 Keke Wang1
Keke Wang1 Zhikun Guo
Zhikun Guo
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 30 November 2021
Sec. Cell Adhesion and Migration
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.596702
As the primary component of elastic fibers, elastin plays an important role in maintaining the elasticity and tensile ability of cardiovascular, pulmonary and many other tissues and organs. Studies have shown that elastin expression is regulated by a variety of molecules that have positive and negative regulatory effects. However, the specific mechanism is unclear. Moreover, elastin is reportedly involved in the development and progression of many cardiovascular diseases through changes in its expression and structural modifications once deposited in the extracellular matrix. This review article summarizes the role of elastin in myocardial ischemia-reperfusion, atherosclerosis, and atrial fibrillation, with emphasis on the potential molecular regulatory mechanisms.
Elastin is an extracellular matrix (ECM) protein responsible for the extensibility and elastic recoil of many vertebrate tissues, such as large arteries, heart valves, pulmonary tissues, skin, and certain ligaments and cartilages (Reichheld et al., 2019). Elastic fibers, which are the products of enzymatically cross-linked tropoelastin monomers and microfibrillar proteins, are composed of an insoluble polymerized elastin (ELN) core and microfibers located at the periphery (Yeo et al., 2011; Shin and Yanagisawa, 2019). Elastic fiber networks play an essential role in maintaining normal physiological functions, conferring elasticity, and recoiling to tissues and organs. In fact, both genetic and acquired cardiovascular diseases are associated with the insufficiency or disorganization of elastin and the breakage of elastic fibers (Cocciolone et al., 2018).
To better understand the relationship between elastin and the pathogenesis of cardiovascular diseases, we explored the structure, biosynthesis, and molecular regulation mechanism of elastin. Furthermore, we aim to provide new ideas for the treatment of these diseases.
Elastin is a resilient connective tissue protein found in the ECM of most vertebrate tissues, and it is an important part in the interstitium of tissues that undergo repeated physical deformations in the human body (Swee et al., 1995). Owing to its extensive crosslinked structures, elastin is a long-lived protein that degrades slowly in healthy tissues, with a half-life of about 70 years (Shapiro et al., 1991). As a polymeric molecule, elastin has hydrophobic and insoluble properties and can resist acid and alkali. In the presence of water, elastin exists in the form of a rubbery extension with a low elastic modulus (Partridge, 1969). In addition, over 75% of the sequence of elastin consists of just four non-polar amino acids, namely, glycine, valine, alanine, and proline (Vrhovski and Weiss, 1998). Like hydroxyproline-rich collagen, elastin contains about one-third glycine and approximately one-ninth proline. Tropoelastin, the soluble precursor of elastin with a molecular weight of about 60 kDa, is only encoded by a single gene, ELN, which is located on chromosome 7q11.1-21.1 with a size of 45 kb (Partridge, 1969; Swee et al., 1995; Tarakanova et al., 2018). The human ELN gene has 34 exons while the bovine gene contains 36 exons. Among them, exon 36 is highly conserved and codes for a hydrophilic C-terminus as well as a large 3′-untranslated region, in addition, the human gene does not have bovine exons 34 and 35 (Figure 1; Vrhovski and Weiss, 1998). Recently, alternate splicing variants of human tropoelastin mRNA transcripts have been reported. However, the specific generation mechanism of tropoelastin mRNA isoforms is still unclear (Reichheld et al., 2019).
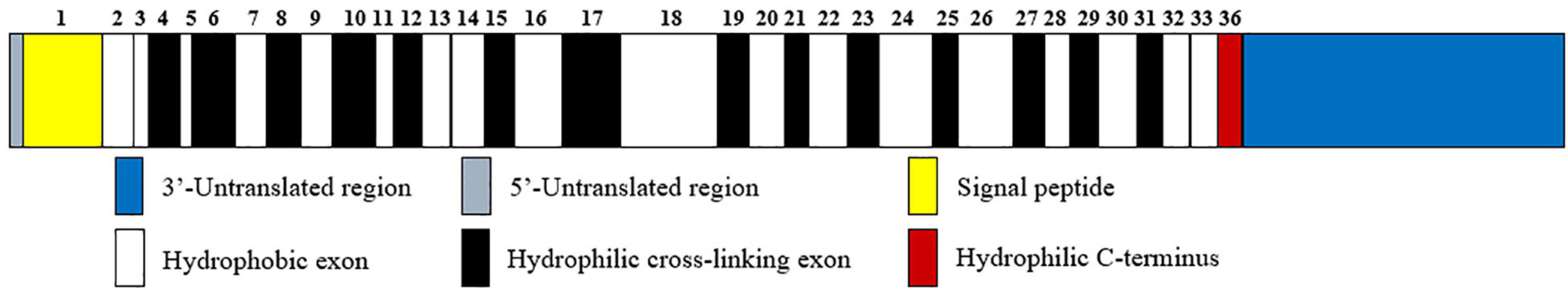
Figure 1. Human tropoelastin domain structure. Tropoelastin contains alternating hydrophobic (white rectangle) and hydrophilic cross-linking domains (black rectangle), and the width of the rectangles corresponds to the length of the domains. The number above the domains represents the sequence of exons that encode them. Among exons, exon 36 encodes a hydrophilic C-terminus (red rectangle) in addition to a 3′-untranslated region (blue rectangle).
Two major domains are found in tropoelastin: alternating hydrophobic regions and hydrophilic cross-link domains (Figure 1). The hydrophobic domains of tropoelastin are rich in non-polar amino acids, particularly glycine, valine, proline, and alanine, which often appear in repeats of 3–6 peptides, such as GVGVP, GGVP, and GVAP. The composition of this amino acid forms a hydrophobic interaction, which is required for the elastic property of the fiber. The hydrophilic cross-link domains are rich in alanine and lysine and are required for the lysyl oxidase-mediated formation of desmosine cross-links and are necessary to form insoluble elastin (Boyd et al., 1991; Vrhovski and Weiss, 1998).
Tropoelastin is known to be the most elastic and expansive monomer protein that can extend to eight times its length. Furthermore, the tropoelastin molecular shape is asymmetric. It extends into a coil-like structure in the N-terminal region of the molecule. Between molecules are supported by hinges, which act as a bridge to connect the N-terminal region to the C-terminal region of intercellular interaction, and then cross-linked by lysine oxidase (LOX) to form mature fibers (Baldock et al., 2011; Tarakanova et al., 2018).
Elastin is abundant in elastic tissues, and tissues rich in elastin include the aorta and major blood vessels (28–32% dry mass), the lungs (3–7%), elastic ligaments (50%), tendons (4%), and the skin (2–3%) (Uitto, 1979). Elastin and elastic fibers are also found within liver and myocardial tissues. Structurally, elastic fibers are primarily composed of extensively crosslinked elastin (>90%) and microfibrils rich in acidic glycoproteins and are organized into 8–16 nm bead-like fibrils of beaded appearance (Vrhovski and Weiss, 1998).
The elastic fibers in a healthy heart are mainly distributed in endocardium, visceral pericardium (epicardium), arterial wall, and cardiomyocytes and interweave with collagen fibers supporting elastic properties and strength. However, elastic fibers seldom form large fiber bundles like collagen fibers (Cunningham et al., 2006; Tsamis et al., 2013; Rodriguez and Tan, 2017). Studies showed that the length of mature elastic fibers was present at birth and maintain until adulthood in human heart parenchyma. The elastic fibers formed an irregular network in the myocardial interstitium in adults and a similar phenomenon was also observed in infants and children with thinner elastic fibers, however, in the elderly hearts the elastic fibers were thicker. What’s more, they also found the average length of elastic fiber was increased beyond the third decade of life (de Carvalho Filho et al., 1996).
