 Antionette L. Williams
Antionette L. Williams Brenda L. Bohnsack
Brenda L. Bohnsack
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 23 December 2020
Sec. Cell Growth and Division
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.595896
This article is part of the Research TopicCellular Mechanisms during Normal and Abnormal Craniofacial DevelopmentView all 22 articles
During vertebrate embryonic development, a population of dorsal neural tube-derived stem cells, termed the neural crest (NC), undergo a series of morphogenetic changes and extensive migration to become a diverse array of cell types. Around the developing eye, this multipotent ocular NC cell population, called the periocular mesenchyme (POM), comprises migratory mesenchymal cells that eventually give rise to many of the elements in the anterior of the eye, such as the cornea, sclera, trabecular meshwork, and iris. Molecular cell biology and genetic analyses of congenital eye diseases have provided important information on the regulation of NC contributions to this area of the eye. Nevertheless, a complete understanding of the NC as a contributor to ocular development remains elusive. In addition, positional information during ocular NC migration and the molecular pathways that regulate end tissue differentiation have yet to be fully elucidated. Further, the clinical challenges of ocular diseases, such as Axenfeld-Rieger syndrome (ARS), Peters anomaly (PA) and primary congenital glaucoma (PCG), strongly suggest the need for better treatments. While several aspects of NC evolution have recently been reviewed, this discussion will consolidate the most recent current knowledge on the specification, migration, and contributions of the NC to ocular development, highlighting the anterior segment and the knowledge obtained from the clinical manifestations of its associated diseases. Ultimately, this knowledge can inform translational discoveries with potential for sorely needed regenerative therapies.
The neural crest (NC) is an embryonic population of multipotent cells that are extremely important for vertebrate body development. Concomitant with gastrulation, induction at the neural plate border leads to the delamination of cells from the neural ectoderm during primary neurulation (Williams and Bohnsack, 2015, 2019; Gandhi and Bronner, 2018; Pla and Monsoro-Burq, 2018; Rogers and Nie, 2018; Szabo and Mayor, 2018; Prasad et al., 2019). These early NC cells then undergo an epithelial-to-mesenchymal transition (EMT) and subsequently migrate throughout the body to form a diverse set of cells and tissues, including bone, cartilage, peripheral neurons, and melanocytes. NC cells derived from the prosencephalon, mesencephalon, and rhombencephalon comprise the cranial subpopulation that contributes to the frontonasal process, periocular mesenchyme (POM) and pharyngeal arches. Through these embryonic structures and cell populations, NC cells then give rise to craniofacial connective, skeletal and neuronal tissues (Cordero et al., 2011; Kish et al., 2011). Furthermore, cranial NC cells within the POM enter the anterior segment of the eye and form parts of the cornea, iris, sclera, ciliary body, and aqueous outflow pathways (Whikehart, 2010; Cordero et al., 2011; Kish et al., 2011; Williams and Bohnsack, 2015).
As an extremely dynamic cell population, the NC is of high clinical interest, as disruption of this cell population results in a wide range of congenital abnormalities (Bohnsack and Kahana, 2013; Williams and Bohnsack, 2015, 2019; Akula et al., 2019). Since described by Wilheim His in 1868, our understanding of NC biology has grown, and the unique characteristics of NC cells have been an interesting and well-studied topic. Comparative analyses across multiple diverse vertebrate model organisms have elucidated the progressive and conserved steps involved in NC development from induction to cell specification, delamination, emigration, and eventual differentiation (Simoes-Costa and Bronner, 2013; Green et al., 2015). Diligent and systematic work in this field has revealed a host of regulatory genes and interactions to further tease out the gene regulatory network underlying complex NC development (Simoes-Costa and Bronner, 2015; Martik and Bronner, 2017). However, much of the evidence pertains to the genetic networks responsible for ectodermal and neurectodermal patterning, and little is known about the specification of the ocular NC. Indeed, the continued requirement of NC stem cells for the maintenance of adult eyes (Jinno et al., 2010; Green et al., 2015; Chawla et al., 2018) emphasizes the importance of this cell population over the vertebrate lifetime. This review will provide an update of the morphogenetic, gene regulation and signaling events controlling ocular NC migration and differentiation, with particular emphasis on the ocular anterior segment. The current challenge is to determine the molecular events that accompany distinct positional stages, as fate maps of the various ocular NC populations are lacking, and there is little to no information on the biology underlying end tissue differentiation in the eye. An adequate understanding of ocular evolution, particularly anterior segment development, and the pathogenesis of its associated disorders will bring us closer to deciphering the mechanisms that participate in the normal development, maturation and maintenance of the eye and the potential therapeutics thereof.
Central to understanding eye development are the concurrent events intertwining cranial NC development and optic cup morphogenesis. During late gastrulation, the developing eye is established as a single eye field located in the medial anterior neural plate at the boundary between the telencephalon and diencephalon (reviewed in Miesfeld and Brown, 2019; Figure 1A). Concurrent with the delamination of NC cells from the neural plate border, the eye field undergoes bilateral bifurcation to cause the evagination of the left and right optic vesicles (Figure 1B). Upon contact with the overlying surface ectoderm, the optic vesicles invaginate to form bilayer optic cups (Figure 1C). The interaction between the optic vesicle and the lens placode of the surface ectoderm induces lens vesicle evagination and establishes early ocular anterior segment formation (Figures 1C,D). During this process, the optic fissure is formed along the inferonasal aspect of the cup as it envelops the hyaloid vasculature (Figure 1C). Meanwhile, cranial NC cells that have undergone EMT migrate into the developing head (Figure 2). Cells from the diencephalon and anterior mesencephalon divide into two waves that move either dorsal or ventral to the optic cup. A subpopulation of these cells remains within the POM, while additional cells travel anteriorly into the frontonasal process. NC cells from the posterior mesencephalon and rhombencephalon take a more direct route toward the pharyngeal arches on the ventral side of the embryo.
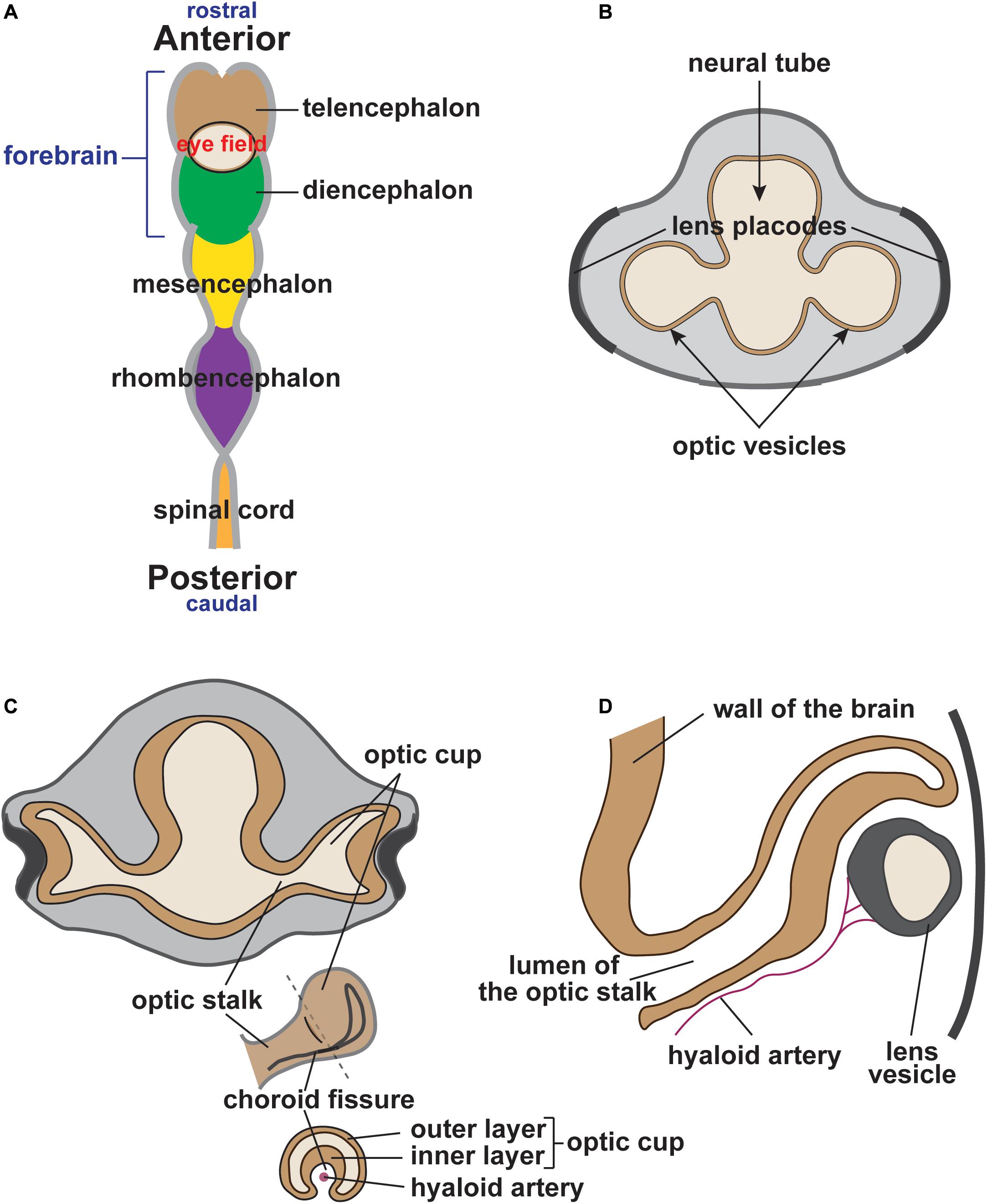
Figure 1. Vertebrate eye development (human eye). (A) During early vertebrate development, the eye field is established at the boundary between the telencephalon (brown) and the diencephalon (green). (B) Optic vesicles bilaterally protrude from either side of the forebrain approaching the thickened surface ectoderm (lens placodes). (C) The interaction between the optic vesicle and the lens placode of the surface ectoderm results in optic vesicle invagination, optic cup formation and lens placode evagination (lens pit). Simultaneously, the optic fissure is formed along the inferonasal aspect of the optic cup, which surrounds the hyaloid artery. (D) Continued evagination of surface ectoderm leads to the formation of an independent lens vesicle.
