
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 30 November 2020
Sec. Mitochondrial Research
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.604377
 Jacopo Junio Valerio Branca1
Jacopo Junio Valerio Branca1 Alessandra Pacini1
Alessandra Pacini1 Massimo Gulisano1Niccolò Taddei2
Massimo Gulisano1Niccolò Taddei2 Claudia Fiorillo2*
Claudia Fiorillo2* Matteo Becatti2
Matteo Becatti2Cadmium (Cd) is a well-known heavy metal and environmental toxicant and pollutant worldwide, being largely present in every kind of item such as plastic (toys), battery, paints, ceramics, contaminated water, air, soil, food, fertilizers, and cigarette smoke. Nowadays, it represents an important research area for the scientific community mainly for its effects on public health. Due to a half-life ranging between 15 and 30 years, Cd owns the ability to accumulate in organs and tissues, exerting deleterious effects. Thus, even at low doses, a Cd prolonged exposure may cause a multiorgan toxicity. Mitochondria are key intracellular targets for Cd-induced cytotoxicity, but the underlying mechanisms are not fully elucidated. The present review is aimed to clarify the effects of Cd on mitochondria and, particularly, on the mitochondrial electron transport chain.
Cadmium (Cd) is a toxic heavy metal without known biological function in humans. It can be easily found in house dust and tobacco smoke (Hogervorst et al., 2007), and for this reason, the airways are considered among the primary routes for the entry of Cd. As recently demonstrated in a human airway tissue model (Xiong et al., 2019), Cd impairs cilia functions and enters the lung, where it can induce apoptosis by reactive oxygen species (ROS) production and by altering the reduced glutathione (GSH)/oxidized glutathione (GSSG) redox balance. Once in the lung alveoli and diffused in the bloodstream, Cd is transported to the whole body and, due to its low rate of excretion, can deeply affect kidney functional integrity (Gonick, 2008). As demonstrated in rats (Patra et al., 1999), Cd accumulation in the proximal tubular epithelium of the nephron directly compromises mitochondrial functions (Gobe and Crane, 2010), increasing mitochondrial permeability and swelling, thus inhibiting respiration. These alterations are responsible for an increase in ROS production, ultimately leading to renal cell apoptosis via cytochrome c and caspase pathways (Gobe and Crane, 2010; Mao et al., 2011).
Other organs and cellular compartments such as testes (i.e., Sertoli, Leydig, and germ cells) (Zhu et al., 2020), osteocytes (Uwagie-Ero et al., 2019), cardiomyocytes (Chen et al., 2015), and peripheral and central nervous system (CNS; Wang and Du, 2013) also represent main targets of Cd toxicity. A recent meta-analysis has suggested that increased levels of Cd in human serum might play a pivotal role in neurodegenerative disorder progression (Xu et al., 2018). In vitro and in vivo studies demonstrated a direct role of Cd in ROS production and endoplasmic reticulum stress (Shukla et al., 1996; Branca et al., 2019a), ultimately leading to the impairment of blood–brain barrier (BBB) integrity. Indeed, once the BBB integrity is compromised, heavy metals and other toxicants can enter the CNS and activate glial cells, causing brain inflammation and neural degeneration (Branca et al., 2020). Once inside the brain parenchyma, Cd can induce mitochondrial dysfunction and subsequent cytochrome c release in neural cells (Branca et al., 2018, 2019b).
Mitochondria are organelles of bacterial origin involved in several cellular processes, such as energy homeostasis, cell proliferation, metabolism, and cell death. Mitochondria play a crucial role in the metabolism of eukaryotic cells, producing energy through aerobic respiration. The metabolic energy derived from oxidation of carbohydrates, lipids, and amino acids is used to generate adenosine triphosphate (ATP) through a process called oxidative phosphorylation (OXPHOS) (Milenkovic et al., 2017). This series of oxidation–reduction reactions involves the electron transfer from NADH (nicotinamide adenine dinucleotide) or FADH2 (flavin adenine dinucleotide) to molecular oxygen through a series of specific electron carriers that constitute the electron transport chain (ETC)—or respiratory chain—of the inner mitochondrial membrane (IMM). ETC consists of four complexes: Complex I (NADH-coenzyme Q reductase), Complex II (succinate dehydrogenase), Complex III (coenzyme Q-cytochrome c reductase), and Complex IV (cytochrome c oxidase). Complexes I, III, and IV establish the proton gradient across the IMM for ATP synthesis at ATP synthase (Figure 1). Ten protons are transported from the matrix across the IMM for every electron pair transferred from NADH2 to O2. These protons are utilized by ATP synthase to synthetize 2.5 ATP molecules from ADP and inorganic phosphate (Papa et al., 2006).
