 Hiroki Yoshioka
Hiroki Yoshioka Yurie Mikami
Yurie Mikami Sai Shankar Ramakrishnan
Sai Shankar Ramakrishnan Akiko Suzuki
Akiko Suzuki Junichi Iwata
Junichi Iwata
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 09 June 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.621045
This article is part of the Research Topic Genetic, Environmental and Synergistic Gene-Environment Contributions to Craniofacial Defects View all 11 articles
Cleft lip with/without cleft palate (CL/P) is one of the most common congenital birth defects, showing the complexity of both genetic and environmental contributions [e.g., maternal exposure to alcohol, cigarette, and retinoic acid (RA)] in humans. Recent studies suggest that epigenetic factors, including microRNAs (miRs), are altered by various environmental factors. In this study, to investigate whether and how miRs are involved in cleft palate (CP) induced by excessive intake of all-trans RA (atRA), we evaluated top 10 candidate miRs, which were selected through our bioinformatic analyses, in mouse embryonic palatal mesenchymal (MEPM) cells as well as in mouse embryos treated with atRA. Among them, overexpression of miR-27a-3p, miR-27b-3p, and miR-124-3p resulted in the significant reduction of cell proliferation in MEPM cells through the downregulation of CP-associated genes. Notably, we found that excessive atRA upregulated the expression of miR-124-3p, but not of miR-27a-3p and miR-27b-3p, in both in vivo and in vitro. Importantly, treatment with a specific inhibitor for miR-124-3p restored decreased cell proliferation through the normalization of target gene expression in atRA-treated MEPM cells and atRA-exposed mouse embryos, resulting in the rescue of CP in mice. Taken together, our results indicate that atRA causes CP through the induction of miR-124-3p in mice.
Cleft lip with/without cleft palate (CL/P) is the second most common congenital birth defect worldwide, with a prevalence of as high as 1 in 700 live births (IPDTOC Working Group, 2011). CL/P impacts on various physiological functions such as swallowing, feeding, speech, and hearing, even after multiple surgical corrections, orthodontic treatment, and speech therapy (Ferguson, 1988); therefore, the quality of life of both patients and their families is strongly diminished (Sischo et al., 2017; Kummer, 2018; De Cuyper et al., 2019). Palate development is regulated through fine-tuned spatiotemporal gene regulatory networks that control the growth, elevation, and fusion of the palatal shelves through cell migration, proliferation, apoptosis, differentiation, and extracellular matrix secretion and arrangement. Dysregulation of each step of palatogenesis results in a failure in normal palate development and causes cleft palate (CP). Our previous studies showed that at least 223 genes [a total of 198 genes related to cleft palate only (CPO), including cleft in the secondary palate, primary palate, soft palate, and submucous CP, and a total of 45 genes related to cleft lip with cleft palate (CLP), with 20 genes duplicated in CPO and CLP adjusted] in mice and 185 genes (a total of 27 genes related to CPO and a total of 177 genes related to CL/P) in humans are associated with CP (Suzuki et al., 2018a,b, 2019a). Thus, a large number of genes play crucial roles in palate development.
The etiology of CL/P is further complicated with interactions between genetic and environmental factors (Murray, 2002; Beaty et al., 2016; Gonseth et al., 2019). As for environmental factors, maternal exposures to smoking and alcohol consumption are known to be a risk for CL/P (Ericson et al., 1979; Munger et al., 1996; Romitti et al., 1999; Garland et al., 2020). In addition, several chemicals are known to be teratogens that cause CL/P [e.g., dexamethasone, dioxins, and heavy metals (Bove et al., 1995; Buser and Pohl, 2015; Suhl et al., 2018; Pi et al., 2019)]. Malnutrition and mutations in genes related to nutritional metabolic/signaling pathways are also associated with CL/P in humans and animal models. For example, retinoic acid (RA), a metabolite of vitamin A, plays important roles in cell fate determination, cellular patterning, and cell differentiation in development (Rhinn and Dolle, 2012; Roberts, 2020). Excessive RA intake causes CPO in mice (Zhang et al., 2003; Kuriyama et al., 2008; Wang et al., 2019). Mice with deletion in Cyp26b1, a key enzyme for RA degradation, cause CPO, micrognathia, truncation of the fore/hind limbs, and ossification defects in calvaria bones (Maclean et al., 2009). In addition, mice with a mutation in retinol dehydrogenase 10, a key enzyme for RA synthesis (Rdh10m366Asp mice), show reduced RA levels and exhibit midline cleft, syndactyly, and a malformed forebrain (Ashique et al., 2012). Mice with dominant negative mutations in retinoic acid receptor alpha (Rara403*) and mice with deficiency of both RA receptors alpha and gamma (Rara–/–;Rarg–/–) exhibit midline cleft (Damm et al., 1993; Lohnes et al., 1994; Mark et al., 1995). Thus, an appropriate amount of RA is crucial for normal embryonic development, with either too much or too low RA levels causing CP in mice. In humans, decreased serum levels of vitamin A and RA binding protein 4 (RBP4), a RA translocator, have been reported in non-syndromic CL/P patients (Zhang et al., 2014). In addition, excessive vitamin A intake is known to be associated with multiple birth defects (Lammer et al., 1985; Martinez-Frias and Salvador, 1990; Werler et al., 1990); however, the minimum teratogenic dose appears to be well above the level consumed by most women through multivitamin and vitamin A supplements during pregnancy (Mills et al., 1997; Skare et al., 2012).
A number of microRNAs (miRs), which are small non-coding RNAs (21–25 nucleotides) that regulate the expression of target genes at the post-transcriptional level (Hudder and Novak, 2008; Hou et al., 2011), play important roles in a wide array of cellular functions during the development of various tissues, including the upper lip and the palate (Shin et al., 2012; Seelan et al., 2014; Warner et al., 2014; Mukhopadhyay et al., 2019). For instance, loss of a miR-processing enzyme, such as DROSHA and DICER, results in craniofacial developmental defects in mice (Zehir et al., 2010; Nie et al., 2011; Schoen et al., 2017), and polymorphisms in DROSHA are associated with risk of CL/P in humans (Xu et al., 2018). In addition, mice with a deletion of miR-17-92 cluster, which is located on chromosome 14 in mice and chromosome 13 in humans, exhibit either bilateral or unilateral CLP and delayed endochondral ossification, hypoplastic lung, and cardiac ventricular septal defect (Ventura et al., 2008; de Pontual et al., 2011; Wang et al., 2013). By contrast, overexpression of miR-17-92 in mouse palatal mesenchymal cells results in increased cell proliferation (Li et al., 2012). Thus, miRs can regulate cellular functions through the regulation of their target’s gene expression. Currently, while the importance of miRs in development is well recognized through loss-of-function studies for miRs, it remains largely unknown which miRs are elevated by environmental factors to suppress genes that are crucial for palate development.
Our previous bioinformatic studies showed that 18 miRs are possibly involved in the regulation of CP-associated genes in mice (Suzuki et al., 2018a). Recent studies show that all-trans RA (atRA) modulates miR expressions in human cancer cell lines (Liu et al., 2018, 2019). However, it is still unclear which miRs are functionally relevant in palate development and whether expression of these miRs is altered by atRA exposure. In this study, we investigated the mechanism of how miRs contribute to pathogenesis of CPO induced by excessive atRA intake.
MEPM cells were isolated from the palatal shelves of E13.5 C57BL/6J mice, as previously described (Iwata et al., 2014). The palatal shelves from one embryo were used for each cell culture, and three independent experiments were conducted using cells from different litters. MEPM cells were maintained under Dulbecco’s modified Eagle’s medium (DMEM; Sigma Aldrich, St. Louis, MO, United States) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin (Sigma Aldrich), 2-mercaptoethanol (Gibco, Waltham, MA, United States), and non-essential amino acids (Sigma Aldrich) at 37°C in a humidified atmosphere with 5% CO2.
C57BL/6J mice were obtained from The Jackson Laboratory, Bar Harbor, ME, United States. Pregnant female mice were orally administered 40–70 mg/kg atRA (R2625, Sigma-Aldrich) suspended in 10% ethanol and 90% corn oil emulsion at E11.5. Control mice received an equivalent amount of emulsion without atRA (0.1 ml/10 g body weight). For the rescue experiments, 50 mg/kg atRA was orally administered at E11.5, and then the miR-124-3p inhibitor (Integrated DNA Technologies, Coralville, IA, United States) was intraperitoneally injected at 5 mg/kg at E12.5 and E13.5. The protocol was approved by the Animal Welfare Committee (AWC) and the Institutional Animal Care and Use Committee (IACUC) of UTHealth (AWC 19-0079). All mice were maintained at the animal facility of UTHealth.
MEPM cells were plated onto 96-well cell culture plates at a density of 5,000 cells per well and treated with a mimic for negative control (4464061), miR-21a-5p (4464066; MC10206), miR-27a-3p (4464066; MC10939), miR-27b-3p (4464066; MC10750), miR-30a-5p (4464066; MC11062), miR-124-3p (4464066; MC10691), miR-141-3p (4464066; MC10860), miR-200a-3p (4464066; MC10991), miR-203-3p (4464066; MC10152), miR-320-3p (4464066; MC11621), and miR-381-3p (4464066; MC10242) [mirVana miRNA mimic (chemically modified double-stranded RNA molecules), ThermoFisher Scientific, Waltham, MA, United States], or an inhibitor for negative control (4464079), miR-27a-3p (4464084; MH10939), miR-27b-3p (4464084; MH10750), and miR-124-3p (4464084; MH10691) [mirVana miRNA inhibitor (chemically modified, single-stranded oligonucleotides with patented secondary structure), Thermo Fisher Scientific], using the Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific), according to the manufacturer’s protocol (3 pmol of the mimic or the inhibitor and 0.3 μl of the transfection reagent in 100 μl DMEM per well). Cell proliferation was measured using the Cell Counting Kit 8 (Dojindo Molecular Technologies Inc., Gaithersburg, MD, United States) 24, 48, or 72 h after treatments (n = 6 per group). For the atRA exposure experiments, MEPM cells were plated onto 96-well cell culture plates at a density of 5,000 cells per well and treated with 30 μM atRA. After 24, 48, or 72 h of treatment, cell numbers were determined as described above.
MEPM cells were plated onto 35-mm dishes at a density of 25,000/dish and treated with 30 μM atRA or control vehicle (dimethyl sulfoxide). After 24 h, the cells were incubated with 100 μg/ml BrdU (Sigma Aldrich) for 1 h. Incorporated BrdU was stained with a rat monoclonal antibody against BrdU (ab6326; Abcam, Cambridge, MA, United States 1:1,000), as previously described (Iwata et al., 2014). A total of six fields, which were randomly selected from three independent experiments, were used for the quantification of BrdU-positive cells.