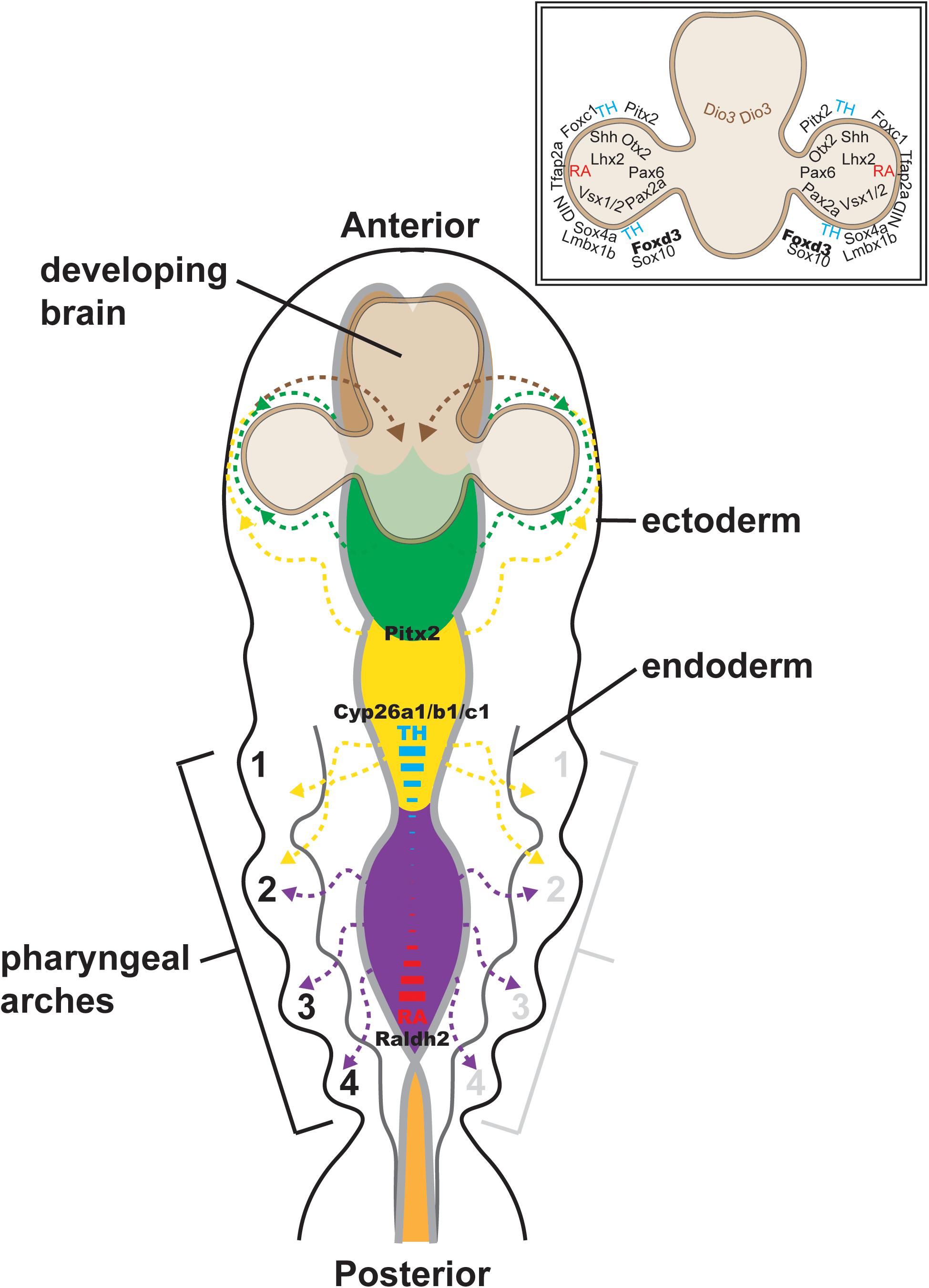
Figure 2. Cranial neural crest migration into the head. Time-lapse imaging of NC cell migration in zebrafish has shown that cranial NC cells from the diencephalon (green dashed lines and arrows) and anterior mesencephalon (anterior yellow dashed lines and arrows) travel either dorsal or ventral to the optic cup to establish the POM. A subpopulation of these cells migrates anteriorly into the frontonasal process (brown dashed lines and arrows). NC cells from the posterior mesencephalon (posterior yellow dashed lines and arrows) and rhombencephalon (purple dashed lines and arrows) migrate toward the pharyngeal arches on the ventral side of the embryo. Some of the factors implicated in the regulation of NC migration (Pax6, Pitx2, Foxd3, Sox10, Foxc1, etc.) are shown (inset, upper right). The signaling gradient that maintains the integrity of cranial NC cell migration [i.e., thyroid hormone (TH; blue) and retinoic acid (RA; red)] into the posterior pharyngeal arches is also shown.
In zebrafish, there is interplay between thyroid hormone and retinoic acid in regulating the different streams of cranial NC (reviewed in Williams and Bohnsack, 2019; Figure 2). NC cell migration into the posterior pharyngeal arches requires a low level of thyroid hormone, but a high level of retinoic acid. This requirement corresponds with the high levels of retinoic acid centered within the posterior rhombencephalon by the spatial expression of retinoic acid synthesis (retinaldehyde dehydrogenase 2; Raldh2) and degradation (cytochrome P450 family 26; Cyp26a1, Cyp26b1, and Cyp26c1) enzymes. In contrast, NC cell migration into the first pharyngeal arch, which most prominently gives rise to the maxilla and mandible bones, requires high thyroid hormone and low retinoic acid levels. The reciprocal effects of thyroid hormone and retinoic acid may be mediated by their shared retinoid X receptor (RXR). In anterior cranial NC cells, thyroid hormone is required for initiation of the migratory stream ventral to the eye, but then localized degradation by iodothyronine deiodinase 3 (Dio3) is necessary for completion of their migratory arc from the POM into the frontonasal process. On the other hand, the dorsal NC migratory stream requires a high level of thyroid hormone for both the initiation and completion of migration. In addition to its location at the posterior rhombencephalon, retinoic acid is also concentrated at the dorsal and ventral optic cup. This high level of retinoic acid is required for the proper migration of NC streams both dorsal and ventral to the eye as well as the migration of the POM NC population (Bohnsack and Kahana, 2013). The transcription factor paired like homeodomain 2 (Pitx2), well known for its association with Axenfeld-Rieger syndrome (ARS) (discussed below), also regulates these early stages of cranial NC migration. In zebrafish, morpholino oligonucleotide knockdown of Pitx2 expression inhibits migration and induces the apoptosis of NC cells derived from the mesencephalon, thereby resulting in decreased NC populations within the first pharyngeal arch and the ventral POM (Bohnsack et al., 2012; Ji et al., 2016).
Following migration, the NC cells within the POM surround the optic vesicle and play an important but ill-defined role in establishing the optic stalk and cup (Figure 2). Indeed, Pitx2 gene knockout mice show severe eye phenotypes, including anophthalmia (lack of eye), microphthalmia (small disorganized eye) and optic nerve defects, such as eyes that attach directly to the ventral hypothalamus (Evans and Gage, 2005). Importantly, NC-targeted Pitx2 conditional gene knockouts show a similar phenotype (Liu and Semina, 2012), indicating that these eye abnormalities reflect the cell non-autonomous effects of Pitx2 in NC cells. However, a direct connection between Pitx2 and known regulators of optic stalk and cup development, such as sonic hedgehog (Shh), paired box protein 2 (Pax2), Pax6, retinal homeobox protein (Rax), visual system homeobox 1 (Vsx1), Vsx2, orthodenticle homeobox 2 (Otx2) and LIM homeobox protein 2 (Lhx2), has yet to be established. Further, in zebrafish, loss of NC cells within the POM through genetic targeting of the NC specifiers, transcription factor AP-2 alpha (Tfap2a) and forkhead box D3 (Foxd3), disrupts optic cup invagination and optic fissure formation (Wang et al., 2011). NC cell production of the extracellular matrix protein, Nidogen (Nid), contributes to basement membrane formation around the optic vesicle and is critical for retinal pigment epithelial cell movements during cup invagination (Bryan et al., 2020). In addition, NC cells help to establish patterning of the optic cup, which is critical for optic fissure closure. The optic fissure extends from the optic stalk to the distal edge of the optic cup and transmits the transient hyaloid vasculature that nourishes the developing lens and retina. Closure of the optic fissure is a complex process that has not been fully elucidated at the molecular level, and the failure of fissure closure results in optic nerve, chorioretinal and iris colobomas. Dorsal-ventral patterning of the optic cup by a similar set of transcription factors important for optic stalk and cup development is critical for setting up fissure closure. Mutant animal models of these retinal genes are characterized by anophthalmia, microphthalmia, and coloboma. Similarly, knockdown of the NC-specific transcription factor, SRY-box transcription factor 4a (Sox4a), also causes coloboma, as decreased Sox4 gene expression results in the mislocalization of Pax2a, Vsx1, and Vsx2 within the retina (Wen et al., 2015). Thus, early eye morphogenesis and cranial NC development intersect at multiple points, with important crosstalk between the tissues that regulate optic cup formation and direct NC migration.
Following optic cup morphogenesis, including the establishment of the optic fissure and anterior segment, NC cells within the POM begin migration into the eye (Figure 3). The patterns of migration vary between species. Classic anatomic studies in human embryos suggest three distinct waves of NC cells that contribute first to the corneal stroma and endothelium, second to the iris stroma and third to the trabecular meshwork (Williams and Bohnsack, 2015; Miesfeld and Brown, 2019). In contrast, there are two NC cell waves in mice and a single continuous migratory mesenchymal wave in chick (reviewed in Miesfeld and Brown, 2019). Fate mapping studies in mice have shown that both NC and mesoderm contribute to the corneal stroma and corneal endothelium, while in chick, both of these corneal structures as well as the iris stroma are NC derived (Gage et al., 2005). Time-lapse imaging of pre- and post-migratory NC cells in zebrafish embryos has shown at least three waves, including two distinct Sox10-positive and one Foxd3-positive cell populations (Eason et al., 2017; Takamiya et al., 2020). Sox10-positive cells migrate early after optic cup formation and preliminary fate mapping studies show minimal contributions to the adult eye (unpublished data). Foxd3-positive NC cells migrate into the anterior segment via two pathways: (1) adjacent to the hyaloid vasculature within the optic fissure and (2) between the surface ectoderm and the optic cup (Mork and Crump, 2015; Williams and Bohnsack, 2017; Van Der Meulen et al., 2020; Figure 3). Foxd3-positive cells have a continued presence within the corneal endothelium and iris in zebrafish larvae and young juveniles. However, Foxd3 expression is lost by adulthood and fate mapping of this population of cells has yet to be accomplished.