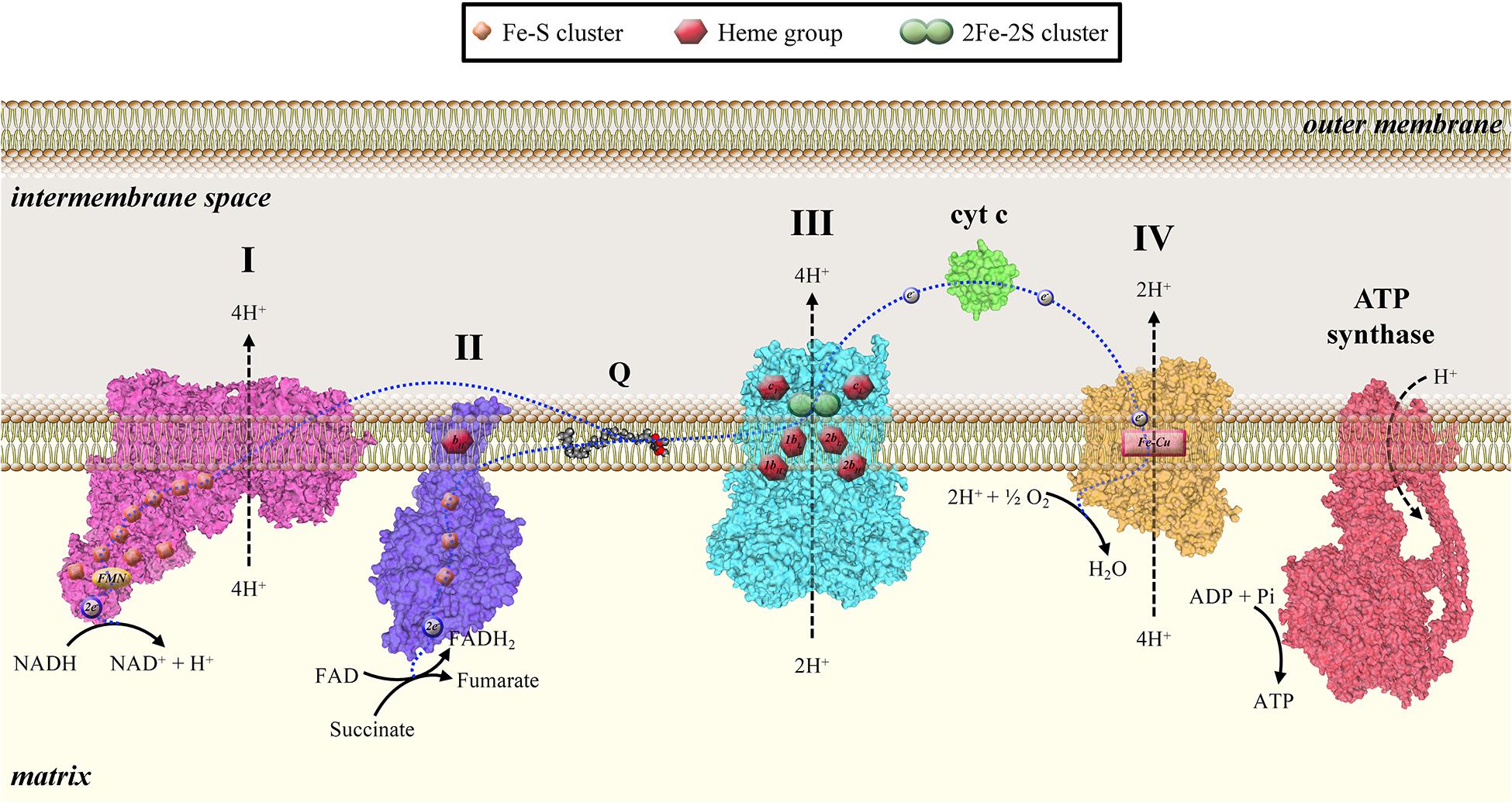
Figure 1. Mitochondrial electron transport chain consists of four complexes: Complex I (NADH-coenzyme Q reductase), Complex II (succinate dehydrogenase), Complex III (coenzyme Q-cytochrome c reductase), and Complex IV (cytochrome c oxidase). Complexes I, III, and IV establish the proton gradient across the IMM for ATP synthesis by ATP synthase.
Under physiological conditions, the IMM is selectively permeable to solutes and, in mammalian mitochondria, has a cutoff of 1.5 kDa. Apoptotic stimuli, ROS, and mitochondrial Ca2+ overload can induce IMM to undergo permeability transition. This sudden increase in the IMM permeability leads to mitochondrial depolarization, inhibition of respiration and ATP synthesis, Ca2+ release, osmotic pressure increase, and mitochondrial swelling (Bernardi et al., 2006). Matrix swelling alters the outer mitochondrial membrane (OMM) integrity resulting in apoptotic factor mobilization [cytochrome c (cyt c), apoptosis-inducing factor (AIF), endonuclease G], mitochondrial dysfunction, and cell death (Bernardi and Di Lisa, 2015). The mitochondrial permeability transition is due to opening of the permeability transition pore (PTP), a high conductance channel at contact sites between the IMM and OMM that is crucially regulated by a variety of pathophysiological effectors. However, the molecular structure of the PTP is controversial. It is widely accepted that the voltage-dependent anion channel (VDAC), ATP synthase subunits, and adenine nucleotide translocase (ANT) are components of the PTP complex (Karch et al., 2019).
Mitochondria are among the major sources of endogenous ROS that are formed as natural by-products of physiological ETC activity and participate in cellular signaling (Brand, 2016; Zhang et al., 2016). Reactive oxygen species overproduction can damage cellular lipids, proteins, or DNA, inhibiting their normal function (Valko et al., 2007). Reactive oxygen species exposure has led cells to develop a series of defense mechanisms, called antioxidants (Cadenas, 1997). Under physiological conditions, a balance between ROS production and the intracellular antioxidant levels exists. When this equilibrium is disturbed either by an increase in ROS production or a decrease in antioxidant levels, a condition called oxidative stress, resulting in cell dysfunction or cell death, occurs (Valko et al., 2007).
