 Ying Ye
Ying Ye Xiaoting Sun
Xiaoting Sun Yongtian Lu
Yongtian Lu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 27 November 2020
Sec. Molecular and Cellular Oncology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.600350
This article is part of the Research TopicThe Adipose Tissue Microenvironment in Cancer: Molecular Mechanisms and Targets for TreatmentView all 14 articles
Obesity-derived disturbances in fatty acid and cholesterol metabolism are linked to numerous diseases, including various types of malignancy. In tumor cells, metabolic alterations have been long recognized and intensively studied. However, metabolic changes in host cells in the tumor microenvironment and their contribution to tumor development have been largely overlooked. During the last decade, research advances show that fatty acid oxidation, cholesterol metabolism, and lipid accumulation play critical roles in cancer-associated host cells such as endothelial cells, lymph endothelial cells, cancer-associated fibroblasts, tumor-associated myeloid cells, and tumor-associated lymphocytes. In addition to anti-angiogenic therapies and immunotherapy that have been practiced in the clinic, metabolic regulation is considered another promising cancer therapy targeting non-tumor host cells. Understanding the obesity-associated metabolism changes in cancer-associated host cells may ultimately be translated into therapeutic options that benefit cancer patients. In this mini-review, we briefly summarize the lipid metabolism associated with obesity and its role in host cells in the tumor microenvironment. We also discuss the current understanding of the molecular pathways involved and future perspectives to benefit from this metabolic complexity.
The global obesity pandemic affects most high-income and middle-income countries, and it is associated with an increased incidence of certain types of cancer (Swinburn et al., 2011; Demark-Wahnefried et al., 2012). Obese adipocytes could release fatty acids (FAs), lipoproteins, hormones, and growth factors into extracellular space and circulation. It is recognized that adipocytes provide fuel and triggers for the metabolic reprogramming in changing tumor behaviors (Nieman et al., 2013).
Although aerobic glycolysis is the dominant metabolic paradigm, cancer cells exploit lipid and cholesterol to meet their unlimited energy demands. In some types of cancer, lipid-dependent metabolism becomes a prominent pathway for energy production (Caro et al., 2012). Cancer cells obtain lipids by taking up the exogenous lipids and de novo synthesis of endogenous lipids. Free fatty acids (FFAs) are taken up through FA translocase CD36, FA transport proteins (FATPs)/SLC27A, and fatty acid-binding proteins (FABPs) for mitochondrial oxidation and energy production, while cholesterol-rich lipoproteins are taken up by receptors such as the low-density lipoprotein receptor (LDLR). Cholesterol can facilitate membrane microdomain formation, which can initiate tumor growth (Oneyama et al., 2009). Other than membrane composition, cholesterol is a precursor for bile acids and steroid hormones which can initiate cancer progression (Attard et al., 2009). For de novo lipogenesis, citrate is exported from the mitochondrion as a substrate, and ATP citrate lyase, acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN) sequentially promote FA production for further synthesis of triacylglycerols, cholesterol esters, and phospholipids. Compared with healthy tissues that prefer to use circulating lipids, tumor cells express a significant amount of FASN protein (Cai et al., 2015). Clinically, the first-in-class FASN inhibitor currently is under phase II trial and shows antitumor potential (Falchook et al., 2017). The lipid metabolism pathway is summarized in Figure 1.
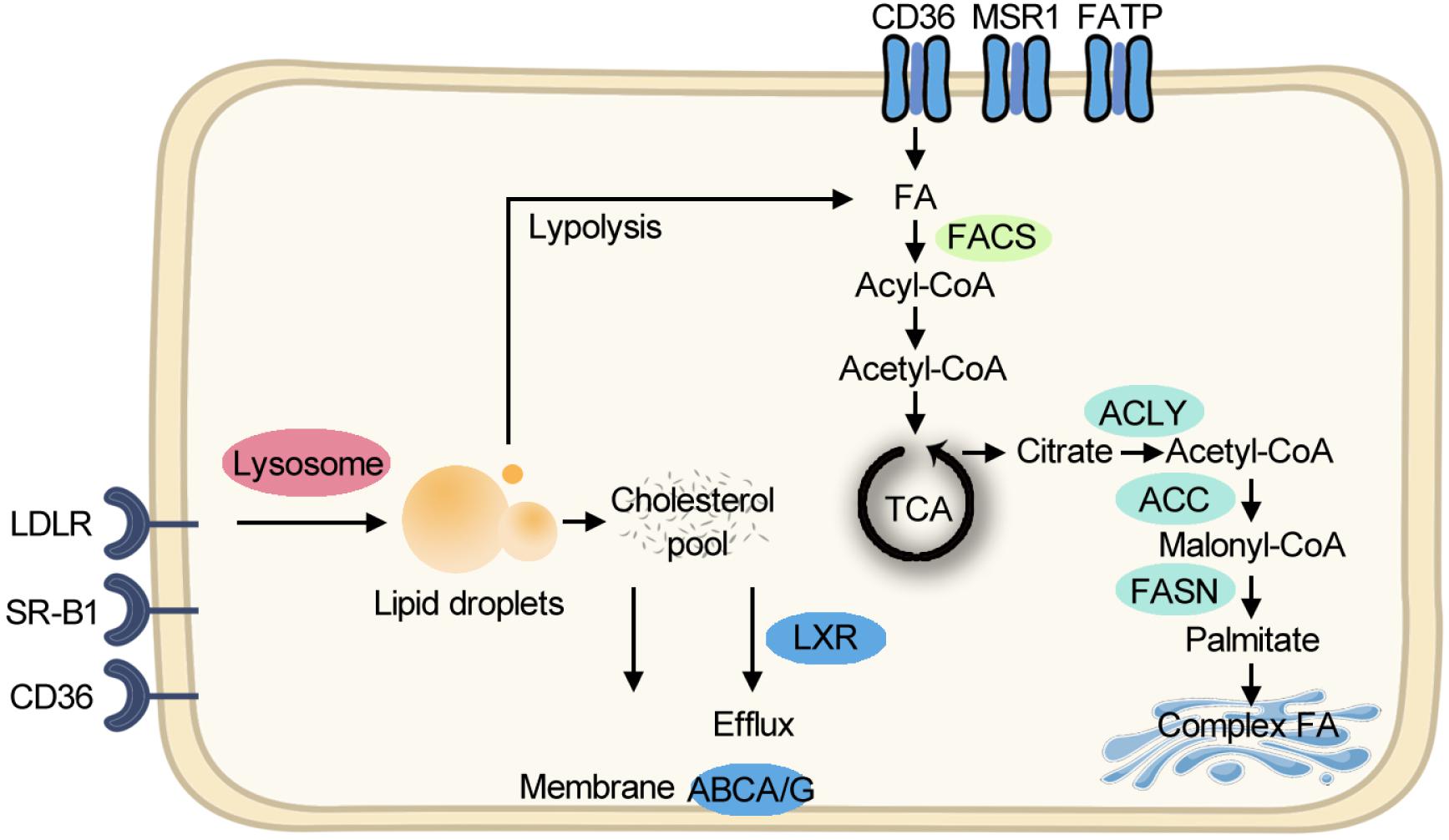
Figure 1. Simplified scheme of major lipid metabolic pathways. Exogenous FAs are taken up for FAO and energy production. Exogenous cholesterol-rich lipoproteins are imported for building membranes. De novo lipogenesis starts from exported citrate and makes complex FA in the Golgi body. FA, fatty acid; FAO, fatty acid oxidation; TCA, tricarboxylic acid cycle; CoA, coenzyme A; ACLY, ATP citrate lyase; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; FACS, fatty acyl-CoA synthetase; ABCA/G, ATP-Binding Cassette A/G; LXR, liver X receptor; LDLR, low-density lipoprotein receptor; SR-B1, the scavenger receptor, class B type 1; MSR1, macrophage scavenger receptor 1; FATP, fatty acid transport proteins.