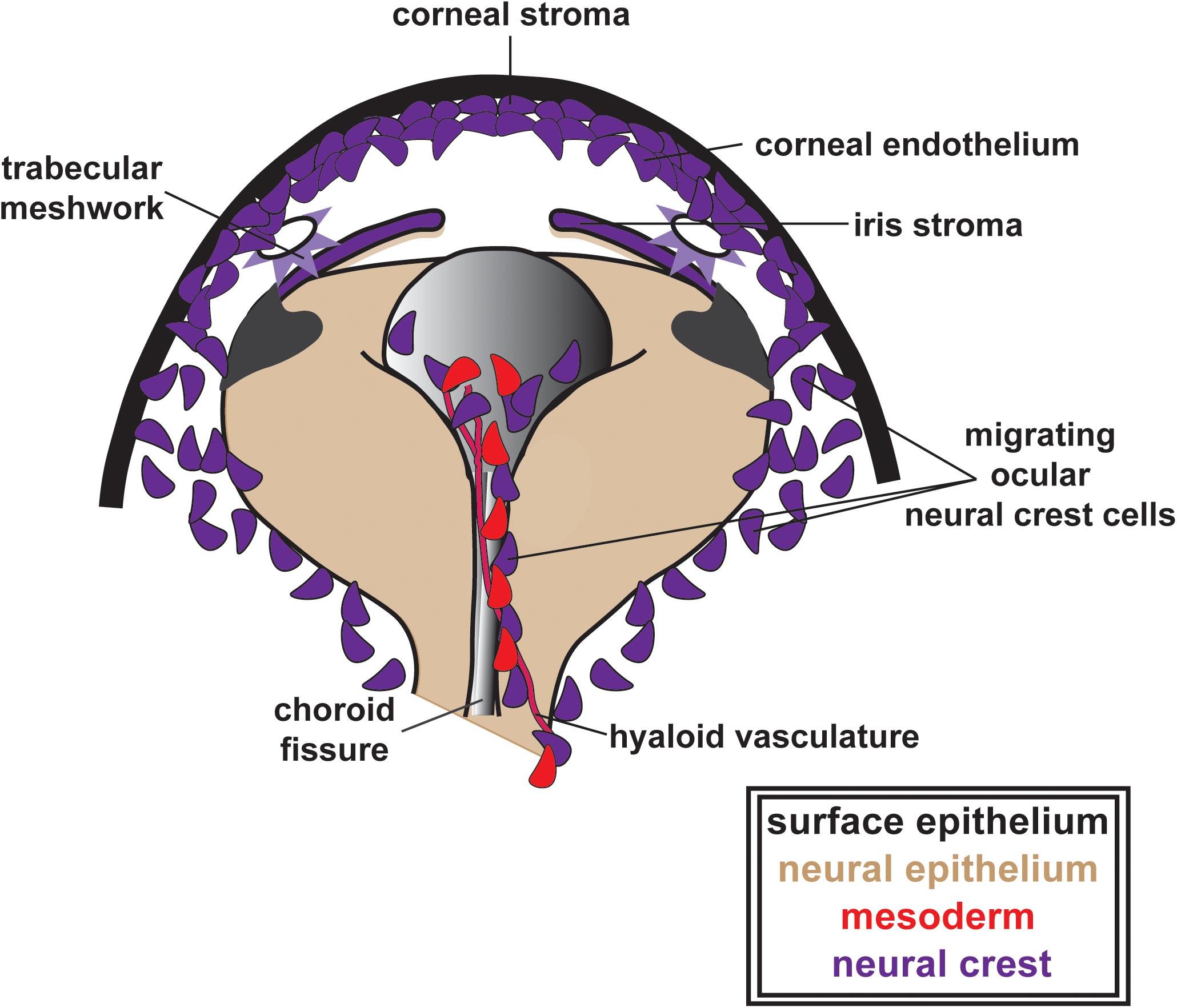
Figure 3. Ocular neural crest migration and establishment of the ocular anterior segment. POM NC cells (purple), along with mesoderm cells (red), migrate adjacent to the hyaloid vasculature within the optic fissure and between the surface ectoderm and the optic cup contributing to the corneal stroma and endothelium, the iris stroma and the trabecular meshwork. In this figure, these structures are all indicated in purple to illustrate their ocular NC derivation. Notably, as previously discussed in the text above, the patterns of ocular NC cell migration vary between species (three distinct waves of NC cells in humans, two NC cell waves in mice and a single continuous migratory wave in chick). However, for simplicity, these patterns are not depicted here.
With the loss of early markers, such as Sox10 and Crestin, NC cells adopt the expression of other transcription factors, namely Pitx2, Foxc1, Lmx1b, and Eya2 (Van Der Meulen et al., 2020). Further, signaling molecules, such as retinoic acid, and interactions between NC cells and the adjacent ocular tissues have continued effects on the ocular migration and end differentiation of NC cells. To date, much of the knowledge regarding NC cell contributions to the anterior segment of the eye derives from clinical and basic science studies focused on the pathogenesis of congenital eye diseases.
Axenfeld-Rieger syndrome is characterized by congenital malformations affecting NC-derived craniofacial and ocular structures. Most prominent are the ocular abnormalities that combine Axenfeld anomaly (posterior embryotoxon) and Rieger anomaly (iris hypoplasia), resulting in corectopia (irregular pupil), and pseudopolycoria (multiple pupils) (Figure 4A). Over 50% of affected individuals have glaucoma, which due to iris and trabecular meshwork abnormalities, often requires multiple surgeries to preserve vision (Zepeda et al., 2020). In addition, individuals with ARS may have a distinctive craniofacial structure that includes telecanthus, maxillary hypoplasia, and broad flat nasal bridge, teeth abnormalities, such as oligodontia and microdontia, and congenital heart abnormalities.
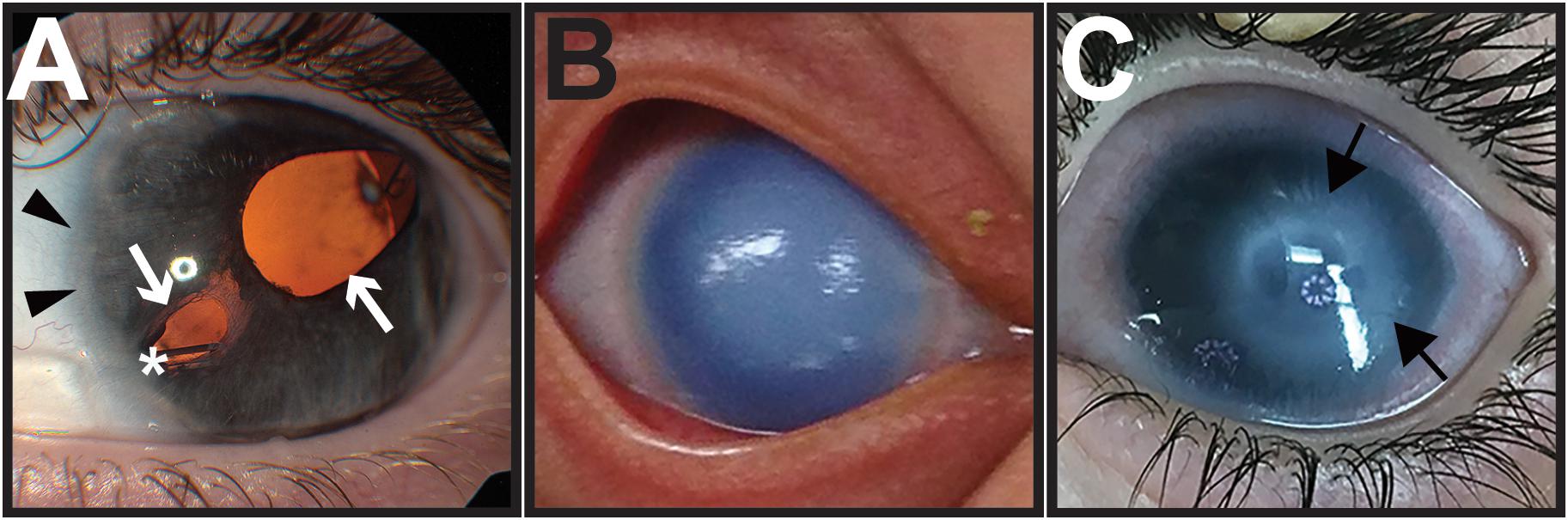
Figure 4. Congenital eye diseases associated with neural crest defects in the anterior segment. (A) Axenfeld-Rieger syndrome is characterized by anteriorization of Schwalbe’s line (posterior embryotoxon, black arrowheads) and iris hypoplasia, causing corectopia and pseudopolycoria (white arrows). Over half of affected individuals develop glaucoma, and many require placement of a glaucoma drainage device (asterisk) to control intraocular pressure. (B) Primary congenital glaucoma is due to developmental abnormalities in the trabecular meshwork and aqueous outflow tracts. As a result, elevated intraocular pressures in infants cause corneal edema and buphthalmos (increased eye size). (C) Peters anomaly shows central corneal opacification (black arrows) reflecting the abnormal separation of the lens vesicle from the surface ectoderm.
ARS shows genetic heterogeneity, and autosomal dominant mutations in Pitx2 and Foxc1 genes account for up to 70% of cases (Reis et al., 2012; Seifi et al., 2017; Zamora and Salini, 2020). Based on the clinical correlation with ARS, these two homeobox transcription factors play critical roles in craniofacial and ocular NC development. In mice and zebrafish embryos, Pitx2 is expressed in NC cells soon after delamination, and as discussed above, this factor regulates migration into the pharyngeal arches and POM (Chawla et al., 2016). In zebrafish, Pitx2 knockdown causes decreased NC cell migration within the pharyngeal arches, and this effect accounts for the maxillary hypoplasia and cardiac outflow tract and teeth anomalies seen in human disease. Further, in mice, complete knockout of Pitx2 results in severe heart defects and embryonic lethality that precludes full craniofacial and ocular analysis (Kitamura et al., 1999; Evans and Gage, 2005). In contrast, mice heterozygous for the Pitx2 null allele and those in which the Pitx2 knockout was limited to NC cells were viable and displayed corneal, iris, and scleral defects similar to ARS in humans (Chen and Gage, 2016). Retinoic acid is a known regulator of Pitx2 expression (Bohnsack et al., 2012; Williams and Bohnsack, 2015; Chawla et al., 2016; Hebert et al., 2017; Draut et al., 2019; Williams and Bohnsack, 2019), but has differential effects depending on embryonic stage. During the early stages, retinoic acid decreases Pitx2 expression within NC cells migrating toward the pharyngeal arches. However, during later NC cell migration, retinoic acid increases Pitx2 expression within the periocular region in both mice and zebrafish (Gage et al., 2008; Duester, 2009; Kumar and Duester, 2010; Zacharias and Gage, 2010; Bohnsack et al., 2012). Additional studies in mice have shown that paracrine retinoic acid signaling in the POM, mediated via nuclear retinoic acid receptors RXR alpha, RAR beta and RAR gamma, not only regulates the remodeling of the POM, but also controls the expression of both Foxc1 and Pitx2 (Matt et al., 2005; reviewed in Williams and Bohnsack, 2019). Moreover, defects in retinoic acid signaling in NC cells are sufficient to completely reshape eye development and mediate patterning of the optic vesicle (Matt et al., 2008). Lymphoid enhancer-binding factor 1 (Lef-1) and β-catenin bind the Pitx2 gene promotor in mice, indicating a role for the canonical Wnt signaling pathway in regulating Pitx2 expression. Notably, Pitx2 regulates the Wnt antagonist, Dkk and thus forms a regulatory feedback loop within NC cells.
