 Christina Doxaki
Christina Doxaki Konstantinos Palikaras
Konstantinos Palikaras- 1General University Hospital of Heraklion, Crete, Greece
- 2Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology-Hellas, Crete, Greece
Maintenance of neuronal homeostasis is a challenging task, due to unique cellular organization and bioenergetic demands of post-mitotic neurons. It is increasingly appreciated that impairment of mitochondrial homeostasis represents an early sign of neuronal dysfunction that is common in both age-related neurodegenerative as well as in neurodevelopmental disorders. Mitochondrial selective autophagy, known as mitophagy, regulates mitochondrial number ensuring cellular adaptation in response to several intracellular and environmental stimuli. Mounting evidence underlines that deregulation of mitophagy levels has an instructive role in the process of neurodegeneration. Although mitophagy induction mediates the elimination of damaged mitochondria and confers neuroprotection, uncontrolled runaway mitophagy could reduce mitochondrial content overstressing the remaining organelles and eventually triggering neuronal cell death. Unveiling the molecular mechanisms of neuronal mitophagy and its intricate role in neuronal survival and cell death, will assist in the development of novel mitophagy modulators to promote cellular and organismal homeostasis in health and disease.
Introduction
Mitochondria are remarkably dynamic organelles that divide, fuse and migrate in different cellular compartments. The processes of mitochondrial fission and fusion ensure metabolite and mitochondrial DNA (mtDNA) exchange for dilution of dysfunctional elements as well as dictate organelle shape, number and bioenergetic functionality. Neurons require high energy levels and depend on mitochondrial homeostasis to carry out their functions and sustain neuronal circuit formation, communication and activity (Misgeld and Schwarz, 2017; Palikaras and Tavernarakis, 2020). Hence, neuronal cells are equipped with specialized molecular mechanisms for efficient distribution of mitochondria. Mitochondria are typically localized to areas of high-energy demand including the distal portion of the initial segment of axons, the nodes of Ranvier, growth cones, presynaptic buttons and postsynaptic densities (Steketee et al., 2012; Smith et al., 2016; Misgeld and Schwarz, 2017; Garcia et al., 2019; Verreet et al., 2019). While progress has been made in identifying the proteins involved in mitochondrial transport within neurons, the significance of localizing mitochondria in neuronal compartments, where they can respond to local changes in neuronal activity and energy metabolism, is largely unknown and only starting to be exploited (Sheng and Cai, 2012; Misgeld and Schwarz, 2017; Garcia et al., 2019; Palikaras and Tavernarakis, 2020).
It becomes increasingly appreciated that the removal of misfolded proteins, protein aggregates and damaged mitochondria, is of crucial importance for the proper function and long-term survival of neurons. The detailed molecular mechanisms that govern mitochondrial selective autophagy in such a highly differentiated and compartmentalized cell, as well as their relevance to neuronal physiology only now begin to be elucidated. During the last decade, mitophagy was widely studied in the context of stress and pathological conditions (Palikaras et al., 2018; Lou et al., 2020; Yan et al., 2020). A growing body of evidence highlights mitophagy as a physiological process that occurs constitutively at baseline levels in the nervous system. Interestingly, basal mitophagy levels differ between brain regions and neuronal sub-populations. The dentate gyrus, lateral ventricle and Purkinje cells display high levels of mitophagy, whereas mitophagy is low in the neurons of the striatum, cortex and substantia nigra (Sun et al., 2015). Furthermore, neuronal mitophagy declines with age leading to accumulation of defective organelles (Palikaras et al., 2015; Sun et al., 2015; Evans and Holzbaur, 2020a). Increased mitochondrial damage is a hallmark of aging and age-associated neurodegeneration highlighting that neuronal cells are particularly sensitive to age-dependent mitophagy impairment.
While the role of mitophagy in cellular and organismal physiology is essential, several pathological conditions, such as mitochondrial disorders, ischemic stroke, chronic cerebral hypoperfusion, and diabetes, are shown to stimulate uncontrolled mitochondrial elimination that subsequently leads to neuronal cell death (Shi et al., 2014; Su et al., 2018; Devi et al., 2019; Park et al., 2019; Zaninello et al., 2020). These findings underline mitophagy as a “double-edged sword” for neuronal homeostasis and viability. Here, we survey recent advances toward the elucidation of the intricate role of mitophagy in neuronal survival and cell death.
Molecular Mechanisms of Mitophagy in Neuronal Cells
The maintenance of a healthy mitochondrial pool is pivotal for cellular and organismal homeostasis (Figure 1). Post-mitotic neuronal cells are more susceptible to mitochondrial damage due to their increased energetic demands. Therefore, aged or dysfunctional mitochondria need either to be repaired through mitochondrial surveillance quality mechanisms, including proteasome system, mitochondrial proteases, mitochondrial derived vesicles (MDVs), fission-fusion machinery and mitochondrial unfolded protein response (UPRmt), or be eliminated by selective mitochondrial autophagy (Figure 1) (Palikaras et al., 2018).
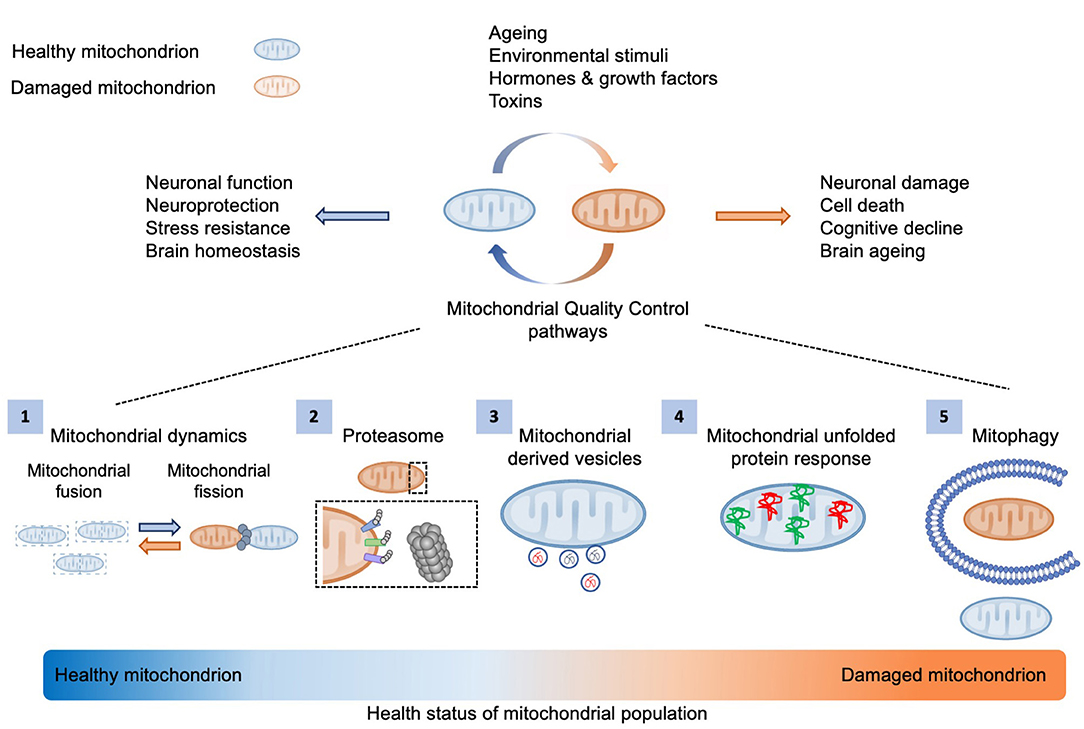
Figure 1. Mitochondrial quality control mechanisms. Several environmental and intracellular stimuli influence mitochondrial activity and integrity. The maintenance of a healthy mitochondrial population is essential for cellular metabolism and brain function. Thus, aged or defective organelles need either to be restored via mitochondrial quality control mechanisms, including (1) mitochondrial dynamics, (2) proteasome system, (3) mitochondrial derived vesicles (MDVs), and (4) mitochondrial unfolded protein response (UPRmt), or (5) be degraded by mitophagy.
