 Hyunsu Jung
Hyunsu Jung Su Yeon Kim
Su Yeon Kim Fatma Sema Canbakis Cecen
Fatma Sema Canbakis Cecen Yongcheol Cho
Yongcheol Cho Seok-Kyu Kwon
Seok-Kyu Kwon
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 18 December 2020
Sec. Molecular and Cellular Pathology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.599792
This article is part of the Research Topic Molecular Links between Mitochondrial Damage and Parkinson's Disease and Related Disorders View all 12 articles
Calcium ions (Ca2+) play critical roles in neuronal processes, such as signaling pathway activation, transcriptional regulation, and synaptic transmission initiation. Therefore, the regulation of Ca2+ homeostasis is one of the most important processes underlying the basic cellular viability and function of the neuron. Multiple components, including intracellular organelles and plasma membrane Ca2+-ATPase, are involved in neuronal Ca2+ control, and recent studies have focused on investigating the roles of mitochondria in synaptic function. Numerous mitochondrial Ca2+ regulatory proteins have been identified in the past decade, with studies demonstrating the tissue- or cell-type-specific function of each component. The mitochondrial calcium uniporter and its binding subunits are major inner mitochondrial membrane proteins contributing to mitochondrial Ca2+ uptake, whereas the mitochondrial Na+/Ca2+ exchanger (NCLX) and mitochondrial permeability transition pore (mPTP) are well-studied proteins involved in Ca2+ extrusion. The level of cytosolic Ca2+ and the resulting characteristics of synaptic vesicle release properties are controlled via mitochondrial Ca2+ uptake and release at presynaptic sites, while in dendrites, mitochondrial Ca2+ regulation affects synaptic plasticity. During brain aging and the progress of neurodegenerative disease, mitochondrial Ca2+ mishandling has been observed using various techniques, including live imaging of Ca2+ dynamics. Furthermore, Ca2+ dysregulation not only disrupts synaptic transmission but also causes neuronal cell death. Therefore, understanding the detailed pathophysiological mechanisms affecting the recently discovered mitochondrial Ca2+ regulatory machineries will help to identify novel therapeutic targets. Here, we discuss current research into mitochondrial Ca2+ regulatory machineries and how mitochondrial Ca2+ dysregulation contributes to brain aging and neurodegenerative disease.
Mitochondria affect cellular functions via their roles in ATP production, lipid synthesis, reactive oxygen species (ROS) generation, and Ca2+ regulation. Recent studies of mitochondria-dependent Ca2+ handling have revealed the molecular identities of Ca2+-control components, including the mitochondrial calcium uniporter (MCU) and its auxiliary subunits (Mammucari et al., 2017). Furthermore, the development of enhanced Ca2+ sensors has enabled subcellular investigations of how mitochondria contribute to synaptic transmission. In addition, mitochondrial matrix-targeting sequence-tagged genetically encoded calcium indicators (GECIs) have allowed direct monitoring of mitochondrial Ca2+ dynamics (Kwon et al., 2016a).
In aged animals and humans, mitochondrial functional impairment is a key hallmark of brain aging (Grimm and Eckert, 2017; Mattson and Arumugam, 2018; Muller et al., 2018). Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and other aging-related neurodegenerative diseases also show mitochondrial defects. However, the detailed molecular mechanisms underlying these defects, particularly those related to mitochondrial Ca2+, have not yet been studied in depth.
Here, we describe mitochondrial Ca2+-related features and unveiled mitochondrial Ca2+ regulatory molecular mechanisms of brain aging and neurodegenerative disease models, and discuss experimental methods and controversies within the current research.
Ca2+ ions enter neurons through ionotropic glutamate receptors and voltage-dependent Ca2+ channels, with the imported Ca2+ then affecting various cellular processes, including the modulation of synaptic strength and Ca2+-mediated cell death (Ghosh and Greenberg, 1995). At the presynapse, Ca2+ triggers synaptic vesicle exocytosis, and residual Ca2+ alters synaptic release properties toward asynchronous release. Moreover, short-term synaptic plasticity can be controlled by presynaptic Ca2+ dynamics, while in dendrites, Ca2+ influences various signaling cascades involved in long-term synaptic plasticity and gene transcription (Hayashi and Majewska, 2005; Higley and Sabatini, 2008; Sudhof, 2012; Kaeser and Regehr, 2014; Kwon et al., 2016a).
Cytosolic Ca2+ is controlled by plasma membrane Ca2+ pumps and intracellular organelles, including mitochondria and the endoplasmic reticulum (ER). The ER imports Ca2+ through sarco/endoplasmic Ca2+ ATPase and releases it via ryanodine receptors or IP3 receptors (IP3Rs) (Verkhratsky, 2005). ER is partially tethered to mitochondria by mitochondria-associated membrane (MAM) or mitochondria-ER contact sites (MERCs) proteins such as Mitofusin 2, Sigma-1 receptor, vesicle-associated membrane protein-associated protein B (VAPB)/protein tyrosine phosphatase-interacting protein 51 (PTPIP51), IP3R/glucose-regulated protein (Grp75)/Voltage-dependent anion-selective channel 1 (VDAC1), and PDZ domain containing 8 (PDZD8), enabling ER-to-mitochondria Ca2+ transfer (Rapizzi et al., 2002; Szabadkai et al., 2006; Hayashi and Su, 2007; De Vos et al., 2012; Hirabayashi et al., 2017). The involvement of MAM in neurodegenerative diseases is a major topic in the field, but previous reviews wonderfully covered this scope (Paillusson et al., 2016; Liu and Zhu, 2017; Area-Gomez et al., 2018; Bernard-Marissal et al., 2018; Gomez-Suaga et al., 2018; Lau et al., 2018). Therefore, only a part of studies using direct observation of mitochondrial Ca2+ dynamics will be discussed here.
Several Ca2+ regulatory proteins have been identified in the outer and inner mitochondrial membranes. VDACs located in the outer mitochondrial membrane (OMM) are responsible for importing various ions and metabolites (Colombini, 2016). In the inner mitochondrial membrane, MCU mediates mitochondrial membrane potential-dependent Ca2+ influx into the mitochondrial matrix (Kirichok et al., 2004; Baughman et al., 2011; De Stefani et al., 2011). Reduced MCU-dependent Ca2+ uptake at presynaptic sites elevates cytosolic Ca2+ and alters short-term synaptic plasticity and synchronous release (Kang et al., 2008; Kwon et al., 2016b). In addition, a recent study showed upregulation of mitochondrial fission and dendritic mitochondrial Ca2+ transients following chemically induced long-term potentiation (LTP), with the interference of fission impairing mitochondrial Ca2+ uptake and LTP (Divakaruni et al., 2018).
Mitochondrial calcium uniporter forms complexes with other proteins, which regulate its opening dynamics (Mammucari et al., 2017; Pallafacchina et al., 2018). Mitochondrial calcium uptake protein 1/2/3 (MICU1/2/3) are the first MCU binding proteins to be characterized. MICU1 and MICU2 serve as molecular gatekeepers that negatively regulate MCU under low Ca2+ but positively regulate it under high cytosolic Ca2+ (Csordas et al., 2013; Patron et al., 2014; Liu et al., 2016). MICU3 is abundant in the brain and enhances mitochondrial Ca2+ uptake, with silencing of MICU3 in cortical neurons causing a reduction in stimulation-induced mitochondrial Ca2+ levels (Patron et al., 2019). Furthermore, the presynaptic MICU3-dependent increase in Ca2+ sensitivity allows MCU to open without Ca2+ release from ER and facilitates Ca2+-mediated mitochondrial ATP production and synaptic vesicle endocytosis (Ashrafi et al., 2020).
