
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 25 February 2021
Sec. Signaling
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.613336
This article is part of the Research TopicOrganelles Relationships and Interactions: a Cancer PerspectiveView all 9 articles
 Paula Díaz1,2†Alejandra Sandoval-Bórquez1,2†
Paula Díaz1,2†Alejandra Sandoval-Bórquez1,2† Roberto Bravo-Sagua1,3
Roberto Bravo-Sagua1,3 Andrew F. G. Quest1,2,4*
Andrew F. G. Quest1,2,4* Sergio Lavandero1,2,4,5*
Sergio Lavandero1,2,4,5*In recent decades, compelling evidence has emerged showing that organelles are not static structures but rather form a highly dynamic cellular network and exchange information through membrane contact sites. Although high-throughput techniques facilitate identification of novel contact sites (e.g., organelle-organelle and organelle-vesicle interactions), little is known about their impact on cellular physiology. Moreover, even less is known about how the dysregulation of these structures impacts on cellular function and therefore, disease. Particularly, cancer cells display altered signaling pathways involving several cell organelles; however, the relevance of interorganelle communication in oncogenesis and/or cancer progression remains largely unknown. This review will focus on organelle contacts relevant to cancer pathogenesis. We will highlight specific proteins and protein families residing in these organelle-interfaces that are known to be involved in cancer-related processes. First, we will review the relevance of endoplasmic reticulum (ER)-mitochondria interactions. This section will focus on mitochondria-associated membranes (MAMs) and particularly the tethering proteins at the ER-mitochondria interphase, as well as their role in cancer disease progression. Subsequently, the role of Ca2+ at the ER-mitochondria interphase in cancer disease progression will be discussed. Members of the Bcl-2 protein family, key regulators of cell death, also modulate Ca2+ transport pathways at the ER-mitochondria interphase. Furthermore, we will review the role of ER-mitochondria communication in the regulation of proteostasis, focusing on the ER stress sensor PERK (PRKR-like ER kinase), which exerts dual roles in cancer. Second, we will review the relevance of ER and mitochondria interactions with other organelles. This section will focus on peroxisome and lysosome organelle interactions and their impact on cancer disease progression. In this context, the peroxisome biogenesis factor (PEX) gene family has been linked to cancer. Moreover, the autophagy-lysosome system is emerging as a driving force in the progression of numerous human cancers. Thus, we will summarize our current understanding of the role of each of these organelles and their communication, highlighting how alterations in organelle interfaces participate in cancer development and progression. A better understanding of specific organelle communication sites and their relevant proteins may help to identify potential pharmacological targets for novel therapies in cancer control.
Higher organisms are characterized by the cooperation between cell populations, which carry out different, complementary functions. This entails the existence of various gene expression programs encoded in the same genome, and processes such as cell differentiation and proliferation. All these processes must be dynamic and tightly regulated to maintain homeostasis in response to the ever-changing external and internal environments. However, multicellularity comes at a price: dysregulation of the aforementioned processes can lead to tumorigenesis and cancer (Trigos et al., 2018).
Cancer cells not only proliferate uncontrollably, but also they avoid differentiation and attain several special traits, such as the ability to increase the supply of nutrients, become invisible to the immune system, change their metabolism and adapt to surroundings that vary as the tumor progresses, among others (Fouad and Aanei, 2017). This transformation requires extensive genetic remodeling, which radically changes the intra and intercellular landscape.
Within the eukaryotic cell, organelles compartmentalize specific processes, which determines the cell phenotype and thus, have an impact on tumorigenesis. Relevant for this review, mitochondria play a key role in regulating cell death, as well as providing energy, which is, in turn, a major player in cell adaption and survival (Bravo-Sagua et al., 2017). The endoplasmic reticulum (ER), on the other hand, synthesizes large amounts of proteins and governs intracellular Ca2+ signaling, two activities crucial for cell viability (Oakes and Papa, 2015). Lysosomes, in turn, possess lytic enzymes required for the degradation of damaged organelles and other intracellular structures, serving as a quality control mechanism (Lawrence and Zoncu, 2019). This function involves a group of processes collectively termed autophagy (Lawrence and Zoncu, 2019). Finally, peroxisomes represent a set of vesicles that participate in lipid and oxidative metabolism, and are emerging players in cancer development (Islinger et al., 2018).
Since organelles harbor distinct and fundamental activities, they are meticulously modulated to preserve tissue physiology and avoid perturbations in homeostasis. Furthermore, organelles communicate with each other to coordinate stress responses, which entails damage-sensing at the ER surface by ER stress sensors, and increased ATP generation by mitochondria, fueled by metabolites generated through lysosome- and peroxisome-mediated degradation processes. One means of communication between them are interorganelle contacts, which permit direct exchange of signaling molecules and scaffolding of key regulatory complexes, thereby avoiding the fusion of their membranes and luminal continuity as a result (Lopez-Crisosto et al., 2015). Such is the importance of these contacts that their alteration is associated with various pathologies, especially cardiometabolic diseases (Lopez-Crisosto et al., 2017). ER-mitochondria contacts are the most widely studied organelle contacts, and, interestingly decline in most pathologies. This is consistent with the notion that lacking a coordinated response leads to maladaptation and, ultimately, cell dysfunction (Lopez-Crisosto et al., 2015).
Processes associated with cancer development and progression correlate with alterations in organelle communication (Boroughs and Deberardinis, 2015), although the nature of such dysregulation is far more complex and, in some cases, contradictory. This ambiguity in cancer may arise because its cause is not the lack of cell adaptation, but rather the opposite, namely the exacerbated capacity to overcome adversities at any cost (Fouad and Aanei, 2017). Thus, it follows that cancer cells have altered organelle interfaces, especially those regulating cell death and survival, adjustment to stress and removal, or even preservation of dysfunctional structures. However, while an intriguing concept, the evidence available supporting this notion is still rather scarce. Here, we provide a revision of the current literature, with the objective of revealing the relevance of organelle (mis)communication in the genesis of cancer disease.
Mitochondria are double-membrane bound organelles, responsible for oxidative cell metabolism and reactive oxygen species (ROS) generation, Ca2+ homeostasis and apoptosis, among others (Wallace, 2012). Besides their own regulation, mitochondria interact and communicate with other organelles (Nunnari and Suomalainen, 2012). However, dysregulation in such communication may trigger aberrant mitochondrial function resulting in impaired energy metabolism and ion buffering. As a result, mitochondrial dysfunction plays an important role in cancer (Doghman-Bouguerra and Lalli, 2019).
Mitochondria interact with the ER mainly through membrane structures referred to as mitochondria-associated ER membranes (MAMs). MAMs are well-characterized, 10–30 nm organelle contact sites (Bernhard and Rouiller, 1956; Wieckowski et al., 2009) that are rich in Ca2+ transporters, enzymes participating in lipid synthesis and transport, as well as tumor suppressors and proteins encoded by oncogenes that regulate cell signaling pathways (Lee and Min, 2018). Thus, MAM-associated protein dysfunction is involved in tumorigenesis and tumor progression.
Mitofusins (MFN) are GTPases embedded in the ER surface and outer mitochondrial membrane (OMM), with a key role in mitochondrial dynamics. MFN regulates ER-mitochondria contacts between Ca2+-transfer sites (De Brito and Scorrano, 2008; Filadi et al., 2015; Naon et al., 2016). ER-resident MFN2 interacts with mitochondrial MFN1/MFN2 and regulates cell survival by playing anti-proliferative and pro-apoptotic roles (Ma et al., 2015; Wang et al., 2018b; Liu et al., 2019), as well as by participating in autophagy (Xue et al., 2018). MFN2 is considered a tumor suppressor (Figure 1) and is silenced in many malignant tumors (Zhang et al., 2013; Li et al., 2018). Thus, dysregulation of MFN2 function as a tethering protein at the ER-mitochondria interphase (De Brito and Scorrano, 2008, 2009) is likely to participate in cancer progression.