MEPM cells were plated onto a 60-mm dish at a density of 50,000 cells per dish and treated with either 30 μM atRA or vehicle for 72 h, or with each miR mimic or control miR for 48 h. The treated cells were lysed with RIPA buffer (Cell Signaling Technology, Danvers, MA, United States) with a protease inhibitor cocktail (Roche, Indianapolis, IN, United States). The cells were harvested and centrifuged at 21,130 × g for 10 min at 4°C. The supernatant of each sample was collected, and protein concentration was determined using the BCA protein kit (Pierce). Protein samples were applied to Mini-PROTEAN TGX Gels (Bio-Rad, Hercules, CA, United States) and transferred to a polyvinylidene difluoride (PVDF) membrane. A mouse monoclonal antibody against GAPDH (MAB374, Millipore, Burlington, MA, United States, 1:6,000), a rabbit monoclonal antibody against CCND1 (2978, Cell Signaling Technology, 1:1,000), and a rabbit polyclonal antibody against cleaved caspase 3 (9661, Cell Signaling Technology, 1:1,000) were used. Peroxidase-conjugated anti-mouse IgG (7076, Cell Signaling Technology, 1:100,000) and anti-rabbit IgG (7074, Cell Signaling Technology, 1:100,000) were used as secondary antibodies.
MEPM cells were plated onto a 60-mm dish at a density of 40,000 cells per dish. When the cells reached 80% confluence, they were treated with a mimic or an inhibitor for miR-27a-3p, miR-27b-3p, miR-124-3p, or a negative control, at 3 pmol in 6 μl of transfection reagent (Lipofectamine RNAiMAX transfection reagent in 4 ml DMEM per dish). After 24 h of treatment, total RNA was extracted with the QIAshredder and miRNeasy Mini Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. For the atRA experiments, the cells were plated onto a 60-mm dish at a density of 50,000 cells per dish and treated with 30 μM atRA for 24 h, and total RNA from MEPM cells (n = 6 per group) was isolated as described above. For the animal experiments, palatal shelves were microdissected at E13.5 and E14.5. Total RNA (1 μg) from each sample was reverse-transcribed using iScript Reverse Transcription Supermix for qRT-PCR (Bio-Rad), and the cDNA was amplified with iTaq Universal SYBR Green Supermix (Bio-Rad) using the CFX96 Touch Real-Time PCR Detection system (Bio-Rad). The PCR primers used in this study are listed in Supplementary Table 1. The amount of each mRNA was normalized by Gapdh. miR expression was measured with Taqman Fast Advanced Master Mix and Taqman Advanced miR cDNA Synthesis Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Probes for miR-27a-3p (mmu478384_mir), miR-27b-3p (mmu478270_mir), miR-124-3p (mmu480901_mir), and miR-26a-5p (477995_mir) were purchased from Thermo Fisher Scientific.
The embryos’ heads were collected at E13.5, E14.5, and E18.5 and fixed with 4% paraformaldehyde overnight. After decalcification with 10% ethylenediaminetetraacetic acid-2Na-2H2O (EDTA), all samples were dehydrated and embedded in paraffin. Paraffin-embedded tissues were sectioned at 4-μm thickness and stained with hematoxylin and eosin (H&E). For immunohistochemistry, paraffin sections were deparaffinized and rehydrated. After antigen retrieval treatment with citrate buffer (pH 6.0) and blocking of endogenous peroxidase with 0.3% hydrogen peroxide in methanol, the sections were incubated with anti-cytokeratin 14 mouse monoclonal antibody (Abcam, ab7800, 1:200 dilution), anti-Ki-67 rabbit monoclonal antibody (Abcam, ab16667, 1:200 dilution), anti-VCAN rabbit polyclonal antibody (Novus Biologicals, Centennial, CO, United States, NBP1-85432, 1:200 dilution), or anti-CDC42 rabbit polyclonal antibody (Proteintech, Rosemont, IL, United States, 10155-1-AP, 1:50 dilution) at 4°C overnight. The sections were then incubated with a secondary antibody, goat anti-rabbit IgG-Alexa Fluor 488 (Thermo Fisher Scientific; A-11008; 1:500 dilution) or goat anti-rabbit IgG (H + L), biotinylated (Vector Laboratories, Burlingame, CA, United States; BA-1000; 1:500 dilution) for 1 h at room temperature. Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for nuclear staining for fluorescent imaging and methylene blue for bright field imaging. Azan staining was performed as previously described (Iwata et al., 2013). A total of six fields, which were randomly selected from three independent experiments, were used for the quantification of Ki-67-positive cells. Fluorescence images were obtained using a confocal microscope (Ti-C2, Nikon, Melville, NY, United States), and color images were obtained using a light microscope (BX43, Olympus, Center Valley, PA, United States); n = 6 per group in each experiment.
Timed-pregnant mice were euthanized at E13.5 and decapitated in PBS. The mandible and tongue were removed from the embryos, and each explant, including the upper half of the head, was placed in a glass tube containing BGJb medium (Gibco, 12591) supplemented with 50% fetal bovine serum, 0.1% ascorbic acid, and antibiotics. The tubes were placed in a rotary apparatus rotating at 50 rpm in an incubator at 37°C and 5% CO2. After 3 days in culture with/without 30 μM atRA, the explants were fixed in 4% PFA and processed.
MEPM cells were plated onto 60-mm dishes at a density of 50,000/dish and treated with 30 μM atRA or control vehicle (dimethyl sulfoxide). After 24 h, the cells were harvested by trypsin and washed twice with cold BioLegend’s Cell Staining Buffer (BioLegend, San Diego, CA, United States; 420201). Cells were centrifuged at 500 g for 5 min at 4°C. The cell pellets were resuspended with Annexin V Binding Buffer (BioLegend; 422201) at a concentration of 1.5 × 106 cells/ml. Resuspended cells (100 μl) were transferred into a Falcon tube and incubated with 5 μl of Annexin V (BioLegend: 421301) and 10 μl of propidium iodide (BioLegend: 421301) for 15 min at room temperature in the dark. A volume of 400 μl of Annexin V Binding Buffer was added to the Falcon tube, and the samples were analyzed with FACS Aria II (BD Biosciences, San Jose, CA, United States).
All experiments were performed independently three times. All statistical analyses were performed using the SPSS software (version 26.0, IBM, Armonk, NY, United States). The statistical significance of the differences between two groups (control and treated groups) was evaluated using independent t-tests. The statistical significance for multiple two groups was evaluated using multiple t-tests after Bonferroni correction. An adjusted p-value after Bonferroni correction (equivalent to non-adjusted p < 0.05) was considered to be statistically significant. For a comparison among multiple groups (e.g., control, treated, and rescued groups) with one factor such as gene or positive cell, a one-way analysis of variance (ANOVA) with Tukey’s honest significant difference test was used for assessment. For a comparison among multiple groups (e.g., control, treated, and rescued groups) with multiple factors, a two-way ANOVA with Tukey’s honest significant difference test was used for assessment. Cell proliferation assays were analyzed using a two-way ANOVA with Dunnett’s (vs. control) or Tukey’s (between all groups) honest significant difference test. A p < 0.05 was considered to be statistically significant. Data are represented as mean ± standard deviation in the graphs.
To evaluate the effect of overexpression of miRs, which were predicted through our bioinformatic analyses (Suzuki et al., 2018a,b), MEPM cells were treated with each miR mimic (miR-21a-5p, miR-27a-3p, miR-27b-3p, miR-30a-5p, miR-124-3p, miR-141-3p, miR-200a-3p, miR-203-3p, miR-320-3p, and miR-381-3p) and analyzed for their effect on cell proliferation. Among them, miR-27a-3p, miR-27b-3p, miR-30a-5p, and miR-124-3p mimics significantly inhibited cell proliferation in MEPM cells, while the mimic of either miR-21a-5p, miR-141-3p, miR-200a-3p, miR-203-3p, miR-320-3p, or miR-381-3p did not affect cell proliferation (Figure 1A). We confirmed that miR-27a-3p, miR-27b-3p, and miR-124-3p mimics did not induce apoptosis (Figure 1B). To identify CP-associated genes targeted by either the miR-27a-3p, miR-27b-3p, or miR-124-3p mimic, we conducted quantitative RT-PCR analysis for the predicted target genes (38 CP-associated genes in miR-27a-3p, 37 CP-associated genes in miR-27b-3p, and 55 CP-associated genes in miR-124-3p) in MEPM cells after treatment with each miR mimic (Supplementary Table 2). Among them, the expression of four genes (Bmi1, Dicer1, Pds5b, and Tgfbr3) in the miR-27a-3p mimic, four genes (Bmi1, Eya1, Gab1, and Spry2) in the miR-27b-3p mimic, and nine genes (Alx1, Axin1, Fst, Hic1, Sp8, Tm7sf2, Tshz1, Vcan, and Zeb1) in the miR-124-3p mimic were significantly downregulated in candidate target genes downregulated with treatment of each miR mimic (Figures 2A–C). To further evaluate the role of each miR in cell proliferation and gene regulation, we treated MEPM cells with a specific inhibitor for either miR-27a-3p, miR-27b-3p, or miR-124-3p. We found that inhibitors of miR-27a-3p, miR-27b-3p, and miR-124-3p failed to change cell proliferation activity (Supplementary Figure 1A). We then performed quantitative RT-PCR analysis for the predicted target genes and found that the expression of 17 genes (Acvr2a, Bmi1, Cdc42, Chd7, Ephb2, Eya4, Gab1, Pax9, Pdgfra, Prdm16, Runx1, Six1, Sox11, Spry1, Spry2, Tgfbr3, and Zeb1) in the miR-27a-3p inhibitor, 6 genes (Apaf1, Eya4, Pds5d, Sos1, Spry2, and Sumo1) in the miR-27b-3p inhibitor, and 9 genes (Axin1, Cdc42, Esrp1, Fst, Gas1, Mmp16, Pbx3, Vcan, and Zeb1) in the miR-124-3p inhibitor were significantly upregulated in candidate target genes upregulated with treatment of each miR inhibitor (Supplementary Figures 1B–D). Therefore, these results suggest that Bmi1 and Tgfbr3 in miR-27a-3p, Spry2 in miR-27b-3p, and Axin1, Fst, Vcan, and Zeb1 in miR-124-3p were strong candidates regulated by the miRs in a dose-dependent manner.