An additional target of Pitx2 is the LIM-homeodomain transcription factor, Lmx1b, which is initially expressed in POM NC cells and then subsequently in the iris, cornea, trabecular meshwork, and hyaloid vasculature. Lmx1b plays a role in NC cell migration and survival, such that homozygous mutant mice have microphthalmia with iris and ciliary body hypoplasia (Pressman et al., 2000). Autosomal dominant Lmx1b gene mutations cause Nail-Patella Syndrome (OMIM 161200), in which affected individuals are at risk for open angle glaucoma, but do not exhibit congenital eye abnormalities (Vollrath et al., 1998). This suggests that heterozygosity in the Lmx1b gene is adequate for ocular NC cell development, but not for postnatal maintenance of the structure and function of the trabecular meshwork. In mice, Pitx2 also regulates Tfap2b, which is expressed in NC cells in the POM and regulates anterior chamber development via a Lmbx1b-independent pathway (Chen and Gage, 2016). Human Tfap2b gene mutations are associated with autosomal dominant Char syndrome (OMIM 169100), which is characterized by craniofacial abnormalities, but not eye defects. However, NC-specific Tfap2b knockout mice had closed iridocorneal angles due to abnormal iris, cornea, trabecular meshwork, and ciliary body development (Martino et al., 2016). In addition, Tfap2b regulates Zp4, Col8a2, and N-Cadherin in the corneal endothelium (Martino et al., 2016).
Fewer studies have focused on Foxc1 and NC cell development. In mice and zebrafish, genetic knockout or morpholino knockdown of Foxc1 are also embryonic lethal, while heterozygous animals show ocular abnormalities consistent with ARS (Smith et al., 2000). Foxc1 mediates the development of the corneal endothelium and maintains the avascular nature of the cornea, which is necessary for the transparency of this tissue and normal vision (Seo et al., 2012; Koo and Kume, 2013). Additionally, Foxc2 mutations hamper the specification of corneal epithelial cells and leads to ectopic corneal neovascularization and corneal conjunctivalization in mice (Seo et al., 2017), suggesting that both Foxc1 and Foxc2 collectively ensure precise ocular surface development. Pitx2 and Foxc1 colocalize within cells, and each of their genes affects the activity of the other. Indeed, in mice, Pitx2 interacts with the activation domain and negatively regulates the activity of Foxc1 (Berry et al., 2006). In contrast, decreased levels of Foxc1 and Foxc2 activity within NC cells have been associated with significant declines in Pitx2 expression and a more severe phenotype, including microphthalmia, corneal opacification, and eyelid fusion in these animals (Seo et al., 2017). In animal models, Eya2, Fgf19, Foxo1a, and Galnt4 have also been identified as downstream targets of Foxc1 in NC cells. However, unlike the Pitx2 targets, the clinical significance of these genes is not understood, as none of these genes are associated with cranial NC or eye abnormalities.
Taken together, the regulation and interaction between Pitx2 and Foxc1 in the NC are essential for craniofacial and ocular (corneal) development, and mutations in either of their genes manifests as the rare congenital disease, ARS. Nonetheless, additional studies are required to identify further downstream targets of Pitx2 and Foxc1 and determine their roles in anterior segment development.
Primary congenital glaucoma (PCG) is due to abnormal formation of the trabecular meshwork and aqueous outflow tracts resulting in increased intraocular pressure. PCG presents between birth and 2 years of age, but most commonly in the first 6 months with the classic triad of photophobia (light sensitivity), epiphora (tearing), and blepharospasm (frequent blinking/eye closure) (Figure 4B). These symptoms are due to corneal edema and Haab’s striae (breaks in Descemets layer), which are a result of elevated intraocular pressure. Further, PCG is characterized by buphthalmos (enlarged eye) with subsequent axial lengthening and myopic shift as well as glaucomatous optic neuropathy. Children with PCG require urgent surgery to lower the intraocular pressure and preserve vision.
Primary congenital glaucoma is most commonly associated with autosomal recessive mutations in the Cyp1b1 gene (Li et al., 2011). Independent of the retinoic acid catalysis enzymes Raldh2, Raldh3, and Raldh4, Cyp1b1 regulates the two-step conversion of vitamin A first to retinaldehyde and then to retinoic acid (Chambers et al., 2007). However, endogenous in vivo targets of Cyp1b1 have yet to be verified. In zebrafish embryos, Cyp1b1 is expressed within the retina in the inferior optic fissure and lesser-known superior optic fissure in correlation with optic fissure patency (Chambers et al., 2007; Williams and Bohnsack, 2017; Hocking et al., 2018). Indeed, the overexpression of Cyp1b1 prevents fissure closure, resulting in colobomas in zebrafish (Williams and Bohnsack, 2017), and a handful of case reports of superior coloboma have been associated with Cyp1b1 gene mutations (Hocking et al., 2018). In contrast, knockdown of Cyp1b1 causes premature closure and inhibits later NC migration through the inferior optic fissure, and this affect is independent of retinoic acid (Williams and Bohnsack, 2017). The sequence of NC differentiation in the anterior segment is first cornea, then iris and subsequently aqueous outflow tracts. Thus, one hypothesis is that the lack of later NC migration due to decreased Cyp1b1 specifically causes abnormalities in aqueous outflow, resulting in PCG. Additional studies in mice have demonstrated expression of Cyp1b1 in the trabecular meshwork (Zhao et al., 2015), and heterozygosity of Cyp1b1 gene mutations has been associated with the presentation of juvenile onset glaucoma after the age of 2 years. This finding suggests that Cyp1b1 plays a role in trabecular meshwork formation and maintenance. However, the molecular targets and cellular functions of Cyp1b1 within this tissue are unknown.
Peters anomaly (PA) is a congenital eye disease characterized by central corneal opacification due to the abnormal separation of the lens from the overlying surface ectoderm. The resulting abnormal migration and differentiation of ocular NC cells leads to iris-corneal adhesions and absence of corneal endothelium and Descemets membrane (Figure 4C). In severe cases, the lens is also adhered to the cornea, which can cause corneal staphylomas (Alallah et al., 2020; Chang et al., 2020; Katz et al., 2020; Kletke et al., 2020; Samara and Eldaya, 2020). In addition, cataract, microphthalmia, and aniridia (iris absence or hypoplasia) are commonly associated with PA. Glaucoma complications due to a closed angle configuration and abnormal trabecular meshwork and aqueous outflow tract formation are observed in approximately two-thirds of cases and often requires multiple surgeries to obtain intraocular pressure control (Ozeki et al., 2000; Gould and John, 2002; Sowden, 2007; Harissi-Dagher and Colby, 2008; Dolezal et al., 2019). Although most cases are isolated to the eye, systemic findings can include craniofacial anomalies, congenital heart defects, and developmental delay (Dolezal et al., 2019).
Genetic mutations in or chromosomal deletions involving known genes that regulate anterior segment development have also been associated with PA. While Pitx2 and Foxc1 are typically affiliated with ARS and Cyp1b1 is associated with PCG, mutations in these genes have been linked to rare cases of PA. Notably, the underlying molecular pathogenesis of the mutations in these genes that lead to PA versus their more commonly associated congenital diseases is not well understood (Honkanen et al., 2003; Weisschuh et al., 2008; Berker et al., 2009; Arikawa et al., 2010; Gage et al., 2014; Hassed et al., 2017).
Pax6 gene mutations have also been linked to PA. Notably, mutations in Pax6 are more typically associated with aniridia, a panocular congenital disease characterized by foveal hypoplasia, optic nerve hypoplasia, limbal stem cell deficiency, and varying degrees of iris hypoplasia (reviewed in Lim et al., 2017; Sannan et al., 2017; Syrimis et al., 2018; Wawrocka and Krawczynski, 2018; Lima Cunha et al., 2019; Lee et al., 2020; Tripathy and Salini, 2020). However, phenotypic variation of the same Pax6 gene mutation has shown both aniridia and PA within one family (Wang et al., 2018), and PA has been associated with aniridia in more than 10% of cases (Dolezal et al., 2019).
Animal models have shown that Pax6 targets essential extracellular signaling molecules that control multiple steps in eye morphogenesis (Cvekl and Callaerts, 2017). Pax6 is initially expressed in the optic pit and subsequently in the optic vesicle, optic stalk and overlying surface ectoderm (Enwright and Grainger, 2000; Grocott et al., 2011; Cvekl and Callaerts, 2017; Takamiya et al., 2020). Pax6 is important for optic stalk formation and retinal differentiation, which correlates with optic nerve and foveal hypoplasia observed with human Pax6 gene mutations. Further, during cup morphogenesis, Pax6 expression is primarily restricted to the surface ectoderm and the distal end of the optic cup. This expression underpins formation of the lens placode and subsequent detachment of the lens from the cornea (Takamiya et al., 2020) and helps to explain the relationship between Pax6 mutations and PA. Notably, Pax6 gene mutations and deletions yield phenotypic differences between humans and animal models. While Pax6 gene mutations in humans are most commonly associated with aniridia (Lee et al., 2020; Tripathy and Salini, 2020), in mice and zebrafish, microphthalmia and severe anterior segment dysgenesis are observed (Takamiya et al., 2015; Yasue et al., 2017; Grant et al., 2020). Although not well understood, these phenotypic differences among species demonstrate the multiple essential roles of Pax6 in eye development.
Mutations within Pitx2, Foxc1, Cyp1b1, and Pax6 only account for a small percentage of cases of PA. Further clinical genetic analyses of PA cases have revealed the importance of additional transcription factors, including Pitx3 and Foxe3 for ocular development. Clinical and histological analyses revealed adhesions between either the iris or lens and the cornea, with the anterior dislocation of Schwalbe’s line and cataracts in families with a 17-bp insertion mutation in the Pitx3 gene (Summers et al., 2008). In addition, targeted sequencing has revealed other known and novel mutations in Pitx3 in PA patients (Zazo Seco et al., 2018). In mice, Pitx3 is expressed in the lens, and homozygous mouse mutants with lenticular expression of truncated Pitx3 presented with microphthalmia and aphakia (Wada et al., 2014). Further, a homozygous guanine insertion (c.415_416insG) mutation in the Pitx3 gene in mice resulted in an eyeless phenotype characterized by closed lids, corneal thickening, and corneolenticular adhesion (Rosemann et al., 2010). In addition, Pitx3 positively regulates Foxe3 expression in the anterior lens epithelium, and although these two genes do not interact directly, Pitx3 knockdown in mice and zebrafish eliminates Foxe3 expression. While Foxe3 mutations are typically associated with congenital aphakia (absence of lens), there have been reports of PA (Garcia-Montalvo et al., 2014). In mice, Foxe3 mutations result in corneolenticular adhesions, abnormally shaped lens fiber cell nuclei, chorioretinal folds, missing corneal endothelium, disarrayed corneal stroma, and reduced corneal epithelium thickness (Medina-Martinez et al., 2005). Further, a significant reduction in lens epithelial cell proliferation has been observed, and the differentiation of these cells occurred appreciably earlier than that in wild-type mice (Medina-Martinez et al., 2005).