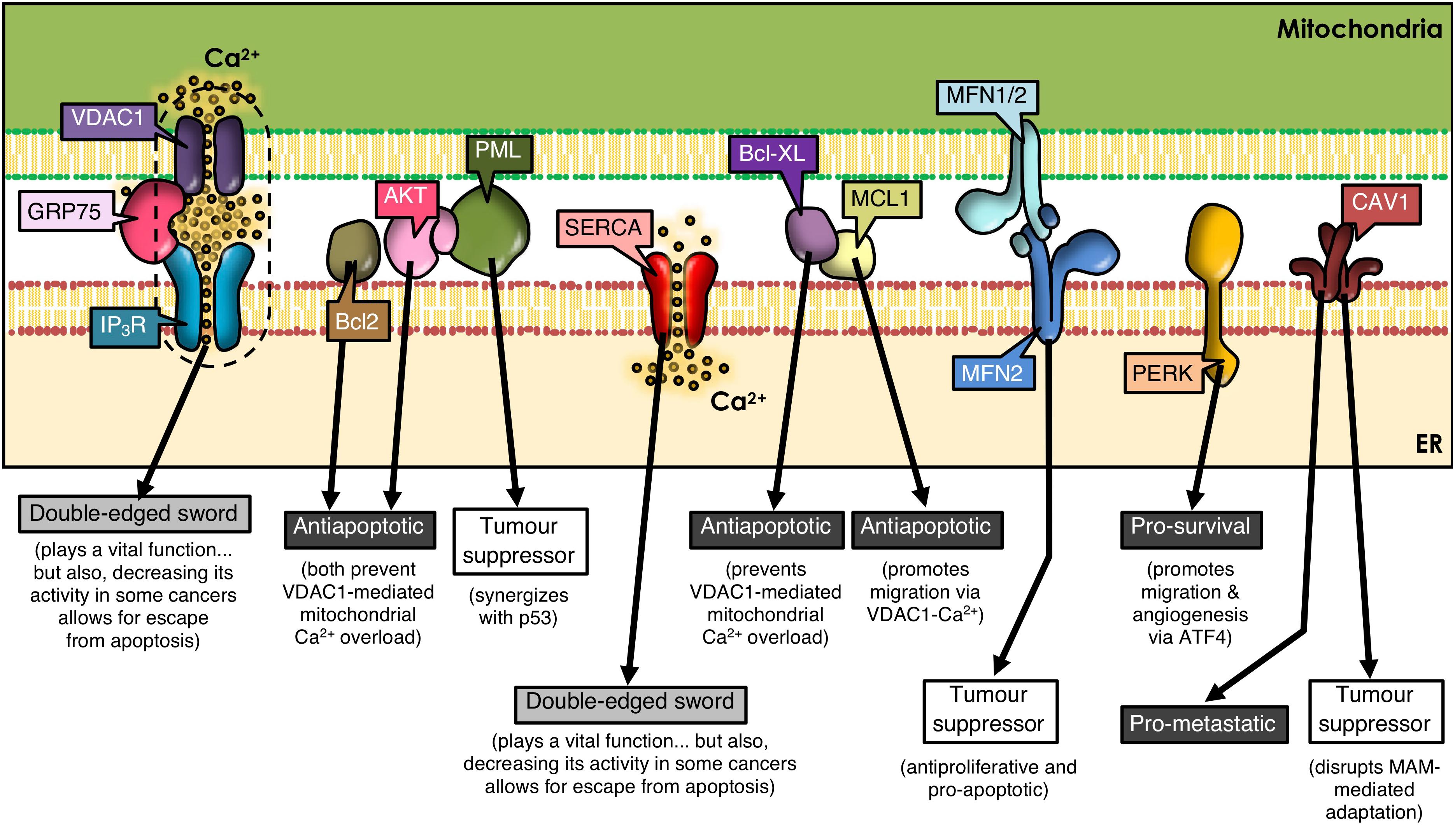
Figure 1. ER-mitochondria contacts and cancer development. Many proteins present at MAMs determine the outcome of tumorigenesis. VDAC1, GRP75, IP3R, and SERCA play a dual role, as they are essential for cell viability by mediating ER-to-mitochondria Ca2+ transfer; but, paradoxically, a decrease in their function prevents apoptosis in some cancer types. Many other MAM-residing proteins regulate IP3R-VDAC1 interplay, tilting the balance to one side or the other. Additional subsets of proteins are shown that either reside in or translocate to MAMs, thereby controlling signaling networks that modulate cell fate.
Caveolin-1 (CAV1), a membrane-associated scaffolding protein, that acts both as a tumor suppressor and a promoter of metastasis depending on the type of cancer and stage (Campos et al., 2019; Simon et al., 2020), is enriched in MAMs (Sala-Vila et al., 2016; Bravo-Sagua et al., 2019; Figure 1). There CAV1 plays a controversial role; on the one hand, CAV1 reportedly limits the adaptation to stress in tumor cells through impairment of ER-mitochondria contacts (Bravo-Sagua et al., 2019); on the other hand, using livers from wild-type and CAV1-deficient mice, it was shown that CAV1 promotes ER-mitochondria contacts, thereby contributing to the recruitment and regulation of intracellular steroids and lipoprotein metabolism (Sala-Vila et al., 2016). Whether these opposite effects may be related to the subcellular localization of CAV1 or the specific tumor cell-type, remains to be explored.
Ca2+ enters the mitochondria from the ER through MAMs where it regulates bioenergetics and metabolism by controling the activity of key enzymes of the tricarboxylic acid cycle and fatty acid (FA) oxidation (Glancy and Balaban, 2012). In addition, Ca2+ is important in mitochondrial fission and control of apoptosis. The low-affinity mitochondrial Ca2+ uniporter (MCU) receptor on the inner mitochondrial membrane (IMM) participates in Ca2+ uptake from the mitochondrial matrix and Ca2+ permeates to the OMM through the voltage-dependent anion channel 1 (VDAC1) (Baughman et al., 2011; Chaudhuri et al., 2013; Morciano et al., 2018). To do so, high local concentrations of Ca2+ need to be generated at MAMs, which are enriched in the Ca2+-sensitive inositol 1,4,5-trisphosphate receptor (IP3R). Ca2+ in the ER is rapidly released to the surrounding cytoplasm through IP3Rs, exposing mitochondria to higher concentrations of Ca2+ (Csordas et al., 2006, 2010). Alterations in the expression and/or function of these Ca2+-transport/binding systems have been implicated in oncogenesis and cancer progression (Marchi and Pinton, 2016; Bultynck and Campanella, 2017).
Several types of cancer cells undergo extensive reorganization of Ca2+ signaling to promote tumor progression. This reorganization of Ca2+-dependent mechanisms (i.e., Ca2+ transport pathways, Ca2+-dependent signaling) has a direct impact on cell survival by altering proliferation, migration, invasion, and metastasis (Chen et al., 2013; Monteith et al., 2017). Tumor cells display a dependency on constitutive Ca2+ transfer to maintain viability (Cardenas et al., 2016). For instance, the chaperone glucose regulatory protein 75 (GRP75), a protein expressed at MAMs, allows efficient IP3R-mediated Ca2+ transfer into mitochondria through VDAC on the OMM. However, upregulation of GRP75 in cancer cells has been associated with increased susceptibility to cell death (Wadhwa et al., 2006; Deocaris et al., 2007; Figure 1).