Belyaeva et al. (2004b) reported that both mitochondrial ETC and membrane permeability are the primary targets of Cd-induced mitochondrial dysfunction. On the other hand, Ciapaite et al. (2009) elegantly demonstrated, in mitochondria isolated from the liver of male Wistar rats, that Cd interferes with mitochondrial respiration solely through the inhibitory effect downstream Q, resulting in respiratory and phosphorylation rate reduction. Therefore, in the presence of a high inorganic phosphate concentration, Cd can induce uncoupling (Koike et al., 1991; Korotkov et al., 1998). However, the effect of Cd on mitochondrial respiration is related to concentration: while a high Cd concentration inhibits basal respiration, a low dose stimulates resting-state respiration (Belyaeva and Korotkov, 2003). Moreover, in the MDA-MB231 human breast cancer cell line, Cd long-term treatment induces an increase in the rate of respiration (Cannino et al., 2008), while short- and long-term Cd treatment in HB2 mammary luminal immortalized epithelial cells decreases respiratory activity and promotes mitochondrial membrane polarization (Cannino et al., 2009).
Many years ago, it was proposed that the toxic effect of Cd could be mainly due to ETC enzyme inhibition (Miccadei and Floridi, 1993) and to proton flux through Pi/H+ symporter leading to an uncoupling effect (Koike et al., 1991). In particular, it has been suggested that Cd, reacting with SH groups from the matrix side of the mitochondrial membrane, leads to Pi/H+ symporter conformational change and penetrates into the mitochondria via the Ca2+ uniporter. This conformational change results in the stimulation of H+ and Pi influxes, and the transported H+ acts as a trigger for uncoupling (Koike et al., 1991). More recently, new data have highlighted the strict dependence of ETC function on Cd-induced toxicity (Belyaeva et al., 2004a, 2011; Wang et al., 2004; Ivanina et al., 2008; Ciapaite et al., 2009; Adiele et al., 2012; Belyaeva, 2018). Cysteine residues, Fe–S clusters, thiol and binding sites for divalent metals are potential Cd target sites (Kurochkin et al., 2011), leading to ETC inhibition, proton motive force dissipation, and cell dysfunction (Wang et al., 2004; Kurochkin et al., 2011). Moreover, it has been demonstrated that a high respiration rate (expressed in terms of oxygen consumption; nmol O2/mg protein/min) promotes Cd accumulation (Belyaeva et al., 2002; Adiele et al., 2010), suggesting that cell energy state influences Cd accumulation. Here, we reviewed the main effects of Cd on mitochondrial ETC complexes.
Mammalian Complex I (NADH-coenzyme Q reductase), with an estimated mass of about 1,000 kDa, is a crucial enzyme in mitochondrial respiration. This giant complex transfers a pair of electrons from NADH to Q to establish the proton motive force across the IMM required for ATP synthesis. Four protons are transported from the matrix across the IMM per two electrons passed from NADH to Q (Sazanov, 2015). It contains 44 polypeptide chains, one molecule of flavin mononucleotide (FMN), and nine Fe-S clusters, together containing a total of 20–26 iron atoms (Agip et al., 2019). On the matrix side of the IMM, the two-electron donor NADH transfers electrons to FMN. Then, the reduced FMN (FMNH2) transfers electrons to a series of Fe-S proteins. Fe–S proteins are one-electron transfer agents; therefore, they can transfer one electron at a time to substrate. At this purpose, the flavin of FMN can act as either a one-electron or a two-electron transfer agent linking NADH and Fe–S proteins. Finally, two electrons are transferred from Fe–S cluster to Q.
Cadmium is a powerful uncoupling agent and inhibits the succinate- and malate/pyruvate-stimulated respiration (Müller and Ohnesorge, 1984). Moreover, in rat hepatic mitochondria, it has been suggested that Cd interacts with Complex I at Q site and NADH site levels, resulting in Complex I inhibition (Cameron et al., 1986). The importance of Fe-S clusters for Cd interaction was confirmed by Kurochkin et al. (2011) who demonstrated that the substrate oxidation and proton leak subsystems are the main targets for Cd toxicity in oyster mitochondria. Moreover, Adiele et al. (2012) confirmed the importance of Fe–S clusters for Cd interaction showing, in isolated rainbow trout hepatic mitochondria, the inhibitory effect of Cd on Complexes I, II, and III; the nonresponse of Complex IV could be due to the absence of Fe-S clusters in this ETC complex. On the contrary, no significant inhibition of Complex I activity was observed in Cd-treated mitochondria isolated from the liver, brain, and heart of the guinea pig (Wang et al., 2004). Furthermore, many studies support the involvement of Complex I in mitochondrial PTP and cell death, supporting the direct involvement of the ETC complexes in PTP formation and regulation (Grivennikova et al., 2001; Batandier et al., 2004; Belyaeva et al., 2004a, 2012; García et al., 2005; Belyaeva, 2010). Monteiro et al. (2018) demonstrated, in mitochondria isolated from human osteoblast MG-63 cell line, decreased Complex I and IV activities, leading to an impairment of mitochondria ATP production as a result of Cd treatment. Recently, it was demonstrated that Cd treatment induces ROS production at Complex I flavin site and inhibits respiration in rainbow trout liver mitochondria, confirming the key role of Complex I in Cd-induced ROS production (Okoye et al., 2019).