Several molecular mechanisms have been uncovered to mediate mitochondrial removal to date, highlighting that mitophagy could be triggered in response to diverse stimuli via multiple signaling pathways, in distinct cellular contexts. Accumulating evidence demonstrates an intricate coordination between mitophagy regulatory mechanisms and highlights their conservation from yeast to mammals (Palikaras et al., 2018; Pickles et al., 2018; Lou et al., 2020). The PINK1 (PTEN-induced putative kinase 1)/Parkin pathway is the most well-characterized signaling cascade that orchestrates mitochondrial degradation in response to stress (Harper et al., 2018; Palikaras et al., 2018; Montava-Garriga and Ganley, 2020). PINK1 is a mitochondrial protein kinase that is stabilized on the outer mitochondrial membrane upon challenged conditions. PINK1 activity is induced upon its auto-phosphorylation leading to the recruitment of Parkin on mitochondrial surface (Hasson et al., 2013; Lazarou et al., 2015; Khaminets et al., 2016; Pickles et al., 2018; Sekine and Youle, 2018). In turn, Parkin ubiquitinates various outer mitochondrial membrane proteins promoting either their degradation or their association with autophagy adaptors and general autophagic machinery (Hasson et al., 2013; Heo et al., 2015; Lazarou et al., 2015; Khaminets et al., 2016; Gatica et al., 2018; Pickles et al., 2018; Sekine and Youle, 2018). Several studies underlie the causative interplay between PINK1 and Parkin activation in response to mitochondrial dysfunction. Systematic proteomic analysis both in non-neuronal and neuronal cells suggest the stimulation of a feed-forward mechanism that involves the PINK1-dependent phosphorylation of ubiquitin (Ub) and poly-Ub chains on damaged organelles to enhance mitophagy signal upon stress (Harper et al., 2018; Ordureau et al., 2018, 2020). Furthermore, several PINK1- and Parkin-independent molecular pathways have been identified implicating the significance of multiple mitochondrial proteins or lipids, including FUNDC1 (FUN14 Domain Containing 1), BNIP3 (BCL2/adenovirus E1B 19-kDa-interacting protein 3), NIX/BNIP3L (BCL2/adenovirus E1B 19-kDa-interacting protein 3-Like), BCL2L13 (BCL2- Like 13), FKBP8 (FK506 binding protein 8), PHB2, cardiolipin, and ceramide among others, which act as receptors and facilitate mitophagy (Sentelle et al., 2012; Harper et al., 2018; Palikaras et al., 2018; Montava-Garriga and Ganley, 2020).
The removal of neuronal mitochondria is a challenging cellular event since the majority of mitochondrial population is located at the distal neuronal compartments, far away from the cell body of the neuron, where mature acidic lysosomes mainly present (Holtzman and Novikoff, 1965; Cai et al., 2010; Evans and Holzbaur, 2020b; Han et al., 2020). Despite the spatial limitations, timely degradation of impaired mitochondria is essential for neuronal protection against cell death. Recent studies have suggested that Parkin-mediated mitophagy is limited in a small subset of mature neurons and takes place much more slowly than in other cell types (Cai et al., 2012a,b; Lin et al., 2017; Puri et al., 2019). These results support the notion that alternative molecular mechanisms sustain mitochondrial homeostasis upon mild stress, before the stimulation of Parkin-mediated mitophagy. Indeed, a very recent study has demonstrated that the mitochondrial E3 ubiquitin ligase 1 (Mul1) facilitates an early checkpoint to preserve mitochondrial integrity and restrains neuronal mitophagy under mild mitochondrial stress (Puri et al., 2019). Interestingly, Mul1 affects the stability of Mitofusin 2 (MFN2) preserving mitochondrial homeostasis. Mul1 deficiency increases MFN2 protein levels promoting hyperfusion of the mitochondrial network. Notably, MFN2 is also located on the endoplasmic reticulum (ER)-mitochondrial contact sites where it regulates calcium homeostasis (De Brito and Scorrano, 2008; Filadi et al., 2015; Mclelland et al., 2018). Therefore, depletion of Mul1 in neurons results in impaired ER-mitochondrial tethering due to MFN2 upregulation, which in turn triggers the elevation of cytoplasmic calcium levels, calcineurin activation, DRP1 (Dynamin-related protein 1)-mediated mitochondrial fragmentation and eventually mitophagy (Puri et al., 2019). Studies in non-neuronal cells have shown that MFN2 is phosphorylated by PINK1, and thereby promotes Parkin recruitment on the outer mitochondrial membrane (Chen and Dorn, 2013). In turn, Parkin ubiquitinates MFN2 to induce its degradation, which results in the subsequent release of mitochondria from the ER, thereby enhancing mitophagy (Chen and Dorn, 2013; Mclelland et al., 2018). Although the core mitophagy constituents are conserved in both neurons and non-neuronal cells, the kinetics of mitochondrial elimination varies not only between different cell types, but also between neuronal sub-populations. These results indicate the existence of a multi-step, highly organized and precise mitochondrial quality control system.
A recent study reported that the degradation of defective organelles is a rate-limiting event in neuronal cells (Evans and Holzbaur, 2020a). Mild stress conditions trigger mitochondrial depolarization, resulting in their subsequent sequestration by autophagosomes and their delivery from the distal neuronal compartments to the soma. Although mitophagy in non-neuronal cells is taking place rapidly, neuronal mitoautophagosomes remain intact in non-acidified organelles, whereby their elimination is a very slow process (Evans and Holzbaur, 2020a). Optineurin (OPTN) and TANK-binding kinase 1 (TBK1) act downstream of Parkin to recognize defective organelles and promote their autophagosomal engulfment and retrograde transportation under antioxidant deprivation. Interestingly, OPTN-mediated mitophagy is spatially-restricted in neuronal cell bodies, with only few mitophagic events to be detected in axons or dendrites (Evans and Holzbaur, 2020a). These results further support the notion that mitophagy is differentially regulated across neuronal compartments, as it is previously demonstrated both in vitro and in vivo (Ashrafi et al., 2014; Devireddy et al., 2015; Mcwilliams et al., 2016; Sung et al., 2016; Puri et al., 2019; Zaninello et al., 2020). Despite the fact that several adaptor molecules, such as NDP52, OPTN, p62 and TAX1BP1 (Tax1 binding protein 1), have been identified to facilitate mitophagy in non-neuronal cells, it remains still elusive whether these adaptor proteins might participate and/or co-regulate the highly compartmentalized nature of neuronal mitophagy.