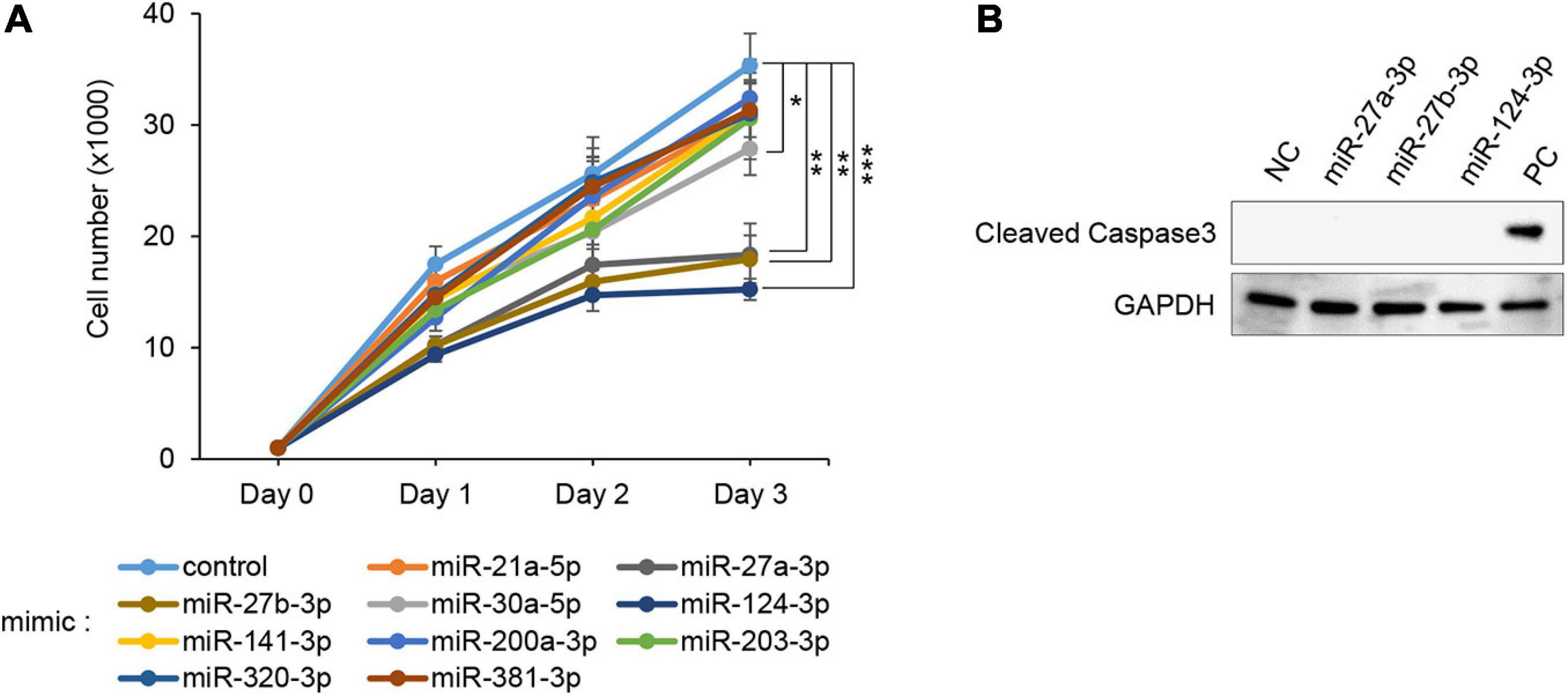
Figure 1. Effect of overexpression of the predicted miRs on cell proliferation in MEPM cells. (A) Cell proliferation assays using MEPM cells from E13.5 palatal shelves treated with the indicated miR mimic; control, miR-21a-5p, miR-27a-3p, miR-27b-3p, miR-30a-5p, miR-124-3p, miR-141-3p, miR-200a-3p, miR-203-3p, miR-320, and miR-381-3p mimic. Two-way ANOVA with Dunnett’s test (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001. Each treatment group was compared with the control. (B) Immunoblotting analysis for cleaved caspase 3 in MEPM cells treated with control, miR-27a-3p, miR-27b-3p, or miR-124-3p mimic for 48 h. Intestine was used as a positive control (PC). GAPDH was used as an internal control. Representative images from two independent experiments are shown.
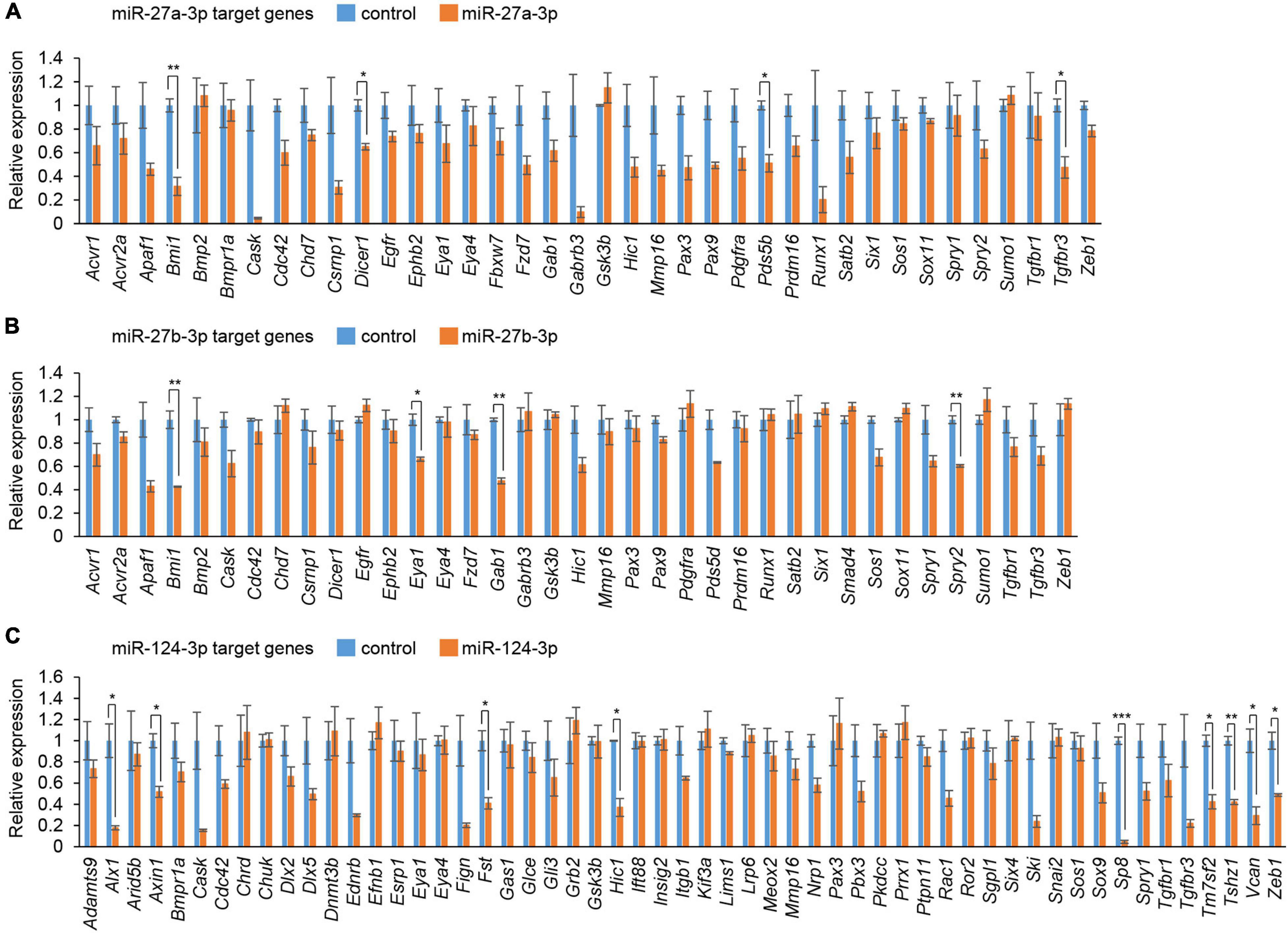
Figure 2. Cleft palate-associated genes suppressed by overexpression of miR-27a-3p, miR-27b-3p, or miR-124-3p in MEPM cells. (A–C) Quantitative RT-PCR for the indicated genes after treatment with control or miR-27a-3p mimic (A), control or miR-27b-3p mimic (B), and control or miR-124a-3p mimic (C). Multiple t-tests adjusted by Bonferroni (n = 3). *Adjusted p < 0.00132 in A (38 genes), adjusted p < 0.00135 in B (37 genes), adjusted p < 0.000909 in C (55 genes), **adjusted p < 0.000263 in A (38 genes), adjusted p < 0.000270 in B (37 genes), adjusted p < 0.000182 in C (55 genes), ***adjusted p < 0.0000182 in C (55 genes).
We conducted cell proliferation assays using MEPM cells treated with atRA. To determine the atRA concentration that affects cell proliferation, the cells were treated with 10 and 30 μM atRA. We found that 30 μM atRA significantly inhibited cell proliferation in MEPM cells (Figure 3A and Supplementary Figure 2A). The reduced cell proliferation by atRA was confirmed with BrdU incorporation assays (Figure 3B) and expression of Cyclin D1 (CCND1), a cell cycle accelerator, in MEPM cells treated with atRA (Figure 3C). In addition, cleaved caspase 3, an indicator of apoptosis, was undetectable in cells treated and untreated with atRA (Supplementary Figure 2B). Moreover, flow cytometry analysis showed no significant change in the profile (cell death vs. healthy cells) of cells treated with atRA (Supplementary Figure 2C). Taken together, these data indicate that atRA inhibits the proliferation of MEPM cells. Interestingly, the expression of miR-124-3p was specifically induced by atRA treatment (Figure 3D); by contrast, expression of miR-27a-3p and miR-27b-3p was not altered by atRA treatment. As expected, the miR-124-3p target genes (Axin1, Fst, Vcan, and Zeb1) were significantly downregulated with atRA treatment (Figure 3E). Taken together, these observations suggest that the expression of these genes was downregulated by atRA through miR-124-3p. We confirmed that the expression of all the other predicted genes regulated by miR-124-3p (additional 39 genes) was not correlated with the atRA condition (Supplementary Figure 3). For instance, although three genes (Bmpr1a, Gli3, and Snal2) were significantly downregulated and Six4 was significantly upregulated under atRA treatment, the expression of these genes was not altered with treatment with the miR-124-3p mimic. Therefore, the use of both a mimic and an inhibitor for the identification of genes regulated by miR-124-3p was helpful to identify genes directly regulated by miR-124-3p.