In addition to genetic alterations, in utero exposure to environmental toxins, such as alcohol, can disrupt anterior segment development and lead to PA. Fetal alcohol spectrum disorders encompass a wide range of behavioral and physical defects that result from alcohol consumption during pregnancy. Fetal alcohol syndrome (FAS) is the most severe form, and this disease is characterized by neurologic and cognitive disabilities and typical craniofacial abnormalities, such as a thinned vermillion, shortened palpebral fissures, a cleft palate, and/or cleft lip (Vorgias and Bernstein, 2020). In addition, the eyes and vision are often affected, most commonly showing neuroepithelial-derived optic cup abnormalities, such as microphthalmia and optic nerve hypoplasia. In rare cases, FAS is also associated with PA, suggesting that the steps in eye development are sensitive to alcohol exposure. Interestingly, despite the almost universal craniofacial features in FAS, the NC-derived anterior segment is less commonly affected (Brennan and Giles, 2014). This finding suggests molecular distinctions between different NC cell populations, i.e., cranial NC cells that give rise to craniofacial structures versus those that give rise to the ocular structures. Interestingly, compared to Sox10-expressing NC cells fated to craniofacial structures, the survival and migration of ocular Foxd3-positive NC cells were less sensitive to the effects of alcohol. To some extent, this difference was mediated by a higher basal level of mitochondrial superoxide dismutase (Sod2) in the anterior segment of the eye compared to that in the craniofacial NC, resulting in a greater capacity to activate oxidative stress in response to ethanol exposure in the eye (Eason et al., 2017). These results are consistent with the propensity for craniofacial defects versus the rarity of ocular anomalies in FAS. A recent study showed that miRNA-135a overexpression markedly decreased ethanol-induced apoptosis in NC cells in zebrafish embryos, and the microinjection of miRNA-135a mimics ameliorated growth retardation and craniofacial defects (Yuan et al., 2020). These results suggest that microRNAs play a role in NC-mediated ocular and craniofacial development.
Taken together, anterior segment development requires the coordinated movement of cells and tissues derived from the surface ectoderm, neural epithelium and NC. Genetic or toxic disruption of lens formation and subsequent NC cell migration and differentiation results in a common phenotype of PA. However, the identification of genetic mutations is lacking in a majority of cases, indicating that many other yet to be identified genes are important during this process. Additional studies are required not only to fully elucidate this disease pathogenesis but also to understand the molecular regulation of anterior segment development.
Most studies on the NC have focused on characterizing the genes that coordinate the development of the cranial NC and its multiple derivatives. However, as the ocular NC appears to be the source of the tissues involved in rare congenital diseases, it is also important to understand how specific cellular defects and implicated genes ultimately lead to phenotypic abnormalities, as this information could considerably increase our understanding of the end tissue differentiation of this NC cell population and improve the management and prevention of ocular diseases. In particular, disorders of the ocular anterior segment, commonly referred to as anterior segment dysgenesis, result from dynamic interactions between embryological, genetic, and developmental factors with frequent phenotypic overlap and extensive mutations at more than one genetic locus, and, for the most part, the mechanisms by which these gene mutations result in disease are not precisely known, but often relate to dosage sensitivity or alterations in protein function.
Alterations (either increased or decreased) in the levels of functional Pitx2 and Foxc1 cause disease. Without question, both proteins show dosage sensitivity, as minute deletions of either Foxc1 or Pitx2 lead to ARS by way of haploinsufficiency, and a previous study reported an ARS phenotype that may also result from the genetic duplication of Foxc1 (Berry et al., 2006). In addition, zebrafish carrying a heterozygous Foxc1 gene deletion mutation showed ocular defects from haploinsufficiency consistent with the ocular manifestations in their human counterparts (Ferre-Fernández et al., 2020). Similarly, an assessment of the requirements for Pitx2 showed that heterozygotes for either hypomorphic or null alleles in the Pitx2 gene have eye abnormalities consistent with ARS, indicating a Pitx2 dosage requirement for eye development in mice (Gage et al., 1999a,b). In zebrafish, morpholino knockdown of all known alternative Pitx2 proteins resulted in abnormal craniofacial and ocular development (Liu and Semina, 2012). Notably, hypermorphic alleles of the Pitx2 gene have been identified in rare cases of ARS (Priston et al., 2001), and a phenotype consistent with ARS was demonstrated in zebrafish following the microinjection of mRNA transcribed from plasmid DNA carrying a dominant negative gene mutation (K50E) in the human Pitx2 gene (Bohnsack et al., 2012). These data further reinforce the idea that Pitx2 levels must be strictly regulated for normal ocular development and function.
Notably, in vertebrates, Pax6 function is also hallmarked by gene dosage, and aniridia in humans is linked with mutations that lead to Pax6 haploinsufficiency. Indeed, the high and continuous expression of Pax6 in ectodermal tissues, such as the cornea, corneal epithelium, ciliary epithelium iris, and lens, is indispensable for the expression of structural genes (crystallins and cell adhesion molecules), signaling molecules affecting ocular NC migration (reviewed in Cvekl and Callaerts, 2017) and auxiliary transcription factors, including Six3, cellular musculoaponeurotic fibrosarcoma (c-Maf) and prospero homeobox 1 (Prox1) (Cvekl et al., 2004). Moreover, low transitory levels of Pax6 gene expression are necessary for the establishment of the aqueous outflow system (i.e., trabecular meshwork differentiation) and the generation of the corneal endothelium (Baulmann et al., 2002).
Missense mutations in the genes implicated in ocular diseases result in alterations in protein functions. For example, although Pitx2 haploinsufficiency has been demonstrated as a mechanism of ARS, a majority of the reported Pitx2 gene mutations indicate reduced or abolished protein function resulting from defective DNA binding, decreased target gene induction or both (Cox et al., 2002; Tumer and Bach-Holm, 2009). Similarly, biochemical analyses of human ARS-causing mutations revealed severe ARS mutations that completely disrupted DNA binding activity and significantly decreased the transactivation activity of Pitx2 (Kozlowski and Walter, 2000; Priston et al., 2001; Espinoza et al., 2002). These results showed that variations in Pitx2 activity, and thereby function, underlie the range of phenotypes observed in disorders affecting the anterior segment in ocular development. In addition, missense mutations of Foxc1 also show variable effects on its nuclear localization, DNA-binding ability and transactivation activity (Kozlowski and Walter, 2000; Priston et al., 2001; Espinoza et al., 2002; Seifi et al., 2017). The glaucomatous mutations of Cyp1b1 also reportedly decrease the stability, abundance, or catalytic activity of this enzyme (Jansson et al., 2001; Chavarria-Soley et al., 2008; Choudhary et al., 2008; Campos-Mollo et al., 2009; Lopez-Garrido et al., 2010, 2013; Medina-Trillo et al., 2016). Notably, recent studies have demonstrated the significance of Cyp1b1 for the establishment and functional viability of the trabecular meshwork (Mookherjee et al., 2012; Zhao et al., 2013). Thus, mutations affecting the function of the Cyp1b1 gene may also disrupt the function of the trabecular meshwork, resulting in dysregulated intraocular pressure and glaucomatous optic nerve damage. Similarly, investigations of the functional consequences of dominant and recessive Foxe3 gene mutations have shown altered and loss-of-function transcriptional activity, respectively (Islam et al., 2015).
The normal formation, migration and differentiation of NC cells is key to the evolution of various tissues and systems. Studying the signaling pathways important for the development of the NC may increase progress in the management of degenerative diseases affecting NC-derived craniofacial and ocular structures. Notably, the extensive migration and differentiation of NC cells into diverse cell types is reminiscent of the migration, invasion and proliferation of metastatic cancer cells and the multipotency of stem cells (Kerosuo et al., 2015; Maguire et al., 2015; Gallik et al., 2017), suggesting similar pathways and molecular mechanisms between these cell types. Additionally, the signals that are important during embryogenesis have recently been highlighted as key factors in the maintenance of adult tissues, and new therapeutic targets of adulthood diseases may be discovered through enhancements or modifications of these pathways. Although much is known on the level of the cranial NC, inadequate knowledge of a NC-derived stem cell population with regenerative properties to reestablish the structures in the anterior eye (cornea, iris, lens, or aqueous tract) is a notable therapeutic challenge. Recent studies have provided information concerning a NC-derived limbal stem cell niche that holds promise for regenerative therapies (Rama et al., 2010; Utheim et al., 2013; Sasamoto et al., 2018; Yazdanpanah et al., 2019). However, many unresolved issues remain. Determining the molecular markers that differentiate cranial from ocular NC, establishing the differences between the NC cell populations that enter the eye via the ocular fissure versus those that migrate between the optic cup and surface ectoderm and characterizing differences between NC cells that form the iris versus the cornea are paramount to achieving a wholistic understanding of the various genes and pathways that will lead to novel and innovative treatment options.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Akula, M., Park, J. W., and West-Mays, J. A. (2019). Relationship between neural crest cell specification and rare ocular diseases. J. Neurosci. Res. 97, 7–15. doi: 10.1002/jnr.24245
Alallah, J., Mohtisham, F., and Khan, Z. (2020). Spontaneous bilateral corneal perforation in a neonate: a case report. Int. J. Pediatr. Adolesc. Med. 7, 53–55.