Factors that increase Ca2+ concentrations have been reported to upregulate VDAC expression and subsequently, the release of cytochrome C and Smac/Diablo, leading to apoptosis (Shoshan-Barmatz et al., 2017). Mcl-1, an anti-apoptotic member of the Bcl-2 family, binds with high affinity to VDAC1 and promotes lung cancer cell migration by a mechanism that involves Ca2+-dependent ROS production (Huang et al., 2014; Figure 1). Moreover, the BH4 domain of Bcl-XL, selectively targets VDAC1 and inhibits apoptosis by decreasing VDAC1-mediated Ca2+ influx into the mitochondria (Monaco et al., 2015; Figure 1). These reports underscore the relevance of highly regulated MAM proteins in ER-mitochondrial Ca2+ transfer, and implicate their dysregulation in tumorigenesis.
The activity of the sarco/ER Ca2+-ATPase (SERCA) pump is, among others, regulated by proteins encoded by oncogenes and tumor suppressor proteins in MAMs (Vandecaetsbeek et al., 2011). Particularly, the SERCA2b subtype is highly abundant in MAMs (Lynes et al., 2012; Chemaly et al., 2018) and the tumor suppressor p53 controls SERCA2b activation to promote Ca2+-dependent apoptosis (Mcdonnell et al., 2019). Thus, the ER releases increased amounts of Ca2+, which enters the mitochondria causing Ca2+ overload and apoptosis. However, in cancer cells, p53 at MAMs is mutated or inactivated, and thus, the ER cannot maintain elevated Ca2+ levels, which allows cancer cells to escape apoptosis contributing to tumor progression (Giorgi et al., 2015; Figure 1).
The complete inhibition of IP3R activity results in markedly compromised mitochondria bioenergetics and increased vulnerability to cell death and reduced melanoma tumor growth (Cardenas et al., 2016). Moreover, proteins encoded by oncogenes and tumor suppressors modulate ER IP3R activity in MAMs, thus altering Ca2+ signaling in cancer cells (Fan et al., 2017).
For instance, IP3R phosphorylation is markedly increased in cancer cells by hyperactive Akt (Szado et al., 2008), thereby reducing Ca2+ release from the ER to mitochondria, and promoting tumor survival (Szado et al., 2008). Other studies have shown that promyelocytic leukemia protein (PML), a tumor suppressor which localizes to MAMs (Giorgi et al., 2010), forms a complex with Akt, and decreases binding of protein phosphatase 2A (PP2A) to IP3Rs, suggesting that PP2A no longer dephosphorylates and inactivates Akt. This leads to phosphorylation of Akt and IP3Rs, decreasing Ca2+ release and further protecting the mitochondria from Ca2+-mediated apoptosis (Marchi et al., 2012). Moreover, PML physically interacts and synergizes with tumor suppressor protein p53 during apoptosis (Bernardi et al., 2008; Figure 1). Deletion of PML is associated with pleural mesothelioma and breast cancer, among others (Plevova et al., 2007a, b; Wang et al., 2018c). Additionally, Bcl-2 family members in the ER regulate IP3R activity (Lian et al., 2018) and, consequently apoptosis, by controlling the release of cytochrome C, the integrity of mitochondrial membranes and the activation of caspases (Youle and Strasser, 2008). Other evidence indicates that Bcl-2 binds to and inhibits IP3Rs, which reduces Ca2+ release, leading to decreased apoptosis (Rong et al., 2009). In summary, the dysregulation of proteins involved in Ca2+ transport at MAMs or in oncogenes and tumor suppressor proteins in cancer, alters Ca2+ transfer from the ER to the mitochondria, and, whether the outcome is the inhibition of Ca2+ transfer or Ca2+ overload, it will contribute directly to cancer disease progression by modulating cell death and/or survival.
ER stress is emerging as an important modulator of different pathologies and as a mechanism contributing to cancer cell death (Oakes and Papa, 2015). PERK (PRKR-like ER kinase), a key ER stress sensor of the unfolded protein response (UPR), is uniquely enriched at MAMs (Verfaillie et al., 2012). As a large number of molecular chaperones assist in the folding of unfolded proteins during the UPR, they consume large amounts of ATP. In order to increase ATP generation, cells usually increase the contact area between ER and mitochondria, which in turn increases Ca2+ release from the ER to the mitochondria (Bravo et al., 2012). However, if ER stress becomes chronic, ER-mitochondrial contacts and ER Ca2+ release increases, which, together with mitochondrial Ca2+ influx, leads to apoptosis. Moreover in cancer cells, the UPR is constitutively activated. It has been reported that PERK plays a critical role in tumor invasion and metastasis (Jamison et al., 2015; Pytel et al., 2016; Rozpedek et al., 2016; Feng et al., 2017). PERK signaling, which is activated downstream of the UPR and the integrated stress response (ISR), is triggered in response to a range of pathophysiological changes, and enables cancer cells to survive the adverse conditions typically observed in the tumor microenvironment. The ISR can be induced by both, intrinsic (i.e., ER stress) and extrinsic factors. The latter include hypoxia, amino acid and glucose deprivation, among others (Pakos-Zebrucka et al., 2016). Within the ISR, different protein kinases phosphorylate the α-subunit of eukaryotic initiation factor-2 (eIF2α), including heme-regulated eIF2α kinase (HRI), general control non-depressible protein 2 (GCN2), double stranded RNA dependent protein kinase (PKR) and PERK (Pakos-Zebrucka et al., 2016; Wang et al., 2018a). Phosphorylation of eIF2α inhibits global protein translation and synthesis, thereby reducing the number of proteins entering the ER (Baird and Wek, 2012), but increasing the cap-independent translation of other mRNAs, such as activating transcription factor 4 (ATF4) (Vattem and Wek, 2004). In addition to enabling cell survival, PERK-ATF4 signaling triggers multiple steps in the metastatic cascade, including angiogenesis, migration, survival, and colonization of secondary organ sites (Feng et al., 2017). PERK is also required for the metastatic dissemination of cancer cells that have undergone epithelial-to-mesenchymal transition (Feng et al., 2017; Figure 1). Thus, while PERK contributes to tighter MAMs during ER stress-induced apoptosis (Verfaillie et al., 2012), other roles of MAM resident-PERK and their contribution to cancer disease progression remain unclear.