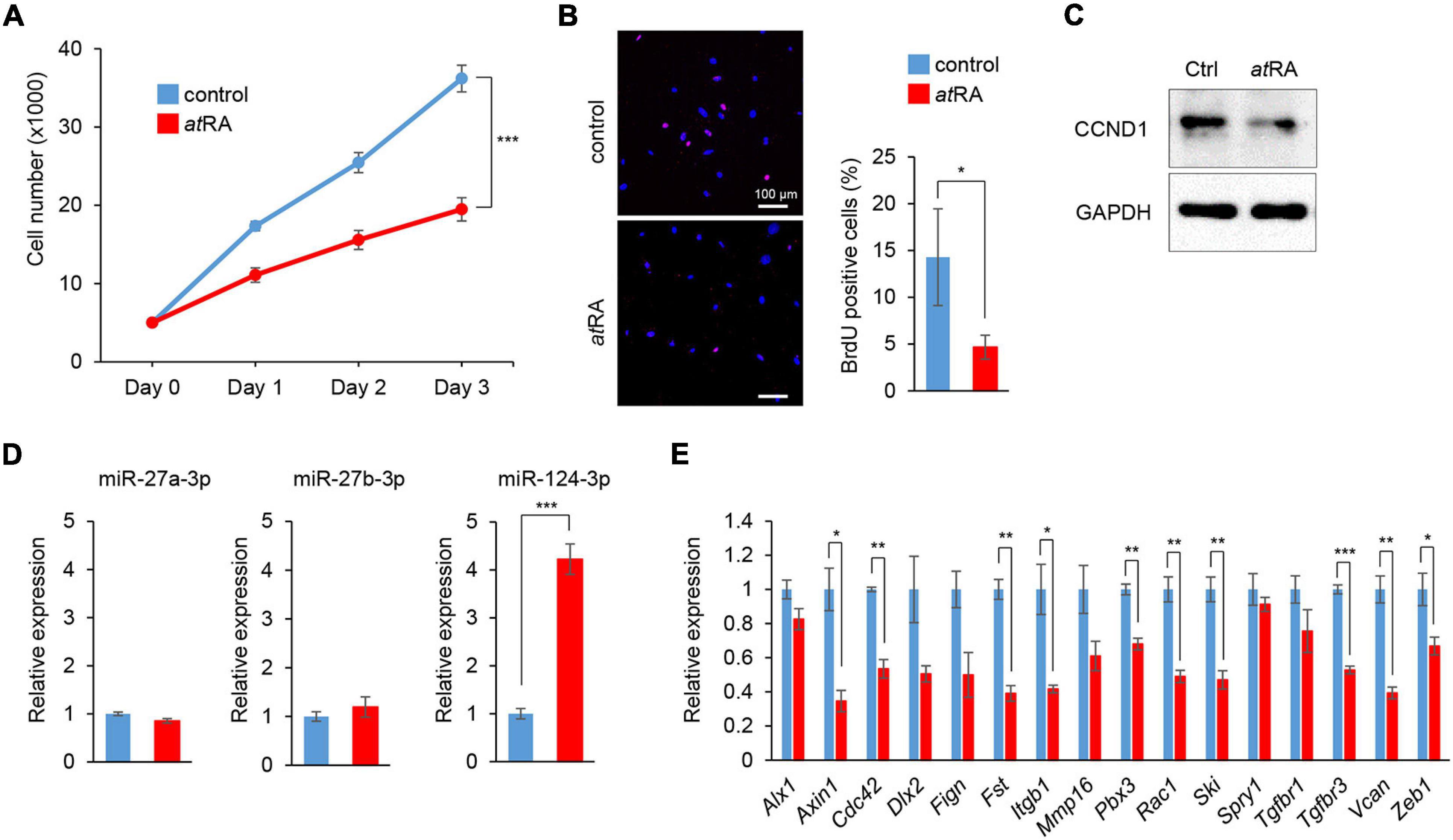
Figure 3. Influence of atRA treatment on cell proliferation and gene expression in MEPM cells. (A) Cell proliferation assays in MEPM cells treated with 30 μM atRA for 24, 48, and 72 h. Two-way ANOVA with Dunnett’s test (n = 6). ***p < 0.001 vs. control. (B) BrdU staining (red) in MEPM cells after treatment with 30 μM atRA for 72 h. Nuclei were counterstained with DAPI (blue). Representative images from two independent experiments are shown. Graph shows the quantification of BrdU-positive cells. Independent t-test. *p < 0.05 vs. control. Bars, 50 μm. (C) Immunoblotting analysis of CCND1 and GAPDH in MEPM cells treated with 30 μM atRA for 72 h. Representative images from two independent experiments are shown. (C) Immunoblotting analysis for CCND1 in MEPM cells treated with 30 μM atRA for 72 h. GAPDH was used as an internal control. (D) Quantitative RT-PCR for miR-27a-3p, miR-27b-3p, or miR-124-3p after treatment with atRA for 24 h in MEPM cells. Multiple t-tests adjusted by Bonferroni (n = 3). ***Adjusted p < 0.00033 (three miRs). (E) Quantitative RT-PCR for the indicated genes after treatment with atRA for 24 h in MEPM cells. Multiple t-tests adjusted by Bonferroni (n = 3–6). *Adjusted p < 0.00313 (16 genes), **adjusted p < 0.000625 (16 genes), ***adjusted p < 0.0000625 (16 genes) vs. control.
To evaluate the contribution of miR-124-3p to atRA-induced cell proliferation inhibition, we treated MEPM cells with a miR-124-3p inhibitor under atRA treatment. The miR-124-3p inhibitor specifically suppressed miR-124-3p expression for treatment at 24 and 48 h (Supplementary Figures 4A,B). The miR-124-3p inhibitor could partially rescue reduced cell proliferation (Figure 4A). As expected, both the number of BrdU-positive cells and CCND1 expression were normalized with treatment with the miR-124-3p inhibitor (Figures 4B,C). In addition, the expression of miR-124-3p target genes (Fst, Vcan, and Zeb1) was partially normalized with the miR-124-3p inhibitor under atRA conditions (Figure 4D). Taken together, our results indicate that atRA inhibits cell proliferation through miR-124-3p expression in MEPM cells.
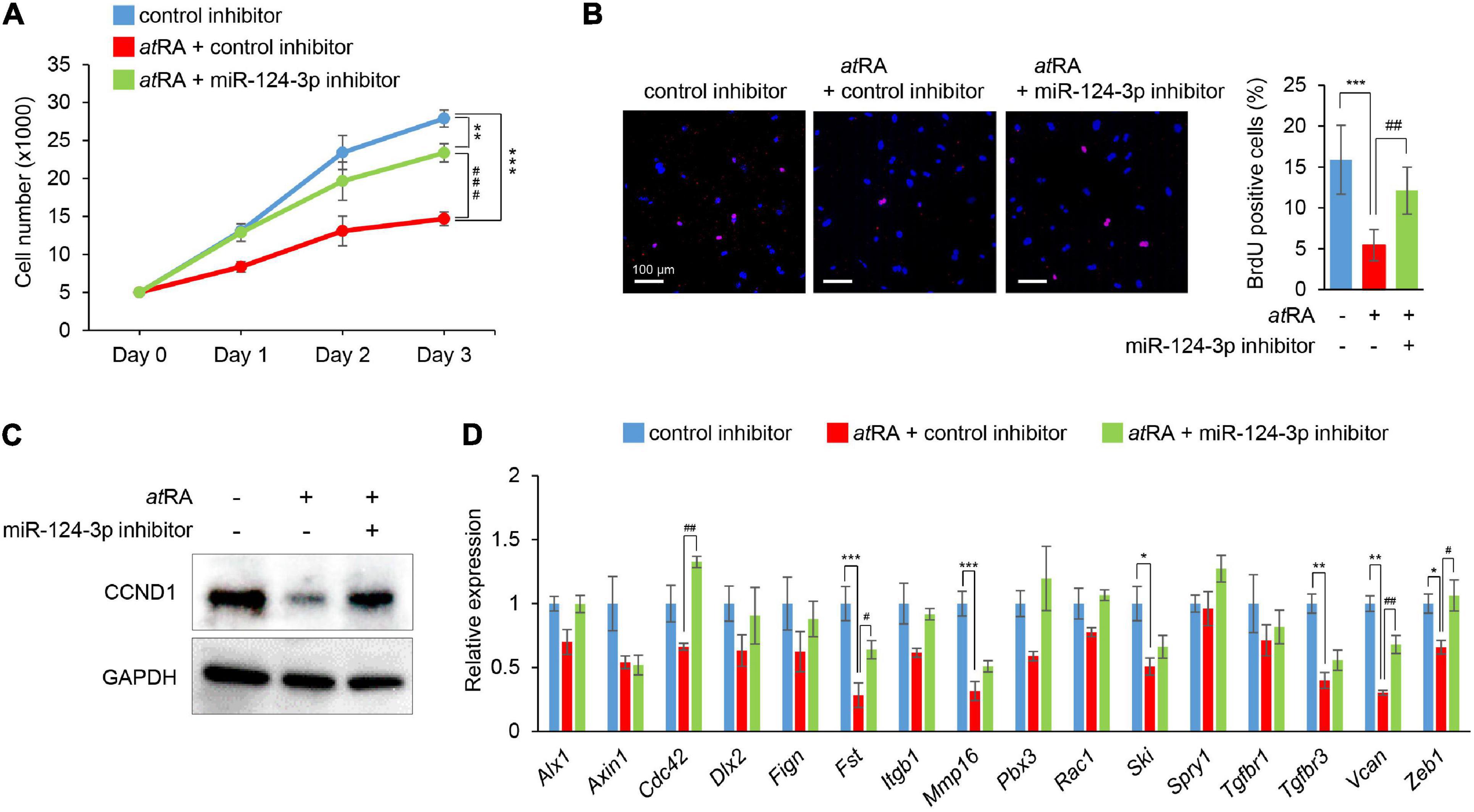
Figure 4. Normalization of miR-124-3p expression restores atRA-induced cell proliferation defect in MEPM cells. (A) Cell proliferation assays in MEPM cells treated with a control or a miR-124-3p inhibitor under the 30 μM atRA condition for 0, 24, 48, or 72 h (Days 0–3). Two-way ANOVA with Tukey’s honest significant difference test (n = 6 per group). **p < 0.01, ***p < 0.001, ###p < 0.001. (B) BrdU staining (red) in MEPM cells after treatment with/without a miR-124-3p inhibitor under the 30 μM atRA condition for 72 h. Nuclei were counterstained with DAPI (blue). Representative images from two independent experiments are shown. Graph shows the quantification of BrdU-positive cells. One-way ANOVA with Tukey’s honest significant difference test. ***p < 0.001 vs. control inhibitor + vehicle, ##p < 0.01 vs. control inhibitor + atRA. Bars, 50 μm. (C) Immunoblotting analysis of CCND1 and GAPDH in MEPM cells treated with 30 μM atRA under the effect of a miR-124-3p inhibitor for 72 h. Representative images from two independent experiments are shown. (D) Quantitative RT-PCR for the indicated genes in MEPM cells after treatment with atRA under the effect of a miR-124-3p inhibitor for 24 h. *Adjusted p < 0.00313 (16 genes), **adjusted p < 0.000625 (16 genes), and ***adjusted p < 0.0000625 (16 genes) vs. control inhibitor + vehicle. #Adjusted p < 0.00313 (16 genes), ##adjusted p < 0.000625 (16 genes), and ###adjusted p < 0.0000625 (16 genes). Two-way ANOVA with Tukey’s honest significant difference test (n = 3–6).
The oral administration of atRA to pregnant mice is known to induce CP (Abbott et al., 1989; Ross et al., 2000; Wang et al., 2002; Lai et al., 2003; Yao et al., 2011; Havasi et al., 2013; Hu et al., 2013; Hou et al., 2019; Roberts, 2020). However, the amount and timing of atRA administration that induces CP may differ in each mouse strain and protocol. Therefore, we tested various doses (40, 50, 60, and 70 mg/kg) of oral atRA in C57BL/6J mice. We found that atRA administration at 40 mg/kg induced CP with 57% penetrance (12/21 embryos), while more than 50 mg/kg atRA induced CP with 100% penetrance (Table 1). Therefore, we administered a single dose of 50 mg/kg atRA at E11.5 in this study (Figure 5A). The palatal shelves of embryos treated with vehicle were normally elevated and fused at E14.5, while the palatal shelves of embryos treated with atRA failed to elevate at E14.5, causing CP at E18.5 (Figure 5B). To confirm the reduction of cell proliferation in the developing palates of mice treated with atRA, we evaluated cell proliferation by Ki-67 immunostaining. As expected, the number of Ki-67-positive cells (i.e., proliferating cells) was significantly reduced in the palatal shelves of atRA-treated embryos (Figure 5C). Next, we microdissected the palatal shelves at E13.5 and E14.5 from mice treated with either vehicle or atRA and measured the miR expression. In the vehicle control group, the expression of miR-27a-3p and miR-27b-3p was not changed between E13.5 and E14.5, while the expression of miR-124-3p at E14.5 was upregulated compared to that of E13.5 (Figure 5D). The expression of miR-124-3p was significantly upregulated with atRA administration at both E13.5 and E14.5 compared to controls, while the expression of miR-27a-3p and miR-27b-3p was comparable to the controls (Figure 5D). Furthermore, quantitative RT-PCR analysis for genes targeted by miR-124-3p confirmed that a total of three genes (Fst, Vcan, and Zeb1) were significantly downregulated in the palatal shelves of mice given atRA compared to controls (Figure 5E). To confirm that the hypoplastic mandible secondarily caused CP in mice treated with atRA, we cultured E13.5 craniofacial explants, which were extracted from the mandible and tongue, with/without atRA for 3 days (Figure 5F). In controls, the palatal shelves were elevated and almost completely fused. By contrast, the explants treated with atRA showed a widely opened palate. These results indicate that atRA primarily causes CP in mice treated with atRA.