The differential kinetic patterns of mitochondrial turnover under both basal and challenged conditions could explain the enhanced vulnerability of neurons upon mitochondrial dysfunction. Additionally, age-dependent perturbations in cellular homeostasis, including proteostasis collapse, lysosomal dysfunction, mitochondrial impairment, increased oxidative stress and genomic instability among others, could further impede the efficiency of mitophagy exacerbating neuronal susceptibility to degeneration (Hou et al., 2018; Palikaras et al., 2018; Hipp et al., 2019; Lie and Nixon, 2019).
Neuroprotective Role of Mitophagy
Defective mitochondrial turnover results in the progressive accumulation of damaged organelles and is characterized as a hallmark of aging and age-related neurodegenerative pathologies (Figure 2) (Hou et al., 2019; Lautrup et al., 2019; Cai and Jeong, 2020; Lou et al., 2020). Post-mortem Alzheimer's disease (AD) hippocampal samples display smaller mitochondria with altered cristae formation and impaired function compared with their counterparts in age-matched healthy controls (Fang et al., 2019). Further evidence stemming from human induced pluripotent stem cells (iPSCs)-derived AD neurons indicated significantly decreased phosphorylation levels of ULK1 (Unc-51 Like Autophagy Activating Kinase 1) and TBK1, thereby suggesting that the initiation of the mitophagy process is impaired (Fang et al., 2019). Congruently, levels of other essential autophagy/mitophagy regulators, such as Parkin, OPTN, Mul1, BECN1 (Beclin 1), AMBRA1 (Autophagy And Beclin 1 Regulator 1), FUNDC1, are diminished in AD samples (Pickford et al., 2008; Martin-Maestro et al., 2016; Fang et al., 2019). Moreover, a recent study demonstrated that overexpression of wild type and disease-associated tau results in reduced Parkin recruitment onto mitochondrial surface and mitophagy inhibition in both nematodes and neuroblastoma cells (Cummins et al., 2019). These findings suggest that the impairment of basal mitophagy is an early event in AD human brain and a causative factor in the development and progression of disease pathophysiology (Kerr et al., 2017; Fang et al., 2019).
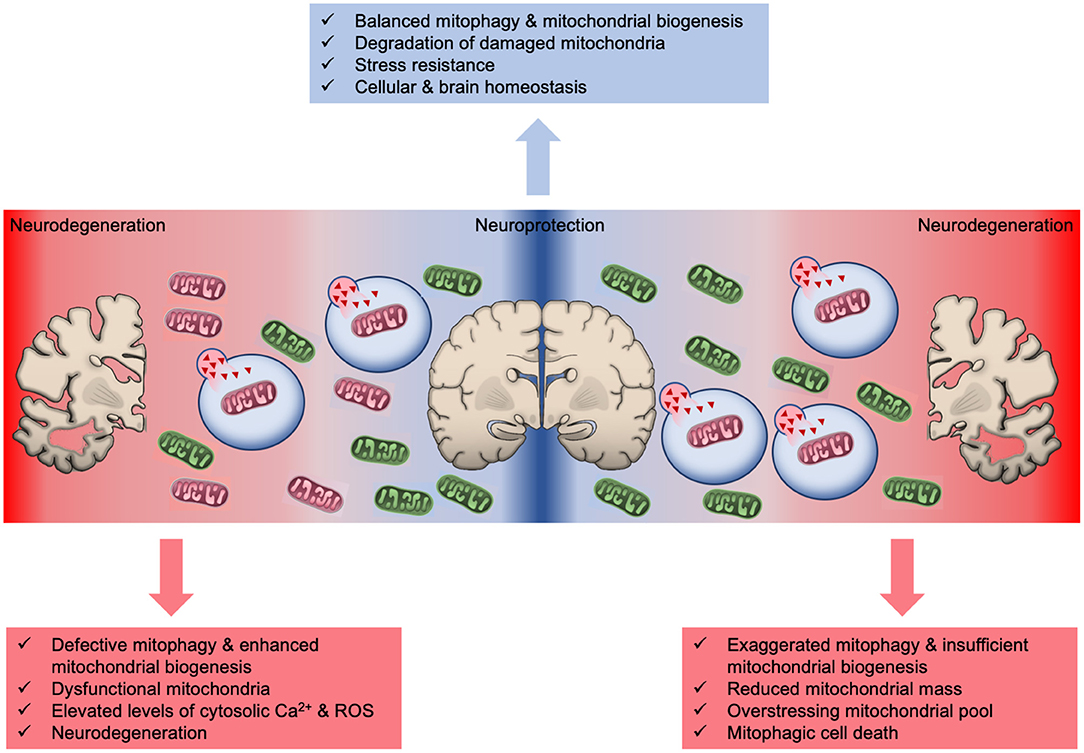
Figure 2. The intricate role of mitophagy in neuroprotection and neurodegeneration. Basal mitophagy eliminates defective mitochondria and simultaneously new organelles are generated through mitochondrial biogenesis to sustain mitochondrial population. Excessive mitophagy significantly reduces mitochondrial number overstressing the remaining organelles and eventually leads to energetic crisis and cell death. Similarly, mitophagy defects results in the accrual of damaged mitochondria promoting neurodegeneration and brain aging.
In addition to AD, mitochondrial abnormalities and mitophagy defects have also been associated with the pathogenesis of Parkinson's disease (PD) (Liu et al., 2019). Loss of function mutations of PARK6 and PARK2 genes, which encode PINK1 and Parkin respectively, are linked to familial form of parkinsonism (Chu, 2019). Although the PINK1/Parkin pathway has been extensively studied in both neuronal and non-neuronal cell cultures, its role in mitophagy regulation and neuronal survival in vivo remains still controversial. Studies in flies and rats demonstrated that Parkin deficiency leads to altered mitochondrial morphology and function accompanied by dopaminergic neuronal loss and motility defects (Kitada et al., 1998; Whitworth et al., 2005; Yang et al., 2006; Dave et al., 2014; Pickrell and Youle, 2015). However, PINK1 and Parkin deficiency does not trigger any severe PD-associated phenotype in mice, suggesting the existence of compensatory mitochondrial quality control mechanisms (Goldberg et al., 2003; Perez and Palmiter, 2005). Further supporting this notion, transgenic flies and mice expressing mitophagy reporter identified that PINK1 and Parkin are dispensable for basal neuronal mitophagy (Lee et al., 2018; Mcwilliams et al., 2018). Interestingly, a recent study in mice identified that exhaustive exercise and increased mtDNA damage stimulate dopaminergic neuronal loss and locomotion defects in both PINK1 and Parkin mutant mice (Sliter et al., 2018). These findings underscore that acute and/or chronic organismal stress might contribute to the development and progression of diverse pathological conditions, especially in sensitive genetic backgrounds.