Elastin gene expression and protein synthesis occur within a narrow time frame from late embryonic development to the end of adolescence, producing no de novo elastin throughout the adult life of animals. Considering the elasticity and the approximately 70-year half-life of elastin, the limited time of expression and synthesis is enough for the protein to last a lifetime in most species (Cocciolone et al., 2018). In a stable state, the basal levels of elastin expression and breakdown remain low in adult tissues and cells isolated from adult tissues. In vivo, elastogenic cells including smooth muscle cells, fibroblasts, and endothelial cells can produce and secrete tropoelastin (Wise et al., 2009), moreover, we found that the co-expression of cardiac troponin T and elastin were detected in rat cardiomyocytes and rat myocardial tissues. Thus we speculate that the rat cardiomyocytes can secrete elastin and it may be involved in the potential energy of myocardial cells and the construction of intercellular elastic fibers (Han et al., 2019). In addition, elastogenesis begins with the transcription of ELN; subsequently, the mature mRNA of tropoelastin moves from the nucleus to the cytoplasm and then translated into tropoelastin in the rough endoplasmic reticulum (ER) (Swee et al., 1995). In the ER, tropoelastin interacts with elastin-binding protein (EBP) and protects tropoelastin from intracellular degradation and coacervation. Subsequently, the complex of EBP and tropoelastin is secreted to the cell surface to self-assembly. Tropoelastin monomers form multimers through the aggregation of their hydrophobic domains, and then deposit onto the microfibril scaffold. Under the modification of LOX, tropoelastin undergo extensive cross-linking and eventually form mature elastic fibers (Figure 2; Ozsvar et al., 2021). Recent studies suggest tropoelastin may follow an unconventional secretory pathway before transport to the cell surface. The assembly of elastin is thought to occur within the cell or at unique assembly sites on the plasma membrane, and after the self-aggregation, tropoelastin and other necessary assembly proteins are transported from the Golgi into an endosomal-like compartment along with LOX for crosslinking (Rabkin, 2017; Schrader et al., 2018; Kozel and Mecham, 2019; Shin and Yanagisawa, 2019). This may be a new perspective for better understanding the assembly mechanism of elastin. In the ER, tropoelastin also binds to several molecular chaperones, such as BIP and FKBP65 to assist with protein folding (Davis et al., 1998). The role of BIP is primarily to bind synthesized proteins, whereas FKBP65, an ER-resident peptidyl-prolyl cis-trans isomerase, modifies the kinetics of tropoelastin self-assembly in attenuating premature intracellular self-aggregation. The inherent ability of elastin to self-aggregate was described as a coacervation phenomenon and coacervation might play a critical role in the elastogenesis process which once impaired may induce elastin haploinsufficiency disorders including supravalvular aortic stenosis (Yeo et al., 2011). Studies showed that coacervation is an endothermic, entropically-driven process in which hydrophobic proteins separate from the solution as a second phase when the temperature is increased, and perhaps assisting in the alignment of lysine residues for crosslinking (Vrhovski et al., 1997). It has been reported that coacervation might order and align tropoelastin for crosslinking into the polymeric elastin matrix. And even if the lysine residues were modified the elastin polypeptides still were soluble without coacervation. Therefore, coacervation is important for the alignment of lysine residues present in the crosslinked region and the subsequent cross-link formation (Debelle and Tamburro, 1999; Keeley et al., 2002; Bellingham et al., 2003; Ostuni et al., 2007).
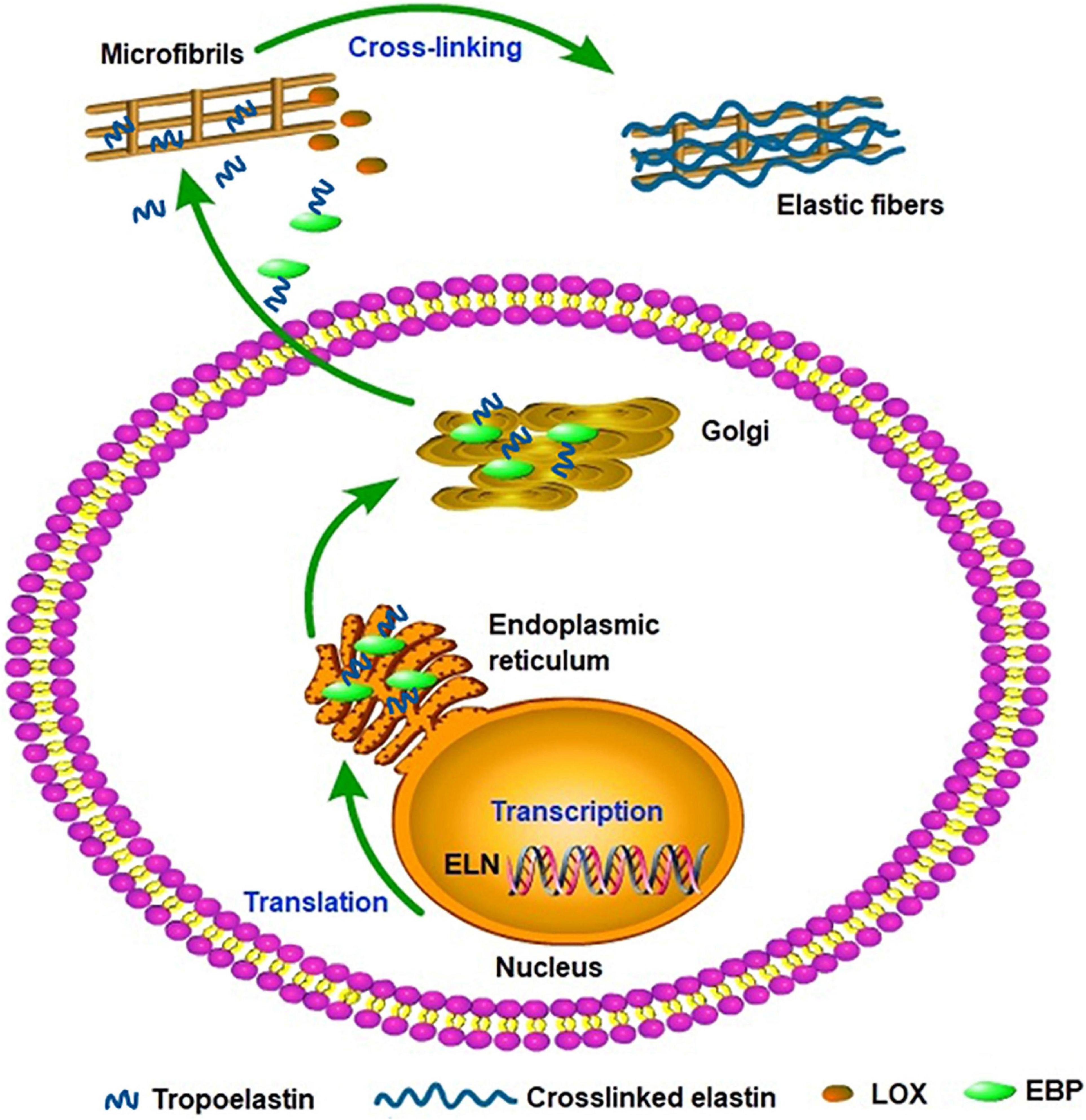
Figure 2. Tropoelastin secretory/assembly pathway. The synthesis of tropoelastin begins with the transcription of the ELN gene in the nucleus. In the endoplasmic reticulum, tropoelastin interacts with EBP and folds in structure. After the storage and transport of Golgi, the complex of EBP and tropoelastin is secreted to the cell surface for self-assembly. Then, tropoelastin dissociates from EBP and deposits onto the microfibril scaffold. In the presence of LOX, tropoelastin cross-links and eventually forms mature elastic fibers.
