
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 22 March 2021
Sec. Molecular and Cellular Pathology
Volume 9 - 2021 | https://doi.org/10.3389/fcell.2021.552549
This article is part of the Research TopicIntracellular Mechanisms of α-Synuclein ProcessingView all 18 articles
 Michele Perni1†
Michele Perni1† Annemieke van der Goot2†
Annemieke van der Goot2† Ryan Limbocker1,3†
Ryan Limbocker1,3† Tjakko J. van Ham4
Tjakko J. van Ham4 Francesco A. Aprile1‡Catherine K. Xu1
Francesco A. Aprile1‡Catherine K. Xu1 Patrick Flagmeier1Karen Thijssen2
Patrick Flagmeier1Karen Thijssen2 Pietro Sormanni1
Pietro Sormanni1 Giuliana Fusco1Serene W. Chen1
Giuliana Fusco1Serene W. Chen1 Pavan K. Challa1Julius B. Kirkegaard5
Pavan K. Challa1Julius B. Kirkegaard5 Romain F. Laine6
Romain F. Laine6 Kai Yu Ma1,2
Kai Yu Ma1,2 Martin B. D. Müller1,2Tessa Sinnige1
Martin B. D. Müller1,2Tessa Sinnige1 Janet R. Kumita1Samuel I. A. Cohen1
Janet R. Kumita1Samuel I. A. Cohen1 Renée Seinstra2
Renée Seinstra2 Gabriele S. Kaminski Schierle7Clemens F. Kaminski7Denise Barbut8
Gabriele S. Kaminski Schierle7Clemens F. Kaminski7Denise Barbut8 Alfonso De Simone9
Alfonso De Simone9 Tuomas P. J. Knowles1
Tuomas P. J. Knowles1 Michael Zasloff8
Michael Zasloff8 Ellen A. A. Nollen2*
Ellen A. A. Nollen2* Michele Vendruscolo1*Christopher M. Dobson1§
Michele Vendruscolo1*Christopher M. Dobson1§The aggregation of α-synuclein is a hallmark of Parkinson's disease (PD) and a variety of related neurological disorders. A number of mutations in this protein, including A30P and A53T, are associated with familial forms of the disease. Patients carrying the A30P mutation typically exhibit a similar age of onset and symptoms as sporadic PD, while those carrying the A53T mutation generally have an earlier age of onset and an accelerated progression. We report two C. elegans models of PD (PDA30P and PDA53T), which express these mutational variants in the muscle cells, and probed their behavior relative to animals expressing the wild-type protein (PDWT). PDA30P worms showed a reduced speed of movement and an increased paralysis rate, control worms, but no change in the frequency of body bends. By contrast, in PDA53T worms both speed and frequency of body bends were significantly decreased, and paralysis rate was increased. α-Synuclein was also observed to be less well localized into aggregates in PDA30P worms compared to PDA53T and PDWT worms, and amyloid-like features were evident later in the life of the animals, despite comparable levels of expression of α-synuclein. Furthermore, squalamine, a natural product currently in clinical trials for treating symptomatic aspects of PD, was found to reduce significantly the aggregation of α-synuclein and its associated toxicity in PDA53T and PDWT worms, but had less marked effects in PDA30P. In addition, using an antibody that targets the N-terminal region of α-synuclein, we observed a suppression of toxicity in PDA30P, PDA53T and PDWT worms. These results illustrate the use of these two C. elegans models in fundamental and applied PD research.
α-Synuclein (α-syn) is an intrinsically disordered protein expressed at high levels in the human brain, which in Parkinson's disease (PD) and related disorders aggregates to form Lewy bodies (Gómez Tortosa et al., 1998; Spillantini et al., 1998; Dawson and Dawson, 2003; Chiti and Dobson, 2006, 2017; Knowles et al., 2014; Dettmer et al., 2016). Because the aberrant assembly of α-syn is a common feature in the development of these diseases (Chiti and Dobson, 2006), intense efforts have been devoted toward understanding and inhibiting this phenomenon (Lee and Trojanowski, 2006; Tóth et al., 2014). Growing evidence shows that the formation of α-syn aggregates may be induced by aberrant protein-protein or protein-membrane interactions (Auluck et al., 2010; Galvagnion et al., 2015; Dettmer et al., 2016), by malfunctions of molecular chaperones (Witt, 2013; Cox et al., 2014), and by the effects of post-translational modifications (Fujiwara et al., 2002; Hasegawa et al., 2002; Saito et al., 2003; Bendor et al., 2013) and familial mutations in the α-syn gene (Li et al., 2001; Zarranz et al., 2004; Emmer et al., 2011; Sacino et al., 2013). The pathological phenotype of non-heritable idiopathic PD has been shown to be close to that associated with familial PD. However, familial forms of PD, which account for a 10–15% of all PD cases, can have a different age of onset, severity of the disease, and resistance to treatments (Kasten and Klein, 2013).
Among the disease-associated mutations (Li et al., 2001; Zarranz et al., 2004; Emmer et al., 2011; Sacino et al., 2013), the amino acid substitutions A30P and A53T (Li et al., 2001) have been shown to be linked with familial PD (Thomas and Beal, 2007). It has been observed that patients carrying the A30P mutation typically exhibit a similar age of onset and symptoms as sporadic PD, while those carrying the A53T mutation have an earlier age of onset and an accelerated progression of the disease (Polymeropoulos et al., 1997; Krüger et al., 2001; Schiesling et al., 2008). Biophysical studies have shown that these mutations significantly affect the in vitro mechanism of aggregation of α-syn (Flagmeier et al., 2016), and in particular, A53T α-syn was shown to aggregate more rapidly than the A30P or wild-type α-syn (Narhi et al., 1999; Li et al., 2001, 2002). Less agreement, however, exists as to whether the A30P variant aggregates more rapidly (Narhi et al., 1999; Li et al., 2001), at a similar rate (Lemkau et al., 2012) or more slowly (Conway et al., 2000), than the wild-type protein. Recently, we utilized a three-pronged strategy to characterize the influence of these mutations on the mechanism of the aggregation of α-syn in vitro (Flagmeier et al., 2016) and found that the rates of fibril amplification, but not of lipid-induced nucleation, were slightly enhanced in the case of the A30P variant, and were markedly increased in the case of the A53T mutant compared with the wild-type protein (Flagmeier et al., 2016). The importance of studying these mutational variants in animal models has been investigated using a variety of different animal models such as mice, fish or flies (Dehay et al., 2015; Jagmag et al., 2016; Visanji et al., 2016). In several transgenic mice lines, overexpressing human wild-type, A53T, or A30P α-synuclein showed high correlation with transgene expression, in combination with toxic gain of function mechanism for α-synuclein pathogenesis (Visanji et al., 2016). Overexpression of these genes can indeed lead to neurodegeneration, loss of striatal dopamine, and locomotors dysfunction (Dehay et al., 2015). Nevertheless, invertebrates such as Drosophila have also proven powerful very tools to investigate the molecular mechanisms of toxicity associated with α-syn aggregation (Mizuno et al., 2010) due to their 75% homology with human disease genes, rapid generation cycle (10–14 days), short life span and cost-effectiveness to maintain (Mizuno et al., 2010). α-Syn expression in Drosophila can cause dopaminergic neuron loss, Lewy body-like inclusion body formation and locomotor dysfunction (Feany and Bender, 2000) making this invertebrate an attractive model to study PD.
