
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 18 December 2020
Sec. Molecular and Cellular Pathology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.611757
This article is part of the Research Topic Ectopic Mineralization of Tissues: Mechanisms, Risk Factors, Diseases and Prevention View all 29 articles
 Driss Ehirchiou1†
Driss Ehirchiou1† Ilaria Bernabei1†
Ilaria Bernabei1† Véronique Chobaz1
Véronique Chobaz1 Mariela Castelblanco1‡Thomas Hügle1Alexander So1
Mariela Castelblanco1‡Thomas Hügle1Alexander So1 Li Zhang2
Li Zhang2 Nathalie Busso1*
Nathalie Busso1* Sonia Nasi1
Sonia Nasi1Osteoarthritis (OA) is a progressive joint disease that is strongly associated with calcium-containing crystal formation (mineralization) by chondrocytes leading ultimately to cartilage calcification. However, this calcification process is poorly understood and treatments targeting the underlying disease mechanisms are lacking. The CD11b/CD18 integrin (Mac-1 or αMβ2), a member of the beta 2 integrin family of adhesion receptors, is critically involved in the development of several inflammatory diseases, including rheumatoid arthritis and systemic lupus erythematosus. We found that in a collagen-induced arthritis, CD11b-deficient mice exhibited increased cartilage degradation compared to WT control animals. However, the functional significance of CD11b integrin signaling in the pathophysiology of chondrocytes remains unknown. CD11b expression was found in the extracellular matrix and in chondrocytes in both healthy and damaged human and murine articular cartilage. Primary murine CD11b KO chondrocytes showed increased mineralization when induced in vitro by secondary calciprotein particles (CPP) and quantified by Alizarin Red staining. This increased propensity to mineralize was associated with an increased alkaline phosphatase (Alp) expression (measured by qRT-PCR and activity assay) and an enhanced secretion of the pro-mineralizing IL-6 cytokine compared to control wild-type cells (measured by ELISA). Accordingly, addition of an anti-IL-6 receptor antibody to CD11b KO chondrocytes reduced significantly the calcification and identified IL-6 as a pro-mineralizing factor in these cells. In the same conditions, the ratio of qRT-PCR expression of collagen X over collagen II, and that of Runx2 over Sox9 (both ratio being indexes of chondrocyte hypertrophy) were increased in CD11b-deficient cells. Conversely, the CD11b activator LA1 reduced chondrocyte mineralization, Alp expression, IL-6 production and collagen X expression. In the meniscectomy (MNX) model of murine knee osteoarthritis, deficiency of CD11b led to more severe OA (OARSI scoring of medial cartilage damage in CD11b: 5.6 ± 1.8, in WT: 1.2 ± 0.5, p < 0.05, inflammation in CD11b: 2.8 ± 0.2, in WT: 1.4 ± 0.5). In conclusion, these data demonstrate that CD11b signaling prevents chondrocyte hypertrophy and chondrocyte mineralization in vitro and has a protective role in models of OA in vivo.
Osteoarthritis (OA) is a long-term chronic degenerative and disabling joint disease (Sen and Hurley, 2020) for which no effective curative disease modifying treatment is available. A wide range of current treatments only lessen the symptom but not the underlying condition. Understanding the pathological mechanisms and the signaling pathways controlling OA development and progression could provide novel targets that would improve the treatment of this disorder (Huang et al., 2018; Kim et al., 2018).
OA is characterized by progressive loss of articular cartilage, osteophyte formation, synovial inflammation and calcification in joint structures such as cartilage (Ea et al., 2011; Loeser et al., 2012; Frallonardo et al., 2018; Yan et al., 2020). Calcium-containing crystals, which encompass basic calcium phosphate (BCP) and calcium pyrophosphate dihydrate crystals (CPPD), have been demonstrated to be of etiologic importance in OA. Indeed they were shown to actively trigger inflammatory, catabolic and oxidant responses in joint cells such as synovial fibroblasts, macrophages, osteoclasts and chondrocytes (Fuerst et al., 2009; Nasi et al., 2016b; Bertrand et al., 2020; Yan et al., 2020). However, the receptors and signaling pathways modulating ectopic calcification in OA joints only start to be elucidated (Castelblanco et al., 2020).
Cell-surface receptors, such as integrins, mediate interaction between chondrocytes and the extracellular matrix (ECM) components, orchestrating its physiological turn-over. Integrins are composed of different combinations of α- and β-subunits, most of which have been found expressed in chondrocytes. In joint physiology, integrins have been demonstrated to play key roles in chondrogenic differentiation (Varas et al., 2007; LaPointe et al., 2013; Song and Park, 2014) and chondrocyte survival (Loeser, 2014). In joint pathology, lack of integrin signaling has shown to mediate the catabolic reactions responsible for cartilage matrix degradation, ultimately leading to OA (Lapadula et al., 1997; Forsyth et al., 2002; Almonte-Becerril et al., 2014; Loeser, 2014; Tian et al., 2015). For instance, mice deficient in β1 integrins exhibit disorganized articular cartilage, reduced mobility and increased OA changes (Raducanu et al., 2009). On the other hand, cartilage ECM components such as fibronectin and collagens, are known ligands of integrins. In fact, Forsyth et al. have reported that α5β1 integrin mediates matrix degradation induced by fibronectin fragments in cultured chondrocytes (Forsyth et al., 2002).
CD11b/CD18 (Mac-1 or αMβ2) is a member of the beta 2 integrin family of adhesion receptors (Rosetti and Mayadas, 2016; Schittenhelm et al., 2017). CD11b serves as a signaling regulator and its deficiency has been implicated in the development of inflammatory diseases, such as systemic lupus erythematosus (SLE), lupus nephritis and rheumatoid arthritis (Ehirchiou et al., 2007; Reed et al., 2013; Stevanin et al., 2017; Khan et al., 2018). In SLE, genetic variants in the human ITGAM gene, which encodes for CD11b, have been strongly associated with susceptibility to the disease (Nath et al., 2008). Furthermore, Dahdal et al. (2018) have reported a positive correlation between disease activity and serum calcification-propensity, as measured by a novel functional blood test (T50) that is able to measure the time for spontaneous formation of crystalline calcium phosphate deposits. It has also been revealed that CD11b is involved in MyD88/TRIF pathway in SLE, by decreasing IFN-α,β and NF-κB transcribed cytokines, namely interleukin-6 (IL-6) (Faridi et al., 2017), which can promote calcium-containing crystal formation (Nasi et al., 2016b). An additional evidence of the effect of CD11b on reducing the pro-mineralizing IL-6, is in a mouse model of collagen-induced arthritis, where our laboratory reported that CD11b KO mice exhibited higher IL-6 serum levels as compared with those of WT control animals (Stevanin et al., 2017).
However, the role of CD11b integrin in calcification has not yet been examined. Based on these observations, we hypothesized that CD11b integrin signaling might be involved in the process of cartilage calcification. Here, we found that both murine and human chondrocytes expressed CD11b integrin. Furthermore, our results indicate that CD11b deficiency in chondrocytes disrupted articular cartilage homeostasis by enhancing chondrocytes calcification, via increased IL-6 production and a switch toward hypertrophic cell differentiation, overall leading to cartilage degradation. Finally, our results identified a newly CD11b-dependent pathway that may be a target in the future development of a successful therapeutic approach in OA.
Articular chondrocytes were isolated from C57BL/6J and CD11b KO mice as described previously (Nasi et al., 2016a). Cells were cultured at a density of 3.5 × 104 cells/cm2 for 7 days in complete DMEM (10% fetal bovine serum (FBS), 1% Penicillin Streptomycin). Medium was changed every 2–3 days. Cells were detached with trypsin-EDTA (Amimed) and counted for stimulation experiments, all performed in complete DMEM medium, with LA1 20uM (ChemBridge) dissolved in dimethyl sulfoxide (DMSO) or equivalent quantity of the vehicle as control, added 30 min before the mineralization experiment. Chondrocytes were stimulated with secondary calciprotein particles (CPP) for 4, 6, or 24 h, or in BGjb medium (10% FBS, 1% Penicillin Streptomycin) with 50 μg/mL ascorbic acid and 20 mM β-glycerol phosphate for 14 days. Cell monolayers were stained with Mmp13 rabbit polyclonal antibody (Abcam 39012) and quantified by ImageJ as percentage of positively stained cells over total cells in each field. Chondrocytes were stimulated with 10 μg/ml rat anti-mouse IL-6R (15A7, Bio X Cell) monoclonal antibody or irrelevant IgG2b antibody control.
CPP were generated by preparing 20 ml DMEM containing 10% FBS, 1 mmol/L CaCl2, 3.5 mmol/L inorganic phosphate (1.36 mmol/L NaH2PO4 and 2.14 mmol/L Na2HPO4) (Aghagolzadeh et al., 2016). After 7 days incubation at 37°C, the suspension was centrifuged for 3 h at 4500 rpm and 4°C, and the pellet containing CPP was resuspended in 1 ml DMEM and stored at −20°C.
Measurement of lactate dehydrogenase (LDH) as a means of cytotoxicity assay was performed with using CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega) according to the manufacturer’s instructions. Values were calculated as percentage of cytotoxicity using the following formula: LDH release (%) = [(value in sample) — (background)]/[(value in Triton X-100-treated sample) — (background)] x100.
Chondrocytes were treated with CPP (50 μg/ml calcium) for 24 h or calcifying medium for 24 h, then washed in PBS and fixed in 10% formaldehyde. Crystals were quantified with Alizarin Red staining with Adobe® Photoshop® after image binarization and related pixel counts. Calcium content was quantified by the QuantiChromTM Calcium Kit (BioAssay Systems) by reading absorbance at 612 nm using the Spectramax M5e reader (Molecular Devices).
Supernatants were collected from stimulation experiments, and assayed using murine or human IL-6 and murine MCP-1 ELISA kits (eBioscience) following the manufacturer’s protocol. Results were read at 450 nm by Spectramax M5e (Molecular devices). Synovial fluid was obtained from 24 OA patients at time of joint replacement (average age 69 years old ± 10), and CD11b evaluated in these biological samples by human CD11b ELISA (LSBio, LifeSpan BioSciences, Inc.).
Cells were treated with TRIzol (500 μl for 1 million cells) and RNA was extracted (RNA Clean and Concentrator 5-Zymoresearch) and reverse transcribed (Superscript II- InvitrogenTM). Quantitative Real Time PCR (qRT-PCR) was performed with gene specific primers (Table 1) using the LightCycler® 480 system (Roche Applied Science). Normalization was performed against Gapdh reference gene, and fold increase of transcripts was calculated against control cells.
