
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 12 January 2021
Sec. Cellular Biochemistry
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.624216
This article is part of the Research TopicMitochondrial Remodeling and Dynamic Inter-Organellar Contacts in Cardiovascular PhysiopathologyView all 12 articles
 Daniela Ramaccini1,2,3
Daniela Ramaccini1,2,3 Vanessa Montoya-Uribe1
Vanessa Montoya-Uribe1 Femke J. Aan1
Femke J. Aan1 Lorenzo Modesti2,3
Lorenzo Modesti2,3 Yaiza Potes4
Yaiza Potes4 Mariusz R. Wieckowski4
Mariusz R. Wieckowski4 Irena Krga5
Irena Krga5 Marija Glibetić5
Marija Glibetić5 Paolo Pinton2,3,6
Paolo Pinton2,3,6 Carlotta Giorgi2,3
Carlotta Giorgi2,3 Michelle L. Matter1*
Michelle L. Matter1*Cardiac tissue requires a persistent production of energy in order to exert its pumping function. Therefore, the maintenance of this function relies on mitochondria that represent the “powerhouse” of all cardiac activities. Mitochondria being one of the key players for the proper functioning of the mammalian heart suggests continual regulation and organization. Mitochondria adapt to cellular energy demands via fusion-fission events and, as a proof-reading ability, undergo mitophagy in cases of abnormalities. Ca2+ fluxes play a pivotal role in regulating all mitochondrial functions, including ATP production, metabolism, oxidative stress balance and apoptosis. Communication between mitochondria and others organelles, especially the sarcoplasmic reticulum is required for optimal function. Consequently, abnormal mitochondrial activity results in decreased energy production leading to pathological conditions. In this review, we will describe how mitochondrial function or dysfunction impacts cardiac activities and the development of dilated cardiomyopathy.
Mitochondria are highly dynamic organelles, universally recognized as the “powerhouse” of eukaryotic cells, especially in those that require high-energy demand such as cardiomyocytes (Nan et al., 2017). In these cells mitochondria occupy 30% of the total volume of the cell and supply, through oxidative phosphorylation (OXPHOS), approximately 6 kg of adenosine triphosphate (ATP) per day that is required to sustain cardiac function (Cao and Zheng, 2019). In addition to their pivotal role in energy production, mitochondria are the central hub of cellular metabolism providing metabolites for biosynthesis and also producing reactive oxygen species (ROS). Under physiological conditions ROS act as second messengers that are maintained at low concentrations by the scavenging system present in the cell. However, ROS are hyper-produced in many cardiovascular diseases (CVDs), which impairs heart function (Murphy et al., 2016).
It is well established that mitochondrial calcium (Ca2+) fluxes are a key regulator of cardiac function, controlling not only ATP production and mitochondrial metabolism, but also playing a pivotal role in the modulation of muscle contraction (Walsh et al., 2009). In cardiomyocytes mitochondria are well organized and in close proximity to the sarcoplasmatic reticulum (SR), where most cellular Ca2+ is stored (Frederick and Shaw, 2007). Therefore, mitochondria are highly sensitive to Ca2+ oscillations. The release of Ca2+ from SR to mitochondria ensures a balanced activation of SR ATPase and mitochondrial ATP synthesis; all of which contribute to controlling the energy metabolism within a cell (Balaban et al., 2003). Hence, the maintenance of Ca2+ homeostasis is a fundamental requirement for optimal mitochondrial function as mitochondria are a key checkpoint regulating cell survival and cell death.
It is thus not surprising that the maintenance of efficient inter-organelle-communication as well as a conserved “mitochondrial quality control” system (MQC), are fundamental for sustaining mitochondrial bioenergetics demand and metabolic functions (Campos et al., 2016). The term MQC refers to mitochondrial fusion and fission machinery (also called mitochondrial dynamics) and autophagy (called mitophagy when pertaining to mitochondria) (Fan et al., 2020). As we will explain in detail in this review, mitochondrial fusion has the ability to respond to high-energy demand conditions by recovering mitochondria that have been damaged and creating elongated interconnected mitochondrial networks. Fission, however, is the process by which dysfunctional mitochondria are separated and segregated away from healthy ones. These dysfunctional mitochondria may be subsequently either recovered or eliminated through mitophagy (Murphy et al., 2016; Fan et al., 2020; Forte et al., 2020; Oh et al., 2020). These complex processes provide the balance for maintaining proper mitochondrial dynamics through regulation of mitochondrial size, shape and number (Youle and Karbowski, 2005; Piquereau et al., 2013).
An increasing number of studies on cardiac mitochondria have determined that dysfunction in their structure and function contributes to the pathogenesis of CVD including dysrhythmias, ischemia-reperfusion injury (IRI) and cardiomyopathies (CMPs); all of which culminate in end-stage heart failure (HF) (Brown and O’Rourke, 2010; Cadenas et al., 2010; Rosca and Hoppel, 2010; Verdejo et al., 2012).
In this review, we will provide an overview of the main functions of mitochondria within cardiac tissue. Furthermore, we will discuss the involvement of mitochondrial impairment in CVD, focusing our attention on dilated cardiomyopathy (DCM) leading to heart failure. Dilated cardiomyopathy is associated with decreased mitochondrial biogenesis and we will examine DCM subtypes and how mitochondria are dysregulated in these conditions. We will highlight the paucity of targeted treatments for DCM and the necessity for understanding the molecular mechanisms involved in DCM onset and progression. Finally, we will the need for new methods to tease out the complexities of dilated cardiomyopathy, such as the potential use of cardiac organoids to investigate the underlying molecular mechanisms of cardiac function and to develop new targeted therapies for dilated cardiomyopathy.
In heart mitochondria, the primary source of carbons for ATP production relies on fatty acid oxidation (FAO) (Figure 1). Products of beta-oxidation are directed into the tricarboxylic acid cycle (TCA): the starting compound acetyl-CoA enters the cycle and undergoes a series of reactions where electrons are extracted from TCA intermediates in the form of the reducing equivalents NADH and FADH2 and in turn fueling the electron transport chain (ETC) for ATP synthesis (Murphy et al., 2016; Martínez-Reyes and Chandel, 2020). The ETC creates an electrochemical gradient (ΔΨm is −180 mV) along the intermitochondrial membrane (IMM) interface which acts as a driving force for mitochondrial Ca2+ uptake (Giorgi et al., 2012, 2018b; Figure 1). Mitochondria are calcium-buffering organelles in which under resting conditions mitochondrial Ca2+ concentrations are kept low, but after a stimulus Ca2+ is transferred from the SR into the mitochondria that transiently and rapidly takes up large quantities of Ca2+ (Giorgi et al., 2018a,b). Lastly, Ca2+ is extruded from mitochondria by the Na+/ Ca2+ antiporter (NCLX) (Giorgi et al., 2018a; Figure 1).
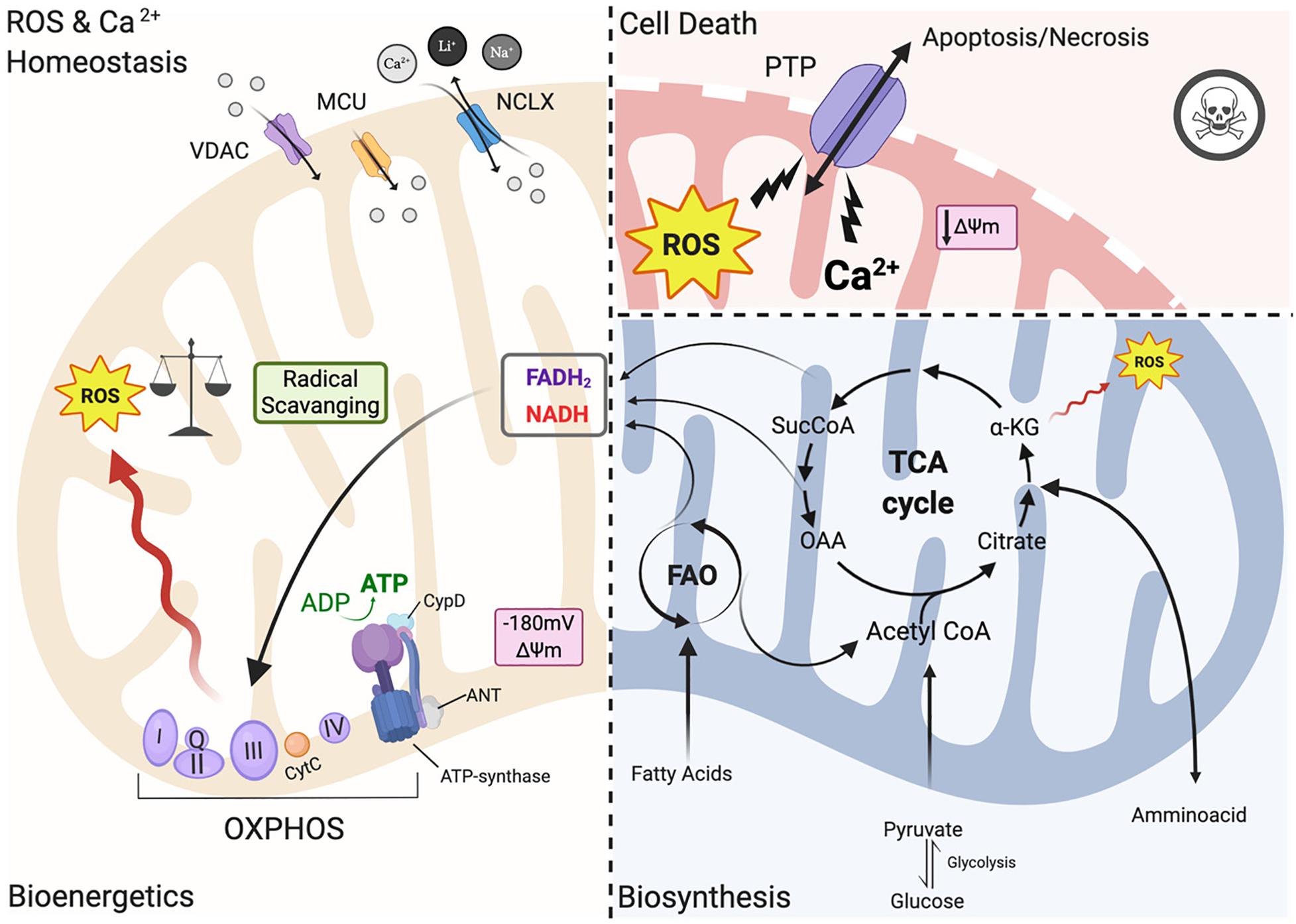
Figure 1. Mitochondrial functions. Left panel: under physiological conditions, mitochondria functions are the core of bioenergetics activities, providing ATP throughout the OXPHOS, which is also an important source of ROS. Basal ROS levels are maintained by the radical scavenging network. Additionally, mitochondria are calcium-buffering organelles. Ca2+ homeostasis is finely controlled by its uptake through voltage-dependent anion-selective channel proteins (VDACs) and the mitochondrial Ca2+ uniporter (MCU) complex, Ca2+ efflux is controlled by NCLX. Right top panel: pathological conditions, ROS burst and mitochondrial Ca2+ overload activate regulated cell death (RCD) inducing either apoptosis or necrosis pathway through the PTPC opening. Right-bottom panel: Ca2+ uptake activates mitochondrial metabolism. Fatty acids are metabolized via FAO toward the TCA cycle providing energy as FADH2 and NADPH are building blocks for biosynthesis. Voltage-dependent anion-selective channel proteins (VDAC), Mitochondrial Calcium Uniporter Complex (MCUC), Mitochondrial Na+/Ca2+ exchanger (NCLX), oxidative phosphorylation (OXPHOS), Permeability transition pore complex (PTPC), ADP/ATP translocase (ANT) and peptidyl-prolyl cis-trans isomerase Cyclophilin D (CypD), cytochrome C (cyt C), adenosine triphosphate (ATP), reactive oxygen species (ROS), tricarboxylic acid cycle (TCA), fatty acid oxidation (FAO), α -ketoglutarate dehydrogenase (α-KG), oxaloacetate (OAA), Acetyl coenzyme A (Acetyl CoA) mitochondrial membrane potential (Δψm) (Created with BioRender.com).
However, under pathological conditions, a cytosolic Ca2+ overload initiates a large and persistent Ca2+ uptake by mitochondria, which triggers the opening of the mitochondrial permeability transition pore (mPTP; a nonspecific pore) (Figure 1; Giorgi et al., 2012; Morganti et al., 2018). mPTP allows for free passage of small molecules and ions (<1.5 kDa) across the IMM, leading to membrane potential dissipation, and consequent imbalance in ATP production, mitochondrial swelling and mitochondrial outer membrane (OMM) rupture all of which cause regulated cell death (RCD) through either apoptosis or necrosis (Figure 1; Bonora et al., 2015). The activation of one of these cell death pathways depends upon the severity of the damage and the kinetics of the pore opening (Javadov and Karmazyn, 2007). mPTP is also involved in the pathogenesis of Ischemia/reperfusion injury (IRI) (Morciano et al., 2015). For example, a moderate injury, which occurs in the case of a short ischemic period, may lead to a short pore opening time thereby triggering apoptosis. On the other hand, a more severe and persistent insult such as a longer hypoxic event may lead to persistent pore opening inducing cell death through necrosis. Myocardial infarction exhibits a necrotic area at the core of the ischemic zone that is surrounded by apoptotic markers, indicating decreased cardiomyocyte survival upon an ischemic event (Javadov and Karmazyn, 2007; Morciano et al., 2015). The mPTP structure remains an area of intense study; the latest findings have been reviewed recently by Bonora et al. (2020).
In the past few years, the mitochondrial F1/F0 ATP Synthase (ATP synthase) has been recognized as a key component of pore formation along with ADP/ATP translocase (ANT) and peptidyl-prolyl cis-trans isomerase Cyclophilin D (CypD) (Figure 1; Bonora et al., 2017; Morciano et al., 2017; Bonora and Pinton, 2019) that together regulate the opening of the permeability transition pore complex (PTPC) (Bonora et al., 2020). It has been demonstrated that dissociation of ATP synthase dimers upon mitochondrial permeability transition (MPT) induction, in particular the C subunit of the F0 part (in its c-ring form), is a key component of the pore (Bonora et al., 2013, 2015, 2017). Mitochondria isolated from Ppif-null mice strongly validates the role of CypD as a pore regulator (Basso et al., 2005) because these mitochondria are unresponsive to mitochondrial Ca2+ overload. Moreover, upon knocking out all three ANT isoforms simultaneously MPT is inhibited (Karch et al., 2019). It remains controversial whether ANT represents a key pore regulator or is a part of the pore, and futher studies are needed to understand its role in PTPC opening (Bonora and Pinton, 2019).
Thus, in order to avoid mitochondrial Ca2+ overload and consequent activation of regulated cell death (RCD), Ca2+ uptake has to be finely controlled. Ca2+ released by the ER rapidly enters the mitochondrial intermembrane space (IMS) by Voltage-dependent anion channels (VDACs), which are localized at the OMM (Figure 1; Giorgi et al., 2018b). The channel exists in three isoforms (VDAC1, VDAC2, VDAC3) expressed almost ubiquitously among tissues with different sub-mitochondrial ratios (Messina et al., 2012). It exhibits two conformations: the open pore conformation with a low transmembrane potential, showing high-conductance and weak anionselective; whereas increasing potential leads to a closed state conformation characterized by cation selectivity and impermeable to nucleotide passage (Giorgi et al., 2018b). In recent years, the mitochondrial Ca2+ uniporter (MCU) was identified as a major player in the regulation of mitochondrial Ca2+ homeostasis (Figure 1; Kirichok et al., 2004). MCU is a multiprotein complex (MCUC) situated at the IMM, which has a low affinity for Ca2+ ions but is highly selective and regulated by auxiliar proteins that make up part of the MCUC (Marchi and Pinton, 2014). MCUC involvement in cardioprotection has been widely studied in recent years: several mouse models with MCU deletions have been developed including a cardiac-specific dominant-negative MCU mouse that is expressed in neonates (Rasmussen et al., 2015), a cardiac conditional MCU-KO mouse (Luongo et al., 2015), and a tamoxifen-inducible cardiac-specific loss of MCU in adult mice (Kwong et al., 2015). Mitochondria isolated from the hearts of these mice are characterized by reduced mitochondrial Ca2+ influx with subsequent reduced susceptibility to mPTP opening and loss of mPTP-related cardioprotection (Kwong et al., 2015; Luongo et al., 2015). On the contrary, deletion of NCLX, a key component of Ca2+ release, is lethal to cells because it induces mitochondrial Ca2+ overload and consequent PTP opening (Luongo et al., 2015).
Mitochondria contribute to cell metabolism by providing building blocks for the synthesis of macromolecules necessary for the maintenance of cellular homeostasis and cell growth. As mentioned above, the TCA cycle represents a metabolic engine in mitochondria where these catabolic and anabolic reactions intersect (Martínez-Reyes and Chandel, 2020). As the cycle runs, metabolic intermediates may be utilized for different biosynthetic reactions (Figure 1; Spinelli and Haigis, 2018). These biosynthetic reactions not only consume the TCA cycle intermediates and direct them away from ATP production but also require substantial energy input (Ritterhoff et al., 2020). Thus, whether the intermediates will be used for synthetic purposes is dependent on the energy state of the cell. Energy requirements for sustained cardiac contractile function are high and most of the cardiac metabolism is directed toward the production of ATP (Doenst et al., 2013; Ritterhoff and Tian, 2017; Figure 1). Conversely, biosynthetic demands in non-proliferative cardiomyocytes of the adult heart are rather low, especially compared to highly proliferative cells such as cancer cells (Karlstaedt et al., 2018). However, biosynthesis increases considerably during cardiac hypertrophy (Karlstaedt et al., 2018; Ritterhoff et al., 2020).