Peroxisomes are metabolic organelles that carry out vital cell functions in lipid metabolism and synthesis, as well as maintenance of the redox balance (Islinger et al., 2018). Furthermore, non-metabolic roles of peroxisomes have been discovered, relating to cellular stress responses and the regulation of immune responses (Dahabieh et al., 2018). Disturbances between membrane contact sites (MCSs), areas of close proximity between the membranes of two organelles, can cause diseases (Scorrano et al., 2019). Most of the pathways that involve peroxisomes require communication with other organelles, such as the ER, mitochondria, lipid droplets and lysosomes, whereby close proximity represents a prerequisite for the efficient transport of metabolites (Sargsyan and Thoms, 2020). The main metabolic pathways shared between peroxisomes and the ER are: peroxisomal β-oxidation, implicated in the degradation of FA and biosynthesis of docosahexaenoic acid and the bile acids, ether lipid synthesis; and membrane lipid transfer (Ferdinandusse et al., 2018). Those shared between peroxisomes and mitochondria are: glyoxylate detoxification, redox exchange, ROS metabolism (Meo-Evoli et al., 2015) and α-oxidation (Michiels et al., 2016). Other MCSs exist that are implicated in the transfer of hydrophobic molecules, such as FA, between lipid droplets and peroxisomes (Valm et al., 2017), as well as cholesterol from or to lysosomes (Chu et al., 2015). The interaction of peroxisomes with the surrounding organelles begins with their biogenesis. Peroxisome formation involves generating a lipid membrane, followed by the acquisition of peroxisome membrane proteins, and subsequently organelle expansion (Farre et al., 2019). The peroxisomes originate from ER-derived vesicles containing the biogenesis factors (PEX) PEX3 and PEX16, or by fusion of PEX-bound vesicles from both, mitochondria (PEX3 and PEX14 factors), and the ER (Sugiura et al., 2017).
In recent years, considerable information has emerged suggesting that dysregulation of peroxisomes is implicated in tumorigenesis (Dahabieh et al., 2018). Enzymes implicated in peroxisomal metabolic processing are dysregulated in numerous neoplasms, including gastric adenocarcinoma (Jindal et al., 2016), cervical cancer (Wu et al., 2018), prostate cancer (Valenca et al., 2015), ovarian cancer (Sun et al., 2018), colorectal neoplasia (Shukla et al., 2017), liver cancer (Chen et al., 2018) and glioblastomas (Ruokun et al., 2016). A recent study reports that silencing PEX2, a peroxin involved in autophagosomal degradation of peroxisomes (pexophagy), reduced tumor growth in liver cancer (Cai et al., 2018). These results suggest that PEX2 and other peroxins, such as PEX5, 10 and 12, are essential for the viability of tumor cells and, thus, represent potential therapeutic targets for the treatment of cancer (Islinger et al., 2018; Figure 2). Another study revealed that PEX3, PEX16, and PEX19 protect lymphoma cells against histone deacetylase inhibitor induced-cell death, thereby promoting tumorigenesis (Dahabieh et al., 2017; Figure 2). These findings highlight a possible contribution of peroxisomes in chemotherapy resistance. Moreover, peroxisomes participate in the secretion of immune system modulators (e.g., interleukin-1, tumor necrosis factor) and lipids with pro- and anti-inflammatory characteristics, all of which are connected to tumor-promoting inflammation (Di Cara et al., 2019).
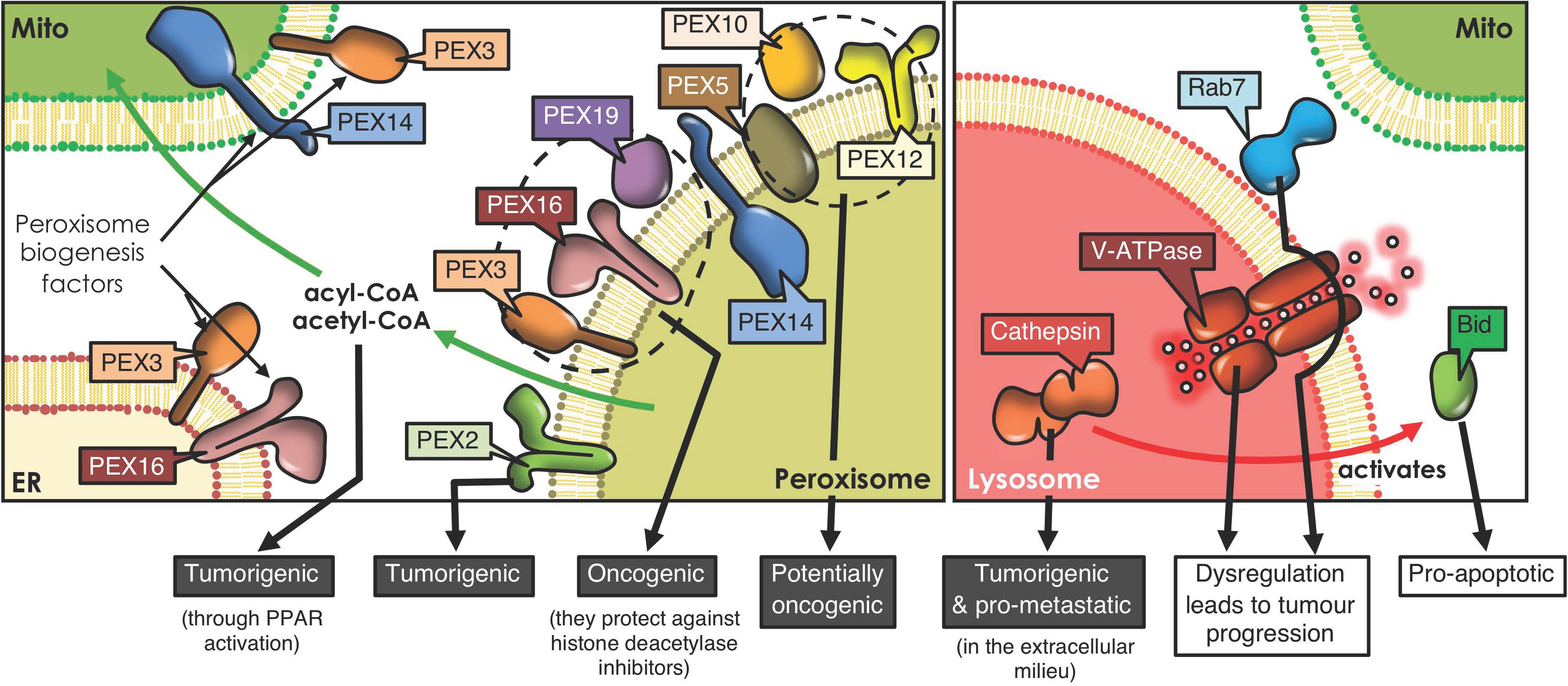
Figure 2. Peroxisome and lysosome interactions and cancer development. Peroxisomes derive from ER and mitochondrial membranes, and the proteins involved in peroxisome biogenesis and their regulation reportedly have tumorigenic potential. Moreover, peroxisome-mitochondria metabolic coupling favors cell proliferation and tumorigenesis through fatty acid transfer. Lysosomes form contacts with mitochondria, orchestrated by proteins, such as Rab7. Lysosomes play an ambiguous role in cancer. For instance, cathepsins, the proteolytic lysosomal enzymes, promote tumorigenesis when released to the extracellular milieu, but trigger Bid-mediated apoptosis upon accessing the cytoplasm. Also, dysregulation of either V-ATPase, the complex that acidifies lysosomes, or Rab7, the GTPase that orchestrates mitochondria-lysosome contact sites, both promote tumor progression (PPAR, Peroxisome proliferator-activated receptor).
Another relevant role of peroxisomes in malignancy is attributable to their crosstalk with mitochondria. Following peroxisomal β-oxidation, the final products, acyl-CoA and acetyl CoA, can enter the mitochondrial pathway and activate peroxisome proliferator-activated receptors, which are implicated in tumorigenesis (Dahabieh et al., 2018; Figure 2). In addition, dysfunction of peroxisomes leads to other metabolic alterations, through deficiency of peroxins (Faust and Kovacs, 2014). PEX2-depleted cells show elevated oxidative stress and elevated ROS causing inhibition of mechanistic target of rapamycin complex 1 (mTORC1) signaling, thereby promoting autophagy (Cai et al., 2018). This mechanism limits cancer initiation in some neoplasms; but in others, cancer cells use recycled metabolites to preserve organelle function and energy homeostasis to meet increased metabolic requirements. Thus, in these cells, autophagy is an essential survival mechanism to maintain cellular growth and proliferation (White et al., 2015).