In order to extend these analyses further to another animal model of α-syn aggregation, we have used the nematode worm Caenorhabditis elegans (C. elegans), which is characterized by a simple anatomy, short lifespan, and well-established genetics. For these reasons, this system has become a powerful tool in biomedical research, in particular for genetic (Dillin et al., 2002; Jorgensen and Mango, 2002; Morley et al., 2002; Lee et al., 2003; Nollen et al., 2004; Hamilton et al., 2005; Kim and Sun, 2007; Sarin et al., 2008; Van Ham et al., 2008, 2010; Van der Goot et al., 2012) and drug (Wu et al., 2006; Alavez et al., 2012; Habchi et al., 2016; Perni et al., 2017a, 2018c; Limbocker et al., 2019). In particular, worms expressing the A30P and A53T variants in dopaminergic neurons have been reported in a previous study (Kuwahara et al., 2006) exhibiting accumulation of α-syn in the cell bodies and neurites of dopaminergic neurons, failure in modulation of locomotory rate in response to food, and reduction in neuronal dopamine content. These cell-specific dysfunctions caused by accumulation of α-syn appear relevant to the genetic and compound screenings aiming at the elucidation of pathological cascade and therapeutic strategies for PD. Further models were developed to evaluate the effect of the α-syn overexpression in other cell tissues, such as the muscle cells (Van Ham et al., 2008), and have been widely used for genetic screenings (Van der Goot et al., 2012).
Building on this evidence, we aimed to create a worm transgenic model expressing A30P and A53T variants that could be applied also in high-throughput drug screening studies. To achieve this goal, we chose to overexpress the A30P and A53T variants in the big muscle cells of the worms to affect directly the worms motility. We were then able to directly monitor the impact of the α-syn mutational variants on the worm fitness by using our recently developed high-throughput machine vision system (Perni et al., 2018a,b). We describe here the creation of two C. elegans models of familial PD that express the human α-syn gene carrying the A30P and the A53T mutations, indicated here as PDA30P and PDA53T, respectively. We used for comparison a well-characterized PD worm model, which is based on the overexpression of wild type α-syn tagged with the yellow fluorescent protein (YFP) in the muscle cells of the worms (Van Ham et al., 2008), indicated here as PDWT. In order to facilitate a direct comparison between the variants and the wild-type worms, we also generated a fusion construct of YFP with the A30P and A53T variants. The control healthy worms, which express only YFP in the big muscle cells, are indicated here as the YFP strain. The PDWT reference model, in which the presence of α-syn causes characteristic phenotypic changes (Van Ham et al., 2008), has been used successfully to probe the nature of a range of neurodegenerative conditions and has been employed in high-throughput screens to identify genes and to search for α-syn-related phenotypes (Van Ham et al., 2010; Van der Goot et al., 2012).
The aggregation of α-syn has been shown to be enhanced dramatically by its binding to lipid membranes (Flagmeier et al., 2016), and we recently showed that disrupting this interactions can be achieved with small molecules (Perni et al., 2017b; Limbocker et al., 2020). We reported in particular that the aminosterol squalamine (Moore et al., 1993; Rao et al., 2000; Zasloff et al., 2001, 2011), and related compounds (Perni et al., 2018c) can inhibit the binding of α-syn to membranes, reduce the initiation of its aggregation in vitro, and decrease its toxicity in human neuroblastoma cells and in a C. elegans model of PD (Perni et al., 2017b). Squalamine is currently in clinical trials for the treatment of symptoms associated with PD (ClinicalTrials.gov Identifier: NCT03781791). In order to explore the value of these worm models in the context of familial forms of PD, we used our recently developed high-throughput screening strategy (Perni et al., 2017b, 2018a,b) to investigate the effects of squalamine on the A30P and A53T worm variants developed in this study. We complemented these studies by also administering to our worm models an antibody that binds to a region of α-syn that has previously been identified to play a key structural role in its membrane-associated aggregation (Fusco et al., 2016) and to mitigate the toxicity of α-syn oligomers (Fusco et al., 2017).
We first characterized the behavior of the PDA30P and PDA53T worms in combination with the definition of the aggregation profile of α-syn in these two strains, and compared the results with the corresponding data for PDWT worms. We observed that well-established behavioral characteristics, such as body bends per minute (BPMs) (Van Ham et al., 2008; Gidalevitz et al., 2009; Van der Goot et al., 2012; Habchi et al., 2016, 2017; Aprile et al., 2017; Perni et al., 2017a,b), speed of movement (Morley et al., 2002; Van Ham et al., 2008; Gidalevitz et al., 2009) and paralysis rate (Link, 1995; Lublin and Link, 2013; Perni et al., 2017b), were all affected to different extents by the overexpression of the A30P and A53T variants (Figure 1). In particular, the PDA30P worms showed reduced speed of movement and an increased paralysis rate, but no relevant change in the frequency of body bends (BPMs) (Figure 1A). By contrast, both the frequency of body bends and speed of movement were found to be significantly decreased (P < 0.005) in the PDA53T worms relative to the YFP and PDWT worms. PDA53T worms also showed a higher level of reduction in bend frequency and speed of movement, and higher paralysis rate, when compared with the PDA30P and PDWT worms (Figure 1A). These results suggest that the observed effects of the modified protein are related to different mechanisms of induced dysfunction compared to wild-type protein. Despite the observed phenotypical differences, the levels of expression of α-syn present in the PDWT, PDA30P and PDA53T worms were found to be similar (Supplementary Figure 1).
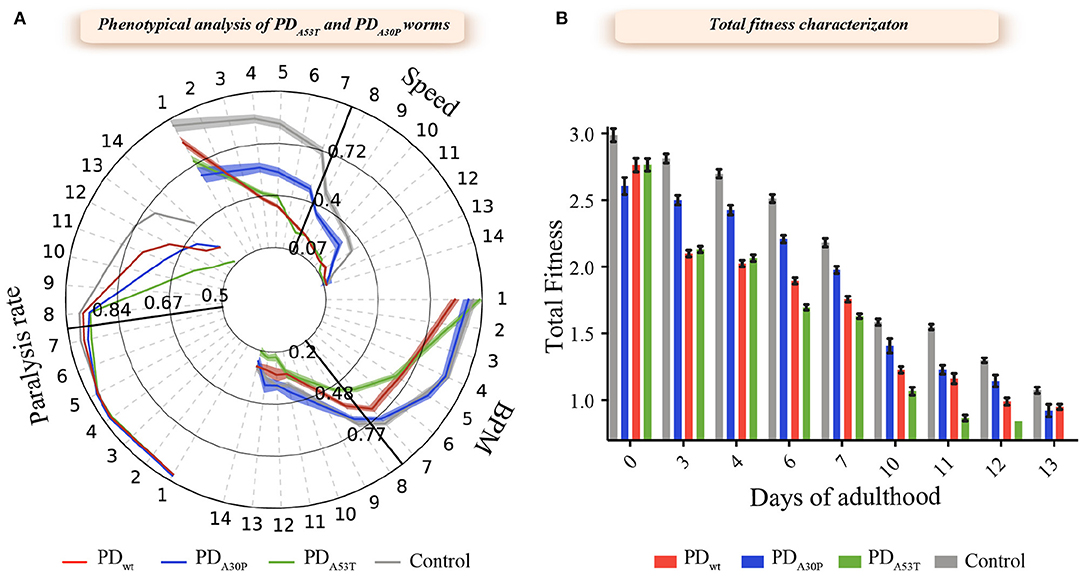
Figure 1. Behavioral characterization of the PDA30P and PDA53T worm strains. (A) Three common readouts of worm fitness were investigated for each strain using an automated worm tracking procedure (Perni et al., 2017b). The results are presented with a behavioral time-course map of PDA30P and PDA53T worms over 14 days of adulthood. The speed of movement, number of body bends per minute (BPMs), and the rate of paralysis were followed during aging; data are normalized with respect to day 1 to illustrate the progressive decline in all readouts. PD worms overexpressing wild-type α-syn:YFP in the body-wall muscle cells (PDWT) were used as a positive control, while worms expressing only YFP were used as negative healthy controls (Control). Shadowed areas represent standard errors of the mean (SEM). (B) The rate of body bends, speed and paralysis rate were combined into a single score of total fitness and evaluated during the duration of the experiment. For each experiment, about 1,000 worms were analyzed and each experiment was carried out in triplicate; one representative experiment of three experiments is shown. At each time point, the mutant worms exhibited lower fitness (p < 0.0005) when compared to healthy worms; error bars represent the standard error of the mean (SEM); the statistical significance was assessed using the 2-way ANOVA method with Dunnett's multiple comparison test.