However, the precise molecular assembly pathway of the mechanical properties of elastin remains poorly understood.
The dynamic regulation of elastin expression and formation in the development of various diseases involves a variety of molecules through transcriptional and post-transcriptional mechanisms, and it has engaged the interest of many researchers.
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidases that degrade ECM components, including elastin. MMPs can be activated by their proteolysis or other MMPs and the cleavage in amino-terminal prepeptide domains of serine proteases. Among MMPs, MMP-2 (gelatinase A), MMP-7 (matrilysin), MMP-9 (gelatinase B), and MMP-12 (macrophage elastase) have been shown to degrade elastin and its precursor tropoelastin. Moreover, recent studies reported that MMP-14 (MT1-MMP), has a similar cleavage effect on elastin, and MMP-14 also mediates MMP-2 activation (Rabkin, 2017; Miekus et al., 2019). By contrast, tissue inhibitors of MMP (TIMP), as endogenous inhibitors, prevent excessive ECM degradation caused by MMPs. Several studies have shown that the expression of MMP-2, MMP-7, MMP-9, MMP-12, and MMP-14 is significantly up-regulated in many cardiovascular diseases. However, changes in TIMPs levels are not entirely consistent (Johnson et al., 2014; Arvidsson et al., 2019; Deng et al., 2019; Kanagala et al., 2020; Merico et al., 2021; Morishita et al., 2021).
Cytokine-controlled elastin regulatory axes are involved in elastin expression regulation. As upstream regulators of MMPs and TIMPs, cytokines may cause an imbalance of the MMP/TIMP ratio resulting in altered myocardial (Kwak, 2013). Tumor necrosis factor-alpha (TNF-α), which is a pro-inflammatory cytokine, inhibits tropoelastin mRNA levels in skin fibroblasts, aortic smooth muscle cells, and lung fibroblasts. A previous study showed that TNF-α may induce vascular smooth muscle cell activation to produce MMP-2 and MMP-9 as well as inhibits TIMP activity and leads to elastin degradation in vitro (Kothapalli and Ramamurthi, 2010). TNF-α induced elastin degradation is associated with vasculitis caused by Kawasaki disease and vascular aneurysms, and the TNF-α deficiency prevents elastin degradation and aneurysm formation (Hui-Yuen et al., 2006). Like TNF-α, interleukin-1β (IL-1β) is a pro-inflammatory cytokine as well. In neonatal rat lung fibroblasts, IL-1β enhanced the release of elastases and down-regulated elastin transcription (Kuang et al., 2002). In the adult murine lungs, the production of IL-1β increased MMP-9 and MMP-12 expression and caused the disruption of elastin fibers in alveolar septa and airway fibrosis (Lappalainen et al., 2005). Conversely, transforming growth factor β (TGFβ), a potent profibrotic cytokine multiple, may have positive effects on tropoelastin transcription and stabilization and enhance tropoelastin mRNA expression. TGFβ1 is a member of the TGFβ superfamily, and several studies demonstrated that TGFβ1 inhibits MMP-2 and MMP-9 activation and stimulates TIMP-1 expression, thereby suppressing elastin degradation (Alvira et al., 2011; Sproul and Argraves, 2013). In addition, the overexpression of TGFβ1 stabilizes abdominal aortic aneurysms injured by inflammation and proteolysis to restore their ability to withstand arterial pressure (Dai et al., 2005). Heparin-binding epidermal growth factor-like growth factor (HB-EGF) is an EGF receptor ligand that downregulated elastin mRNA via activation of epidermal growth factor receptor and reduces matrix accumulation of elastin (Liu et al., 2003; Kwak, 2013). Furthermore, on the aging process of human mammary epithelial cells, a co-localization of MMP-7 and HB-EGF was demonstrated, and tropoelastin expression increased is associated with impaired HB-EGF and MMP-7 down-modulation. These observations suggest that MMP-7 and HB-EGF are interrelated in elastin regulation, but the specific mechanism remains unclear (Bertram and Hass, 2009).
MicroRNAs (miRNAs) are small non-coding RNAs primarily involved in post-transcriptional regulation of gene expression by predominantly targeting the 3′UTR of mRNAs through destabilizing the transcript of mRNA or interfering in the translation process. Previous research has found that elastin expression is mainly regulated by the miR-29 family members (particularly miR-29b) and the miR-15 family members (particularly miR-195). Studies have demonstrated that miR-195 and miR-29b overexpression leads to a decrease in elastin. Using vectors carrying putative binding sites of miR-195 and miR-29b transfected into smooth muscle cells, previous works have shown that elastin is a direct target for both miRNAs. In in vivo experiments, miR-195 inhibited elastin and collagen expression and also restrained MMP-2 and MMP-9 activity, a degrading enzyme of ECM (Zampetaki et al., 2014; Kowara et al., 2017). In the heart, miR-29b is a key regulator of aortic dilatation, and its dysbiosis is also consistent with ECM remodeling after acute myocardial infarction (MI) (Kowara et al., 2017). Moreover, miR-29a inhibition can remarkably increase the ELN expression level in human cells. Therefore, the antagonistic effect of miR-29 may upregulate the ELN level in the case of elastically enhanced elastin dissolution or deficiency (Zhang et al., 2012). miR-181b is another epigenetic regulator of elastin gene expression. Using the mouse atherosclerosis models of ApoE–/– and LDLR–/–, a study found that miR-181b inhibition substantially increased elastin and collagen expression levels through negative regulation of downstream target TIMP-3 expression and promoted a fibrotic response and consequent stabilization of existing plaques or aneurysms (Di Gregoli et al., 2017). Specific molecular regulatory mechanisms of elastin synthesis are shown in Figure 3.