Lysosomes, together with the ER and Golgi apparatus, are members of a network of intracellular membranous organelles whose functions are essential to maintain cell homeostasis (Buratta et al., 2020). Lysosomes degrade and recycle macromolecules via endocytosis, phagocytosis, and autophagy (Piao and Amaravadi, 2016; Lawrence and Zoncu, 2019). Moreover, they participate in extracellular events by secreting their contents through fusion with the plasma membrane (Davidson and Vander Heiden, 2017). Thus, lysosomes are extremely dynamic organelles, essential in a variety of cellular processes, such as cell signaling, death, immunity, and stress responses (Piao and Amaravadi, 2016). The dysregulation of these functions plays an important role in tumorigenesis, suggesting that lysosomal function is critical in this context. In fact, numerous hallmarks of cancer may be acquired as a result of lysosomal dysfunction (Hanahan and Weinberg, 2011). Specifically, the release of lysosomal proteases may activate caspases and lead to cell death; however, inhibition of lysosomal function, which hinders clearance of dead cells, contributes to inflammation and promotes tumorigenesis (Davidson and Vander Heiden, 2017). In addition, changes in the interactions between lysosomes and other organelles can limit organellophagy, thereby affecting the recycling of macromolecules and consequently, altering cellular energetics (Boroughs and Deberardinis, 2015). These changes include, the dysregulation of specific receptors, such as members of the FAM134 reticulon protein family in the ER (Hanahan and Weinberg, 2011; Khaminets et al., 2015). Overall, numerous hallmarks of cancer may be acquired as a result of lysosomal dysfunction and changes in the communication between lysosomes and other organelles (Hanahan and Weinberg, 2011).
Lysosomal and mitochondrial functions are connected (Plotegher and Duchen, 2017), since damaged mitochondria are engulfed by autophagosomes (Lazarou et al., 2015), which then fuse with the lysosome/late endosome to produce autolysosomes where the mitochondria are degraded (mitophagy) (Wong et al., 2019). In addition, mitochondria-derived vesicles may fuse with lysosomes to eliminate their contents (Plotegher and Duchen, 2017). In doing so, toxic accumulation of damaged proteins, especially in the mitochondria, which augment oxidative stress and activate oncogenic signaling, can be avoided (White, 2015). On the other hand, lysosome-mitochondria interactions may also contribute to cancer initiation (Ferguson, 2015). Cell transformation results in an elevated demand for nutrients, associated with the increased production of cell mass, and lysosomes may provide the required molecules together with autophagy, to preserve mitochondrial functions and energy homeostasis (White et al., 2015). Additionally, lysosomes and mitochondria are connected via non-degradative processes involving the formation of dynamic interorganelle MCSs (Wong et al., 2019). These processes are regulated by Rab7, which controls this organelle network, as well as processes including mitochondrial fission (Yamano et al., 2018; Figure 2). Changes in interorganelle transfer of metabolites may alter cell homeostasis and, consequently, favor the development of diseases linked to dysfunction of both organelles, such as cancer (Wong et al., 2019). For instance, it has been reported that cytosolic cathepsins (lysosomal hydrolases) may repress tumor growth by activation of an intrinsic apoptotic pathway. Contrarily, extracellular cathepsins (e.g., B, S, and E) may stimulate tumor growth, progression and metastasis in different neoplasms by aiding in permeating the basement membrane and activating pro-tumorigenic proteins (Piao and Amaravadi, 2016; Figure 2). Additionally, the V-ATPase (lysosomal membrane protein), a proton pump that modulates intravesicular acidification in lysosomes, is an important regulator of endocytic trafficking (Plotegher and Duchen, 2017). A recent study reveals that V-ATPase is a master effector of transcription factor E2F1-mediated lysosomal trafficking, essential in mTORC1 activation and suppression of autophagy, which promotes tumor progression (Meo-Evoli et al., 2015; Figure 2).
Furthermore, lysosomal membrane permeabilization (LMP; loss of membrane integrity), causes the release of lysosomal enzymes into the cytosol, triggering apoptosis, autophagy and necroptosis (Piao and Amaravadi, 2016). LMP can accelerate apoptosis through cathepsin-mediated cleavage of Bid, to promote the release of cytochrome C via Bax (intrinsic pathway activation) (Wang, 2015; Piao and Amaravadi, 2016; Figure 2).
A number of cellular processes in which lysosomes participate have been identified as potential therapeutic targets in cancer. These include drugs that permeabilize lysosomal membranes, cathepsin inhibitors, and molecules that become protonated in order to increase the concentration of specific drugs inside the organelle. However, until now, related clinical data is scarce (Davidson and Vander Heiden, 2017). Thus, future studies on lysosomes and their interaction with other organelles will be critical to understanding the role that lysosome-communication plays in cancer genesis and progression.
Organelles are the major players in intracellular communication networks. Multiple diseases have been studied by analyzing organelles individually, as is the case for mitochondria or the ER. Advances in microscopy have provided further insights to the nature of organelle communication; revealing both, physical and functional interactions at MCSs (Csordas et al., 2010; Valm et al., 2017; Shai et al., 2018).
Currently there is a consensus that organelles contact one another and communicate in a dynamic fashion. Over the last decade, evidence highlighting the importance of organelle communication in disease has become available (Bravo-Sagua et al., 2014; Lopez-Crisosto et al., 2015, 2017; Plotegher and Duchen, 2017; Torres et al., 2017). Moreover, organelle communication impacts on various cancer hallmarks and therefore plays a critical role in oncogenesis. This is the case of CAV1, a protein located at MAMs that acts both as a tumor suppressor and a promoter of metastasis (Campos et al., 2019; Simon et al., 2020). On the other hand, the ER stress sensor PERK, also present in MAMs, has been associated with apoptotic cell death and the promotion of more aggressive/migratory phenotypes. Additionally, the main peroxisome-organelle interactions so far studied involve peroxins, which also behave as tumorigenic factors. Lastly, cathepsins have been described in lysosomes as proteins that contribute to tumor progression and cell death. Overall, the details of how such communication between organelles contributes to cell homeostasis often still remain poorly understood.
The development of molecules that target organelle interactions at a sub-cellular level, represents a promising field in cancer treatment (Piao and Amaravadi, 2016; Kerkhofs et al., 2018). However, to exploit this potential a better understanding of the differences between interorganelle contacts in healthy and tumor cells is required.
PD, AS-B, and RB-S drafted the manuscript with support from AFGQ and SL. RB-S drafted the figures. PD, AS-B, RB-S, AFGQ, and SL critically revised the article. All authors read and approved the final article.