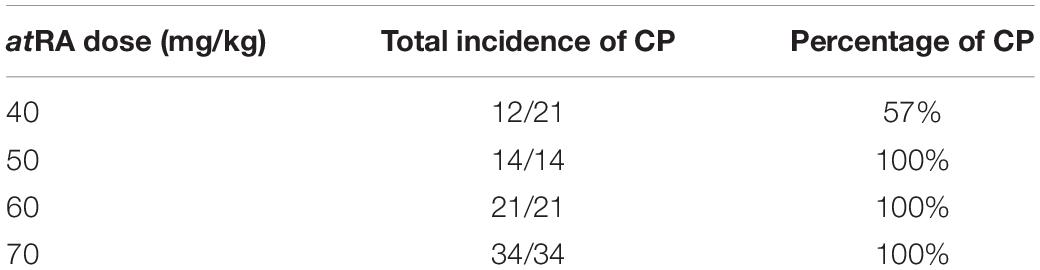
Table 1. Incidence of cleft palate (CP) by atRA administration at different doses.
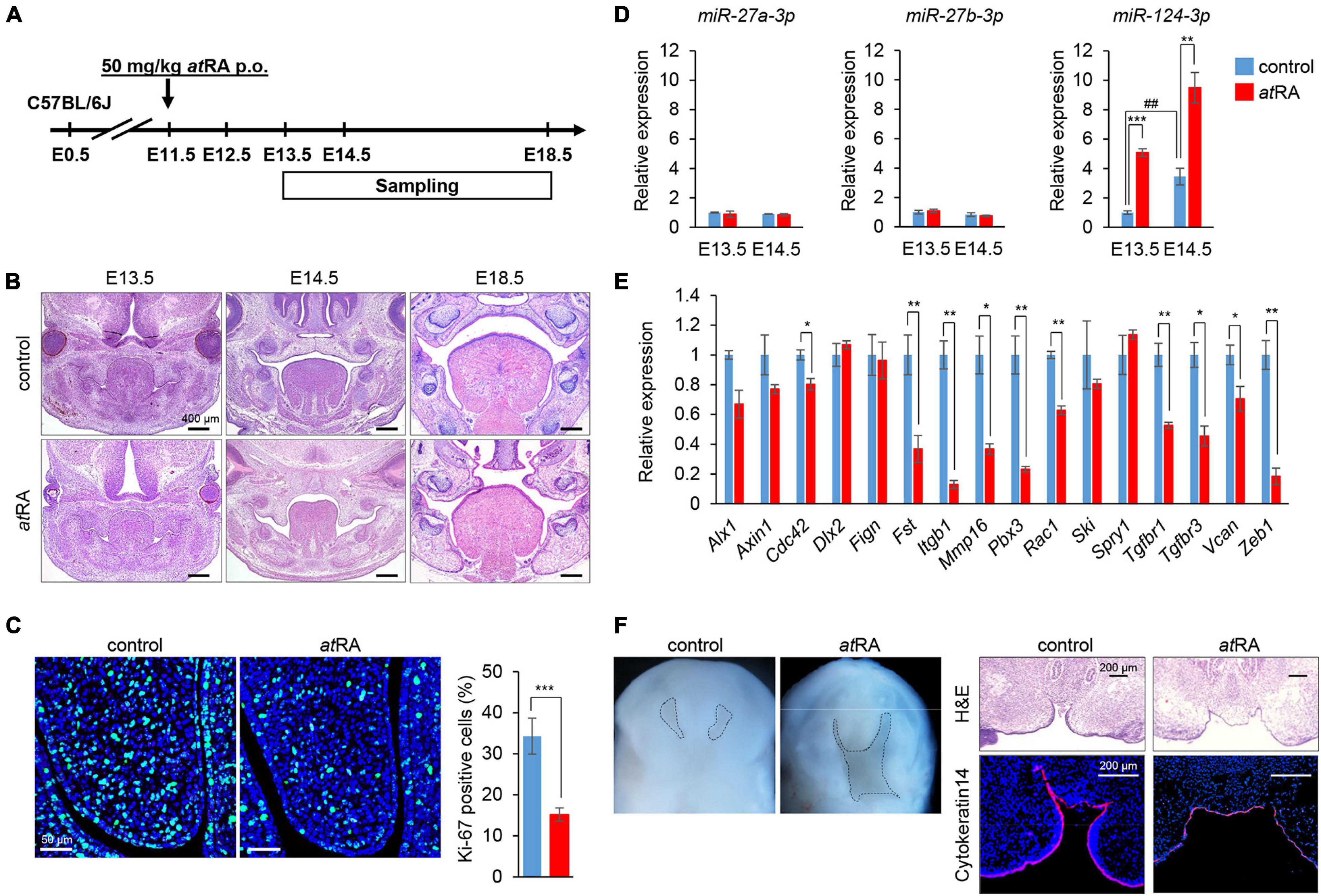
Figure 5. Influence of atRA administration on palatal development and gene expression in mice. (A) Schematic experimental design. (B) H&E staining of face at E13.5, E14.5, and E18.5. Bars, 400 μm. (C) Ki-67 staining (green) in the developing palate of E13.5 mice treated with control vehicle or atRA (50 mg/kg p.o. at E11.5). Nuclei were counterstained with DAPI (blue). Representative images from two independent experiments are shown. Graph shows the quantification of Ki-67-positive cells. Independent t-tests. ***p < 0.001 vs. control. Bars, 50 μm. (D) Quantitative RT-PCR for miR-27a-3p, miR-27b-3p, or miR-124-3p after administration of atRA in palatal shelves at E13.5 and E14.5. Two-way ANOVA with Tukey’s honest significant difference test (n = 3). **p < 0.01, ***p < 0.001 vs. control at each indicated day and ##p < 0.01 vs. E13.5 control. (E) Quantitative RT-PCR for the indicated genes after treatment with atRA for 24 h in palatal shelves at E14.5. Multiple t-tests adjusted by Bonferroni (n = 3–6). *Adjusted p < 0.00313 (16 genes), **adjusted p < 0.000625 (16 genes) vs. control. (F) Intraoral views of heads (left panels) from E13.5 embryos, from which the mandible and tongue were removed and cultured for 3 days with control vehicle or atRA (n = 4 per group). Dashed lines delineate the palatal shelves. H&E staining and K14 immunostaining (red) (right panels) of coronal sections of 3-day cultured explants with control vehicle or atRA. The nuclei were counterstained with DAPI (blue). Bars, 200 μm.
Finally, we attempted to rescue CP induced by atRA in mice through treatment with an inhibitor specific for miR-124-3p (Figure 6A). The administration of a miR-124-3p inhibitor reduced CP penetrance from 100 to 35% in the atRA-induced CPO mouse model (Figures 6B,C). The reduced cell proliferation was partially normalized in mice injected with a miR-124-3p inhibitor under atRA-treated conditions (Figure 6D). Moreover, the expression of Vcan and Zeb1 was partially normalized with a miR-124-3p inhibitor upon atRA exposure (Figure 6E). To analyze the expression pattern of extracellular matrix (ECM) formation in mice treated with atRA, we performed Azan staining for the visualization of overall ECM production and pattern. We did not detect any alteration in ECM patterning. Furthermore, to confirm that there was alteration in the patterning, but not in the level, of expression of the molecules, we conducted immunohistochemical analyses for VCAN and CDC42 in mice treated with atRA and confirmed that there was no change in the expression pattern (Figure 6F). Taken together, our results indicate that atRA induces CPO through the upregulation of miR-124-3p expression in mice.