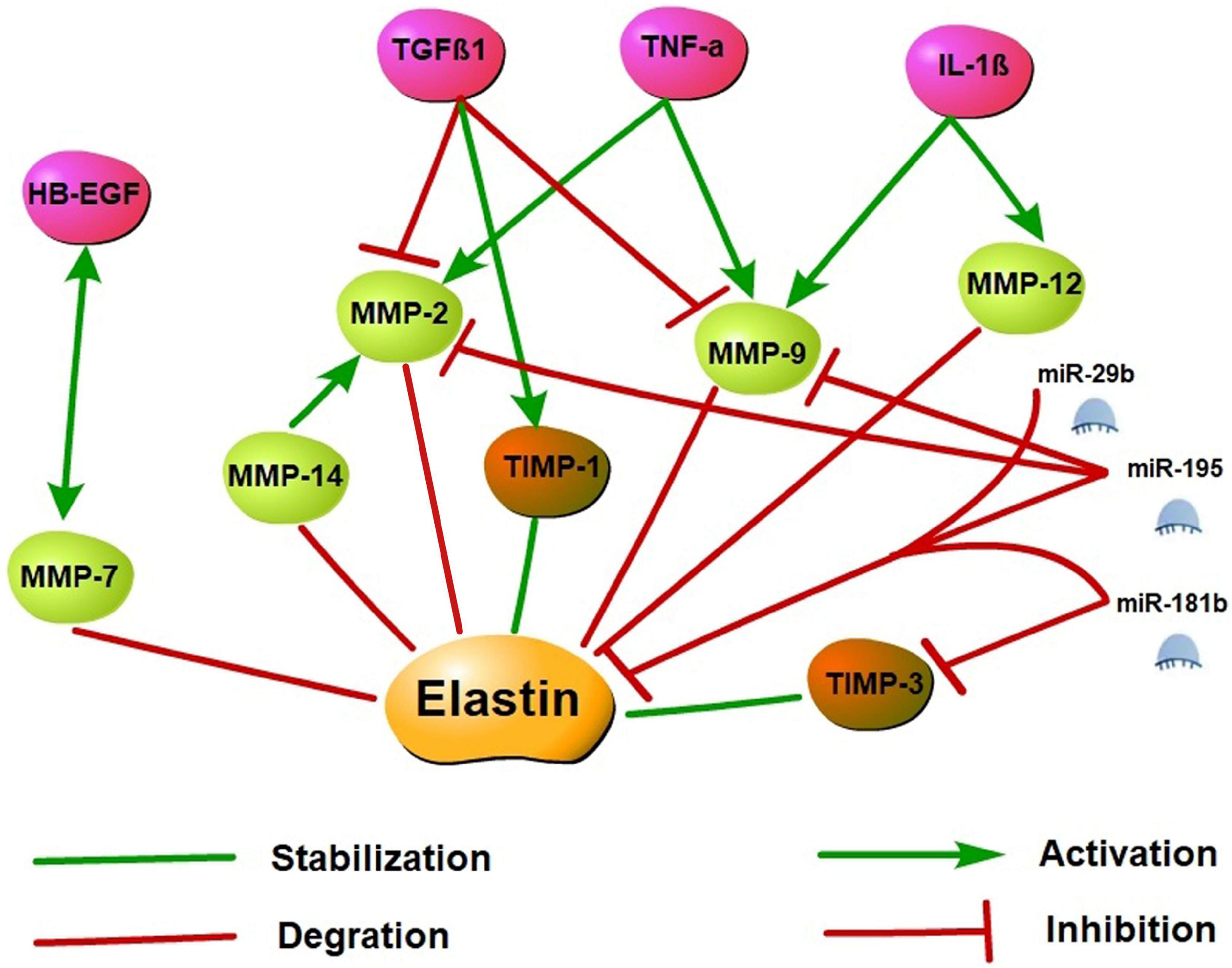
Figure 3. Elastin synthesis molecular regulatory networks. Elastin synthesis is dynamically regulated by a variety of molecules. Activated MMP may promote the degradation of elastin and its precursor tropoelastin, while the increase of TIMP may stabilize the synthesis of elastin. TNF-α, IL-1β, and HB-EGF may stimulate MMP activation and inhibit TIMP to induce elastin degradation. However, TGFβ1 may inhibit MMP activation and stimulate TIMP expression to suppress elastin degradation. Additionally, miR-29b, miR-195, and miR-181b are epigenetic regulatory molecules of elastin. These miRNAs may inhibit elastin expression by targeting the 3′ UTR of ELN mRNA, and restrained MMP and TIMP activity to involve in the regulation of elastin synthesis.
Among other exogenous factors regulating elastin synthesis, vitamin C inhibits elastin gene expression and tropoelastin synthesis by changing mRNA stability and post-translational mechanism. As an ATP-dependent K+ channel opener, minoxidil can promote the expression of elastin mRNA in skin fibroblasts and smooth muscle cells. The regulatory mechanism of minoxidil on these cells is completed through Ca2+-ERK-dependent pathway in vitro (Daamen and Quaglino, 2019). Studies have shown that the blood pressure and functional arterial stiffness of ELN± mice reduced to wild-type (WT) levels and the carotid and cerebral blood flows were restored when treated with minoxidil. Moreover, the expression of ELN was increased and 126 other matrix-associated genes were altered after the minoxidil treatment in ELN± mice (Knutsen et al., 2018). Another research in aged mice found that the mRNA levels of tropoelastin, fibulin-5, and lysyl-oxidase were increased, and the expression of elastin was reinduced accompanied by the formation of newly synthesized elastic fibers. At the same time, the levels of advanced glycation end products were reduced with the chronic treatment of minoxidil (Coquand-Gandit et al., 2017). These results suggest that minoxidil may be involved in arterial remodeling and the improvement of cardiovascular disease by regulating the expression of elastin. And the treatment of minoxidil could be a potential strategy to improve arterial aging.
Atherosclerosis is a disease leading to the formation of plaques consisting of lipids, calcified regions, and immune and foam cells, along with vascular smooth muscles and endothelial cells in large- and medium-sized arteries (Fhayli et al., 2019). The altered homeostasis between synthesis and proteolysis of ECM proteins contributes to the development of atherosclerosis, and the alteration of elastin amounts, incorrect assembly, elastic fiber modification, and elastin fragments are associated with atherosclerosis. In vivo, damaged or degenerated elastic fibers are usually not repaired but are replaced by collagen and proteoglycan, making the arterial wall stiffer (Wahart et al., 2019). Moreover, a decrease in the ratio of elastin and collagen leads to arterial stiffness and arterial dilatation, further negatively affecting the normal function of blood vessels.
Calcification is a hallmark of late atherosclerosis (Speer and Giachelli, 2004). In the early stages, calcification is diffuse or punctate, but as the process progresses, calcium deposition, aggregation, and consolidation eventually form large flake calcium deposits. In addition, the diffuse calcification of vascular intima is also related to some components of ECM, especially matrix vesicles. Matrix vesicles come from osteoblast, cartilage cell, odontoblast and budded from cell, then apart from the cell to form self-governed organelle. Matrix vesicles provide suitable nucleation and micro-environment to calcium crystal, and the elastin in the vascular wall provides a network scaffold for calcium crystal deposition. Moreover, the elastic fibers in the ECM may take part to the arterial wall calcification. Electron microscopy and histochemistry revealed two different types of elastic fiber calcification in human atherosclerotic plaques. No structural changes in elastin are detected in type I calcification, whereas structural changes in elastin occur earlier in type II calcification (Bobryshev, 2005). Khavandgar et al. (2014) demonstrated that in matrix Gla protein (MGP)-deficient and ELN haploinsufficient (MGP–/–; ELN±) mice, the severity of medial calcification is elastin dependent, and elastin haploinsufficiency has significantly reduced arterial mineral deposition compared with MGP–/– mice. This experiment suggests that the content of elastin might be an important determinant of medial calcification. Degradation of elastin in the arterial wall is also a typical feature of atherosclerosis. Elastin-derived peptides (EDPs), the degradation products of elastin, display a wide range of biological activities, and EDPs have previously been shown to be related with atherosclerosis. Studies have shown that serum concentrations of EDP (S-EDP) are higher in atherosclerotic patients compared to healthy subjects, and concentrations of elastin-derived peptides were statistically significant higher in children from families at high risk for atherosclerosis (Gmiński et al., 1991). However, Petersen et al. (2002) suggested that increased concentrations of S-EDP were associated with aneurysmal and ulcerative, but not occlusive, manifestations of atherosclerosis. The presence of EDPs in serum may reflect the intensity of the elastic dissolution process in atherosclerotic lesions. Inflammatory cytokines, growth factors, oxidative stress, and hypoxia produced in the process of atherosclerosis lead to the breakdown of elastin and the production of elastokines (Bobryshev, 2005; Wahart et al., 2019). EDPs can dissolve in the blood and spread out in the whole body from disrupted plaques, which may cause harmful complications such as plaque instability. Therefore, we speculate that increased concentrations of EDPs may be a potential risk factor for atherosclerotic complications. From the results, we speculate that the structural alterations of elastin and elastic fibers occurring in the early stages of atherosclerosis can induce the calcified deposits formed on the base of pre-existing structures with the deposition of unesterified cholesterol. Besides that, EDPs, the degradation products of elastin actively participate in the pathogenesis of atherosclerosis such as low-density lipoprotein oxidation and vascular calcification and accelerate the progression of the disease.