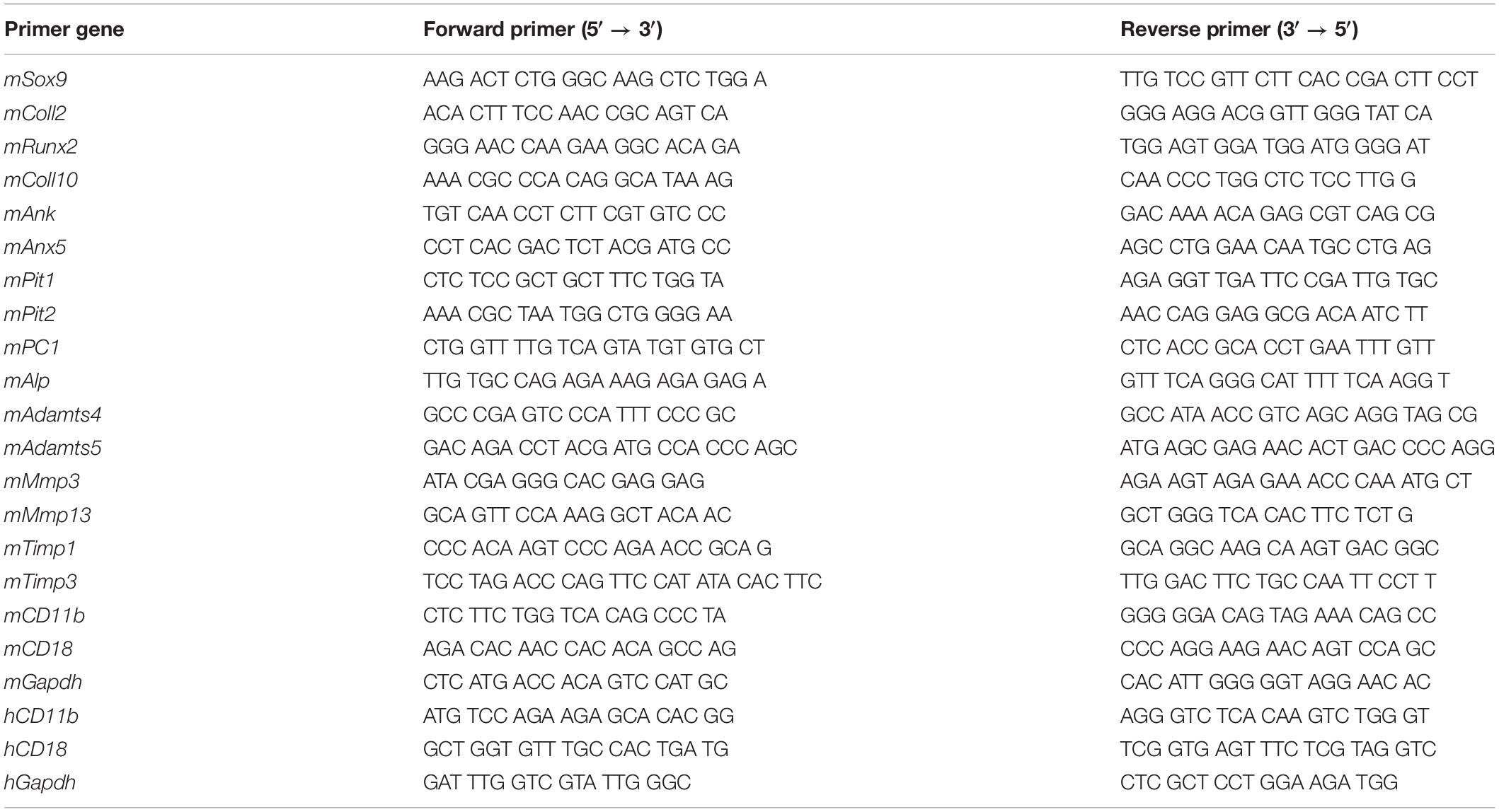
Table 1. Gene primers (forward and reverse) sequences for RT-qPCR analysis (m, murine; h, human).
Cells were lysed in 0.01% sodium dodecyl sulfate (SDS) and alkaline phosphatase (Alp) activity was measured by use of p-Nitrophenyl Phosphate assay (Alkaline Phosphatase Assay Kit, Abcam, ab83369). Lysate was read at 405 nm by spectrophotometric measurement (Spectramax M5e plate reader).
Flow cytometry was used to assess CD11b expression on chondrocytes. Cells were harvested from confluent cultures by treatment with a non-enzymatic cell dissociation buffer (5 mM EDTA, 20 mM HEPES in PBS). Cells were re-suspended in fluorescence-activated cell sorting (FACS) buffer (3% fetal calf serum, 5 mM EDTA in PBS) and incubated with PE anti-CD11b (M1/70, eBioscience), FITC anti-CD45 (30-F11, eBioscience) and FITC anti-F4/80 (BM8, eBioscience) for 30 min at 4°C in the dark. For intracellular staining, cells were permeabilized with Cytofix/Cytoperm solution (BD Biosciences) for 20 min and then incubated with biotin anti-Collagen II (2B1.5, Abcam) followed by a streptavidin APC conjugate antibody. Flow cytometric analyses were performed on LSRII cytometer using FACS Diva6 (Becton Dickinson) and FlowJoX software for data processing. The Imaging flow cytometry was carried out with the ImageStream®X Mark II Imaging Flow Cytometer (Merck Millipore, Billerica, MA, United States) using INSPIRE software. For data evaluation, IDEAS software version 6.0 was used.
Proliferation rate of WT and CD11b KO chondrocyte were analyzed by FACS after 3 days of culture by using Cell Proliferation Dye eFluorTM 670 (Invitrogen).
CD11b KO mice (C57BL/6J background) were provided by Prof. Britta Engelhardt (Theodor Kocher Institute, University of Bern). C57BL/6J purchased from Charles River were used as control mice. All animals were kept in a temperature-controlled environment in a ventilated rack with a 12:12-h light:dark cycle. Food and water were given ad libitum.
Control wild-type (WT) and CD11b KO female mice were anesthetized at 12 weeks of age and knee joint instability was induced surgically by partial medial meniscectomy (MNX) of the right knee, and the contralateral knee was sham-operated as control (Kamekura et al., 2005). Animals were sacrificed 2 months after surgery. For the aging model, control WT and CD11b KO mice were sacrificed at 37 weeks of age. For all experimental settings, knees dissected and fixed in 10% formaldehyde.
Scanning of mice knees was performed with a SkyScan 1076 X-ray μCT scanning system (SkyScan, Belgium), as previously described (Nasi et al., 2016b). New crystal formations were quantified by tissue volume (mm3), its bone mineral density (BMD, g/cm3) and its crystal content (μg). Tibial subchondral bone was analyzed as means of bone mineral density (BMD, g/cm3) and trabecular thickness (Tb.Th, mm).
Fixed knees were decalcified in 5% formic acid, dehydrated, and embedded in paraffin. Frontal sections of 6 μm at different deepness of cartilage were stained with Safranin-O-fast green. OARSI scoring of medial tibial plateau and medial femoral condyle was performed (score from 0 to 6 for each, in which 0 represents a healthy cartilage; 0.5 is some loss of safranin-O staining, meaning loss of proteoglycans; 1 some fibrillations in the superficial layer and no loss of cartilage; 2 erosion of the surface; 3 to 5 represent various percentages of the deepness of the cartilage affected by vertical clefts and erosion) (Glasson et al., 2010). Furthermore, inflammation of the synovial membrane was scored (each deepness of the sections was scored on a 0 to 3 scale of inflammation, then the average was calculated for one sample). Paraffin-embedded knee sections were stained with CD11b antibody (133357, Abcam). MMP13 was detected with anti-MMP13 (Ab39012 from Abcam), and quantified by calculating positive cells by use of Image J (U. S. National Institutes of Health, Bethesda, MD, United States).
Cartilage explants were obtained from 13 human OA patients undergoing total knee replacement surgery from Lausanne University Hospital, Switzerland (average age 71 years old ± 11). Sections of macroscopically undamaged and damaged cartilage were dissected from the femoral condyles and processed for histological analysis from 3 patients. The rest of undamaged and damaged cartilage was cut in small pieces, washed twice with PBS and twice with DMEM. Cartilage was degraded for 10 h in 10% Liberase (Roche) DMEM, and digested tissue passed through a 70 μm filter (BD biosciences) to obtain articular chondrocytes. Cells were plated and cultured as described above.
All values are expressed as the mean ± SD. Variation between data sets was evaluated using the Student’s t-test or one-way or two-way ANOVA test, where appropriate. Differences were considered statistically significant for a value of p < 0.05 (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ****p < 0.0001). Data was analyzed with GraphPad Prism software (GraphPad software), San Diego, CA.
Experiments on mice were performed in strict accordance to the Swiss Federal Regulations. The protocol was approved by the “Service de la consommation et des affaires vétérinaires du Canton de Vaud,” Switzerland. All efforts were made to minimize suffering and minimize the number of mice needed to assess statistical significance and experimental reproducibility. Human samples were obtained with the approval of the Centre Hospitalier Universitaire Vaudois ethical committee and patients’ written informed consent.
To determine the distribution of CD11b expression in the mouse cartilage, CD11b immunostaining was performed on knee sections from WT and CD11b KO mice. As seen in Figure 1A (left panels), positive signal was primarily detected in chondrocytes in the superficial layer of cartilage both in healthy and meniscectomized WT knee joints (MNX). Verifying specificity of the antibody, no CD11b staining was detected in articular chondrocytes of CD11b KO mice (Figure 1A, top right panel).