This work was supported by the FONDECYT 1200490 to SL, FONDECYT 1170925 to AFGQ, FONDECYT 3180783 to AS-B, and FONDAP 15130011 to AFGQ, RB-S, and SL from Agencia Nacional de Investigación y Desarrollo (ANID), Chile.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Baird, T. D., and Wek, R. C. (2012). Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 3, 307–321. doi: 10.3945/an.112.002113
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Bernardi, R., Papa, A., and Pandolfi, P. P. (2008). Regulation of apoptosis by PML and the PML-NBs. Oncogene 27, 6299–6312. doi: 10.1038/onc.2008.305
Bernhard, W., and Rouiller, C. (1956). Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Biophys. Biochem. Cytol. 2, 73–78. doi: 10.1083/jcb.2.4.73
Boroughs, L. K., and Deberardinis, R. J. (2015). Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 17, 351–359. doi: 10.1038/ncb3124
Bravo-Sagua, R., Parra, V., Lopez-Crisosto, C., Diaz, P., Quest, A. F., and Lavandero, S. (2017). Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 7, 623–634. doi: 10.1002/cphy.c160013
Bravo-Sagua, R., Parra, V., Ortiz-Sandoval, C., Navarro-Marquez, M., Rodriguez, A. E., Diaz-Valdivia, N., et al. (2019). Caveolin-1 impairs PKA-DRP1-mediated remodelling of ER-mitochondria communication during the early phase of ER stress. Cell Death Differ. 26, 1195–1212. doi: 10.1038/s41418-018-0197-1
Bravo-Sagua, R., Torrealba, N., Paredes, F., Morales, P. E., Pennanen, C., Lopez-Crisosto, C., et al. (2014). Organelle communication: signaling crossroads between homeostasis and disease. Int. J. Biochem. Cell Biol. 50, 55–59. doi: 10.1016/j.biocel.2014.01.019
Bravo, R., Gutierrez, T., Paredes, F., Gatica, D., Rodriguez, A. E., Pedrozo, Z., et al. (2012). Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int. J. Biochem. Cell Biol. 44, 16–20. doi: 10.1016/j.biocel.2011.10.012
Bultynck, G., and Campanella, M. (2017). Tumor suppressive Ca(2+) signaling is driven by IP3 receptor fitness. Cell Stress 1, 73–78.
Buratta, S., Tancini, B., Sagini, K., Delo, F., Chiaradia, E., Urbanelli, L., et al. (2020). Lysosomal Exocytosis, Exosome Release and Secretory Autophagy: The Autophagic- and Endo-Lysosomal Systems Go Extracellular. Int. J. Mol. Sci. 21:2576. doi: 10.3390/ijms21072576
Cai, M., Sun, X., Wang, W., Lian, Z., Wu, P., Han, S., et al. (2018). Disruption of peroxisome function leads to metabolic stress, mTOR inhibition, and lethality in liver cancer cells. Cancer Lett. 421, 82–93. doi: 10.1016/j.canlet.2018.02.021
Campos, A., Burgos-Ravanal, R., Gonzalez, M. F., Huilcaman, R., Lobos Gonzalez, L., and Quest, A. F. G. (2019). Cell Intrinsic and Extrinsic Mechanisms of Caveolin-1-Enhanced Metastasis. Biomolecules 9:314. doi: 10.3390/biom9080314
Cardenas, C., Muller, M., Mcneal, A., Lovy, A., Jana, F., Bustos, G., et al. (2016). Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 14, 2313–2324. doi: 10.1016/j.celrep.2016.02.030
Chaudhuri, D., Sancak, Y., Mootha, V. K., and Clapham, D. E. (2013). MCU encodes the pore conducting mitochondrial calcium currents. Elife 2:e00704.
Chemaly, E. R., Troncone, L., and Lebeche, D. (2018). SERCA control of cell death and survival. Cell Calcium. 69, 46–61. doi: 10.1016/j.ceca.2017.07.001
Chen, X. F., Tian, M. X., Sun, R. Q., Zhang, M. L., Zhou, L. S., Jin, L., et al. (2018). SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 19, e45124.
Chen, Y. F., Chen, Y. T., Chiu, W. T., and Shen, M. R. (2013). Remodeling of calcium signaling in tumor progression. J. Biomed. Sci. 20:23. doi: 10.1186/1423-0127-20-23
Chu, B. B., Liao, Y. C., Qi, W., Xie, C., Du, X., Wang, J., et al. (2015). Cholesterol transport through lysosome-peroxisome membrane contacts. Cell 161, 291–306. doi: 10.1016/j.cell.2015.02.019
Csordas, G., Renken, C., Varnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Csordas, G., Varnai, P., Golenar, T., Roy, S., Purkins, G., Schneider, T. G., et al. (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39, 121–132. doi: 10.1016/j.molcel.2010.06.029
Dahabieh, M. S., Di Pietro, E., Jangal, M., Goncalves, C., Witcher, M., Braverman, N. E., et al. (2018). Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. Biochim. Biophys. Acta Rev. Cancer 1870, 103–121. doi: 10.1016/j.bbcan.2018.07.004
Dahabieh, M. S., Ha, Z., Di Pietro, E., Nichol, J. N., Bolt, A. M., Goncalves, C., et al. (2017). Peroxisomes protect lymphoma cells from HDAC inhibitor-mediated apoptosis. Cell Death Differ. 24, 1912–1924. doi: 10.1038/cdd.2017.115
Davidson, S. M., and Vander Heiden, M. G. (2017). Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 57, 481–507. doi: 10.1146/annurev-pharmtox-010715-103101
De Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
De Brito, O. M., and Scorrano, L. (2009). Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9, 222–226. doi: 10.1016/j.mito.2009.02.005
Deocaris, C. C., Widodo, N., Shrestha, B. G., Kaur, K., Ohtaka, M., Yamasaki, K., et al. (2007). Mortalin sensitizes human cancer cells to MKT-077-induced senescence. Cancer Lett. 252, 259–269. doi: 10.1016/j.canlet.2006.12.038
Di Cara, F., Andreoletti, P., Trompier, D., Vejux, A., Bulow, M. H., Sellin, J., et al. (2019). Peroxisomes in Immune Response and Inflammation. Int. J. Mol. Sci. 20, 3877. doi: 10.3390/ijms20163877
Doghman-Bouguerra, M., and Lalli, E. (2019). ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 1866, 650–662. doi: 10.1016/j.bbamcr.2019.01.009
Fan, C., Tang, Y., Wang, J., Xiong, F., Guo, C., Wang, Y., et al. (2017). Role of long non-coding RNAs in glucose metabolism in cancer. Mol. Cancer 16:130.
Farre, J. C., Mahalingam, S. S., Proietto, M., and Subramani, S. (2019). Peroxisome biogenesis, membrane contact sites, and quality control. EMBO Rep. 20:e46864.
Faust, P. L., and Kovacs, W. J. (2014). Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 98, 75–85. doi: 10.1016/j.biochi.2013.10.019
Feng, Y. X., Jin, D. X., Sokol, E. S., Reinhardt, F., Miller, D. H., and Gupta, P. B. (2017). Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 8:1079.
Ferdinandusse, S., Denis, S., Van Roermund, C. W. T., Preece, M. A., Koster, J., Ebberink, M. S., et al. (2018). A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl-CoA oxidase. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 952–958. doi: 10.1016/j.bbadis.2017.12.032
Ferguson, S. M. (2015). Beyond indigestion: emerging roles for lysosome-based signaling in human disease. Curr. Opin. Cell Biol. 35, 59–68. doi: 10.1016/j.ceb.2015.04.014
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U S A 112, E2174–E2181.
Fouad, Y. A., and Aanei, C. (2017). Revisiting the hallmarks of cancer. Am. J. Cancer Res. 7, 1016–1036.
