036
Ornello, R., Degan, D., Tiseo, C., Di Carmine, C., Perciballi, L., Pistoia, F., et al. (2018). Distribution and temporal trends from 1993 to 2015 of ischemic stroke subtypes: a systematic review and meta-analysis. Stroke 49, 814–819. doi: 10.1161/STROKEAHA.117.020031
Parhami, F., Basseri, B., Hwang, J., Tintut, Y., and Demer, L. L. (2002). High-density lipoprotein regulates calcification of vascular cells. Circ. Res. 91, 570–576. doi: 10.1161/01.RES.0000036607.05037.DA
Pi, X., Lockyer, P., Dyer, L. A., Schisler, J. C., Russell, B., Carey, S., et al. (2012). Bmper inhibits endothelial expression of inflammatory adhesion molecules and protects against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 2214–2222. doi: 10.1161/ATVBAHA.112.252015
Proudfoot, D., Davies, J. D., Skepper, J. N., Weissberg, P. L., and Shanahan, C. M. (2002). Acetylated low-density lipoprotein stimulates human vascular smooth muscle cell calcification by promoting osteoblastic differentiation and inhibiting phagocytosis. Circulation 106, 3044–3050. doi: 10.1161/01.CIR.0000041429.83465.41
Proudfoot, D., Skepper, J. N., Hegyi, L., Bennett, M. R., Shanahan, C. M., and Weissberg, P. L. (2000). Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 87, 1055–1062. doi: 10.1161/01.RES.87.11.1055
Qiao, J. H., Fishbein, M. C., Demer, L. L., and Lusis, A. J. (1995). Genetic determination of cartilaginous metaplasia in mouse aorta. Arterioscler. Thromb. Vasc. Biol. 15, 2265–2272. doi: 10.1161/01.ATV.15.12.2265
Rasheed, A., Robichaud, S., Nguyen, M. A., Geoffrion, M., Wyatt, H., Cottee, M. L., et al. (2020). Loss of MLKL (Mixed Lineage Kinase Domain-Like Protein) decreases necrotic core but increases macrophage lipid accumulation in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 40, 1155–1167. doi: 10.1161/ATVBAHA.119.313640
Rattazzi, M., Bennett, B. J., Bea, F., Kirk, E. A., Ricks, J. L., Speer, M., et al. (2005). Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arterioscler. Thromb. Vasc. Biol. 25, 1420–1425. doi: 10.1161/01.ATV.0000166600.58468.1b
Richardson, P. D., Davies, M. J., and Born, G. V. (1989). Influence of plaque configuration and stress distribution on fissuring of coronary atherosclerotic plaques. Lancet 2, 941–944. doi: 10.1016/S0140-6736(89)90953-7
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131. doi: 10.1056/NEJMoa1707914
Roberts, S., Narisawa, S., Harmey, D., Millán, J. L., and Farquharson, C. (2007). Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J. Bone Miner. Res. 22, 617–627. doi: 10.1359/jbmr.070108
Roijers, R. B., Debernardi, N., Cleutjens, J. P., Schurgers, L. J., Mutsaers, P. H., and van der Vusse, G. J. (2011). Microcalcifications in early intimal lesions of atherosclerotic human coronary arteries. Am. J. Pathol. 178, 2879–2887. doi: 10.1016/j.ajpath.2011.02.004
Romero-Garcia, S., and Prado-Garcia, H. (2019). Mitochondrial calcium: transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 54, 1155–1167. doi: 10.3892/ijo.2019.4696
Rosenfeld, M. E., Polinsky, P., Virmani, R., Kauser, K., Rubanyi, G., and Schwartz, S. M. (2000). Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler. Thromb. Vasc. Biol. 20, 2587–2592. doi: 10.1161/01.ATV.20.12.2587
Roth, G. A., Johnson, C., Abajobir, A., Abd-Allah, F., Abera, S. F., Abyu, G., et al. (2017). Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J. Am. Coll. Cardiol. 70, 1–25. doi: 10.1016/j.jacc.2017.04.052
Rutsch, F., Ruf, N., Vaingankar, S., Toliat, M. R., Suk, A., Höhne, W., et al. (2003). Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 34, 379–381. doi: 10.1038/ng1221
Sage, A. P., Lu, J., Tintut, Y., and Demer, L. L. (2011). Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 79, 414–422. doi: 10.1038/ki.2010.390
Schrijvers, D. M., De Meyer, G. R., Kockx, M. M., Herman, A. G., and Martinet, W. (2005). Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 25, 1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7
Sheen, C. R., Kuss, P., Narisawa, S., Yadav, M. C., Nigro, J., Wang, W., et al. (2015). Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J. Bone Miner. Res. 30, 824–836. doi: 10.1002/jbmr.2420
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shioi, A., Katagi, M., Okuno, Y., Mori, K., Jono, S., Koyama, H., et al. (2002). Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ. Res. 91, 9–16. doi: 10.1161/01.RES.0000026421.61398.F2
Song, Y., Hou, M., Li, Z., Luo, C., Ou, J. S., Yu, H., et al. (2017). TLR4/NF-κB/Ceramide signaling contributes to Ox-LDL-induced calcification of human vascular smooth muscle cells. Eur. J. Pharmacol. 794, 45–51. doi: 10.1016/j.ejphar.2016.11.029