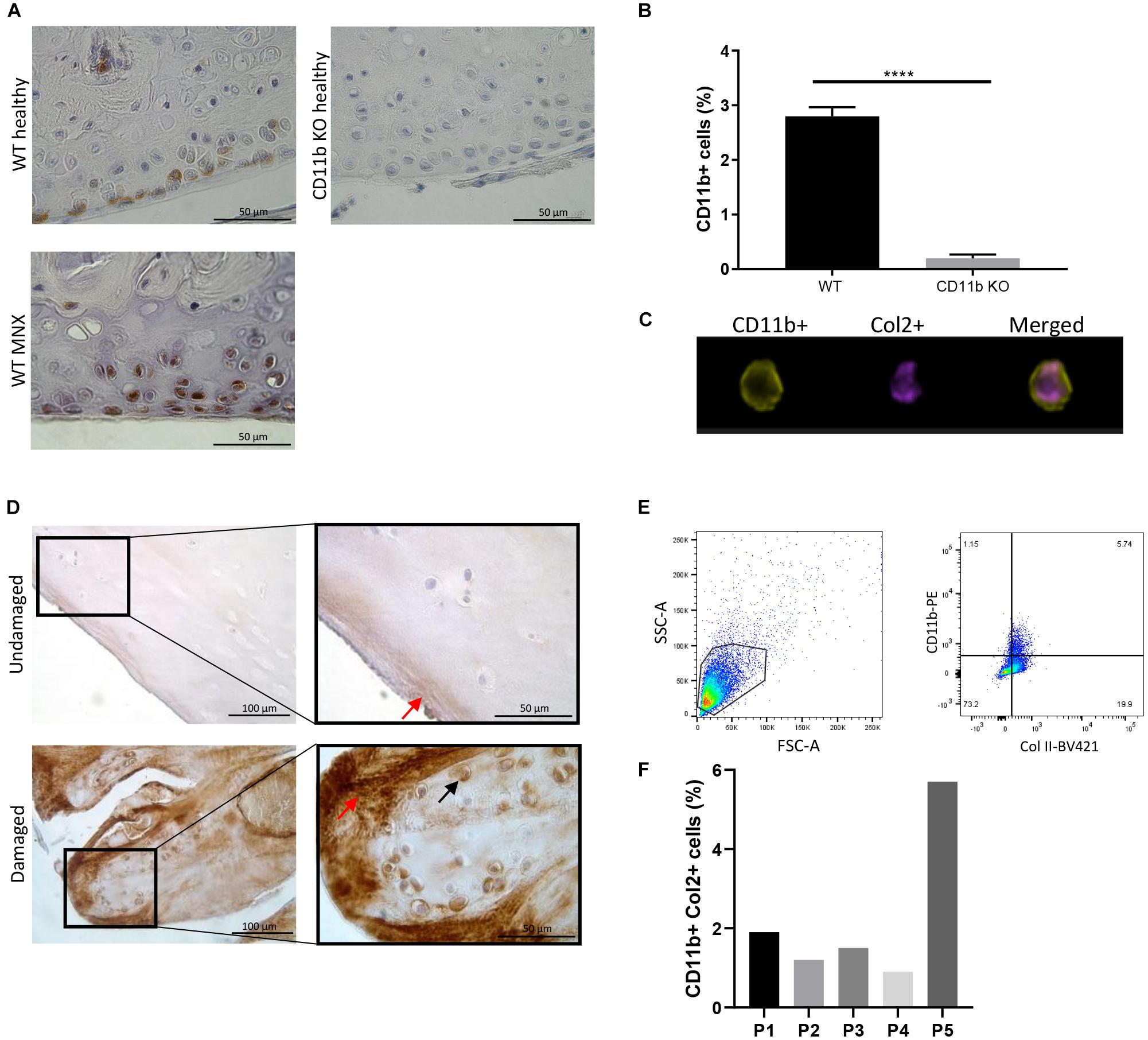
Figure 1. CD11b integrin is expressed in murine and human articular cartilage. (A) CD11b immunohistochemical staining (brown) in knee cartilage from healthy WT and CD11b KO mice, and meniscectomized (MNX) WT mice. For each group, one representative picture from one out of 3 mice is shown. Scale bars 50 μm. (B) FACS analysis and quantification of CD11b positive and Col2 positive primary murine chondrocytes, and (C) Amnis FACS analysis of CD11b positive (green) and Col2 positive (purple) staining; ****p < 0.0001. (D) CD11b immunohistochemical staining (brown) in undamaged and damaged femoral knee cartilage from OA patients undergoing joint replacement (black arrows indicate intracellular CD11b expression, red arrows indicate extracellular CD11b expression). For each group, one representative picture from one (P13) out of 3 patients is shown. Scale bars 100 and 50 μm. (E,F) Flow cytometry analysis of CD11b and Col2 expression on primary human chondrocytes obtained from cartilage of 5 osteoarthritis patients.
This data was confirmed by flow cytometry analysis. Indeed, 3–4% of WT primary chondrocytes extracted from articular cartilage expressed the CD11b integrin subunit (Figure 1B and Supplementary Figure 1D). Combining high-resolution microscopy and FACS (Amnis analysis), we found that CD11b positive cells also expressed collagen II, a specific marker of chondrocytes (Figure 1C). In addition, the chondrocytes showed no expression of F4/80, a macrophage marker (Supplementary Figure 1D) and were negative for CD45 (Supplementary Figure 1E), an antigen used to define leukocyte populations. Furthermore, CD11b expression in murine chondrocytes was demonstrated by q-PCR (Supplementary Figure 1A).
We next analyzed if CD11b was expressed in human cartilage by CD11b immunostaining analysis. Cartilage was obtained from the knee joint of three OA patients and separated into macroscopically healthy and damaged cartilages. The results in Figure 1D and Supplementary Figure 1C showed that CD11b was present in chondrocytes in the superficial layer of human articular cartilage (black arrows). Interestingly, a diffuse expression pattern of CD11b at the extracellular matrix was also observed in the damaged cartilage (red arrows). In Figures 1E,F, CD11b expression in human chondrocytes was confirmed by flow cytometry analysis, with an expression level varying from 2 to 6% of the cells. Further proof of CD11b expression by human chondrocytes was obtained by qPCR (Supplementary Figure 1B), showing both CD11b and CD18 expression, with a predominant expression of CD18. We hypothesized that extracellular matrix deposited CD11b could result from shedding of the integrin (Gomez et al., 2012). Indeed, we detected by ELISA measurement soluble CD11b in human OA synovial fluid (24 patients, average 4.5 ng/ml ± 0.4).
We then evaluated the effect of CD11b deficiency on chondrocyte mineralization. Primary murine chondrocytes were allowed to mineralize in the presence of calciprotein particles (CPP) and mineralization was assessed after 24 h by Alizarin Red. As shown in Figure 2A, CD11b KO chondrocyte culture exhibited massive calcium containing crystal deposits compared to WT control (in red). Quantification of Alizarin Red staining showed a 3-fold significant increase of crystals in CD11b KO cells (Figure 2B). The increased calcifying ability of CD11b KO chondrocytes was not restricted to the model used, as we obtained similar results with another model of calcification (complete medium with 50 μg/mL ascorbic acid and 20 mM β-glycerol phosphate for 14 days), i.e., CD11b KO chondrocytes mineralized more compared to WT control (Supplementary Figure 2A). This effect was not due to cytotoxicity, as measured by lactate dehydrogenase (LDH) activity (Supplementary Figure 2B).
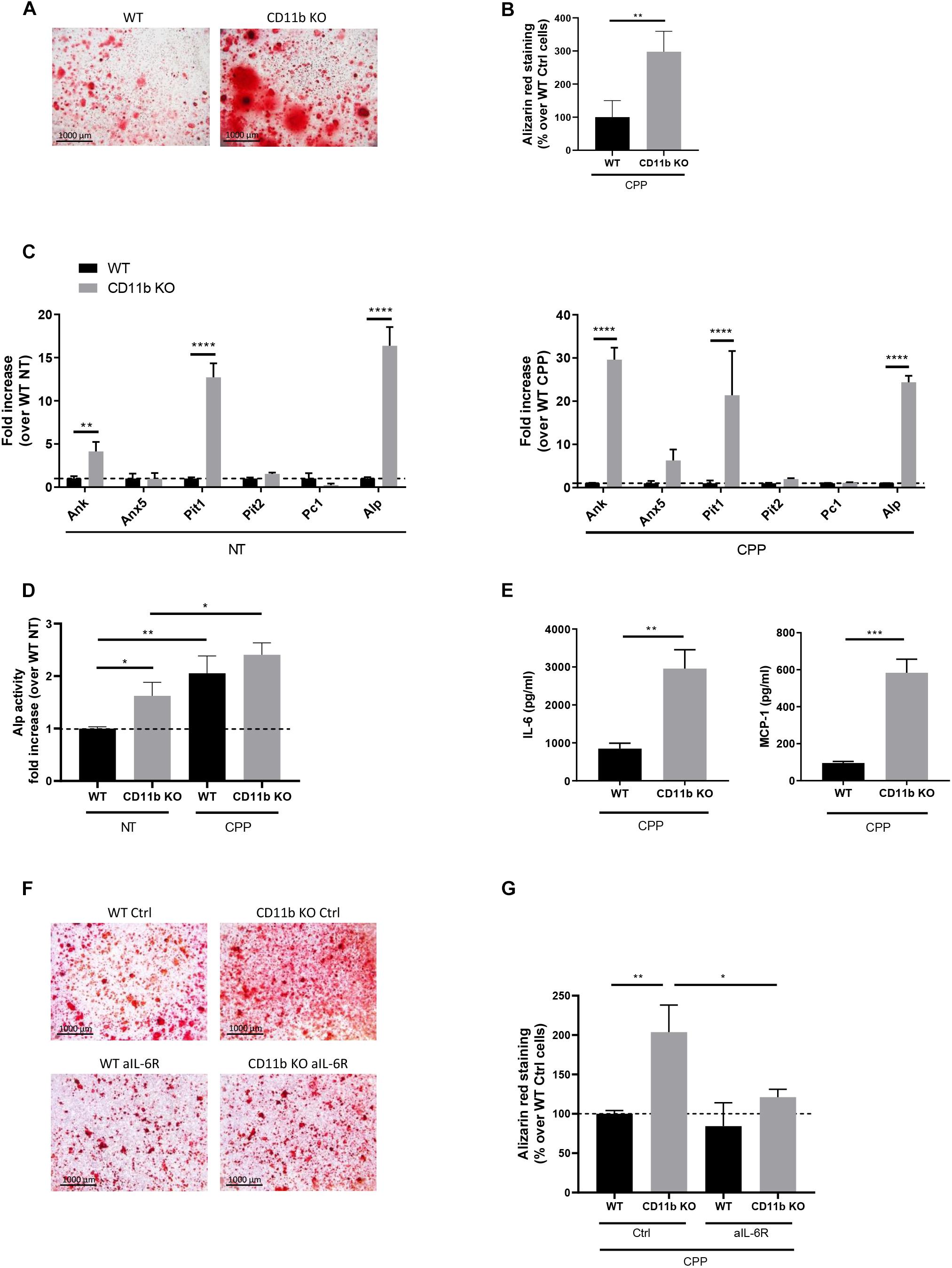
Figure 2. CD11b deficiency increases mineralization by murine articular chondrocytes. (A) Alizarin Red staining of WT and CD11b KO primary murine chondrocytes stimulated with secondary CPP for 24 h in DMEM + 10% FBS. Pictures show one representative culture well of one experiment out of three independent experiments. Scale bars 1000 μm. (B) The graph shows the percentage of Alizarin Red positive surface over total surface, calculated in Adobe® Photoshop® and normalized on WT chondrocytes. Values represent means ± SD of triplicates samples from one representative experiment out of three independent experiment; **p < 0.01. (C) qRT-PCR of the indicated genes in primary murine chondrocytes stimulated or not with secondary CPP for 4 h. Values represent fold increase of CD11b KO chondrocytes over their WT control cells (dotted line), shown in means ± SD of triplicates samples; **p < 0.01, ****p < 0.0001. (D) Quantification of Alkaline phosphatase activity in WT and CD11b KO primary murine chondrocytes treated or not with secondary CPP for 6 h. Values represent fold increase of Alp activity in CD11b KO over WT NT cells (dotted line) as means ± SD of triplicates samples; *p < 0.05, **p < 0.01. (E) IL-6 and MCP-1 concentration in cell supernatants of WT and CD11b KO primary murine chondrocytes of point (A). Values represent means ± SD of triplicates samples; **p < 0.01, ***p < 0.001. (F) Alizarin Red staining of WT and CD11b KO murine chondrocytes stimulated with secondary CPP for 24 h in DMEM + 10% FBS and treated or not with an anti-IL-6 receptor (aIL-6R, 10 μg/ml) or its IgG control (Ctrl, 10 μg/ml). Scale bars 1000 μm. (G) The graph shows the percentage of Alizarin Red positive surface over the total surface, calculated in Adobe® Photoshop® and normalized on control WT chondrocytes. Values represent means ± SD of triplicates samples; *p < 0.05, **p < 0.01.