Complex II (succinate dehydrogenase) is the smallest mitochondrial ETC complex that links directly the ETC to the tricarboxylic acid (TCA) cycle. Unlike other respiratory complexes, Complex II is totally encoded by nuclear DNA and does not move protons to the IMM space (Bezawork-Geleta et al., 2017). This complex has a mass of 124 kDa and is composed of two hydrophilic subunits, three Fe-S centers, a flavoprotein, and two hydrophobic subunits, which contain one heme b and the binding site for Q (Sun et al., 2005). In the TCA cycle, the conversion of succinate to fumarate is coupled by the reduction of FAD to FADH2 in Complex II. FADH2 transfers two electrons to Fe–S clusters, which pass them to Q (Bezawork-Geleta et al., 2017). Despite many studies that investigated the effects of Cd on ETC Complex II (Liu and Liun, 1990; Fariss, 1991; Jay et al., 1991; Korotkov et al., 1998; Belyaeva et al., 2001, 2011; Casalino et al., 2002; Wang et al., 2004; Modi and Katyare, 2009), the mechanisms of Cd-induced Complex II injury are not fully elucidated. Many years ago, Toury et al. (1985) demonstrated a fall in Complex II activity in chronically treated rats exposed to Cd. In the same study, it was shown that isolated mitochondria from liver, kidney, and muscle of Cd-treated rats maintained partial energy coupling but displayed a rapid early fall in Complex IV activity followed by a partial restoration after 6 months of treatment and a progressively decreasing Complex II (Toury et al., 1985). Complex II inhibition blocks electron transfer and electron accumulation at Complex II, resulting in superoxide anion production. The key role of Complex II in ROS production was confirmed by Quinlan et al. (2012), showing that ROS from Complex II originates from the flavin site of the enzyme. Moreover, it has been demonstrated that a high succinate concentration associated with a high membrane potential-induced reverse electron transfer from Complex II into Complex I with superoxide production (Muller et al., 2008). Complex II inhibition by Cd was confirmed in mitochondria isolated from the liver, brain, and heart, even though only Complex III was demonstrated responsible for ROS production (Wang et al., 2004). It has been recently demonstrated that malonate (Complex II inhibitor) exerted protective effects against Cd-induced necrosis in rat hepatocellular carcinoma AS-30D cells and reduced ROS production induced by Cd in PC12 cells, suggesting the key role of Complex II in Cd-induced mitochondrial alterations (Belyaeva, 2018).
Complex III (coenzyme Q-cytochrome c reductase, also known as the cytochrome bc1 complex) mediates electron transport from Q to cyt c via a peculiar pathway known as the Q cycle. Complex III is a pear-shaped dimer, with 11 subunits per monomer. Cyt b, cyt c1, and 2Fe–2S cluster are the catalytical subunits. The cyt b contains two Q binding sites, Qo near the intermembrane space and Qi at the matrix side of the membrane (Wu et al., 2016). QH2 is oxidized to semi-ubiquinone (Q•–) after transferring an electron to the 2Fe–2S cluster; simultaneously, two protons are transported from the matrix across the IMM. Then, the 2Fe–2S cluster transfers the electron to cyt c via cyt c1. At the Qo site, the formed Q•– transfers the electron to another Q molecule via cyt b, forming Q•– at the Qi site. Finally, a second QH2 molecule is oxidized at the Qo site, and two other protons are released across the IMM. This second QH2 molecule transfers one electron to the 2Fe–2S cluster and the other to Q•– at Qi site via cyt b producing QH2 (Crofts et al., 2017).
It has been shown that the toxic effect of Cd has been ascribable to mitochondrial ETC inhibition by impairing electron flow through Complex III (Miccadei and Floridi, 1993). Complex III has shown a high sensitivity to Cd that leads to around 77% inhibition and, as reported by electron spin resonance (ESR) experiments, Complex III might be the only site of ROS production induced by Cd (Wang et al., 2004). Cd may bind between Q•– and cyt b of the Qo site of Complex III, resulting in Q•– accumulation at the Qo site. Q•– is very unstable and can transfer one electron to molecular oxygen forming superoxide (Wang et al., 2004). The protective effect against Cd-induced cytotoxicity of 2,6-dichloroindophenol (DCIP), a complex III bypass agent, confirms these data (Monroe and Halvorsen, 2009). Moreover, it has been suggested that ROS produced by Complex III were functionally linked to the mitochondrial PTP (Armstrong et al., 2004) and that Complex III and Complex I might be involved in the mitochondrial PTP promoted by Cd (Belyaeva et al., 2012).
Complex IV, also called cyt c oxidase (COX), transfers electrons from cyt c to reduce oxygen. Simultaneously, Complex IV also drives four protons across the IMM. Mammalian Complex IV has a mass of 204 kDa and contains two hemes (a and a3) and three copper ions, two in the CuA center and one in the CuB site.