Although the lipid metabolism and cholesterol metabolism in cancer cells per se have received substantial amount of interest over the past two decades, it needs to be considered that the tumor microenvironment (TME) is modulated by complex signaling networks from malignant cells and multiple other components, including vascular cells, stromal fibroblasts, inflammatory cells, and blood cells. Constantly exposed to various growth factors controlling tumor angiogenesis, lymphatic growth, stromal fibroblast expansion, and inflammation, TME becomes favorable for tumor growth, metastasis, and drug resistance. Whether obesity-derived FA and cholesterols can stimulate these host non-malignant cells is largely overlooked. There are still challenges to be faced in understanding how host cell lipid metabolism facilitates tumor development and in bringing the drugs that target cancer-associated host cell metabolism to the clinic.
In 1960, researchers found that most obese individuals have elevated FFA levels in serum (Gordon, 1960). FFAs from highly saturated fat consumption or dysregulated lipolysis can induce various responses, including insulin resistance, inflammation, lipotoxicity, and endothelial dysfunction (Pleiner et al., 2002; Jiao et al., 2011; Oh et al., 2018). Furthermore, FFAs exert pathophysiological functions through free fatty acid receptors (Vangaveti et al., 2010).
During obesity, dyslipidemia is a classic hallmark with reduced high-density lipoprotein cholesterol (HDL-C) levels and increased levels of small, dense low-density lipoprotein (LDL) particles, circulating triglycerides (TGs), or both (Kathiresan et al., 2006). Besides genetic dyslipidemia, lifestyle with excessive dietary intake of total calories, saturated fat, cholesterol, and trans fats are the leading cause for the increased rates of cholesterol synthesis and reduced expression of LDLR (Glatz and Katan, 1993).
Tumor cell-derived angiogenic factors induce neovascularization in TME. Although tumors utilize similar mechanisms as normal growing tissues, tumor vasculatures are generally malformed with a high degree of disorganization, lack of clear separation between arterioles and venules, lack of pericyte coverage, and high permeability (Cao, 2009). Compared with the quiescent state of endothelial cells (EC) in healthy tissues, the angiogenic features of tumor vessels require a proliferative and migratory EC state, which is the basis of antiangiogenic drugs, such as bevacizumab. This sprouting state is recently shown driven by metabolic switches in EC.
It is reported that glycolytic flux is doubled under proliferative status in EC, and the glycolysis activator 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3) is the key enzyme for EC glycolysis (De Bock et al., 2013). Under physiological status, shear stress blocks PFKFB3-related metabolism in EC via flow-sensitive transcription factor (Doddaballapur et al., 2015). In contrast, PFKFB3 is upregulated in pathological angiogenesis and PFKFB3 blockade shows the antiangiogenic effect (Schoors et al., 2014). Although proliferating EC is considered to rely on glycolysis, it exerts a metabolic paradigm shift to fatty acid oxidation (FAO) upon glucose deprivation (Dagher et al., 2001). Indeed, CPT1α-guided FAO stimulates EC proliferation (Schoors et al., 2015), and inhibition of CPT1α in ECs impairs vessel stability (Patella et al., 2015). Interestingly, in proliferating EC, FAO is not providing additional ATP but is used for de novo synthesis of nucleotides (Schoors et al., 2015). It is reported that obesity-related hormone leptin promotes FAO in ECs by increasing CPT1α activity, suggesting obesity not only provides the fuel but also is capable of triggering host cell metabolism (Yamagishi et al., 2001). Moreover, under proliferation, EC increases FA uptake and the expression of FABP4 (Elmasri et al., 2009). FABP4 inhibition leads to a marked increase of FAO in tumoral EC and decreases tumor angiogenesis (Harjes et al., 2017). It is reported that vascular endothelial growth factor (VEGF)-B is involved in modulating FA uptake in EC (Hagberg et al., 2010). Whether VEGF-B blockade inhibits tumor angiogenesis requires further validation.
Emerging evidence suggests that cholesterol levels may regulate angiogenesis. Elevated circulating cholesterol level promotes tumor angiogenesis, and cholesterol uptake-blocking agent ezetimibe significantly inhibits tumor angiogenesis (Solomon et al., 2009). Cholesterol efflux from ECs to HDL reduces lipid rafts, interferes with VEGFR2 signaling, and inhibits angiogenesis (Fang et al., 2013). Activation of endothelial liver X receptors (LXRs), regulators of cholesterol homeostasis, reduces tumor angiogenesis by impairing the compartmentation of VEGFR2 (Noghero et al., 2012). Moreover, cholesterol trafficking is a potential target for blocking angiogenesis in TME (Lyu et al., 2017). Of note, VEGF-B is reported to impair LDLR recycling and reduce cholesterol uptake in EC. Its role in TME needs additional investigation (Moessinger et al., 2020).