Mounting evidence suggests that mutant huntingtin (mHtt) influences directly mitochondrial homeostasis contributing to Huntington's disease (HD) pathogenesis (Orr et al., 2008; Shirendeb et al., 2011; Yan et al., 2020). Furthermore, autophagy-mediated degradation of both cytoplasmic and mitochondrial constituents is perturbed in HD leading to the progressive accumulation of protein aggregates and dysfunctional mitochondria (Martinez-Vicente et al., 2010). Indeed, microscopic examination of the dentate gyrus region revealed that basal mitophagy levels are diminished in a mouse model of HD compared to age-matched control animals (Sun et al., 2015). Supporting the neuroprotective role of mitophagy, genetic studies in flies and rodents uncovered that mHtt is associated directly with mitochondrial membranes and impairs mitochondrial integrity and mitophagy execution leading to neurodegeneration (Hwang et al., 2015; Khalil et al., 2015; Guo et al., 2016; Franco-Iborra et al., 2020).
Pharmacological upregulation of mitophagy is shown to promote neuroprotection against several neurodegenerative pathologies (Georgakopoulos et al., 2017; Palikaras et al., 2018). Indeed, dietary supplementation with urolithin A (UA), actinonin and NAD+ (Nicotinamide Adenine Dinucleotide)-precursor molecules, restores multiple pathological features of AD, including mitochondrial damage, neuronal defects, Aβ aggregation, aberrant tau phosphorylation levels, neuroinflammation, and cognitive dysfunction among others (Hou et al., 2018; Fang, 2019; Lautrup et al., 2019; Gilmour et al., 2020). Moreover, nicotinamide riboside (NR; NAD+-precursor molecules) administration preserves mitochondrial function and diminishes degeneration of dopaminergic neurons through mitophagy induction in flies, mouse and iPSCs models of PD (Schondorf et al., 2018). Recent clinical trials suggest that administration of both NR and UA is safe without demonstrating any adverse effect in compound-treated compared to placebo-treated group and highlight their therapeutic potential (Dollerup et al., 2018; Andreux et al., 2019; Conze et al., 2019; Radenkovic et al., 2020). Interestingly, NAD+-precursor molecules confer neuroprotection through the activation of BNIP3-mediated mitophagy in both AD and PD models (Schondorf et al., 2018; Fang et al., 2019). These results underline the requirement of personalized pharmacological approaches that depend on the patient genetic background to overcome mitophagy pathway deficiencies in pathological conditions (Xie et al., 2019).
Spermidine, a natural occurring compound, is shown to induce autophagy and mitophagy promoting cytoprotection, stress resistance and longevity in yeast cells, nematodes, flies, and mice (Eisenberg et al., 2009, 2016; Madeo et al., 2018). Furthermore, spermidine-rich feeding reverses the age-induced decline of polyamines in brain tissue and subsequently improves synaptic impairment and memory loss in an autophagy-dependent manner (Gupta et al., 2013, 2016; Maglione et al., 2019). A randomized clinical trial was recently conducted to evaluate the impact of spermidine supplementation on memory performance in humans (Wirth et al., 2018). Spermidine-treated aged-individuals displayed moderate improvement in the memory performance and enhanced mnemonic discrimination abilities compared to placebo-treated group (Wirth et al., 2018). The decreased levels of spermidine in blood serum of AD patients compared to healthy individuals, further support the therapeutic potential of polyamine-rich food supplementation against age-related dementia in humans (Madeo et al., 2018; Joaquim et al., 2019).
Taken together, the central role of mitophagy in neuronal communication, activity and survival is steadily emerging. It is increasingly appreciated that impairment of mitochondrial metabolism represents an early sign of neuronal deficits that is common in both age-related deterioration of brain function as well as in neurodegenerative disorders. Therefore, the development of novel therapeutic interventions to modulate mitochondrial turnover and preserve healthy mitochondrial population could confer neuroprotection against brain aging before any irreversible impairment.
Mitophagic Cell Death in Neurons
The tight coordination between mitochondrial biogenesis and degradation is essential for cellular, tissue and organismal physiology (Palikaras and Tavernarakis, 2014; Palikaras et al., 2015). Insufficient mitochondrial biogenesis and excessive mitophagy diminish mitochondrial population that burdens the remaining organelles, which subsequently promote mitophagy-mediated cell death (Figure 2) (Palikaras and Tavernarakis, 2014; Subramaniam, 2020). Indeed, excessive mitochondrial elimination has been indicated as a cause of cell death in several disease models (Subramaniam, 2020). Notably, BNIP3-mediated mitophagy is shown to promote neuronal death both in vitro and in vivo upon ischemic stroke (Shi et al., 2014). BNIP3 deficiency prevents neuronal loss in neonatal brains upon ischemia/hypoxia (I/H) treatment. Although BNIP3 and NIX/BNIP3L share high degree of sequence homology, elevated NIX/BNIP3L levels could not compensate BNIP3 depletion to trigger mitophagy and cell death in response to ischemic stroke. Therefore, it is suggested that mitophagic cell death is mainly regulated by BNIP3, whereas NIX/BNIP3L sustains basal mitophagy levels in neurons (Shi et al., 2014). Further supporting the pro-death activities of BNIP3, a recent study demonstrated increased protein levels of BNIP3 and mitophagy upregulation in a rat model of chronic cerebral hyperfusion (CCH). Chronic state of reduced cerebral blood flow leads to CCH that is linked to neurological damage and cognitive decline (Somredngan and Thong-Asa, 2018). Interestingly, administration of URB597, a fatty acid amide hydrolase inhibitor, decreases BNIP3 and Parkin protein levels, which confers neuroprotection due to mitophagy inhibition (Somredngan and Thong-Asa, 2018).
The brain region hippocampus retains its neurogenic capacities throughout adulthood and is severely affected during brain aging or injury. Evidently, neuronal stem cell plasticity, survival and differentiation are under constant and tight proteostatic and metabolic regulation (Garcia-Prat et al., 2017; Khacho et al., 2019). Moreover, transgenic mice expressing mitophagy reporter display increased basal mitophagy levels in the hippocampal dentate gyrus, highlighting the pivotal role of mitochondrial quality control in neurogenesis (Sun et al., 2015; Khacho et al., 2019). Hippocampal neuronal stem cells (HNSCs) differentiation and viability depends on extracellular stimuli, including insulin/insulin-like growth factors (IGFs). It is already documented that insulin depletion triggers autophagy-dependent cell death in HNSCs (Aberg et al., 2000; Lichtenwalner et al., 2001). Parkin stimulation deregulates ER-mitochondrial tethering promoting cytoplasmic calcium elevation and mitophagic cell death in hippocampal neurons upon insulin withdrawal (Park et al., 2019). Altered insulin signaling interferes with hippocampal neuronal function and is associated with several neurodegenerative and psychiatric disorders (Bernstein et al., 2020; Palikaras and Tavernarakis, 2020). Taken together these results underline the impact of imbalanced mitochondrial biogenesis and mitophagy in neuronal stem cell survival.