Essential MCU regulator (EMRE) is another MCU complex protein that bridges MCU and MICU1 and regulates the level of Ca2+ in the mitochondrial matrix. In addition, recent unveiled structural features of EMRE show that it triggers dimerization of MCU-EMRE complex and controls pore opening (Sancak et al., 2013; Vais et al., 2016; Wang et al., 2019). The MCU paralog MCUb exerts an inhibitory effect on MCU, with overexpression of MCUb completely abolishing MCU currents (Raffaello et al., 2013; Mammucari et al., 2017). Mitochondrial calcium uniporter regulator 1 (MCUR1) is a scaffold factor, whose absence results in the failure of MCU to form a complex (Tomar et al., 2016; Supplementary Figure 1).
The mitochondrial Na+/Ca2+ exchanger (NCLX) is one of the primary Ca2+ efflux units in mitochondria (Palty et al., 2010). Genetic ablation of NCLX increases Ca2+ retention in mitochondria and causes mitochondria-dependent cell death (Luongo et al., 2017). Also, H+/Ca2+ exchanger is considered as a Ca2+ efflux component, although its molecular identity is arguable (Jiang et al., 2009; De Marchi et al., 2014). Additional Ca2+ release-associated protein, mitochondrial permeability transition pore (mPTP), is activated by Ca2+ overload and ROS, leading to apoptosis or necrosis (Giorgi et al., 2018). This pore has been mainly studied under pathological conditions, including aging and neurodegenerative diseases, and the roles of core components including ATP synthase, cyclophilin D (CyPD), and the adenine nucleotide translocators (ANTs), are recently updated, although there are debates (Kokoszka et al., 2004; Bonora et al., 2013; Alavian et al., 2014; Karch and Molkentin, 2014; Raffaello et al., 2016; He et al., 2017; Rottenberg and Hoek, 2017; Zhou et al., 2017; Bernardi, 2018; Muller et al., 2018; Carroll et al., 2019; Karch et al., 2019).
Dysregulation of Ca2+ homeostasis is one of the hallmarks of brain aging (Mattson and Arumugam, 2018), with impaired Ca2+ control in aged brains resulting in various cellular and physiological deficits. Hippocampal CA1 pyramidal neurons in aged animals show elevated Ca2+ currents, as confirmed by Ca2+ imaging using multiple Ca2+ fluorophores (Landfield and Pitler, 1984; Disterhoft et al., 1996; Verkhratsky et al., 1998; Thibault et al., 2001; Lessmann et al., 2003). Age-associated Ca2+ changes have also been observed in other brain regions and in peripheral nerves (Verkhratsky et al., 1998).
Age-dependent dysregulation of Ca2+ results from various molecular changes, including increased voltage-gated Ca2+ channel expression, reduced Ca2+ binding protein expression, and impaired mitochondrial and ER Ca2+ handling (Mattson and Arumugam, 2018). Ca2+ isotope uptake by isolated synaptosomal mitochondria is significantly reduced in aged rat brains (Leslie et al., 1985). In addition, cytosolic Ca2+ dynamics in aged rodent brain slices or acutely dissociated neurons have been monitored using chemical Ca2+ dyes, such as Fura-2. Use of this dye in combination with a mitochondrial membrane potential indicator or mitochondrial uncoupler has revealed that the potential is disrupted in aged neurons, resulting in a decrease in mitochondrial Ca2+ uptake and an elevation of cytosolic Ca2+ upon stimulation (Xiong et al., 2002; Murchison et al., 2004). Mitochondrial Ca2+ buffering is also reduced in aged Rhesus monkeys, shown using isolated putamen mitochondria (Pandya et al., 2015).
Ca2+ dysregulation is a common feature of several neurodegenerative diseases, including AD and PD (Beal, 1998; Zundorf and Reiser, 2011; Liao et al., 2017; Pchitskaya et al., 2018). Disruption of Ca2+ homeostasis and mitochondrial Ca2+ overload have been observed before pathological features of these diseases appear, highlighting the importance of neuronal Ca2+ regulation (Lesne et al., 2008; Surmeier et al., 2017).
AD is characterized by the accumulation of amyloid beta (Aβ) peptide, which is produced by abnormal proteolytic cleavage of amyloid precursor protein (APP), as well as by the formation of neurofibrillary tangles composed of tau protein and by neuronal loss, leading to learning and memory impairment (Oddo et al., 2003; Mattson et al., 2008). Mutations in APP or in the γ-secretase components presenilin1 and 2 (PSEN1/2) are the most well-characterized alterations contributing to dominantly inherited familial AD (FAD) (Chen et al., 2017; Muller et al., 2018).
Increased Aβ expression in FAD models or exogenous application of Aβ leads to elevated cytosolic Ca2+. In the past two decades, multiple underlying mechanisms have been suggested, including mitochondrial Ca2+ dysregulation (Du et al., 2008; Supnet and Bezprozvanny, 2010; Jadiya et al., 2019; Calvo-Rodriguez et al., 2020). In-vivo Ca2+ imaging with mitochondria-targeted Förster resonance energy transfer (FRET)-based GECI has directly demonstrated an Aβ-dependent mitochondrial Ca2+ increase in mouse cortex. This upregulation was observed prior to neuronal death, with blockade of MCU restoring the mitochondrial Ca2+ level in the APP/PS1 mutant mouse model (Calvo-Rodriguez et al., 2020).
Brain levels of NCLX protein are significantly reduced in human AD patients and in 3xTg-AD triple mutant mice (expressing mutations in APP, presenilin 1, and tau). Mutant APP-expressing-N2a cells also show decreased NCLX expression, resulting in impaired mitochondrial Ca2+ extrusion, consistent with the increased mitochondrial Ca2+ transients revealed by the mitochondria-localized GECI mito-R-GECO1. Alleviation of mitochondrial Ca2+ overload by NCLX expression in 3xTg-AD mice rescues cognitive decline and AD-related pathology (Jadiya et al., 2019). Another Ca2+ extrusion-related molecule, CypD, which is part of mPTP, is known to interact with mitochondrially transported Aβ. Inhibition or genetic ablation of CypD protects neurons from Aβ-triggered cell death and rescues impaired LTP and deficits in spatial learning and memory (Du et al., 2008).
Mitochondrial Ca2+ overload in AD can also result from impaired ER–mitochondria communication. Previous studies suggest that ER–mitochondria contacts are increased in AD models, promoting Ca2+ transfer to mitochondria (Zampese et al., 2011; Area-Gomez et al., 2012; Hedskog et al., 2013; Calvo-Rodriguez et al., 2019). ER-to-mitochondria Ca2+ transfer has been monitored using the mitochondria-localized chemical dye Rhod-5N or a mitochondrial matrix- or OMM-targeted protein Ca2+ sensors (Zampese et al., 2011; Hedskog et al., 2013; Calvo-Rodriguez et al., 2019). Opposite to these, some studies using electron microscopy (EM) and fluorescent imaging have reported reduced ER-mitochondria contacts in AD animal models and patients (Sepulveda-Falla et al., 2014; Martino Adami et al., 2019; Lau et al., 2020; Figure 1).