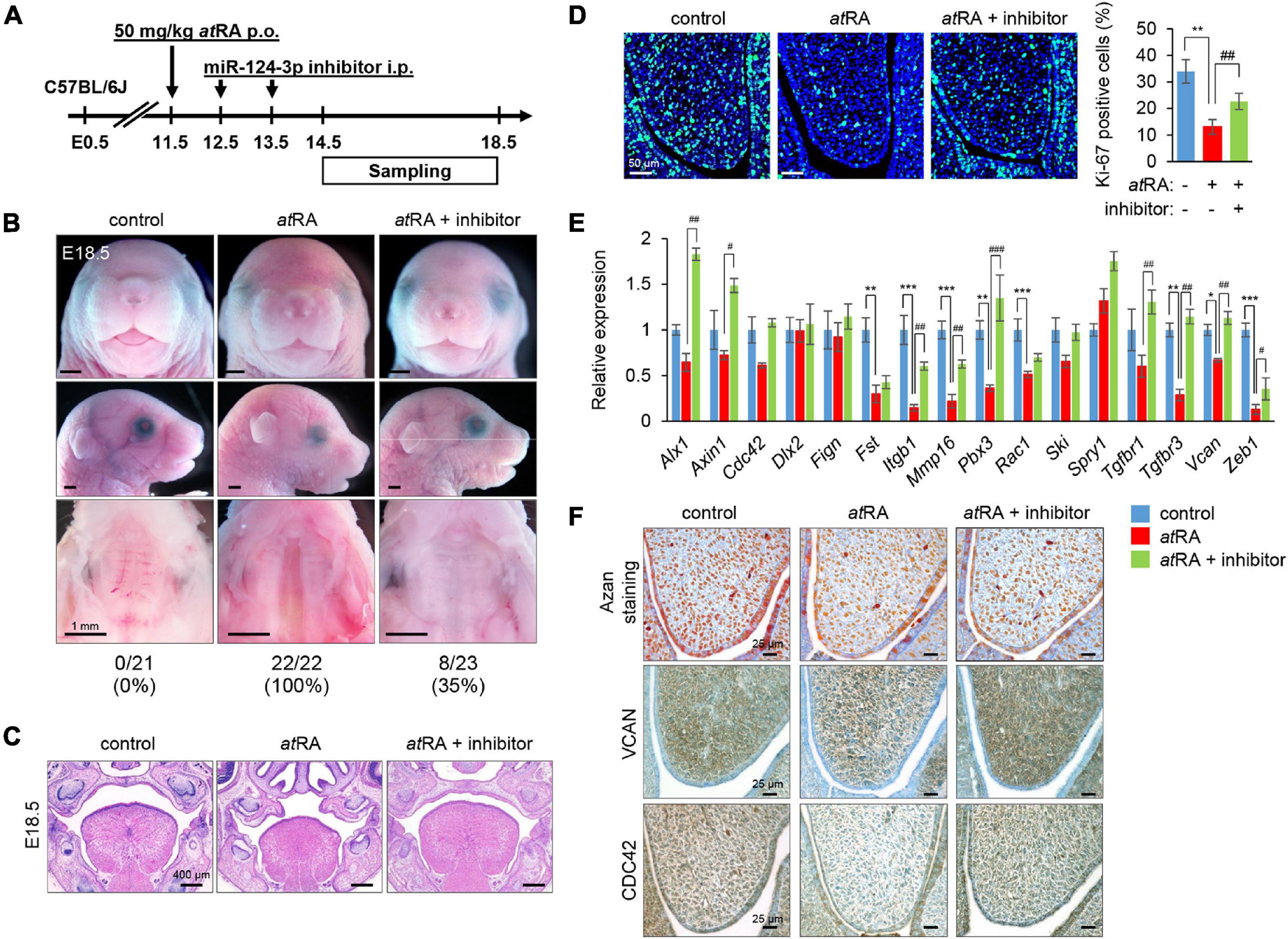
Figure 6. Inhibition of miR-124-3p expression rescues atRA-induced CP in mice. (A) Schematic experimental design. (B) Gross picture of E18.5 C57BL/6J mice treated with control vehicle, atRA, or atRA + miR-124-3p inhibitor. Bars, 1 mm. (C) H&E staining of the face of E18.5 C57BL/6J mice treated with control vehicle, atRA, or atRA + miR-124-3p inhibitor. Bars, 400 μm. (D) Ki-67 staining (green) in the developing palate of E13.5 C57BL/6J mice treated with/without a miR-124-3p inhibitor under atRA condition. Nuclei were counterstained with DAPI (blue). Representative images from two independent experiments are shown. Graph shows the quantification of Ki-67 positive cells. One-way ANOVA with Tukey’s honest significant difference test. **p < 0.01 vs. control. ##p < 0.01 vs. atRA. Bars, 50 μm. (E) Quantitative RT-PCR for the indicated genes after treatment with/without a miR-124-3p inhibitor under atRA condition in the palatal shelves of E14.5 C57BL/6J mice. Two-way ANOVA with Tukey’s honest significant difference test (n = 3). *Adjusted p < 0.00313 (16 genes), **adjusted p < 0.000625 (16 genes), and ***adjusted p < 0.0000625 (16 genes) vs. control. #Adjusted p < 0.00313 (16 genes), ##adjusted p < 0.000625 (16 genes), and ###adjusted p < 0.0000625 (16 genes) vs. atRA. (F) AZAN staining (upper panels) and immunohistochemical staining for VCAN (middle panels; brown) and CDC42 (bottom panels; brown) in E13.5 C57BL/6J mice treated with control vehicle, atRA, or atRA + miR-124-3p inhibitor. Bars, 25 μm.
miRs have been implicated in developmental processes and various types of cancers (Mukhopadhyay et al., 2010; Chen et al., 2012), and dysregulation of miR expression has been identified in a variety of human diseases (Mendell and Olson, 2012). Recent studies show that miRs play important roles in craniofacial development (Shin et al., 2012; Seelan et al., 2014; Warner et al., 2014; Mukhopadhyay et al., 2019); for example, administration of miR-23b and miR-133 duplex results in facial defects in zebrafish (Ding et al., 2016). A bioinformatic study suggested that miR-199a-5p may be associated with CL/P through the modulation of transforming growth factor (TGF)-α expression (Chen et al., 2018). miR-140-5p overexpression inhibits cell proliferation through the suppression of genes associated with CP, Fgf9, and Pdgfra in MEPM cells (Li et al., 2020). Mice with a deletion of the miR-17-92 cluster (includes six miRs: miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a, and miR-92a-1) exhibit CP through altered bone morphogenetic protein (BMP) signaling (Wang et al., 2013).
In this study, we found that the overexpression of miR-27a-3p, miR-27b-3p, and miR-124-3p could inhibit cell proliferation through the suppression of genes associated with mouse CP. miR-27 is a small somatic-enriched miR family comprising two paralogous members, miR-27a and miR-27b, in vertebrates (Kozomara and Griffiths-Jones, 2014). In zebrafish, miR-27 is highly expressed in the pharyngeal arches, and knockdown of miR-27 results in a complete loss of pharyngeal cartilage due to downregulated cell proliferation (Kara et al., 2017). miR-27a-3p, a member of the miR-23a/27a/24-2 cluster, is significantly upregulated in laryngeal carcinoma (Tian et al., 2014) and plays important roles in cell proliferation and migration in human gastric and breast cancers (Mertens-Talcott et al., 2007; Zhou et al., 2016). miR-27b-3p, a member of the miR-23b/27b/24-1 cluster, promotes cell proliferation and invasion in breast cancers (Jin et al., 2013), while miR-27b-3p inhibits cell growth and tumor progression in neuroblastoma (Lee et al., 2012), suggesting that miR-27b-3p functions are dependent on cancer types. In addition, a recent study showed that circulating miR-27b might be a potential biomarker for non-alcoholic fatty liver disease (Tan et al., 2014), and another study showed that miR-27b-3p is associated with CP in non-syndromic CL/P (Wang et al., 2017).
miR-124-3p is abundant in the human and mouse brain (Lagos-Quintana et al., 2002; Landgraf et al., 2007). In addition, miR-124-3p acts as a tumor suppressor against breast cancer and hepatocellular carcinoma (Liang et al., 2013; Long et al., 2018). Our previous study showed that the overexpression of miR-124-3p suppresses cell proliferation in primary mouse lip mesenchymal cells (Suzuki et al., 2019b). miR-124-3p regulates cell cycle by targeting integrin subunit beta 1 (ITGB1) in oral squamous cell carcinoma cells (Hunt et al., 2011), suggesting that miR-124-3p expression might remain at lower levels during normal palate development. Therefore, miR-124-3p may be a potential biomarker and a therapeutic target to prevent and diagnose CP.
In this study, we found that Bmi1 and Tgfbr3 in miR-27a-3p, Spry2 in miR-27b-3p, and Axin1, Fst, Vcan, and Zeb1 in miR-124-3p were regulated in MEPM cells through both gain-of-function and loss-of-function assays for miR in gene regulation. Because these genes are involved in various signaling pathways, including Wnt/β-catenin, BMP, TGF-β, and epidermal growth factor (EGF) signaling (Iwata et al., 2011; Reynolds et al., 2020), the overexpression of these miRs might lead to the suppression of multiple CP-associated genes through the dysregulation of these signaling pathways. In addition, we found that atRA specifically induced miR-124-3p, which suppressed the expression of genes associated with CP. While the expression of some predicted genes (Dlx2, Fign, Ski, Spry1, Tgfbr1, and Vcan) targeted by miR-124-3p was not changed by atRA in MEPM cells and mouse palatal shelves, these genes may be regulated by a combination of other miRs or through feedback loops. In addition, since four genes (Axin1, Fst, Vcan, and Zeb1) were downregulated in both miR-124-3p overexpression and atRA treatment conditions, these genes may be closely associated with atRA-induced CP in mice. AXIN1 (a WNT signaling negative regulator), FST (a BMP signaling inhibitor), and VCAN (an extracellular matrix) regulate growth factor signaling, which plays crucial roles in embryonic development; therefore, the fine-tuning regulation of the signaling pathways is important for proper embryogenesis. Homeobox gene ZEB1 regulates the epithelial-mesenchymal transition (EMT) in cancer and embryonic development (Schmalhofer et al., 2009; Gheldof et al., 2012); miR-200 family plays a role in EMT by regulating the ZEB1 expression in the developing palate in mice (Carpinelli et al., 2020).
Our rescue experiments show that a miR-124-3p inhibitor can reduce CP penetrance in the atRA-induced CP mouse model. Thus, our study is useful to identify the causative molecular mechanism of CP. However, there are some limitations in this study. First, it is unclear whether any combination of miR inhibitors can achieve better outcomes. In a future study, we will investigate which miRs are the most significantly associated with atRA-induced CP. Second, the expression of most genes was tested only by quantitative RT-PCR analysis. A future study may include not only the level but also the patterning of gene expression associated with atRA-induced CP. Finally, atRA is known to induce tongue abnormalities (Cong et al., 2014), and this study did not exclude the possibility that atRA-induced CP was secondarily caused by tongue abnormalities. A future study will test whether or not tongue anomalies due to atRA administration cause CP using a tissue explant culture system. Despite these limitations, the understanding of miR dysregulation by atRA will shed light on the link between environmental and genetic factors in CP.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The animal study was reviewed and approved by the Animal Welfare Committee (AWC) and the Institutional Animal Care and Use Committee (IACUC) of UTHealth (AWC19-0079).
AS and JI designed the research. HY, YM, and SR performed the experiments. HY, AS, and JI wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
This study was supported by grants from the National Institute of Dental and Craniofacial Research, the NIH (R03DE026208, R03DE026509, R01DE029818, and R01DE026767 to JI), and UTHealth School of Dentistry faculty funding to JI.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.621045/full#supplementary-material
Abbott, B. D., Harris, M. W., and Birnbaum, L. S. (1989). Etiology of retinoic acid-induced cleft palate varies with the embryonic stage. Teratology 40, 533–553. doi: 10.1002/tera.1420400602
Ashique, A. M., May, S. R., Kane, M. A., Folias, A. E., Phamluong, K., Choe, Y., et al. (2012). Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis 50, 415–423. doi: 10.1002/dvg.22002
Beaty, T. H., Marazita, M. L., and Leslie, E. J. (2016). Genetic factors influencing risk to orofacial clefts: today’s challenges and tomorrow’s opportunities. F1000Research 5:2800. doi: 10.12688/f1000research.9503.1
Bove, F. J., Fulcomer, M. C., Klotz, J. B., Esmart, J., Dufficy, E. M., and Savrin, J. E. (1995). Public drinking water contamination and birth outcomes. Am. J. Epidemiol. 141, 850–862. doi: 10.1093/oxfordjournals.aje.a117521
Buser, M. C., and Pohl, H. R. (2015). Windows of sensitivity to toxic chemicals in the development of cleft palates. J. Toxicol. Environ. Health Part B Crit. Rev. 18, 242–257. doi: 10.1080/10937404.2015.1068719
Carpinelli, M. R., de Vries, M. E., Auden, A., Butt, T., Deng, Z., Partridge, D. D., et al. (2020). Inactivation of Zeb1 in GRHL2-deficient mouse embryos rescues mid-gestation viability and secondary palate closure. Dis. Models Mech. 13:dmm042218.
Chen, B., Li, H., Zeng, X., Yang, P., Liu, X., Zhao, X., et al. (2012). Roles of microRNA on cancer cell metabolism. J. Transl. Med. 10:228. doi: 10.1186/1479-5876-10-228
Chen, G., Li, M. X., Wang, H. X., Hong, J. W., Shen, J. Y., Wang, Q., et al. (2018). Identification of key genes in cleft lip with or without cleft palate regulated by miR-199a-5p. Int. J. Pediatr. Otorhinolaryngol. 111, 128–137. doi: 10.1016/j.ijporl.2018.06.005
Cong, W., Liu, B., Liu, S., Sun, M., Liu, H., Yang, Y., et al. (2014). Implications of the Wnt5a/CaMKII pathway in retinoic acid-induced myogenic tongue abnormalities of developing mice. Sci. Rep. 4:6082.