To assess the influence of the amino acid substitutions on the behavior of the different worm strains, we first calculated the total fitness values, in both cases defined by a sum of the behavioral parameters, and compared these values to these of the PDWT worms (Perni et al., 2017b, 2018b). The total fitness score is calculated as the sum of the frequency of body bend, speed of movement, and paralysis rate, normalized by the value at day 1. In the case of the PDA30P worms, we observed a moderate reduction in the fitness value compared to the control YFP worms (Figure 1). A comparison of PDA53T worms with control YFP worms after day 6 of adulthood, however, demonstrated a significantly increased level of dysfunction that correlates with the higher degree of formation of inclusions in the former model (Figures 2A,B). This dysfunction appeared also more extensive than the one observed in the case of PDWT and YFP control worms. This observation is consistent with the reported effect of the A53T mutation, which is to increase the aggregation of α-syn in vitro. In particular, in these latter experiments we found that the lipid-induced nucleation and fibril amplification steps that result in the formation of an increased number of new aggregates, are accelerated for the A53T variant compared to the wild-type protein (Flagmeier et al., 2016), in accord with the in vivo findings.
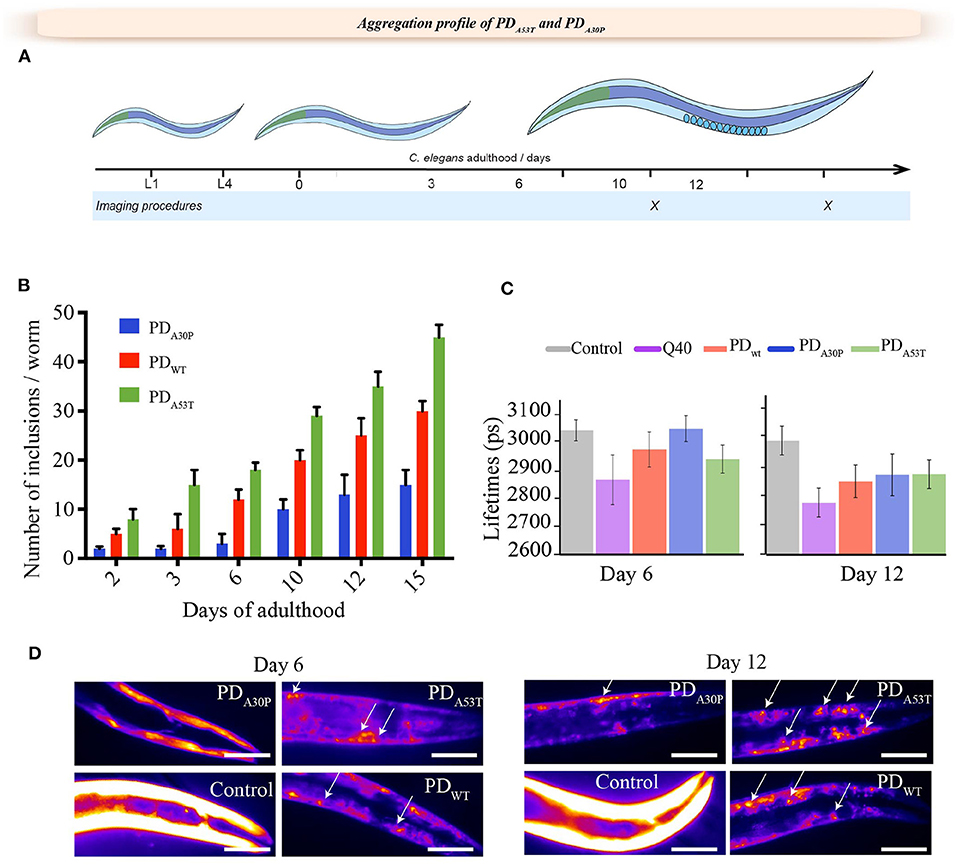
Figure 2. Age-dependent formation of inclusions in PDA30P and PDA53T worms. (A) Schematic description of the lifespan of C. elegans. Days 6 and 12 of adulthood are reported here as representative for the formation of α-syn inclusions in vivo. (B) Quantification of the number of α-syn inclusions in PDA30P and PDA53T worms, compared to PDWT worms (Van Ham et al., 2008). (C) PDA30P and PDA53T amyloid-like aggregation profiles measured using TG-FLIM (Laine et al., 2019), (left) day 6 and (right) day 12. The average fluorescence lifetime (in ps) was measured for PDA30P (blue) and PDA53T (green) worms, and compared to control worms (gray) and PDWT worms (red) (Van Ham et al., 2008), and to Q40 (pink) polyglutamine worms, which have a high propensity to form amyloid-like aggregates (Morley et al., 2002). The FLIM analysis shows no amyloid-like features in the α-syn inclusions in PDA30P worms at day 6 of adulthood, unlike PDA53T, PDWT and Q40 worms. A statistically significant increase in amyloid-like aggregation was observed in all the strains after day 12 of adulthood (insets) (p < 0.05). For each experiment, 25 worms were analyzed. (D) Representative images showing the inclusion profile in PDA30P and PDA53T worms and compared to PDWT worms and YFP controls at day 6 and 12 (Van Ham et al., 2008).
Protein aggregation can be studied in vitro by means of a range of well-established biophysical techniques (Arosio et al., 2014; Buell et al., 2014; Galvagnion et al., 2015; Flagmeier et al., 2016; Habchi et al., 2016). As direct observations of the nature and kinetics of the aggregation processes taking place in vivo provide opportunities to extend such findings to physiological conditions (Morley et al., 2002; Nollen et al., 2004; Van Ham et al., 2008; Van der Goot et al., 2012; Habchi et al., 2016), we investigated here the development of aggregates in both PDA30P and PDA53T worms, and compared their aggregation profiles with those of PDWT worms (Van Ham et al., 2008) (Figure 2). We observed that until day 6 of adulthood, inclusions in PDWT and PDA30P worms showed a diffused fluorescence intensity pattern similar to that of the control worms expressing only YFP, indicating that they are largely unstructured and diffuse (Figures 2B,D). After that, we could observe the presence of more well-defined aggregates (Figures 2B,D).