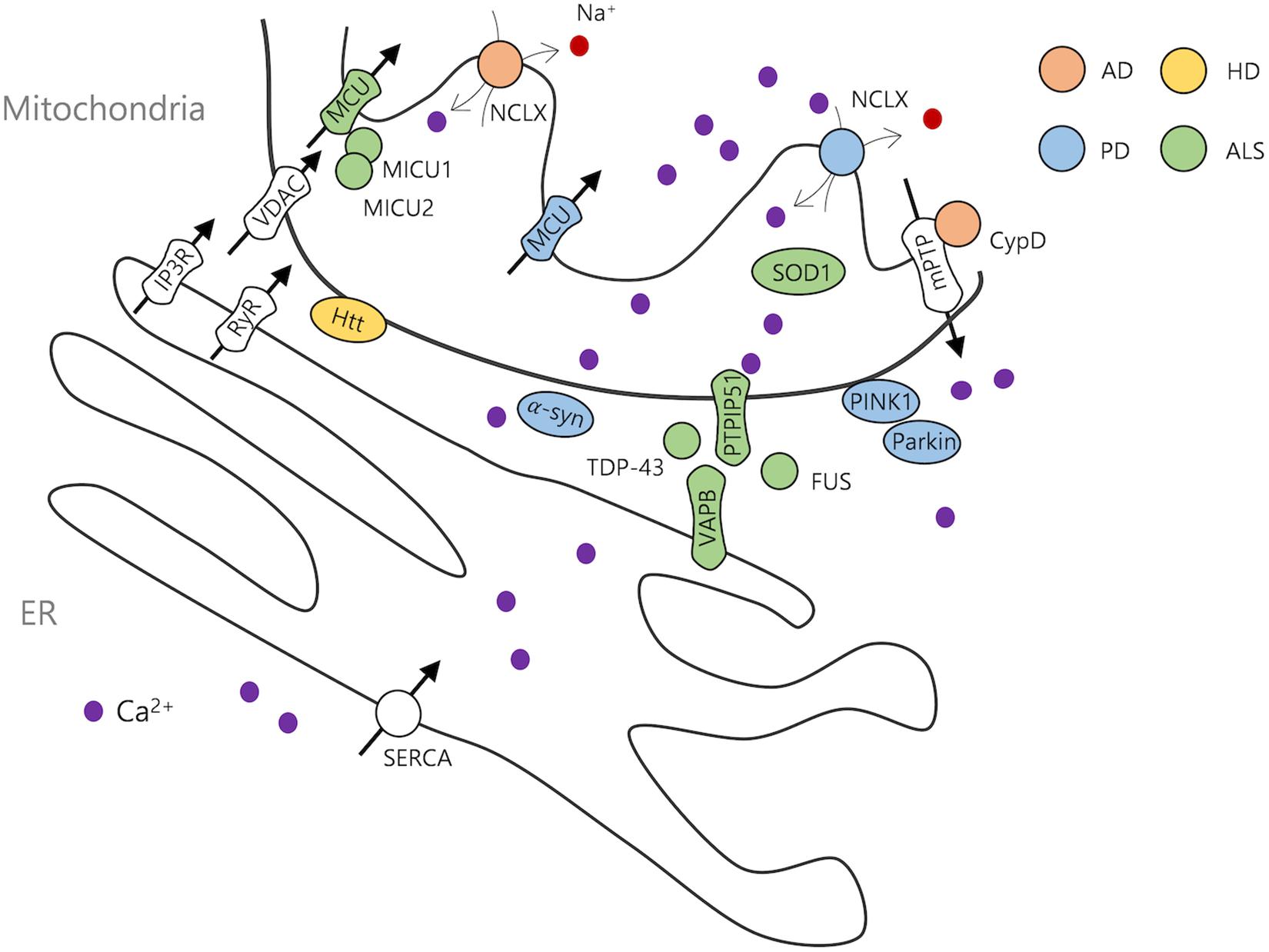
Figure 1. Neurodegenerative disease-related ER and mitochondrial proteins. AD, Alzheimer’s disease; PD, Parkinson’s disease; HD, Huntington’s disease; ALS, amyotrophic lateral sclerosis.
Apolipoprotein E4 (ApoE4) is the major risk factor for sporadic AD and it can increase ER–mitochondria contacts (Tambini et al., 2016; Orr et al., 2019). ApoE4-expressing cells show higher cytosolic and mitochondrial Ca2+, and given that ApoE4 expression alters neuronal MAM-tethering protein composition, this could explain enhanced MAM activity in sporadic AD (Orr et al., 2019; Table 1).
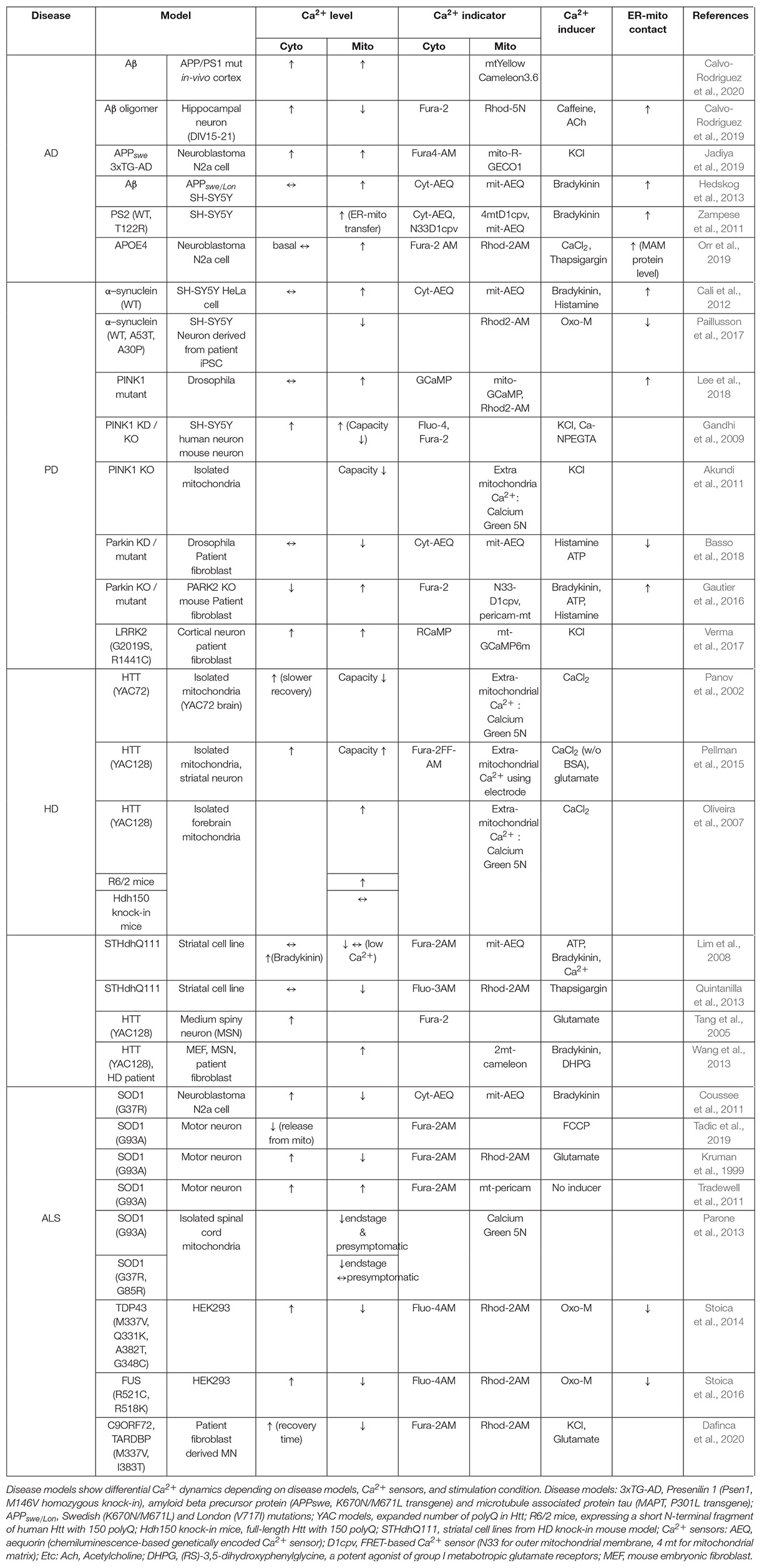
Table 1. Ca2+ dynamics in neurodegenerative disease models.