In addition, elastin is also affected by glycosylation and carbamylation (Gorisse et al., 2016) which may exacerbate the progression of chronic kidney diseases by modulating the hardness of elastin. However, whether these modifications have a direct effect on atherosclerosis progression remains to be demonstrated.
As we summarized above elastin can be cleaved by elastinases and MMPs, and the excessive production of elastin fragmentation may have a deleterious effect on atherosclerosis progression. Thus, chemical compounds designed to bind to elastin and simultaneously inhibit elastolytic activities might be new therapeutical strategies. Selective murine MMP-12 inhibitor has been reported by the group of Dive, MMP-12 is associated with plaque progression and instability. The MMP-12 inhibitor significantly reduced atherosclerotic plaque area and delay atherosclerosis development in ApoE–/– mice (Quillard et al., 2011).
Atherosclerosis can cause progressive stenosis of the coronary artery, which makes the blood and oxygen supply of the heart insufficient, resulting in ischemia of downstream tissues. Generally, ischemia-reperfusion can only restore the blood flow of ischemic tissue, cannot restore tissue, organ function, on the contrary, aggravate tissue, organ dysfunction, and structural damage called ischemia-reperfusion injury. The mechanisms involved in myocardial ischemia-reperfusion injury may include reperfusion triggered oxidative burst, calcium overload, and mitochondrial damage, which together induce cardiomyocyte apoptosis and necrosis, leading to irreversible damage (Yellon and Hausenloy, 2007).
When ischemia-reperfusion occurs, myocardial interstitial components are reconstructed while cardiomyocytes are damaged. However, such reconstruction is rarely reported in recent literature. Myocardial tissues on normal state of physiology contain a small number of elastic fibers, which are primarily derived from fibroblasts and smooth muscle cells. Research in rats shows that fibrosis occurs in the infarcted area after suffering myocardial ischemia (MI), forming cicatricial tissues, which decrease the elasticity of the ventricular wall and influence heart function. And the expression of recombinant elastin contributes to maintaining the elasticity of MI tissues (Mizuno et al., 2005). Our studies have proved that the increase in collagen fiber content and the decrease in elastic fiber content are the pathological basis of the enhanced tissue hardness and the decrease of elasticity in myocardial scar tissues located in the infarcted area in Sprague–Dawley rats (Yu et al., 2018). Therefore, elastin is believed to play an important role in preventing regional expansion of cicatricial tissues in the infarcted area, left ventricular enlargement, and improvement of ejection fraction (Mizuno et al., 2005).
Researchers examined the images of elastin and mature elastin fibers using an elastin/tropoelastin-specific contrast agent (i.e., Gd-ESMA) by constructing myocardial ischemia model in mice, and found the presence of tropoelastin and mature elastin in the infarct scar regions, and this presence was associated with improved ejection fraction of heart after MI in vivo (Protti et al., 2015). Mizuno demonstrated that increasing the expression of recombinant elastin within the myocardial scar by transfecting elastin gene into rat endothelial cells can reduce scar expansion and preserve heart function after MI (Mizuno et al., 2005). Lichtenauer et al. (2011) showed that in the rat myocardial infarction model, the scar tissue gradually formed after myocardial infarction, in which the expression of elastin protein increased, but this myocardial remodeling can be blocked by the intravenous or intramyocardial injection of apoptotic leukocyte suspension. Compared with collagen in fibrotic tissues, a high proportion of elastin can change the composition of myocardial scar to maintain the elasticity of the infarcted heart and improve cardiac function, thereby avoiding progressive heart failure. Therefore, the increased synthesis and composition of elastin fibers is a decisive mechanism for maintaining infarct scar structure and elasticity to avoid the gradual loss of cardiac biomechanical functions. However, none of these studies has involved the specific molecular regulatory mechanism of elastin in the disease process, which needs further study.
The fragmentation of elastin by elastinolytic enzymes named EDP is a hallmark of aging tissues. This aging marker was also observed in myocardial tissue during ischemia. EDPs were considered to have detrimental effects on the cardiovascular system, showing chemotactic activity against neutrophils, inducing T-Helper-type1 polarization in human blood lymphocytes, secretion of cytokines and enzymes in fibroblasts and aortic smooth muscle cells (Debret et al., 2005). Robinet et al. (2007) have shown that EDPs interaction with S-Gal (elastin binding protein) can protect the heart against ischemia/reperfusion injury by triggering nitric oxide (NO) release and activating PI3-kinase/Akt and ERK1/2 in human coronary endothelial cells (HCAECs) and rat neonatal cardiomyocytes (RCs). But researchers later found that such protective effect might be lost with aging in rats. The elastin receptor complex (ERC) is a heterotrimer that is composed of elastin binding protein, protective protein or cathepsin A (PPCA), and membrane-bound neuraminidase 1 (Neu-1) and derived from the lysosomal β-galactosidase(β-Gal) complex. S-Gal is a spliced variant of β-Gal and this splicing form, which retains the ability to bind galactose or lactose and lacks enzyme activity, can bind to EDPs as antagonists (Maurice et al., 2013).
At present, blocking the combination of elastic factor and ERC may be a new treatment strategy. For example, a peptide that mimics the S-Gal sequence is designed to limit the harmful effects of elastin fragmentation at the molecular level.
Beyond that, both NO and reactive oxygen species (ROS) are involved in the myocardial ischemia-reperfusion injury or neurovascular protection after ischemic stroke, depending on their concentrations. Elastin-Derived Peptide VGVAPG decreased the expression of eNos, iNos, and nNos mRNA and proteins in mouse astrocytes in vitro. The VGVAPG peptide also reduces the production of NO in cells and increases the production of ROS. Therefore, it is speculated that VGVAPG peptide is involved in activating the survival/healing pathway of astrocytes (Szychowski and Gmiński, 2019).
Reactive oxygen species are generally considered as toxic by-products of aerobic metabolism and the main cause of macromolecular damage, and reperfusion is associated with the outbreak of ROS production. Previous studies have shown that ischemic oxidative stress is a precursor of cell death in the process of myocardial cell reperfusion (Robin et al., 2007). Therefore, whether EDPs generation plays a beneficial protective role with myocardial ischemia-reperfusion injury is still an unsolved problem.
Atrial fibrillation (AF) is a common clinical arrhythmia that affects atrial cardiomyocytes and interstitials. One of the generally recognized factors that affect the development and maintenance of AF is cardiac remodeling. Experimental models showed that AF leads to a variable degree of structural remodeling of atrial tissues that in turn promotes further maintenance of arrhythmias (Wakili et al., 2011). However, limited clinical evidence is available to support this view.
Tissue interstitial fibrosis, the important morphological basis for AF, is also an important marker for structural reconstruction. According to morphological observation, myocardial fibrosis is the increase of ECM proportion, especially collagen (protein) fiber deposition. As the important components of the atrial wall, collagen, elastin, and reticular fibers determine its mechanical properties, such as compliance (Fomovsky et al., 2010). Smorodinova et al. (2015) showed that the contents of interstitial elastin, collagen I, and collagen III in atrial tissues of patients in the AF and SR groups were similar, and no remarkable difference was observed in vascular endothelial growth factor (VEGF) expression and microvascular density. However, the expression of elastin, collagen I, collagen III, and VEGF in the right atrium tissue was remarkably higher than that in the left atrium tissue, and the microvascular density of the left atrium myocardium was higher than that of the right atrium. Therefore, interstitial fibrosis and other morphological changes in atrial tissues are associated with structural heart diseases but not with AF. However, regional differences in these institutions in the left and right atria warrant further study. Recent research showed that in animal models, atrial stretch leads to increased atrial fibrosis, thereby inducing regional remodeling and slow conduction. Atrial stretch causes ANP release, calcium overload, calcineurin activation, AT1 receptor involvement, and altered levels of MMP and TIMP. Moreover, atrial stretch is associated with tissue turnover and remodeling. Furthermore, atrial distraction can induce inflammation and fibroblast proliferation, collagen elaboration, and scar formation, and this process is TGF-β1 mediated (Yang et al., 2017). However, the specific mechanism of elastin and AF is not clear, so it is necessary to explore further.