We further analyzed the nature of the aggregates using fluorescence lifetime imaging (FLIM), a technique that enables the specific kinetics of protein aggregation to be followed in vivo (Schierle et al., 2011; Laine et al., 2019). This methodology is based on a fluorophore covalently linked to the amyloidogenic protein of interest (Schierle et al., 2011). We have previously shown that a reduction in the fluorescence lifetime of a reporter fluorophore, such as YFP, correlates with the degree of aggregation of the protein to which it is attached, and that this effect provides a quantitative measure of the degree of protein aggregation in vitro, in live cells and in C. elegans (Schierle et al., 2011). This decrease in lifetime is thought to be associated with the fluorescence energy transfer to electronic states associated with the amyloid structure (Schierle et al., 2011). Taken together, these results indicate that the process of aggregation in vitro and the ability of A53T to induce dysfunction in nematode worms from day 6 of adulthood is significantly faster than that of A30P and that of the wild-type protein, as also observed in vitro (Flagmeier et al., 2016).
The aminosterol squalamine (Rao et al., 2000; Zasloff et al., 2001, 2011) was shown to be an effective inhibitor of in vitro (Perni et al., 2017b), and to suppress α-syn-mediated toxicity in neuronal cells and in a C. elegans model of PD (Perni et al., 2017b). The primary mode of action of this compound is the displacement of monomeric and oligomeric forms of α-syn from lipid membranes both in lipid vesicles and in cell membranes.
In order to investigate the use of the PDA30P and PDA53T worm models and obtain insights into the nature of familial forms of PD, we administered squalamine to both PDA30P and PDA53T worms by evaluating its effect on the rate and degree of aggregation of the α-syn variants within the worms. We observed that squalamine had a smaller effect on the behavior of the PDA30P compared to PDWT worms, but increased substantially the rate of body bends, speed of movement and the paralysis rate of the PDA53T worms, as found with PDWT worms, and effectively restored their behavior to that of the control YFP worms (Figure 3A). These results are illustrated further by comparison of the values of the total fitness in each case (Figure 3B).
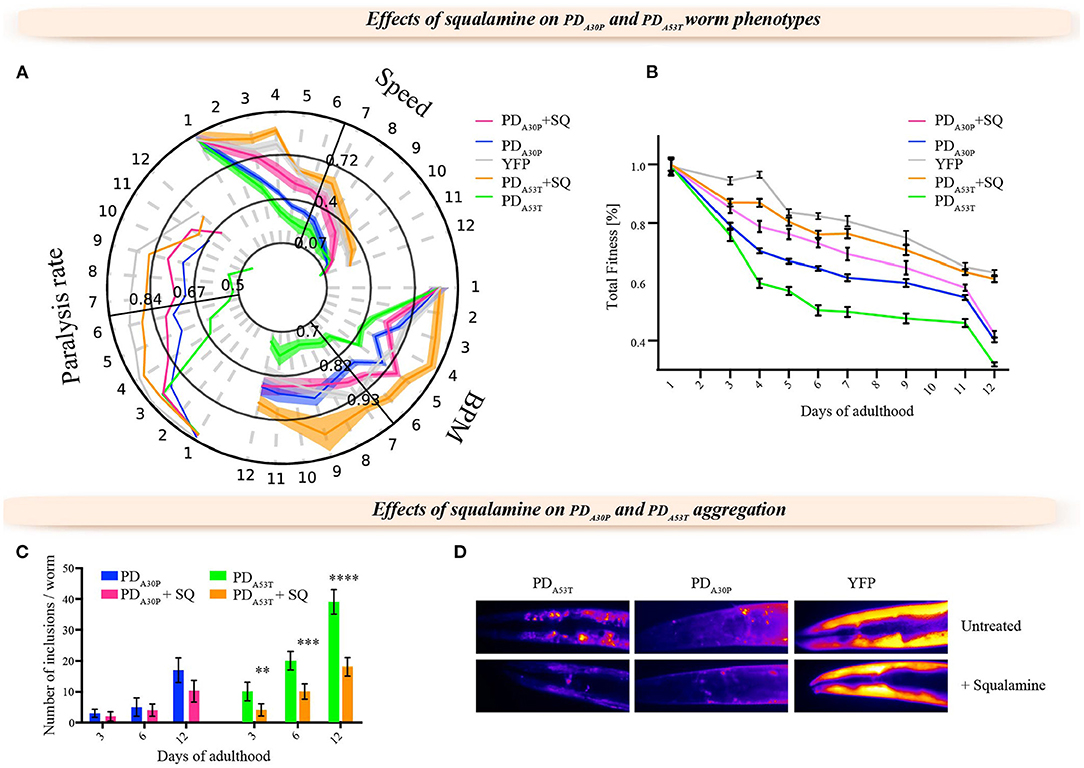
Figure 3. Effects of squalamine on the formation of α-syn inclusions and the fitness of PDA30P and PDA53T worms. (A) Behavioral time-course map reporting thee rate of body bends, swimming speed and paralysis rate for PDA30P and PDA53T worms exposed to 10 μM squalamine. (B) Calculation of the total fitness corresponding to the three readouts in (A) (Perni et al., 2017b, 2018b). The effects of squalamine on control worms were negligible (Perni et al., 2017b), and only a small protective effect could be observed in PDA30P, while strong protective effects were observed in PDA53T worms consistently with the effects on the PDWT worms (Perni et al., 2017b). Each experiment was carried out in triplicate, and about 1,000 worms were analyzed in each replicate; one representative experiment of the three is shown. (C) Quantification of the number of inclusions in PDA30P and PDA53T worms with or without squalamine; squalamine reduced significantly the number of inclusions in PDA53T worms **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001. (D) Representative images showing a substantial decrease in the number of α-syn inclusions in the presence of squalamine in PDA53T worms. In order to show the inclusions more clearly, the image focus on only the heads of the worms, although the quantification was carried out using the whole worms. This result is consistent with previous observations for PDWT worms (Perni et al., 2017b). In the YFP worms, the expression pattern is not significantly affected while in PDA30P worms the number of inclusions appears mildly reduced. All measurements were carried out at day 12 of adulthood.
We next investigated the effects of squalamine on the formation of aggregates of α-syn in the PDA53T and PDA30P worms (Figures 3C,D). In the presence of squalamine, the number of α-syn inclusions was reduced in the PDA53T worms, but less so in the PDA30P worms, despite the fact that the levels of α-syn expression in PDA30P and PDA53T worms in the presence of squalamine were similar to that of the PDWT animals (Supplementary Figure 1). We also found that squalamine did not significantly affect the lipid-induced aggregation process of the A30P variant in vitro (Supplementary Figure 2), while it did so for the wild-type protein (Perni et al., 2017b). As the A30P variant has been shown to have reduced binding to cell membranes (Jo et al., 2002), the observation of the reduced effects of squalamine in the A30P variant compared to the A53T and wild-type variants further supports the conclusion that the mechanism of action of this small molecule in vivo is mediated by its competitive binding to cell membranes (Perni et al., 2017b).