St Hilaire, C., Ziegler, S. G., Markello, T. C., Brusco, A., Groden, C., Gill, F., et al. (2011). NT5E mutations and arterial calcifications. N. Engl. J. Med. 364, 432–442. doi: 10.1056/NEJMoa0912923
Stewart, A. J., Leong, D. T. K., and Farquharson, C. (2018). PLA2 and ENPP6 may act in concert to generate phosphocholine from the matrix vesicle membrane during skeletal mineralization. FASEB J. 32, 20–25. doi: 10.1096/fj.201700521r
Sun, Y., Byon, C. H., Yuan, K., Chen, J., Mao, X., Heath, J. M., et al. (2012). Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 111, 543–552. doi: 10.1161/CIRCRESAHA.112.267237
Tintut, Y., Patel, J., Parhami, F., and Demer, L. L. (2000). Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 102, 2636–2642. doi: 10.1161/01.CIR.102.21.2636
Tyson, K. L., Reynolds, J. L., McNair, R., Zhang, Q., Weissberg, P. L., and Shanahan, C. M. (2003). Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vasc. Biol. 23, 489–494. doi: 10.1161/01.ATV.0000059406.92165.31
van der Heijden, T., Kritikou, E., Venema, W., van Duijn, J., van Santbrink, P. J., Slütter, B., et al. (2017). NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler. Thromb. Vasc. Biol. 37, 1457–1461. doi: 10.1161/ATVBAHA.117.309575
Vengrenyuk, Y., Carlier, S., Xanthos, S., Cardoso, L., Ganatos, P., Virmani, R., et al. (2006). A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. U.S.A. 103, 14678–14683. doi: 10.1073/pnas.0606310103
Vesey, A. T., Jenkins, W. S., Irkle, A., Moss, A., Sng, G., Forsythe, R. O., et al. (2017). 18 F-fluoride and 18 F-fluorodeoxyglucose positron emission tomography after transient ischemic attack or minor ischemic stroke: case-control study. Circ. Cardiovasc. Imaging 10:e004976. doi: 10.1161/CIRCIMAGING.116.004976
Wen, C., Yang, X., Yan, Z., Zhao, M., Yue, X., Cheng, X., et al. (2013). Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int. J. Cardiol. 168, 2242–2247. doi: 10.1016/j.ijcard.2013.01.211
Wiedermann, C. J., Kiechl, S., Dunzendorfer, S., Schratzberger, P., Egger, G., Oberhollenzer, F., et al. (1999). Association of endotoxemia with carotid atherosclerosis and cardiovascular disease: prospective results from the Bruneck Study. J. Am. Coll. Cardiol. 34, 1975–1981. doi: 10.1016/S0735-1097(99)00448-9
Wu, L. N., Yoshimori, T., Genge, B. R., Sauer, G. R., Kirsch, T., Ishikawa, Y., et al. (1993). Characterization of the nucleational core complex responsible for mineral induction by growth plate cartilage matrix vesicles. J. Biol. Chem. 268, 25084–25094. doi: 10.1016/S0021-9258(19)74574-8
Yadav, M. C., Bottini, M., Cory, E., Bhattacharya, K., Kuss, P., Narisawa, S., et al. (2016). Skeletal mineralization deficits and impaired biogenesis and function of chondrocyte-derived matrix vesicles in phospho1(-/-) and phospho1/Pi t1 double-knockout mice. J. Bone Miner. Res. 31, 1275–1286. doi: 10.1002/jbmr.2790
Yan, J., Stringer, S. E., Hamilton, A., Charlton-Menys, V., Götting, C., Müller, B., et al. (2011). Decorin GAG synthesis and TGF-β signaling mediate Ox-LDL-induced mineralization of human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 31, 608–615. doi: 10.1161/ATVBAHA.110.220749
Yao, Y., Bennett, B. J., Wang, X., Rosenfeld, M. E., Giachelli, C., Lusis, A. J., et al. (2010). Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ. Res. 107, 485–494. doi: 10.1161/CIRCRESAHA.110.219071
Yin, Y., Li, X., Sha, X., Xi, H., Li, Y. F., Shao, Y., et al. (2015). Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler. Thromb. Vasc. Biol. 35, 804–816. doi: 10.1161/ATVBAHA.115.305282
Zebboudj, A. F., Imura, M., and Boström, K. (2002). Matrix GLA protein, a regulatory protein for bone morphogenetic protein-2. J. Biol. Chem. 277, 4388–4394. doi: 10.1074/jbc.M109683200
Zhao, G., Xu, M. J., Zhao, M. M., Dai, X. Y., Kong, W., Wilson, G. M., et al. (2012). Activation of nuclear factor-kappa B accelerates vascular calcification by inhibiting ankylosis protein homolog expression. Kidney Int. 82, 34–44. doi: 10.1038/ki.2012.40
Keywords: atherosclerosis, calcification, chondrocyte, inflammation, cardiovascular morbidity and mortality
Citation: Canet-Soulas E, Bessueille L, Mechtouff L and Magne D (2021) The Elusive Origin of Atherosclerotic Plaque Calcification. Front. Cell Dev. Biol. 9:622736. doi: 10.3389/fcell.2021.622736
Received: 29 October 2020; Accepted: 08 February 2021;
Published: 09 March 2021.
Edited by:
Monzur Murshed, McGill University, CanadaReviewed by:
Colin Farquharson, University of Edinburgh, United KingdomLeon J. Schurgers, Maastricht University, Netherlands
Elena Aikawa, Harvard Medical School, United States
Copyright © 2021 Canet-Soulas, Bessueille, Mechtouff and Magne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Magne, ZGF2aWQubWFnbmVAdW5pdi1seW9uMS5mcg==