Arikawa, A., Yoshida, S., Yoshikawa, H., Ishikawa, K., Yamaji, Y., Arita, R. I., et al. (2010). Case of novel PITX2 gene mutation associated with Peters’ anomaly and persistent hyperplastic primary vitreous. Eye 24, 391–393. doi: 10.1038/eye.2009.114
Baulmann, D. C., Ohlmann, A., Flugel-Koch, C., Goswami, S., Cvekl, A., and Tamm, E. R. (2002). Pax6 heterozygous eyes show defects in chamber angle differentiation that are associated with a wide spectrum of other anterior eye segment abnormalities. Mech. Dev. 118, 3–17. doi: 10.1016/s0925-4773(02)00260-5
Berker, N., Alanay, Y., Elgin, U., Volkan-Salanci, B., Simsek, T., Akarsu, N., et al. (2009). A new autosomal dominant Peters’ anomaly phenotype expanding the anterior segment dysgenesis spectrum. Acta Ophthalmol. 87, 52–57. doi: 10.1111/j.1600-0420.2007.01082.x
Berry, F. B., Lines, M. A., Oas, J. M., Footz, T., Underhill, D. A., Gage, P. J., et al. (2006). Functional interactions between FOXC1 and PITX2 underlie the sensitivity to FOXC1 gene dose in Axenfeld-Rieger syndrome and anterior segment dysgenesis. Hum. Mol. Genet. 15, 905–919. doi: 10.1093/hmg/ddl008
Bohnsack, B. L., and Kahana, A. (2013). Thyroid hormone and retinoic acid interact to regulate Zebrafish craniofacial neural crest development. Dev. Biol. 373, 300–309. doi: 10.1016/j.ydbio.2012.11.005
Bohnsack, B. L., Kasprick, D. S., Kish, P. E., Goldman, D., and Kahana, A. (2012). A Zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Invest. Ophthalmol. Vis. Sci. 53, 7–22. doi: 10.1167/iovs.11-8494
Brennan, D., and Giles, S. (2014). Ocular involvement in fetal alcohol spectrum disorder: a review. Curr. Pharm. Des. 20, 5377–5387. doi: 10.2174/1381612820666140205144114
Bryan, C. D., Casey, M. A., Pfeiffer, R. L., Jones, B. W., and Kwan, K. M. (2020). Optic cup morphogenesis requires neural crest-mediated basement membrane assembly. Development 147:dev181420. doi: 10.1242/dev.181420
Campos-Mollo, E., Lopez-Garrido, M. P., Blanco-Marchite, C., Garcia-Feijoo, J., Peralta, J., Belmonte-Martinez, J., et al. (2009). CYP1B1 mutations in Spanish patients with primary congenital glaucoma: phenotypic and functional variability. Mol. Vis. 15, 417–431.
Chambers, D., Wilson, L., Maden, M., and Lumsden, A. (2007). RALDH-independent generation of retinoic acid during vertebrate embryogenesis by CYP1B1. Development 134, 1369–1383. doi: 10.1242/dev.02815
Chang, Y. L., Chao, A. S., Chou, C. Y., Chang, S. D., Chiang, M. C., and Lee, Y. S. (2020). A donor twin discordant with Peters anomaly in a twin-twin transfusion syndrome case: a case report. BMC Pregnancy Childbirth 20:558. doi: 10.1186/s12884-020-03269-0
Chavarria-Soley, G., Sticht, H., Aklillu, E., Ingelman-Sundberg, M., Pasutto, F., Reis, A., et al. (2008). Mutations in CYP1B1 cause primary congenital glaucoma by reduction of either activity or abundance of the enzyme. Hum. Mutat. 29, 1147–1153. doi: 10.1002/humu.20786
Chawla, B., Schley, E., Williams, A. L., and Bohnsack, B. L. (2016). Retinoic acid and Pitx2 regulate early neural crest survival and migration in craniofacial and ocular development. Birth Defects Res. B Dev. Reprod. Toxicol. 107, 126–135. doi: 10.1002/bdrb.21177
Chawla, B., Swain, W., Williams, A. L., and Bohnsack, B. L. (2018). Retinoic acid maintains function of neural crest-derived ocular and craniofacial structures in adult Zebrafish. Invest. Ophthalmol. Vis. Sci. 59, 1924–1935. doi: 10.1167/iovs.17-22845
Chen, L., and Gage, P. J. (2016). Heterozygous Pitx2 null mice accurately recapitulate the ocular features of axenfeld-rieger syndrome and congenital glaucoma. Invest. Ophthalmol. Vis. Sci. 57, 5023–5030. doi: 10.1167/iovs.16-19700
Choudhary, D., Jansson, I., Sarfarazi, M., and Schenkman, J. B. (2008). Characterization of the biochemical and structural phenotypes of four CYP1B1 mutations observed in individuals with primary congenital glaucoma. Pharmacogenet. Genomics 18, 665–676. doi: 10.1097/fpc.0b013e3282ff5a36
Cordero, D. R., Brugmann, S., Chu, Y., Bajpai, R., Jame, M., and Helms, J. A. (2011). Cranial neural crest cells on the move: their roles in craniofacial development. Am. J. Med. Genet. A 155A, 270–279. doi: 10.1002/ajmg.a.33702
Cox, C. J., Espinoza, H. M., McWilliams, B., Chappell, K., Morton, L., Hjalt, T. A., et al. (2002). Differential regulation of gene expression by PITX2 isoforms. J. Biol. Chem. 277, 25001–25010. doi: 10.1074/jbc.m201737200
Cvekl, A., and Callaerts, P. (2017). PAX6: 25th anniversary and more to learn. Exp. Eye Res. 156, 10–21. doi: 10.1016/j.exer.2016.04.017
Cvekl, A., Yang, Y., Chauhan, B. K., and Cveklova, K. (2004). Regulation of gene expression by Pax6 in ocular cells: a case of tissue-preferred expression of crystallins in lens. Int. J. Dev. Biol. 48, 829–844. doi: 10.1387/ijdb.041866ac
Dolezal, K. A., Besirli, C. G., Mian, S. I., Sugar, A., Moroi, S. E., and Bohnsack, B. L. (2019). Glaucoma and cornea surgery outcomes in peters anomaly. Am. J. Ophthalmol. 208, 367–375. doi: 10.1016/j.ajo.2019.08.012
Draut, H., Liebenstein, T., and Begemann, G. (2019). New insights into the control of cell fate choices and differentiation by retinoic acid in cranial, axial and caudal structures. Biomolecules 9:860. doi: 10.3390/biom9120860
Duester, G. (2009). Keeping an eye on retinoic acid signaling during eye development. Chem. Biol. Interact. 178, 178–181. doi: 10.1016/j.cbi.2008.09.004
Eason, J., Williams, A. L., Chawla, B., Apsey, C., and Bohnsack, B. L. (2017). Differences in neural crest sensitivity to ethanol account for the infrequency of anterior segment defects in the eye compared with craniofacial anomalies in a Zebrafish model of fetal alcohol syndrome. Birth Defects Res. 109, 1212–1227. doi: 10.1002/bdr2.1069
Enwright, J. F. III, and Grainger, R. M. (2000). Altered retinoid signaling in the heads of small eye mouse embryos. Dev. Biol. 221, 10–22. doi: 10.1006/dbio.2000.9652
Espinoza, H. M., Cox, C. J., Semina, E. V., and Amendt, B. A. (2002). A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome. Hum. Mol. Genet. 11, 743–753. doi: 10.1093/hmg/11.7.743
Evans, A. L., and Gage, P. J. (2005). Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum. Mol. Genet. 14, 3347–3359. doi: 10.1093/hmg/ddi365
Ferre-Fernández, J. J., Thompson, S., Collery, R. F., Nordquist, E., Lincoln, J., and Semina, E. V. (2020). Disruption of foxc1 genes in Zebrafish results in dosage-dependent phenotypes overlapping Axenfeld-Rieger syndrome. Hum. Mol. Genet. 29, 2723–2735. doi: 10.1093/hmg/ddaa163
Gage, P. J., Kuang, C., and Zacharias, A. L. (2014). The homeodomain transcription factor PITX2 is required for specifying correct cell fates and establishing angiogenic privilege in the developing cornea. Dev. Dyn. 243, 1391–1400. doi: 10.1002/dvdy.24165
Gage, P. J., Qian, M., Wu, D., and Rosenberg, K. I. (2008). The canonical Wnt signaling antagonist DKK2 is an essential effector of PITX2 function during normal eye development. Dev. Biol. 317, 310–324. doi: 10.1016/j.ydbio.2008.02.030
Gage, P. J., Rhoades, W., Prucka, S. K., and Hjalt, T. (2005). Fate maps of neural crest and mesoderm in the mammalian eye. Investig. Ophthalmol. Vis. Sci. 46, 4200–4208. doi: 10.1167/iovs.05-0691
Gage, P. J., Suh, H., and Camper, S. A. (1999a). Dosage requirement of Pitx2 for development of multiple organs. Development 126, 4643–4651.
Gage, P. J., Suh, H., and Camper, S. A. (1999b). The bicoid-related Pitx gene family in development. Mamm. Genome 10, 197–200. doi: 10.1007/s003359900970
Gallik, K. L., Treffy, R. W., Nacke, L. M., Ahsan, K., Rocha, M., Green-Saxena, A., et al. (2017). Neural crest and cancer: divergent travelers on similar paths. Mech. Dev. 148, 89–99. doi: 10.1016/j.mod.2017.08.002
Gandhi, S., and Bronner, M. E. (2018). Insights into neural crest development from studies of avian embryos. Int. J. Dev. Biol. 62, 183–194. doi: 10.1387/ijdb.180038sg
Garcia-Montalvo, I. A., Pelcastre-Luna, E., Nelson-Mora, J., Buentello-Volante, B., Miranda-Duarte, A., and Zenteno, J. C. (2014). Mutational screening of FOXE3, GDF3, ATOH7, and ALDH1A3 in congenital ocular malformations. Possible contribution of the FOXE3 p.VAL201MET variant to the risk of severe eye malformations. Ophthalmic Genet. 35, 190–192. doi: 10.3109/13816810.2014.903983
Gould, D. B., and John, S. W. (2002). Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum. Mol. Genet. 11, 1185–1193. doi: 10.1093/hmg/11.10.1185
Grant, M. K., Bobilev, A. M., Rasys, A. M., Branson Byers, J., Schriever, H. C., Hekmatyar, K., et al. (2020). Global and age-related neuroanatomical abnormalities in a Pax6-deficient mouse model of aniridia suggests a role for Pax6 in adult structural neuroplasticity. Brain Res. 1732:146698. doi: 10.1016/j.brainres.2020.146698
Green, S. A., Simoes-Costa, M., and Bronner, M. E. (2015). Evolution of vertebrates as viewed from the crest. Nature 520, 474–482. doi: 10.1038/nature14436
Grocott, T., Johnson, S., Bailey, A. P., and Streit, A. (2011). Neural crest cells organize the eye via TGF-beta and canonical Wnt signalling. Nat. Commun. 2:265.