To characterize how CD11b might interfere with calcification in chondrocyte cultures, we assessed changes in the expression of genes involved in the calcification process (Ea et al., 2011) (Ank, Anx5, Pit1, Pit2, Pc1 and Alp) by qRT-PCR. Results in Figure 2C show that Ank, Pit1 and Alp expression were significantly increased in CD11b KO chondrocytes both in unstimulated or calcifying conditions (CPP). Regarding Pit2 and Pc1, no significant differences were observed. We provided further confirmation of the calcifying phenotype of CD11b KO cells by measuring the activity of alkaline phosphatase (Alp) in the cell lysate in non-treated and CPP treated chondrocytes. The Alp specific activity was higher in CD11b KO cells compared to that of WT control (Figure 2D). Altogether, these results demonstrate that CD11b deficiency in chondrocytes enhanced the expression of calcification genes.
We previously demonstrated that IL-6 had pro-mineralizing effects in chondrocytes and, reciprocally, BCP crystals increased IL-6, thus leading to a vicious circle (Nasi et al., 2016b). Hence, we speculated that increased IL-6 secretion by CD11b-deficient chondrocytes could favor mineralization. Indeed, we detected by ELISA increased IL-6 levels in the medium from CD11b KO chondrocytes stimulated with CPP (Figure 2E). Additionally, CPP stimulation significantly increased MCP-1 secretion in CD11b deficient chondrocytes compared to WT control. Next, to understand if IL-6 secretion by CD11b KO chondrocytes was the main calcification inducer in these cells, we added anti IL-6 receptor antibody and CPP on WT and CD11b KO chondrocytes (Figure 2F). Indeed, by blocking IL-6 signaling in CD11b KO cells, the effect on calcification was abrogated and restored to WT levels, as confirmed by quantification in Figure 2G. Therefore, deficiency of CD11b in chondrocytes enhances the pro-inflammatory molecules IL-6 and MCP-1, sustaining the loop between inflammation and calcification.
In order to investigate the mechanisms underlying the effects of CD11b integrin on extracellular matrix degradation in vitro, we analyzed the gene expression level of key catabolic factors in WT and CD11b KO chondrocytes after 4 h exposure to CPP and their non-treated controls. We focused our analysis on metalloproteases (Adamts4, Adamts5, Mmp3, Mmp13) and inhibitors of metalloproteases (Timp1, Timp3), whose expression plays a major role in chondrocyte extracellular matrix degradation during OA (Tseng et al., 2009; Troeberg and Nagase, 2012; Roughley and Mort, 2014). As shown in Figure 3A, CD11b KO chondrocytes, with or without stimulation with CPP, showed significantly higher levels of Adamts4 and Timp3 mRNA than in control WT chondrocytes. While Timp1, Adamts5, and Mmp13 mRNA had no significant changes, there was lower Mmp3 in CD11b KO chondrocytes.
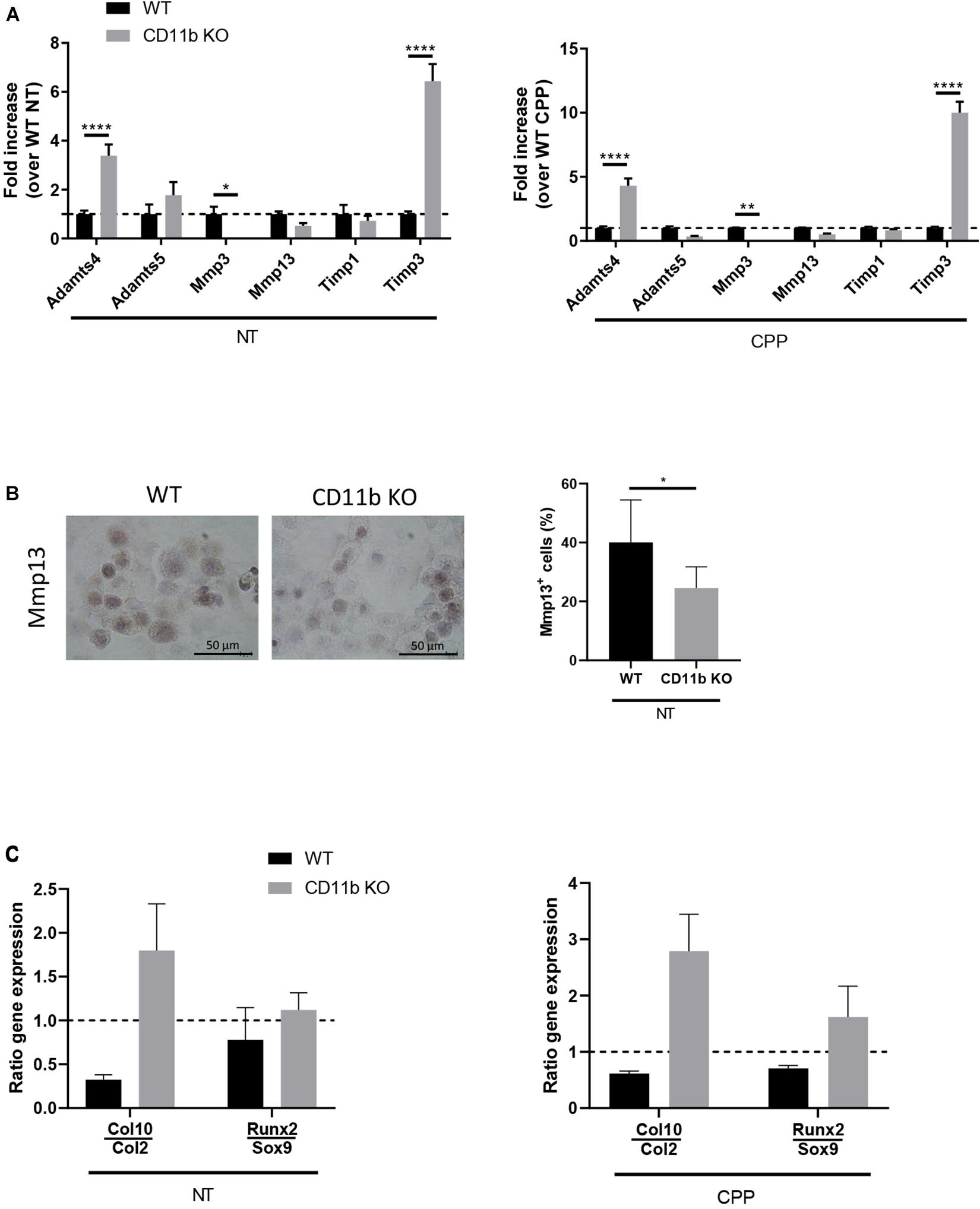
Figure 3. CD11b deficiency in chondrocytes alters the expression of matrix-degrading enzymes and is associated with a pro-hypertrophic expression profile. (A) qRT-PCR of the indicated genes in WT and CD11b KO primary murine chondrocytes at basal level (left graph) or in cell stimulated with CPP for 4 h (right graph). Values represent fold increase of CD11b KO chondrocytes over their WT control cells, shown in means ± SD of triplicates samples; *p < 0.05, **p < 0.01, ****p < 0.0001. (B) Mmp13 immunohistochemical staining (brown) in primary murine chondrocytes from WT and CD11b mice. For each group, one representative picture out of triplicates wells is shown. Scale bars 50 μm. The graph shows quantification of Mmp13 expression calculated as the percentage of positive cells in each field. Three fields were scored for each triplicate wells and means ± SD plotted; *p < 0.05. (C) qRT-PCR of the indicated genes in WT and CD11b KO primary murine chondrocytes at basal level (left graph) or in cells stimulated with CPP for 4 h (right graph). Values represent the ratio between late-stage hypertrophic differentiation markers over early-stage differentiation genes.
Following expression level analysis, we investigated the presence of Mmp13 by immunohistochemistry (Figure 3B, left). Mmp13 was significantly lowered in CD11b deficient chondrocytes, as confirmed by Image J quantification (Figure 3B, graph on the right).
We then analyzed, by qPCR, markers of articular cartilage hypertrophy including early differentiation Sox9 and Col2, and hypertrophy markers Runx2 and Col10 (Figure 3C). The ratio calculated on Col10 over Col2 and Runx2 over Sox9 revealed an increase in hypertrophic phenotype in CD11b KO chondrocytes, both at basal level and in CPP treated condition. Finally we ruled out that chondrocyte proliferation was affected by CD11b deficiency, as the proliferation response of isolated WT and CD11b KO chondrocytes by FACS using dye dilution assays for cell proliferation was similar (Supplementary Figure 2C).
It has been reported that treatment with CD11b agonist LA1 led to integrin activation and suppression of inflammation in macrophages (Faridi et al., 2017). Given the relationship between inflammation and chondrocyte calcification, we hypothesized that CD11b activation may play a protective role during the process of calcification. To assess the hypothesis, primary WT murine chondrocytes were allowed to calcify for 24 h with CPP in the presence or absence of LA1, and assessed by Alizarin Red staining. We found that LA1 treatment significantly reduced chondrocyte mineralization (Figure 4A), without any associated in vitro cytotoxicity as measured by LDH activity (Supplementary Figure 3A). Importantly, we also found that LA1 significantly inhibited IL-6 and MCP-1 secretion in a dose dependent manner confirming the anti-inflammatory role of LA1-mediated integrin activation in chondrocytes (Figure 4B).