Recent findings demonstrated that excessive mitophagy eliminates mitochondrial content in striatal neurons and triggers cell death upon mitochondrial damage (Sharma et al., 2019). The small GTPase Rhes (Ras homolog enriched in striatum) is highly expressed in striatum brain region and is associated with lysosomal and mitochondrial cellular compartments. Interestingly, Rhes induces NIX-mediated runaway mitophagy and reduces mitochondrial content in response to 3-Nitropropionic acid (3-NP) mitochondrial toxicant (Sharma et al., 2019). Evidently, 3-NP supplementation promotes striatal lesions and causes severe pathological symptoms in mammals reminiscent of HD (Brouillet et al., 1993; Fu et al., 1995; He et al., 1995; Guyot et al., 1997). Therefore, it is tempting to speculate that imbalance of mitophagy might be a contributing factor to striatal neurodegeneration and pathogenesis of HD.
Several studies in mammals and nematodes demonstrated that disruption of the continual redistribution of mitochondria compromises axonal stability, synaptic integrity and neuropeptide release leading to neurodegeneration (Misko et al., 2012; Cherra et al., 2013; Rawson et al., 2014; Zhao et al., 2018; Han et al., 2020; Zaninello et al., 2020). A very recent study reported that exaggerated autophagy/mitophagy depletes axonal mitochondria in retinal ganglion cells (RGCs) and mediates the development of autosomal optic atrophy (ADOA) (Zaninello et al., 2020). ADOA is an incurable genetic disorder caused by mutations in the OPA1 gene (Alexander et al., 2000; Delettre et al., 2000). Although, the main pathological feature of ADOA is optic nerve degeneration and visual loss during early childhood, several patients develop multi-systemic deteriorations, including deafness, ataxia, myopathies and paraplegia among others, underlining the pivotal role of OPA1 activity in the maintenance of mitochondrial metabolism and tissue homeostasis (Yu-Wai-Man et al., 2010; Belenguer and Pellegrini, 2013). Further elaborating on the contribution of mitophagy to ADOA pathogenesis, it is documented that OPA1 mutation stimulates AMPK (AMP-activated protein kinase) activity in axonal hillock and mediates mitochondrial degradation in nematode and mouse neurons. Notably, autophagy deficiency restores mitochondrial content in axons and inhibits visual defects in an ADOA mouse model highlighting the detrimental effect of uncontrolled mitophagy in disease development and progression (Zaninello et al., 2020).
Conclusion
Mitophagy preserves energy homeostasis facilitating the elimination of defective organelles and, thereby, preventing the release of various harmful byproducts of mitochondrial activity. Hence, mitophagy stimulation exerts cytoprotection against premature aging and neuronal death (Palikaras et al., 2018; Bakula and Scheibye-Knudsen, 2020). Although the molecular mechanisms of mitochondrial selective autophagy have been extensively studied, several controversial questions remain to be addressed about neuronal mitophagy. The stimulus and the molecular mechanisms, which regulate autophagosomal formation in axons, cargo recognition and sequestration, transport to the soma, fusion with lysosomes and degradation are still obscure. Furthermore, several gaps remain in our understanding of major aspects of mitochondrial contributions to neuronal vulnerability and aging that might trigger cellular and/or tissue damage.
Aging represents the greatest risk factor for the onset of degenerative diseases of the nervous system. These include the very common Alzheimer's, Parkinson's, and Huntington's diseases among others, all of which are tightly associated with mitochondrial and autophagic defects (Chu, 2019; Bakula and Scheibye-Knudsen, 2020; Cai and Jeong, 2020; Evans and Holzbaur, 2020b; Lou et al., 2020; Yan et al., 2020). Additionally, the molecular pathways that orchestrate the crosstalk between mitophagy, apoptosis and necrotic cell death in axonal and dendritic degeneration remain unknown. Unraveling potent chemical modulators of mitophagy and understanding how mitochondrial elimination promotes either neuroprotection or neuronal cell death would be essential for the development of novel and context-specific pharmacological interventions against neurodegenerative disorders.
Author Contributions
CD and KP wrote and edited the manuscript. KP supervised manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
KP was funded by a grant from the Hellenic Foundation for Research and Innovation (HFRI) and the General Secretariat for Research and Technology (GSRT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the work of many investigators whose studies were not cited due to space limitation. We thank Dr. Yahyah Aman and Dr. Andrea Princz for critical reading of the manuscript.
References
Aberg, M. A., Aberg, N. D., Hedbacker, H., Oscarsson, J., and Eriksson, P. S. (2000). Peripheral infusion of IGF-I selectively induces neurogenesis in the adult rat hippocampus. J. Neurosci. 20, 2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi: 10.1038/79944
Andreux, P. A., Blanco-Bose, W., Ryu, D., Burdet, F., Ibberson, M., Aebischer, P., et al. (2019). The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 1, 595–603. doi: 10.1038/s42255-019-0073-4
Ashrafi, G., Schlehe, J. S., Lavoie, M. J., and Schwarz, T. L. (2014). Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 206, 655–670. doi: 10.1083/jcb.201401070
Bakula, D., and Scheibye-Knudsen, M. (2020). MitophAging: mitophagy in aging and disease. Front. Cell Dev. Biol. 8:239. doi: 10.3389/fcell.2020.00239
Belenguer, P., and Pellegrini, L. (2013). The dynamin GTPase OPA1: more than mitochondria? Biochim. Biophys. Acta 1833, 176–183. doi: 10.1016/j.bbamcr.2012.08.004
Bernstein, H. G., Keilhoff, G., Dobrowolny, H., and Steiner, J. (2020). Enhanced mitochondrial autophagy (mitophagy) in oligodendrocytes might play a role in white matter pathology in schizophrenia. Med. Hypotheses 134:109443. doi: 10.1016/j.mehy.2019.109443
Brouillet, E., Jenkins, B. G., Hyman, B. T., Ferrante, R. J., Kowall, N. W., Srivastava, R., et al. (1993). Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J. Neurochem. 60, 356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x
Cai, Q., and Jeong, Y. Y. (2020). Mitophagy in Alzheimer's disease and other age-related neurodegenerative diseases. Cells 9:150. doi: 10.3390/cells9010150