It has been shown in male Sprague-Dawley rats that Cd at low doses decreases the activity of Complex IV in liver mitochondria, but not in those of the kidney (Müller and Stacey, 1988). These data are confirmed, in mitochondria isolated from the liver of male Wistar rats, by the observation that Cd inhibited the ETC downstream Q (Ciapaite et al., 2009), supporting the results of Toury et al. (1985) that showed in mitochondria from the liver, kidney, and muscle of Cd-treated rats a significant decrease in cyt c oxidase activity. The reduced cyt c oxidase activity was also highlighted in the proximal tubular epithelial cells of rats subjected to prolonged Cd intoxication (Takaki et al., 2004). Moreover, in MDA-MB231 tumor cells, it was found that mRNA and protein levels of COXII and COXIV subunits decreased after long-term treatment with Cd, while short Cd exposure showed no effects (Cannino et al., 2008). More recently, Monteiro et al. (2018) demonstrated the Complex I and IV inhibition following Cd treatment in human MG-63 osteoblast cell line, confirming the key role of Cd in cyt c oxidase inhibition.
Cd is an environmental pollutant shown to display several undesirable effects on human health, targeting numerous organs and tissues. The molecular mechanisms responsible for most of the biological effects of Cd are not fully elucidated, and the toxicity targets have not been completely identified. Several studies focused their attention on mitochondria, which seem to represent the main intracellular targets of Cd. In particular, the different four complexes present in ETC have been suggested to play a key role in Cd-induced cytotoxicity, which is mainly displayed by mitochondrial ETC enzyme inhibition, enhanced ROS production, and uncoupling effect. However, other studies are needed to ascertain the dose- and exposure-dependent effects of Cd on in vivo experimental models.
JJVB, MB, and CF conceived the structure of the manuscript and drafted the manuscript. AP, MG, and NT critically revised the manuscript. All authors approved the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Adiele, R. C., Stevens, D., and Kamunde, C. (2010). Reciprocal enhancement of uptake and toxicity of cadmium and calcium in rainbow trout (Oncorhynchus mykiss) liver mitochondria. Aquat. Toxicol. 96, 319–327. doi: 10.1016/j.aquatox.2009.11.019
Adiele, R. C., Stevens, D., and Kamunde, C. (2012). Differential inhibition of electron transport chain enzyme complexes by cadmium and calcium in isolated rainbow trout (Oncorhynchus mykiss) hepatic mitochondria. Toxicol. Sci. 127, 110–119. doi: 10.1093/toxsci/kfs091
Agip, A.-N. A., Blaza, J. N., Fedor, J. G., and Hirst, J. (2019). Mammalian respiratory complex I through the lens of Cryo-EM. Annu. Rev. Biophys. 48, 165–184. doi: 10.1146/annurev-biophys-052118-115704
Armstrong, J. S., Yang, H., Duan, W., and Whiteman, M. (2004). Cytochrome bc 1 regulates the mitochondrial permeability transition by two distinct pathways. J. Biol. Chem. 279, 50420–50428. doi: 10.1074/jbc.M408882200
Batandier, C., Leverve, X., and Fontaine, E. (2004). Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 279, 17197–17204. doi: 10.1074/jbc.M310329200
Belyaeva, E. A. (2010). Mitochondrial respiratory chain inhibitors modulate the metal-induced inner mitochondrial membrane permeabilization. Acta Biochim. Pol. 57, 435–441. doi: 10.18388/abp.2010_2430
Belyaeva, E. A. (2018). Respiratory complex II in mitochondrial dysfunction-mediated cytotoxicity: insight from cadmium. J. Trace Elements Med. Biol. 50, 80–92. doi: 10.1016/j.jtemb.2018.06.009
Belyaeva, E. A., Glazunov, V. V., and Korotkov, S. M. (2002). Cyclosporin A-sensitive permeability transition pore is involved in Cd2-induced dysfunction of isolated rat liver mitochondria: doubts no more. Arch. Bioche. Biophys. 405, 252–264. doi: 10.1016/s0003-9861(02)00400-9
Belyaeva, E. A., Glazunov, V. V., and Korotkov, S. M. (2004a). Cd2+ versus Ca2+-produced mitochondrial membrane permeabilization: a proposed direct participation of respiratory complexes I and III. Chem. Biol. Interact. 150, 253–270. doi: 10.1016/j.cbi.2004.09.019
Belyaeva, E. A., Glazunov, V. V., and Korotkov, S. M. (2004b). Cd2+ -promoted mitochondrial permeability transition: a comparison with other heavy metals. Acta Biochim. Pol. 51, 545–551.