Whenever metabolic intermediates are removed from the TCA cycle for biosynthetic reactions, they need to be restored to ensure the cycle’s continued running (Martínez-Reyes and Chandel, 2020). This replenishment of the intermediate pool is named anaplerosis. Increased anaplerotic flux through carboxylation of glycolysis-derived pyruvate to malate was previously reported in hypertrophied rat hearts and paralleled by elevated expression of malic enzyme, which catalyzes this reaction (Sorokina et al., 2007; Pound et al., 2009). During cardiac hypertrophy, there is an energy source switch from FAO to increased glucose utilization, with a general reduction in oxidative metabolism (Doenst et al., 2013; Ritterhoff and Tian, 2017; Ritterhoff et al., 2020). Taken together, increasing the use of pyruvate for anaplerosis reduces its accessibility for oxidation and may lead to energy inefficiency of the TCA cycle (Sorokina et al., 2007; Pound et al., 2009), which contributes to contractile dysfunction and subsequent heart failure. A better understanding of how these mechanisms are regulated is needed for potential targeted treatments of cardiac dysfunction.
Mitochondria are one of the important sources of ROS production within most mammalian cells, including cardiomyocytes (Figure 1; Chen and Zweier, 2014). Moreover, interspecies comparisons performed in recent years show that ROS regulatory systems are dependent on organism, type of tissue, physiological state, age and pathological conditions to finely tune the underlying responses (Barja, 1999). The primary ROS generated in cardiac mitochondria is superoxide radical anion (O2⋅–), which can be reduced through dismutation to hydrogen peroxide (H2O2). Hydroxyl radicals (OH⋅) are also generated from the decomposition of hydroperoxides, or by the reaction of excited atomic oxygen with water. The mitochondrial respiratory chain is a powerful endogenous source of O2⋅–, which is a toxic by-product of oxidative phosphorylation (Figure 1). Electrons from NADH and FADH2 flow through the electron chain to reduce oxygen to form H2O (Figure 1). Large amounts of O2⋅– are generated when oxygen is incompletely reduced due to electron leaking at complexes I and III (Kussmaul and Hirst, 2006; Bleier and Dröse, 2013; Vinogradov and Grivennikova, 2016). Apart from the sites of ROS production within the mitochondrial respiratory chain there are other mitochondrial enzymes that generate either O2⋅– or H2O2. For example, NADPH oxidase 4 (Nox4) is an important source of ROS in heart mitochondria. Nox4 expression is upregulated in failing cardiomyocytes and contributes to the increase of mitochondrial O2⋅– levels that drives oxidative stress (Kuroda et al., 2010). α-ketoglutarate dehydrogenase (α-KGDH) is one of the TCA enzymes that is the most vulnerable to environmental changes (Figure 1; Tretter, 2004). α-KGDH generates O2⋅– during its catalytic function upon excessive NADH levels (Tretter and Adam-Vizi, 2005), making α-KGDH an important mitochondrial site for ROS production.
The ROS scavenging network coordinately works to maintain proper basal ROS levels and redox signaling in cells to control mitochondrial oxidative stress (Figure 1). The mitochondrial antioxidant defense system includes endogenous antioxidant enzymes such as superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase, and the peroxiredoxin/thioredoxin system (discussed in greater detail in Peoples et al., 2019). ROS are not only byproducts of mitochondrial metabolism (Figure 1), but are commonly involved as second messengers in cellular signaling impacting both adaptive and maladaptive cardiomyocytes responses (Cave et al., 2006). The redox-sensitive cellular processes are involved in cardiac development and differentiation, angiogenesis, cardiac regeneration and cardiomyocyte apoptosis (Tretter, 2004). Indeed, basal levels of ROS are required for human embryonic stem cells (ESCs) to differentiate into cardiomyocytes (Ji et al., 2010). In particular, accumulating evidence points to mitochondrial-mediated ROS generation as having a key role in cardiomyocyte differentiation. Nox4-dependent mitochondrial oxidative stress is one of the major pathways activated in undifferentiated ESCs, driving their differentiation into cardiomyocytes (Murray et al., 2013). Accordingly, Nox4 depletion in ESCs impairs cardiomyocyte differentiation (Li et al., 2006). Another study demonstrated Nox4 expression was significantly reduced in differentiated cardiomyocytes (Crespo et al., 2010).
Oxidative stress signaling also orchestrates angiogenesis in cardiomyocytes through Nox4 regulation. Nox4 is involved in the stimulation of angiogenesis, protecting against contractile dysfunction and hypertrophy under situations of chronic load-induced stress (Zhang et al., 2010). Furthermore, functional alterations of mitochondria and the subsequent increase of ROS production are critical in cardiac repair and regeneration. Postnatal heart maturation is associated with the transition from glycolytic to oxidative metabolism, which drives an increase in ROS production derived from the ETC and reduces cardiomyocyte regeneration capacity (Puente et al., 2014). In general, ROS levels are increased in response to heart damage including ischemic injury (Chouchani et al., 2014; Puente et al., 2014). Blocking ROS production and activation of scavenger systems promotes heart regeneration after cardiac injury (Tao et al., 2016). Conversely, mitochondrial dysfunction and the subsequent increase of mitochondrial ROS cause cardiomyocytes cell cycle arrest and activates apoptotic responses resulting in lethal dilated cardiomyopathy (Yang et al., 2018; Zhang et al., 2018). Overall, further insight into cellular mechanisms by which mitochondrial redox signaling disturbs physiological oxidative stress may uncover novel CVD therapeutic targets.
In adult cardiomyocytes mitochondria mobility is limited with mitochondria moving along microtubule networks (Frederick and Shaw, 2007). In most mammalian cells mitochondria generally cluster around the nucleus (Yoon, 2004), but mitochondria can be at different cytoplasmic locations leading to mitochondrial heterogeneity within different cell types (Kuznetsov et al., 2009; Piquereau et al., 2013). In cardiomyocytes, this heterogenous population can be divided up into three separate populations, characterized by their location within the cardiomyofibers: subsarcolemmal mitochondria (SSM), intermyofibrillar mitochondria (IFM) or perinuclear mitochondria (PNM) (Shimada et al., 1984). Electron microscopy and transmission electron microscopy show these distinct populations of mitochondria as the morphology is unique for both their location and function (Shimada et al., 1984; Manneschi and Federico, 1995; Vendelin et al., 2005; Wikstrom et al., 2009). SSM are located just under the surface sarcolemma and possess closely packed cristae. Holmuhamedov and colleagues characterized and assessed SSM, finding that SSM have a high sensitivity to Ca2+ overload-mediated inhibition of ATP synthesis (Holmuhamedov et al., 2012). PNM are clustered at nuclear pores between and around the two nuclei commonly found in cardiomyocytes. Due to their well-developed curved cristae, PNM have little matrix area that allows for higher ATP production (Hackenbrock, 1966). Lu and coworkers provide us with one of the few studies on PNM and found that PNM morphology is more spherical than IFM and SSM, where the lack of myofibrillar constraints allows for the PNM spherical shape and its high mobility. This group also determined that PNM are physically closer to protein synthesis sites for perinuclear mitochondrial biogenesis, indicating that PNM are involved in transcription and translation processes (Piquereau et al., 2013; Lu et al., 2019). Lastly, IFM are very well organized, as they lay closely parallel to contractile myofilaments. This highly organized structure may cause IFMs to be restricted in their position and mobility; however it also provides bioenergetic support needed for contraction and mitochondrial interaction with the cytoskeleton and the SR (Wilding et al., 2006). IFM form an interface with the SR, which allows molecules to be transported between the SR and mitochondria for effective signal transduction (Eisner et al., 2013).
The SR is of extreme importance in cardiomyocytes, as it critically regulates excitation-contraction coupling by releasing its stored Ca2+ via the type 2 ryanodine receptor (RyR2) (Fu et al., 2006; Figure 2). To understand the release of Ca2+ by the SR, we will discuss the SR compartments. Structurally, the SR is a diverse organelle, consisting of junctional, corbular, and network SR. These components of the SR form a complex tubular network where the network SR is formed by a series of interconnected tubules that are located in the region between the transverse-tubules (T-tubules) (Figure 2). The junctional SR is the domain where specialized junctions are formed with the sarcolemma T-tubules, allowing the SR to bring its ryanodine sensitive Ca2+ channels (RyRs) in close range with the sarcolemma voltage-gated L-type Ca2+ channels (Franzini-Armstrong and Protasi, 1997; Franzini-Armstrong et al., 1999; Figure 2). The corbular SR expresses RyRs as well, however it does not form junctions with the sarcolemma (Vega et al., 2011). The membrane depolarization, as a result of excitation-contraction (EC), causes the L-type Ca2+ channels to open up close to the junctional SR (jSR). This results in a small amount of Ca2+ entering the limited cytosolic space that separates the SR and the T-tubule sarcolemma. This increase in Ca2+ concentration exceeds the threshold for the RyR2 to be activated through a mechanism of Ca2+-induced Ca2+ release (CICR) (Figure 2; Fabiato, 1983). This activation of a small number of clustered RyR2 affects the concentration of intracellular calcium, inducing “calcium sparks” (Cheng et al., 1993). When multiple “sparks” are activated by the EC, a rise of intracellular calcium can be detected in the dyadic cleft (Sharma et al., 2000), thereby initiating myocardial contractions.
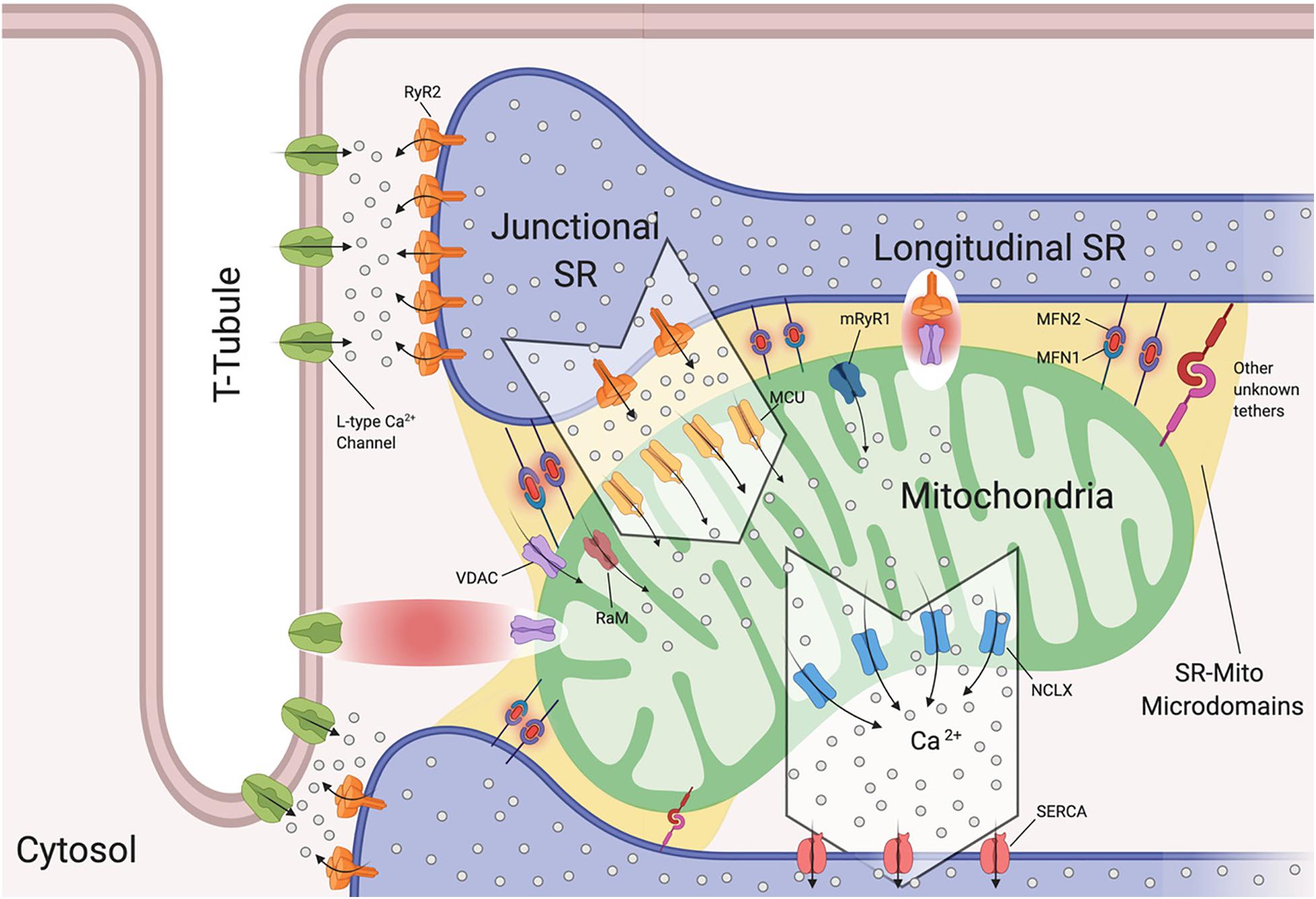
Figure 2. Sarcoplasmic reticulum-mitochondria communications. Sarcoplasmic reticulum (SR) is in close proximity to T-tubules and mitochondria. Depicted are the channels involved in Ca2+ flux by which SR regulates excitation-contraction coupling and Ca2+ signaling with mitochondria. Microdomains where Ca2+ exchanges occur are shown. Ryanodine Receptor 2 (RyR2) and L-Type Ca2+ Channels located at the t-tubule-SR interface; voltage-dependent anion-selective channel proteins (VDAC) colocalize with RyR2 and also with L-Type Ca2+ channels, Mitochondrial Calcium Uniporter Complex (MCUC) is mainly located at the SR-Mito interface, Rapid modes of Ca2+ uptake (RaM), ryanodyne receptor type 1 (mRyR1), Mitochondrial Na+/Ca2+ exchanger (NCLX) is located at the opposite side of MCUC near the sarcoplasmic/endoplasmic reticulum Ca2+ ATPases (SERCA). Mitofusin1/2 (MFN1/2) function as SR-mitochondrial tethers (Created with BioRender.com).
Interestingly, it has been reported that L-type Ca2+ channels regulate mitochondrial functions through actin filaments (Viola and Hool, 2010). Viola and colleagues demonstrated that Ca2+ influx through this channel increases superoxide production, NADH levels, metabolism and also mitochondrial membrane potential in a calcium independent pathway (Viola et al., 2009). The cytoplasmic β-subunit of the L-type Ca2+ channel is anchored to the actin cytoskeleton. Disruption of this tether decreases Ca2+ flux, leading to poor oxygen consumption and ATP production by the mitochondria (Viola et al., 2009). Moreover, this group found that cardiomyocytes isolated from mdx mice (a mouse model of Duschennes muscular dystrophy that in patients causes dilated cardiomyopathy) had impaired communication between L-type Ca2+ and mitochondria through an alteration in the cytoskeletal network that led to a decrease in metabolic functions (Viola et al., 2014). They were the first to show a physical and functional association between L-type Ca2+ and VDAC through F-actin (Viola et al., 2013, 2014; Figure 2). They further reported that mdx cardiomyocytes maintain a higher level of resting calcium and L-type Ca2+ channel activation plays a role in the observed mitochondrial calcium changes all of which may promote DCM (Viola et al., 2013).
Interactions between the SR and mitochondria play a key role in cardiomyocyte contraction and multiple studies have provided us with evidence of mitochondrial Ca2+ uptake, and thus increased mitochondrial Ca2+ levels, in response to SR-mediated Ca2+ release (Bassani et al., 1992; Negretti et al., 1993; Szalai et al., 2000). As mentioned previously, cardiac mitochondria uptake cytosolic Ca2+ through MCUC. However, this channel requires at least a concentration of 2–5 μM of free Ca2+ in the SR’s bulk in order to be activated (Kirichok et al., 2004). This Ca2+ concentration is reached only within specific microdomains at the SR-mitochondria interface (Figure 2). Specifically, in such extremely structured cells, MCUC is expressed more in areas in close contact with the jSR, which contain Ca2+-releasing RyR2 channels (Figure 2; De La Fuente et al., 2016). Moreover, Ca2+ fluxes must be tightly regulated. De la Fuente and colleagues demonstrated that MCUC and NCLX are spatially excluded in cardiac mitochondria (Figure 2) in order to optimize Ca2+ signals and sustain mitochondrial metabolism required for cardiomyocyte contraction, while also reducing the energy required for mitochondrial membrane potential depolarization (De La Fuente et al., 2016, 2018). It remains controversial whether mitochondrial Ca2+ uptake occurs quickly and synchronously with the cytosolic Ca2+ fluctuations in a beat to beat model (García-Pérez et al., 2008) or if it increases slowly (Griffiths et al., 1997). The differences between these two models rely on the experimental approach used in terms of probes, stimulation and species (De la Fuente and Sheu, 2019).
It should be noted that at the microdomain level, the molecular bridge permitting Ca2+ exchange between SR-Mitochondria is formed by RyR2 and VDAC2 channels (Figure 2; Min et al., 2012). Moreover, in aged cardiomyoctes the physical interaction between RyR2 and VDAC is significantly reduced, leading to lower mitochondrial Ca2+ uptake, thereby promoting oxidative stress and energy impairment (Fernandez-Sanz et al., 2014). However, this event is independent of RyR2 and VDAC expression levels and does not correlate with MFN2 levels (Fernandez-Sanz et al., 2014).
It has become widely accepted that mitochondrial dysfunction is associated with heart disease (Bonora et al., 2019). Pathological SR-dependent Ca2+ leak through RyR2 channels is involved in excessive mitochondrial Ca2+ uptake (Santulli et al., 2015; Ruiz-Meana et al., 2019). Santulli et al. were the first to show using a murine model that a feedback loop exists between SR and mitochondria where Ca2+ leak through RyR2 channels causes mitochondrial Ca2+ overload and ROS burst that enhances Ca2+ leak and thereby worsening mitochondrial dysfunction (Santulli et al., 2015). Moreover, in human and murine senescent cardiomyocytes, the SR-mitochondria Ca2+ exchange is significantly impaired due to RyR2 glycation (Ruiz-Meana et al., 2019). These aged cardiomyocytes display a deficient dicarbonyl detoxification pathway initiating Ca2+ leak through RyR2 channels and further increasing mitochondrial Ca2+ uptake. Taken together, this mechanism is involved in the transition from a healthy cardiomyocyte to a failing cardiomyocyte, as it may induce bioenergetic deficit through mitochondrial damage leading to mitochondrial dysfunction (Ruiz-Meana et al., 2019).