In order to probe further the behavior of the various α-syn forms in C. elegans, we administered to the PDA30P worms a previously described antibody (Fusco et al., 2016) that binds to the N-terminal region of the α-syn sequence (residues 1–25). This region was found to play a key structural role in the membrane-associated aggregation of α-syn (Fusco et al., 2016) and in the toxicity of α-syn oligomers (Fusco et al., 2017). When the antibody was incubated with PDA30P, a reduction in the toxicity that resulted from the overexpression of α-syn was observed, to an extent similar to that observed in the case of PDWT (Perni et al., 2017b). The effect of the antibody on PDA30P appeared to be slightly greater than that induced by squalamine (Figure 4), which could be a result of the more specific action of the antibody in suppressing the toxicity associated with overexpression of α-syn molecules in the worms, particularly showing a direct interaction with the exposed N-terminal region of α-syn in the oligomeric species. By contrast, the antibody was observed to exert effects similar to those resulting from the addition of squalamine on the toxicity observed in PDA53T worms, as shown by an increase in the rate of body bends and in the speed of movement, and by a decrease in the paralysis rates (Figure 4).
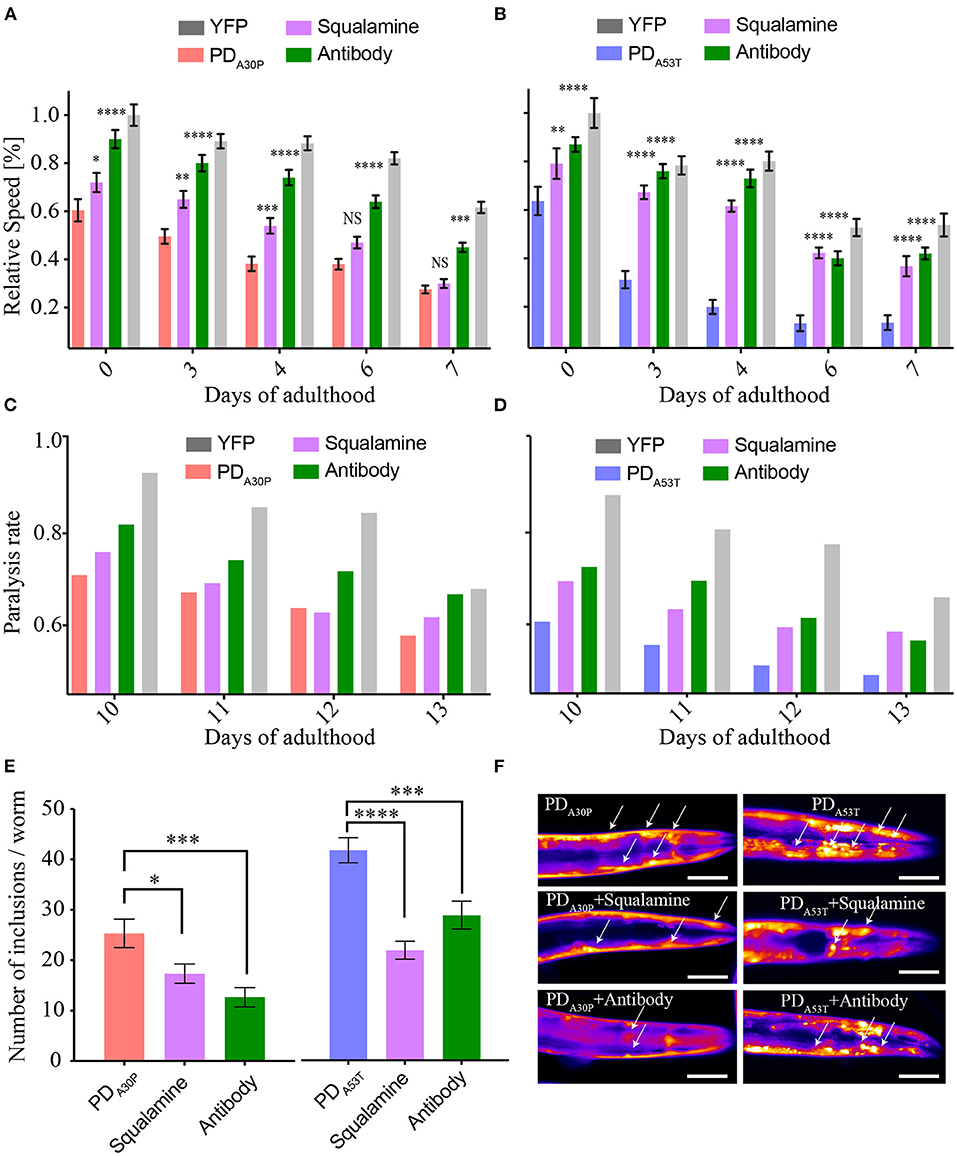
Figure 4. Comparison of the effects of the antibody and squalamine administration to PDA30P and PDA53T worms. (A,B) At a concentration of 10 μM, the antibody rescues the motility dysfunction (relative speed) induced by the over-expression of A30P, with squalamine having a slightly smaller effect. The worm motility is also rescued in the A53T worms by the antibody and squalamine to similar extents. Errors represent the standard error on the mean (SEM) *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 and ****P ≤ 0.0001. (C,D) Paralysis rate, reported as the fraction of worms that are mobile, corresponding to the time points shown in (A,B); the variability between biological replicates is in the 2–9% range, and the variability between technical replicates is in the 1–4% range. (E,F) Number of inclusions at day 12 of adulthood *P ≤ 0.05, ***P ≤ 0.001 and ****P ≤ 0.0001. The scale bar indicates 80 μm.
We have created and characterized two C. elegans strains, PDA30P and PDA53T, expressing the A30P and A53T mutational variants, respectively, which are associated with familial forms of PD. We have then demonstrated that these two mutational variants affect the worms in different ways and to different extents. The expression of the A30P species was shown to reduce specifically certain aspects of worm behavior, notably speed of swimming, compared with the wild-type protein. Overexpression of the A53T mutation, however, had a more dramatic effect than that found for the wild-type protein, and the worms expressing this variant behaved in a dysfunctional manner at a significantly younger age than did those expressing the A30P or the wild-type forms. Overall, the expression of the A53T variant resulted in a more significant decrease in the bends, and speed of movement compared with the A30P and wild-type proteins. We note that worms expressing the A30P and A53T variants in dopaminergic neurons exhibited a less severe phenotype (Kuwahara et al., 2006), suggesting that the overexpression of α-syn in muscle cells may lead to increased toxicity through additional mechanisms with respect to those involved in PD.
These findings are broadly consistent with the measurement and analysis of the kinetics of aggregation in vitro. In particular, the observation that the expression of the A30P variant alters the phenotype of the worms moderately compared to the dysfunction associated with the expression of the wild-type protein, is in agreement with the findings that the overall rate of aggregation is only mildly affected for the A30P variants when compared to wild-type in vitro (Conway et al., 2000; Flagmeier et al., 2016). Initiation of the in vitro aggregation process, however, has been found to be faster for the A53T variant than for A30P or wild-type protein (Flagmeier et al., 2016), an observation consistent with more rapid decline of the fitness of the PDA53T related to the PDA30P or PDWT worms. Taken together, these results are particularly interesting in the context of the clinical manifestations of the A30P and A53T mutations, where patients with the A30P mutation appear generally to exhibit similar age of onset and rate of disease progression to those suffering of sporadic PD, while patients carrying the A53T mutation generally exhibit an earlier age of onset and have a more rapid rate of progression of the disease (Polymeropoulos et al., 1997; Krüger et al., 2001; Schiesling et al., 2008).