Belyaeva, E. A., Glazunov, V. V., Nikitina, E. R., and Korotkov, S. M. (2001). Bivalent metal ions modulate Cd2+ effects on isolated rat liver mitochondria. J. Bioenerget. Biomembr. 33, 303–318. doi: 10.1023/a:1010607307099
Belyaeva, E. A., and Korotkov, S. M. (2003). Mechanism of primary Cd2+-induced rat liver mitochondria dysfunction: discrete modes of Cd2+ action on calcium and thiol-dependent domains. Toxicol. Appl. Pharmacol. 192, 56–68. doi: 10.1016/S0041-008X(03)00255-2
Belyaeva, E. A., Korotkov, S. M., and Saris, N.-E. (2011). In vitro modulation of heavy metal-induced rat liver mitochondria dysfunction: a comparison of copper and mercury with cadmium. J. Trace Elements Med. Biol. 25, S63–S73. doi: 10.1016/j.jtemb.2010.10.007
Belyaeva, E. A., Sokolova, T. V., Emelyanova, L. V., and Zakharova, I. O. (2012). Mitochondrial electron transport chain in heavy metal-induced neurotoxicity: effects of cadmium. Mercury, and Copper. Sci. World J. 2012, 1–14. doi: 10.1100/2012/136063
Bernardi, P., and Di Lisa, F. (2015). The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J. Mol. Cel. Cardiol. 78, 100–106. doi: 10.1016/j.yjmcc.2014.09.023
Bernardi, P., Krauskopf, A., Basso, E., Petronilli, V., Blalchy-Dyson, E., Di Lisa, F., et al. (2006). The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 273, 2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x
Bezawork-Geleta, A., Rohlena, J., Dong, L., Pacak, K., and Neuzil, J. (2017). Mitochondrial complex II: at the crossroads. Trends Biochem. Sci. 42, 312–325. doi: 10.1016/j.tibs.2017.01.003
Branca, J. J. V., Fiorillo, C., Carrino, D., Paternostro, F., Taddei, N., Gulisano, M., et al. (2020). Cadmium-induced oxidative stress: focus on the central nervous system. Antioxidants 9:492. doi: 10.3390/antiox9060492
Branca, J. J. V., Maresca, M., Morucci, G., Mello, T., Becatti, M., Pazzagli, L., et al. (2019a). Effects of cadmium on ZO-1 tight junction integrity of the blood brain barrier. Int. J. Mol. Sci. 20:6010. doi: 10.3390/ijms20236010
Branca, J. J. V., Morucci, G., Becatti, M., Carrino, D., Ghelardini, C., Gulisano, M., et al. (2019b). Cannabidiol protects dopaminergic neuronal cells from cadmium. Int. J. Environ. Res. Public Health 16:4420. doi: 10.3390/ijerph16224420
Branca, J. J. V., Morucci, G., Maresca, M., Tenci, B., Cascella, R., Paternostro, F., et al. (2018). Selenium and zinc: two key players against cadmium-induced neuronal toxicity. Toxicol. Vitro 48, 159–169. doi: 10.1016/j.tiv.2018.01.007
Brand, M. D. (2016). Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 100, 14–31. doi: 10.1016/j.freeradbiomed.2016.04.001
Cadenas, E. (1997). Basic mechanisms of antioxidant activity. BioFactors 6, 391–397. doi: 10.1002/biof.5520060404
Cameron, I., McNamee, P. M., Markham, A., Morgan, R. M., and Wood, M. (1986). The effects of cadmium on succinate and NADH-linked substrate oxidations in rat hepatic mitochondria. J. Appl. Toxicol. 6, 325–330. doi: 10.1002/jat.2550060505
Cannino, G., Ferruggia, E., Luparello, C., and Rinaldi, A. M. (2008). Effects of cadmium chloride on some mitochondria-related activity and gene expression of human MDA-MB231 breast tumor cells. J. Inorg. Biochem. 102, 1668–1676. doi: 10.1016/j.jinorgbio.2008.04.002
Cannino, G., Ferruggia, E., Luparello, C., and Rinaldi, A. M. (2009). Mitochondrial compartment: a possible target of cadmium effects on breast epithelial cells. Mol. Cell Biochem. 328, 75–84. doi: 10.1007/s11010-009-0076-7
Casalino, E., Calzaretti, G., Sblano, C., and Landriscina, C. (2002). Molecular inhibitory mechanisms of antioxidant enzymes in rat liver and kidney by cadmium. Toxicology 179, 37–50. doi: 10.1016/S0300-483X(02)00245-7
Chen, C., Zhang, S., Liu, Z., Tian, Y., and Sun, Q. (2015). Cadmium toxicity induces ER stress and apoptosis via impairing energy homoeostasis in cardiomyocytes. Biosci. Rep. 35:e00214. doi: 10.1042/BSR20140170
Ciapaite, J., Nauciene, Z., Baniene, R., Wagner, M. J., Krab, K., and Mildaziene, V. (2009). Modular kinetic analysis reveals differences in Cd2+ and Cu2+ ion-induced impairment of oxidative phosphorylation in liver: metal-induced impairment of mitochondrial function. FEBS J. 276, 3656–3668. doi: 10.1111/j.1742-4658.2009.07084.x
Crofts, A. R., Rose, S. W., Burton, R. L., Desai, A. V., Kenis, P. J. A., and Dikanov, S. A. (2017). The Q-cycle mechanism of the bc 1 complex: a biologist’s perspective on atomistic studies. J. Phys. Chem. B 121, 3701–3717. doi: 10.1021/acs.jpcb.6b10524
Fariss, M. W. (1991). Cadmium toxicity: unique cytoprotective properties of alpha tocopheryl succinate in hepatocytes. Toxicology 69, 63–77. doi: 10.1016/0300-483X(91)90154-S
García, N., Correa, F., and Chávez, E. (2005). On the role of the respiratory complex I on membrane permeability transition. J. Bioenerg. Biomembr. 37, 17–23. doi: 10.1007/s10863-005-4119-9