It is important to mention that MCUC is not the only manner in which mitochondrial Ca2+ uptake occurs in cardiomyocytes. Three different approaches of MCU-knockout mice (global constitutive, cardiac-specific, dominant negative overexpression) have been developed (Pan et al., 2013; Kwong et al., 2015; Luongo et al., 2015). These models demonstrate that MCUC is dispensable for heart function in basal cardiac activity, while under “fight-or-flight” conditions MCU deletion shows inhibition of acute mitchondrial Ca2+ uptake. Moreover, these mice are highly protected from mPTP opening during ischemia-reperfusion injury. Therefore, in basal resting conditions, mitochondrial Ca2+ influx for maintaining ATP production and cardiac metabolism occurs through other channels such as rapid modes of Ca2+ uptake (RaM) (Buntinas et al., 2001) and ryanodyne receptor type 1 (mRyR1) (Beutner et al., 2001, 2005; Figure 2) both of which are located in the IMM. RaM displays a faster Ca2+ uptake compared to MCUC (Buntinas et al., 2001), while mRyR1 opens at lower cytosolic Ca2+ concentrations (Beutner et al., 2001, 2005).
As discussed above, in cardiomyocytes SR-mitochondria Ca2+ transfer occurs mainly in areas of direct physical contact. However, the proteins involved in the tethering have been poorly investigated in the heart. Currently, mitofusin 2 (MFN2) is the protein that has been suggested to tether this physical interaction (Figure 2; Papanicolaou et al., 2011; Chen et al., 2012). It remains a matter of discussion whether MFN2 acts as a tether or a spacer at the ER/SR-mito interface. Two different MFN2-KO mouse models have been generated showing opposite results. When the gene deletion is made after birth the distance of SR-mitochondria increased leading to a rise of Ca2+ concentration in the SR (Chen et al., 2012). Moreover, isopronenterol stimulation of cardiomyocytes, display an increase in cytosolic Ca2+ concentrations and lower mitochondrial Ca2 uptake. Of note, this model does not show mitochondrial bioenergetic impairment (Chen et al., 2012). On the other hand, if the gene is deleted at the embryonic stage, there are no differences in the SR-mitochondria distance and therefore, no alterations in Ca2+ fluxes. This result may be due to compensatory remodeling (Papanicolaou et al., 2012). Each of these mouse models demonstrates mitochondrial morphology alterations, contractile depression and cardiac hypertrophy in the adults. However, more studies are needed to investigate the role of MFN2 in tethering SR-mitochondria and also to determine whether other proteins may be involved in this tethering (Figure 2).
Mitochondrial health is tightly correlated to the ability of these dynamic organelles to move and change their morphology in response to the surrounding environment (Ong et al., 2017). The term “mitochondrial dynamics” refers to fusion and fission processes of mitochondrial structures within a living cell. Mitochondria constantly shape themselves through fusion and fission in response to changes in energy requirements (Hu et al., 2019). The balance between mitochondrial fusion and fission determines the number (biogenesis), morphology and activity of these multifunctional organelles (Marín-García and Akhmedov, 2016). In the heart, rapid responses to body demands depend on this balance of fusion and fission that modulate multiple mitochondrial functions such as energy production, ROS generation, Ca2+ homeostasis and cell death (Marín-García and Akhmedov, 2016).
Several GTPases are involved in the fission and fusion processes, which utilize GTP energy to guide conformational changes (Urbani and Babu, 2019). Mitochondrial fission is driven by dynamin-related protein-1 (Drp1) that is recruited to mitochondria by human fission protein-1 (hFis1), mitochondrial fission factor (Mff) and mitochondrial dynamics proteins 49 and 51 (MiD49 and 51) (Figure 3; Ong et al., 2017). This process, characterized by the fragmentation of mitochondria into more restricted and rounded organelles (Sharp and Archer, 2015; Sciarretta et al., 2018), is essential for mitosis and is required for the specific clearance of injured mitochondria through mitophagy (Ong et al., 2017). Specifically, in the heart, mitophagy is used to preserve a healthy pool of mitochondria under stress conditions (Nan et al., 2017). Mitochondrial outer membrane fusion is directed by mitofusin 1 (MFN1) and MFN2 and mitochondrial inner membrane fusion is executed by optic atrophy 1(Opa1), leading to the formation of functional elongated organelles (Figure 3; Ong et al., 2017). This process is fundamental for mitochondrial DNA (mtDNA) maintenance and inheritance, membrane potential transmission and Ca2+ signaling within the mitochondrial machinery (Westermann, 2010; Archer, 2013; Elgass et al., 2013; Hoppins, 2014). Of note, in stress-free conditions neonatal and adult cardiomyocytes demonstrate differences in the rate of mitochondrial dynamics. Indeed, in neonatal cardiomyocytes fusion and fission processes are more frequent and rapid compared to the levels observed in adult cardiomyocytes (Forte et al., 2020). Mitochondria in neonatal cardiomyocytes are highly mobile and distributed throughout the cytoplasm within a filamentous network, while adult cardiomyocyte mitochondria are more static and spatially arranged into three subpopulations, as described above, which constrains their movements (Vásquez-Trincado et al., 2016). The homeostasis of mitochondrial dynamism is ensured by the interaction and cooperation of the aforementioned proteins. Moreover, alterations in the balance of mitochondrial dynamics are correlated to cardiac disorders (Marín-García and Akhmedov, 2016; Hu et al., 2019) leading to aberrant mitochondrial network morphology.
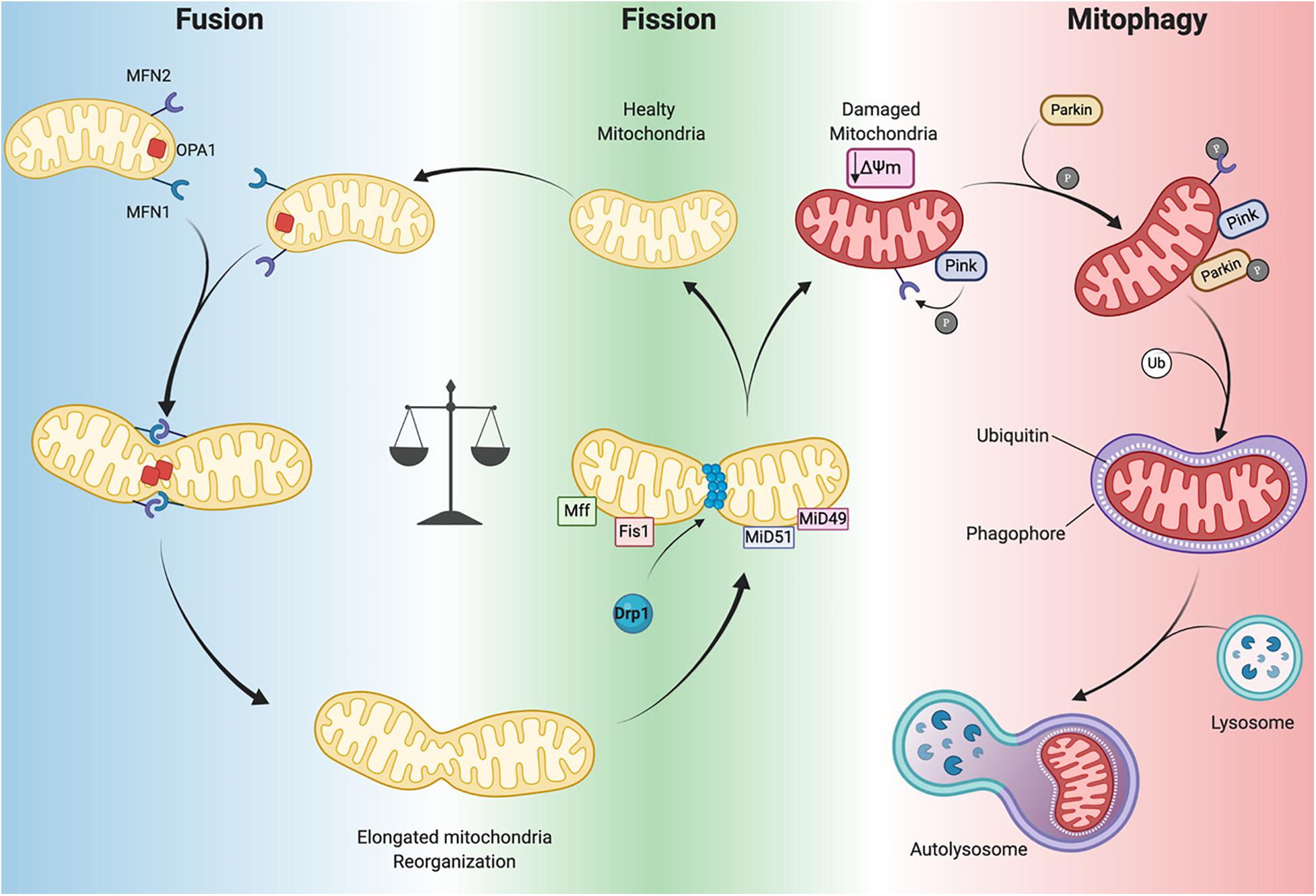
Figure 3. Mitochondrial dynamics. Mitochondrial neworks are mantained by the balance betwen fusion (left panel) and fission (central panel). Damaged mitochondria are cleared by mitophagy pathway (right panel). Dynamin-related protein-1 (Drp1), fission protein-1 (Fis1), mitochondrial fission factor (Mff), mitochondrial dynamics proteins 49 and 51 (MiD49 and MiD51), mitofusin 1/2 (MFN1 and MFN2) optic atrophy 1(Opa1), PTEN-induced kinase 1 (PINK1) ubiquitin ligase Parkin, phosphorylation (P), Ubiquitin (Ub), mitochondrial membrane potential (Δψm) (Created with BioRender.com).
Mitophagy is a specific form of autophagy exploited by the cellular machinery to digest dysfunctional and senescent mitochondria through autophagosomes, under basal and stress conditions (Shires and Gustafsson, 2015). This process is tightly regulated by the mitochondrial PTEN-induced kinase 1 (PINK1) and the cytosolic ubiquitin ligase Parkin. In Parkin-mediated mitophagy, upon loss of ΔΨm, PINK1 accumulates in the OMM where it recruits Parkin (Figure 3). This may occur directly by PINK1-mediated phosphorylation of Parkin (Koyano et al., 2014) or indirectly by phosphorylation of MFN2 (Figure 3; Chen and Dorn, 2013). Indeed, Matsua and colleagues reported that upon a ΔΨm decrease, cytosolic Parkin is recruited to mitochondria by PINK1 through Parkin’s phosphorylation at Ser65 within its ubiquitin-like domain. This phosphorylation event is necessary for the efficient translocation of Parkin to mitochondria (an initial step of mitophagy). Upon activation parkin initiates ubiquitination of mitochondrial proteins to promote phagasome recruitment and subsequent degradation of mitochondrial proteins by the lysosome (Figure 3; Kondapalli et al., 2012; Shiba-Fukushima et al., 2012; Koyano et al., 2014).
It has also been observed that the phosphorylation of MFN2 by PINK1 is essential for Parkin recruitment to damaged mitochondria, thus suggesting a connection between mitochondrial dynamics and mitophagy (Figure 3; Chen and Dorn, 2013). A detailed discussion of mitophagy is reviewed elsewhere (Shires and Gustafsson, 2015; Sciarretta et al., 2018). Mitochondrial autophagy plays a critical cardioprotective role; although when impaired it is detrimental to the heart (Saito and Sadoshima, 2015; Morciano et al., 2020). In mouse hearts lacking Mfn2 expression there is a reduction in Parkin-mediated mitophagy and contractility and increased hypertrophy leading to heart failure by 30 weeks of age (Song et al., 2014). As mentioned previously, during ischemia/reperfusion (I/R) mitophagy appears to protect the heart. Indeed, in a cardiac-specific conditional Drp1 knockout mouse the inhibition of the mitophagic flux causes accumulation of injured and dysfunctional mitochondria, leading to cardiomyocyte death during reperfusion (Ikeda et al., 2015). In addition, ablation of Drp1 in adult mouse cardiomyocytes dampens mitochondrial fission and significantly upregulates Parkin, which leads to mitophagy and lethal cardiomyopathy (Song et al., 2015). Notably, loss of Parkin in adult mouse hearts did not affect function. However, in neonates a lethal cardiomyopathy due to defective mitophagy clearance of fetal mitochondria was observed in cardiomyocyte-specific Parkin ablation within three weeks after birth (Gong et al., 2015). Moreover, PINK1 deficiency in mice leads to cardiac mitochondrial dysfunction and excessive oxidative stress (Billia et al., 2011). These findings highlight that PINK1/Parkin and the mitophagy machinery are crucial for cardiac homeostasis. Moreover, mitophagy impairment results in defective cellular homeostasis, leading to cardiomyopathy and ultimately heart failure (Nan et al., 2017; Bonora et al., 2019).
Cardiomyopathies are a heterogeneous condition, which effect myocardial structure and function culminating in heart failure. Due to the complexity of this disease its classification continues to evolve. However, the scenario is not simple, since any attempt at classification is limited by the criterion choice of classification itself (phenotype, etiology, clinical, morphological, functional). Additionally, the heterogeneity and all the overlapping forms of cardiomyopathy make this work more complex (Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies; WHO/ISFC, 1980; Maron et al., 2006; Thiene et al., 2007; Arbustini et al., 2014).
Among all cardiomyopathies, dilated cardiomyopathy (DCM) continues to lack proper characterization and understanding (Merlo et al., 2019). It is presented with a mixed etiology and high incidence of genetic mutations. Patients with dilated cardiomyopathy may present with ventricular arrhythmias in the absence of signs of heart failure. An arrhythmogenic indication may be found in arrhythmogenic right ventricular (RV) cardiomyopathy (ARVC) and left-dominant arrhythmogenic cardiomyopathy, hypertrophic cardiomyopathy and LV noncompaction all of which are associated with increased risk of sudden cardiac death. Furthermore, DCM may also occur in patients with mitochondrial cardiomyopathy and metabolic disorders (Thiene et al., 2007; Merlo et al., 2019). Moreover, new genetic mutations continue to be identified for DC, its related peripartum cardiomyopathy and those of the arrhythmogenic cardiomyopathies, including ARVC, left-dominant arrhythmogenic cardiomyopathy, and channelopathies (Sen-Chowdhry et al., 2008, 2010; Spezzacatene et al., 2015).
In this section we will focus on how changes in mitochondrial health are associated with dilated cardiomyopathy onset and progression (Figure 4). Dilated cardiomyopathy characteristics resulting from gene mutations have been identified in patients with peripartum cardiomyopathy, metabolic disorders, mitochondrial dynamics, OXPHOS dysfunction, Fatty acid and cardiolipin metabolism (Barth’s syndrome), all of which we will touch on here (Figure 4). In each of these syndromes echocardiographic data points to dilated cardiomyopathy presenting with left ventricular systolic dysfunction (left ventricular ejection fraction of <45% or fractional shortening of <30%) and cardiac hypertrophy. A number of mitochondrial diseases including OXPHOS disorders (Marin-Garcia et al., 2000; Alston et al., 2012, 2015; Jain-Ghai et al., 2013) and Barth syndrome (Hoch, 1992; Spencer et al., 2006; Gebert et al., 2009; Wang H.-Y.J. et al., 2014) correlate with the incidence of DCM, suggesting mitochondrial function as a key prognostic in the pathogenesis of DCM.