Many studies on elastin structure and biosynthesis have been conducted, but its specific method of assembly has not been determined thus far. At present, studies on elastin expression regulation often tend to focus exclusively on cell experiments or animal models and thus cannot accurately simulate the complex internal environmental homeostasis of humans. This homeostasis is often accompanied by alterations of elastin expression levels and structures in many cardiovascular diseases. However, its accurate mechanism of action is complex and remains inconclusive. It’s necessary for us to better understand how enzymes associated with inflammation lead to structural remodeling and loss of functional integrity of elastic fibers in heart diseases. And it’s also important to seek biomarkers of elastic fiber injury to realize the diagnosis and treatment in the early stages of diseases. Therefore, the comprehensive action of elastin and its various regulatory molecules must be explored to establish strategies for the prevention and treatment of related diseases.
ZG conceived the study. XM prepared the figures. KW drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Nature Science Foundation of China (81570268).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alvira, C. M., Guignabert, C., Kim, Y. M., Chen, C., Wang, L., Duong, T. T., et al. (2011). Inhibition of transforming growth factor β worsens elastin degradation in a murine model of Kawasaki disease. Am. J. Pathol. 178, 1210–1220. doi: 10.1016/j.ajpath.2010.11.054
Arvidsson, M., Ahmed, A., Bouzina, H., and Rådegran, G. (2019). Matrix metalloproteinase 7 in diagnosis and differentiation of pulmonary arterial hypertension. Pulm. Circ. 9:2045894019895414. doi: 10.1177/2045894019895414
Baldock, C., Oberhauser, A. F., Ma, L., Lammie, D., Siegler, V., Mithieux, S. M., et al. (2011). Shape of tropoelastin, the highly extensible protein that controls human tissue elasticity. Proc. Natl. Acad. Sci. U. S. A. 108, 4322–4327. doi: 10.1073/pnas.1014280108
Bellingham, C. M., Lillie, M. A., Gosline, J. M., Wright, G. M., Starcher, B. C., Bailey, A. J., et al. (2003). Recombinant human elastin polypeptides self-assemble into biomaterials with elastin-like properties. Biopolymers 70, 445–455. doi: 10.1002/bip.10512
Bertram, C., and Hass, R. (2009). Cellular senescence of human mammary epithelial cells (HMEC) is associated with an altered MMP-7/HB-EGF signaling and increased formation of elastin-like structures. Mech. Ageing Dev. 130, 657–669. doi: 10.1016/j.mad.2009.08.001
Bobryshev, Y. V. (2005). Calcification of elastic fibers in human atherosclerotic plaque. Atherosclerosis 180, 293–303. doi: 10.1016/j.atherosclerosis.2005.01.024
Boyd, C. D., Christiano, A. M., Pierce, R. A., Stolle, C. A., and Deak, S. B. (1991). Mammalian tropoelastin: multiple domains of the protein define an evolutionarily divergent amino acid sequence. Matrix 11, 235–241. doi: 10.1016/s0934-8832(11)80230-1
Cocciolone, A. J., Hawes, J. Z., Staiculescu, M. C., Johnson, E. O., Murshed, M., and Wagenseil, J. E. (2018). Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 315, H189–H205. doi: 10.1152/ajpheart.00087.2018
Coquand-Gandit, M., Jacob, M. P., Fhayli, W., Romero, B., Georgieva, M., Bouillot, S., et al. (2017). Chronic Treatment with Minoxidil Induces Elastic Fiber Neosynthesis and Functional Improvement in the Aorta of Aged Mice. Rejuvenation Res. 20, 218–230. doi: 10.1089/rej.2016.1874
Cunningham, K. S., Veinot, J. P., and Butany, J. (2006). An approach to endomyocardial biopsy interpretation. J. Clin. Pathol. 59, 121–129. doi: 10.1136/jcp.2005.026443
Daamen, W. F., and Quaglino, D. (2019). Signaling pathways in elastic tissues. Cell. Signal. 63:109364. doi: 10.1016/j.cellsig.2019.109364
Dai, J., Losy, F., Guinault, A. M., Pages, C., Anegon, I., Desgranges, P., et al. (2005). Overexpression of transforming growth factor-beta1 stabilizes already-formed aortic aneurysms: a first approach to induction of functional healing by endovascular gene therapy. Circulation 112, 1008–1015. doi: 10.1161/circulationaha.104.523357
Davis, E. C., Broekelmann, T. J., Ozawa, Y., and Mecham, R. P. (1998). Identification of tropoelastin as a ligand for the 65-kD FK506-binding protein, FKBP65, in the secretory pathway. J. Cell Biol. 140, 295–303. doi: 10.1083/jcb.140.2.295
de Carvalho Filho, E. T., de Carvalho, C. A., and de Souza, R. R. (1996). Age-related changes in elastic fibers of human heart. Gerontology 42, 211–217. doi: 10.1159/000213795
Debelle, L., and Tamburro, A. M. (1999). Elastin: molecular description and function. Int. J. Biochem. Cell Biol. 31, 261–272. doi: 10.1016/s1357-2725(98)00098-3
Debret, R., Antonicelli, F., Theill, A., Hornebeck, W., Bernard, P., Guenounou, M., et al. (2005). Elastin-derived peptides induce a T-helper type 1 polarization of human blood lymphocytes. Arterioscler. Thromb. Vasc. Biol. 25, 1353–1358. doi: 10.1161/01.ATV.0000168412.50855.9f
Deng, X. S., Meng, X., Li, F., Venardos, N., Fullerton, D., and Jaggers, J. (2019). MMP-12-Induced Pro-osteogenic Responses in Human Aortic Valve Interstitial Cells. J. Surg. Res. 235, 44–51. doi: 10.1016/j.jss.2018.09.005
Di Gregoli, K., Mohamad Anuar, N. N., Bianco, R., White, S. J., Newby, A. C., George, S. J., et al. (2017). MicroRNA-181b Controls Atherosclerosis and Aneurysms Through Regulation of TIMP-3 and Elastin. Circ. Res. 120, 49–65. doi: 10.1161/circresaha.116.309321
Fhayli, W., Boete, Q., Harki, O., Briancon-Marjollet, A., Jacob, M. P., and Faury, G. (2019). Rise and fall of elastic fibers from development to aging. Consequences on arterial structure-function and therapeutical perspectives. Matrix Biol. 84, 41–56. doi: 10.1016/j.matbio.2019.08.005
Fomovsky, G. M., Thomopoulos, S., and Holmes, J. W. (2010). Contribution of extracellular matrix to the mechanical properties of the heart. J. Mol. Cell. Cardiol. 48, 490–496. doi: 10.1016/j.yjmcc.2009.08.003
Gmiński, J., Drózdz, M., Ulfig-Maślanka, R., and Najda, J. (1991). Evaluation of elastin metabolism in children from families with high risk of atherosclerosis. Atherosclerosis 91, 185–189. doi: 10.1016/0021-9150(91)90166-z
Gorisse, L., Pietrement, C., Vuiblet, V., Schmelzer, C. E., Kohler, M., Duca, L., et al. (2016). Protein carbamylation is a hallmark of aging. Proc. Natl. Acad. Sci. U. S. A. 113, 1191–1196. doi: 10.1073/pnas.1517096113
Han, B., Ren, M. F., Chang, Y. Q., and Guo, Z. K. (2019). Expression of elastin in rat cardiomyocytes and cardiomyocyte cell line H9C2. Acta Antomica Sin. 50, 292–296.