Gobe, G., and Crane, D. (2010). Mitochondria, reactive oxygen species and cadmium toxicity in the kidney. Toxicol. Lett. 198, 49–55. doi: 10.1016/j.toxlet.2010.04.013
Grivennikova, V. G., Kapustin, A. N., and Vinogradov, A. D. (2001). Catalytic activity of NADH-ubiquinone oxidoreductase (complex I) in intact mitochondria. evidence for the slow active/inactive transition. J. Biol. Chem. 276, 9038–9044. doi: 10.1074/jbc.M009661200
Hogervorst, J., Plusquin, M., Vangronsveld, J., Nawrot, T., Cuypers, A., Van Hecke, E., et al. (2007). House dust as possible route of environmental exposure to cadmium and lead in the adult general population. Environ. Res. 103, 30–37. doi: 10.1016/j.envres.2006.05.009
Ivanina, A. V., Habinck, E., and Sokolova, I. M. (2008). Differential sensitivity to cadmium of key mitochondrial enzymes in the eastern oyster, Crassostrea virginica Gmelin (Bivalvia: Ostreidae). Comp. Biochem. Physiol. C Toxicol. Pharmacol. 148, 72–79. doi: 10.1016/j.cbpc.2008.03.009
Jay, D., Zamorano, R., Muñoz, E., Gleason, R., and Boldu, J. L. (1991). Study of the interaction of cadmium with membrane-bound succinate dehydrogenase. J. Bioenerg. Biomembr. 23, 381–389. doi: 10.1007/BF00762229
Karch, J., Bround, M. J., Khalili, H., Sargent, M. A., Latchman, N., Terada, N., et al. (2019). Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 5:eaaw4597. doi: 10.1126/sciadv.aaw4597
Koike, H., Shinohara, Y., and Terada, H. (1991). Why is inorganic phosphate necessary for uncoupling of oxidative phosphorylation by Cd2+ in rat liver mitochondria? Biochim. Biophys. Acta 1060, 75–81. doi: 10.1016/S0005-2728(05)80121-5
Korotkov, S. M., Skulskii, I. A., and Glazunov, V. V. (1998). Cd2+ effects on respiration and swelling of rat liver mitochondria were modified by monovalent cations. J. Inorg. Biochem. 70, 17–23. doi: 10.1016/s0162-0134(98)00008-7
Kurochkin, I. O., Etzkorn, M., Buchwalter, D., Leamy, L., and Sokolova, I. M. (2011). Top-down control analysis of the cadmium effects on molluscan mitochondria and the mechanisms of cadmium-induced mitochondrial dysfunction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R21–R31. doi: 10.1152/ajpregu.00279.2010
Liu, R. M., and Liun, Y. G. (1990). Effects of cadmium on the energy metabolism of isolated hepatocytes: its relationship with the nonviability of isolated hepatocytes caused by cadmium. Biomed. Environ. Sci. 3, 251–261.
Mao, W., Zhang, N., Zhou, F., Li, W., Liu, H., Feng, J., et al. (2011). Cadmium directly induced mitochondrial dysfunction of human embryonic kidney cells. Hum. Exp. Toxicol. 30, 920–929. doi: 10.1177/0960327110384286
Miccadei, S., and Floridi, A. (1993). Sites of inhibition of mitochondrial electron transport by cadmium. Chem. Biol. Interact. 89, 159–167. doi: 10.1016/0009-2797(93)90006-K
Milenkovic, D., Blaza, J. N., Larsson, N.-G., and Hirst, J. (2017). The enigma of the respiratory chain supercomplex. Cell Metab. 25, 765–776. doi: 10.1016/j.cmet.2017.03.009
Modi, H. R., and Katyare, S. S. (2009). Effect of treatment with cadmium on structure-function relationships in rat liver mitochondria: studies on oxidative energy metabolism and lipid/phospholipids profiles. J. Membrane Biol. 232, 47–57. doi: 10.1007/s00232-009-9217-x
Monroe, R. K., and Halvorsen, S. W. (2009). Environmental toxicants inhibit neuronal Jak tyrosine kinase by mitochondrial disruption. Neurotoxicology 30, 589–598. doi: 10.1016/j.neuro.2009.03.007
Monteiro, C., Ferreira de Oliveira, J. M. P., Pinho, F., Bastos, V., Oliveira, H., Peixoto, F., et al. (2018). Biochemical and transcriptional analyses of cadmium-induced mitochondrial dysfunction and oxidative stress in human osteoblasts. J. Toxicol. Environ. Health A 81, 705–717. doi: 10.1080/15287394.2018.1485122
Muller, F. L., Liu, Y., Abdul-Ghani, M. A., Lustgarten, M. S., Bhattacharya, A., Jang, Y. C., et al. (2008). High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochem. J. 409, 491–499. doi: 10.1042/BJ20071162
Müller, L., and Ohnesorge, F. K. (1984). Cadmium-induced alteration of the energy level in isolated hepatocytes. Toxicology 31, 297–306. doi: 10.1016/0300-483X(84)90110-0
Müller, L., and Stacey, N. H. (1988). Subcellular toxicity of low level cadmium in rats: effect on cytochrome c oxidase. Toxicology 51, 25–34. doi: 10.1016/0300-483X(88)90077-7
Okoye, C. N., MacDonald-Jay, N., and Kamunde, C. (2019). Effects of bioenergetics, temperature and cadmium on liver mitochondria reactive oxygen species production and consumption. Aquat. Toxicol. 214:105264. doi: 10.1016/j.aquatox.2019.105264
Papa, S., Lorusso, M., and Di Paola, M. (2006). Cooperativity and flexibility of the protonmotive activity of mitochondrial respiratory chain. Biochim. Biophys. Acta 1757, 428–436. doi: 10.1016/j.bbabio.2006.03.015
Patra, R. C., Swarup, D., and Senapati, S. K. (1999). Effects of cadmium on lipid peroxides and superoxide dismutase in hepatic, renal and testicular tissue of rats. Vet. Hum. Toxicol. 41, 65–67.