The role of FA synthesis in tumor ECs remains incompletely understood. It is reported ACC regulates EC migration, and FASN is critical for vessel sprouting. ACC inhibition shifted the phospholipid composition of EC membranes and reduced membrane fluidity, filopodia formation, and migratory capacity (Glatzel et al., 2018). FASN knockout elevates malonyl-CoA levels, causing malonylation of mTOR and impairment of pathological angiogenesis (Bruning et al., 2018).
Intratumoral and peritumoral lymphatics are essential for lymph metastasis of tumor cells. However, the role of metabolism in lymphatic endothelial cells (LECs) is largely unknown. It is reported that FAO is crucial for lymphatic development through epigenetic regulation of lymphatic transcription factor PROX1 (Wong et al., 2017). The role of FA synthesis on the lymphatic network and tumor-induced lymphangiogenesis is still unknown. It seems like FASN is needed for the growth and maintenance of LECs, and FASN inhibitor reduces LEC migration and tumor lymph metastasis (Bastos et al., 2017).
It is reasonable to speculate that EC metabolism in TME is context-dependent (Li et al., 2019a). Single-cell RNA sequencing studies revealed extensive heterogeneity of metabolic gene expression signatures between ECs from different tissues (Li et al., 2019b). Tumor type may determine the metabolism pattern of ECs. Moreover, in TME, malignancy-derived various cytokines may disrupt EC metabolic switch. To target EC metabolism as an antiangiogenic therapy in TME, it is necessary to consider the impact of tumor type and tumor-derived cytokines. Hypoxia may be another parameter regulating EC metabolism. ECs, especially sprouting ECs, have to establish functional vessels for avascularized tissues. Under this status, it requires ECs to have a functional metabolism under hypoxia. In tumors, this need becomes even stronger. Whether obesity-derived FA and LDL can affect this unique metabolism requires further investigation. For LECs metabolism in TME, limited studies prevent us from understanding it in detail. We do know FAO and FASN are involved in the regulation of lymphangiogenesis. Further studies will be required to explore the mechanism and to understand the significance.
Healthy fibroblasts can become activated during tumorigenesis. In general, cancer-associated fibroblasts (CAFs) promote tumor progression by secreting tumor-supporting factors, acting as a barrier to immune surveillance, and facilitate tumor cell migration (Yang et al., 2016b; Sahai et al., 2020). Of note, fibroblasts can exhibit phenotype heterogeneity; depletion of fibroblasts accelerates pancreatic cancer, suggesting CAFs may restrain tumor growth (Ozdemir et al., 2014). Multiple studies demonstrate fibroblast subsets in TME with differential abilities to affect tumor progression (Sahai et al., 2020). Understanding the metabolism of CAFs has potential importance for revealing the complexity of CAFs and their interactions with cell compartments in TME.
Several studies described metabolic features of CAFs, and majority of them focus on glucose and glutamine metabolism. Transforming growth factor (TGF)-β- or platelet-derived growth factor (PDGF)-induced CAFs switch from oxidative phosphorylation to aerobic glycolysis to meet the requirements of extracellular matrix (ECM) production (Zhang et al., 2015). It should be noted that studies on CAF lipid metabolism are quite limited. However, the lipid metabolism and fate determination of fibroblasts in other tissues can provide us with enlightenment. In TME, ECs may convert into a fibroblast-like state, known as endothelial-to-mesenchymal transition (EndoMT) (Platel et al., 2019). It is reported that FAO is a negative regulator for EndoMT (Xiong et al., 2018). In skin fibroblasts, peroxisome proliferator-activated receptors (PPARs) signaling that regulates FA uptake and oxidation promotes a catabolic phenotype by enhancing ECM internalization and lysosomal degradation (Zhao et al., 2019). The CD36-expressing fibroblasts transplantation improved skin elasticity and reduced ECM deposition in mice (Zhao et al., 2019). In general, FA uptake and FAO reduce fibrogenesis and proliferation in fibroblasts. This view warrants validation in CAFs.
Cholesterol levels may also regulate fibrogenesis and fibroblast activation. However, the existing evidence is insufficient and contradictory. In cardiac fibroblasts, LXR agonist prevents TGF-β-induced collagen synthesis and α-smooth muscle actin expression (Cannon et al., 2015). Another group reported that LXR inverse agonist suppresses fibrosis in non-alcoholic steatohepatitis in vivo, suggesting opposing effects of LXR signaling on regulating fibrogenesis (Griffett et al., 2015).
Interestingly, CAFs are capable of synthesizing and transferring lipids to neighboring cells (Santi et al., 2015). Higher FASN activity in CAF identified its role as FAs supplier in breast cancer (Lopes-Coelho et al., 2018). A recent study using lipidomic profiling showed that CAFs provide fatty acyl, long-chain, and unsaturated FAs for colorectal cancer tumor cell development (Gong et al., 2020). There is still a lack of evidence of lipid synthesis changing CAF phenotypes.
In other types of fibroblasts, FAO may reduce fibrosis and fibrogenesis. If this result can be replicated in TME, metabolic regulation in CAF may have antitumor potential. It is worth noting that upon CAF activation, ECM overexpression is not completely negative for tumor treatment. Type I collagen reduction, together with a significant decrease in tumor tissue stiffness, promotes tumor metastasis (Ozdemir et al., 2014). This work raises a warning for blunting CAF for fibrogenesis using FAO. The other point is that in skin fibroblast, FAO did not downregulate glycolysis in vivo, which means regulation is not made through a classic metabolic switching. It is necessary to verify this phenomenon in the TME. In general, due to the lack of understanding of CAF metabolism and the essential impact of CAF on the TME, research on CAF metabolism should become the next focus in this field. Once enough evidence is provided, it may present exciting new therapeutic opportunities for the management of cancer.
Understanding immune cell metabolism in cancer is of growing significance in the past decade with the success of immunotherapy. In TME, infiltrated immune cells include tumor-associated macrophages (TAMs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), and several T cell subpopulations. Understanding the differential metabolic requirements of diverse immune cells helps us regulate the complex immune response in TME. Among various kinds of tumor-associated host cell types, the role of lipid metabolism in regulating immune cells has been most intensively studied. Knowledge from classic immunology and evidence collected in TME indicates the importance of immune cell lipid metabolism in tumor growth and provides novel therapeutics for cancer. Evidence has been well-documented in several comprehensive reviews (Geeraerts et al., 2017; Le Bourgeois et al., 2018; Giovanelli et al., 2019; Leone and Powell, 2020). For the balance of this article, here we briefly discuss lipid metabolism in immune cells as below.