The degree of dysfunction of the C. elegans model expressing human α-syn has recently been shown to be reduced substantially by the administration of squalamine (Perni et al., 2017b), a naturally active aminosterol, and we have shown here that this small molecule decreases the amount of fitness reduction and aggregation to a lower extent in the PDA30P than in the PDWT worms, but has a more substantial effect in the PDA53T worms, which is similar to that observed in PDWT worms. These results are consistent with the finding that squalamine reduces the membrane-associated initiation of the aggregation of α-syn by displacing it from the surfaces of lipid bilayers (Perni et al., 2017b). In addition, we observed that an antibody targeting the N-terminal region of the protein, which plays a key role in both the aggregation process and the induction of cellular toxicity by α-syn oligomers, was also protective in PDWT and PDA53T worms, while less so in PDA30P worms. Overall, this analysis provides support to the strategy of reducing the binding of α-syn to lipid membranes as a potential therapeutics strategy for PD.
Extended experimental procedures are described in SI Materials and Methods. In vitro kinetic experiments and purifications of wild type and mutant α-syn were carried out as previously indicated (Flagmeier et al., 2016). TG-FLIM imaging was carried out on a home-built microscopy platform described elsewhere (Schierle et al., 2011; Laine et al., 2019). In vivo experiments were carried out by using a well-studied C. elegans model of PD (Link, 1995) and custom made A53T and A30P strains. Microinjection was used to create new transgenic strains and standard conditions were used for the propagation of C. elegans (Brenner, 1974). Squalamine was synthesized as previously described (Zhang et al., 1998) and automated behavioral assays were carried out as previously described (Perni et al., 2017b, 2018a,b). Measurements on inclusions in vivo were performed using ImageJ software as previously described (Van der Goot et al., 2012; Perni et al., 2017b). Western blot analysis was carried out as previously described (Limbocker et al., 2019). The transduction of the antibody was carried out as previously reported (Aprile et al., 2017; Perni et al., 2017a).
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethical review and approval was not required for the animal study because C. elegans does not require ethical review and approval.
AG, TH, KT created the C. elegans strains. MP, RLi, and MM characterized the C. elegans strains. MP and RLi carried out the measurements of the measurements of the effects of squalamine. PF carried out the in vitro experiments. RLa and MP carried out the FLIM experiments. MP carried out the antibodies testing. RLi and FA carried out the western blotting and subsequent analysis. GF, SCh, and AD were involved in the antibody design. MP, FA, PF, RLi, MM, EN, MV, and CD designed the study. All the authors were involved and contributed in the writing and revision of the manuscript.
CK acknowledged funding from the UK Engineering and Physical Sciences Research Council, EPSRC (grants EP/L015889/1 and EP/H018301/1), the Wellcome Trust (grants 3-3249/Z/16/Z and 089703/Z/09/Z) and the UK Medical Research Council, MRC (grants MR/K015850/1 and MR/K02292X/1), MedImmune, and Infinitus (China) Ltd. This project has received funding from the European Union's Horizon 2020 research and innovation programme under Grant Agreement No. 722380, and from the Cambridge Centre for Misfolding Diseases. FA thanks UK Research and Innovation (Future Leaders Fellowship MR/S033947/1) and the Alzheimer's Society, United Kingdom (317, 511) for support. EAAN acknowledged support from a European Research Council (ERC) starting grant (281622 PDControl) and a RIDE grant from NWO (948-00-017).
The views expressed herein are those of the authors and do not reflect the position of the United States Military Academy, the Department of the Army, or the Department of Defense.
MZ and DB are inventors in a patent for the use of squalamine in the treatment of PD. CD, MV, SCo, and TK are co-founders, and MP is an employee of Wren Therapeutics, which is independently pursuing inhibitors of protein misfolding and aggregation.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.552549/full#supplementary-material
Alavez, S., Vantipalli, M. C., Zucker, D. J. S., Klang, I. M., and Lithgow, G. J. (2012). Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472, 226–229. doi: 10.1038/nature09873
Aprile, F. A., Sormanni, P., Perni, M., Arosio, P., Linse, S., Knowles, T. P. J., et al. (2017). Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Sci. Adv. 3:e1700488. doi: 10.1126/sciadv.1700488
Arosio, P., Vendruscolo, M., Dobson, C. M., and Knowles, T. P. J. (2014). Chemical kinetics for drug discovery to combat protein aggregation diseases. Trends Pharmacol. Sci. 35, 127–135. doi: 10.1016/j.tips.2013.12.005
Auluck, P. K., Caraveo, G., and Lindquist, S. (2010). α-Synuclein: membrane interactions and toxicity in Parkinson's disease. Annu. Rev. Cell Dev. Biol. 26, 211–233. doi: 10.1146/annurev.cellbio.042308.113313
Bendor, J. T., Logan, T. P., and Edwards, R. H. (2013). The function of α-synuclein. Neuron 79, 1044–1066. doi: 10.1016/j.neuron.2013.09.004
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. doi: 10.1093/genetics/77.1.71
Buell, A. K., Galvagnion, C., Gaspar, R., Sparr, E., Vendruscolo, M., Knowles, T. P. J., et al. (2014). Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A. 111, 7671–7676. doi: 10.1073/pnas.1315346111
Chiti, F., and Dobson, C. M. (2006). Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366. doi: 10.1146/annurev.biochem.75.101304.123901
Chiti, F., and Dobson, C. M. (2017). Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 86, 27–68. doi: 10.1146/annurev-biochem-061516-045115
Conway, K. A., Lee, S. J., and Rochet, J. C. (2000). Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U. S. A. 97, 571–576. doi: 10.1073/pnas.97.2.571
Cox, D., Carver, J. A., and Ecroyd, H. (2014). Preventing alpha-synuclein aggregation: the role of the small heat-shock molecular chaperone proteins. Biochim. Biophys. Acta 1842, 1830–1843. doi: 10.1016/j.bbadis.2014.06.024
Dawson, T. M., and Dawson, V. L. (2003). Molecular pathways of neurodegeneration in Parkinson's disease. Science 302, 819–822. doi: 10.1126/science.1087753
Dehay, B., Vila, M., Bezard, E., Brundin, P., and Kordower, J. H. (2015). Alpha-synuclein propagation: new insights from animal models. Mov. Disord. 31, 161–168. doi: 10.1002/mds.26370
Dettmer, U., Selkoe, D., and Bartels, T. (2016). New insights into cellular α-synuclein homeostasis in health and disease. Curr. Opin. Neurobiol. 36, 15–22. doi: 10.1016/j.conb.2015.07.007
Dillin, A., Hsu, A.-L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. doi: 10.1126/science.1077780