Quinlan, C. L., Orr, A. L., Perevoshchikova, I. V., Treberg, J. R., Ackrell, B. A., and Brand, M. D. (2012). Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 287, 27255–27264. doi: 10.1074/jbc.M112.374629
Sazanov, L. A. (2015). A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 16, 375–388. doi: 10.1038/nrm3997
Shukla, A., Shukla, G. S., and Srimal, R. (1996). Cadmium-induced alterations in blood- brain barrier permeability and its possible correlation with decreased microvessel antioxidant potential in rat. Hum. Exp. Toxicol. 15, 400–405. doi: 10.1177/096032719601500507
Sun, F., Huo, X., Zhai, Y., Wang, A., Xu, J., Su, D., et al. (2005). Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121, 1043–1057. doi: 10.1016/j.cell.2005.05.025
Takaki, A., Jimi, S., Segawa, M., Hisano, S., Takebayashi, S., and Iwasaki, H. (2004). Long-term cadmium exposure accelerates age-related mitochondrial changes in renal epithelial cells. Toxicology 203, 145–154. doi: 10.1016/j.tox.2004.06.005
Toury, R., Boissonneau, E., Stelly, N., Dupuis, Y., Berville, A., and Perasso, R. (1985). Mitochondria alterations in Cd2+-treated rats: general regression of inner membrane cristae and electron transport impairment. Biol. Cell 55, 71–85. doi: 10.1111/j.1768-322X.1985.tb00411.x
Uwagie-Ero, E. A., Abiaezute, C. N., Nwaehujor, C. O., Nnaji, T. O., Odigie, E. A., Ekeolu, O. K., et al. (2019). Osteocyte viability and bone density in cadmium chloride-induced osteoporosis ameliorated with Pilostigma thonningii stem bark-extracted D-3-O-methy-chiroinositol. Anim. Models Exp. Med. 2, 25–33. doi: 10.1002/ame2.12054
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T. D., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. doi: 10.1016/j.biocel.2006.07.001
Wang, B., and Du, Y. (2013). Cadmium and its neurotoxic effects. Oxid. Med. Cell Longev. 2013, 1–12. doi: 10.1155/2013/898034
Wang, Y., Fang, J., Leonard, S. S., and Krishna Rao, K. M. (2004). Cadmium inhibits the electron transfer chain and induces reactive Oxygen species. Free Radic. Biol. Med. 36, 1434–1443. doi: 10.1016/j.freeradbiomed.2004.03.010
Wu, M., Gu, J., Guo, R., Huang, Y., and Yang, M. (2016). Structure of mammalian respiratory supercomplex I 1 III 2 IV 1. Cell 167, 1598.e10–1609.e10. doi: 10.1016/j.cell.2016.11.012
Xiong, R., Wu, Q., Trbojevich, R., Muskhelishvili, L., Davis, K., Bryant, M., et al. (2019). Disease-related responses induced by cadmium in an in vitro human airway tissue model. Toxicol. Lett. 303, 16–27. doi: 10.1016/j.toxlet.2018.12.009
Xu, L., Zhang, W., Liu, X., Zhang, C., Wang, P., and Zhao, X. (2018). Circulatory levels of toxic metals (Aluminum, Cadmium, Mercury, Lead) in patients with Alzheimer’s Disease: a quantitative meta-analysis and systematic review. J. Alzheimers Dis. 62, 361–372. doi: 10.3233/JAD-170811
Zhang, J., Wang, X., Vikash, V., Ye, Q., Wu, D., Liu, Y., et al. (2016). ROS and ROS-mediated cellular signaling. Oxid. Med. Cell Longev. 2016, 1–18. doi: 10.1155/2016/4350965
Keywords: cadmium, cytotoxicity, mitochondria, mitochondrial electron transport chain, mitochondrial complexes
Citation: Branca JJV, Pacini A, Gulisano M, Taddei N, Fiorillo C and Becatti M (2020) Cadmium-Induced Cytotoxicity: Effects on Mitochondrial Electron Transport Chain. Front. Cell Dev. Biol. 8:604377. doi: 10.3389/fcell.2020.604377
Received: 09 September 2020; Accepted: 05 November 2020;
Published: 30 November 2020.
Edited by:
Vito De Pinto, University of Catania, ItalyCopyright © 2020 Branca, Pacini, Gulisano, Taddei, Fiorillo and Becatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Fiorillo, Y2xhdWRpYS5maW9yaWxsb0B1bmlmaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.