Damm, K., Heyman, R. A., Umesono, K., and Evans, R. M. (1993). Functional inhibition of retinoic acid response by dominant negative retinoic acid receptor mutants. Proc. Natl. Acad. Sci. U.S.A. 90, 2989–2993. doi: 10.1073/pnas.90.7.2989
De Cuyper, E., Dochy, F., De Leenheer, E., and Van Hoecke, H. (2019). The impact of cleft lip and/or palate on parental quality of life: a pilot study. Int. J. Pediatr. Otorhinolaryngol. 126:109598. doi: 10.1016/j.ijporl.2019.109598
de Pontual, L., Yao, E., Callier, P., Faivre, L., Drouin, V., Cariou, S., et al. (2011). Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat. Genet. 43, 1026–1030. doi: 10.1038/ng.915
Ding, H. L., Hooper, J. E., Batzel, P., Eames, B. F., Postlethwait, J. H., Artinger, K. B., et al. (2016). MicroRNA profiling during craniofacial development: potential roles for Mir23b and Mir133b. Front. Physiol. 7:281. doi: 10.3389/fphys.2016.00281
Ericson, A., Kallen, B., and Westerholm, P. (1979). Cigarette smoking as an etiologic factor in cleft lip and palate. Am. J. Obstetr. Gynecol. 135, 348–351. doi: 10.1016/0002-9378(79)90703-8
Garland, M. A., Sun, B., Zhang, S., Reynolds, K., Ji, Y., and Zhou, C. J. (2020). Role of epigenetics and miRNAs in orofacial clefts. Birth Defects Res. 112, 1635–1659. doi: 10.1002/bdr2.1802
Gheldof, A., Hulpiau, P., van Roy, F., De Craene, B., and Berx, G. (2012). Evolutionary functional analysis and molecular regulation of the ZEB transcription factors. Cell. Mol. Life Sci.: CMLS 69, 2527–2541. doi: 10.1007/s00018-012-0935-3
Gonseth, S., Shaw, G. M., Roy, R., Segal, M. R., Asrani, K., Rine, J., et al. (2019). Epigenomic profiling of newborns with isolated orofacial clefts reveals widespread DNA methylation changes and implicates metastable epiallele regions in disease risk. Epigenetics 14, 198–213. doi: 10.1080/15592294.2019.1581591
Havasi, A., Haegele, J. A., Gall, J. M., Blackmon, S., Ichimura, T., Bonegio, R. G., et al. (2013). Histone acetyl transferase (HAT) HBO1 and JADE1 in epithelial cell regeneration. Am. J. Pathol. 182, 152–162. doi: 10.1016/j.ajpath.2012.09.017
Hou, L., Wang, D., and Baccarelli, A. (2011). Environmental chemicals and microRNAs. Mutation Res. 714, 105–112. doi: 10.1016/j.mrfmmm.2011.05.004
Hou, L., Zhu, L., Li, H., Jiang, F., Cao, L., Hu, C. Y., et al. (2019). MiR-501-3p forms a feedback loop with FOS, MDFI, and MyoD to regulate C2C12 myogenesis. Cells 8:573. doi: 10.3390/cells8060573
Hu, X., Gao, J., Liao, Y., Tang, S., and Lu, F. (2013). Retinoic acid alters the proliferation and survival of the epithelium and mesenchyme and suppresses Wnt/beta-catenin signaling in developing cleft palate. Cell. Death Dis. 4:e898. doi: 10.1038/cddis.2013.424
Hudder, A., and Novak, R. F. (2008). miRNAs: effectors of environmental influences on gene expression and disease. Toxicol. Sci. : Off. J. Soc. Toxicol. 103, 228–240. doi: 10.1093/toxsci/kfn033
Hunt, S., Jones, A. V., Hinsley, E. E., Whawell, S. A., and Lambert, D. W. (2011). MicroRNA-124 suppresses oral squamous cell carcinoma motility by targeting ITGB1. FEBS Lett. 585, 187–192. doi: 10.1016/j.febslet.2010.11.038
IPDTOC Working Group (2011). Prevalence at birth of cleft lip with or without cleft palate: data from the International Perinatal Database of Typical Oral Clefts (IPDTOC). Cleft Palate-Craniofacial J. : Off. Publ. Am. Cleft Palate-Craniofacial Assoc. 48, 66–81. doi: 10.1597/09-217
Iwata, J., Parada, C., and Chai, Y. (2011). The mechanism of TGF-beta signaling during palate development. Oral Dis. 17, 733–744. doi: 10.1111/j.1601-0825.2011.01806.x
Iwata, J., Suzuki, A., Pelikan, R. C., Ho, T. V., and Chai, Y. (2013). Noncanonical transforming growth factor beta (TGFbeta) signaling in cranial neural crest cells causes tongue muscle developmental defects. J. Biol. Chem. 288, 29760–29770. doi: 10.1074/jbc.m113.493551
Iwata, J., Suzuki, A., Pelikan, R. C., Ho, T. V., Sanchez-Lara, P. A., and Chai, Y. (2014). Modulation of lipid metabolic defects rescues cleft palate in Tgfbr2 mutant mice. Hum. Mol. Genet. 23, 182–193. doi: 10.1093/hmg/ddt410
Jin, L., Wessely, O., Marcusson, E. G., Ivan, C., Calin, G. A., and Alahari, S. K. (2013). Prooncogenic factors miR-23b and miR-27b are regulated by Her2/Neu, EGF, and TNF-alpha in breast cancer. Cancer Res. 73, 2884–2896. doi: 10.1158/0008-5472.can-12-2162
Kara, N., Wei, C., Commanday, A. C., and Patton, J. G. (2017). miR-27 regulates chondrogenesis by suppressing focal adhesion kinase during pharyngeal arch development. Dev. Biol. 429, 321–334. doi: 10.1016/j.ydbio.2017.06.013
Kozomara, A., and Griffiths-Jones, S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73.
Kummer, A. W. (2018). A Pediatrician’s guide to communication disorders secondary to cleft lip/palate. Pediatr. Clin. North Am. 65, 31–46. doi: 10.1016/j.pcl.2017.08.019
Kuriyama, M., Udagawa, A., Yoshimoto, S., Ichinose, M., Sato, K., Yamazaki, K., et al. (2008). DNA methylation changes during cleft palate formation induced by retinoic acid in mice. Cleft Palate-Craniofacial J. : Off. Publ. Am. Cleft Palate-Craniofacial Assoc. 45, 545–551. doi: 10.1597/07-134.1
Lagos-Quintana, M., Rauhut, R., Yalcin, A., Meyer, J., Lendeckel, W., and Tuschl, T. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol.: CB 12, 735–739. doi: 10.1016/s0960-9822(02)00809-6
Lai, L., Bohnsack, B. L., Niederreither, K., and Hirschi, K. K. (2003). Retinoic acid regulates endothelial cell proliferation during vasculogenesis. Development 130, 6465–6474. doi: 10.1242/dev.00887
Lammer, E. J., Chen, D. T., Hoar, R. M., Agnish, N. D., Benke, P. J., Braun, J. T., et al. (1985). Retinoic acid embryopathy. New England J. Med. 313, 837–841.
Landgraf, P., Rusu, M., Sheridan, R., Sewer, A., Iovino, N., Aravin, A., et al. (2007). A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129, 1401–1414.
Lee, J. J., Drakaki, A., Iliopoulos, D., and Struhl, K. (2012). MiR-27b targets PPARgamma to inhibit growth, tumor progression and the inflammatory response in neuroblastoma cells. Oncogene 31, 3818–3825. doi: 10.1038/onc.2011.543
Li, A., Jia, P., Mallik, S., Fei, R., Yoshioka, H., Suzuki, A., et al. (2020). Critical microRNAs and regulatory motifs in cleft palate identified by a conserved miRNA-TF-gene network approach in humans and mice. Briefings Bioinform. 21, 1465–1478. doi: 10.1093/bib/bbz082
Li, L., Shi, J. Y., Zhu, G. Q., and Shi, B. (2012). MiR-17-92 cluster regulates cell proliferation and collagen synthesis by targeting TGFB pathway in mouse palatal mesenchymal cells. J. Cell. Biochem. 113, 1235–1244. doi: 10.1002/jcb.23457
Liang, Y. J., Wang, Q. Y., Zhou, C. X., Yin, Q. Q., He, M., Yu, X. T., et al. (2013). MiR-124 targets Slug to regulate epithelial-mesenchymal transition and metastasis of breast cancer. Carcinogenesis 34, 713–722. doi: 10.1093/carcin/bgs383
Liu, D., Zhong, L., Yuan, Z., Yao, J., Zhong, P., Liu, J., et al. (2019). miR-382-5p modulates the ATRA-induced differentiation of acute promyelocytic leukemia by targeting tumor suppressor PTEN. Cell Signal 54, 1–9. doi: 10.1016/j.cellsig.2018.11.012
Liu, W., Song, Y., Zhang, C., Gao, P., Huang, B., and Yang, J. (2018). The protective role of all-transretinoic acid (ATRA) against colorectal cancer development is achieved via increasing miR-3666 expression and decreasing E2F7 expression. Biomed. Pharmacother. 104, 94–101. doi: 10.1016/j.biopha.2018.05.015
Lohnes, D., Mark, M., Mendelsohn, C., Dolle, P., Dierich, A., Gorry, P., et al. (1994). Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 120, 2723–2748. doi: 10.1242/dev.120.10.2723
Long, H. D., Ma, Y. S., Yang, H. Q., Xue, S. B., Liu, J. B., Yu, F., et al. (2018). Reduced hsa-miR-124-3p levels are associated with the poor survival of patients with hepatocellular carcinoma. Mol. Biol. Rep. 45, 2615–2623. doi: 10.1007/s11033-018-4431-1
Maclean, G., Dolle, P., and Petkovich, M. (2009). Genetic disruption of CYP26B1 severely affects development of neural crest derived head structures, but does not compromise hindbrain patterning. Dev. Dynamics : Off. Publ. Am. Assoc. Anatomists 238, 732–745. doi: 10.1002/dvdy.21878
Mark, M., Lohnes, D., Mendelsohn, C., Dupe, V., Vonesch, J. L., Kastner, P., et al. (1995). Roles of retinoic acid receptors and of Hox genes in the patterning of the teeth and of the jaw skeleton. Int. J. Dev. Biol. 39, 111–121.
Martinez-Frias, M. L., and Salvador, J. (1990). Epidemiological aspects of prenatal exposure to high doses of vitamin A in Spain. Eur. J. Epidemiol. 6, 118–123. doi: 10.1007/bf00145783
Mendell, J. T., and Olson, E. N. (2012). MicroRNAs in stress signaling and human disease. Cell 148, 1172–1187. doi: 10.1016/j.cell.2012.02.005
Mertens-Talcott, S. U., Chintharlapalli, S., Li, X., and Safe, S. (2007). The oncogenic microRNA-27a targets genes that regulate specificity protein transcription factors and the G2-M checkpoint in MDA-MB-231 breast cancer cells. Cancer Res. 67, 11001–11011. doi: 10.1158/0008-5472.can-07-2416
Mills, J. L., Simpson, J. L., Cunningham, G. C., Conley, M. R., and Rhoads, G. G. (1997). Vitamin A and birth defects. Am. J. Obstetr. Gynecol. 177, 31–36.