Harissi-Dagher, M., and Colby, K. (2008). Anterior segment dysgenesis: peters anomaly and sclerocornea. Int. Ophthalmol. Clin. 48, 35–42. doi: 10.1097/iio.0b013e318169526c
Hassed, S. J., Li, S., Xu, W., and Taylor, A. C. (2017). A novel mutation in PITX2 in a patient with axenfeld-rieger syndrome. Mol. Syndromol. 8, 107–109. doi: 10.1159/000454963
Hebert, S. L., Fitzpatrick, K. R., McConnell, S. A., Cucak, A., Yuan, C., and McLoon, L. K. (2017). Effects of retinoic acid signaling on extraocular muscle myogenic precursor cells in vitro. Exp. Cell. Res. 361, 101–111. doi: 10.1016/j.yexcr.2017.10.007
Hocking, J. C., Famulski, J. K., Yoon, K. H., Widen, S. A., Bernstein, C. S., Koch, S., et al. (2018). Morphogenetic defects underlie Superior Coloboma, a newly identified closure disorder of the dorsal eye. PLoS Genet. 14:e1007246. doi: 10.1371/journal.pgen.1007246
Honkanen, R. A., Nishimura, D. Y., Swiderski, R. E., Bennett, S. R., Hong, S., Kwon, Y. H., et al. (2003). A family with Axenfeld-Rieger syndrome and peters anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene. Am. J. Ophthalmol. 135, 368–375. doi: 10.1016/s0002-9394(02)02061-5
Islam, L., Kelberman, D., Williamson, L., Lewis, N., Glindzicz, M. B., Nischal, K. K., et al. (2015). Functional analysis of FOXE3 mutations causing dominant and recessive ocular anterior segment disease. Hum. Mutat. 36, 296–300. doi: 10.1002/humu.22741
Jansson, I., Stoilov, I., Sarfarazi, M., and Schenkman, J. B. (2001). Effect of two mutations of human CYP1B1, G61E and R469W, on stability and endogenous steroid substrate metabolism. Pharmacogenetics 11, 793–801. doi: 10.1097/00008571-200112000-00007
Ji, Y., Buel, S. M., and Amack, J. D. (2016). Mutations in Zebrafish pitx2 model congenital malformations in Axenfeld-Rieger syndrome but do not disrupt left-right placement of visceral organs. Dev. Biol. 416, 69–81. doi: 10.1016/j.ydbio.2016.06.010
Jinno, H., Morozova, O., Jones, K. L., Biernaskie, J. A., Paris, M., Hosokawa, R., et al. (2010). Convergent genesis of an adult neural crest-like dermal stem cell from distinct developmental origins. Stem Cells 28, 2027–2040. doi: 10.1002/stem.525
Katz, B., Enright, J., Couch, S., Harocopos, G., and Lee, A. R. (2020). Atypical presentation of Cat Eye Syndrome in an infant with Peters anomaly and microphthalmia with cyst. Ophthalmic Genet. 41, 645–649. doi: 10.1080/13816810.2020.1814346
Kerosuo, L., Nie, S., Bajpai, R., and Bronner, M. E. (2015). Crestospheres: long-term maintenance of multipotent, premigratory neural crest stem cells. Stem Cell Rep. 5, 499–507. doi: 10.1016/j.stemcr.2015.08.017
Kish, P. E., Bohnsack, B. L., Gallina, D., Kasprick, D. S., and Kahana, A. (2011). The eye as an organizer of craniofacial development. Genesis 49, 222–230. doi: 10.1002/dvg.20716
Kitamura, K., Miura, H., Miyagawa-Tomita, S., Yanazawa, M., Katoh-Fukui, Y., Suzuki, R., et al. (1999). Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development 126, 5749–5758.
Kletke, S. N., Vincent, A., Maynes, J. T., Elbaz, U., Mireskandari, K., Lam, W. C., et al. (2020). A de novo mutation in PITX2 underlies a unique form of Axenfeld-Rieger syndrome with corneal neovascularization and extensive proliferative vitreoretinopathy. Ophthalmic Genet. 41, 358–362. doi: 10.1080/13816810.2020.1768556
Koo, H. Y., and Kume, T. (2013). FoxC1-dependent regulation of vascular endothelial growth factor signaling in corneal avascularity. Trends Cardiovasc. Med. 23, 1–4. doi: 10.1016/j.tcm.2012.08.002
Kozlowski, K., and Walter, M. A. (2000). Variation in residual PITX2 activity underlies the phenotypic spectrum of anterior segment developmental disorders. Hum. Mol. Genet. 9, 2131–2139. doi: 10.1093/hmg/9.14.2131
Kumar, S., and Duester, G. (2010). Retinoic acid signaling in perioptic mesenchyme represses Wnt signaling via induction of Pitx2 and Dkk2. Dev. Biol. 340, 67–74. doi: 10.1016/j.ydbio.2010.01.027
Lee, J., Suh, Y., Jeong, H., Kim, G. H., Byeon, S. H., Han, J., et al. (2020). Aberrant expression of PAX6 gene associated with classical aniridia: identification and functional characterization of novel noncoding mutations. J. Hum. Genet. [Epub ahead of print].
Li, N., Zhou, Y., Du, L., Wei, M., and Chen, X. (2011). Overview of cytochrome p450 1B1 gene mutations in patients with primary congenital glaucoma. Exp. Eye Res. 93, 572–579. doi: 10.1016/j.exer.2011.07.009
Lim, H. T., Kim, D. H., and Kim, H. (2017). PAX6 aniridia syndrome: clinics, genetics, and therapeutics. Curr. Opin. Ophthalmol. 28, 436–447. doi: 10.1097/icu.0000000000000405
Lima Cunha, D., Arno, G., Corton, M., and Moosajee, M. (2019). The spectrum of PAX6 mutations and genotype-phenotype correlations in the eye. Genes 10:1050. doi: 10.3390/genes10121050
Liu, Y., and Semina, E. V. (2012). pitx2 Deficiency results in abnormal ocular and craniofacial development in Zebrafish. PLoS One 7:e30896. doi: 10.1371/journal.pone.0030896
Lopez-Garrido, M. P., Blanco-Marchite, C., Sanchez-Sanchez, F., Lopez-Sanchez, E., Chaques-Alepuz, V., Campos-Mollo, E., et al. (2010). Functional analysis of CYP1B1 mutations and association of heterozygous hypomorphic alleles with primary open-angle glaucoma. Clin. Genet. 77, 70–78. doi: 10.1111/j.1399-0004.2009.01284.x
Lopez-Garrido, M. P., Medina-Trillo, C., Morales-Fernandez, L., Garcia-Feijoo, J., Martinez-de-la-Casa, J. M., Garcia-Anton, M., et al. (2013). Null CYP1B1 genotypes in primary congenital and nondominant juvenile glaucoma. Ophthalmology 120, 716–723. doi: 10.1016/j.ophtha.2012.09.016
Maguire, L. H., Thomas, A. R., and Goldstein, A. M. (2015). Tumors of the neural crest: common themes in development and cancer. Dev. Dyn. 244, 311–322. doi: 10.1002/dvdy.24226
Martik, M. L., and Bronner, M. E. (2017). Regulatory logic underlying diversification of the neural crest. Trends Genet 33, 715–727. doi: 10.1016/j.tig.2017.07.015
Martino, V. B., Sabljic, T., Deschamps, P., Green, R. M., Akula, M., Peacock, E., et al. (2016). Conditional deletion of AP-2β in mouse cranial neural crest results in anterior segment dysgenesis and early-onset glaucoma. Dis. Model. Mech. 9, 849–861. doi: 10.1242/dmm.025262
Matt, N., Dupe, V., Garnier, J. M., Dennefeld, C., Chambon, P., Mark, M., et al. (2005). Retinoic acid-dependent eye morphogenesis is orchestrated by neural crest cells. Development 132, 4789–4800. doi: 10.1242/dev.02031
Matt, N., Ghyselinck, N. B., Pellerin, I., and Dupe, V. (2008). Impairing retinoic acid signalling in the neural crest cells is sufficient to alter entire eye morphogenesis. Dev. Biol. 320, 140–148. doi: 10.1016/j.ydbio.2008.04.039
Medina-Martinez, O., Brownell, I., Amaya-Manzanares, F., Hu, Q., Behringer, R. R., and Jamrich, M. (2005). Severe defects in proliferation and differentiation of lens cells in Foxe3 null mice. Mol. Cell Biol. 25, 8854–8863. doi: 10.1128/mcb.25.20.8854-8863.2005
Medina-Trillo, C., Ferre-Fernandez, J. J., Aroca-Aguilar, J. D., Bonet-Fernandez, J. M., and Escribano, J. (2016). Functional characterization of eight rare missense CYP1B1 variants involved in congenital glaucoma and their association with null genotypes. Acta Ophthalmol. 94, e555–e560.
Miesfeld, J. B., and Brown, N. L. (2019). Eye organogenesis: a hierarchical view of ocular development. Curr. Top. Dev. Biol. 132, 351–393. doi: 10.1016/bs.ctdb.2018.12.008
Mookherjee, S., Acharya, M., Banerjee, D., Bhattacharjee, A., and Ray, K. (2012). Molecular basis for involvement of CYP1B1 in MYOC upregulation and its potential implication in glaucoma pathogenesis. PLoS One 7:e45077. doi: 10.1371/journal.pone.0045077
Mork, L., and Crump, G. (2015). Zebrafish craniofacial development: a window into early patterning. Curr. Top. Dev. Biol. 115, 235–269. doi: 10.1016/bs.ctdb.2015.07.001
Ozeki, H., Shirai, S., Nozaki, M., Sakurai, E., Mizuno, S., Ashikari, M., et al. (2000). Ocular and systemic features of Peters’ anomaly. Graefes Arch. Clin. Exp. Ophthalmol. 238, 833–839. doi: 10.1007/s004170000177
Pla, P., and Monsoro-Burq, A. H. (2018). The neural border: induction, specification and maturation of the territory that generates neural crest cells. Dev. Biol. 444(Suppl. 1), S36–S46.