Emmer, K. L., Waxman, E. A., Covy, J. P., and Giasson, B. I. (2011). E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J. Biol. Chem. 286, 35104–35118. doi: 10.1074/jbc.M111.247965
Feany, M. B., and Bender, W. W. (2000). A Drosophila model of Parkinson's disease. Nature 404, 394–398. doi: 10.1038/35006074
Flagmeier, P., Meisl, G., Vendruscolo, M., Knowles, T. P. J., Dobson, C. M., Buell, A. K., et al. (2016). Mutations associated with familial Parkinson's disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A. 113, 10328–10333. doi: 10.1073/pnas.1604645113
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell. Biol. 4, 160–164. doi: 10.1038/ncb748
Fusco, G., Chen, S. W., Williamson, P. T. F., Cascella, R., Perni, M., Jarvis, J. A., et al. (2017). Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 358, 1440–1443. doi: 10.1126/science.aan6160
Fusco, G., De Simone, A., Arosio, P., Vendruscolo, M., Veglia, G., and Dobson, C. M. (2016). Structural ensembles of membrane-bound α-synuclein reveal the molecular determinants of synaptic vesicle affinity. Sci. Rep. 6:27125. doi: 10.1038/srep27125
Galvagnion, C., Buell, A. K., Meisl, G., Michaels, T. C. T., Vendruscolo, M., Knowles, T. P. J., et al. (2015). Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234. doi: 10.1038/nchembio.1750
Gidalevitz, T., Krupinski, T., Garcia, S., and Morimoto, R. I. (2009). Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 5:e1000399. doi: 10.1371/journal.pgen.1000399
Gómez Tortosa, E., Ingraham, A. O., Irizarry, M. C., and Hyman, B. T. (1998). Dementia with Lewy bodies. J. Am. Geriatr. Soc. 46, 1449–1458. doi: 10.1111/j.1532-5415.1998.tb06016.x
Habchi, J., Arosio, P., Perni, M., Costa, A. R., Yagi-Utsumi, M., Joshi, P., et al. (2016). An anticancer drug suppresses the primary nucleation reaction that initiates the production of the toxic A 42 aggregates linked with Alzheimers disease. Sci. Adv. 2:e1501244. doi: 10.1126/sciadv.1501244
Habchi, J., Chia, S., Limbocker, R., Mannini, B., Ahn, M., Perni, M., et al. (2017). Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 114, E200–E208. doi: 10.1073/pnas.1615613114
Hamilton, B., Dong, Y., Shindo, M., Liu, W., Odell, I., Ruvkun, G., et al. (2005). A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 19, 1544–1555. doi: 10.1101/gad.1308205
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M. Y., et al. (2002). Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 277, 49071–49076. doi: 10.1074/jbc.M208046200
Jagmag, S. A., Tripathi, N., Shukla, S. D., Maiti, S., and Khurana, S. (2016). Evaluation of models of Parkinson's disease. Front. Neurosci. 9:503. doi: 10.3389/fnins.2015.00503
Jo, E., Fuller, N., Rand, R. P., St George-Hyslop, P., and Fraser, P. E. (2002). Defective membrane interactions of familial Parkinson's disease mutant A30P alpha-synuclein. J. Mol. Biol. 315, 799–807. doi: 10.1006/jmbi.2001.5269
Jorgensen, E. M., and Mango, S. E. (2002). The art and design of genetic screens: caenorhabditis elegans. Nat. Rev. Genet. 3, 356–369. doi: 10.1038/nrg794
Kasten, M., and Klein, C. (2013). The many faces of alpha-synuclein mutations. Mov. Disord. 28, 697–701. doi: 10.1002/mds.25499
Kim, Y., and Sun, H. (2007). Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell 6, 489–503. doi: 10.1111/j.1474-9726.2007.00302.x
Knowles, T. P. J., Vendruscolo, M., and Dobson, C. M. (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396. doi: 10.1038/nrm3810
Krüger, R., Kuhn, W., Leenders, K. L., Sprengelmeyer, R., Müller, T., Woitalla, D., et al. (2001). Familial parkinsonism with synuclein pathology: clinical and PET studies of A30P mutation carriers. Neurology 56, 1355–1362. doi: 10.1212/WNL.56.10.1355
Kuwahara, T., Koyama, A., Gengyo-Ando, K., Masuda, M., Kowa, H., Tsunoda, M., et al. (2006). Familial Parkinson mutant α-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J. Biol. Chem. 281, 334–340. doi: 10.1074/jbc.M504860200
Laine, R. F., Sinnige, T., Ma, K. Y., Haack, A. J., Poudel, C., Gaida, P., et al. (2019). Fast fluorescence lifetime imaging reveals the aggregation processes of α-synuclein and polyglutamine in aging Caenorhabditis elegans. ACS Chem. Biol. 14, 1628–1636. doi: 10.1021/acschembio.9b00354
Lee, S. S., Kennedy, S., Tolonen, A. C., and Ruvkun, G. (2003). DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644–647. doi: 10.1126/science.1083614
Lee, V. M. Y., and Trojanowski, J. Q. (2006). Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52, 33–38. doi: 10.1016/j.neuron.2006.09.026
Lemkau, L. R., Comellas, G., Kloepper, K. D., Woods, W. S., George, J. M., and Rienstra, C. M. (2012). Mutant protein A30P α-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J. Biol. Chem. 287, 11526–11532. doi: 10.1074/jbc.M111.306902
Li, J., Uversky, V. N., and Fink, A. L. (2001). Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 40, 11604–11613. doi: 10.1021/bi010616g
Li, J., Uversky, V. N., and Fink, A. L. (2002). Conformational behavior of human a-synuclein is modulated by familial Parkinson's disease point mutations A30P and A53T. Neurotoxicology 23, 553–567. doi: 10.1016/S0161-813X(02)00066-9
Limbocker, R., Chia, S., Ruggeri, F. S., Perni, M., Cascella, R., Heller, G. T., et al. (2019). Trodusquemine enhances Aβ42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 10:225. doi: 10.1038/s41467-018-07699-5
Limbocker, R., Mannini, B., Ruggeri, F. S., Cascella, R., Xu, C. K., Perni, M., et al. (2020). Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 3:435. doi: 10.1038/s42003-020-01140-8
Link, C. D. (1995). Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 92, 9368–9372. doi: 10.1073/pnas.92.20.9368
Lublin, A. L., and Link, C. D. (2013). Alzheimer's disease drug discovery: in vivo screening using Caenorhabditis elegans as a model for β-amyloid peptide-induced toxicity. Drug Discov. Today Technol. 10, e115–e119. doi: 10.1016/j.ddtec.2012.02.002
Mizuno, H., Fujikake, N., Wada, K., and Nagai, Y. (2010). α-Synuclein transgenic Drosophila as a model of Parkinson's disease and related synucleinopathies. Parkinsons Dis. 2011:212706. doi: 10.4061/2011/212706
Moore, K. S., Wehrli, S., Roder, H., Rogers, M., Forrest, J. N., McCrimmon, D., et al. (1993). Squalamine: an aminosterol antibiotic from the shark. Proc. Natl. Acad. Sci. U. S. A. 90, 1354–1358. doi: 10.1073/pnas.90.4.1354
Morley, J. F., Brignull, H. R., Weyers, J. J., and Morimoto, R. I. (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 99, 10417–10422. doi: 10.1073/pnas.152161099
Narhi, L., Wood, S. J., Steavenson, S., Jiang, Y., Wu, G. M., Anafi, D., et al. (1999). Both familial Parkinson's disease mutations accelerate -synuclein aggregation. J. Biol. Chem. 274, 9843–9846. doi: 10.1074/jbc.274.14.9843
Nollen, E. A. A., Garcia, S. M., van Haaften, G., Kim, S., Chavez, A., Morimoto, R. I., et al. (2004). Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U. S. A. 101, 6403–6408. doi: 10.1073/pnas.0307697101