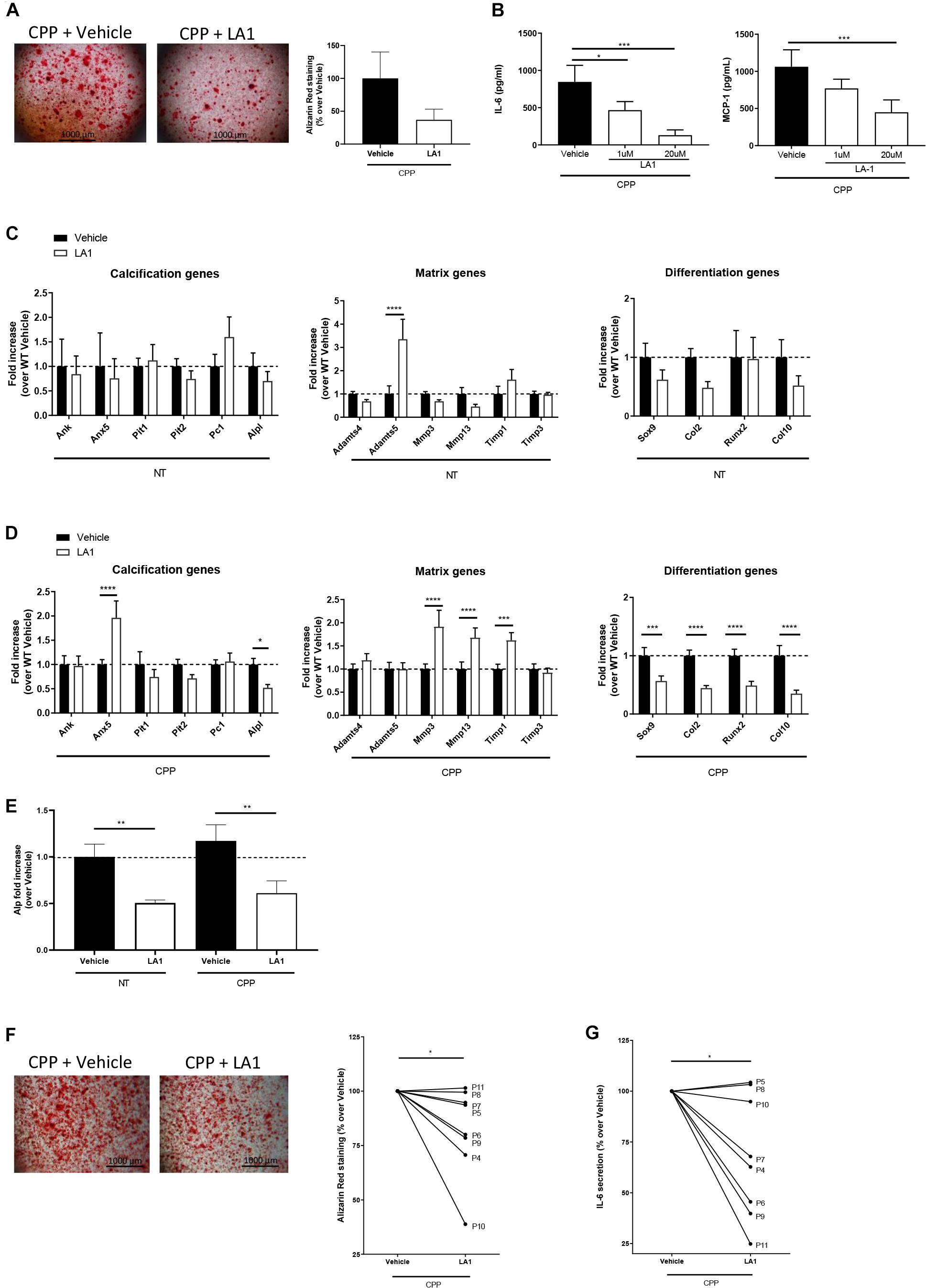
Figure 4. CD11b agonist reduces murine and human chondrocytes calcification in vitro. (A) Alizarin Red staining of WT primary murine chondrocytes stimulated with secondary CPP ± LA1 (20 μM) for 24 h in DMEM + 10% FBS. Pictures show one representative culture well of one experiment out of three independent experiments. The graph shows the percentage of Alizarin Red positive surface over the total surface, normalized on Vehicle treated cells. Values represent means ± SD of triplicates samples. (B) IL-6 and MCP-1 concentration in cell supernatants of WT primary chondrocytes stimulated with secondary CPP and LA1 at different concentration (0 μM - Vehicle-, 1 μM and 20 μM). Values represent means ± SD of triplicates samples; *p < 0.05, ***p < 0.001. (C) qRT-PCR of the indicated genes in murine chondrocytes stimulated with LA1 (20 μM) or its vehicle for 4 h. Values represent fold increase of LA1 treated chondrocytes over their Vehicle-treated cells, shown in means ± SD of triplicates samples; ****p < 0.0001. (D) qRT-PCR of the indicated genes in WT primary murine chondrocytes stimulated with secondary CPP ± LA1 (20 μM) for 4 h. Values represent fold increase in LA1 treated chondrocytes over Vehicle-treated cells, shown as means ± SD of triplicates samples; *p < 0.05, ***p < 0.001, ****p < 0.0001. (E) Quantification of Alkaline phosphatase activity in WT chondrocytes treated with secondary CPP ± LA1 for 6 h. Values represent fold increase of Alp activity in LA1 treated chondrocytes over Vehicle-treated cells treated, shown as means ± SD of triplicates samples; **p < 0.01. (F) Alizarin Red staining of human primary chondrocytes stimulated with secondary CPP and treated with LA1 or its Vehicle. Pictures show one representative culture well of one OA patient (P9) out of eight patients. The graph shows the corresponding Alizarin Red positive surface over the total surface, calculated in Adobe® Photoshop®, and then normalized. Lines connect the two conditions for each patient, values are calculated over CPP-Vehicle and represent means of triplicates samples; *p < 0.05. (G) IL-6 concentration in supernatants of human primary chondrocytes at point (F). Values represent means of triplicates samples; *p < 0.05.
To further demonstrate that CD11b activation on chondrocytes prevents cartilage calcification in vitro, we performed RT-qPCR analysis of the previously investigated genes related to calcification, differentiation and extracellular matrix turn-over. In Figure 4D, Anx5 was significantly increased in WT chondrocytes treated with LA1, compared to control, while Alp was decreased. This latter modulation is in line with the previous result in which CD11b KO cells had increased Alp. The genes Sox9, Col2, Runx2, Col10, Mmp3, Mmp13 and Timp1 were all decreased with LA1 stimulation. In LA1 treated chondrocytes without CPP, Adamts5 was significantly increased compared to control cells (Figure 4C). Furthermore, in Figure 4E, we investigated Alp activity in WT chondrocytes treated with LA1 or its control, and found that LA1 decreased Alp both with CPP and without.
To further evaluate the anti-calcifying effect of LA1 on human cells, we performed Alizarin Red staining on human primary chondrocytes extracted from damaged cartilage (8 OA patients). LA1 reduced mineralization in six out of eight primary chondrocyte cultures (Figure 4F). Moreover, in the same conditions LA1 significantly reduced IL-6 levels in the supernatant (Figure 4G). Finally, the anti-calcifying property of LA1 was not restricted to chondrocytes, as LA1 was also able to inhibit crystal deposition in human primary synoviocytes purified from OA patient’s synovial membrane (Supplementary Figure 3B), after calcification induction.
Based on the increased propensity of CD11b-deficient chondrocytes to mineralize and to produce IL-6 and on the increased levels of IL-6 produced in vivo (Stevanin et al., 2017), we hypothesized genetic ablation of CD11b in mice would predispose these animals to OA.
We analyzed the development of instability-induced OA changes in CD11b deficient and WT mice using the knee meniscectomy (MNX) model as previously described (Nasi et al., 2016b), and sacrifice after 8 weeks from the knee surgery. Cartilage damage, as evidenced by OARSI scoring of medial tibial plateau and medial femoral condyle, was significantly increased in CD11b-deficient mice, with decreased proteoglycans content (less Safranin-O staining), more fissurations and fibrillations compared to WT control mice (Figures 5A,B). Moreover, the joints of CD11b KO mice exhibited significantly increased synovial inflammation (Figure 5C), and increased, but not significant, osteophyte formation (Figure 5D) in CD11b KO mice, with respect to their WT control.