Furthermore, mitochondria contribute to the presynaptic defects observed in AD. Increased insulin-like growth factor-1 receptor (IGF-1R) levels are found in AD patient and mouse model brain samples (Moloney et al., 2010; Zhang et al., 2013). IGF-1R regulates synaptic transmission by modulating presynaptic mitochondrial Ca2+ buffering and ATP production, as measured using the mitochondria-targeted GECI and the ATP sensor, although detailed mechanisms are not known. Interestingly, inhibition of IGF-1R reverses altered synaptic release in an APP/PS1 mutant model (Gazit et al., 2016).
Parkinson’s disease is characterized at the cellular level by dopaminergic neuron loss in the substantia nigra. Several genes contributing to PD pathogenesis have been identified, including α-synuclein, leucine-rich repeat kinase 2 (LRRK2), PTEN-induced kinase 1 (PINK1), and parkin (Abou-Sleiman et al., 2006; Ferreira and Massano, 2017). Ca2+ regulation is especially important for dopaminergic neurons because of their steady and autonomous pacemaker function (Chan et al., 2007; Guzman et al., 2010).
Mutation of α-synuclein and its aggregation into Lewy bodies are well-known pathological processes in PD. Interestingly, α-synuclein is localized to ER, mitochondria, and MAM and contributes to regulating ER–mitochondria communication (Li et al., 2007; Cali et al., 2012; Guardia-Laguarta et al., 2014). In one study, overexpression of WT or mutant α-synuclein in HeLa and SH-SY5Y cells was found to increase mitochondrial Ca2+ by enhancing ER–mitochondria interaction (Cali et al., 2012, 2019). However, another study using mutant α-synuclein-overexpressing cells produced conflicting results in terms of ER–mitochondria interactions (Guardia-Laguarta et al., 2014). Furthermore, overexpression of α-synuclein (WT/mutant) in SH-SY5Y cells disturbed the interaction between VAPB and PTPIP51, and this was accompanied by reduced ER-to-mitochondria Ca2+ transfer (Paillusson et al., 2017). High dose of WT/mutant α-synuclein can form the aggregates, and this in turn reduces ER-mitochondria contacts (Cali et al., 2012, 2019). Therefore, the dose-dependent effect could be the possible cause of discrepancies (Figure 1).
PINK1, a mitochondrial serine/threonine kinase, and parkin, an E3 ubiquitin ligase, are proposed to underlie mitochondrial quality control, with mutations in either gene highly related to PD (Narendra and Youle, 2011; Pickrell and Youle, 2015). Dopaminergic neuron-specific mitochondrial Ca2+ imaging with mito-GCaMP, a mitochondria-targeted GECI, in Drosophila PD models revealed elevated mitochondrial Ca2+. Pharmacological and genetic inhibition of IP3R and MCU restore mitochondrial Ca2+ and dopaminergic neuron loss (Lee et al., 2018). In contrast, due to negative regulation of NCLX, PINK1-deficient cortical neurons show reduced mitochondrial Ca2+ capacity and higher mitochondrial Ca2+ accumulation (Gandhi et al., 2009). Similarly, purified mitochondria from PINK1–/– mouse brain show a significantly decreased mitochondrial Ca2+ buffering capacity (Akundi et al., 2011).
Primary fibroblasts from PD patients with parkin mutations had reduced mitochondrial Ca2+ uptake due to loosened ER–mitochondria connectivity (Basso et al., 2018). Contrary to these results, other studies found that fibroblasts from Parkin-deficient mice or from PD patients with a parkin mutation show increased ER–mitochondria contacts (Gautier et al., 2016). In addition, OMM- and matrix-targeted Ca2+ sensors revealed higher ER-mitochondrial Ca2+ transfer (Gautier et al., 2016).
Dendrite shortening in LRRK2 mutant models is a well-known change related to mitochondrial dysfunction (Cherra et al., 2013). Abnormal mitochondrial function results from the stimulation-induced increase in mitochondrial Ca2+, which is accompanied by upregulated MCU expression. Interestingly, chemical inhibition or knockdown of MCU successfully restores neurite length (Verma et al., 2017; Table 1).
HD is a hereditary neurodegenerative disease characterized by involuntary movements, psychiatric abnormalities, and dementia. The major pathogenic features of the disease are progressive striatal neuronal loss, particularly of GABAergic medium spiny neurons, and extension of the N-terminal polyglutamine (polyQ) stretch of the huntingtin protein (Walker, 2007; Brustovetsky, 2016).
In the mutant huntingtin transgenic mouse brain, polyQ-stretched huntingtin is associated with the mitochondrial membrane (Choo et al., 2004). Interestingly, deficits in mitochondrial Ca2+ buffering have been observed after applying Calcium Green-5N to isolated mitochondria from HD mouse brain to monitor extramitochondrial Ca2+ (Panov et al., 2002). Live imaging of HD model striatal cell lines with mitochondria-targeted aequorin or Rhod-2AM indicates that mitochondria are able to handle a low Ca2+ challenge, but that a higher Ca2+ concentration disrupts their buffering ability (Lim et al., 2008; Quintanilla et al., 2013).
However, other studies demonstrated increased Ca2+ uptake capacity in isolated mitochondria from HD model mouse forebrains (Oliveira et al., 2007; Pellman et al., 2015). Furthermore, mitochondrial Ca2+ influx is higher in primary medium spiny neurons of HD model mice (Tang et al., 2005) and in fibroblasts from HD patients (Wang et al., 2013), which leads to cell death or mitochondrial DNA damage. Interestingly, this excitotoxicity is prevented by MCU or mPTP inhibition (Tang et al., 2005; Figure 1 and Table 1).
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by progressive muscle paralysis resulting from the degeneration of upper and lower motor neurons. ALS exhibits multiple pathogenic features, including oxidative stress, mitochondrial dysfunction, and protein dysfunction of 43-kDa transactivating response region binding protein (TDP-43) and cytoplasmic Cu2+/Zn2+-superoxide dismutase 1 (SOD1) (Ferraiuolo et al., 2011; Lee et al., 2011; Tadic et al., 2014).
Monitoring with mitochondria-/ER-targeted ratiometric sensor proteins and Fura-2 identified elevated levels of mitochondrial, ER, and cytosolic Ca2+ in the motor neurons of ALS mutant transgenic mice (SOD1G93A) (Tradewell et al., 2011). However, in another study, Rhod-2- and Fura-2-based Ca2+ imaging showed significantly decreased mitochondrial Ca2+ uptake and increased cytosolic Ca2+ in SOD1G93A mice motor neurons (Kruman et al., 1999). Decreased mitochondrial Ca2+ buffering capacity in SOD1G93A-expressing mice can be restored by CypD deletion, which regulates mPTP opening, suppressing cell death (Parone et al., 2013). Other ALS mutant (SOD1G37R-overexpressing) N2a cells also show significantly reduced mitochondrial Ca2+ uptake and elevated cytosolic Ca2+ (Coussee et al., 2011).