TAMs account for the largest fraction of the myeloid infiltrate in solid malignancies, and they display heterogeneous transcriptional programs and phenotype plasticity (Cassetta et al., 2019). At the beginning of this century, alternatively activated macrophages are found to be prone to increase FAO (Vats et al., 2006). Lipids uptake via CD36 and upregulation of FAO supports reactive oxygen species (ROS), JAK1-STAT6 activation, and hence the protumor function (Zhang et al., 2018; Su et al., 2020). DCs regulate the balance between immunity and tolerance through selective activation by the triggers in TME. Generally, tolerogenic DCs rely on FAO for their energetic demands, increasing the tumor burden in mice (Malinarich et al., 2015). Msr1-mediated FA uptake reduces DCs’ capacity to process antigens (Herber et al., 2010). MDSCs play vital roles in TME, and their inhibition is critical for successful cancer immunotherapy. MDSCs prefer FAO over glycolysis as a primary source of energy, while treatment with FAO inhibitors improved antitumor immunity (Hossain et al., 2015). In general, induction of FAO primes myeloid cells in TME for an immunosuppressive phenotype.
For cholesterol metabolism, TAMs readily take up lipoproteins from dying cells and developed mechanisms for eliminating cholesterol from the cell. Cholesterol efflux in TAMs supports IL-4 signaling and promotes tumor progression (Goossens et al., 2019). For other myeloid cells, cholesterol metabolism promotes a protumor phenotype. LXR signaling inhibits CCR7 expression in DCs and their migration to tumor-draining lymph nodes (Villablanca et al., 2010). In contrast, cholesterol loading in LXR knockout CD11c+ cells promotes the antigen presentation (Ito et al., 2016). Under a high-fat diet, cholesterol acts on neutrophils via its metabolite, 27-hydroxycholesterol, and priming an immune-suppressive environment for tumor metastasis (Baek et al., 2017).
Dysregulation of key enzymes in lipid metabolism may lead to various problems such as lipid accumulation. FASN upregulation in tumor-associated myeloid cells stimulates PPARβ/δ and supports tumor cell invasion (Park et al., 2015). In support of this view, various lipolytic enzymes involved in intracellular lipid metabolism, including monoacylglycerol lipase, AB-hydrolase containing 5, epidermal fatty acid-binding protein, and adipocyte/macrophage fatty acid-binding protein, strongly affect TAM function and tumor progression (Rao et al., 2015; Miao et al., 2016; Hao et al., 2018; Xiang et al., 2018). In DCs, chaperone-binding oxidatively truncated lipids prevent the translocation of pMHC to the surface of DC and partially explained the DC attenuation (Veglia et al., 2017). In MDSCs, lipid accumulation reprograms MDSC to be highly immunosuppressive cells (Al-Khami et al., 2017). In general, a number of studies indicate that lipid synthesis and accumulation play an important role in the function of myeloid cells.
T cells can be divided into many subtypes and kill tumors directly or indirectly by synthesizing various biological molecules. In T cells, glycolysis is observed upon activation by the T-cell receptors (TCR) and costimulatory signals in TME (Chang et al., 2013). In contrast, FAO mainly provides energy for Treg and memory T cells (O’Sullivan et al., 2014; Miska et al., 2019). FAO blockade suppresses Treg population, while the addition of FAs promotes their differentiation (Michalek et al., 2011). Interestingly, PD-1 inhibits glycolysis and promoting FAO in T cells, and PD-1 blockade recovers T cell capacity (Patsoukis et al., 2015), suggesting the link between lipid metabolism and immune checkpoint. Of note, linoleic acid, a type of FA accumulated in fatty liver disease, causes more oxidative damage and mediates selective loss of intrahepatic CD4+ T lymphocytes (Ma et al., 2016), suggesting obesity triggers specific T cell response.
The role of cholesterol metabolism in T cells is controversial. T cell activation triggers simultaneous suppression of the LXR pathway for cholesterol transport and induction of the sterol regulatory element-binding protein (SREBP) pathway for cholesterol synthesis (Bensinger et al., 2008). In TME, increasing the cholesterol level in CD8+ T cells may induce exhaustion by endoplasmic reticulum (ER) stress (Ma et al., 2019). However, another group reported that cholesterol causes enhanced T-cell receptor clustering in CD8+ T cells and antitumor effect (Yang et al., 2016a).
FA synthesis supplies membrane materials for activated effector T cells. For example, SREBPs are crucial for CD8+ T cell expansion (Kidani et al., 2013), while blocking ACC1 restrains the formation of TH17 cells and promotes the development of Treg cells (Berod et al., 2014). In general, lipid metabolism is also important for maintaining the balance between effector T cells and Treg cells; studies on the effect of lipid metabolism on T cell function need to focus on the different T cell types.
As a cell group with direct or indirect killing effect in TME, immune cells are attractive therapeutic targets for metabolic regulation. However, similar metabolism in activated antitumoral immune cells and tumor cells leads to competition between tumor development and antitumor immunity. In TME, glucose competition, hypoxia, and lactic acid secretion promote immunosuppressive phenotype in TAMs, DCs, and T cells. The accumulation of FAs caused by obesity may facilitate this process. The prerequisite of targeting lipid metabolism of immune cells to treat tumors is a better understanding of lipid metabolism in different cell types and its overall consequences. The effects of lipid metabolism on various types of cancer-associated host cells are summarized in Figures 2A,B.