Cai, Q., Lu, L., Tian, J. H., Zhu, Y. B., Qiao, H., and Sheng, Z. H. (2010). Snapin-regulated late endosomal transport is critical for efficient autophagy-lysosomal function in neurons. Neuron 68, 73–86. doi: 10.1016/j.neuron.2010.09.022
Cai, Q., Zakaria, H. M., and Sheng, Z. H. (2012a). Long time-lapse imaging reveals unique features of PARK2/Parkin-mediated mitophagy in mature cortical neurons. Autophagy 8, 976–978. doi: 10.4161/auto.20218
Cai, Q., Zakaria, H. M., Simone, A., and Sheng, Z. H. (2012b). Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr. Biol. 22, 545–552. doi: 10.1016/j.cub.2012.02.005
Chen, Y., and Dorn, G. W. II. (2013). PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Cherra, S. J. III, Steer, E., Gusdon, A. M., Kiselyov, K., and Chu, C. T. (2013). Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 182, 474–484. doi: 10.1016/j.ajpath.2012.10.027
Chu, C. T. (2019). Multiple pathways for mitophagy: a neurodegenerative conundrum for Parkinson's disease. Neurosci. Lett. 697, 66–71. doi: 10.1016/j.neulet.2018.04.004
Conze, D., Brenner, C., and Kruger, C. L. (2019). Safety and metabolism of long-term administration of NIAGEN (Nicotinamide Riboside Chloride) in a randomized, double-blind, placebo-controlled clinical trial of healthy overweight adults. Sci. Rep. 9:9772. doi: 10.1038/s41598-019-46120-z
Cummins, N., Tweedie, A., Zuryn, S., Bertran-Gonzalez, J., and Gotz, J. (2019). Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 38:e99360. doi: 10.15252/embj.201899360
Dave, K. D., De Silva, S., Sheth, N. P., Ramboz, S., Beck, M. J., Quang, C., et al. (2014). Phenotypic characterization of recessive gene knockout rat models of Parkinson's disease. Neurobiol. Dis. 70, 190–203. doi: 10.1016/j.nbd.2014.06.009
De Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1038/nature07534
Delettre, C., Lenaers, G., Griffoin, J. M., Gigarel, N., Lorenzo, C., Belenguer, P., et al. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet. 26, 207–210. doi: 10.1038/79936
Devi, T. S., Yumnamcha, T., Yao, F., Somayajulu, M., Kowluru, R. A., and Singh, L. P. (2019). TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 8:bio038521. doi: 10.1242/bio.038521
Devireddy, S., Liu, A., Lampe, T., and Hollenbeck, P. J. (2015). The organization of mitochondrial quality control and life cycle in the nervous system in vivo in the absence of PINK1. J. Neurosci. 35, 9391–9401. doi: 10.1523/JNEUROSCI.1198-15.2015
Dollerup, O. L., Christensen, B., Svart, M., Schmidt, M. S., Sulek, K., Ringgaard, S., et al. (2018). A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin. Nutr. 108, 343–353. doi: 10.1093/ajcn/nqy132
Eisenberg, T., Abdellatif, M., Schroeder, S., Primessnig, U., Stekovic, S., Pendl, T., et al. (2016). Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438. doi: 10.1038/nm.4222
Eisenberg, T., Knauer, H., Schauer, A., Buttner, S., Ruckenstuhl, C., Carmona-Gutierrez, D., et al. (2009). Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 11, 1305–1314. doi: 10.1038/ncb1975
Evans, C. S., and Holzbaur, E. L. (2020a). Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife 9:e50260. doi: 10.7554/eLife.50260
Evans, C. S., and Holzbaur, E. L. F. (2020b). Quality control in neurons: mitophagy and other selective autophagy mechanisms. J. Mol. Biol. 432, 240–260. doi: 10.1016/j.jmb.2019.06.031
Fang, E. F. (2019). Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy 15, 1112–1114. doi: 10.1080/15548627.2019.1596497
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U. S. A. 112, E2174–2181. doi: 10.1073/pnas.1504880112
Franco-Iborra, S., Plaza-Zabala, A., Montpeyo, M., Sebastian, D., Vila, M., and Martinez-Vicente, M. (2020). Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy. 1–18. doi: 10.1080/15548627.2020.1728096
Fu, Y., He, F., Zhang, S., and Jiao, X. (1995). Consistent striatal damage in rats induced by 3-nitropropionic acid and cultures of arthrinium fungus. Neurotoxicol. Teratol. 17, 413–418. doi: 10.1016/0892-0362(94)00078-R
Garcia, G. C., Bartol, T. M., Phan, S., Bushong, E. A., Perkins, G., Sejnowski, T. J., et al. (2019). Mitochondrial morphology provides a mechanism for energy buffering at synapses. Sci. Rep. 9:18306. doi: 10.1038/s41598-019-54159-1
Garcia-Prat, L., Sousa-Victor, P., and Munoz-Canoves, P. (2017). Proteostatic and metabolic control of stemness. Cell Stem Cell 20, 593–608. doi: 10.1016/j.stem.2017.04.011
Gatica, D., Lahiri, V., and Klionsky, D. J. (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242. doi: 10.1038/s41556-018-0037-z
Georgakopoulos, N. D., Wells, G., and Campanella, M. (2017). The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 13, 136–146. doi: 10.1038/nchembio.2287
Gilmour, B. C., Gudmundsrud, R., Frank, J., Hov, A., Lautrup, S., Aman, Y., et al. (2020). Targeting NAD(+) in translational research to relieve diseases and conditions of metabolic stress and ageing. Mech. Ageing Dev. 186:111208. doi: 10.1016/j.mad.2020.111208
Goldberg, M. S., Fleming, S. M., Palacino, J. J., Cepeda, C., Lam, H. A., Bhatnagar, A., et al. (2003). Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 278, 43628–43635. doi: 10.1074/jbc.M308947200
Guo, X., Sun, X., Hu, D., Wang, Y. J., Fujioka, H., Vyas, R., et al. (2016). VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease. Nat. Commun. 7:12646. doi: 10.1038/ncomms12646
Gupta, V. K., Pech, U., Bhukel, A., Fulterer, A., Ender, A., Mauermann, S. F., et al. (2016). Spermidine suppresses age-associated memory impairment by preventing adverse increase of presynaptic active zone size and release. PLoS Biol. 14:e1002563. doi: 10.1371/journal.pbio.1002563
Gupta, V. K., Scheunemann, L., Eisenberg, T., Mertel, S., Bhukel, A., Koemans, T. S., et al. (2013). Restoring polyamines protects from age-induced memory impairment in an autophagy-dependent manner. Nat. Neurosci. 16, 1453–1460. doi: 10.1038/nn.3512
Guyot, M. C., Hantraye, P., Dolan, R., Palfi, S., Maziere, M., and Brouillet, E. (1997). Quantifiable bradykinesia, gait abnormalities and Huntington's disease-like striatal lesions in rats chronically treated with 3-nitropropionic acid. Neuroscience 79, 45–56. doi: 10.1016/S0306-4522(96)00602-1
Han, S., Jeong, Y. Y., Sheshadri, P., Su, X., and Cai, Q. (2020). Mitophagy regulates integrity of mitochondria at synapses and is critical for synaptic maintenance. EMBO Rep. 21:e49801. doi: 10.15252/embr.201949801
Harper, J. W., Ordureau, A., and Heo, J. M. (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108. doi: 10.1038/nrm.2017.129