Prasad, M. S., Charney, R. M., and Garcia-Castro, M. I. (2019). Specification and formation of the neural crest: perspectives on lineage segregation. Genesis 57:e23276. doi: 10.1002/dvg.23276
Pressman, C. L., Chen, H., and Johnson, R. L. (2000). LMX1B, a LIM homeodomain class transcription factor, is necessary for normal development of multiple tissues in the anterior segment of the murine eye. Genesis 26, 15–25. doi: 10.1002/(sici)1526-968x(200001)26:1<15::aid-gene5>3.0.co;2-v
Priston, M., Kozlowski, K., Gill, D., Letwin, K., Buys, Y., Levin, A. V., et al. (2001). Functional analyses of two newly identified PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome. Hum. Mol. Genet. 10, 1631–1638. doi: 10.1093/hmg/10.16.1631
Rama, P., Matuska, S., Paganoni, G., Spinelli, A., De Luca, M., and Pellegrini, G. (2010). Limbal stem-cell therapy and long-term corneal regeneration. N. Engl. J. Med. 363, 147–155. doi: 10.1056/nejmoa0905955
Reis, L. M., Tyler, R. C., Volkmann Kloss, B. A., Schilter, K. F., Levin, A. V., Lowry, R. B., et al. (2012). PITX2 and FOXC1 spectrum of mutations in ocular syndromes. Eur. J. Hum. Genet. 20, 1224–1233. doi: 10.1038/ejhg.2012.80
Rogers, C. D., and Nie, S. (2018). Specifying neural crest cells: from chromatin to morphogens and factors in between. Wiley Interdiscip. Rev. Dev. Biol. 7:e322. doi: 10.1002/wdev.322
Rosemann, M., Ivashkevich, A., Favor, J., Dalke, C., Holter, S. M., Becker, L., et al. (2010). Microphthalmia, parkinsonism, and enhanced nociception in Pitx3 (416insG) mice. Mamm. Genome 21, 13–27. doi: 10.1007/s00335-009-9235-0
Samara, A., and Eldaya, R. W. (2020). Ocular and brain imaging findings in Peters’ anomaly: a case report and literature review. Radiol. Case Rep. 15, 863–866. doi: 10.1016/j.radcr.2020.04.011
Sannan, N. S., Gregory-Evans, C. Y., Lyons, C. J., Lehman, A. M., Langlois, S., Warner, S. J., et al. (2017). Correlation of novel PAX6 gene abnormalities in aniridia and clinical presentation. Can. J. Ophthalmol. 52, 570–577. doi: 10.1016/j.jcjo.2017.04.006
Sasamoto, Y., Ksander, B. R., Frank, M. H., and Frank, N. Y. (2018). Repairing the corneal epithelium using limbal stem cells or alternative cell-based therapies. Expert Opin. Biol. Ther. 18, 505–513. doi: 10.1080/14712598.2018.1443442
Seifi, M., Footz, T., Taylor, S. A., and Walter, M. A. (2017). Comparison of bioinformatics prediction, molecular modeling, and functional analyses of foxc1 mutations in patients with axenfeld-rieger syndrome. Hum. Mutat. 38, 169–179. doi: 10.1002/humu.23141
Seo, S., Chen, L., Liu, W., Zhao, D., Schultz, K. M., Sasman, A., et al. (2017). Foxc1 and Foxc2 in the neural crest are required for ocular anterior segment development. Invest. Ophthalmol. Vis. Sci. 58, 1368–1377. doi: 10.1167/iovs.16-21217
Seo, S., Singh, H. P., Lacal, P. M., Sasman, A., Fatima, A., Liu, T., et al. (2012). Forkhead box transcription factor FoxC1 preserves corneal transparency by regulating vascular growth. Proc. Natl. Acad. Sci. U.S.A. 109, 2015–2020. doi: 10.1073/pnas.1109540109
Simoes-Costa, M., and Bronner, M. E. (2013). Insights into neural crest development and evolution from genomic analysis. Genome Res. 23, 1069–1080. doi: 10.1101/gr.157586.113
Simoes-Costa, M., and Bronner, M. E. (2015). Establishing neural crest identity: a gene regulatory recipe. Development 142, 242–257. doi: 10.1242/dev.105445
Smith, R. S., Zabaleta, A., Kume, T., Savinova, O. V., Kidson, S. H., Martin, J. E., et al. (2000). Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum. Mol. Genet. 9, 1021–1032. doi: 10.1093/hmg/9.7.1021
Sowden, J. C. (2007). Molecular and developmental mechanisms of anterior segment dysgenesis. Eye 21, 1310–1318. doi: 10.1038/sj.eye.6702852
Summers, K. M., Withers, S. J., Gole, G. A., Piras, S., and Taylor, P. J. (2008). Anterior segment mesenchymal dysgenesis in a large Australian family is associated with the recurrent 17 bp duplication in PITX3. Mol. Vis. 14, 2010–2015.
Syrimis, A., Nicolaou, N., Alexandrou, A., Papaevripidou, I., Nicolaou, M., Loukianou, E., et al. (2018). Molecular analysis of Cypriot families with aniridia reveals a novel PAX6 mutation. Mol. Med. Rep. 18, 1623–1627.
Takamiya, M., Stegmaier, J., Kobitski, A. Y., Schott, B., Weger, B. D., Margariti, D., et al. (2020). Pax6 organizes the anterior eye segment by guiding two distinct neural crest waves. PLoS Genet. 16:e1008774. doi: 10.1371/journal.pgen.1008774
Takamiya, M., Weger, B. D., Schindler, S., Beil, T., Yang, L., Armant, O., et al. (2015). Molecular description of eye defects in the Zebrafish Pax6b mutant, sunrise, reveals a Pax6b-dependent genetic network in the developing anterior chamber. PLoS One 10:e0117645. doi: 10.1371/journal.pone.0117645
Tumer, Z., and Bach-Holm, D. (2009). Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur. J. Hum. Genet. 17, 1527–1539. doi: 10.1038/ejhg.2009.93
Utheim, T. P., Lyberg, T., and Raeder, S. (2013). The culture of limbal epithelial cells. Methods Mol. Biol. 1014, 103–129.
Van Der Meulen, K. L., Vocking, O., Weaver, M. L., Meshram, N. N., and Famulski, J. K. (2020). Spatiotemporal characterization of anterior segment mesenchyme heterogeneity during Zebrafish ocular anterior segment development. Front. Cell. Dev. Biol. 8:379. doi: 10.3389/fcell.2020.00379
Vollrath, D., Jaramillo-Babb, V. L., Clough, M. V., McIntosh, I., Scott, K. M., Lichter, P. R., et al. (1998). Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Hum. Mol. Genet. 7, 1091–1098. doi: 10.1093/hmg/7.7.1091
Wada, K., Matsushima, Y., Tada, T., Hasegawa, S., Obara, Y., Yoshizawa, Y., et al. (2014). Expression of truncated PITX3 in the developing lens leads to microphthalmia and aphakia in mice. PLoS One 9:e111432. doi: 10.1371/journal.pone.0111432
Wang, G. M., Prasov, L., Al-Hasani, H., Marrs, C., Tolia, S., Wiinikka-Buesser, L., et al. (2018). Phenotypic variation in a four-generation family with aniridia carrying a novel PAX6 mutation. J. Ophthalmol. 2018:5978293.
Wang, W. D., Melville, D., Montero-Balaguer, M., Hatzopoulos, A., and Knapik, E. (2011). Tfap2a and Foxd3 regulate early steps in the development of the neural crest progenitor population. Dev. Biol. 360, 173–185. doi: 10.1016/j.ydbio.2011.09.019
Wawrocka, A., and Krawczynski, M. R. (2018). The genetics of aniridia - simple things become complicated. J. Appl. Genet. 59, 151–159. doi: 10.1007/s13353-017-0426-1
Weisschuh, N., Wolf, C., Wissinger, B., and Gramer, E. (2008). A novel mutation in the FOXC1 gene in a family with Axenfeld-Rieger syndrome and Peters’ anomaly. Clin. Genet. 74, 476–480. doi: 10.1111/j.1399-0004.2008.01025.x
Wen, W., Pillai-Kastoori, L., Wilson, S. G., and Morris, A. C. (2015). Sox4 regulates choroid fissure closure by limiting Hedgehog signaling during ocular morphogenesis. Dev. Biol. 399, 139–153. doi: 10.1016/j.ydbio.2014.12.026
Whikehart, D. R. (2010). “Corneal endothelium: overview,” in Encyclopedia of the Eye, eds R. Dana, D. A. Dartt, and J. C. Besharse (Amsterdam: Elsevier), 424–434. doi: 10.1016/b978-0-12-374203-2.00074-9
Williams, A. L., and Bohnsack, B. L. (2015). Neural crest derivatives in ocular development: discerning the eye of the storm. Birth Defects Res. C Embryo Today 105, 87–95. doi: 10.1002/bdrc.21095
Williams, A. L., and Bohnsack, B. L. (2017). Multi-photon time lapse imaging to visualize development in real-time: visualization of migrating neural crest cells in Zebrafish embryos. J. Vis. Exp. 126:56214.
Williams, A. L., and Bohnsack, B. L. (2019). What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 57:e23308. doi: 10.1002/dvg.23308
Yasue, A., Kono, H., Habuta, M., Bando, T., Sato, K., Inoue, J., et al. (2017). Relationship between somatic mosaicism of Pax6 mutation and variable developmental eye abnormalities-an analysis of CRISPR genome-edited mouse embryos. Sci. Rep. 7:53.
Yazdanpanah, G., Haq, Z., Kang, K., Jabbehdari, S., Rosenblatt, M. L., and Djalilian, A. R. (2019). Strategies for reconstructing the limbal stem cell niche. Ocul. Surf. 17, 230–240. doi: 10.1016/j.jtos.2019.01.002
Yuan, F. Y. Y., Fan, H., Li, Y., Lu, L., Liu, J., Feng, W., et al. (2020). MicroRNA-135a protects against ethanol-induced apoptosis in neural crest cells and craniofacial defects in Zebrafish by modulating the Siah1/p38/p53 pathway. Front. Cell. Dev. Biol. 8:583959. doi: 10.3389/fcell.2020.583959
Zacharias, A. L., and Gage, P. J. (2010). Canonical Wnt/beta-catenin signaling is required for maintenance but not activation of Pitx2 expression in neural crest during eye development. Dev. Dyn. 239, 3215–3225. doi: 10.1002/dvdy.22459
Zazo Seco, C., Plaisancie, J., Lupasco, T., Michot, C., Pechmeja, J., Delanne, J., et al. (2018). Identification of PITX3 mutations in individuals with various ocular developmental defects. Ophthalmic Genet. 39, 314–320. doi: 10.1080/13816810.2018.1430243
Zepeda, E. M., Branham, K., Moroi, S. E., and Bohnsack, B. L. (2020). Surgical outcomes of Glaucoma associated with Axenfeld-Rieger syndrome. BMC Ophthalmol. 20:172. doi: 10.1186/s12886-020-01417
Zhao, Y., Sorenson, C. M., and Sheibani, N. (2015). Cytochrome P450 1B1 and primary congenital glaucoma. J. Ophthalmic Vis. Res. 10, 60–67. doi: 10.4103/2008-322x.156116
Keywords: neural crest, periocular mesenchyme, ocular development, anterior segment, ocular diseases
Citation: Williams AL and Bohnsack BL (2020) The Ocular Neural Crest: Specification, Migration, and Then What? Front. Cell Dev. Biol. 8:595896. doi: 10.3389/fcell.2020.595896
Received: 17 August 2020; Accepted: 04 December 2020;
Published: 23 December 2020.
Edited by:
Poongodi Geetha-Loganathan, SUNY Oswego, United StatesReviewed by:
Laura Kerosuo, National Institutes of Health (NIH), United StatesCopyright © 2020 Williams and Bohnsack. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brenda L. Bohnsack, YmJvaG5zYWNrQGx1cmllY2hpbGRyZW5zLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.