Giorgi, C., Bonora, M., Sorrentino, G., Missiroli, S., Poletti, F., Suski, J. M., et al. (2015). p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. U S A 112, 1779–1784. doi: 10.1073/pnas.1410723112
Giorgi, C., Ito, K., Lin, H. K., Santangelo, C., Wieckowski, M. R., Lebiedzinska, M., et al. (2010). PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 330, 1247–1251. doi: 10.1126/science.1189157
Glancy, B., and Balaban, R. S. (2012). Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. doi: 10.1021/bi2018909
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Huang, H., Shah, K., Bradbury, N. A., Li, C., and White, C. (2014). Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 5:e1482. doi: 10.1038/cddis.2014.419
Islinger, M., Voelkl, A., Fahimi, H. D., and Schrader, M. (2018). The peroxisome: an update on mysteries 2.0. Histochem. Cell Biol 150, 443–471. doi: 10.1007/s00418-018-1722-5
Jamison, S., Lin, Y., and Lin, W. (2015). Pancreatic endoplasmic reticulum kinase activation promotes medulloblastoma cell migration and invasion through induction of vascular endothelial growth factor A. PLoS One 10:e0120252. doi: 10.1371/journal.pone.0120252
Jindal, Y., Singh, A., Kumar, R., Varma, K., Misra, V., Misra, S. P., et al. (2016). Expression of Alpha Methylacyl CoA Racemase (AMACR) in Gastric Adenocarcinoma and Its Correlation with Helicobacter pylori Infection. J. Clin. Diagn Res. 10, EC10–EC12.
Kerkhofs, M., Bittremieux, M., Morciano, G., Giorgi, C., Pinton, P., Parys, J. B., et al. (2018). Emerging molecular mechanisms in chemotherapy: Ca(2+) signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis. 9:334.
Khaminets, A., Heinrich, T., Mari, M., Grumati, P., Huebner, A. K., Akutsu, M., et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358.
Lawrence, R. E., and Zoncu, R. (2019). The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142. doi: 10.1038/s41556-018-0244-7
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, S., and Min, K. T. (2018). The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 41, 1000–1007.
Li, Y., Dong, W., Shan, X., Hong, H., Liu, Y., Liu, X., et al. (2018). The anti-tumor effects of Mfn2 in breast cancer are dependent on promoter DNA methylation, the P21(Ras) motif and PKA phosphorylation site. Oncol Lett. 15, 8011–8018.
Lian, Y., Xiong, F., Yang, L., Bo, H., Gong, Z., Wang, Y., et al. (2018). Long noncoding RNA AFAP1-AS1 acts as a competing endogenous RNA of miR-423-5p to facilitate nasopharyngeal carcinoma metastasis through regulating the Rho/Rac pathway. J. Exp. Clin. Cancer Res. 37:253.
Liu, X., Sun, J., Yuan, P., Shou, K., Zhou, Y., Gao, W., et al. (2019). Mfn2 inhibits proliferation and cell-cycle in Hela cells via Ras-NF-kappaB signal pathway. Cancer Cell Int. 19:197.
Lopez-Crisosto, C., Bravo-Sagua, R., Rodriguez-Pena, M., Mera, C., Castro, P. F., Quest, A. F., et al. (2015). ER-to-mitochondria miscommunication and metabolic diseases. Biochim. Biophys. Acta 1852, 2096–2105. doi: 10.1016/j.bbadis.2015.07.011
Lopez-Crisosto, C., Pennanen, C., Vasquez-Trincado, C., Morales, P. E., Bravo-Sagua, R., Quest, A. F. G., et al. (2017). Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14, 342–360. doi: 10.1038/nrcardio.2017.23
Lynes, E. M., Bui, M., Yap, M. C., Benson, M. D., Schneider, B., Ellgaard, L., et al. (2012). Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 31, 457–470. doi: 10.1038/emboj.2011.384
Ma, L. I., Chang, Y., Yu, L., He, W., and Liu, Y. (2015). Pro-apoptotic and anti-proliferative effects of mitofusin-2 via PI3K/Akt signaling in breast cancer cells. Oncol Lett. 10, 3816–3822. doi: 10.3892/ol.2015.3748
Marchi, S., Marinello, M., Bononi, A., Bonora, M., Giorgi, C., Rimessi, A., et al. (2012). Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis. 3, e304. doi: 10.1038/cddis.2012.45
Marchi, S., and Pinton, P. (2016). Alterations of calcium homeostasis in cancer cells. Curr. Opin. Pharmacol. 29, 1–6. doi: 10.1016/j.coph.2016.03.002
Mcdonnell, S. J., Spiller, D. G., White, M. R. H., Prior, I. A., and Paraoan, L. (2019). ER stress-linked autophagy stabilizes apoptosis effector PERP and triggers its co-localization with SERCA2b at ER-plasma membrane junctions. Cell Death Discov. 5:132.
Meo-Evoli, N., Almacellas, E., Massucci, F. A., Gentilella, A., Ambrosio, S., Kozma, S. C., et al. (2015). V-ATPase: a master effector of E2F1-mediated lysosomal trafficking, mTORC1 activation and autophagy. Oncotarget 6, 28057–28070. doi: 10.18632/oncotarget.4812
Michiels, C., Tellier, C., and Feron, O. (2016). Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta 1866, 76–86. doi: 10.1016/j.bbcan.2016.06.004
Monaco, G., Decrock, E., Arbel, N., Van Vliet, A. R., La Rovere, R. M., De Smedt, H., et al. (2015). The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 290, 9150–9161. doi: 10.1074/jbc.m114.622514
Monteith, G. R., Prevarskaya, N., and Roberts-Thomson, S. J. (2017). The calcium-cancer signalling nexus. Nat. Rev. Cancer 17, 367–380.
Morciano, G., Marchi, S., Morganti, C., Sbano, L., Bittremieux, M., Kerkhofs, M., et al. (2018). Role of Mitochondria-Associated ER Membranes in Calcium Regulation in Cancer-Specific Settings. Neoplasia 20, 510–523. doi: 10.1016/j.neo.2018.03.005
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U S A 113, 11249–11254. doi: 10.1073/pnas.1606786113
Nunnari, J., and Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Oakes, S. A., and Papa, F. R. (2015). The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 10, 173–194. doi: 10.1146/annurev-pathol-012513-104649
Pakos-Zebrucka, K., Koryga, I., Mnich, K., Ljujic, M., Samali, A., and Gorman, A. M. (2016). The integrated stress response. EMBO Rep. 17, 1374–1395.
Piao, S., and Amaravadi, R. K. (2016). Targeting the lysosome in cancer. Ann. N Y Acad. Sci. 1371, 45–54. doi: 10.1111/nyas.12953
Plevova, P., Bouchal, J., Fiuraskova, M., Foretova, L., Navratilova, M., Zapletalova, J., et al. (2007a). PML protein expression in hereditary and sporadic breast cancer. Neoplasma 54, 263–268.
Plevova, P., Bouchal, J., Fiuraskova, M., Papezova, M., Krepelova, A., Curik, R., et al. (2007b). PML and TRF2 protein expression in hereditary and sporadic colon cancer. Neoplasma 54, 269–277.