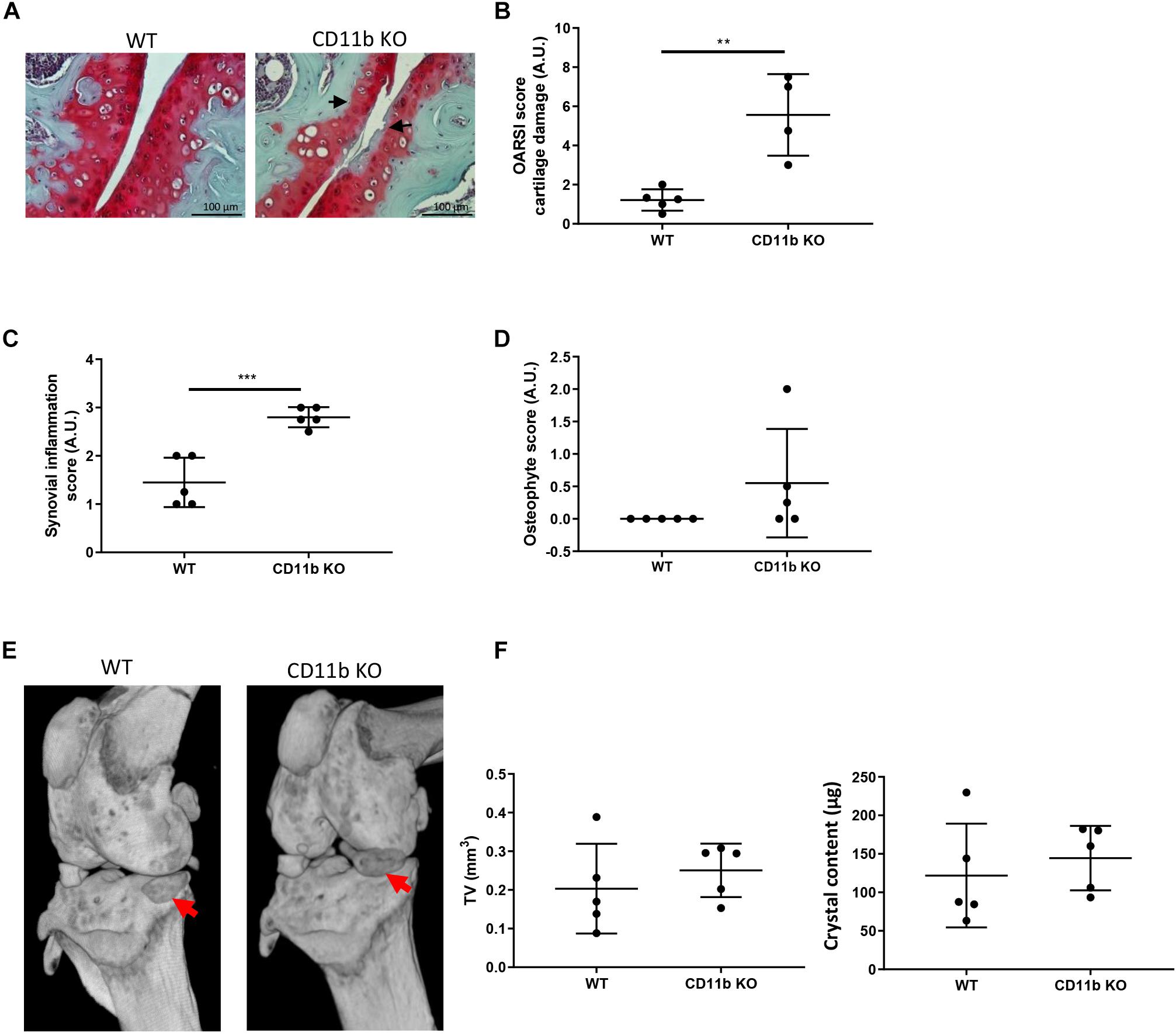
Figure 5. Cartilage damage and periarticular crystal deposits are exacerbated in CD11b deficient mice following experimental OA. (A) Representative Safranin-O histological staining of knee joints from WT and CD11b KO mice 8 weeks after MNX. Black arrows show degenerative OA changes in CD11b KO mice. Scale bars 100 μm. (B) Corresponding total OARSI score for cartilage damage in medial compartment of WT and CD11b KO knees. Values represent means ± SD; **p < 0.01. (C) Synovial inflammation scoring in WT and CD11b KO MNX mice knees; ***p < 0.001. (D) Osteophyte scoring in WT and CD11b KO MNX mice knees. (E) Representative micro-CT scan images of murine knee joints from WT and CD11b KO mice of point (A). Red arrows show calcified deposits in meniscectomized knees. (F) Corresponding CT Analyzer quantitative analysis of new formation volume (TV: tissue volume, mm3) and new formation crystal content (μg) in WT and CD11b KO menisectomized mice. Data are expressed as the mean ± SD.
In addition, microCT-scan examination was conducted to analyze the crystal deposition (Figure 5E). By CT Analyzer quantification, both the volume of newly formed crystals as well as the overall crystal content, were slightly increased in CD11b KO mice compared to WT control, although these differences did not reach statistical significance (Figure 5F).
In order to rule out an underlying cartilage or bone phenotype in CD11b KO mice, that may predispose them to OA, we analyzed cartilage damage and subchondral bone structure in unchallenged WT and CD11b KO mice. In the first experimental setting we considered knees from sham-operated 20 weeks old WT and CD11b KO mice. OARSI scoring revealed no difference in cartilage degradation between WT and CD11b KO mice (Supplementary Figures 4A,B). Moreover, no difference was seen in subchondral bone mineral density and subchondral bone trabecular thickness (Supplementary Figures 4C,D) between the two different genotypes. Similar results were obtained in aged WT and CD11b KO mice (37 weeks old), where no difference in cartilage damage (Supplementary Figure 4E,F) nor in subchondral bone parameters was seen (Supplementary Figures 4G,H).
Integrin CD11b/CD18 is a receptor originally described on neutrophils and macrophages, known to support adhesion and molecular cross-talk of these cells with the extracellular matrix proteins. It regulates many functions including phagocytosis, migration and immune tolerance (Ehirchiou et al., 2007; Rosetti and Mayadas, 2016), but it can also bind to a great number of different ligands, among which fibrinogen (Rosetti and Mayadas, 2016), fibronectin and laminin (Bohnsack and Zhou, 1992) can also be found in cartilage. CD11b/CD18 is generally thought to be exclusively expressed on leukocytes. In this work, we showed that CD11b is also expressed on chondrocytes, and importantly, established a novel role of CD11b integrin signaling in cartilage calcification and degradation in a mouse model of OA.
We first demonstrated the expression of the CD11b integrin subunit on primary murine and human chondrocytes and in articular cartilage of both murine and human OA patients by combining flow cytometry, qRT-PCR analysis and immunohistochemistry. CD11b is expressed only on a subset of chondrocytes in the superficial zone of the articular cartilage, and by FACS we confirmed 3–4% of chondrocytes expressing this integrin subunit. In line, in an open access database of single cell RNA sequencing on human OA chondrocytes (Ji et al., 2019), the gene encoding for CD11b was expressed in 3.15% of cells (analysis not shown), and these CD11b expressing cells also expressed Col2 as a marker of chondrocytes. Our results are also in agreement with a previous study reporting a very low expression of β2 integrin on chondrocytes (Iannone et al., 2001). Surprisingly, we also found CD11b in the extracellular matrix of cartilage from OA patients. This could be accounted for by cleavage of the extracellular domain of CD11b from the cell surface after cell activation under inflammatory conditions. This cleavage involves matrix metalloproteases and leads to subsequent binding of cleaved CD11b to extracellular matrix proteins (Zen et al., 2011). The presence of soluble complexes of CD11 and CD18 have been reported in human plasma and in synovial fluid from patients with arthritis (Gjelstrup et al., 2010). We confirmed the presence of soluble CD11b in the synovial fluid of OA patients that we analyzed, although cellular sources and targets of soluble CD11b remain to be determined. In OA joints, we postulate that the main source could be synovial cells and infiltrating inflammatory cells such as macrophages and neutrophils. Indeed, these cells express high levels of CD11b and inflammation-induced matrix metalloproteases which could contribute to CD11b shedding (Gjelstrup et al., 2010; Manferdini et al., 2016). The pathological relevance of extracellular CD11b is unclear and further investigations will be needed to determine the contribution of the truncated form of CD11b on OA progression.
We demonstrated that CD11b KO chondrocytes mineralized more than WT chondrocytes and that this could be via multiple mechanisms. First, CD11b KO chondrocytes showed more active calcification machinery compared to WT cells. Calcium-containing crystals, and in particular BCP crystals, are formed when excess of calcium (Ca2+) and inorganic phosphate (Pi) are uptaken into matrix vesicles (Anderson et al., 2004; Ea et al., 2011). Crystal formation starts when inorganic pyrophosphate (PPi) is extruded extracellularly via the multipass transmembrane transporter Ank. Extracellular PPi is then hydrolyzed to Pi by the enzyme tissue-non-specific alkaline phosphatases (Alp). Finally, Pi is concentrated in matrix vesicles through the cotransporters Pit1/Pit2 and Ca2+ through annexin 5 (Anx5). Here we demonstrated that CD11b KO chondrocytes, both at basal- and CPP-condition, express higher levels of the pro-mineralizing genes Ank, Alp and Pit1, and have more active Alp enzyme. Down-regulation of these calcification factors could be a potential mechanism by which CD11b exerts its anti-mineralizing properties in chondrocytes. From the literature, we can speculate possible cross-talks between CD11b-dependent signaling pathways such as JNK and NF-kB and these calcification players. Activation of the JNK (Suzuki et al., 2006; Wu et al., 2016) or of the NF-kB (Koumakis et al., 2019) pathway has been demonstrated to induce Pit1 expression and subsequent cell matrix mineralization or inflammation respectively. JNK and NF-kB activation were also associated to increased Alp expression and osteoblastic differentiation of arterial smooth muscle cells (Chang et al., 2020). As a second calcification mechanism, in our study we found that CD11b deficient chondrocytes secreted increased amounts of pro-inflammatory and pro-mineralizing IL-6 and MCP-1 when stimulated with CPP. More importantly, by blocking the receptor of IL-6 on CD11b KO chondrocytes we were able to greatly decrease calcification. Another characteristic of mineralizing chondrocytes is hypertrophy. Indeed, CD11b deficiency induced considerable upregulation of Col10 and downregulation of Col2, as shown by the ratio, indicating that CD11b deficient chondrocytes are in a hypertrophic state. In summary, we demonstrated that CD11b is an important negative regulator of mineralization in chondrocytes and most if not all of its anti-mineralizing effects could be accounted by decreased pro-mineralizing IL-6 cytokine levels (Nasi et al., 2016b). To address the underlying mechanisms involved in enhanced cartilage degradation of CD11b-deficient mice after MNX, we analyzed the key ECM markers under this disease setting. Surprisingly, deletion of the CD11b gene did not cause a coordinated increased expression of enzymes related to ECM degradation, as the pro-catabolic aggrecanase Adamts4 was upregulated in CD11b KO chondrocytes concomitantly with its inhibitor Timp3. Another pro-catabolic metalloprotease Mmp13 was downregulated in CD11b KO chondrocytes. Upon stimulation with the CD11b agonist LA1, both Adamts5 and its inhibitor Timp1 were increased. Given these expression patterns of ECM enzymes, we ruled out their early modulation as a crucial mechanism explaining the more severe phenotype observed in CD11b KO cells and mice, and the in vitro protective effect of LA1.
However, as mentioned above, measurement of the supernatant from stimulated CD11b KO chondrocytes showed an increase in both IL-6 and MCP-1 levels, which are known players in cartilage catabolism, alongside cartilage calcification (Kapoor et al., 2011; Nasi et al., 2016b).