Multiple studies suggest that specific molecular processes underlie ALS progression, but their findings are contentious. Hypoglossal motor neurons of SOD1G93A-transgenic mice show upregulated MCU and MICU1 expression at the end stage of the disease (P115–140) (Fuchs et al., 2013). However, in symptomatic cervical spinal cord motor neurons, MCU level is significantly decreased (Tadic et al., 2019).
Other ALS-associated genes have also been identified, including TDP-43, fused in sarcoma (FUS), VAPB, and expanded hexanucleotide repeats in intron 1 of the encoding chromosome 9 open reading frame 72 (C9ORF72). The OMM protein PTPIP51 is a known binding partner of the ER protein VAPB. VAPB–PTPIP51 interaction in mouse motor neurons is disrupted by overexpression of ALS mutant or wild-type TDP-43 and FUS, also leading to disruption of Ca2+ homeostasis in HEK293T cells (Stoica et al., 2014, 2016). ALS patient fibroblast-derived motor neurons with C9ORF72 and TDP-43 mutations show delayed clearance of cytosolic Ca2+, lower mitochondrial buffering capacity, and imbalance of MICU1 and MICU2 expression (Dafinca et al., 2020) (Figure 1 and Table 1).
In summary, brain aging and neurodegenerative diseases involve mitochondria- and ER-mitochondria contact-related Ca2+ regulatory defects. These alterations have been revealed using various experimental methods, including electrophysiological recording and live imaging. However, large part of in-vitro studies for neurodegenerative diseases have performed using cell lines and patient-derived fibroblasts rather than neurons. In addition, Ca2+ signals were triggered by various chemicals, and most conditions are not neurophysiological (Table 1). Depending on tissues and brain regions, mitochondrial Ca2+ uptake capacity and regulatory components can be different (Markus et al., 2016; Vecellio Reane et al., 2016; Patron et al., 2019). Therefore, application of recently advanced genetically encoded Ca2+ sensors for specific organelles will provide more precise neuron type-specific data (Kwon et al., 2016a).
Finally, mitochondrial Ca2+ regulatory molecular mechanisms have recently been revealed, with some studies showing that the composition of the MCU complex can change during disease progress and differ between mutant types (Table 1). Thus, revealing the detailed pathophysiological mechanisms of mitochondrial defects at the molecular level could lead to novel therapeutic targets that are specific for particular mutations and disease stages.
HJ, SYK, FC, and S-KK wrote the manuscript and created the figures and table. S-KK and YC provided guidance and edited the manuscript. All authors contributed to the article and approved the submitted version.
S-KK was supported by grants from the Brain Research Program and the Bio and Medical Technology Development Program of the National Research Foundation funded by the Korean Government (nos. NRF2017M3C7A1043838, NRF2019M3E5D2 A01063794, NRF2019M3F3A1A02072175, and NRF2020R1C1C1 006386), and the Korea Institute of Science and Technology Institutional Program (no. 2E30070). YC was supported by a Korea University Grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
A draft of this manuscript was edited by the Doran Amos, Ph.D., and Julia Slone-Murphy, Ph.D., ELS, of NeuroEdit Ltd.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.599792/full#supplementary-material
Supplementary Figure 1 | MCU complex machinery. When the cytosolic Ca2+ level is low, MICU1/MICU2 heterodimer keeps MCU as a closed-form, whereas in high Ca2+ concentration, their conformational change helps MCU allow Ca2+ influx toward mitochondrial matrix. Otherwise, MICU1/MICU3 heterodimer has less gatekeeping function than MICU1/MICU2 dimer, which leads to opening the MCU complex in lower Ca2+ condition.
Abou-Sleiman, P. M., Muqit, M. M., and Wood, N. W. (2006). Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat. Rev. Neurosci. 7, 207–219. doi: 10.1038/nrn1868
Akundi, R. S., Huang, Z., Eason, J., Pandya, J. D., Zhi, L., Cass, W. A., et al. (2011). Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One 6:e16038. doi: 10.1371/journal.pone.0016038
Alavian, K. N., Beutner, G., Lazrove, E., Sacchetti, S., Park, H. A., Licznerski, P., et al. (2014). An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 111, 10580–10585. doi: 10.1073/pnas.1401591111
Area-Gomez, E., de Groof, A., Bonilla, E., Montesinos, J., Tanji, K., Boldogh, I., et al. (2018). A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 9:335. doi: 10.1038/s41419-017-0215-0
Area-Gomez, E., Del Carmen Lara Castillo, M., Tambini, M. D., Guardia-Laguarta, C., de Groof, A. J., Madra, M., et al. (2012). Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 31, 4106–4123. doi: 10.1038/emboj.2012.202
Ashrafi, G., de Juan-Sanz, J., Farrell, R. J., and Ryan, T. A. (2020). Molecular tuning of the axonal mitochondrial Ca(2+) uniporter ensures metabolic flexibility of neurotransmission. Neuron 105, 678–687.e5. doi: 10.1016/j.neuron.2019.11.020
Basso, V., Marchesan, E., Peggion, C., Chakraborty, J., von Stockum, S., Giacomello, M., et al. (2018). Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 138, 43–56. doi: 10.1016/j.phrs.2018.09.006
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Beal, M. F. (1998). Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis. Ann. Neurol. 44(Suppl. 1), S110–S114. doi: 10.1002/ana.410440716
Bernardi, P. (2018). Why F-ATP synthase remains a strong candidate as the mitochondrial permeability transition pore. Front. Physiol. 9:1543. doi: 10.3389/fphys.2018.01543
Bernard-Marissal, N., Chrast, R., and Schneider, B. L. (2018). Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: a broken relationship? Cell Death Dis. 9:333. doi: 10.1038/s41419-017-0125-1
Bonora, M., Bononi, A., De Marchi, E., Giorgi, C., Lebiedzinska, M., Marchi, S., et al. (2013). Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12, 674–683. doi: 10.4161/cc.23599
Brustovetsky, N. (2016). Mutant huntingtin and elusive defects in oxidative metabolism and mitochondrial calcium handling. Mol. Neurobiol. 53, 2944–2953. doi: 10.1007/s12035-015-9188-0
Cali, T., Ottolini, D., Negro, A., and Brini, M. (2012). alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 287, 17914–17929. doi: 10.1074/jbc.M111.302794
Cali, T., Ottolini, D., Vicario, M., Catoni, C., Vallese, F., Cieri, D., et al. (2019). splitGFP technology reveals dose-dependent ER-mitochondria interface modulation by alpha-Synuclein A53T and A30P mutants. Cells 8:1072. doi: 10.3390/cells8091072
Calvo-Rodriguez, M., Hernando-Perez, E., Nunez, L., and Villalobos, C. (2019). Amyloid beta oligomers increase ER-Mitochondria Ca(2+) cross talk in young hippocampal neurons and exacerbate aging-induced intracellular Ca(2+) remodeling. Front. Cell Neurosci. 13:22. doi: 10.3389/fncel.2019.00022
Calvo-Rodriguez, M., Hou, S. S., Snyder, A. C., Kharitonova, E. K., Russ, A. N., Das, S., et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 11:2146. doi: 10.1038/s41467-020-16074-2