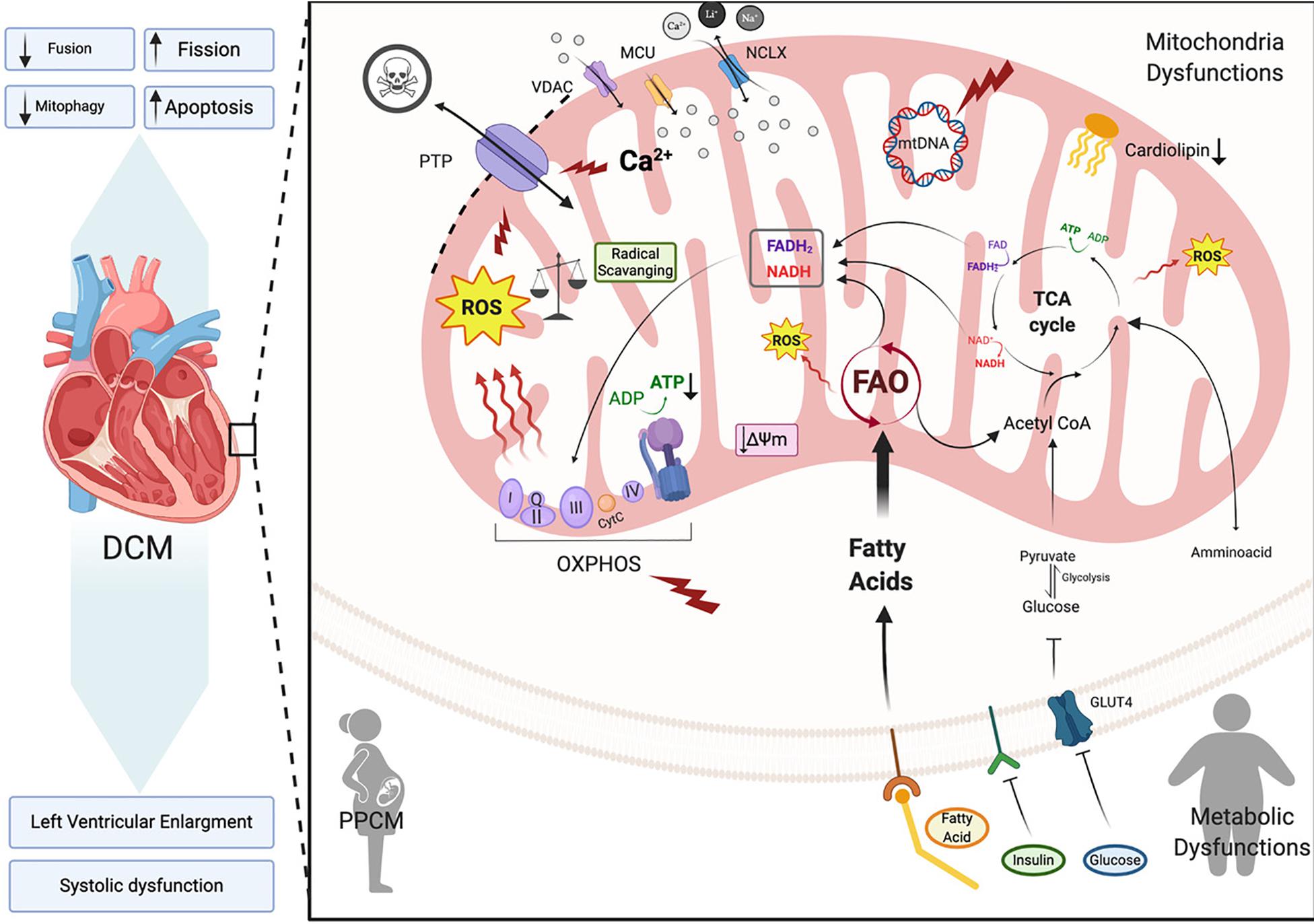
Figure 4. Dilated cardiomyopathy. Schematic representation of the primary pathway of cardiomyocyte mitochondrial dysfunction, which induces dilated cardiomyopathy (DCM). Metabolic imbalance, ROS overproduction and dysregulation of Ca2+ homeostasis are key changes inducing dilated cardiomyopathy. These overall changes cause increased mitochondrial fission events and activation of cardiomyocyte apoptosis. Voltage-dependent anion-selective channel proteins (VDAC), Mitochondrial Calcium Uniporter Complex (MCUC), Mitochondrial Na+/Ca2+ exchanger (NCLX), oxidative phosphorylation (OXPHOS), adenosine triphosphate (ATP), reactive oxigen species (ROS), tricarboxylic acid cycle (TCA), fatty acid oxidation (FAO), Glucose transporter type 4 (GLUT-4), mitochondrial membrane potential (Δψm), cytochrome C (cyt C) (Created with BioRender.com).
Dilated cardiomyopathy (DCM) is a leading cause of heart failure worldwide with a higher incidence in underdeveloped countries, however its prevalence varies due to geographic and socioeconomic conditions (Bozkurt et al., 2016). It is a form of heart disease that presents with dilation of the left or both ventricles and leads to systolic dysfunction and subsequent heart failure (Figure 4; Dellefave and McNally, 2010; De Paris et al., 2019). DCM is also considered idiopathic if no other vascular conditions such as hypertension are detected (Mestroni et al., 1999). Between 20–35% of idiopathic DCM cases may be linked to a family history with an inherited gene defect (Hershberger et al., 2010). Primarily mutations in genes that are involved in sarcomere structure and contractility, cytoskeletal arrangement, electrolytes balance and mitochondrial function (De Paris et al., 2019) are linked to DCM. In DCM, the cardiac muscle becomes thin and weakened causing the open area of the chamber to become enlarged. As a result, the heart is unable to pump blood efficiently (NIH, 2020). In order to maintain cardiac output, ventricular volumes increase and sarcomere contractility is reduced thereby, producing the thin-walled dilated appearance that characterizes DCM (De Paris et al., 2019). This ventricular remodeling is driven by irregular cardiomyocyte pathophysiology encompassing cardiomyocyte hypertrophy, impaired calcium cycling, apoptosis and fibrosis. A decrease in cardiac efficiency, as measured by myocardial oxygen consumption, leads to a progressive weakening of energy-starved cardiac myocytes pushing the heart toward heart failure (De Paris et al., 2019; Chen, 2020).
Because the heart is an organ requiring a high energy demand, regulation of mitochondrial metabolism plays an important role in the pathogenesis of this and other CVDs (Rosca and Hoppel, 2010). During the first stages of DCM, increased mitochondrial number (Figure 4) acts as a compensating mechanism for maintaining energy supply (Zak et al., 1980). However, the number of mitochondria declines during DCM progression leading to a reduction in ATP, decreased contractility and increased ROS all of which results in diastolic dysfunction and heart failure (Flarsheim et al., 1996; Goffart et al., 2004; Figure 4). Oxidative stress that presents as a consequence of increased ROS production is a key part of the pathogenesis of DCM. In vitro studies on isolated rat cardiomyocytes determined that physiologic stretch induces a Ca2+ spike through activation of NADPH Oxidase 2 (NOX2) and subsequently the ryanodine receptors in the SR. The authors found that in healthy cardiomyocytes, NOX regulation of ROS production plays a beneficial role through oxidation of the RyR2 channel, which mediates cardiac Ca2+-induced Ca2+ release (Figure 2). However, in muscular dystrophy mdx diseased cardiomyocytes these Ca2+ sparks induce arrhythmogenic Ca2+ waves (Prosser et al., 2011). Moreover, in an mdx mouse model of Duchenne muscular dystrophy Ca2+ release led to hyperactive ROS and subsequent cardiomyopathy. Whether this occurs in DCM remains to be determined. However, in patients with DCM, NADPH-upregulation of NOX increases ROS production through elevated rac1-GTPase activity (Maack et al., 2003). Furthermore, isolated ventricular cardiomyocytes from a rabbit heart failure model display decreased Ca2+ uptake resulting in reduced mitochondrial NADPH availability (Despa et al., 2002). Decreased NADPH subsequently increases ROS (Kohlhaas et al., 2010; Figure 4). Taken together, these findings point to Ca2+ controlled uptake at the mitochondria as a key regulator of ROS and an imbalance in NADPH-ROS causes disturbances in excitation-contraction coupling leading to cardiac dysfunction (Sag et al., 2013) and heart failure. ROS overproduction may also induce myocardial fibrosis, which is a common factor in DCM patients presenting with diastolic and systolic dysfunction (Assomull et al., 2006; Herpel et al., 2006). Cardiac stress, in general, plays a key role in initiating intrinsic apoptotic mechanisms in cardiomyocytes through mitochondrial dysfunction (Green and Reed, 1998). As detailed in previous sections dysfunctional mitochondria are efficiently removed by mitophagy in cardiomyocytes for cell maintenance and survival (Figure 3; Vásquez-Trincado et al., 2016). However, during cardiac stress, autophagy flux is reduced and damaged mitochondria accumulate resulting in enhanced oxidative stress and cardiomyocyte apoptosis (Figure 4; Campos et al., 2016). Uncontrolled autophagy is a component of the pathogenesis of DCM, cardiac hypertrophy and ischemic heart disease (Chistiakov et al., 2018).
The role of mitochondrial morphological alterations in the physiopathology of cardiomyopathies have become more apparent in recent years with changes in mitochondrial fission and fusion being at the forefront. Mitochondrial fission and fusion proteins are essential for normal cardiac remodeling and homeostasis (Chen et al., 2011). Impairments in mitochondrial morphology and function due to genetic deletion of fission and fusion proteins and their interacting partners may lead to dilated cardiomyopathy and heart failure (Nan et al., 2017; Ong et al., 2017; Hernandez-Resendiz et al., 2020). Dynamin-related protein 1 (DRP1) is involved in controlling mitochondrial fission. Genetic ablation of the Drp1 gene is embryonically lethal at day E12.5 (Manczak et al., 2012), however cardiac-specific deletion of Drp1 in the murine adult heart triggers mitochondrial elongation and mitophagy suppression leading to a higher susceptibility to ischemia/reperfusion and cardiomyopathy (Ikeda et al., 2015). Further studies demonstrated that Python mutant mice with a Drp1 gene point mutation (C425F) develop mitochondrial defects and DCM, as a result of diminished mitochondrial fission and mitophagy (Cahill et al., 2015). This C452F mutation is within the M domain, which is highly conserved and involved in protein-protein interactions (Cahill et al., 2015). Moreover, mice deficient in the mitochondrial fission regulator Mff also develop DCM leading to heart failure and death. Interestingly, the simultaneous deletion of mitochondrial fission and fusion regulator genes MFF (mitofission) and MFN (mitofusin) rescued Mff knockout mice, with improved cardiac function, enhanced mitochondrial oxidative capacity and increased survival (Chen et al., 2015).
In regards to fusion, cardiac-specific ablation of mitofusins Mfn1 and Mfn2 in mouse embryos causes death at days E9.5–10.5 (Chen et al., 2011). At late embryonic stage, the genetic inactivation of both these mitofusins promotes mitochondrial dysfunction leading to the development of a lethal cardiomyopathy; possibly due to biogenesis alterations, diminished mtDNA and enhanced mitochondrial fragmentation (Papanicolaou et al., 2012). Furthermore, conditional cardiac Mfn1/Mfn2 gene deletions in adult mouse hearts present with mitochondrial fragmentation, mitochondrial respiratory chain deterioration and develop a lethal DCM. Interestingly, loss of the Mfn1 gene alone is well tolerated in mice (Papanicolaou et al., 2011; Chen et al., 2012), whereas Mfn2-null mice display mitochondrial enlargement (Chen and Dorn, 2013), increased ROS production (Song et al., 2014) and cardiac hypertrophy (Papanicolaou et al., 2011). Proteolytic processing of fusion protein Opa1 also plays a critical role in the regulation of mitochondrial fusion. Opa1 proteolysis by stress-activated OMA1 peptidase induces mitochondrial fragmentation and DCM onset in a cardiac-specific Yme1l peptidase-null mouse (Wai et al., 2015). Taken together, alterations in mitochondrial fusion and fission machinery in the heart promotes mitochondrial metabolic impairments that induce dilated cardiomyopathy.
Peripartum cardiomyopathy (PPCM) is a rare form of DCM that develops in the last month of pregnancy or within five months postpartum. It presents with left ventricular systolic dysfunction (left ventricular ejection fraction of < 45% or fractional shortening of <30%) and cardiac hypertrophy (Satpathy et al., 2008). PPCM occurs in the absence of any identifiable cause and is exclusive to patients with no prior history of heart disease (Crane, 1976; Hibbard, 1999). PPCM occurs worldwide with an incidence of 1:1,000 births (Sliwa et al., 2017; Cunningham et al., 2019) with its incidence varying geographically being the highest in the United States, South Africa, Nigeria, and Haiti. In the US, its incidence is 1:2,230 live births (Mielniczuk et al., 2006; Brar et al., 2007; Gunderson et al., 2011; Kolte et al., 2014; Cunningham et al., 2019). Mortality also varies depending location with an estimate of 3–40% of patients diagnosed with PPCM Although it is a condition of unknown etiology, it also occurs in women with a previous history of pre-eclampsia or as a result of multiple pregnancies (Kolte et al., 2014; Arany and Elkayam, 2016).
PPCM shares many similarities with DCM including clinical symptoms such as ventricular dilation and systolic dysfunction (Figure 4; Pearson et al., 2000; Elliott et al., 2007). However, both diseases differ in progression and outcome and there are molecular differences. Similar to DCM, oxidative stress plays a key role in the pathogenesis of PPCM in patients and in mouse models in which ROS levels are highly increased compared to controls. According to Hilfiker-Kleiner and colleagues (Hilfiker-Kleiner et al., 2007), one possible reason for this increase is due to a cardiomyocyte-specific deletion of signal transducer and activator of transcription 3 (STAT3) as studied in a mouse model of PPCM. In these mice downregulation of the antioxidant Manganese superoxide dismutase (MnSOD) increases ROS production within mitochondria, which activates Cathepsin D that cleaves prolactin (PRL, 23 kDa) into a smaller 16 kDa piece. This negatively affects cardiomyocyte microvasculature and metabolism in these mice (Hilfiker-Kleiner et al., 2007, 2012; Hilfiker-Kleiner and Sliwa, 2014). STAT3 levels are downregulated in PPCM patient hearts suggesting that its expression may be cardioprotective during pregnancy (Ricke-Hoch et al., 2013). Importantly, PLR levels increase and remain high toward the end of pregnancy and after delivery which is in accord with the development of PPCM (Grattan et al., 2008). PPCM patients present with changes in PRL as well (Hilfiker-Kleiner et al., 2007; Haghikia et al., 2013) suggesting an oxidative stress plays a key role in PPCM.
A different mouse model of PPCM containing a cardiac specific deletion of PPARγ coactivator-1α (PGC-1α) showed that MnSOD was also reduced in the heart thus increasing ROS production and resulting in disturbed mitochondrial metabolism (Patten et al., 2012). Although research in cardiomyopathy-related genes have begun to elucidate the pathogenesis of PPCM, the molecular mechanisms underlying the development and progression of PPCM and development of targeted therapies have yet to be elucidated.
Early case studies in cardiomyopathy-related genes identified a clinical overlap between PPCM and DCM, however the extent of this interconnection remains unknown (Lee and Judge, 2017). More recent studies have reported that mutations in cardiac sarcomere proteins are a pathogenic cause of PPCM. These mutations include deleterious truncations in the titin encoding TTN gene that is a clinical feature shared with idiopathic DCM (van Spaendonck-Zwarts et al., 2014; Ware et al., 2016; Ballard, 2019). Titin is part of the structural organization and assembly of the sarcomere from the Z-disc to the M-line along with other developmental, regulatory and mechanical functions in cardiac and skeletal muscle (Lee and Judge, 2017). Mutations in many sarcomeric proteins located at the Z-disc may also lead to cardiomyopathies (Knöll et al., 2002; Frank et al., 2006; Sheikh et al., 2007). For example, mutations in the myosin heavy chain 7 (MYH7) gene that encodes the sarcomeric protein β-Myosin Heavy Chain (β-MHC) cause PPCM (Walsh et al., 2010; Fiorillo et al., 2016; Bollen and van der Velden, 2017). β-MHC forms the heavy chain structure of type II myosin in sarcomeres, which in a sliding mechanism with actin filaments, generates the mechanical forces needed for muscle contraction. Mutations in the STAT3 gene have also been found to contribute to PPCM (Ballard, 2019; Harhous et al., 2019). Other genes in which mutations have been reported to be associated with PPCM onset and progression include truncations in DMD (dystrophin that causes Duchenne’s Muscular Dystrophy) (Cheng and Prior, 2013; Ahmed et al., 2016), DSP (desmoplakin) (Ware et al., 2016), TPM1 (α-tropomyosin)(Ware et al., 2016), and missense mutations in MYBPC3 (cardiac myosin binding protein C) (Morales et al., 2010), TNNC1 (cardiac troponin C) (Mestroni et al., 1994), TNNT2 (cardiac troponin T) (Morales et al., 2010), and LAMP2 (lysosome-associated membrane protein) (Ware et al., 2016). This list of gene mutations causing PPCM continues to grow as more genes are identified (Lee and Judge, 2017; Ballard, 2019). Additional evidence supporting an involvement of gene mutations in PPCM includes familial incidence, genome-wide association studies and variability of occurrence of PPCM among women from different regions and ethnicities (Lee and Judge, 2017).
Metabolic cardiomyopathy is a heart muscle disorder that primarily develops in the presence of chronic metabolic conditions, such as type 2 diabetes, obesity, and insulin resistance (Figure 4; Nishida and Otsu, 2017). These conditions are frequently overlapping, resulting in similar metabolic-related structural and functional cardiac alterations, independent of hypertension or coronary artery disease and are collectively referred to as diabetic cardiomyopathy (Nakamura and Sadoshima, 2020). During the early stage of this disorder, metabolic disturbances do not cause significant structural changes in the heart, but result in other cellular abnormalities (e.g., impaired mitochondrial function, oxidative and ER stress and altered Ca2+ handling) all of which contribute to changes in diastolic function (Figure 4; Nishida and Otsu, 2017). However, as the disease progresses, these abnormalities accumulate, culminating in cardiomyocyte death, hypertrophy, fibrosis, and diastolic and systolic dysfunction (Riehle and Bauersachs, 2019; Tan et al., 2020; Figure 4). The etiology of metabolic cardiomyopathy is multifactorial and has been previously reviewed (Nishida and Otsu, 2017; Riehle and Bauersachs, 2019; Nakamura and Sadoshima, 2020; Tan et al., 2020).
In the presence of obesity, type 2 diabetes, or insulin resistance, the heart functions with a dysregulated energy metabolism. More specifically, impaired insulin-receptor signaling leads to reduced translocation of glucose transporter 4 to the cell membrane resulting in reduced glucose uptake and availability for oxidation (Figure 4; Cook et al., 2010; Tan et al., 2020). On the other hand, increased fatty acid uptake (Figure 4) and utilization occur due to increased membrane localization of fatty acid translocase (FAT/CD36) and higher PPAR-α activity (Coort et al., 2004; Nakamura et al., 2019; Riehle and Bauersachs, 2019), which induces the expression of genes involved in fatty acid uptake and oxidation and further prevents glucose oxidation through stimulation of pyruvate dehydrogenase kinase 4 expression (Nakamura and Sadoshima, 2020). Consequently, high rates of FAO and loss of glucose availability increase oxygen consumption, impair cardiac efficiency, and induce mitochondrial ROS production (Figure 4; Boudina et al., 2007; Borghetti et al., 2018). This imbalance between fatty acid uptake and oxidation leads to excessive accumulation of lipids and lipotoxic intermediates (e.g., ceramides) in cardiomyocytes, which has been associated with increased ROS levels, ER stress, mitochondrial membrane remodeling, and cardiomyocyte apoptosis (Figure 4; Bikman and Summers, 2011; Wende et al., 2012; Riehle and Bauersachs, 2019). Under hyperglycemic conditions, toxic glucose intermediates may contribute to the generation of advanced-glycosylation end products (AGEs) that trigger enhanced proinflammatory and profibrotic signaling in the heart (Singh et al., 2001; Nakamura and Sadoshima, 2020). These pathways promote increased extracellular matrix (ECM) protein production and reduced activity of ECM-degrading enzymes, both of which contribute to cardiac fibrosis and contractile dysfunction (Westermann et al., 2007; D’Souza et al., 2011; Borghetti et al., 2018). Additionally, activation of the renin-angiotensin-aldosterone system increases angiotensin II that stimulates cardiac fibrosis and hypertrophy (Kumar et al., 2012).