Hui-Yuen, J. S., Duong, T. T., and Yeung, R. S. (2006). TNF-alpha is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J. Immunol. 176, 6294–6301. doi: 10.4049/jimmunol.176.10.6294
Johnson, J. L., Jenkins, N. P., Huang, W. C., Di Gregoli, K., Sala-Newby, G. B., Scholtes, V. P., et al. (2014). Relationship of MMP-14 and TIMP-3 expression with macrophage activation and human atherosclerotic plaque vulnerability. Mediators Inflamm. 2014:276457. doi: 10.1155/2014/276457
Kanagala, P., Arnold, J. R., Singh, A., Chan, D. C. S., Cheng, A. S. H., Khan, J. N., et al. (2020). Characterizing heart failure with preserved and reduced ejection fraction: an imaging and plasma biomarker approach. PLoS One 15:e0232280. doi: 10.1371/journal.pone.0232280
Keeley, F. W., Bellingham, C. M., and Woodhouse, K. A. (2002). Elastin as a self-organizing biomaterial: use of recombinantly expressed human elastin polypeptides as a model for investigations of structure and self-assembly of elastin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 357, 185–189. doi: 10.1098/rstb.2001.1027
Khavandgar, Z., Roman, H., Li, J., Lee, S., Vali, H., Brinckmann, J., et al. (2014). Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. 29, 327–337. doi: 10.1002/jbmr.2039
Knutsen, R. H., Beeman, S. C., Broekelmann, T. J., Liu, D., Tsang, K. M., Kovacs, A., et al. (2018). Minoxidil improves vascular compliance, restores cerebral blood flow, and alters extracellular matrix gene expression in a model of chronic vascular stiffness. Am. J. Physiol. Heart Circ. Physiol. 315, H18–H32. doi: 10.1152/ajpheart.00683.2017
Kothapalli, C. R., and Ramamurthi, A. (2010). Induced elastin regeneration by chronically activated smooth muscle cells for targeted aneurysm repair. Acta Biomater. 6, 170–178. doi: 10.1016/j.actbio.2009.06.006
Kowara, M., Cudnoch-Jedrzejewska, A., Opolski, G., and Wlodarski, P. (2017). MicroRNA regulation of extracellular matrix components in the process of atherosclerotic plaque destabilization. Clin. Exp. Pharmacol. Physiol. 44, 711–718. doi: 10.1111/1440-1681.12772
Kozel, B. A., and Mecham, R. P. (2019). Elastic fiber ultrastructure and assembly. Matrix Biol. 84, 31–40. doi: 10.1016/j.matbio.2019.10.002
Kuang, P. P., Berk, J. L., Rishikof, D. C., Foster, J. A., Humphries, D. E., Ricupero, D. A., et al. (2002). NF-kappaB induced by IL-1beta inhibits elastin transcription and myofibroblast phenotype. Am. J. Physiol. Cell Physiol. 283, C58–C65. doi: 10.1152/ajpcell.00314.2001
Kwak, H. B. (2013). Aging, exercise, and extracellular matrix in the heart. J. Exerc. Rehabil. 9, 338–347. doi: 10.12965/jer.130049
Lappalainen, U., Whitsett, J. A., Wert, S. E., Tichelaar, J. W., and Bry, K. (2005). Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 32, 311–318. doi: 10.1165/rcmb.2004-0309OC
Lichtenauer, M., Mildner, M., Baumgartner, A., Hasun, M., Werba, G., Beer, L., et al. (2011). Intravenous and intramyocardial injection of apoptotic white blood cell suspensions prevents ventricular remodelling by increasing elastin expression in cardiac scar tissue after myocardial infarction. Basic Res. Cardiol. 106, 645–655. doi: 10.1007/s00395-011-0173-0
Liu, J., Rich, C. B., Buczek-Thomas, J. A., Nugent, M. A., Panchenko, M. P., and Foster, J. A. (2003). Heparin-binding EGF-like growth factor regulates elastin and FGF-2 expression in pulmonary fibroblasts. Am. J. Physiol. Lung. Cell. Mol. Physiol. 285, L1106–L1115. doi: 10.1152/ajplung.00180.2003
Maurice, P., Blaise, S., Gayral, S., Debelle, L., Laffargue, M., Hornebeck, W., et al. (2013). Elastin fragmentation and atherosclerosis progression: the elastokine concept. Trends Cardiovasc. Med. 23, 211–221. doi: 10.1016/j.tcm.2012.12.004
Merico, V., Imberti, J. F., Zanoni, M., Boriani, G., Garagna, S., and Imberti, R. (2021). Inhibition of lysyl oxidase stimulates TGF-β signaling and metalloproteinases-2 and -9 expression and contributes to the disruption of ascending aorta in rats: protection by propylthiouracil. Heart Vessels 36, 738–747. doi: 10.1007/s00380-020-01750-6
Miekus, N., Luise, C., Sippl, W., Baczek, T., Schmelzer, C. E. H., and Heinz, A. (2019). MMP-14 degrades tropoelastin and elastin. Biochimie 165, 32–39. doi: 10.1016/j.biochi.2019.07.001
Mizuno, T., Yau, T. M., Weisel, R. D., Kiani, C. G., and Li, R. K. (2005). Elastin stabilizes an infarct and preserves ventricular function. Circulation 112, I81–I88. doi: 10.1161/01.circulationaha.105.523795
Morishita, T., Uzui, H., Sato, Y., Mitsuke, Y., and Tada, H. (2021). Associations between cachexia and metalloproteinases, haemodynamics and mortality in heart failure. Eur. J. Clin. Invest. 51:e13426. doi: 10.1111/eci.13426
Ostuni, A., Bochicchio, B., Armentano, M. F., Bisaccia, F., and Tamburro, A. M. (2007). Molecular and supramolecular structural studies on human tropoelastin sequences. Biophys. J. 93, 3640–3651. doi: 10.1529/biophysj.107.110809
Ozsvar, J., Yang, C., Cain, S. A., Baldock, C., Tarakanova, A., and Weiss, A. S. (2021). Tropoelastin and Elastin Assembly. Front. Bioeng. Biotechnol. 9:643110. doi: 10.3389/fbioe.2021.643110
Partridge, S. M. (1969). Elastin, biosynthesis and structure. Gerontologia 15, 85–100. doi: 10.1159/000211679
Petersen, E., Wågberg, F., and Angquist, K. A. (2002). Serum concentrations of elastin-derived peptides in patients with specific manifestations of atherosclerotic disease. Eur. J. Vasc. Endovasc. Surg. 24, 440–444. doi: 10.1053/ejvs.2002.1750
Protti, A., Lavin, B., Dong, X., Lorrio, S., Robinson, S., Onthank, D., et al. (2015). Assessment of Myocardial Remodeling Using an Elastin/Tropoelastin Specific Agent with High Field Magnetic Resonance Imaging (MRI). J. Am. Heart Assoc. 4:e001851. doi: 10.1161/jaha.115.001851
Quillard, T., Croce, K., Jaffer, F. A., Weissleder, R., and Libby, P. (2011). Molecular imaging of macrophage protease activity in cardiovascular inflammation in vivo. Thromb. Haemost. 105, 828–836. doi: 10.1160/th10-09-0589
Rabkin, S. W. (2017). The Role Matrix Metalloproteinases in the Production of Aortic Aneurysm. Prog. Mol. Biol. Transl. Sci. 147, 239–265. doi: 10.1016/bs.pmbts.2017.02.002