Plotegher, N., and Duchen, M. R. (2017). Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol. Med. 23, 116–134. doi: 10.1016/j.molmed.2016.12.003
Pytel, D., Majsterek, I., and Diehl, J. A. (2016). Tumor progression and the different faces of the PERK kinase. Oncogene 35, 1207–1215. doi: 10.1038/onc.2015.178
Rong, Y. P., Bultynck, G., Aromolaran, A. S., Zhong, F., Parys, J. B., De Smedt, H., et al. (2009). The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. U S A 106, 14397–14402. doi: 10.1073/pnas.0907555106
Rozpedek, W., Pytel, D., Mucha, B., Leszczynska, H., Diehl, J. A., and Majsterek, I. (2016). The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during Endoplasmic Reticulum stress. Curr. Mol. Med. 16, 533–544. doi: 10.2174/1566524016666160523143937
Ruokun, C., Yake, X., Fengdong, Y., Xinting, W., Laijun, S., and Xianzhi, L. (2016). Lentivirus-mediated silencing of HSDL2 suppresses cell proliferation in human gliomas. Tumour Biol. 37, 15065–15077. doi: 10.1007/s13277-016-5402-6
Sala-Vila, A., Navarro-Lerida, I., Sanchez-Alvarez, M., Bosch, M., Calvo, C., Lopez, J. A., et al. (2016). Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 6:27351.
Sargsyan, Y., and Thoms, S. (2020). Staying in Healthy Contact: How Peroxisomes Interact with Other Cell Organelles. Trends Mol. Med. 26, 201–214. doi: 10.1016/j.molmed.2019.09.012
Scorrano, L., De Matteis, M. A., Emr, S., Giordano, F., Hajnoczky, G., Kornmann, B., et al. (2019). Coming together to define membrane contact sites. Nat. Commun. 10:1287.
Shai, N., Yifrach, E., Van Roermund, C. W. T., Cohen, N., Bibi, C., Cavellini, L., et al. (2018). Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 9:1761.
Shoshan-Barmatz, V., De, S., and Meir, A. (2017). The Mitochondrial Voltage-Dependent Anion Channel 1, Ca(2+) Transport, Apoptosis, and Their Regulation. Front. Oncol. 7:60. doi: 10.3389/fonc.2017.00060
Shukla, N., Adhya, A. K., and Rath, J. (2017). Expression of Alpha - Methylacyl - Coenzyme A Racemase (AMACR) in Colorectal Neoplasia. J. Clin. Diagn Res. 11, EC35–EC38.
Simon, L., Campos, A., Leyton, L., and Quest, A. F. G. (2020). Caveolin-1 function at the plasma membrane and in intracellular compartments in cancer. Cancer Metastasis Rev. 39, 435–453. doi: 10.1007/s10555-020-09890-x
Sugiura, A., Mattie, S., Prudent, J., and Mcbride, H. M. (2017). Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 542, 251–254. doi: 10.1038/nature21375
Sun, Q., Zhang, Y., Su, J., Li, T., and Jiang, Y. (2018). Role of Hydroxysteroid Dehydrogenase-Like 2 (HSDL2) in Human Ovarian Cancer. Med. Sci. Monit. 24, 3997–4008. doi: 10.12659/msm.909418
Szado, T., Vanderheyden, V., Parys, J. B., De Smedt, H., Rietdorf, K., Kotelevets, L., et al. (2008). Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. U S A 105, 2427–2432. doi: 10.1073/pnas.0711324105
Torres, S., Balboa, E., Zanlungo, S., Enrich, C., Garcia-Ruiz, C., and Fernandez-Checa, J. C. (2017). Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 8:982. doi: 10.3389/fphys.2017.00982
Trigos, A. S., Pearson, R. B., Papenfuss, A. T., and Goode, D. L. (2018). How the evolution of multicellularity set the stage for cancer. Br. J. Cancer 118, 145–152. doi: 10.1038/bjc.2017.398
Valenca, I., Pertega-Gomes, N., Vizcaino, J. R., Henrique, R. M., Lopes, C., Baltazar, F., et al. (2015). Localization of MCT2 at peroxisomes is associated with malignant transformation in prostate cancer. J. Cell Mol. Med. 19, 723–733. doi: 10.1111/jcmm.12481
Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., et al. (2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167. doi: 10.1038/nature22369
Vandecaetsbeek, I., Vangheluwe, P., Raeymaekers, L., Wuytack, F., and Vanoevelen, J. (2011). The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb. Perspect. Biol. 3:a004184. doi: 10.1101/cshperspect.a004184
Vattem, K. M., and Wek, R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U S A 101, 11269–11274. doi: 10.1073/pnas.0400541101
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891. doi: 10.1038/cdd.2012.74
Wadhwa, R., Takano, S., Kaur, K., Deocaris, C. C., Pereira-Smith, O. M., Reddel, R. R., et al. (2006). Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 118, 2973–2980. doi: 10.1002/ijc.21773
Wang, C., Tan, Z., Niu, B., Tsang, K. Y., Tai, A., Chan, W. C. W., et al. (2018a). Inhibiting the integrated stress response pathway prevents aberrant chondrocyte differentiation thereby alleviating chondrodysplasia. Elife 7:e37673.
Wang, K. (2015). Autophagy and apoptosis in liver injury. Cell Cycle 14, 1631–1642. doi: 10.1080/15384101.2015.1038685
Wang, W., Liu, X., Guo, X., and Quan, H. (2018b). Mitofusin-2 Triggers Cervical Carcinoma Cell Hela Apoptosis via Mitochondrial Pathway in Mouse Model. Cell Physiol. Biochem. 46, 69–81. doi: 10.1159/000488410
Wang, Y. A., Li, X. L., Mo, Y. Z., Fan, C. M., Tang, L., Xiong, F., et al. (2018c). Effects of tumor metabolic microenvironment on regulatory T cells. Mol. Cancer 17, 168.
White, E., Mehnert, J. M., and Chan, C. S. (2015). Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 21, 5037–5046. doi: 10.1158/1078-0432.ccr-15-0490
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J., and Pinton, P. (2009). Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590. doi: 10.1038/nprot.2009.151
Wong, Y. C., Kim, S., Peng, W., and Krainc, D. (2019). Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 29, 500–513. doi: 10.1016/j.tcb.2019.02.004
Wu, W., Liu, F., Wu, K., Chen, Y., Wu, H., Dai, G., et al. (2018). Lon Peptidase 2, Peroxisomal (LONP2) Contributes to Cervical Carcinogenesis via Oxidative Stress. Med. Sci. Monit. 24, 1310–1320. doi: 10.12659/msm.908966
Xue, R., Meng, Q., Lu, D., Liu, X., Wang, Y., and Hao, J. (2018). Mitofusin2 Induces Cell Autophagy of Pancreatic Cancer through Inhibiting the PI3K/Akt/mTOR Signaling Pathway. Oxid Med. Cell Longev. 2018:2798070.
Yamano, K., Wang, C., Sarraf, S. A., Munch, C., Kikuchi, R., Noda, N. N., et al. (2018). Endosomal Rab cycles regulate Parkin-mediated mitophagy. Elife 7:e31326.
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. doi: 10.1038/nrm2308
Keywords: interorganelle communication, cancer, mitochondria, endoplasmic reticulum, lysosome, peroxisome
Citation: Díaz P, Sandoval-Bórquez A, Bravo-Sagua R, Quest AFG and Lavandero S (2021) Perspectives on Organelle Interaction, Protein Dysregulation, and Cancer Disease. Front. Cell Dev. Biol. 9:613336. doi: 10.3389/fcell.2021.613336
Received: 02 October 2020; Accepted: 01 February 2021;
Published: 25 February 2021.
Edited by:
Simone Patergnani, University of Ferrara, ItalyReviewed by:
Gerardo Z. Lederkremer, Tel Aviv University, IsraelCopyright © 2021 Díaz, Sandoval-Bórquez, Bravo-Sagua, Quest and Lavandero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew F. G. Quest, YXF1ZXN0QG1lZC51Y2hpbGUuY2w=; Sergio Lavandero, c2xhdmFuZGVyQHVjaGlsZS5jbA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.