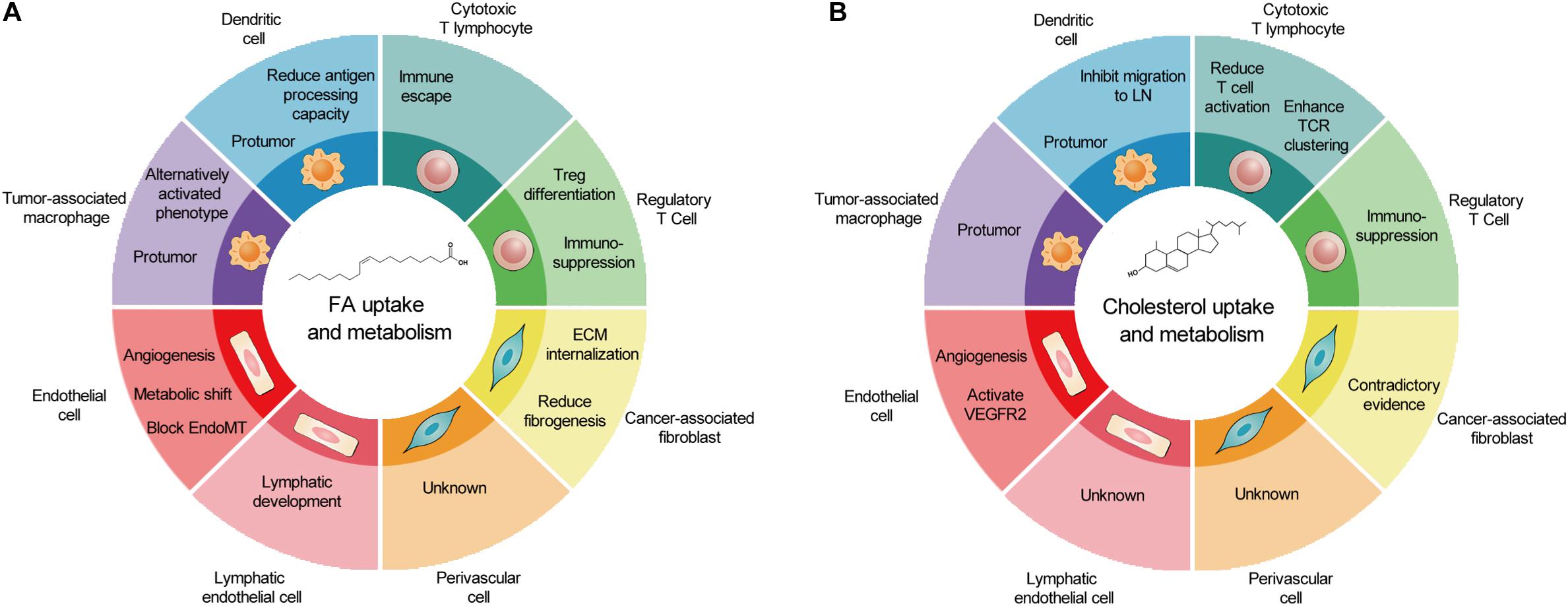
Figure 2. The outcomes of fatty acid and cholesterol metabolism in cancer-associated host cells. (A) In TME, FA metabolism promotes metabolic shift and angiogenesis in EC and LEC, reduces fibrogenesis, and generally induces protumor phenotype in immune cells. (B) In contrast, cholesterol metabolism promotes EC proliferation, induces protumor phenotype in immune cells, and produces contradictory effects in CAFs and cytotoxic T lymphocytes. Of note, several host cell types remain un-investigated. TME, tumor microenvironment; FA, fatty acid; EC, endothelial cell; LEC, lymphatic endothelial cell; CAFs, cancer-associated fibroblasts.
Targeting non-tumor host cells, such as antiangiogenic therapy and immunotherapy, has achieved significant effects in the clinical practice of cancer treatment. However, metabolic regulation for combating tumors has not yet become mainstream. Host cell metabolism in TME is significantly different from those in healthy tissues, and these differences provide opportunities to target host cell metabolism for treating tumors. Among the host cell populations in TME, immune cells are undoubtedly the most promising and the most studied cell group. Targeting the metabolism of suppressive immune cells or targeting effector cell metabolism to enhance tumor killing gives promising results in pre-clinical studies. However, similar evidence is lacking in other host cell types in TME. In this untouched research field, a series of studies are needed to understand obesity-related lipid metabolism and its influence on the host cells in TME. Moreover, future work should focus on the disruption of the TME on the metabolism of these host cells. The change of physical parameters in TME can increase angiogenesis, promote CAF activation, suppress the immune response, modulate drug resistance, and induce certain metabolic programs to support the malignancy. It is necessary to understand the molecular pathways involved in the host cell lipid metabolism to clarify what effect obesity has on this complex metabolic disruption and to benefit from this metabolic complexity.
YY generated the ideas and reviewed publications. YL and XS participated in discussions. YY and XS wrote the manuscript. All authors contributed to the article and approved the submitted version.
YY was supported by the National Natural Science Foundation of China (81600839) and the Shanghai Municipal Health Commission (20164Y0222).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Kaijing Gao for her artistic input.
Al-Khami, A. A., Zheng, L., Del Valle, L., Hossain, F., Wyczechowska, D., Zabaleta, J., et al. (2017). Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 6:e1344804. doi: 10.1080/2162402X.2017.1344804
Attard, G., Cooper, C. S., and de Bono, J. S. (2009). Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell 16, 458–462. doi: 10.1016/j.ccr.2009.11.006
Baek, A. E., Yu, Y. A., He, S., Wardell, S. E., Chang, C. Y., Kwon, S., et al. (2017). The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 8:864. doi: 10.1038/s41467-017-00910-z
Bastos, D. C., Paupert, J., Maillard, C., Seguin, F., Carvalho, M. A., Agostini, M., et al. (2017). Effects of fatty acid synthase inhibitors on lymphatic vessels: an in vitro and in vivo study in a melanoma model. Lab. Invest. 97, 194–206. doi: 10.1038/labinvest.2016.125
Bensinger, S. J., Bradley, M. N., Joseph, S. B., Zelcer, N., Janssen, E. M., Hausner, M. A., et al. (2008). LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134, 97–111. doi: 10.1016/j.cell.2008.04.052
Berod, L., Friedrich, C., Nandan, A., Freitag, J., Hagemann, S., Harmrolfs, K., et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 20, 1327–1333. doi: 10.1038/nm.3704
Bruning, U., Morales-Rodriguez, F., Kalucka, J., Goveia, J., Taverna, F., Queiroz, K. C. S., et al. (2018). Impairment of angiogenesis by fatty acid synthase inhibition involves mTOR malonylation. Cell. Metab. 28, 866.e15–880.e15. doi: 10.1016/j.cmet.2018.07.019
Cai, Y., Wang, J., Zhang, L., Wu, D., Yu, D., Tian, X., et al. (2015). Expressions of fatty acid synthase and HER2 are correlated with poor prognosis of ovarian cancer. Med. Oncol. 32:391. doi: 10.1007/s12032-014-0391-z
Cannon, M. V., Yu, H. J., Candido, W. M., Dokter, M. M., Lindstedt, E. L., Sillje, H. H. W., et al. (2015). The liver X receptor agonist AZ876 protects against pathological cardiac hypertrophy and fibrosis without lipogenic side effects. Eur. J. Heart Fail. 17, 273–282. doi: 10.1002/ejhf.243
Cao, Y. (2009). Tumor angiogenesis and molecular targets for therapy. Front. Biosci. 14, 3962–3973. doi: 10.2741/3504
Caro, P., Kishan, A. U., Norberg, E., Stanley, I. A., Chapuy, B., Ficarro, S. B., et al. (2012). Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 22, 547–560. doi: 10.1016/j.ccr.2012.08.014
Cassetta, L., Fragkogianni, S., Sims, A. H., Swierczak, A., Forrester, L. M., Zhang, H., et al. (2019). Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 35, 588.e10–602.e10. doi: 10.1016/j.ccell.2019.02.009