Carroll, J., He, J., Ding, S., Fearnley, I. M., and Walker, J. E. (2019). Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 116, 12816–12821. doi: 10.1073/pnas.1904005116
Chan, C. S., Guzman, J. N., Ilijic, E., Mercer, J. N., Rick, C., Tkatch, T., et al. (2007). ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 447, 1081–1086. doi: 10.1038/nature05865
Chen, G. F., Xu, T. H., Yan, Y., Zhou, Y. R., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Cherra, S. J. III, Steer, E., Gusdon, A. M., Kiselyov, K., and Chu, C. T. (2013). Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am. J. Pathol. 182, 474–484. doi: 10.1016/j.ajpath.2012.10.027
Choo, Y. S., Johnson, G. V., MacDonald, M., Detloff, P. J., and Lesort, M. (2004). Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 13, 1407–1420. doi: 10.1093/hmg/ddh162
Colombini, M. (2016). The VDAC channel: molecular basis for selectivity. Biochim. Biophys. Acta 1863, 2498–2502. doi: 10.1016/j.bbamcr.2016.01.019
Coussee, E., De Smet, P., Bogaert, E., Elens, I., Van Damme, P., Willems, P., et al. (2011). G37R SOD1 mutant alters mitochondrial complex I activity, Ca(2+) uptake and ATP production. Cell Calcium 49, 217–225. doi: 10.1016/j.ceca.2011.02.004
Csordas, G., Golenar, T., Seifert, E. L., Kamer, K. J., Sancak, Y., Perocchi, F., et al. (2013). MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2)(+) uniporter. Cell Metab. 17, 976–987. doi: 10.1016/j.cmet.2013.04.020
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 14, 892–908. doi: 10.1016/j.stemcr.2020.03.023
De Marchi, U., Santo-Domingo, J., Castelbou, C., Sekler, I., Wiederkehr, A., and Demaurex, N. (2014). NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J. Biol. Chem. 289, 20377–20385. doi: 10.1074/jbc.M113.540898
De Stefani, D., Raffaello, A., Teardo, E., Szabo, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
De Vos, K. J., Morotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Disterhoft, J. F., Thompson, L. T., Moyer, J. R. Jr., and Mogul, D. J. (1996). Calcium-dependent afterhyperpolarization and learning in young and aging hippocampus. Life Sci. 59, 413–420. doi: 10.1016/0024-3205(96)00320-7
Divakaruni, S. S., Van Dyke, A. M., Chandra, R., LeGates, T. A., Contreras, M., Dharmasri, P. A., et al. (2018). Long-term potentiation requires a rapid burst of dendritic mitochondrial fission during induction. Neuron 100, 860–875e7. doi: 10.1016/j.neuron.2018.09.025
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M., and Shaw, P. J. (2011). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 7, 616–630. doi: 10.1038/nrneurol.2011.152
Ferreira, M., and Massano, J. (2017). An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol. Scand. 135, 273–284. doi: 10.1111/ane.12616
Fuchs, A., Kutterer, S., Muhling, T., Duda, J., Schutz, B., Liss, B., et al. (2013). Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Physiol. 591, 2723–2745. doi: 10.1113/jphysiol.2012.247981
Gandhi, S., Wood-Kaczmar, A., Yao, Z., Plun-Favreau, H., Deas, E., Klupsch, K., et al. (2009). PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 33, 627–638. doi: 10.1016/j.molcel.2009.02.013
Gautier, C. A., Erpapazoglou, Z., Mouton-Liger, F., Muriel, M. P., Cormier, F., Bigou, S., et al. (2016). The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 25, 2972–2984. doi: 10.1093/hmg/ddw148
Gazit, N., Vertkin, I., Shapira, I., Helm, M., Slomowitz, E., Sheiba, M., et al. (2016). IGF-1 receptor differentially regulates spontaneous and evoked transmission via mitochondria at hippocampal synapses. Neuron 89, 583–597. doi: 10.1016/j.neuron.2015.12.034
Ghosh, A., and Greenberg, M. E. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science 268, 239–247. doi: 10.1126/science.7716515
Giorgi, C., Marchi, S., and Pinton, P. (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730. doi: 10.1038/s41580-018-0052-8
Gomez-Suaga, P., Bravo-San Pedro, J. M., Gonzalez-Polo, R. A., Fuentes, J. M., and Niso-Santano, M. (2018). ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 9:337. doi: 10.1038/s41419-017-0079-3
Grimm, A., and Eckert, A. (2017). Brain aging and neurodegeneration: from a mitochondrial point of view. J. Neurochem. 143, 418–431. doi: 10.1111/jnc.14037
Guardia-Laguarta, C., Area-Gomez, E., Rub, C., Liu, Y., Magrane, J., Becker, D., et al. (2014). alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Guzman, J. N., Sanchez-Padilla, J., Wokosin, D., Kondapalli, J., Ilijic, E., Schumacker, P. T., et al. (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468, 696–700. doi: 10.1038/nature09536
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
Hayashi, Y., and Majewska, A. K. (2005). Dendritic spine geometry: functional implication and regulation. Neuron 46, 529–532. doi: 10.1016/j.neuron.2005.05.006
He, J., Carroll, J., Ding, S., Fearnley, I. M., and Walker, J. E. (2017). Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc. Natl. Acad. Sci. U.S.A. 114, 9086–9091. doi: 10.1073/pnas.1711201114
Hedskog, L., Pinho, C. M., Filadi, R., Ronnback, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Higley, M. J., and Sabatini, B. L. (2008). Calcium signaling in dendrites and spines: practical and functional considerations. Neuron 59, 902–913. doi: 10.1016/j.neuron.2008.08.020
Hirabayashi, Y., Kwon, S. K., Paek, H., Pernice, W. M., Paul, M. A., Lee, J., et al. (2017). ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 358, 623–630. doi: 10.1126/science.aan6009
Jadiya, P., Kolmetzky, D. W., Tomar, D., Di Meco, A., Lombardi, A. A., Lambert, J. P., et al. (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 10:3885. doi: 10.1038/s41467-019-11813-6
Jiang, D., Zhao, L., and Clapham, D. E. (2009). Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147. doi: 10.1126/science.1175145
Kaeser, P. S., and Regehr, W. G. (2014). Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu. Rev. Physiol. 76, 333–363. doi: 10.1146/annurev-physiol-021113-170338
Kang, J. S., Tian, J. H., Pan, P. Y., Zald, P., Li, C., Deng, C., et al. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. doi: 10.1016/j.cell.2007.11.024
Karch, J., Bround, M. J., Khalil, H., Sargent, M. A., Latchman, N., Terada, N., et al. (2019). Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci. Adv. 5:eaaw4597. doi: 10.1126/sciadv.aaw4597
Karch, J., and Molkentin, J. D. (2014). Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 111, 10396–10397. doi: 10.1073/pnas.1410104111
Kirichok, Y., Krapivinsky, G., and Clapham, D. E. (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. doi: 10.1038/nature02246
Kokoszka, J. E., Waymire, K. G., Levy, S. E., Sligh, J. E., Cai, J., Jones, D. P., et al. (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427, 461–465. doi: 10.1038/nature02229
Kruman, I. I., Pedersen, W. A., Springer, J. E., and Mattson, M. P. (1999). ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp. Neurol. 160, 28–39. doi: 10.1006/exnr.1999.7190