Despite the significant body of research reporting various possible mechanisms contributing to metabolic cardiomyopathy, the pathology of this disorder is still not entirely understood. Several studies have proposed that diabetes-induced changes in mitochondrial function lead to cardiomyopathy. A detailed review by Schilling (2015) discusses the hypothesis that diabetes promotes mitochondrial dynamic dysregulation, which is a trigger in the development of diabetic-induced cardiomyopathy progression and heart failure. However, more mechanistic studies are needed to confirm this hypothesis and understand the underlying pathobiology to advance treatment with targeted therapeutics. It should be noted that in a broader context, metabolic cardiomyopathy can also develop as a consequence of different inherited metabolic storage disorders that manifest during childhood (Albakri, 2019). However, their pathology differs from systemic disease-related cardiomyopathy and involves altered energy production due to deficiencies in certain enzymes regulating glycogen, glycolipid, and glycosaminoglycan metabolism (Guertl et al., 2001; Albakri, 2019).
Approximately 50% of patients suffering with mitochondrial diseases also present with cardiomyopathy (Florian et al., 2015). In many cases mitochondrial cardiomyopathies have an underlying genetic component resulting in malfunction of mitochondrial respiratory chain, FAO or cardiolipin synthesis and alterations of mitochondrial dynamics (El-Hattab and Scaglia, 2016a; Figure 4).
Taking into account that cardiac muscles are one of the high energy tissues in the body, it is not surprising that mitochondrial disorders associated with OXPHOS dysfunction manifest as cardiomyopathy (Figure 4; Thorburn et al., 2004). Moreover, mitochondrial disorder-related cardiomyopathies may be associated with defects in the synthesis of coenzyme Q10 (Potgieter et al., 2013), synthesis of the OXPHOS Fe–S clusters, transport of adenine nucleotides across IMM maintenance of mtDNA, transfer of mitochondrial RNAs, ribosomal proteins, ribosomal RNAs and translation factors (El-Hattab and Scaglia, 2016b). Mutations in the genes encoding mitochondrial proteins often lead to aberrant OXPHOS machinery resulting in not only an ATP deficiency but also increased ROS production and/or alterations in the antioxidant defense system, nitric oxide (NO) deficiency and dysregulation of Ca2+ homeostasis (El-Hattab and Scaglia, 2016a,b).
The energy substrates primarily used by the heart include fatty acids and carbohydrates; however fatty acids are the main energy substrate for the heart and they provide the majority of cofactors crucial for mitochondrial oxidative phosphorylation. All things considered, it is not surprising that alterations in the mitochondrial FAO pathway lead to the development of heart failure. Fatty acid and glucose metabolism are interconnected to regulate each other in a process referred to as the glucose/fatty acid cycle often called Randle Cycle (Randle et al., 1963; Arslanian and Kalhan, 1994). Interestingly, in the heart an increased rate of FAO decreases glucose oxidation and conversly an increased rate of glucose oxidation inhibits FAO (Figure 4). Alterations in key enzymes involved in FAO lead to mitochondrial cardiomyopathy. These enzymes include very long-chain acyl-CoA dehydrogenase (VLCAD), long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD); trifunctional protein (TFP); carnitine-acylcarnitine translocase (CACT); carnitine palmitoyltransferase type 2 (CPT2); carnitine transporter (CT) and multiple acyl-CoA dehydrogenase (MAD) (Merritt et al., 2018). In the case of diabetic cardiomyopathy (where an oversupply of fatty acids is responsible for the observed cardiac lipotoxicity) excess fatty acids promote accumulation of lipid intermediates and surprisingly, in the case of diabetes, it is accompanied by increased FAO (Figure 4; Fillmore et al., 2014).
Cardiolipin is an essential constituent of IMM and contributes up to 20% of total IMM lipids (Schlame and Greenberg, 2017; Tatsuta and Langer, 2017). Due to the fact that IMM is comprised of approximately 75% protein and 25% lipid, alterations in the cardiolipin content and/or its composition have a direct impact on the structure and properties of the IMM and influence many mitochondrial processes including oxidative phosphorylation and protein translocation to mitochondria. Cardiolipin is essential (other phospholipids cannot substitute for it) for optimal function of the mitochondrial respiratory chain complexes I, III, and IV and is required for the structural integrity and formation of respiratory chain supercomplexes (Figure 4; Pfeiffer et al., 2003; Dudek et al., 2013; Letts et al., 2016). Moreover, changes in cardiolipin content or its’ species composition contribute to higher ROS production (Paradies et al., 2004; Nickel et al., 2014). Interestingly, mutations in the DNAJC19 gene encoding a component of the mitochondrial protein import machinery in the IMM also induces cardiolipin accumulation with altered acyl chain. This is most likely due to DNAJC19’s interaction with prohibitins (PBH) to regulate cardiolipin remodeling (Richter-Dennerlein et al., 2014).
Barth Syndrome (BTHS) is a rare X-linked recessive mitochondrial cardiomyopathy caused by an altered cardiolipin metabolism. BTHS pathology includes changes in mitochondrial membrane phospholipids, lactic acidosis, organic aciduria and skeletal muscle weakness. BTHS is linked to gene mutations in the phospholipid transacylase localized to mitochondria, taffazzin (TAZ) (Dudek and Maack, 2017) and is involved in cardiolipin acyl chain remodeling. In physiological conditions up to 90% of cardiolipin in cardiac mitochondria exist as tetralinoleoylcardiolipin. Decreases in tafazzin activity reduce cardiolipin abundance and increase monolysocardiolipin levels. There are several detailed reviews on BTHS and cardiolipin metabolism in cardiomyopathy (Ikon and Ryan, 2017; see Dudek, 2017).
Overall patients presenting with dilated cardiomyopathy continue to progress to end stage heart failure (1:3 patients) with disease maintenance occurring in 1:4 patients. Currently, a heart transplant is the best treatment option because there are no targeted therapies available due to the high complexity of dilated cardiomyopathy (Begic et al., 2018). Targeted therapies need to be developed that treat this complex disease potentially by targeting the underlying molecular pathways that are dysregulated. For example, targeting the OXPHOS pathway and subsequent mitochondrial dysfunction may be a way to attenuate cardiomyocyte death and maintain cardiac function (Brown et al., 2017). Advances in genetic analysis and molecular pathway alterations due to these gene mutations has paved the way for early detection and focused research on the development of potential preventative therapies (Verdonschot et al., 2019). Currently, mitochondrial-specific targeting therapies for dilated cardiomyopathy have not yet entered the clinic as they are at the basic research level. As research continues it will help determine whether restoring mitochondrial function may be way an efficacious way to treat dilated cardiomyopathies.
One research area that has emerged recently is the development of cardiac-specific organoids to study the complexities of cardiovascular disease. The term organoid is used to describe in vitro 3D multicellular tissues generated from pluripotent and adult stem cells that recapitulate some of the key structural and functional features of their in vivo counterparts (de Souza, 2018). Advances in efficient differentiation of induced-pluripotent cells (iPSC) into several cardiac cell types provide a novel unlimited cell source for developing cardiac organoids, especially due to the difficulties of adult stem cell isolation and ethical concerns regarding embryonic cells (Moretti et al., 2013; Zamani et al., 2018). Unlike traditional 2D cell cultures, cardiac organoids provide more accurate representations of the complexity of cell-cell and cell-extracellular matrix interactions. Moreover, being derived from human stem cells, they reflect cardiac (patho)physiology more appropriately compared with animal models thereby helping to overcome substantial functional between-species differences and animal to human translation issues (Moretti et al., 2013).
To date, iPSC cells and derived 3D organoids are used to model several cardiac disorders caused by cardiomyocyte gene mutations including dilated cardiomyopathy, hypertrophic cardiomyopathy, Barth’s syndrome and glycogen-storage cardiomyopathy (Wang G. et al., 2014; Hinson et al., 2015, 2016; Cashman et al., 2016; Nugraha et al., 2019). In addition to these monogenic diseases, cardiac organoids may provide great promise for modeling complex, lifestyle-related heart pathologies including acute myocardial infarction (Richards et al., 2020) and cardiomyopathy. For instance, relevant in vitro models of metabolic cardiomyopathy should embody all of the key features of the diabetic heart, including metabolic shift, lipotoxicity, insulin resistance and altered functionality (contractility and changes in mitochondrial function). Most of these characteristics have been previously recapitulated in 2D in vitro models that use iPSC-derived cardiomyocytes exposed to a diabetic-like environment (Drawnel et al., 2014). However, iPSC-derived cardiomyocytes are somewhat immature, with a fetal-like profile, which relies primarily on glucose utilization, compared to adult cardiomyocytes where metabolism depends on FAO. Ideally, a model of metabolic cardiomyopathy would provide evidence of this metabolic switch, which may be challenging to achieve using iPSC-derived cardiomyocytes (Jiang et al., 2018; Granéli et al., 2019). Nevertheless, considerable advances have been made in overcoming this issue of immaturity, especially using 3D culture, mechanical, or electrical conditioning techniques that provide a more adult-like cardiomyocyte phenotype in terms of oxidative metabolism, gene expression and calcium handling (Ronaldson-Bouchard et al., 2018; Golforoush and Schneider, 2020). Moreover, combining different cardiac cell types into 3D tissue-like organoids promotes the maturity and function of iPSCs-derived cardiomyocytes (Giacomelli et al., 2017; Golforoush and Schneider, 2020). Recently, cardiac organoids containing four different cell types (cardiomyocytes, epicardial, endothelial cells, and cardiac fibroblast) derived from the same iPSC have been created (Helms et al., 2019). Furthermore, Lee and colleagues developed a 3D heart organoid comprised of cardiomyocytes, conducting tissues, endothelial and smooth muscle cells, with atrial and ventricular parts, myocardial contraction, and gene expression profiles resembling their in vivo counterparts (Lee et al., 2020). Further research is necessary to determine how these organoids may be used to accurately model the complex pathology of dilated cardiomyopathy for a better understanding of underlying molecular mechanisms, mitochondrial function and identification of potential therapeutic targets.
Mitochondria are the central hub of the cell, controlling and regulating several different functions such as bioenergetics, biosynthesis, ROS production and cell death. Considering the high-energy demand of the heart, much research has focused on how mitochondria play a central role in maintaining myocardial homeostasis. Morphological and functional changes of mitochondria are associated with CVD. Therefore, a finely tuned healthy mitochondrial network is required in order to prevent CVDs. Mechanisms of surveillance are also required to conserve mitochondrial fitness and alterations of this quality control system, which requires a delicate balance between fusion-fission machinery and mitophagy and loss of this balance, may lead to the accumulation of damaged and dysfunctional mitochondria. Loss of healthy mitochondria in the heart is linked directly with a decline of energy supply, increased ROS and culminates with cardiomyopathy onset and heart failure.
To date, current therapies focus on reducing heart energy demand and preventing further worsening of cardiac muscle function (Acquatella, 2000; Verdonschot et al., 2019). Continued research into the molecular mechanisms of CVD is needed to develop targeted therapies. Indeed, the continuing development of novel approaches such as organoids to study the aberrant pathology of the heart will further our understanding of how mitochondria are cardioprotective and identify new therapeutic targets for treating and preventing cardiomyopathies.
DR: conceptualization, writing—original draft, writing—review and editing. VM-U, FA, LM, YP, MW, IK, and MG: writing—original draft. PP: review and editing. CG: conceptualization, review, and editing. MM: conceptualization, funding acquisition, review, and editing. All authors have read and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by NIH R01HD091162 (to MM); the Italian Association for Cancer Research (IG-19803 to CG and IG-23670 to PP); Progettidi Rilevante Interesse Nazionale (PRIN20177E9EPY to CG and PRIN2017E5L5P3 to PP); local funds from University of Ferrara and A-ROSE (to PP and CG); CG and PP are grateful to Camilla degli Scrovegni for continuous support. MW and YP are supported by the National Science Center, Poland (UMO02018/29/B/NZ1/00589). MG and IK are supported by Ministry of Education, Science and Technological Development of the Republic of Serbia (contract#451-03-68/2020-14/200015).
Acquatella, H. (2000). [Dilated cardiomyopathy: recent advances and current treatment]. Rev. Esp. Cardiol. 53, (Suppl. 1), 19–27.
Ahmed, A., Spinty, S., Murday, V., Longman, C., and Khand, A. (2016). A de-novo deletion of dystrophin provoking severe ‘peri-partum cardiomyopathy’: the importance of genetic testing in peripartum cardiomyopathy to uncover female carriers. Int. J. Cardiol. 203, 1084–1085. doi: 10.1016/j.ijcard.2015.10.239
Albakri, A. (2019). Metabolic cardiomyopathy: a review and pooled analysis of pathophysiology, diagnosis and clinical management. Med. Clin. Arch. 3, 1–14. doi: 10.15761/MCA.1000152
Alston, C. L., Ceccatelli Berti, C., Blakely, E. L., Oláhová, M., He, L., McMahon, C. J., et al. (2015). A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency. Hum. Genet. 134, 869–879. doi: 10.1007/s00439-015-1568-z
Alston, C. L., Davison, J. E., Meloni, F., van der Westhuizen, F. H., He, L., Hornig-Do, H.-T., et al. (2012). Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J. Med. Genet. 49, 569–577. doi: 10.1136/jmedgenet-2012-101146
Arany, Z., and Elkayam, U. (2016). Peripartum cardiomyopathy. Circulation 133, 1397–1409. doi: 10.1161/CIRCULATIONAHA.115.020491
Arbustini, E., Narula, N., Tavazzi, L., Serio, A., Grasso, M., Favalli, V., et al. (2014). The MOGE(S) classification of cardiomyopathy for clinicians. J. Am. Coll. Cardiol. 64, 304–318. doi: 10.1016/j.jacc.2014.05.027
Archer, S. L. (2013). Mitochondrial dynamics — mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 369, 2236–2251. doi: 10.1056/NEJMra1215233
Arslanian, S. A., and Kalhan, S. C. (1994). Correlations between fatty acid and glucose metabolism: potential explanation of insulin resistance of puberty. Diabetes 43, 908–914. doi: 10.2337/diab.43.7.908
Assomull, R. G., Prasad, S. K., Lyne, J., Smith, G., Burman, E. D., Khan, M., et al. (2006). Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J. Am. Coll. Cardiol. 48, 1977–1985. doi: 10.1016/j.jacc.2006.07.049
Balaban, R. S., Bose, S., French, S. A., and Territo, P. R. (2003). Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. Am. J. Physiol. Physiol. 284, C285–C293. doi: 10.1152/ajpcell.00129.2002
Ballard, C. K. (2019). 乳鼠心肌提取 HHS public access. Curr. Emerg. Hosp. Med. Rep. 7, 127–134. doi: 10.1007/s40138-019-00192-3.Peripartum
Barja, G. (1999). Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 31, 347–366. doi: 10.1023/a:1005427919188
Bassani, R. A., Bassani, J. W., and Bers, D. M. (1992). Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]I during caffeine contractures in rabbit cardiac myocytes. J. Physiol. 453, 591–608. doi: 10.1113/jphysiol.1992.sp019246
Basso, E., Fante, L., Fowlkes, J., Petronilli, V., Forte, M. A., and Bernardi, P. (2005). Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J. Biol. Chem. 280, 18558–18561. doi: 10.1074/jbc.C500089200
Begic, E., Begic, Z., and Naser, N. (2018). Clinical course and treatment of dilated cardiomyopathy during twenty years of follow-up. Med. Arch. 72, 68–70. doi: 10.5455/medarh.2018.72.68-70
Beutner, G., Sharma, V. K., Giovannucci, D. R., Yule, D. I., and Sheu, S.-S. (2001). Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 276, 21482–21488. doi: 10.1074/jbc.M101486200
Beutner, G., Sharma, V. K., Lin, L., Ryu, S.-Y., Dirksen, R. T., and Sheu, S.-S. (2005). Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation–metabolism coupling. Biochim. Biophys. Acta Biomembr. 1717, 1–10. doi: 10.1016/j.bbamem.2005.09.016
Bikman, B. T., and Summers, S. A. (2011). Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Invest. 121, 4222–4230. doi: 10.1172/JCI57144
Billia, F., Hauck, L., Konecny, F., Rao, V., Shen, J., and Mak, T. W. (2011). PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl. Acad. Sci. U.S.A. 108, 9572–9577. doi: 10.1073/pnas.1106291108
Bleier, L., and Dröse, S. (2013). Superoxide generation by complex III: from mechanistic rationales to functional consequences. Biochim. Biophys. Acta Bioenerg. 1827, 1320–1331. doi: 10.1016/j.bbabio.2012.12.002
Bollen, I. A. E., and van der Velden, J. (2017). The contribution of mutations in MYH7 to the onset of cardiomyopathy. Netherlands Hear. J. 25, 653–654.