Hasson, S. A., Kane, L. A., Yamano, K., Huang, C. H., Sliter, D. A., Buehler, E., et al. (2013). High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504, 291–295. doi: 10.1038/nature12748
He, F., Zhang, S., Qian, F., and Zhang, C. (1995). Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid). Neurology 45, 2178–2183. doi: 10.1212/WNL.45.12.2178
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435. doi: 10.1038/s41580-019-0101-y
Holtzman, E., and Novikoff, A. B. (1965). Lysomes in the rat sciatic nerve following crush. J. Cell Biol. 27, 651–669. doi: 10.1083/jcb.27.3.651
Hou, Y., Dan, X., Babbar, M., Wei, Y., Hasselbalch, S. G., Croteau, D. L., et al. (2019). Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581. doi: 10.1038/s41582-019-0244-7
Hou, Y., Lautrup, S., Cordonnier, S., Wang, Y., Croteau, D. L., Zavala, E., et al. (2018). NAD(+) supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. U. S. A. 115, E1876–E1885. doi: 10.1073/pnas.1718819115
Hwang, S., Disatnik, M. H., and Mochly-Rosen, D. (2015). Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington's disease. EMBO Mol. Med. 7, 1307–1326. doi: 10.15252/emmm.201505256
Joaquim, H. P. G., Costa, A. C., Forlenza, O. V., Gattaz, W. F., and Talib, L. L. (2019). Decreased plasmatic spermidine and increased spermine in mild cognitive impairment and Alzheimer's disease patients. Arch. Clin. Psych. 46, 120–124. doi: 10.1590/0101-60830000000209
Kerr, J. S., Adriaanse, B. A., Greig, N. H., Mattson, M. P., Cader, M. Z., Bohr, V. A., et al. (2017). Mitophagy and Alzheimer's disease: cellular and molecular mechanisms. Trends Neurosci. 40, 151–166. doi: 10.1016/j.tins.2017.01.002
Khacho, M., Harris, R., and Slack, R. S. (2019). Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 20, 34–48. doi: 10.1038/s41583-018-0091-3
Khalil, B., El Fissi, N., Aouane, A., Cabirol-Pol, M. J., Rival, T., and Lievens, J. C. (2015). PINK1-induced mitophagy promotes neuroprotection in Huntington's disease. Cell Death Dis. 6:e1617. doi: 10.1038/cddis.2014.581
Khaminets, A., Behl, C., and Dikic, I. (2016). Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. 26, 6–16. doi: 10.1016/j.tcb.2015.08.010
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Lautrup, S., Sinclair, D. A., Mattson, M. P., and Fang, E. F. (2019). NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655. doi: 10.1016/j.cmet.2019.09.001
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, J. J., Sanchez-Martinez, A., Zarate, A. M., Beninca, C., Mayor, U., Clague, M. J., et al. (2018). Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 217, 1613–1622. doi: 10.1083/jcb.201801044
Lichtenwalner, R. J., Forbes, M. E., Bennett, S. A., Lynch, C. D., Sonntag, W. E., and Riddle, D. R. (2001). Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience 107, 603–613. doi: 10.1016/S0306-4522(01)00378-5
Lie, P. P. Y., and Nixon, R. A. (2019). Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 122, 94–105. doi: 10.1016/j.nbd.2018.05.015
Lin, M. Y., Cheng, X. T., Tammineni, P., Xie, Y., Zhou, B., Cai, Q., et al. (2017). Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94, 595–610. doi: 10.1016/j.neuron.2017.04.004
Liu, J., Liu, W., Li, R., and Yang, H. (2019). Mitophagy in Parkinson's disease: from pathogenesis to treatment. Cells 8:712. doi: 10.3390/cells8070712
Lou, G., Palikaras, K., Lautrup, S., Scheibye-Knudsen, M., Tavernarakis, N., and Fang, E. F. (2020). Mitophagy and neuroprotection. Trends Mol. Med. 26, 8–20. doi: 10.1016/j.molmed.2019.07.002
Madeo, F., Eisenberg, T., Pietrocola, F., and Kroemer, G. (2018). Spermidine in health and disease. Science 359:eaan2788. doi: 10.1126/science.aan2788
Maglione, M., Kochlamazashvili, G., Eisenberg, T., Racz, B., Michael, E., Toppe, D., et al. (2019). Spermidine protects from age-related synaptic alterations at hippocampal mossy fiber-CA3 synapses. Sci. Rep. 9:19616. doi: 10.1038/s41598-019-56133-3
Martinez-Vicente, M., Talloczy, Z., Wong, E., Tang, G., Koga, H., Kaushik, S., et al. (2010). Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat. Neurosci. 13, 567–576. doi: 10.1038/nn.2528
Martin-Maestro, P., Gargini, R., Perry, G., Avila, J., and Garcia-Escudero, V. (2016). PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer's disease. Hum. Mol. Genet. 25, 792–806. doi: 10.1093/hmg/ddv616
Mclelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 7:32. doi: 10.7554/eLife.32866.032
Mcwilliams, T. G., Prescott, A. R., Allen, G. F., Tamjar, J., Munson, M. J., Thomson, C., et al. (2016). mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 214, 333–345. doi: 10.1083/jcb.201603039
Mcwilliams, T. G., Prescott, A. R., Montava-Garriga, L., Ball, G., Singh, F., Barini, E., et al. (2018). Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab. 27, 439–449. doi: 10.1016/j.cmet.2017.12.008
Misgeld, T., and Schwarz, T. L. (2017). Mitostasis in neurons: maintaining mitochondria in an extended cellular architecture. Neuron 96, 651–666. doi: 10.1016/j.neuron.2017.09.055
Misko, A. L., Sasaki, Y., Tuck, E., Milbrandt, J., and Baloh, R. H. (2012). Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32, 4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012
Montava-Garriga, L., and Ganley, I. G. (2020). Outstanding questions in mitophagy: what we do and do not know. J. Mol. Biol. 432, 206–230. doi: 10.1016/j.jmb.2019.06.032
Ordureau, A., Paulo, J. A., Zhang, J., An, H., Swatek, K. N., Cannon, J. R., et al. (2020). Global landscape and dynamics of parkin and USP30-dependent ubiquitylomes in ineurons during mitophagic signaling. Mol. Cell 77, 1124–1142. doi: 10.1016/j.molcel.2019.11.013
Ordureau, A., Paulo, J. A., Zhang, W., Ahfeldt, T., Zhang, J., Cohn, E. F., et al. (2018). Dynamics of PARKIN-dependent mitochondrial ubiquitylation in induced neurons and model systems revealed by digital snapshot proteomics. Mol. Cell 70, 211–227. doi: 10.1016/j.molcel.2018.03.012
Orr, A. L., Li, S., Wang, C. E., Li, H., Wang, J., Rong, J., et al. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 28, 2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Palikaras, K., and Tavernarakis, N. (2014). Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 56, 182–188. doi: 10.1016/j.exger.2014.01.021
Palikaras, K., and Tavernarakis, N. (2020). Regulation and roles of mitophagy at synapses. Mech. Ageing Dev. 187:111216. doi: 10.1016/j.mad.2020.111216