Kwon, S. K., Hirabayashi, Y., and Polleux, F. (2016a). Organelle-specific sensors for monitoring Ca(2+) dynamics in neurons. Front. Synaptic Neurosci. 8:29. doi: 10.3389/fnsyn.2016.00029
Kwon, S. K., Sando, R. III, Lewis, T. L., Hirabayashi, Y., Maximov, A., and Polleux, F. (2016b). LKB1 regulates mitochondria-dependent presynaptic calcium clearance and neurotransmitter release properties at excitatory synapses along cortical axons. PLoS Biol. 14:e1002516. doi: 10.1371/journal.pbio.1002516
Landfield, P. W., and Pitler, T. A. (1984). Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 226, 1089–1092. doi: 10.1126/science.6494926
Lau, D. H. W., Hartopp, N., Welsh, N. J., Mueller, S., Glennon, E. B., Morotz, G. M., et al. (2018). Disruption of ER-mitochondria signalling in fronto-temporal dementia and related amyotrophic lateral sclerosis. Cell Death Dis. 9:327. doi: 10.1038/s41419-017-0022-7
Lau, D. H. W., Paillusson, S., Hartopp, N., Rupawala, H., Morotz, G. M., Gomez-Suaga, P., et al. (2020). Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 143:105020. doi: 10.1016/j.nbd.2020.105020
Lee, E. B., Lee, V. M., and Trojanowski, J. Q. (2011). Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50. doi: 10.1038/nrn3121
Lee, K. S., Huh, S., Lee, S., Wu, Z., Kim, A. K., Kang, H. Y., et al. (2018). Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. U.S.A. 115, E8844–E8853. doi: 10.1073/pnas.1721136115
Leslie, S. W., Chandler, L. J., Barr, E. M., and Farrar, R. P. (1985). Reduced calcium uptake by rat brain mitochondria and synaptosomes in response to aging. Brain Res. 329, 177–183. doi: 10.1016/0006-8993(85)90523-2
Lesne, S., Kotilinek, L., and Ashe, K. H. (2008). Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience 151, 745–749. doi: 10.1016/j.neuroscience.2007.10.054
Lessmann, V., Gottmann, K., and Malcangio, M. (2003). Neurotrophin secretion: current facts and future prospects. Prog. Neurobiol. 69, 341–374. doi: 10.1016/s0301-0082(03)00019-4
Li, W. W., Yang, R., Guo, J. C., Ren, H. M., Zha, X. L., Cheng, J. S., et al. (2007). Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18, 1543–1546. doi: 10.1097/WNR.0b013e3282f03db4
Liao, Y., Dong, Y., and Cheng, J. (2017). The function of the mitochondrial calcium uniporter in neurodegenerative disorders. Int. J. Mol. Sci. 18:248. doi: 10.3390/ijms18020248
Lim, D., Fedrizzi, L., Tartari, M., Zuccato, C., Cattaneo, E., Brini, M., et al. (2008). Calcium homeostasis and mitochondrial dysfunction in striatal neurons of Huntington disease. J. Biol. Chem. 283, 5780–5789. doi: 10.1074/jbc.M704704200
Liu, M. L., Zang, T., and Zhang, C. L. (2016). Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 14, 115–128. doi: 10.1016/j.celrep.2015.12.018
Liu, Y., and Zhu, X. (2017). Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 6:21. doi: 10.1186/s40035-017-0092-6
Luongo, T. S., Lambert, J. P., Gross, P., Nwokedi, M., Lombardi, A. A., Shanmughapriya, S., et al. (2017). The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545, 93–97. doi: 10.1038/nature22082
Mammucari, C., Gherardi, G., and Rizzuto, R. (2017). Structure, activity regulation, and role of the mitochondrial calcium uniporter in health and disease. Front. Oncol. 7:139. doi: 10.3389/fonc.2017.00139
Markus, N. M., Hasel, P., Qiu, J., Bell, K. F., Heron, S., Kind, P. C., et al. (2016). Expression of mRNA encoding Mcu and other mitochondrial calcium regulatory genes depends on cell type, neuronal subtype, and Ca2+ signaling. PLoS One 11:e0148164. doi: 10.1371/journal.pone.0148164
Martino Adami, P. V., Nichtova, Z., Weaver, D. B., Bartok, A., Wisniewski, T., Jones, D. R., et al. (2019). Perturbed mitochondria-ER contacts in live neurons that model the amyloid pathology of Alzheimer’s disease. J. Cell Sci. 132:jcs229906. doi: 10.1242/jcs.229906
Mattson, M. P., and Arumugam, T. V. (2018). Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 27, 1176–1199. doi: 10.1016/j.cmet.2018.05.011
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Moloney, A. M., Griffin, R. J., Timmons, S., O’Connor, R., Ravid, R., and O’Neill, C. (2010). Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 31, 224–243. doi: 10.1016/j.neurobiolaging.2008.04.002
Muller, M., Ahumada-Castro, U., Sanhueza, M., Gonzalez-Billault, C., Court, F. A., and Cardenas, C. (2018). Mitochondria and calcium regulation as basis of neurodegeneration associated with aging. Front. Neurosci. 12:470. doi: 10.3389/fnins.2018.00470
Murchison, D., Zawieja, D. C., and Griffith, W. H. (2004). Reduced mitochondrial buffering of voltage-gated calcium influx in aged rat basal forebrain neurons. Cell Calcium 36, 61–75. doi: 10.1016/j.ceca.2003.11.010
Narendra, D. P., and Youle, R. J. (2011). Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 14, 1929–1938. doi: 10.1089/ars.2010.3799
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/s0896-6273(03)00434-3
Oliveira, J. M., Jekabsons, M. B., Chen, S., Lin, A., Rego, A. C., Goncalves, J., et al. (2007). Mitochondrial dysfunction in Huntington’s disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J. Neurochem. 101, 241–249. doi: 10.1111/j.1471-4159.2006.04361.x
Orr, A. L., Kim, C., Jimenez-Morales, D., Newton, B. W., Johnson, J. R., Krogan, N. J., et al. (2019). Neuronal apolipoprotein E4 expression results in proteome-wide alterations and compromises bioenergetic capacity by disrupting mitochondrial function. J. Alzheimers Dis. 68, 991–1011. doi: 10.3233/JAD-181184
Paillusson, S., Gomez-Suaga, P., Stoica, R., Little, D., Gissen, P., Devine, M. J., et al. (2017). alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 134, 129–149. doi: 10.1007/s00401-017-1704-z
Paillusson, S., Stoica, R., Gomez-Suaga, P., Lau, D. H. W., Mueller, S., Miller, T., et al. (2016). There’s Something Wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 39, 146–157. doi: 10.1016/j.tins.2016.01.008
Pallafacchina, G., Zanin, S., and Rizzuto, R. (2018). Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Res. 7::1858. doi: 10.12688/f1000research.15723.1
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U.S.A. 107, 436–441. doi: 10.1073/pnas.0908099107
Pandya, J. D., Grondin, R., Yonutas, H. M., Haghnazar, H., Gash, D. M., Zhang, Z., et al. (2015). Decreased mitochondrial bioenergetics and calcium buffering capacity in the basal ganglia correlates with motor deficits in a nonhuman primate model of aging. Neurobiol. Aging 36, 1903–1913. doi: 10.1016/j.neurobiolaging.2015.01.018
Panov, A. V., Gutekunst, C. A., Leavitt, B. R., Hayden, M. R., Burke, J. R., Strittmatter, W. J., et al. (2002). Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 5, 731–736. doi: 10.1038/nn884
Parone, P. A., Da Cruz, S., Han, J. S., McAlonis-Downes, M., Vetto, A. P., Lee, S. K., et al. (2013). Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J. Neurosci. 33, 4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013
Patron, M., Checchetto, V., Raffaello, A., Teardo, E., Vecellio Reane, D., Mantoan, M., et al. (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. doi: 10.1016/j.molcel.2014.01.013