Bonora, M., Bononi, A., De Marchi, E., Giorgi, C., Lebiedzinska, M., Marchi, S., et al. (2013). Role of the c subunit of the F O ATP synthase in mitochondrial permeability transition. Cell Cycle 12, 674–683. doi: 10.4161/cc.23599
Bonora, M., Morganti, C., Morciano, G., Pedriali, G., Lebiedzinska-Arciszewska, M., Aquila, G., et al. (2017). Mitochondrial permeability transition involves dissociation of F 1 F O ATP synthase dimers and C-ring conformation. EMBO Rep. 18, 1077–1089. doi: 10.15252/embr.201643602
Bonora, M., Patergnani, S., Ramaccini, D., Morciano, G., Pedriali, G., Kahsay, A. E., et al. (2020). Physiopathology of the permeability transition pore: molecular mechanisms in human pathology. Biomolecules 10:998. doi: 10.3390/biom10070998
Bonora, M., and Pinton, P. (2019). A new current for the mitochondrial permeability transition. Trends Biochem. Sci. 44, 559–561. doi: 10.1016/j.tibs.2019.04.009
Bonora, M., Wieckowski, M. R., Chinopoulos, C., Kepp, O., Kroemer, G., Galluzzi, L., et al. (2015). Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34, 1475–1486. doi: 10.1038/onc.2014.96
Bonora, M., Wieckowski, M. R., Sinclair, D. A., Kroemer, G., Pinton, P., and Galluzzi, L. (2019). Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat. Rev. Cardiol. 16, 33–55.
Borghetti, G., von Lewinski, D., Eaton, D. M., Sourij, H., Houser, S. R., and Wallner, M. (2018). Diabetic cardiomyopathy: current and future therapies. Beyond glycemic control. Front. Physiol. 9:1514. doi: 10.3389/fphys.2018.01514
Boudina, S., Sena, S., Theobald, H., Sheng, X., Wright, J. J., Hu, X. X., et al. (2007). Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 56, 2457–2466. doi: 10.2337/db07-0481
Bozkurt, B., Colvin, M., Cook, J., Cooper, L. T., Deswal, A., Fonarow, G. C., et al. (2016). Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement from the American heart association. Circulation 134, e579–e646. doi: 10.1161/CIR.0000000000000455
Brar, S. S., Khan, S. S., Sandhu, G. K., Jorgensen, M. B., Parikh, N., Hsu, J.-W. Y., et al. (2007). Incidence, mortality, and racial differences in peripartum cardiomyopathy. Am. J. Cardiol. 100, 302–304. doi: 10.1016/j.amjcard.2007.02.092
Brown, D. A., and O’Rourke, B. (2010). Cardiac mitochondria and arrhythmias. Cardiovasc. Res. 88, 241–249. doi: 10.1093/cvr/cvq231
Brown, D. A., Perry, J. B., Allen, M. E., Sabbah, H. N., Stauffer, B. L., Shaikh, S. R., et al. (2017). Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 14, 238–250. doi: 10.1038/nrcardio.2016.203
Buntinas, L., Gunter, K. K., Sparagna, G. C., and Gunter, T. E. (2001). The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim. Biophys. Acta Bioenerg. 1504, 248–261.
Cadenas, S., Aragonés, J., and Landázuri, M. O. (2010). Mitochondrial reprogramming through cardiac oxygen sensors in ischaemic heart disease. Cardiovasc. Res. 88, 219–228. doi: 10.1093/cvr/cvq256
Cahill, T. J., Leo, V., Kelly, M., Stockenhuber, A., Kennedy, N. W., Bao, L., et al. (2015). Resistance of dynamin-related protein 1 oligomers to disassembly impairs mitophagy, resulting in myocardial inflammation and heart failure. J. Biol. Chem. 290, 25907–25919. doi: 10.1074/jbc.M115.665695
Campos, J. C., Bozi, L. H. M., Bechara, L. R. G., Lima, V. M., and Ferreira, J. C. B. (2016). Mitochondrial quality control in cardiac diseases. Front. Physiol. 7:479. doi: 10.3389/fphys.2016.00479
Cao, Y.-P., and Zheng, M. (2019). Mitochondrial dynamics and inter-mitochondrial communication in the heart. Arch. Biochem. Biophys. 663, 214–219. doi: 10.1016/j.abb.2019.01.017
Cashman, T. J., Josowitz, R., Johnson, B. V., Gelb, B. D., and Costa, K. D. (2016). Human engineered cardiac tissues created using induced pluripotent stem cells reveal functional characteristics of BRAF-mediated hypertrophic cardiomyopathy. PLoS One 11:e0146697. doi: 10.1371/journal.pone.0146697
Cave, A. C., Brewer, A. C., Narayanapanicker, A., Ray, R., Grieve, D. J., Walker, S., et al. (2006). NADPH oxidases in cardiovascular health and disease. Antioxid. Redox Signal. 8, 691–728. doi: 10.1089/ars.2006.8.691
Chen, H., Ren, S., Clish, C., Jain, M., Mootha, V., McCaffery, J. M., et al. (2015). Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J. Cell Biol. 211, 795–805. doi: 10.1083/jcb.201507035
Chen, Y., Csordás, G., Jowdy, C., Schneider, T. G., Csordás, N., Wang, W., et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca 2+ crosstalk. Circ. Res. 111, 863–875. doi: 10.1161/CIRCRESAHA.112.266585
Chen, Y., and Dorn, G. W. (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Chen, Y., Liu, Y., and Dorn, G. W. (2011). Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331. doi: 10.1161/CIRCRESAHA.111.258723
Chen, Y.-R., and Zweier, J. L. (2014). Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 114, 524–537. doi: 10.1161/CIRCRESAHA.114.300559
Cheng, H., Lederer, W., and Cannell, M. (1993). Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science 262, 740–744. doi: 10.1126/science.8235594
Cheng, V. E., and Prior, D. L. (2013). Peripartum cardiomyopathy in a previously asymptomatic carrier of duchenne muscular dystrophy. Heart Lung Circ. 22, 677–681. doi: 10.1016/j.hlc.2012.11.015
Chistiakov, D. A., Shkurat, T. P., Melnichenko, A. A., Grechko, A. V., and Orekhov, A. N. (2018). The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann. Med. 50, 121–127. doi: 10.1080/07853890.2017.1417631
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijević, D., Sundier, S. Y., Robb, E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi: 10.1038/nature13909
Cook, S. A., Varela-Carver, A., Mongillo, M., Kleinert, C., Khan, M. T., Leccisotti, L., et al. (2010). Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur. Heart J. 31, 100–111. doi: 10.1093/eurheartj/ehp396
Coort, S. L. M., Hasselbaink, D. M., Koonen, D. P. Y., Willems, J., Coumans, W. A., Chabowski, A., et al. (2004). Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese zucker rats. Diabetes 53, 1655–1663. doi: 10.2337/diabetes.53.7.1655
Crespo, F. L., Sobrado, V. R., Gomez, L., Cervera, A. M., and McCreath, K. J. (2010). Mitochondrial reactive oxygen species mediate cardiomyocyte formation from embryonic stem cells in high glucose. Stem Cells 28, 1132–1142. doi: 10.1002/stem.441
Cunningham, F. G., Byrne, J. J., and Nelson, D. B. (2019). Peripartum cardiomyopathy. Obstet. Gynecol. 133, 167–179. doi: 10.1097/AOG.0000000000003011
De La Fuente, S., Fernandez-Sanz, C., Vail, C., Agra, E. J., Holmstrom, K., Sun, J., et al. (2016). Strategic positioning and biased activity of the mitochondrial calcium uniporter in cardiac muscle. J. Biol. Chem. 291, 23343–23362. doi: 10.1074/jbc.M116.755496
De La Fuente, S., Lambert, J. P., Nichtova, Z., Fernandez Sanz, C., Elrod, J. W., Sheu, S.-S., et al. (2018). Spatial separation of mitochondrial calcium uptake and extrusion for energy-efficient mitochondrial calcium signaling in the heart. Cell Rep. 24, 3099–3107.e4. doi: 10.1016/j.celrep.2018.08.040
De la Fuente, S., and Sheu, S.-S. (2019). SR-mitochondria communication in adult cardiomyocytes: a close relationship where the Ca2+ has a lot to say. Arch. Biochem. Biophys. 663, 259–268. doi: 10.1016/j.abb.2019.01.026
De Paris, V., Biondi, F., Stolfo, D., Merlo, M., and Sinagra, G. (2019). “Pathophysiology,” in Dilated Cardiomyopathy: From Genetics to Clinical Management, eds G. Sinagra, M. Merlo, and B. Pinamonti (Cham: Springer International Publishing), 17–25. doi: 10.1007/978-3-030-13864-6_3
Dellefave, L., and McNally, E. M. (2010). The genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 25, 198–204. doi: 10.1097/HCO.0b013e328337ba52
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M., and Bers, D. M. (2002). Intracellular Na + concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105, 2543–2548. doi: 10.1161/01.CIR.0000016701.85760.97
Doenst, T., Nguyen, T. D., and Abel, E. D. (2013). Cardiac metabolism in heart failure. Circ. Res. 113, 709–724. doi: 10.1161/CIRCRESAHA.113.300376
Drawnel, F. M., Boccardo, S., Prummer, M., Delobel, F., Graff, A., Weber, M., et al. (2014). Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep. 9, 810–820. doi: 10.1016/j.celrep.2014.09.055
D’Souza, A., Howarth, F. C., Yanni, J., Dobryznski, H., Boyett, M. R., Adeghate, E., et al. (2011). Left ventricle structural remodelling in the prediabetic Goto-Kakizaki rat. Exp. Physiol. 96, 875–888. doi: 10.1113/expphysiol.2011.058271
Dudek, J. (2017). Role of cardiolipin in mitochondrial signaling pathways. Front. Cell. Dev. Biol. 5:90. doi: 10.3389/fcell.2017.00090
Dudek, J., Cheng, I.-F., Balleininger, M., Vaz, F. M., Streckfuss-Bömeke, K., Hübscher, D., et al. (2013). Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome. Stem Cell Res. 11, 806–819. doi: 10.1016/j.scr.2013.05.005
Dudek, J., and Maack, C. (2017). Barth syndrome cardiomyopathy. Cardiovasc. Res. 113, 399–410. doi: 10.1093/cvr/cvx014
Eisner, V., Csordas, G., and Hajnoczky, G. (2013). Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. 126, 2965–2978. doi: 10.1242/jcs.093609
Elgass, K., Pakay, J., Ryan, M. T., and Palmer, C. S. (2013). Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta Mol. Cell Res. 1833, 150–161. doi: 10.1016/j.bbamcr.2012.05.002
El-Hattab, A. W., and Scaglia, F. (2016a). Mitochondrial cardiomyopathies. Front. Cardiovasc. Med. 3:25. doi: 10.3389/fcvm.2016.00025
El-Hattab, A. W., and Scaglia, F. (2016b). Mitochondrial cytopathies. Cell Calcium 60, 199–206. doi: 10.1016/j.ceca.2016.03.003
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., et al. (2007). Classification of the cardiomyopathies: a position statement from the European society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 29, 270–276. doi: 10.1093/eurheartj/ehm342
Fabiato, A. (1983). Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. Physiol. 245, C1–C14. doi: 10.1152/ajpcell.1983.245.1.C1
Fan, H., He, Z., Huang, H., Zhuang, H., Liu, H., Liu, X., et al. (2020). Mitochondrial quality control in cardiomyocytes: a critical role in the progression of cardiovascular diseases. Front. Physiol. 11:252. doi: 10.3389/fphys.2020.00252
Fernandez-Sanz, C., Ruiz-Meana, M., Miro-Casas, E., Nuñez, E., Castellano, J., Loureiro, M., et al. (2014). Defective sarcoplasmic reticulum–mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 5:e1573. doi: 10.1038/cddis.2014.526
Fillmore, N., Mori, J., and Lopaschuk, G. D. (2014). Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 171, 2080–2090. doi: 10.1111/bph.12475
Fiorillo, C., Astrea, G., Savarese, M., Cassandrini, D., Brisca, G., Trucco, F., et al. (2016). MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J. Rare Dis. 11:91.
Flarsheim, C. E., Grupp, I. L., and Matlib, M. A. (1996). Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am. J. Physiol. Circ. Physiol. 271, H192–H202. doi: 10.1152/ajpheart.1996.271.1.H192
Florian, A., Ludwig, A., Stubbe-Dräger, B., Boentert, M., Young, P., Waltenberger, J., et al. (2015). Characteristic cardiac phenotypes are detected by cardiovascular magnetic resonance in patients with different clinical phenotypes and genotypes of mitochondrial myopathy. J. Cardiovasc. Magn. Reson. 17:40. doi: 10.1186/s12968-015-0145-x
Forte, M., Schirone, L., Ameri, P., Basso, C., Catalucci, D., Modica, J., et al. (2020). The role of mitochondrial dynamics in cardiovascular diseases. Br. J. Pharmacol. doi: 10.1111/bph.15068 [Epub ahead of print].
Frank, D., Kuhn, C., Katus, H. A., and Frey, N. (2006). The sarcomeric Z-disc: a nodal point in signalling and disease. J. Mol. Med. 84, 446–468. doi: 10.1007/s00109-005-0033-1
Franzini-Armstrong, C., and Protasi, F. (1997). Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol. Rev. 77, 699–729. doi: 10.1152/physrev.1997.77.3.699
Franzini-Armstrong, C., Protasi, F., and Ramesh, V. (1999). Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 77, 1528–1539.
Frederick, R. L., and Shaw, J. M. (2007). Moving mitochondria: establishing distribution of an essential organelle. Traffic 8, 1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x
Fu, J., Li, J., Tweedie, D., Yu, H., Chen, L., Wang, R., et al. (2006). Crucial role of the sarcoplasmic reticulum in the developmental regulation of Ca 2+ transients and contraction in cardiomyocytes derived from embryonic stem cells. FASEB J. 20, 181–183. doi: 10.1096/fj.05-4501fje
García-Pérez, C., Hajnóczky, G., and Csordás, G. (2008). Physical coupling supports the local Ca 2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J. Biol. Chem. 283, 32771–32780. doi: 10.1074/jbc.M803385200
Gebert, N., Joshi, A. S., Kutik, S., Becker, T., McKenzie, M., Guan, X. L., et al. (2009). Mitochondrial cardiolipin involved in outer-membrane protein biogenesis: implications for Barth syndrome. Curr. Biol. 19, 2133–2139. doi: 10.1016/j.cub.2009.10.074
Giacomelli, E., Bellin, M., Sala, L., van Meer, B. J., Tertoolen, L. G. J., Orlova, V. V., et al. (2017). Three-dimensional cardiac microtissues composed of cardiomyocytes and endothelial cells co-differentiated from human pluripotent stem cells. Development 144, 1008–1017. doi: 10.1242/dev.143438
Giorgi, C., Agnoletto, C., Bononi, A., Bonora, M., De Marchi, E., Marchi, S., et al. (2012). Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 12, 77–85. doi: 10.1016/j.mito.2011.07.004
Giorgi, C., Danese, A., Missiroli, S., Patergnani, S., and Pinton, P. (2018a). Calcium dynamics as a machine for decoding signals. Trends Cell Biol. 28, 258–273. doi: 10.1016/j.tcb.2018.01.002
Giorgi, C., Marchi, S., and Pinton, P. (2018b). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730.
Goffart, S., Vonkleistretzow, J., and Wiesner, R. (2004). Regulation of mitochondrial proliferation in the heart: power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc. Res. 64, 198–207. doi: 10.1016/j.cardiores.2004.06.030
Golforoush, P., and Schneider, M. D. (2020). Intensive care for human hearts in pluripotent stem cell models. NPJ Regen. Med. 5:4. doi: 10.1038/s41536-020-0090-7
Gong, G., Song, M., Csordas, G., Kelly, D. P., Matkovich, S. J., and Dorn, G. W. (2015). Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350, aad2459. doi: 10.1126/science.aad2459
Granéli, C., Hicks, R., Brolén, G., Synnergren, J., and Sartipy, P. (2019). Diabetic cardiomyopathy modelling using induced pluripotent stem cell derived cardiomyocytes: recent advances and emerging models. Stem Cell Rev. Rep. 15, 13–22.
Grattan, D. R., Steyn, F. J., Kokay, I. C., Anderson, G. M., and Bunn, S. J. (2008). Pregnancy-induced adaptation in the neuroendocrine control of prolactin secretion. J. Neuroendocrinol. 20, 497–507. doi: 10.1111/j.1365-2826.2008.01661.x
Green, D. R., and Reed, J. C. (1998). Mitochondria and apoptosis. Science 281, 1309–1312. doi: 10.1126/science.281.5381.1309
Griffiths, E. J., Stern, M. D., and Silverman, H. S. (1997). Measurement of mitochondrial calcium in single living cardiomyocytes by selective removal of cytosolic indo 1. Am. J. Physiol. Physiol. 273, C37–C44. doi: 10.1152/ajpcell.1997.273.1.C37
Guertl, B., Noehammer, C., and Hoefler, G. (2001). Metabolic cardiomyopathies. Int. J. Exp. Pathol. 81, 349–372. doi: 10.1046/j.1365-2613.2000.00186.x
Gunderson, E. P., Croen, L. A., Chiang, V., Yoshida, C. K., Walton, D., and Go, A. S. (2011). Epidemiology of peripartum cardiomyopathy. Obstet. Gynecol. 118, 583–591. doi: 10.1097/AOG.0b013e318229e6de
Hackenbrock, C. R. (1966). Ultrastructural bases for metabolically linked mechanical activity in mitochondria. J. Cell Biol. 30, 269–297. doi: 10.1083/jcb.30.2.269
Haghikia, A., Podewski, E., Libhaber, E., Labidi, S., Fischer, D., Roentgen, P., et al. (2013). Phenotyping and outcome on contemporary management in a German cohort of patients with peripartum cardiomyopathy. Basic Res. Cardiol. 108:366.