Chang, C. H., Curtis, J. D., Maggi, L. B. Jr., Faubert, B., Villarino, A. V., O’Sullivan, D., et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. doi: 10.1016/j.cell.2013.05.016
Dagher, Z., Ruderman, N., Tornheim, K., and Ido, Y. (2001). Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ. Res. 88, 1276–1282. doi: 10.1161/hh1201.092998
De Bock, K., Georgiadou, M., Schoors, S., Kuchnio, A., Wong, B. W., Cantelmo, A. R., et al. (2013). Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663. doi: 10.1016/j.cell.2013.06.037
Demark-Wahnefried, W., Platz, E. A., Ligibel, J. A., Blair, C. K., Courneya, K. S., Meyerhardt, J. A., et al. (2012). The role of obesity in cancer survival and recurrence. Cancer Epidemiol. Biomarkers Prev. 21, 1244–1259. doi: 10.1158/1055-9965.EPI-12-0485
Doddaballapur, A., Michalik, K. M., Manavski, Y., Lucas, T., Houtkooper, R. H., You, X., et al. (2015). Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler. Thromb. Vasc. Biol. 35, 137–145. doi: 10.1161/ATVBAHA.114.304277
Elmasri, H., Karaaslan, C., Teper, Y., Ghelfi, E., Weng, M., Ince, T. A., et al. (2009). Fatty acid binding protein 4 is a target of VEGF and a regulator of cell proliferation in endothelial cells. FASEB J. 23, 3865–3873. doi: 10.1096/fj.09-134882
Falchook, G., Patel, M., Infante, J., Arkenau, H. T., Dean, E., Brenner, A., et al. (2017). First in human study of the first-in-class fatty acid synthase (FASN) inhibitor TVB-2640. Cancer Res. 77:CT153. doi: 10.1158/1538-7445.Am2017-Ct153
Fang, L., Choi, S. H., Baek, J. S., Liu, C., Almazan, F., Ulrich, F., et al. (2013). Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 498, 118–122. doi: 10.1038/nature12166
Geeraerts, X., Bolli, E., Fendt, S. M., and Van Ginderachter, J. A. (2017). Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front. Immunol. 8:289. doi: 10.3389/fimmu.2017.00289
Giovanelli, P., Sandoval, T. A., and Cubillos-Ruiz, J. R. (2019). Dendritic cell metabolism and function in tumors. Trends Immunol. 40, 699–718. doi: 10.1016/j.it.2019.06.004
Glatz, J. F., and Katan, M. B. (1993). Dietary saturated fatty acids increase cholesterol synthesis and fecal steroid excretion in healthy men and women. Eur. J. Clin. Invest. 23, 648–655. doi: 10.1111/j.1365-2362.1993.tb00726.x
Glatzel, D. K., Koeberle, A., Pein, H., Loser, K., Stark, A., Keksel, N., et al. (2018). Acetyl-CoA carboxylase 1 regulates endothelial cell migration by shifting the phospholipid composition. J. Lipid Res. 59, 298–311. doi: 10.1194/jlr.M080101
Gong, J., Lin, Y., Zhang, H., Liu, C., Cheng, Z., Yang, X., et al. (2020). Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 11:267. doi: 10.1038/s41419-020-2434-z
Goossens, P., Rodriguez-Vita, J., Etzerodt, A., Masse, M., Rastoin, O., Gouirand, V., et al. (2019). Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell. Metab. 29, 1376.e4–1389.e4. doi: 10.1016/j.cmet.2019.02.016
Gordon, E. S. (1960). Non-esterified fatty acids in the blood of obese and lean subjects. Am. J. Clin. Nutr. 8, 740–747.
Griffett, K., Welch, R. D., Flaveny, C. A., Kolar, G. R., Neuschwander-Tetri, B. A., and Burris, T. P. (2015). The LXR inverse agonist SR9238 suppresses fibrosis in a model of non-alcoholic steatohepatitis. Mol. Metab. 4, 353–357. doi: 10.1016/j.molmet.2015.01.009
Hagberg, C. E., Falkevall, A., Wang, X., Larsson, E., Huusko, J., Nilsson, I., et al. (2010). Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464, 917–921. doi: 10.1038/nature08945
Hao, J., Yan, F., Zhang, Y., Triplett, A., Zhang, Y., Schultz, D. A., et al. (2018). Expression of adipocyte/macrophage fatty acid-binding protein in tumor-associated macrophages promotes breast cancer progression. Cancer Res. 78, 2343–2355. doi: 10.1158/0008-5472.CAN-17-2465
Harjes, U., Bridges, E., Gharpure, K. M., Roxanis, I., Sheldon, H., Miranda, F., et al. (2017). Antiangiogenic and tumour inhibitory effects of downregulating tumour endothelial FABP4. Oncogene 36, 912–921. doi: 10.1038/onc.2016.256
Herber, D. L., Cao, W., Nefedova, Y., Novitskiy, S. V., Nagaraj, S., Tyurin, V. A., et al. (2010). Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16, 880–886. doi: 10.1038/nm.2172
Hossain, F., Al-Khami, A. A., Wyczechowska, D., Hernandez, C., Zheng, L., Reiss, K., et al. (2015). Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 3, 1236–1247. doi: 10.1158/2326-6066.CIR-15-0036
Ito, A., Hong, C., Oka, K., Salazar, J. V., Diehl, C., Witztum, J. L., et al. (2016). Cholesterol accumulation in CD11c(+) immune cells is a causal and targetable factor in autoimmune disease. Immunity 45, 1311–1326. doi: 10.1016/j.immuni.2016.11.008
Jiao, P., Ma, J., Feng, B., Zhang, H., Diehl, J. A., Chin, Y. E., et al. (2011). FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKbeta pathways. Obesity 19, 483–491. doi: 10.1038/oby.2010.200
Kathiresan, S., Otvos, J. D., Sullivan, L. M., Keyes, M. J., Schaefer, E. J., Wilson, P. W., et al. (2006). Increased small low-density lipoprotein particle number: a prominent feature of the metabolic syndrome in the Framingham Heart Study. Circulation 113, 20–29. doi: 10.1161/CIRCULATIONAHA.105.567107
Kidani, Y., Elsaesser, H., Hock, M. B., Vergnes, L., Williams, K. J., Argus, J. P., et al. (2013). Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 14, 489–499. doi: 10.1038/ni.2570