Mukhopadhyay, P., Brock, G., Pihur, V., Webb, C., Pisano, M. M., and Greene, R. M. (2010). Developmental microRNA expression profiling of murine embryonic orofacial tissue. Birth Defects Res. Part A, Clin. Mol. Teratol. 88, 511–534. doi: 10.1002/bdra.20684
Mukhopadhyay, P., Smolenkova, I., Warner, D., Pisano, M. M., and Greene, R. M. (2019). Spatio-temporal expression and functional analysis of miR-206 in developing orofacial tissue. MicroRNA 8, 43–60. doi: 10.2174/2211536607666180801094528
Munger, R. G., Romitti, P. A., Daack-Hirsch, S., Burns, T. L., Murray, J. C., and Hanson, J. (1996). Maternal alcohol use and risk of orofacial cleft birth defects. Teratology 54, 27–33. doi: 10.1002/(sici)1096-9926(199607)54:1<27::aid-tera4>3.0.co;2-0
Murray, J. C. (2002). Gene/environment causes of cleft lip and/or palate. Clin. Genet. 61, 248–256. doi: 10.1034/j.1399-0004.2002.610402.x
Nie, X., Wang, Q., and Jiao, K. (2011). Dicer activity in neural crest cells is essential for craniofacial organogenesis and pharyngeal arch artery morphogenesis. Mech. Dev. 128, 200–207. doi: 10.1016/j.mod.2010.12.002
Pi, X., Jin, L., Li, Z., Liu, J., Zhang, Y., Wang, L., et al. (2019). Association between concentrations of barium and aluminum in placental tissues and risk for orofacial clefts. Sci. Tot. Environ. 652, 406–412. doi: 10.1016/j.scitotenv.2018.10.262
Reynolds, K., Zhang, S., Sun, B., Garland, M. A., Ji, Y., and Zhou, C. J. (2020). Genetics and signaling mechanisms of orofacial clefts. Birth Defects Res. 112, 1588–1634. doi: 10.1002/bdr2.1754
Rhinn, M., and Dolle, P. (2012). Retinoic acid signalling during development. Development 139, 843–858. doi: 10.1242/dev.065938
Roberts, C. (2020). Regulating retinoic acid availability during development and regeneration: the role of the CYP26 enzymes. J. Dev. Biol. 8:6. doi: 10.3390/jdb8010006
Romitti, P. A., Lidral, A. C., Munger, R. G., Daack-Hirsch, S., Burns, T. L., and Murray, J. C. (1999). Candidate genes for nonsyndromic cleft lip and palate and maternal cigarette smoking and alcohol consumption: evaluation of genotype-environment interactions from a population-based case-control study of orofacial clefts. Teratology 59, 39–50. doi: 10.1002/(sici)1096-9926(199901)59:1<39::aid-tera9>3.0.co;2-7
Ross, S. A., McCaffery, P. J., Drager, U. C., and De Luca, L. M. (2000). Retinoids in embryonal development. Physiol. Rev. 80, 1021–1054. doi: 10.1152/physrev.2000.80.3.1021
Schmalhofer, O., Brabletz, S., and Brabletz, T. (2009). E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 28, 151–166. doi: 10.1007/s10555-008-9179-y
Schoen, C., Aschrafi, A., Thonissen, M., Poelmans, G., Von den Hoff, J. W., and Carels, C. E. L. (2017). MicroRNAs in palatogenesis and cleft palate. Front. Physiol. 8:165. doi: 10.3389/fphys.2017.00165
Seelan, R. S., Mukhopadhyay, P., Warner, D. R., Appana, S. N., Brock, G. N., Pisano, M. M., et al. (2014). Methylated microRNA genes of the developing murine palate. MicroRNA 3, 160–173. doi: 10.2174/2211536604666150131125805
Shin, J. O., Lee, J. M., Cho, K. W., Kwak, S., Kwon, H. J., Lee, M. J., et al. (2012). MiR-200b is involved in Tgf-beta signaling to regulate mammalian palate development. Histochem. Cell Biol. 137, 67–78. doi: 10.1007/s00418-011-0876-1
Sischo, L., Wilson-Genderson, M., and Broder, H. L. (2017). Quality-of-life in children with orofacial clefts and caregiver well-being. J. Dental Res. 96, 1474–1481. doi: 10.1177/0022034517725707
Skare, O., Jugessur, A., Lie, R. T., Wilcox, A. J., Murray, J. C., Lunde, A., et al. (2012). Application of a novel hybrid study design to explore gene-environment interactions in orofacial clefts. Ann. Hum. Genet. 76, 221–236. doi: 10.1111/j.1469-1809.2012.00707.x
Suhl, J., Leonard, S., Weyer, P., Rhoads, A., Siega-Riz, A. M., Renee Anthony, T., et al. (2018). Maternal arsenic exposure and nonsyndromic orofacial clefts. Birth Defects Res. 110, 1455–1467. doi: 10.1002/bdr2.1386
Suzuki, A., Abdallah, N., Gajera, M., Jun, G., Jia, P., Zhao, Z., et al. (2018a). Genes and microRNAs associated with mouse cleft palate: a systematic review and bioinformatics analysis. Mech. Dev. 150, 21–27. doi: 10.1016/j.mod.2018.02.003
Suzuki, A., Jun, G., Abdallah, N., Gajera, M., and Iwata, J. (2018b). Gene datasets associated with mouse cleft palate. Data Brief 18, 655–673. doi: 10.1016/j.dib.2018.03.010
Suzuki, A., Li, A., Gajera, M., Abdallah, N., Zhang, M., Zhao, Z., et al. (2019a). MicroRNA-374a, -4680, and -133b suppress cell proliferation through the regulation of genes associated with human cleft palate in cultured human palate cells. BMC Med. Genomics 12:93.
Suzuki, A., Yoshioka, H., Summakia, D., Desai, N. G., Jun, G., Jia, P., et al. (2019b). MicroRNA-124-3p suppresses mouse lip mesenchymal cell proliferation through the regulation of genes associated with cleft lip in the mouse. BMC Genomics 20:852. doi: 10.1186/s12864-019-6238-4
Tan, Y., Ge, G., Pan, T., Wen, D., and Gan, J. (2014). A pilot study of serum microRNAs panel as potential biomarkers for diagnosis of nonalcoholic fatty liver disease. PLoS One 9:e105192. doi: 10.1371/journal.pone.0105192
Tian, Y., Fu, S., Qiu, G. B., Xu, Z. M., Liu, N., Zhang, X. W., et al. (2014). MicroRNA-27a promotes proliferation and suppresses apoptosis by targeting PLK2 in laryngeal carcinoma. BMC Cancer 14:678. doi: 10.1186/1471-2407-14-678
Ventura, A., Young, A. G., Winslow, M. M., Lintault, L., Meissner, A., Erkeland, S. J., et al. (2008). Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132, 875–886. doi: 10.1016/j.cell.2008.02.019
Wang, J., Bai, Y., Li, H., Greene, S. B., Klysik, E., Yu, W., et al. (2013). MicroRNA-17-92, a direct Ap-2alpha transcriptional target, modulates T-box factor activity in orofacial clefting. PLoS Genet. 9:e1003785. doi: 10.1371/journal.pgen.1003785
Wang, Q., Kurosaka, H., Kikuchi, M., Nakaya, A., Trainor, P. A., and Yamashiro, T. (2019). Perturbed development of cranial neural crest cells in association with reduced sonic hedgehog signaling underlies the pathogenesis of retinoic-acid-induced cleft palate. Dis. Models Mech. 12:dmm040279.
Wang, S., Sun, C., Meng, Y., Zhang, B., Wang, X., Su, Y., et al. (2017). A pilot study: Screening target miRNAs in tissue of nonsyndromic cleft lip with or without cleft palate. Exp. Therapeutic Med. 13, 2570–2576. doi: 10.3892/etm.2017.4248
Wang, X., Shen, X., Li, X., and Agrawal, C. M. (2002). Age-related changes in the collagen network and toughness of bone. Bone 31, 1–7. doi: 10.1016/s8756-3282(01)00697-4
Warner, D. R., Mukhopadhyay, P., Brock, G., Webb, C. L., Michele Pisano, M., and Greene, R. M. (2014). MicroRNA expression profiling of the developing murine upper lip. Dev. Growth Differ. 56, 434–447. doi: 10.1111/dgd.12140
Werler, M. M., Lammer, E. J., Rosenberg, L., and Mitchell, A. A. (1990). Maternal vitamin A supplementation in relation to selected birth defects. Teratology 42, 497–503. doi: 10.1002/tera.1420420506
Xu, M., Ma, L., Lou, S., Du, Y., Yin, X., Zhang, C., et al. (2018). Genetic variants of microRNA processing genes and risk of non-syndromic orofacial clefts. Oral Dis. 24, 422–428. doi: 10.1111/odi.12741
Yao, Z., Chen, D., Wang, A., Ding, X., Liu, Z., Ling, L., et al. (2011). Folic acid rescue of ATRA-induced cleft palate by restoring the TGF-beta signal and inhibiting apoptosis. J. Oral. Pathol. Med. 40, 433–439. doi: 10.1111/j.1600-0714.2010.00994.x
Zehir, A., Hua, L. L., Maska, E. L., Morikawa, Y., and Cserjesi, P. (2010). Dicer is required for survival of differentiating neural crest cells. Dev. Biol. 340, 459–467. doi: 10.1016/j.ydbio.2010.01.039
Zhang, J., Zhou, S., Zhang, Q., Feng, S., Chen, Y., Zheng, H., et al. (2014). Proteomic analysis of RBP4/vitamin a in children with cleft lip and/or palate. J. Dent. Res. 93, 547–552. doi: 10.1177/0022034514530397
Zhang, Y., Mori, T., Iseki, K., Hagino, S., Takaki, H., Takeuchi, M., et al. (2003). Differential expression of decorin and biglycan genes during palatogenesis in normal and retinoic acid-treated mice. Dev. Dynamics : Off. Publ. Am. Assoc. Anatomists 226, 618–626. doi: 10.1002/dvdy.10267
Keywords: all-trans retinoic acid, environmental factor, cleft palate, microRNA, craniofacial development
Citation: Yoshioka H, Mikami Y, Ramakrishnan SS, Suzuki A and Iwata J (2021) MicroRNA-124-3p Plays a Crucial Role in Cleft Palate Induced by Retinoic Acid. Front. Cell Dev. Biol. 9:621045. doi: 10.3389/fcell.2021.621045
Received: 24 October 2020; Accepted: 05 May 2021;
Published: 09 June 2021.
Edited by:
Sebastian Dworkin, La Trobe University, AustraliaReviewed by:
Rahul N. Kanadia, University of Connecticut, United StatesCopyright © 2021 Yoshioka, Mikami, Ramakrishnan, Suzuki and Iwata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junichi Iwata, SnVuaWNoaS5Jd2F0YUB1dGgudG1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.