Harhous, Z., Booz, G. W., Ovize, M., Bidaux, G., and Kurdi, M. (2019). An update on the multifaceted roles of STAT3 in the heart. Front. Cardiovasc. Med. 6:150. doi: 10.3389/fcvm.2019.00150
Helms, H. R., Jarrell, D. K., and Jacot, J. G. (2019). Generation of cardiac organoids using cardiomyocytes, endothelial cells, epicardial cells, and cardiac fibroblasts derived from human induced pluripotent stem cells. FASEB J. 33:lb170. doi: 10.1096/fasebj.2019.33.1_supplement.lb170
Hernandez-Resendiz, S., Prunier, F., Girao, H., Dorn, G., and Hausenloy, D. J. (2020). Targeting mitochondrial fusion and fission proteins for cardioprotection. J. Cell. Mol. Med. 24, 6571–6585. doi: 10.1111/jcmm.15384
Herpel, E., Pritsch, M., Koch, A., Dengler, T. J., Schirmacher, P., and Schnabel, P. A. (2006). Interstitial fibrosis in the heart: differences in extracellular matrix proteins and matrix metalloproteinases in end-stage dilated, ischaemic and valvular cardiomyopathy. Histopathology 48, 736–747. doi: 10.1111/j.1365-2559.2006.02398.x
Hershberger, R. E., Morales, A., and Siegfried, J. D. (2010). Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet. Med. 12, 655–667. doi: 10.1097/GIM.0b013e3181f2481f
Hibbard, J. (1999). A modified definition for peripartum cardiomyopathy and prognosis based on echocardiography. Obstet. Gynecol. 94, 311–316.
Hilfiker-Kleiner, D., Kaminski, K., Podewski, E., Bonda, T., Schaefer, A., Sliwa, K., et al. (2007). A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 128, 589–600. doi: 10.1016/j.cell.2006.12.036
Hilfiker-Kleiner, D., and Sliwa, K. (2014). Pathophysiology and epidemiology of peripartum cardiomyopathy. Nat. Rev. Cardiol. 11, 364–370. doi: 10.1038/nrcardio.2014.37
Hilfiker-Kleiner, D., Struman, I., Hoch, M., Podewski, E., and Sliwa, K. (2012). 16-kDa prolactin and bromocriptine in postpartum cardiomyopathy. Curr. Heart Fail. Rep. 9, 174–182.
Hinson, J. T., Chopra, A., Lowe, A., Sheng, C. C., Gupta, R. M., Kuppusamy, R., et al. (2016). Integrative analysis of PRKAG2 cardiomyopathy iPS and microtissue models identifies AMPK as a regulator of metabolism, survival, and fibrosis. Cell Rep. 17, 3292–3304. doi: 10.1016/j.celrep.2016.11.066
Hinson, J. T., Chopra, A., Nafissi, N., Polacheck, W. J., Benson, C. C., Swist, S., et al. (2015). Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 349, 982–986. doi: 10.1126/science.aaa5458
Hoch, F. L. (1992). Cardiolipins and biomembrane function. Biochim. Biophys. Acta Rev. Biomembr. 1113, 71–133.
Holmuhamedov, E. L., Oberlin, A., Short, K., Terzic, A., and Jahangir, A. (2012). Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PLoS One 7:e44667. doi: 10.1371/journal.pone.0044667
Hoppins, S. (2014). The regulation of mitochondrial dynamics. Curr. Opin. Cell Biol. 29, 46–52. doi: 10.1016/j.ceb.2014.03.005
Hu, L., Ding, M., Tang, D., Gao, E., Li, C., Wang, K., et al. (2019). Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics 9, 3687–3706. doi: 10.7150/thno.33684
Ikeda, Y., Shirakabe, A., Maejima, Y., Zhai, P., Sciarretta, S., Toli, J., et al. (2015). Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ. Res. 116, 264–278. doi: 10.1161/CIRCRESAHA.116.303356
Ikon, N., and Ryan, R. O. (2017). Barth syndrome: connecting cardiolipin to cardiomyopathy. Lipids 52, 99–108.
Jain-Ghai, S., Cameron, J. M., Al Maawali, A., Blaser, S., MacKay, N., Robinson, B., et al. (2013). Complex II deficiency-A case report and review of the literature. Am. J. Med. Genet. Part A 161, 285–294. doi: 10.1002/ajmg.a.35714
Javadov, S., and Karmazyn, M. (2007). Mitochondrial permeability transition pore opening as an endpoint to initiate cell death and as a putative target for cardioprotection. Cell. Physiol. Biochem. 20, 1–22. doi: 10.1159/000103747
Ji, A.-R., Ku, S.-Y., Cho, M. S., Kim, Y. Y., Kim, Y. J., Oh, S. K., et al. (2010). Reactive oxygen species enhance differentiation of human embryonic stem cells into mesendodermal lineage. Exp. Mol. Med. 42, 175–186. doi: 10.3858/emm.2010.42.3.018
Jiang, Y., Park, P., Hong, S.-M., and Ban, K. (2018). Maturation of cardiomyocytes derived from human pluripotent stem cells: current strategies and limitations. Mol. Cells 41, 613–621. doi: 10.14348/molcells.2018.0143
Karch, J., Bround, M. J., Khalil, H., Sargent, M. A., Latchman, N., Terada, N., et al. (2019). Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 5:eaaw4597. doi: 10.1126/sciadv.aaw4597
Karlstaedt, A., Schiffer, W., and Taegtmeyer, H. (2018). Actionable metabolic pathways in heart failure and cancer—lessons from cancer cell metabolism. Front. Cardiovasc. Med. 5:71. doi: 10.3389/fcvm.2018.00071
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. doi: 10.1038/nature02246
Knöll, R., Hoshijima, M., Hoffman, H. M. M., Person, V., Lorenzen-Schmidt, I., Bang, M.-L. M. L., et al. (2002). The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111, 943–955.
Kohlhaas, M., Liu, T., Knopp, A., Zeller, T., Ong, M. F., Böhm, M., et al. (2010). Elevated cytosolic Na + increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121, 1606–1613. doi: 10.1161/CIRCULATIONAHA.109.914911
Kolte, D., Khera, S., Aronow, W. S., Palaniswamy, C., Mujib, M., Ahn, C., et al. (2014). Temporal trends in incidence and outcomes of peripartum cardiomyopathy in the United States: a nationwide population-based study. J. Am. Heart Assoc. 3:e001056. doi: 10.1161/JAHA.114.001056
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kumar, R., Yong, Q. C., Thomas, C. M., and Baker, K. M. (2012). Review: intracardiac intracellular angiotensin system in diabetes. Am. J. Physiol. Integr. Comp. Physiol. 302, R510–R517. doi: 10.1152/ajpregu.00512.2011
Kuroda, J., Ago, T., Matsushima, S., Zhai, P., Schneider, M. D., and Sadoshima, J. (2010). NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 107, 15565–15570. doi: 10.1073/pnas.1002178107
Kussmaul, L., and Hirst, J. (2006). The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7607–7612. doi: 10.1073/pnas.0510977103
Kuznetsov, A. V., Hermann, M., Saks, V., Hengster, P., and Margreiter, R. (2009). The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 41, 1928–1939. doi: 10.1016/j.biocel.2009.03.007
Kwong, J. Q., Lu, X., Correll, R. N., Schwanekamp, J. A., Vagnozzi, R. J., Sargent, M. A., et al. (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12, 15–22. doi: 10.1016/j.celrep.2015.06.002
Lee, J., Sutani, A., Kaneko, R., Takeuchi, J., Sasano, T., Kohda, T., et al. (2020). In vitro generation of functional murine heart organoids via FGF4 and extracellular matrix. Nat. Commun. 11:4283. doi: 10.1038/s41467-020-18031-5
Lee, Y. Z. J., and Judge, D. P. (2017). The role of genetics in peripartum cardiomyopathy. J. Cardiovasc. Transl. Res. 10, 437–445. doi: 10.1007/s12265-017-9764-y
Letts, J. A., Fiedorczuk, K., and Sazanov, L. A. (2016). The architecture of respiratory supercomplexes. Nature 537, 644–648. doi: 10.1038/nature19774
Li, J., Stouffs, M., Serrander, L., Banfi, B., Bettiol, E., Charnay, Y., et al. (2006). The NADPH oxidase NOX4 drives cardiac differentiation: role in regulating cardiac transcription factors and MAP kinase activation. Mol. Biol. Cell 17, 3978–3988.
Lu, X., Thai, P. N., Lu, S., Pu, J., and Bers, D. M. (2019). Intrafibrillar and perinuclear mitochondrial heterogeneity in adult cardiac myocytes. J. Mol. Cell. Cardiol. 136, 72–84. doi: 10.1016/j.yjmcc.2019.08.013
Luongo, T. S., Lambert, J. P., Yuan, A., Zhang, X., Gross, P., Song, J., et al. (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 12, 23–34. doi: 10.1016/j.celrep.2015.06.017
Maack, C., Kartes, T., Kilter, H., Schäfers, H.-J., Nickenig, G., Böhm, M., et al. (2003). Oxygen free radical release in human failing myocardium is associated with increased activity of Rac1-GTPase and represents a target for statin treatment. Circulation 108, 1567–1574. doi: 10.1161/01.CIR.0000091084.46500.BB
Manczak, M., Sesaki, H., Kageyama, Y., and Reddy, P. H. (2012). Dynamin-related protein 1 heterozygote knockout mice do not have synaptic and mitochondrial deficiencies. Biochim. Biophys. Acta Mol. Basis Dis. 1822, 862–874. doi: 10.1016/j.bbadis.2012.02.017
Manneschi, L., and Federico, A. (1995). Polarographic analyses of subsarcolemmal and intermyofibrillar mitochondria from rat skeletal and cardiac muscle. J. Neurol. Sci. 128, 151–156. doi: 10.1016/0022-510X(94)00227-F
Marchi, S., and Pinton, P. (2014). The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J. Physiol. 592, 829–839. doi: 10.1113/jphysiol.2013.268235
Marín-García, J., and Akhmedov, A. T. (2016). Mitochondrial dynamics and cell death in heart failure. Heart Fail. Rev. 21, 123–136.
Marin-Garcia, J., Goldenthal, M. J., Ananthakrishnan, R., and Pierpont, M. E. (2000). The complete sequence of mtDNA genes in idiopathic dilated cardiomyopathy shows novel missense and tRNA mutations. J. Card. Fail. 6, 321–329. doi: 10.1054/jcaf.2000.19232
Maron, B. J., Towbin, J. A., Thiene, G., Antzelevitch, C., Corrado, D., Arnett, D., et al. (2006). Contemporary definitions and classification of the cardiomyopathies. Circulation 113, 1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287
Martínez-Reyes, I., and Chandel, N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11:102.
Merlo, M., Daneluzzi, C., Mestroni, L., Cannatà, A., and Sinagra, G. (2019). “Historical terminology, classifications, and present definition of DCM,” in Dilated Cardiomyopathy, eds G. Sinagra, M. Merlo, and B. Pinamonti (Cham: Springer International Publishing), 1–9. doi: 10.1007/978-3-030-13864-6_1
Merritt, J. L. II, Norris, M., and Kanungo, S. (2018). Fatty acid oxidation disorders. Ann. Transl. Med. 6, 473–473. doi: 10.21037/atm.2018.10.57
Messina, A., Reina, S., Guarino, F., and De Pinto, V. (2012). VDAC isoforms in mammals. Biochim. Biophys. Acta Biomembr. 1818, 1466–1476. doi: 10.1016/j.bbamem.2011.10.005
Mestroni, L., Krajinovic, M., Severini, G. M., Pinamonti, B., Di Lenarda, A., Giacca, M., et al. (1994). Familial dilated cardiomyopathy. Heart 72, S35–S41. doi: 10.1136/hrt.72.6_Suppl.S35
Mestroni, L., Rocco, C., Gregori, D., Sinagra, G., Di Lenarda, A., Miocic, S., et al. (1999). Familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 34, 181–190.
Mielniczuk, L. M., Williams, K., Davis, D. R., Tang, A. S. L., Lemery, R., Green, M. S., et al. (2006). Frequency of peripartum cardiomyopathy. Am. J. Cardiol. 97, 1765–1768. doi: 10.1016/j.amjcard.2006.01.039
Min, C. K., Yeom, D. R., Lee, K.-E., Kwon, H.-K., Kang, M., Kim, Y.-S., et al. (2012). Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 447, 371–379. doi: 10.1042/BJ20120705
Morales, A., Painter, T., Li, R., Siegfried, J. D., Li, D., Norton, N., et al. (2010). Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation 121, 2176–2182. doi: 10.1161/CIRCULATIONAHA.109.931220
Morciano, G., Bonora, M., Giorgi, C., and Pinton, P. (2017). Other bricks for the correct construction of the mitochondrial permeability transition pore complex. Cell Death Dis. 8:e2698. doi: 10.1038/cddis.2017.96
Morciano, G., Giorgi, C., Bonora, M., Punzetti, S., Pavasini, R., Wieckowski, M. R., et al. (2015). Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 78, 142–153. doi: 10.1016/j.yjmcc.2014.08.015
Morciano, G., Patergnani, S., Bonora, M., Pedriali, G., Tarocco, A., Bouhamida, E., et al. (2020). Mitophagy in cardiovascular diseases. J. Clin. Med. 9:892. doi: 10.3390/jcm9030892
Moretti, A., Laugwitz, K.-L., Dorn, T., Sinnecker, D., and Mummery, C. (2013). Pluripotent stem cell models of human heart disease. Cold Spring Harb. Perspect. Med. 3:a014027. doi: 10.1101/cshperspect.a014027
Morganti, C., Bonora, M., Sbano, L., Morciano, G., Aquila, G., Campo, G., et al. (2018). “The mitochondrial permeability transition pore,” in Mitochondrial Biology and Experimental Therapeutics, ed. P. J. Oliveira (Cham: Springer International Publishing), 47–73. doi: 10.1007/978-3-319-73344-9_5
Murphy, E., Ardehali, H., Balaban, R. S., DiLisa, F., Dorn, G. W., Kitsis, R. N., et al. (2016). Mitochondrial function, biology, and role in disease. Circ. Res. 118, 1960–1991. doi: 10.1161/RES.0000000000000104
Murray, T. V. A., Smyrnias, I., Shah, A. M., and Brewer, A. C. (2013). NADPH oxidase 4 regulates cardiomyocyte differentiation via redox activation of c-Jun protein and the cis- regulation of GATA-4 gene transcription. J. Biol. Chem. 288, 15745–15759. doi: 10.1074/jbc.M112.439844
Nakamura, M., Liu, T., Husain, S., Zhai, P., Warren, J. S., Hsu, C.-P., et al. (2019). Glycogen synthase kinase-3α promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metab. 29, 1119–1134.e12. doi: 10.1016/j.cmet.2019.01.005
Nakamura, M., and Sadoshima, J. (2020). Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 598, 2977–2993. doi: 10.1113/JP276747
Nan, J., Zhu, W., Rahman, M. S., Liu, M., Li, D., Su, S., et al. (2017). Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim. Biophys. Acta Mol. Cell Res. 1864, 1260–1273. doi: 10.1016/j.bbamcr.2017.03.006
Negretti, N., O’Neill, S. C., and Eisner, D. A. (1993). The relative contributions of different intracellular and sarcolemmal systems to relaxation in rat ventricular myocytes. Cardiovasc. Res. 27, 1826–1830. doi: 10.1093/cvr/27.10.1826
Nickel, A., Kohlhaas, M., and Maack, C. (2014). Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 73, 26–33. doi: 10.1016/j.yjmcc.2014.03.011
Nishida, K., and Otsu, K. (2017). Inflammation and metabolic cardiomyopathy. Cardiovasc. Res. 113, 389–398. doi: 10.1093/cvr/cvx012
Nugraha, B., Buono, M. F., Boehmer, L., Hoerstrup, S. P., and Emmert, M. Y. (2019). Human cardiac organoids for disease modeling. Clin. Pharmacol. Ther. 105, 79–85. doi: 10.1002/cpt.1286
Oh, C.-M., Ryu, D., Cho, S., and Jang, Y. (2020). Mitochondrial quality control in the heart: new drug targets for cardiovascular disease. Korean Circ. J. 50, 395–405. doi: 10.4070/kcj.2019.0416
Ong, S.-B., Kalkhoran, S. B., Hernández-Reséndiz, S., Samangouei, P., Ong, S.-G., and Hausenloy, D. J. (2017). Mitochondrial-shaping proteins in cardiac health and disease – the long and the short of it! Cardiovasc. Drugs Ther. 31, 87–107.
Pan, X., Liu, J., Nguyen, T., Liu, C., Sun, J., Teng, Y., et al. (2013). The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 15, 1464–1472. doi: 10.1038/ncb2868
Papanicolaou, K. N., Khairallah, R. J., Ngoh, G. A., Chikando, A., Luptak, I., O’Shea, K. M., et al. (2011). Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 31, 1309–1328.
Papanicolaou, K. N., Kikuchi, R., Ngoh, G. A., Coughlan, K. A., Dominguez, I., Stanley, W. C., et al. (2012). Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ. Res. 111, 1012–1026. doi: 10.1161/CIRCRESAHA.112.274142
Paradies, G., Petrosillo, G., Pistolese, M., Di Venosa, N., Federici, A., and Ruggiero, F. M. (2004). Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart. Circ. Res. 94, 53–59. doi: 10.1161/01.RES.0000109416.56608.64
Patten, I. S., Rana, S., Shahul, S., Rowe, G. C., Jang, C., Liu, L., et al. (2012). Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 485, 333–338. doi: 10.1038/nature11040
Pearson, G. D., Veille, J.-C., Rahimtoola, S., Hsia, J., Oakley, C. M., Hosenpud, J. D., et al. (2000). Peripartum cardiomyopathy. JAMA 283, 1183–1188. doi: 10.1001/jama.283.9.1183
Peoples, J. N., Saraf, A., Ghazal, N., Pham, T. T., and Kwong, J. Q. (2019). Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 51, 1–13. doi: 10.1038/s12276-019-0355-7
Pfeiffer, K., Gohil, V., Stuart, R. A., Hunte, C., Brandt, U., Greenberg, M. L., et al. (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880. doi: 10.1074/jbc.M308366200
Piquereau, J., Caffin, F., Novotova, M., Lemaire, C., Veksler, V., Garnier, A., et al. (2013). Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front. Physiol. 4:102. doi: 10.3389/fphys.2013.00102
Potgieter, M., Pretorius, E., and Pepper, M. S. (2013). Primary and secondary coenzyme Q10 deficiency: the role of therapeutic supplementation. Nutr. Rev. 71, 180–188. doi: 10.1111/nure.12011
Pound, K. M., Sorokina, N., Ballal, K., Berkich, D. A., Fasano, M., LaNoue, K. F., et al. (2009). Substrate–enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content. Circ. Res. 104, 805–812. doi: 10.1161/CIRCRESAHA.108.189951
Prosser, B. L., Ward, C. W., and Lederer, W. J. (2011). X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333, 1440–1445. doi: 10.1126/science.1202768
Puente, B. N., Kimura, W., Muralidhar, S. A., Moon, J., Amatruda, J. F., Phelps, K. L., et al. (2014). The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157, 565–579. doi: 10.1016/j.cell.2014.03.032
Randle, P. J., Garland, P. B., Hales, C. N., and Newsholme, E. A. (1963). The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 281, 785–789.