We found that increased cartilage degradation is associated with synovial inflammation and the formation of osteophytes in CD11b KO mice after meniscectomy, while we could rule out that an underlying pre-existing pathology or bone phenotype exists in sham-operated knees and knees of old CD11b KO mice, which may predispose them to OA. Our data suggest that modulating CD11b could be an effective strategy to protect the cartilage in experimental OA. A growing body of literature demonstrates the role of Leukadherin-1 (LA1) mediated activation of CD11b in regulating inflammation in experimental models of inflammatory disorders. By binding to an allosteric pocket in the CD11b subunit, LA1 changes the conformation of the extracellular domain, thereby preventing infiltration of pro-inflammatory monocytes after organ injury and vascular dysfunction (Maiguel et al., 2011; Schmid et al., 2018). In a study by C. Han et al. (2010), the authors show that CD11b, once activated by Toll-Like Receptor (TLR), triggers spleen tyrosine kinase (Syk) activation and phosphorylation of MyD88 and TRIF, which are then degraded by proteolysis, thereby reducing IL-6, TNF and IFN-β downstream production. Another study (Faridi et al., 2017) demonstrated that LA1 activated CD11b which in turn inhibited MyD88 and TRIF downstream pathway. The effects of LA1 on chondrocytes have not been studied. Our results demonstrated the anti-inflammatory effect of LA1 in chondrocytes. Indeed, we found that WT chondrocytes cultured with LA1 showed a reduction in calcification and a decrease in production of the IL-6 and MCP-1. Additionally, we also obtained significant reduction in calcification and IL-6 release in human chondrocytes treated with LA1. Finally, as human OA synoviocytes can also calcify (Sun et al., 2014), we showed that LA1 can decrease mineralization of these cells, thereby confirming a general anti-mineralizing effect of LA1. On the other hand, we ruled out that the anti-mineralizing effect of LA1 was mediated by reduced chondrocyte hypertrophy, as LA1 suppressed the expression of both early-stage (Coll2, Sox9) and late-stage hypertrophy (Coll10, Runx2) chondrocitic markers.
However, we cannot rule out the possibility that LA1 can block calcification via a CD11b-independent mechanism, which deserves further investigation.
While a direct association between CD11b and calcification has not been reported, genome-wide association studies have established a strong correlation between single-nucleotide polymorphisms (SNPs) in the ITGAM gene coding for CD11b and susceptibility to SLE (Hom et al., 2008; Nath et al., 2008). These SNPs produce a dysfunctional CD11b protein on leukocytes that is deficient in many functions including activation, ligand binding, cell adhesion and phagocytosis (Khan et al., 2018). Moreover, Dahdal et al. reported that SLE patients have significantly elevated calcification propensity in their serum (Dahdal et al., 2018). In addition, earlier research has shown a link between coronary artery calcifications and increased disease activity in young lupus patients (Romero-Diaz et al., 2012). Finally, an association between IL-6 and progression of lupus has been demonstrated using several murine models of SLE (Tang et al., 1991; Alarcon-Riquelme et al., 1992; Kobayashi et al., 1992). However, it is still unknown how the SNPs contribute to the calcification propensity seen in lupus patients. Our data suggest a strong direct link between reduced CD11b function and increased calcification, which can be explained by the inability of CD11b KO chondrocytes to downregulate the pro-inflammatory cytokine and chemokine production.
Although the contribution of CD11b integrin in calcification is clearly established in the present study, we did not investigate which ligands bind to CD11b in the cartilage. Integrin CD11b is a multi-ligand receptor capable of binding ICAM-1, fibrinogen, fibronectin, factor X and complement factor iC3b, among many others (Rosetti and Mayadas, 2016). Integrin ligands have been shown to play a crucial role in OA (Pfander et al., 1999; Pullig et al., 2000). Indeed, previous studies have demonstrated that following cartilage damage, fragmented extracellular matrix molecules can trigger catabolic gene expression in various cells in the joint tissues (Homandberg et al., 2002; Loeser et al., 2003). Further studies will be required to investigate the contribution of CD11b ligands in OA cartilage.
Even though our current work unveils a new pathway in OA, it also presents some limitations that have to be considered. The use of global CD11b KO mice does not allow us to determine whether the observed effects are solely due to lack of CD11b expression in chondrocytes and not due to other cells in the surrounding tissues of the joint. This question will be resolved in future by generating mice with a selective CD11b deficiency in cartilage.
In summary, our study showed for the first time that CD11b signaling plays an important role in regulating cartilage calcification and OA. Deletion of CD11b gene was linked with cartilage degradation in knee joints during OA, mediated by upregulation of the pro-inflammatory IL-6 and MCP-1. Moreover, activation of CD11b with the LA1 agonist decreased the levels of pro-inflammatory cytokines and prevented calcification in vitro. This study offers novel insights into the role of CD11b integrin signaling in regulatory mechanisms that control OA severity and progression, and provides a new target for future therapies, with final aim of improving the quality of life in OA patients.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Centre Hospitalier Universitaire Vaudois ethical committee, Lausanne, Switzerland. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by “Service de la Consommation et des Affaires Vétérinaires du canton de Vaud,” Switzerland.
DE and IB designed, performed and analyzed experiments, and helped with the preparation of the manuscript. VC and MC performed experiments. TH, AS, and LZ provided advice and help with the preparation of the manuscript. NB conceived, supervised the study, and wrote the manuscript. SN performed some experiments, supervised the study and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Fonds National Suisse de la Recherche Scientifique, Switzerland (grant 310030_173134).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Prof. Olivier Guyen (Service d’orthopédie et de traumatologie, CHUV, Switzerland) who kindly provided us with human OA samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.611757/full#supplementary-material
Aghagolzadeh, P., Bachtler, M., Bijarnia, R., Jackson, C., Smith, E. R., Odermatt, A., et al. (2016). Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-alpha. Atherosclerosis 251, 404–414. doi: 10.1016/j.atherosclerosis.2016.05.044
Alarcon-Riquelme, M. E., Moller, G., and Fernandez, C. (1992). Age-dependent responsiveness to interleukin-6 in B lymphocytes from a systemic lupus erythematosus-prone (NZB x NZW)F1 hybrid. Clin. Immunol. Immunopathol. 62, 264–269. doi: 10.1016/0090-1229(92)90101-s
Almonte-Becerril, M., Costell, M., and Kouri, J. B. (2014). Changes in the integrins expression are related with the osteoarthritis severity in an experimental animal model in rats. J. Orthop. Res. 32, 1161–1166. doi: 10.1002/jor.22649
Anderson, H. C., Sipe, J. B., Hessle, L., Dhanyamraju, R., Atti, E., Camacho, N. P., et al. (2004). Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase-deficient mice. Am. J. Pathol. 164, 841–847. doi: 10.1016/s0002-9440(10)63172-0
Bertrand, J., Kraft, T., Gronau, T., Sherwood, J., Rutsch, F., Liote, F., et al. (2020). BCP crystals promote chondrocyte hypertrophic differentiation in OA cartilage by sequestering Wnt3a. Ann. Rheum. Dis. 79, 975–984. doi: 10.1136/annrheumdis-2019-216648
Bohnsack, J. F., and Zhou, X. N. (1992). Divalent cation substitution reveals CD18- and very late antigen-dependent pathways that mediate human neutrophil adherence to fibronectin. J. Immunol. 149, 1340– 1347.
Castelblanco, M., Nasi, S., Pasch, A., So, A., and Busso, N. (2020). The role of the gasotransmitter hydrogen sulfide in pathological calcification. Br. J. Pharmacol. 177, 778–792. doi: 10.1111/bph.14772
Chang, J. F., Hsieh, C. Y., Liou, J. C., Liu, S. H., Hung, C. F., Lu, K. C., et al. (2020). Scavenging intracellular ROS attenuates p-Cresyl sulfate-triggered osteogenesis through MAPK signaling pathway and NF-kappaB activation in human arterial smooth muscle cells. Toxins 12:472. doi: 10.3390/toxins12080472
Dahdal, S., Devetzis, V., Chalikias, G., Tziakas, D., Chizzolini, C., Ribi, C., et al. (2018). Serum calcification propensity is independently associated with disease activity in systemic lupus erythematosus. PLoS One 13:e0188695. doi: 10.1371/journal.pone.0188695
Ea, H. K., Nguyen, C., Bazin, D., Bianchi, A., Guicheux, J., Reboul, P., et al. (2011). Articular cartilage calcification in osteoarthritis: insights into crystal-induced stress. Arthritis Rheum. 63, 10–18. doi: 10.1002/art.27761
Ehirchiou, D., Xiong, Y., Xu, G., Chen, W., Shi, Y., and Zhang, L. (2007). CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J Exp Med. 204, 1519–1524. doi: 10.1084/jem.20062292
Faridi, M. H., Khan, S. Q., Zhao, W., Lee, H. W., Altintas, M. M., Zhang, K., et al. (2017). CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J. Clin. Invest. 127, 1271–1283.
Forsyth, C. B., Pulai, J., and Loeser, R. F. (2002). Fibronectin fragments and blocking antibodies to alpha2beta1 and alpha5beta1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 46, 2368–2376. doi: 10.1002/art.10502
Frallonardo, P., Ramonda, R., Peruzzo, L., Scanu, A., Galozzi, P., Tauro, L., et al. (2018). Basic calcium phosphate and pyrophosphate crystals in early and late osteoarthritis: relationship with clinical indices and inflammation. Clin. Rheumatol. 37, 2847–2853. doi: 10.1007/s10067-018-4166-3
Fuerst, M., Bertrand, J., Lammers, L., Dreier, R., Echtermeyer, F., Nitschke, Y., et al. (2009). Calcification of articular cartilage in human osteoarthritis. Arthritis Rheum. 60, 2694–2703.
Gjelstrup, L. C., Boesen, T., Kragstrup, T. W., Jorgensen, A., Klein, N. J., Thiel, S., et al. (2010). Shedding of large functionally active CD11/CD18 Integrin complexes from leukocyte membranes during synovial inflammation distinguishes three types of arthritis through differential epitope exposure. J. Immunol. 185, 4154–4168. doi: 10.4049/jimmunol.1000952
Glasson, S. S., Chambers, M. G., Van Den Berg, W. B., and Little, C. B. (2010). The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 18(Suppl. 3), S17–S23.
Gomez, I. G., Tang, J., Wilson, C. L., Yan, W., Heinecke, J. W., Harlan, J. M., et al. (2012). Metalloproteinase-mediated Shedding of Integrin beta2 promotes macrophage efflux from inflammatory sites. J. Biol. Chem. 287, 4581–4589. doi: 10.1074/jbc.m111.321182
Han, C., Jin, J., Xu, S., Liu, H., Li, N., and Cao, X. (2010). Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat. Immunol. 11, 734–742. doi: 10.1038/ni.1908
Hom, G., Graham, R. R., Modrek, B., Taylor, K. E., Ortmann, W., Garnier, S., et al. (2008). Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 358, 900–909.
Homandberg, G. A., Costa, V., and Wen, C. (2002). Fibronectin fragments active in chondrocytic chondrolysis can be chemically cross-linked to the alpha5 integrin receptor subunit. Osteoarthritis Cartilage 10, 938–949. doi: 10.1053/joca.2002.0854
Huang, Z., Ding, C., Li, T., and Yu, S. P. (2018). Current status and future prospects for disease modification in osteoarthritis. Rheumatology 57(Suppl._4), iv108–iv123.
Iannone, F., Corrado, A., Grattagliano, V., Cantatore, F. P., Patella, V., and Lapadula, G. (2001). [Phenotyping of chondrocytes from human osteoarthritic cartilage: chondrocyte expression of beta integrins and correlation with anatomic injury]. Reumatismo 53, 122–130.