Perni, M., Aprile, F. A., Casford, S., Mannini, B., Sormanni, P., Dobson, C. M., et al. (2017a). Delivery of native proteins into C. elegans using a transduction protocol based on lipid vesicles. Sci. Rep. 7:7380. doi: 10.1038/s41598-017-13755-9
Perni, M., Casford, S., Aprile, F., Nollen, E. A., Knowles, T., Vendruscolo, M., et al. (2018a). Automated behavioral analysis of large C. elegans populations using a wide field-of-view tracking platform. J. Vis. Exp. e58643. doi: 10.3791/58643
Perni, M., Challa, P. K., Kirkegaard, J. B., Limbocker, R., Koopman, M., Hardenberg, M. C., et al. (2018b). Massively parallel C. elegans tracking provides multi-dimensional fingerprints for phenotypic discovery. J. Neurosci. Methods 306, 57–67. doi: 10.1016/j.jneumeth.2018.02.005
Perni, M., Flagmeier, P., Limbocker, R., Cascella, R., Aprile, F. A., Galvagnion, C., et al. (2018c). Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem. Biol. 13:2308–2319. doi: 10.1021/acschembio.8b00466
Perni, M., Galvagnion, C., Maltsev, A., Meisl, G., Müller, M. B. D., Challa, P. K., et al. (2017b). A natural product inhibits the initiation of α-synuclein aggregation & suppresses its toxicity. Proc. Natl. Acad. Sci. U.S.A. 114, E1009–E1017. doi: 10.1073/pnas.1610586114
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-synuclein gene identified in families with Parkinson's disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Rao, M. N., Shinnar, A. E., Noecker, L. A., Chao, T. L., Feibush, B., Snyder, B., et al. (2000). Aminosterols from the Dogfish Shark Squalus acanthias. J. Nat. Prod. 63, 631–635. doi: 10.1021/np990514f
Sacino, A. N., Thomas, M. A., Ceballos-Diaz, C., Cruz, P. E., Rosario, A. M., Lewis, J., et al. (2013). Conformational templating of α-synuclein aggregates in neuronal-glial cultures. Mol. Neurodegener. 8:17. doi: 10.1186/1750-1326-8-17
Saito, Y., Kawashima, A., Ruberu, N. N., Fujiwara, H., Koyama, S., Sawabe, M., et al. (2003). Accumulation of phosphorylated alpha-synuclein in aging human brain. J. Neuropathol. Exp. Neurol. 62, 644–654. doi: 10.1093/jnen/62.6.644
Sarin, S., Prabhu, S., O'Meara, M. M., Pe'er, I., and Hobert, O. (2008). Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods 5, 865–867. doi: 10.1038/nmeth.1249
Schierle, G. S. K., Bertoncini, C. W., Chan, F. T. S., Van der Goot, A. T., Schwedler, S., Skepper, J., et al. (2011). A FRET sensor for non-invasive imaging of amyloid formation in vivo. Chemphyschem 12, 673–680. doi: 10.1002/cphc.201000996
Schiesling, C., Kieper, N., Seidel, K., and Krüger, R. (2008). Review: familial Parkinson's disease – genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol. Appl. Neurobiol. 34, 255–271. doi: 10.1111/j.1365-2990.2008.00952.x
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. U. S. A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Thomas, B., and Beal, M. F. (2007). Parkinson's disease. Hum. Mol. Genet. 16, R183–R194. doi: 10.1093/hmg/ddm159
Tóth, G., Gardai, S. J., Zago, W., Bertoncini, C. W., Cremades, N., Roy, S. L., et al. (2014). Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson's disease. PLoS ONE 9, e87133. doi: 10.1371/journal.pone.0087133
Van der Goot, A. T., Zhu, W., Vazquez-Manrique, R. P., Seinstra, R. I., Dettmer, K., Michels, H., et al. (2012). Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. U. S. A. 109, 14912–14917. doi: 10.1073/pnas.1203083109
Van Ham, T. J., Holmberg, M. A., Van der Goot, A. T., Teuling, E., Garcia-Arencibia, M., Kim, H.-E., et al. (2010). Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell 142, 601–612. doi: 10.1016/j.cell.2010.07.020
Van Ham, T. J., Thijssen, K. L., Breitling, R., Hofstra, R. M. W., Plasterk, R. H. A., and Nollen, E. A. A. (2008). C. elegans model identifies genetic modifiers of α-synuclein inclusion formation during aging. PLoS Genet. 4:e1000027. doi: 10.1371/journal.pgen.1000027
Visanji, N. P., Brotchie, J. M., Kalia, L. V., Koprich, J. B., Tandon, A., Watts, J. C., et al. (2016). α-Synuclein-based animal models of Parkinson's disease: challenges and opportunities in a new era. Trends Neurosci. 39, 750–762. doi: 10.1016/j.tins.2016.09.003
Witt, S. N. (2013). Molecular chaperones, α-synuclein, and neurodegeneration. Mol. Neurobiol. 47, 552–560. doi: 10.1007/s12035-012-8325-2
Wu, Y., Wu, Z., Butko, P., Christen, Y., Lambert, M. P., Klein, W. L., et al. (2006). Amyloid- -induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and Ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. 26, 13102–13113. doi: 10.1523/JNEUROSCI.3448-06.2006
Zarranz, J. J., Alegre, J., Gomez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zasloff, M., Adams, A. P., Beckerman, B., Campbell, A., Han, Z., Luijten, E., et al. (2011). Squalamine as a broad-spectrum systemic antiviral agent with therapeutic potential. Proc. Natl. Acad. Sci. U. S. A. 108, 15978–15983. doi: 10.1073/pnas.1108558108
Zasloff, M., Williams, J. I., Chen, Q., Anderson, M., Maeder, T., Holroyd, K., et al. (2001). A spermine-coupled cholesterol metabolite from the shark with potent appetite suppressant and antidiabetic properties. Int. J. Obes. Relat. Metab. Disord. 25, 689–697. doi: 10.1038/sj.ijo.0801599
Keywords: C. elegans, Parkinson's disease, alpha-synuclein, drug discovery, protein aggregation, protein misfolding, neurodegenerative diseases, transgenic model
Citation: Perni M, van der Goot A, Limbocker R, van Ham TJ, Aprile FA, Xu CK, Flagmeier P, Thijssen K, Sormanni P, Fusco G, Chen SW, Challa PK, Kirkegaard JB, Laine RF, Ma KY, Müller MBD, Sinnige T, Kumita JR, Cohen SIA, Seinstra R, Kaminski Schierle GS, Kaminski CF, Barbut D, De Simone A, Knowles TPJ, Zasloff M, Nollen EAA, Vendruscolo M and Dobson CM (2021) Comparative Studies in the A30P and A53T α-Synuclein C. elegans Strains to Investigate the Molecular Origins of Parkinson's Disease. Front. Cell Dev. Biol. 9:552549. doi: 10.3389/fcell.2021.552549
Received: 21 April 2020; Accepted: 16 February 2021;
Published: 22 March 2021.
Edited by:
Franziska Richter, University of Veterinary Medicine Hannover, GermanyReviewed by:
Kenya Nishioka, Juntendo University, JapanCopyright © 2021 Perni, van der Goot, Limbocker, van Ham, Aprile, Xu, Flagmeier, Thijssen, Sormanni, Fusco, Chen, Challa, Kirkegaard, Laine, Ma, Müller, Sinnige, Kumita, Cohen, Seinstra, Kaminski Schierle, Kaminski, Barbut, De Simone, Knowles, Zasloff, Nollen, Vendruscolo and Dobson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen A. A. Nollen, ZS5hLmEubm9sbGVuQHVtY2cubmw=; Michele Vendruscolo, bXYyNDVAY2FtLmFjLnVr
†These authors have contributed equally to this work
‡Present address: Francesco A. Aprile, Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, London, United Kingdom
§Deceased
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.