Patron, M., Granatiero, V., Espino, J., Rizzuto, R., and De Stefani, D. (2019). MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 26, 179–195. doi: 10.1038/s41418-018-0113-8
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/j.ceca.2017.06.008
Pellman, J. J., Hamilton, J., Brustovetsky, T., and Brustovetsky, N. (2015). Ca(2+) handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington’s disease. J. Neurochem. 134, 652–667. doi: 10.1111/jnc.13165
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Quintanilla, R. A., Jin, Y. N., von Bernhardi, R., and Johnson, G. V. (2013). Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol. Neurodegener. 8:45. doi: 10.1186/1750-1326-8-45
Raffaello, A., De Stefani, D., Sabbadin, D., Teardo, E., Merli, G., Picard, A., et al. (2013). The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 32, 2362–2376. doi: 10.1038/emboj.2013.157
Raffaello, A., Mammucari, C., Gherardi, G., and Rizzuto, R. (2016). Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem. Sci. 41, 1035–1049. doi: 10.1016/j.tibs.2016.09.001
Rapizzi, E., Pinton, P., Szabadkai, G., Wieckowski, M. R., Vandecasteele, G., Baird, G., et al. (2002). Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624. doi: 10.1083/jcb.200205091
Rottenberg, H., and Hoek, J. B. (2017). The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16, 943–955. doi: 10.1111/acel.12650
Sancak, Y., Markhard, A. L., Kitami, T., Kovacs-Bogdan, E., Kamer, K. J., Udeshi, N. D., et al. (2013). EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342, 1379–1382. doi: 10.1126/science.1242993
Sepulveda-Falla, D., Barrera-Ocampo, A., Hagel, C., Korwitz, A., Vinueza-Veloz, M. F., Zhou, K., et al. (2014). Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J. Clin. Invest. 124, 1552–1567. doi: 10.1172/JCI66407
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Stoica, R., Paillusson, S., Gomez-Suaga, P., Mitchell, J. C., Lau, D. H., Gray, E. H., et al. (2016). ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 17, 1326–1342. doi: 10.15252/embr.201541726
Sudhof, T. C. (2012). The presynaptic active zone. Neuron 75, 11–25. doi: 10.1016/j.neuron.2012.06.012
Supnet, C., and Bezprozvanny, I. (2010). The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 47, 183–189. doi: 10.1016/j.ceca.2009.12.014
Surmeier, D. J., Schumacker, P. T., Guzman, J. D., Ilijic, E., Yang, B., and Zampese, E. (2017). Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 483, 1013–1019. doi: 10.1016/j.bbrc.2016.08.168
Szabadkai, G., Bianchi, K., Varnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Tadic, V., Adam, A., Goldhammer, N., Lautenschlaeger, J., Oberstadt, M., Malci, A., et al. (2019). Investigation of mitochondrial calcium uniporter role in embryonic and adult motor neurons from G93A(hSOD1) mice. Neurobiol. Aging 75, 209–222. doi: 10.1016/j.neurobiolaging.2018.11.019
Tadic, V., Prell, T., Lautenschlaeger, J., and Grosskreutz, J. (2014). The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front. Cell Neurosci. 8:147. doi: 10.3389/fncel.2014.00147
Tambini, M. D., Pera, M., Kanter, E., Yang, H., Guardia-Laguarta, C., Holtzman, D., et al. (2016). ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 17, 27–36. doi: 10.15252/embr.201540614
Tang, T. S., Slow, E., Lupu, V., Stavrovskaya, I. G., Sugimori, M., Llinas, R., et al. (2005). Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. U.S.A. 102, 2602–2607. doi: 10.1073/pnas.0409402102
Thibault, O., Hadley, R., and Landfield, P. W. (2001). Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J. Neurosci. 21, 9744–9756.
Tomar, D., Dong, Z., Shanmughapriya, S., Koch, D. A., Thomas, T., Hoffman, N. E., et al. (2016). MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 15, 1673–1685. doi: 10.1016/j.celrep.2016.04.050
Tradewell, M. L., Cooper, L. A., Minotti, S., and Durham, H. D. (2011). Calcium dysregulation, mitochondrial pathology and protein aggregation in a culture model of amyotrophic lateral sclerosis: mechanistic relationship and differential sensitivity to intervention. Neurobiol. Dis. 42, 265–275. doi: 10.1016/j.nbd.2011.01.016
Vais, H., Mallilankaraman, K., Mak, D. D., Hoff, H., Payne, R., Tanis, J. E., et al. (2016). EMRE is a matrix Ca(2+) sensor that governs gatekeeping of the mitochondrial Ca(2+) uniporter. Cell Rep. 14, 403–410. doi: 10.1016/j.celrep.2015.12.054
Vecellio Reane, D., Vallese, F., Checchetto, V., Acquasaliente, L., Butera, G., De Filippis, V., et al. (2016). A MICU1 splice variant confers high sensitivity to the mitochondrial Ca(2+) uptake machinery of skeletal muscle. Mol. Cell 64, 760–773. doi: 10.1016/j.molcel.2016.10.001
Verkhratsky, A. (2005). Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279. doi: 10.1152/physrev.00004.2004
Verkhratsky, A., Orkand, R. K., and Kettenmann, H. (1998). Glial calcium: homeostasis and signaling function. Physiol. Rev. 78, 99–141. doi: 10.1152/physrev.1998.78.1.99
Verma, M., Callio, J., Otero, P. A., Sekler, I., Wills, Z. P., and Chu, C. T. (2017). Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J. Neurosci. 37, 11151–11165. doi: 10.1523/JNEUROSCI.3791-16.2017
Wang, J. Q., Chen, Q., Wang, X., Wang, Q. C., Wang, Y., Cheng, H. P., et al. (2013). Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 288, 3070–3084. doi: 10.1074/jbc.M112.407726
Wang, Y., Nguyen, N. X., She, J., Zeng, W., Yang, Y., Bai, X. C., et al. (2019). Structural mechanism of EMRE-dependent gating of the human mitochondrial calcium uniporter. Cell 177, 1252–1261.e13. doi: 10.1016/j.cell.2019.03.050
Xiong, J., Verkhratsky, A., and Toescu, E. C. (2002). Changes in mitochondrial status associated with altered Ca2+ homeostasis in aged cerebellar granule neurons in brain slices. J. Neurosci. 22, 10761–10771.
Zampese, E., Fasolato, C., Kipanyula, M. J., Bortolozzi, M., Pozzan, T., and Pizzo, P. (2011). Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. U.S.A. 108, 2777–2782. doi: 10.1073/pnas.1100735108
Zhang, B., Tang, X. C., and Zhang, H. Y. (2013). Alternations of central insulin-like growth factor-1 sensitivity in APP/PS1 transgenic mice and neuronal models. J. Neurosci. Res. 91, 717–725. doi: 10.1002/jnr.23201
Zhou, W., Marinelli, F., Nief, C., and Faraldo-Gomez, J. D. (2017). Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. eLife 6:e23781. doi: 10.7554/eLife.23781
Keywords: mitochondria, calcium regulation, aging, neurodegenerative disease, synaptic regulation
Citation: Jung H, Kim SY, Canbakis Cecen FS, Cho Y and Kwon S-K (2020) Dysfunction of Mitochondrial Ca2+ Regulatory Machineries in Brain Aging and Neurodegenerative Diseases. Front. Cell Dev. Biol. 8:599792. doi: 10.3389/fcell.2020.599792
Received: 28 August 2020; Accepted: 06 November 2020;
Published: 18 December 2020.
Edited by:
Yuzuru Imai, Juntendo University, JapanCopyright © 2020 Jung, Kim, Canbakis Cecen, Cho and Kwon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seok-Kyu Kwon, c2trd29uQGtpc3QucmUua3I=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.