Reichheld, S. E., Muiznieks, L. D., Lu, R., Sharpe, S., and Keeley, F. W. (2019). Sequence variants of human tropoelastin affecting assembly, structural characteristics and functional properties of polymeric elastin in health and disease. Matrix Biol. 84, 68–80. doi: 10.1016/j.matbio.2019.06.010
Robin, E., Guzy, R. D., Loor, G., Iwase, H., Waypa, G. B., Marks, J. D., et al. (2007). Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J. Biol. Chem. 282, 19133–19143. doi: 10.1074/jbc.M701917200
Robinet, A., Millart, H., Oszust, F., Hornebeck, W., and Bellon, G. (2007). Binding of elastin peptides to S-Gal protects the heart against ischemia/reperfusion injury by triggering the RISK pathway. FASEB J. 21, 1968–1978. doi: 10.1096/fj.06-6477com
Rodriguez, E. R., and Tan, C. D. (2017). Structure and Anatomy of the Human Pericardium. Prog. Cardiovasc. Dis. 59, 327–340. doi: 10.1016/j.pcad.2016.12.010
Schrader, C. U., Heinz, A., Majovsky, P., Karaman Mayack, B., Brinckmann, J., Sippl, W., et al. (2018). Elastin is heterogeneously cross-linked. J. Biol. Chem. 293, 15107–15119. doi: 10.1074/jbc.RA118.004322
Shapiro, S. D., Endicott, S. K., Province, M. A., Pierce, J. A., and Campbell, E. J. (1991). Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J. Clin. Invest. 87, 1828–1834. doi: 10.1172/jci115204
Shin, S. J., and Yanagisawa, H. (2019). Recent updates on the molecular network of elastic fiber formation. Essays Biochem. 63, 365–376. doi: 10.1042/ebc20180052
Smorodinova, N., Lantova, L., Blaha, M., Melenovsky, V., Hanzelka, J., Pirk, J., et al. (2015). Bioptic Study of Left and Right Atrial Interstitium in Cardiac Patients with and without Atrial Fibrillation: interatrial but Not Rhythm-Based Differences. PLoS One 10:e0129124. doi: 10.1371/journal.pone.0129124
Speer, M. Y., and Giachelli, C. M. (2004). Regulation of cardiovascular calcification. Cardiovasc. Pathol. 13, 63–70. doi: 10.1016/s1054-8807(03)00130-3
Sproul, E. P., and Argraves, W. S. (2013). A cytokine axis regulates elastin formation and degradation. Matrix Biol. 32, 86–94. doi: 10.1016/j.matbio.2012.11.004
Swee, M. H., Parks, W. C., and Pierce, R. A. (1995). Developmental regulation of elastin production. Expression of tropoelastin pre-mRNA persists after down-regulation of steady-state mRNA levels. J. Biol. Chem. 270, 14899–14906. doi: 10.1074/jbc.270.25.14899
Szychowski, K. A., and Gmiński, J. (2019). The VGVAPG Peptide Regulates the Production of Nitric Oxide Synthases and Reactive Oxygen Species in Mouse Astrocyte Cells In Vitro. Neurochem. Res. 44, 1127–1137. doi: 10.1007/s11064-019-02746-z
Tarakanova, A., Yeo, G. C., Baldock, C., Weiss, A. S., and Buehler, M. J. (2018). Molecular model of human tropoelastin and implications of associated mutations. Proc. Natl. Acad. Sci. U. S. A. 115, 7338–7343. doi: 10.1073/pnas.1801205115
Tsamis, A., Krawiec, J. T., and Vorp, D. A. (2013). Elastin and collagen fibre microstructure of the human aorta in ageing and disease: a review. J. R. Soc. Interface 10:20121004. doi: 10.1098/rsif.2012.1004
Uitto, J. (1979). Biochemistry of the elastic fibers in normal connective tissues and its alterations in diseases. J. Invest. Dermatol. 72, 1–10. doi: 10.1111/1523-1747.ep12530093
Vrhovski, B., Jensen, S., and Weiss, A. S. (1997). Coacervation characteristics of recombinant human tropoelastin. Eur. J. Biochem. 250, 92–98. doi: 10.1111/j.1432-1033.1997.00092.x
Vrhovski, B., and Weiss, A. S. (1998). Biochemistry of tropoelastin. Eur. J. Biochem. 258, 1–18. doi: 10.1046/j.1432-1327.1998.2580001.x
Wahart, A., Hocine, T., Albrecht, C., Henry, A., Sarazin, T., Martiny, L., et al. (2019). Role of elastin peptides and elastin receptor complex in metabolic and cardiovascular diseases. FEBS J. 286, 2980–2993. doi: 10.1111/febs.14836
Wakili, R., Voigt, N., Kaab, S., Dobrev, D., and Nattel, S. (2011). Recent advances in the molecular pathophysiology of atrial fibrillation. J. Clin. Invest. 121, 2955–2968. doi: 10.1172/jci46315
Wise, S. G., Mithieux, S. M., and Weiss, A. S. (2009). Engineered tropoelastin and elastin-based biomaterials. Adv. Protein Chem. Struct. Biol. 78, 1–24. doi: 10.1016/s1876-1623(08)78001-5
Yang, F., Tiano, J., Mittal, S., Turakhia, M., Jacobowitz, I., and Greenberg, Y. (2017). Towards a Mechanistic Understanding and Treatment of a Progressive Disease: atrial Fibrillation. J. Atr. Fibrillation 10:1627. doi: 10.4022/jafib.1627
Yellon, D. M., and Hausenloy, D. J. (2007). Myocardial reperfusion injury. N. Engl. J. Med. 357, 1121–1135. doi: 10.1056/NEJMra071667
Yeo, G. C., Keeley, F. W., and Weiss, A. S. (2011). Coacervation of tropoelastin. Adv. Colloid Interface Sci. 167, 94–103. doi: 10.1016/j.cis.2010.10.003
Yu, Y., Yin, G., Bao, S., and Guo, Z. (2018). Kinetic alterations of collagen and elastic fibres and their association with cardiac function in acute myocardial infarction. Mol. Med. Rep. 17, 3519–3526. doi: 10.3892/mmr.2017.8347
Zampetaki, A., Attia, R., Mayr, U., Gomes, R. S., Phinikaridou, A., Yin, X., et al. (2014). Role of miR-195 in aortic aneurysmal disease. Circ. Res. 115, 857–866. doi: 10.1161/circresaha.115.304361
Zhang, P., Huang, A., Ferruzzi, J., Mecham, R. P., Starcher, B. C., Tellides, G., et al. (2012). Inhibition of microRNA-29 enhances elastin levels in cells haploinsufficient for elastin and in bioengineered vessels–brief report. Arterioscler. Thromb. Vasc. Biol. 32, 756–759. doi: 10.1161/atvbaha.111.238113
Keywords: elastin, matrix metalloproteinase, myocardial ischemia-reperfusion, atherosclerosis, atrial fibrillation
Citation: Wang K, Meng X and Guo Z (2021) Elastin Structure, Synthesis, Regulatory Mechanism and Relationship With Cardiovascular Diseases. Front. Cell Dev. Biol. 9:596702. doi: 10.3389/fcell.2021.596702
Received: 20 August 2020; Accepted: 29 September 2021;
Published: 30 November 2021.
Edited by:
Maurizio Onisto, University of Padua, ItalyReviewed by:
Laurent Martiny, Université de Reims Champagne-Ardenne, FranceCopyright © 2021 Wang, Meng and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhikun Guo, Z3prQHh4bXUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.