Park, H., Chung, K. M., An, H. K., Gim, J. E., Hong, J., Woo, H., et al. (2019). Parkin promotes mitophagic cell death in adult hippocampal neural stem cells following insulin withdrawal. Front. Mol. Neurosci. 12:46. doi: 10.3389/fnmol.2019.00046
Perez, F. A., and Palmiter, R. D. (2005). Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. U. S. A. 102, 2174–2179. doi: 10.1073/pnas.0409598102
Pickford, F., Masliah, E., Britschgi, M., Lucin, K., Narasimhan, R., Jaeger, P. A., et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Invest. 118, 2190–2199. doi: 10.1172/JCI33585
Pickles, S., Vigie, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, 170–185. doi: 10.1016/j.cub.2018.01.004
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Puri, R., Cheng, X. T., Lin, M. Y., Huang, N., and Sheng, Z. H. (2019). Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat. Commun. 10:3645. doi: 10.1038/s41467-019-11636-5
Radenkovic, D., Reason, and Verdin, E. (2020). Clinical evidence for targeting NAD therapeutically. Pharmaceuticals 13:247. doi: 10.3390/ph13090247
Rawson, R. L., Yam, L., Weimer, R. M., Bend, E. G., Hartwieg, E., Horvitz, H. R., et al. (2014). Axons degenerate in the absence of mitochondria in C. elegans. Curr. Biol. 24, 760–765. doi: 10.1016/j.cub.2014.02.025
Schondorf, D. C., Ivanyuk, D., Baden, P., Sanchez-Martinez, A., De Cicco, S., Yu, C., et al. (2018). The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson's disease. Cell Rep. 23, 2976–2988. doi: 10.1016/j.celrep.2018.05.009
Sekine, S., and Youle, R. J. (2018). PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 16:2. doi: 10.1186/s12915-017-0470-7
Sentelle, R. D., Senkal, C. E., Jiang, W., Ponnusamy, S., Gencer, S., Selvam, S. P., et al. (2012). Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8, 831–838. doi: 10.1038/nchembio.1059
Sharma, M., Jarquin, U. N. R., Rivera, O., Kazantzis, M., Eshraghi, M., Shahani, N., et al. (2019). Rhes, a striatal-enriched protein, promotes mitophagy via Nix. Proc. Natl. Acad. Sci. U. S. A. 116, 23760–23771. doi: 10.1073/pnas.1912868116
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shi, R. Y., Zhu, S. H., Li, V., Gibson, S. B., Xu, X. S., and Kong, J. M. (2014). BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci. Ther. 20, 1045–1055. doi: 10.1111/cns.12325
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/hmg/ddr024
Sliter, D. A., Martinez, J., Hao, L., Chen, X., Sun, N., Fischer, T. D., et al. (2018). Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262. doi: 10.1038/s41586-018-0448-9
Smith, H. L., Bourne, J. N., Cao, G., Chirillo, M. A., Ostroff, L. E., Watson, D. J., et al. (2016). Mitochondrial support of persistent presynaptic vesicle mobilization with age-dependent synaptic growth after LTP. Elife 5:14. doi: 10.7554/eLife.15275.014
Somredngan, S., and Thong-Asa, W. (2018). Neurological changes in vulnerable brain areas of chronic cerebral hypoperfusion mice. Ann. Neurosci. 24, 233–242. doi: 10.1159/000481789
Steketee, M. B., Moysidis, S. N., Weinstein, J. E., Kreymerman, A., Silva, J. P., Iqbal, S., et al. (2012). Mitochondrial dynamics regulate growth cone motility, guidance, and neurite growth rate in perinatal retinal ganglion cells in vitro. Invest. Ophthalmol. Vis. Sci. 53, 7402–7411. doi: 10.1167/iovs.12-10298
Su, S. H., Wu, Y. F., Wang, D. P., and Hai, J. (2018). Inhibition of excessive autophagy and mitophagy mediates neuroprotective effects of URB597 against chronic cerebral hypoperfusion. Cell Death Dis. 9:733. doi: 10.1038/s41419-018-0755-y
Subramaniam, S. (2020). Exaggerated mitophagy: a weapon of striatal destruction in the brain? Biochem. Soc. Trans. 48, 709–717. doi: 10.1042/BST20191283
Sun, N., Yun, J., Liu, J., Malide, D., Liu, C., Rovira, I, Holmstrom, K. M., et al. (2015). Measuring in vivo mitophagy. Mol. Cell 60, 685–696. doi: 10.1016/j.molcel.2015.10.009
Sung, H., Tandarich, L. C., Nguyen, K., and Hollenbeck, P. J. (2016). Compartmentalized regulation of Parkin-mediated mitochondrial quality control in the drosophila nervous system in vivo. J. Neurosci. 36, 7375–7391. doi: 10.1523/JNEUROSCI.0633-16.2016
Verreet, T., Weaver, C. J., Hino, H., Hibi, M., and Poulain, F. E. (2019). Syntaphilin-mediated docking of mitochondria at the growth cone is dispensable for axon elongation in vivo. eNeuro 6:ENEURO.0026-19.2019. doi: 10.1523/ENEURO.0026-19.2019
Whitworth, A. J., Theodore, D. A., Greene, J. C., Benes, H., Wes, P. D., and Pallanck, L. J. (2005). Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 102, 8024–8029. doi: 10.1073/pnas.0501078102
Wirth, M., Benson, G., Schwarz, C., Kobe, T., Grittner, U., Schmitz, D., et al. (2018). The effect of spermidine on memory performance in older adults at risk for dementia: a randomized controlled trial. Cortex 109, 181–188. doi: 10.1016/j.cortex.2018.09.014
Xie, C., Aman, Y., Adriaanse, B. A., Cader, M. Z., Plun-Favreau, H., Xiao, J., et al. (2019). Culprit or bystander: defective mitophagy in Alzheimer's disease. Front. Cell Dev. Biol. 7:391. doi: 10.3389/fcell.2019.00391
Yan, X., Wang, B., Hu, Y., Wang, S., and Zhang, X. (2020). Abnormal mitochondrial quality control in neurodegenerative diseases. Front. Cell. Neurosci. 14:138. doi: 10.3389/fncel.2020.00138
Yang, Y., Gehrke, S., Imai, Y., Huang, Z., Ouyang, Y., Wang, J. W., et al. (2006). Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U. S. A. 103, 10793–10798. doi: 10.1073/pnas.0602493103
Yu-Wai-Man, P., Griffiths, P. G., Gorman, G. S., Lourenco, C. M., Wright, A. F., Auer-Grumbach, M., et al. (2010). Multi-system neurological disease is common in patients with OPA1 mutations. Brain 133, 771–786. doi: 10.1093/brain/awq007
Zaninello, M., Palikaras, K., Naon, D., Iwata, K., Herkenne, S., Quintana-Cabrera, R., et al. (2020). Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat. Commun. 11:4029. doi: 10.1038/s41467-020-17821-1
Keywords: aging, cell death, energy metabolism, homeostasis, mitochondria, mitophagy, neurodegeneration, neuroprotection
Citation: Doxaki C and Palikaras K (2021) Neuronal Mitophagy: Friend or Foe? Front. Cell Dev. Biol. 8:611938. doi: 10.3389/fcell.2020.611938
Received: 29 September 2020; Accepted: 22 December 2020;
Published: 18 January 2021.
Edited by:
Shey-Shing Sheu, Thomas Jefferson University, United StatesReviewed by:
Uwe Schlattner, Université Grenoble Alpes, FranceEvandro Fei Fang, University of Oslo, Norway
Copyright © 2021 Doxaki and Palikaras. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konstantinos Palikaras, cGFsaWthcmFza0BpbWJiLmZvcnRoLmdy