Rasmussen, T. P., Wu, Y., Joiner, M. A., Koval, O. M., Wilson, N. R., Luczak, E. D., et al. (2015). Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. U.S.A. 112, 9129–9134. doi: 10.1073/pnas.1504705112
Richards, D. J., Li, Y., Kerr, C. M., Yao, J., Beeson, G. C., Coyle, R. C., et al. (2020). Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat. Biomed. Eng. 4, 446–462. doi: 10.1038/s41551-020-0539-4
Richter-Dennerlein, R., Korwitz, A., Haag, M., Tatsuta, T., Dargazanli, S., Baker, M., et al. (2014). DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 20, 158–171. doi: 10.1016/j.cmet.2014.04.016
Ricke-Hoch, M., Stapel, B., Gorst, I., Haghikia, A., and Hilfiker-Kleiner, D. (2013). “The STAT3 pathway and downstream mechanisms in cardiac remodeling: friend or foe,” in Cardiac Remodeling. Advances in Biochemistry in Health and Disease, eds B. Jugdutt and N. Dhalla, Vol. 5 (New York, NY: Springer). doi: 10.1007/978-1-4614-5930-9_20
Riehle, C., and Bauersachs, J. (2019). Of mice and men: models and mechanisms of diabetic cardiomyopathy. Basic Res. Cardiol. 114:2.
Ritterhoff, J., and Tian, R. (2017). Metabolism in cardiomyopathy: every substrate matters. Cardiovasc. Res. 113, 411–421. doi: 10.1093/cvr/cvx017
Ritterhoff, J., Young, S., Villet, O., Shao, D., Neto, F. C., Bettcher, L. F., et al. (2020). Metabolic remodeling promotes cardiac hypertrophy by directing glucose to aspartate biosynthesis. Circ. Res. 126, 182–196. doi: 10.1161/CIRCRESAHA.119.315483
Ronaldson-Bouchard, K., Ma, S. P., Yeager, K., Chen, T., Song, L., Sirabella, D., et al. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243.
Rosca, M. G., and Hoppel, C. L. (2010). Mitochondria in heart failure. Cardiovasc. Res. 88, 40–50. doi: 10.1093/cvr/cvq240
Ruiz-Meana, M., Minguet, M., Bou-Teen, D., Miro-Casas, E., Castans, C., Castellano, J., et al. (2019). Ryanodine receptor glycation favors mitochondrial damage in the senescent heart. Circulation 139, 949–964. doi: 10.1161/CIRCULATIONAHA.118.035869
Sag, C. M., Wagner, S., and Maier, L. S. (2013). Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic. Biol. Med. 63, 338–349. doi: 10.1016/j.freeradbiomed.2013.05.035
Saito, T., and Sadoshima, J. (2015). Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ. Res. 116, 1477–1490. doi: 10.1161/CIRCRESAHA.116.303790
Santulli, G., Xie, W., Reiken, S. R., and Marks, A. R. (2015). Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. U.S.A. 112, 11389–11394. doi: 10.1073/pnas.1513047112
Satpathy, H. K., Frey, D., Satpathy, R., Satpathy, C., Fleming, A., Mohiuddin, S. M., et al. (2008). Peripartum cardiomyopathy. Postgrad. Med. 120, 28–32. doi: 10.3810/pgm.2008.04.1757
Schilling, J. D. (2015). The mitochondria in diabetic heart failure: from pathogenesis to therapeutic promise. Antioxid. Redox Signal. 22, 1515–1526. doi: 10.1089/ars.2015.6294
Schlame, M., and Greenberg, M. L. (2017). Biosynthesis, remodeling and turnover of mitochondrial cardiolipin. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 3–7. doi: 10.1016/j.bbalip.2016.08.010
Sciarretta, S., Maejima, Y., Zablocki, D., and Sadoshima, J. (2018). The role of autophagy in the heart. Annu. Rev. Physiol. 80, 1–26.
Sen-Chowdhry, S., Syrris, P., Pantazis, A., Quarta, G., McKenna, W. J., and Chambers, J. C. (2010). Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ. Cardiovasc. Genet. 3, 323–330. doi: 10.1161/CIRCGENETICS.109.935262
Sen-Chowdhry, S., Syrris, P., Prasad, S. K., Hughes, S. E., Merrifield, R., Ward, D., et al. (2008). Left-dominant arrhythmogenic cardiomyopathy. J. Am. Coll. Cardiol. 52, 2175–2187. doi: 10.1016/j.jacc.2008.09.019
Sharma, V. K., Ramesh, V., Franzini-Armstrong, C., and Sheu, S. S. (2000). Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 32, 97–104. doi: 10.1023/a:1005520714221
Sharp, W. W., and Archer, S. L. (2015). Mitochondrial dynamics in cardiovascular disease: fission and fusion foretell form and function. J. Mol. Med. 93, 225–228. doi: 10.1007/s00109-015-1258-2
Sheikh, F., Bang, M. L., Lange, S., and Chen, J. (2007). “Z”eroing in on the role of cypher in striated muscle function, signaling, and human disease. Trends Cardiovasc. Med. 17, 258–262. doi: 10.1016/j.tcm.2007.09.002
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2:1002. doi: 10.1038/srep01002
Shimada, T., Horita, K., Murakami, M., and Ogura, R. (1984). Morphological studies of different mitochondrial populations in monkey myocardial cells. Cell Tissue Res. 238, 577–582. doi: 10.1007/BF00219874
Shires, S. E., and Gustafsson, Å. B. (2015). Mitophagy and heart failure. J. Mol. Med. 93, 253–262. doi: 10.1007/s00109-015-1254-6
Singh, R., Barden, A., Mori, T., and Beilin, L. (2001). Advanced glycation end-products: a review. Diabetologia 44, 129–146. doi: 10.1007/s001250051591
Sliwa, K., Mebazaa, A., Hilfiker-Kleiner, D., Petrie, M. C., Maggioni, A. P., Laroche, C., et al. (2017). Clinical characteristics of patients from the worldwide registry on peripartum cardiomyopathy (PPCM). Eur. J. Heart Fail. 19, 1131–1141. doi: 10.1002/ejhf.780
Song, M., Chen, Y., Gong, G., Murphy, E., Rabinovitch, P. S., and Dorn, G. W. (2014). Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ. Res. 115, 348–353. doi: 10.1161/CIRCRESAHA.115.304384
Song, M., Gong, G., Burelle, Y., Gustafsson, Å. B., Kitsis, R. N., Matkovich, S. J., et al. (2015). Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ. Res. 117, 346–351. doi: 10.1161/CIRCRESAHA.117.306859
Sorokina, N., O’Donnell, J. M., McKinney, R. D., Pound, K. M., Woldegiorgis, G., LaNoue, K. F., et al. (2007). Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 115, 2033–2041. doi: 10.1161/CIRCULATIONAHA.106.668665
Spencer, C. T., Bryant, R. M., Day, J., Gonzalez, I. L., Colan, S. D., Thompson, W. R., et al. (2006). Cardiac and clinical phenotype in barth syndrome. Pediatrics 118, e337–e346.
Spezzacatene, A., Sinagra, G., Merlo, M., Barbati, G., Graw, S. L., Brun, F., et al. (2015). Arrhythmogenic phenotype in dilated cardiomyopathy: natural history and predictors of life-threatening arrhythmias. J. Am. Heart Assoc. 4:e002149. doi: 10.1161/JAHA.115.002149
Spinelli, J. B., and Haigis, M. C. (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20, 745–754.
Szalai, G., Csordás, G., Hantash, B. M., Thomas, A. P., and Hajnóczky, G. (2000). Calcium signal transmission between ryanodine receptors and mitochondria. J. Biol. Chem. 275, 15305–15313. doi: 10.1074/jbc.275.20.15305
Tan, Y., Zhang, Z., Zheng, C., Wintergerst, K. A., Keller, B. B., and Cai, L. (2020). Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat. Rev. Cardiol. 17, 585–607.
Tao, G., Kahr, P. C., Morikawa, Y., Zhang, M., Rahmani, M., Heallen, T. R., et al. (2016). Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 534, 119–123. doi: 10.1038/nature17959
Tatsuta, T., and Langer, T. (2017). Intramitochondrial phospholipid trafficking. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 81–89. doi: 10.1016/j.bbalip.2016.08.006
Thiene, G., Corrado, D., and Basso, C. (2007). Revisiting definition and classification of cardiomyopathies in the era of molecular medicine. Eur. Heart J. 29, 144–146. doi: 10.1093/eurheartj/ehm585
Thorburn, D. R., Sugiana, C., Salemi, R., Kirby, D. M., Worgan, L., Ohtake, A., et al. (2004). Biochemical and molecular diagnosis of mitochondrial respiratory chain disorders. Biochim. Biophys. Acta Bioenerg. 1659, 121–128. doi: 10.1016/j.bbabio.2004.08.006
Tretter, L. (2004). Generation of reactive oxygen species in the reaction catalyzed by -ketoglutarate dehydrogenase. J. Neurosci. 24, 7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004
Tretter, L., and Adam-Vizi, V. (2005). Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos. Trans. R. Soc. B Biol. Sci. 360, 2335–2345. doi: 10.1098/rstb.2005.1764
Urbani, A., and Babu, M. (eds). (2019). Mitochondria in Health and in Sickness. Singapore: Springer. doi: 10.1007/978-981-13-8367-0
van Spaendonck-Zwarts, K. Y., Posafalvi, A., van den Berg, M. P., Hilfiker-Kleiner, D., Bollen, I. A. E., Sliwa, K., et al. (2014). Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart J. 35, 2165–2173. doi: 10.1093/eurheartj/ehu050
Vásquez-Trincado, C., García-Carvajal, I., Pennanen, C., Parra, V., Hill, J. A., Rothermel, B. A., et al. (2016). Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 594, 509–525. doi: 10.1113/JP271301
Vega, A. L., Yuan, C., Votaw, V. S., and Santana, L. F. (2011). Dynamic changes in sarcoplasmic reticulum structure in ventricular myocytes. J. Biomed. Biotechnol. 2011:382586. doi: 10.1155/2011/382586
Vendelin, M., Béraud, N., Guerrero, K., Andrienko, T., Kuznetsov, A. V., Olivares, J., et al. (2005). Mitochondrial regular arrangement in muscle cells: a “crystal-like” pattern. Am. J. Physiol. Physiol. 288, C757–C767. doi: 10.1152/ajpcell.00281.2004
Verdejo, H. E., del Campo, A., Troncoso, R., Gutierrez, T., Toro, B., Quiroga, C., et al. (2012). Mitochondria, myocardial remodeling, and cardiovascular disease. Curr. Hypertens. Rep. 14, 532–539.
Verdonschot, J. A. J., Hazebroek, M. R., Ware, J. S., Prasad, S. K., and Heymans, S. R. B. (2019). Role of targeted therapy in dilated cardiomyopathy: the challenging road toward a personalized approach. J. Am. Heart Assoc. 8:e012514. doi: 10.1161/JAHA.119.012514
Vinogradov, A. D., and Grivennikova, V. G. (2016). Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta Bioenerg. 1857, 863–871. doi: 10.1016/j.bbabio.2015.11.004
Viola, H. M., Adams, A. M., Davies, S. M. K., Fletcher, S., Filipovska, A., and Hool, L. C. (2014). Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. U.S.A. 111, E2905–E2914. doi: 10.1073/pnas.1402544111
Viola, H. M., Arthur, P. G., and Hool, L. C. (2009). Evidence for regulation of mitochondrial function by the L-type Ca2+ channel in ventricular myocytes. J. Mol. Cell. Cardiol. 46, 1016–1026. doi: 10.1016/j.yjmcc.2008.12.015
Viola, H. M., Davies, S. M. K., Filipovska, A., and Hool, L. C. (2013). L-type Ca 2+ channel contributes to alterations in mitochondrial calcium handling in the mdx ventricular myocyte. Am. J. Physiol. Circ. Physiol. 304, H767–H775. doi: 10.1152/ajpheart.00700.2012
Viola, H. M., and Hool, L. C. (2010). Cross-talk between L-type Ca 2+ channels and mitochondria. Clin. Exp. Pharmacol. Physiol. 37, 229–235. doi: 10.1111/j.1440-1681.2009.05277.x
Wai, T., García-Prieto, J., Baker, M. J., Merkwirth, C., Benit, P., Rustin, P., et al. (2015). Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350:aad0116. doi: 10.1126/science.aad0116
Walsh, C., Barrow, S., Voronina, S., Chvanov, M., Petersen, O. H., and Tepikin, A. (2009). Modulation of calcium signalling by mitochondria. Biochim. Biophys. Acta Bioenerg. 1787, 1374–1382. doi: 10.1016/j.bbabio.2009.01.007
Walsh, R., Rutland, C., Thomas, R., and Loughna, S. (2010). Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 115, 49–60. doi: 10.1159/000252808
Wang, G., McCain, M. L., Yang, L., He, A., Pasqualini, F. S., Agarwal, A., et al. (2014). Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 20, 616–623. doi: 10.1038/nm.3545
Wang, H.-Y. J., Wu, H.-W., Tsai, P.-J., Liu, C. B., and Zheng, Z.-F. (2014). Matrix-assisted laser desorption/ionization mass spectrometry imaging of cardiolipins in rat organ sections. Anal. Bioanal. Chem. 406, 565–575. doi: 10.1007/s00216-013-7492-y
Ware, J. S., Li, J., Mazaika, E., Yasso, C. M., DeSouza, T., Cappola, T. P., et al. (2016). Shared genetic predisposition in peripartum and dilated cardiomyopathies. N. Engl. J. Med. 374, 233–241. doi: 10.1056/NEJMoa1505517
Wende, A. R., Symons, J. D., and Abel, E. D. (2012). Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 14, 517–531.
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884. doi: 10.1038/nrm3013
Westermann, D., Rutschow, S., Jager, S., Linderer, A., Anker, S., Riad, A., et al. (2007). Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes 56, 641–646. doi: 10.2337/db06-1163
WHO/ISFC (1980). Report of the WHO/ISFC task force on the definition and classification of cardiomyopathies. Heart 44, 672–673. doi: 10.1136/hrt.44.6.672
Wikstrom, J. D., Twig, G., and Shirihai, O. S. (2009). What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int. J. Biochem. Cell Biol. 41, 1914–1927. doi: 10.1016/j.biocel.2009.06.006
Wilding, J. R., Joubert, F., De Araujo, C., Fortin, D., Novotova, M., Veksler, V., et al. (2006). Altered energy transfer from mitochondria to sarcoplasmic reticulum after cytoarchitectural perturbations in mice hearts. J. Physiol. 575, 191–200. doi: 10.1113/jphysiol.2006.114116
Yang, Y.-C., Tsai, C.-Y., Chen, C.-L., Kuo, C.-H., Hou, C.-W., Cheng, S.-Y., et al. (2018). Pkcδ activation is involved in ROS-mediated mitochondrial dysfunction and apoptosis in cardiomyocytes exposed to advanced glycation end products (ages). Aging Dis. 9, 647–663. doi: 10.14336/AD.2017.0924
Yoon, Y. (2004). Sharpening the scissors: mitochondrial fission with aid. Cell Biochem. Biophys. 41, 193–206.
Youle, R. J., and Karbowski, M. (2005). Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 6, 657–663. doi: 10.1038/nrm1697
Zak, R., Rabinowitz, M., Rajamanickam, C., Merten, S., and Kwiatkowska-Patzer, B. (1980). Mitochondrial proliferation in cardiac hypertrophy. Basic Res. Cardiol. 75, 171–178. doi: 10.1007/BF02001410
Zamani, M., Karaca, E., and Huang, N. F. (2018). Multicellular interactions in 3D engineered myocardial tissue. Front. Cardiovasc. Med. 5:147. doi: 10.3389/fcvm.2018.00147
Zhang, D., Li, Y., Heims-Waldron, D., Bezzerides, V., Guatimosim, S., Guo, Y., et al. (2018). Mitochondrial cardiomyopathy caused by elevated reactive oxygen species and impaired cardiomyocyte proliferation. Circ. Res. 122, 74–87. doi: 10.1161/CIRCRESAHA.117.311349
Keywords: mitochondria, cardiomyocytes, cardiomyopathies, organoids model, sarcoplasmic reticulum, Ca ATPase (SERCA) 2+, calcium, heart function
Citation: Ramaccini D, Montoya-Uribe V, Aan FJ, Modesti L, Potes Y, Wieckowski MR, Krga I, Glibetić M, Pinton P, Giorgi C and Matter ML (2021) Mitochondrial Function and Dysfunction in Dilated Cardiomyopathy. Front. Cell Dev. Biol. 8:624216. doi: 10.3389/fcell.2020.624216
Received: 03 November 2020; Accepted: 16 December 2020;
Published: 12 January 2021.
Edited by:
Gaetano Santulli, Columbia University, United StatesReviewed by:
Consolato Sergi, University of Alberta Hospital, CanadaCopyright © 2021 Ramaccini, Montoya-Uribe, Aan, Modesti, Potes, Wieckowski, Krga, Glibetić, Pinton, Giorgi and Matter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michelle L. Matter, bWF0dGVyQGhhd2FpaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.