Ji, Q., Zheng, Y., Zhang, G., Hu, Y., Fan, X., Hou, Y., et al. (2019). Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 78, 100–110. doi: 10.1136/annrheumdis-2017-212863
Kamekura, S., Hoshi, K., Shimoaka, T., Chung, U., Chikuda, H., Yamada, T., et al. (2005). Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthritis Cartilage 13, 632–641. doi: 10.1016/j.joca.2005.03.004
Kapoor, M., Martel-Pelletier, J., Lajeunesse, D., Pelletier, J. P., and Fahmi, H. (2011). Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 7, 33–42. doi: 10.1038/nrrheum.2010.196
Khan, S. Q., Khan, I., and Gupta, V. (2018). CD11b Activity modulates pathogenesis of lupus nephritis. Front. Med. 5:52. doi: 10.3389/fmed.2018.00052
Kim, J. R., Yoo, J. J., and Kim, H. A. (2018). Therapeutics in osteoarthritis based on an understanding of its molecular pathogenesis. Int J Mol Sci. 19:674. doi: 10.3390/ijms19030674
Kobayashi, I., Matsuda, T., Saito, T., Yasukawa, K., Kikutani, H., Hirano, T., et al. (1992). Abnormal distribution of IL-6 receptor in aged MRL/lpr mice: elevated expression on B cells and absence on CD4+ cells. Int. Immunol. 4, 1407–1412. doi: 10.1093/intimm/4.12.1407
Koumakis, E., Millet-Botti, J., Benna, J. E., Leroy, C., Boitez, V., Codogno, P., et al. (2019). Novel function of PiT1/SLC20A1 in LPS-related inflammation and wound healing. Sci. Rep. 9:1808.
Lapadula, G., Iannone, F., Zuccaro, C., Grattagliano, V., Covelli, M., Patella, V., et al. (1997). Integrin expression on chondrocytes: correlations with the degree of cartilage damage in human osteoarthritis. Clin. Exp. Rheumatol. 15, 247–254.
LaPointe, V. L., Verpoorte, A., and Stevens, M. M. (2013). The changing integrin expression and a role for integrin beta8 in the chondrogenic differentiation of mesenchymal stem cells. PLoS One 8:e82035. doi: 10.1371/journal.pone.0082035
Loeser, R. F. (2014). Integrins and chondrocyte-matrix interactions in articular cartilage. Matrix Biol. 39, 11–16. doi: 10.1016/j.matbio.2014.08.007
Loeser, R. F., Forsyth, C. B., Samarel, A. M., and Im, H. J. (2003). Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem. 278, 24577–24585. doi: 10.1074/jbc.m304530200
Loeser, R. F., Goldring, S. R., Scanzello, C. R., and Goldring, M. B. (2012). Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707. doi: 10.1002/art.34453
Maiguel, D., Faridi, M. H., Wei, C., Kuwano, Y., Balla, K. M., Hernandez, D., et al. (2011). Small molecule-mediated activation of the integrin CD11b/CD18 reduces inflammatory disease. Sci. Signal. 4:ra57. doi: 10.1126/scisignal.2001811
Manferdini, C., Paolella, F., Gabusi, E., Silvestri, Y., Gambari, L., Cattini, L., et al. (2016). From osteoarthritic synovium to synovial-derived cells characterization: synovial macrophages are key effector cells. Arthritis Res. Ther. 18:83.
Nasi, S., Ea, H. K., Liote, F., So, A., and Busso, N. (2016a). Sodium thiosulfate prevents chondrocyte mineralization and reduces the severity of murine osteoarthritis. PLoS One 11:e0158196. doi: 10.1371/journal.pone.0158196
Nasi, S., So, A., Combes, C., Daudon, M., and Busso, N. (2016b). Interleukin-6 and chondrocyte mineralisation act in tandem to promote experimental osteoarthritis. Ann. Rheum. Dis. 75, 1372–1379. doi: 10.1136/annrheumdis-2015-207487
Nath, S. K., Han, S., Kim-Howard, X., Kelly, J. A., Viswanathan, P., Gilkeson, G. S., et al. (2008). A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat. Genet. 40, 152–154. doi: 10.1038/ng.71
Pfander, D., Rahmanzadeh, R., and Scheller, E. E. (1999). Presence and distribution of collagen II, collagen I, fibronectin, and tenascin in rabbit normal and osteoarthritic cartilage. J. Rheumatol. 26, 386–394.
Pullig, O., Weseloh, G., Gauer, S., and Swoboda, B. (2000). Osteopontin is expressed by adult human osteoarthritic chondrocytes: protein and mRNA analysis of normal and osteoarthritic cartilage. Matrix Biol. 19, 245–255. doi: 10.1016/s0945-053x(00)00068-8
Raducanu, A., Hunziker, E. B., Drosse, I., and Aszodi, A. (2009). Beta1 integrin deficiency results in multiple abnormalities of the knee joint. J. Biol. Chem. 284, 23780–23792. doi: 10.1074/jbc.m109.039347
Reed, J. H., Jain, M., Lee, K., Kandimalla, E. R., Faridi, M. H., Buyon, J. P., et al. (2013). Complement receptor 3 influences toll-like receptor 7/8-dependent inflammation: implications for autoimmune diseases characterized by antibody reactivity to ribonucleoproteins. J. Biol. Chem. 288, 9077–9083. doi: 10.1074/jbc.m112.403303
Romero-Diaz, J., Vargas-Vorackova, F., Kimura-Hayama, E., Cortazar-Benitez, L. F., Gijon-Mitre, R., Criales, S., et al. (2012). Systemic lupus erythematosus risk factors for coronary artery calcifications. Rheumatology 51, 110–119. doi: 10.1093/rheumatology/ker307
Rosetti, F., and Mayadas, T. N. (2016). The many faces of Mac-1 in autoimmune disease. Immunol. Rev. 269, 175–193. doi: 10.1111/imr.12373
Roughley, P. J., and Mort, J. S. (2014). The role of aggrecan in normal and osteoarthritic cartilage. J. Exp. Orthop. 1:8.
Schittenhelm, L., Hilkens, C. M., and Morrison, V. L. (2017). beta2 Integrins as regulators of dendritic cell, monocyte, and macrophage function. Front Immunol. 8:1866. doi: 10.3389/fimmu.2017.01866
Schmid, M. C., Khan, S. Q., Kaneda, M. M., Pathria, P., Shepard, R., Louis, T. L., et al. (2018). Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun. 9:5379.
Song, E. K., and Park, T. J. (2014). Integrin signaling in cartilage development. Anim. Cells Syst. 18, 365–371. doi: 10.1080/19768354.2014.987319
Stevanin, M., Busso, N., Chobaz, V., Pigni, M., Ghassem-Zadeh, S., Zhang, L., et al. (2017). CD11b regulates the Treg/Th17 balance in murine arthritis via IL-6. Eur. J. Immunol. 47, 637–645. doi: 10.1002/eji.201646565
Sun, Y., Mauerhan, D. R., Franklin, A. M., Zinchenko, N., Norton, H. J., and Hanley, E. N. Jr., et al. (2014). Fibroblast-like synoviocytes induce calcium mineral formation and deposition. Arthritis. 2014:812678.
Suzuki, A., Ghayor, C., Guicheux, J., Magne, D., Quillard, S., Kakita, A., et al. (2006). Enhanced expression of the inorganic phosphate transporter Pit-1 is involved in BMP-2-induced matrix mineralization in osteoblast-like cells. J. Bone. Miner. Res. 21, 674–683. doi: 10.1359/jbmr.020603
Tang, B., Matsuda, T., Akira, S., Nagata, N., Ikehara, S., Hirano, T., et al. (1991). Age-associated increase in interleukin 6 in MRL/lpr mice. Int. Immunol. 3, 273–278. doi: 10.1093/intimm/3.3.273
Tian, J., Zhang, F. J., and Lei, G. H. (2015). Role of integrins and their ligands in osteoarthritic cartilage. Rheumatol. Int. 35, 787–798. doi: 10.1007/s00296-014-3137-5
Troeberg, L., and Nagase, H. (2012). Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 1824, 133–145. doi: 10.1016/j.bbapap.2011.06.020
Tseng, S., Reddi, A. H., and Di Cesare, P. E. (2009). Cartilage Oligomeric Matrix Protein (COMP): a biomarker of arthritis. Biomark Insights. 4, 33–44.
Varas, L., Ohlsson, L. B., Honeth, G., Olsson, A., Bengtsson, T., Wiberg, C., et al. (2007). Alpha10 integrin expression is up-regulated on fibroblast growth factor-2-treated mesenchymal stem cells with improved chondrogenic differentiation potential. Stem Cells Dev. 16, 965–978. doi: 10.1089/scd.2007.0049
Wu, Y., Han, X., Wang, L., Diao, Z., and Liu, W. (2016). Indoxyl sulfate promotes vascular smooth muscle cell calcification via the JNK/Pit-1 pathway. Ren. Fail. 38, 1702–1710. doi: 10.3109/0886022x.2016.1155397
Yan, J. F., Qin, W. P., Xiao, B. C., Wan, Q. Q., Tay, F. R., Niu, L. N., et al. (2020). Pathological calcification in osteoarthritis: an outcome or a disease initiator? Biol. Rev. Camb. Philos. Soc. 95, 960–985. doi: 10.1111/brv.12595
Keywords: CD11b, integrin, calcium-containing crystals, osteoarthritis, animal model, chondrocyte mineralization, cartilage calcification, human cartilage
Citation: Ehirchiou D, Bernabei I, Chobaz V, Castelblanco M, Hügle T, So A, Zhang L, Busso N and Nasi S (2020) CD11b Signaling Prevents Chondrocyte Mineralization and Attenuates the Severity of Osteoarthritis. Front. Cell Dev. Biol. 8:611757. doi: 10.3389/fcell.2020.611757
Received: 29 September 2020; Accepted: 02 December 2020;
Published: 18 December 2020.
Edited by:
Herve Kempf, UMR7365 Ingénierie Moléculaire et Physiopathologie Articulaire (IMOPA), FranceReviewed by:
Frédéric Mallein-Gerin, CNRS UMR 5305, FranceCopyright © 2020 Ehirchiou, Bernabei, Chobaz, Castelblanco, Hügle, So, Zhang, Busso and Nasi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathalie Busso, TmF0aGFsaWUuQnVzc29AY2h1di5jaA==
†These authors have contributed equally to this work and share first authorship
‡Present address: Mariela Castelblanco, Department of Biomedical Sciences, Centre Hospitalier Universitaire Vaudois, University of Lausanne, Lausanne, Switzerland
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.