Le Bourgeois, T., Strauss, L., Aksoylar, H. I., Daneshmandi, S., Seth, P., Patsoukis, N., et al. (2018). Targeting T cell metabolism for improvement of cancer immunotherapy. Front. Oncol. 8:237. doi: 10.3389/fonc.2018.00237
Leone, R. D., and Powell, J. D. (2020). Metabolism of immune cells in cancer. Nat. Rev. Cancer 20, 516–531. doi: 10.1038/s41568-020-0273-y
Li, X., Kumar, A., and Carmeliet, P. (2019a). Metabolic pathways fueling the endothelial cell drive. Annu. Rev. Physiol. 81, 483–503. doi: 10.1146/annurev-physiol-020518-114731
Li, X., Sun, X., and Carmeliet, P. (2019b). Hallmarks of endothelial cell metabolism in health and disease. Cell Metab 30, 414–433. doi: 10.1016/j.cmet.2019.08.011
Lopes-Coelho, F., Andre, S., Felix, A., and Serpa, J. (2018). Breast cancer metabolic cross-talk: fibroblasts are hubs and breast cancer cells are gatherers of lipids. Mol. Cell. Endocrinol. 462(Pt B), 93–106. doi: 10.1016/j.mce.2017.01.031
Lyu, J., Yang, E. J., Head, S. A., Ai, N., Zhang, B., Wu, C., et al. (2017). Pharmacological blockade of cholesterol trafficking by cepharanthine in endothelial cells suppresses angiogenesis and tumor growth. Cancer Lett. 409, 91–103. doi: 10.1016/j.canlet.2017.09.009
Ma, C., Kesarwala, A. H., Eggert, T., Medina-Echeverz, J., Kleiner, D. E., Jin, P., et al. (2016). NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 531, 253–257. doi: 10.1038/nature16969
Ma, X., Bi, E., Lu, Y., Su, P., Huang, C., Liu, L., et al. (2019). Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell. Metab. 30, 143.e5–156.e5. doi: 10.1016/j.cmet.2019.04.002
Malinarich, F., Duan, K., Hamid, R. A., Bijin, A., Lin, W. X., Poidinger, M., et al. (2015). High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 194, 5174–5186. doi: 10.4049/jimmunol.1303316
Miao, H., Ou, J., Peng, Y., Zhang, X., Chen, Y., Hao, L., et al. (2016). Macrophage ABHD5 promotes colorectal cancer growth by suppressing spermidine production by SRM. Nat. Commun. 7:11716. doi: 10.1038/ncomms11716
Michalek, R. D., Gerriets, V. A., Jacobs, S. R., Macintyre, A. N., MacIver, N. J., Mason, E. F., et al. (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303. doi: 10.4049/jimmunol.1003613
Miska, J., Lee-Chang, C., Rashidi, A., Muroski, M. E., Chang, A. L., Lopez-Rosas, A., et al. (2019). HIF-1alpha is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of tregs in glioblastoma. Cell Rep. 27, 226.e4–237.e4. doi: 10.1016/j.celrep.2019.03.029
Moessinger, C., Nilsson, I., Muhl, L., Zeitelhofer, M., Heller Sahlgren, B., Skogsberg, J., et al. (2020). VEGF-B signaling impairs endothelial glucose transcytosis by decreasing membrane cholesterol content. EMBO Rep. 21:e49343. doi: 10.15252/embr.201949343
Nieman, K. M., Romero, I. L., Van Houten, B., and Lengyel, E. (2013). Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim. Biophys. Acta 1831, 1533–1541. doi: 10.1016/j.bbalip.2013.02.010
Noghero, A., Perino, A., Seano, G., Saglio, E., Lo Sasso, G., Veglio, F., et al. (2012). Liver X receptor activation reduces angiogenesis by impairing lipid raft localization and signaling of vascular endothelial growth factor receptor-2. Arterioscler. Thromb. Vasc. Biol. 32, 2280–2288. doi: 10.1161/ATVBAHA.112.250621
Oh, Y. S., Bae, G. D., Baek, D. J., Park, E. Y., and Jun, H. S. (2018). Fatty acid-induced lipotoxicity in pancreatic beta-cells during development of type 2 diabetes. Front. Endocrinol. 9:384. doi: 10.3389/fendo.2018.00384
Oneyama, C., Iino, T., Saito, K., Suzuki, K., Ogawa, A., and Okada, M. (2009). Transforming potential of Src family kinases is limited by the cholesterol-enriched membrane microdomain. Mol. Cell. Biol. 29, 6462–6472. doi: 10.1128/MCB.00941-09
O’Sullivan, D., van der Windt, G. J., Huang, S. C., Curtis, J. D., Chang, C. H., Buck, M. D., et al. (2014). Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88. doi: 10.1016/j.immuni.2014.06.005
Ozdemir, B. C., Pentcheva-Hoang, T., Carstens, J. L., Zheng, X., Wu, C. C., Simpson, T. R., et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. doi: 10.1016/j.ccr.2014.04.005
Park, J., Lee, S. E., Hur, J., Hong, E. B., Choi, J. I., Yang, J. M., et al. (2015). M-CSF from cancer cells induces fatty acid synthase and PPARbeta/delta activation in tumor myeloid cells, leading to tumor progression. Cell Rep. 10, 1614–1625. doi: 10.1016/j.celrep.2015.02.024
Patella, F., Schug, Z. T., Persi, E., Neilson, L. J., Erami, Z., Avanzato, D., et al. (2015). Proteomics-based metabolic modeling reveals that fatty acid oxidation (FAO) controls endothelial cell (EC) permeability. Mol. Cell Proteomics 14, 621–634. doi: 10.1074/mcp.M114.045575
Patsoukis, N., Bardhan, K., Chatterjee, P., Sari, D., Liu, B., Bell, L. N., et al. (2015). PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6:6692. doi: 10.1038/ncomms7692
Platel, V., Faure, S., Corre, I., and Clere, N. (2019). Endothelial-to-mesenchymal transition (EndoMT): roles in tumorigenesis, metastatic extravasation and therapy resistance. J. Oncol. 2019:8361945. doi: 10.1155/2019/8361945
Pleiner, J., Schaller, G., Mittermayer, F., Bayerle-Eder, M., Roden, M., and Wolzt, M. (2002). FFA-induced endothelial dysfunction can be corrected by vitamin C. J. Clin. Endocrinol. Metab. 87, 2913–2917. doi: 10.1210/jcem.87.6.8596
Rao, E., Singh, P., Zhai, X., Li, Y., Zhu, G., Zhang, Y., et al. (2015). Inhibition of tumor growth by a newly-identified activator for epidermal fatty acid binding protein. Oncotarget 6, 7815–7827. doi: 10.18632/oncotarget.3485
Sahai, E., Astsaturov, I., Cukierman, E., DeNardo, D. G., Egeblad, M., Evans, R. M., et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186. doi: 10.1038/s41568-019-0238-1
Santi, A., Caselli, A., Ranaldi, F., Paoli, P., Mugnaioni, C., Michelucci, E., et al. (2015). Cancer associated fibroblasts transfer lipids and proteins to cancer cells through cargo vesicles supporting tumor growth. Biochim. Biophys. Acta 1853, 3211–3223. doi: 10.1016/j.bbamcr.2015.09.013
Schoors, S., Bruning, U., Missiaen, R., Queiroz, K. C., Borgers, G., Elia, I., et al. (2015). Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 520, 192–197. doi: 10.1038/nature14362
Schoors, S., De Bock, K., Cantelmo, A. R., Georgiadou, M., Ghesquiere, B., Cauwenberghs, S., et al. (2014). Partial and transient reduction of glycolysis by PFKFB3 blockade reduces pathological angiogenesis. Cell. Metab. 19, 37–48. doi: 10.1016/j.cmet.2013.11.008
Solomon, K. R., Pelton, K., Boucher, K., Joo, J., Tully, C., Zurakowski, D., et al. (2009). Ezetimibe is an inhibitor of tumor angiogenesis. Am. J. Pathol. 174, 1017–1026. doi: 10.2353/ajpath.2009.080551
Su, P., Wang, Q., Bi, E., Ma, X., Liu, L., Yang, M., et al. (2020). Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res. 80, 1438–1450. doi: 10.1158/0008-5472.CAN-19-2994
Swinburn, B. A., Sacks, G., Hall, K. D., McPherson, K., Finegood, D. T., Moodie, M. L., et al. (2011). The global obesity pandemic: shaped by global drivers and local environments. Lancet 378, 804–814. doi: 10.1016/S0140-6736(11)60813-1
Vangaveti, V., Shashidhar, V., Jarrod, G., Baune, B. T., and Kennedy, R. L. (2010). Free fatty acid receptors: emerging targets for treatment of diabetes and its complications. Ther. Adv. Endocrinol. Metab. 1, 165–175. doi: 10.1177/2042018810381066
Vats, D., Mukundan, L., Odegaard, J. I., Zhang, L., Smith, K. L., Morel, C. R., et al. (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell. Metab. 4, 13–24. doi: 10.1016/j.cmet.2006.05.011
Veglia, F., Tyurin, V. A., Mohammadyani, D., Blasi, M., Duperret, E. K., Donthireddy, L., et al. (2017). Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 8:2122. doi: 10.1038/s41467-017-02186-9
Villablanca, E. J., Raccosta, L., Zhou, D., Fontana, R., Maggioni, D., Negro, A., et al. (2010). Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med. 16, 98–105. doi: 10.1038/nm.2074
Wong, B. W., Wang, X., Zecchin, A., Thienpont, B., Cornelissen, I., Kalucka, J., et al. (2017). The role of fatty acid beta-oxidation in lymphangiogenesis. Nature 542, 49–54. doi: 10.1038/nature21028
Xiang, W., Shi, R., Kang, X., Zhang, X., Chen, P., Zhang, L., et al. (2018). Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 9:2574. doi: 10.1038/s41467-018-04999-8
Xiong, J., Kawagishi, H., Yan, Y., Liu, J., Wells, Q. S., Edmunds, L. R., et al. (2018). A metabolic basis for endothelial-to-mesenchymal transition. Mol. Cell. 69, 689.e7–698.e7. doi: 10.1016/j.molcel.2018.01.010
Yamagishi, S. I., Edelstein, D., Du, X. L., Kaneda, Y., Guzman, M., and Brownlee, M. (2001). Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J. Biol. Chem. 276, 25096–25100. doi: 10.1074/jbc.M007383200
Yang, W., Bai, Y., Xiong, Y., Zhang, J., Chen, S., Zheng, X., et al. (2016b). Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 531, 651–655. doi: 10.1038/nature17412
Yang, Y., Andersson, P., Hosaka, K., Zhang, Y., Cao, R., Iwamoto, H., et al. (2016a). The PDGF-BB-SOX7 axis-modulated IL-33 in pericytes and stromal cells promotes metastasis through tumour-associated macrophages. Nat. Commun. 7:11385. doi: 10.1038/ncomms11385
Zhang, D., Wang, Y., Shi, Z., Liu, J., Sun, P., Hou, X., et al. (2015). Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell. Rep. 10, 1335–1348. doi: 10.1016/j.celrep.2015.02.006
Zhang, Q., Wang, H., Mao, C., Sun, M., Dominah, G., Chen, L., et al. (2018). Fatty acid oxidation contributes to IL-1beta secretion in M2 macrophages and promotes macrophage-mediated tumor cell migration. Mol. Immunol. 94, 27–35. doi: 10.1016/j.molimm.2017.12.011
Keywords: cancer-associated host cells, metabolism, fatty acid oxidation, cholesterol metabolism, tumor microenvironment
Citation: Ye Y, Sun X and Lu Y (2020) Obesity-Related Fatty Acid and Cholesterol Metabolism in Cancer-Associated Host Cells. Front. Cell Dev. Biol. 8:600350. doi: 10.3389/fcell.2020.600350
Received: 29 August 2020; Accepted: 28 September 2020;
Published: 27 November 2020.
Edited by:
Yunlong Yang, Fudan University, ChinaReviewed by:
Hideki Iwamoto, Kurume University School of Medicine, JapanCopyright © 2020 Ye, Sun and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongtian Lu, bHV5b25ndGlhbkAyNjMubmV0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.