 Youssif Ben Zablah
Youssif Ben Zablah Neil Merovitch
Neil Merovitch Zhengping Jia
Zhengping Jia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 12 November 2020
Sec. Cell Adhesion and Migration
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.594998
This article is part of the Research TopicEvolution, Emerging Functions and Structure of Actin-Binding ProteinsView all 32 articles
Actin-depolymerization factor (ADF)/cofilin, a family of actin-binding proteins, are critical for the regulation of actin reorganization in response to various signals. Accumulating evidence indicates that ADF/cofilin also play important roles in neuronal structure and function, including long-term potentiation and depression. These are the most extensively studied forms of long-lasting synaptic plasticity and are widely regarded as cellular mechanisms underlying learning and memory. ADF/cofilin regulate synaptic function through their effects on dendritic spines and the trafficking of glutamate receptors, the principal mediator of excitatory synaptic transmission in vertebrates. Regulation of ADF/cofilin involves various signaling pathways converging on LIM domain kinases and slingshot phosphatases, which phosphorylate/inactivate and dephosphorylate/activate ADF/cofilin, respectively. Actin-depolymerization factor/cofilin activity is also regulated by other actin-binding proteins, activity-dependent subcellular distribution and protein translation. Abnormalities in ADF/cofilin have been associated with several neurodegenerative disorders such as Alzheimer’s disease. Therefore, investigating the roles of ADF/cofilin in the brain is not only important for understanding the fundamental processes governing neuronal structure and function, but also may provide potential therapeutic strategies to treat brain disorders.
Long-lasting changes in the efficacy of synaptic transmission, including long-term potentiation (LTP) and depression (LTD), are widely regarded as the key mechanisms underlying memory storage (Bliss and Collingridge, 1993; Malenka and Bear, 2004; Citri and Malenka, 2008; Neves et al., 2008; Kessels and Malinow, 2009; Kandel et al., 2014; Mateos-Aparicio and Rodríguez-Moreno, 2019). Synaptic plasticity involves changes in postsynaptic reorganization, including glutamate receptor trafficking and morphological remodeling of dendritic spines (Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Collingridge et al., 2004; Lamprecht and LeDoux, 2004; Carlisle and Kennedy, 2005; Segal, 2005; Alvarez and Sabatini, 2007; Ho et al., 2011; Huganir and Nicoll, 2013; Henley and Wilkinson, 2016; Diering and Huganir, 2018), both of which are regulated by the actin cytoskeleton (Cingolani and Goda, 2008; Spence and Soderling, 2015; Nakahata and Yasuda, 2018). Evidence suggests that actin-binding proteins are involved in receptor trafficking as well as morphological changes at the synapse and consequently affect learning and memory (Lamprecht, 2011, 2016; Spence and Soderling, 2015; Borovac et al., 2018). Abnormalities in these proteins are associated with several neurological disorders (Lian and Sheen, 2015). In this review we will focus on the role of actin-depolymerization factor (ADF)/cofilin in the regulation of LTP, LTD, dendritic spines and their dysfunction in Alzheimer’s disease (AD).
Cofilin is a member of the actin-depolymerizing protein family that is important for the regulation of actin cytoskeleton dynamics (Ridley, 2011; Kanellos and Frame, 2016). This family includes cofilin-1 (n-cofilin, non-muscle), cofilin-2 (muscle cofilin) and actin-depolymerization factor (ADF, destrin) and is well conserved among eukaryotes (Maciver and Hussey, 2002). These proteins have a molecular mass of 15–19 kDa and share multiple structural similarities. Each consists of an actin depolymerizing factor homology (ADF-H) domain, which allows for binding to actin subunits, a central alpha helix, a N-terminus extension and a C-terminus helix (Lappalainen et al., 1998; Shishkin et al., 2016). Despite their similarities at the molecular level, these isoforms differ in their degree of affinity for actin (Bamburg, 1999; Vartiainen et al., 2002; Yeoh et al., 2002). Actin-depolymerization factor and cofilin-1 can bind to actin filaments with similar degrees of affinity, whereas cofilin-2 is less efficient at depolymerization (Vartiainen et al., 2002). While ADF is better at sequestering monomeric actin, cofilin-1 is more efficient at nucleation and severing actin filaments (Chin et al., 2016). These biochemical differences reflect variations in the cellular expression between isoforms, where ADF and cofilin-1 are mainly expressed in tissues with higher actin turnover. Specifically, while cofilin-1 is expressed in all cell types, ADF is mainly expressed in neuronal, epithelial, and endothelial cells (Kanellos and Frame, 2016). Cofilin-2 is also expressed in selected tissues including muscles and brain (Thirion et al., 2001; Agrawal et al., 2012; Gurniak et al., 2014). This review will discuss ADF and cofilin-1, which are expressed in neuronal cells. Many studies addressing the roles of ADF/cofilin do not specify which isoform as many of their functions overlap. Also, most antibodies do not differentiate between these isoforms and rescue experiments often use cofilin from lower level eukaryotes that express only one isoform (Moon et al., 1993; Kanellos and Frame, 2016). For these reasons and the sake of simplicity, this group of actin depolymerizing factors will be referred to collectively as ADF/cofilin, except in studies where specific isoforms have been addressed.
The most characterized role of ADF/cofilin is the regulation of actin reorganization and their capacity to increase actin filament turnover (Pollard and Borisy, 2003; Brieher, 2013). Treadmilling is the most accepted model for actin turnover (Figure 1; Wegner, 1982; Blanchoin et al., 2014). In this model, steady state actin filaments preferentially grow at one end, known as the barbed end, by association of ATP-bound actin monomers, whereas actin monomers dissociate at the other end, known as the pointed end (Pollard and Borisy, 2003; Lee and Dominguez, 2010). Following the addition of the ATP-bound actin subunit, ATP undergoes hydrolysis into ADP and Pi, after which Pi is released, leaving ADP-bound subunits at the pointed end. ADP-bound subunits are more prone to dissociate and return to the actin monomers pool. Dissociated ADP-bound subunits then exchange ADP into ATP before entering the cycle again (Carlier and Pantaloni, 1986; Blanchoin and Pollard, 2002; Pollard and Borisy, 2003). The dynamic turnover of actin filaments can be enhanced by an increase in the number of filament ends due to severing of existing filaments (Ichetovkin et al., 2000; Pavlov et al., 2007). It can also be enhanced by an increase in the rate of association (polymerization or nucleation) at the barbed ends and dissociation (depolymerization) at pointed ends (Carlier et al., 1997; Kiuchi et al., 2007).
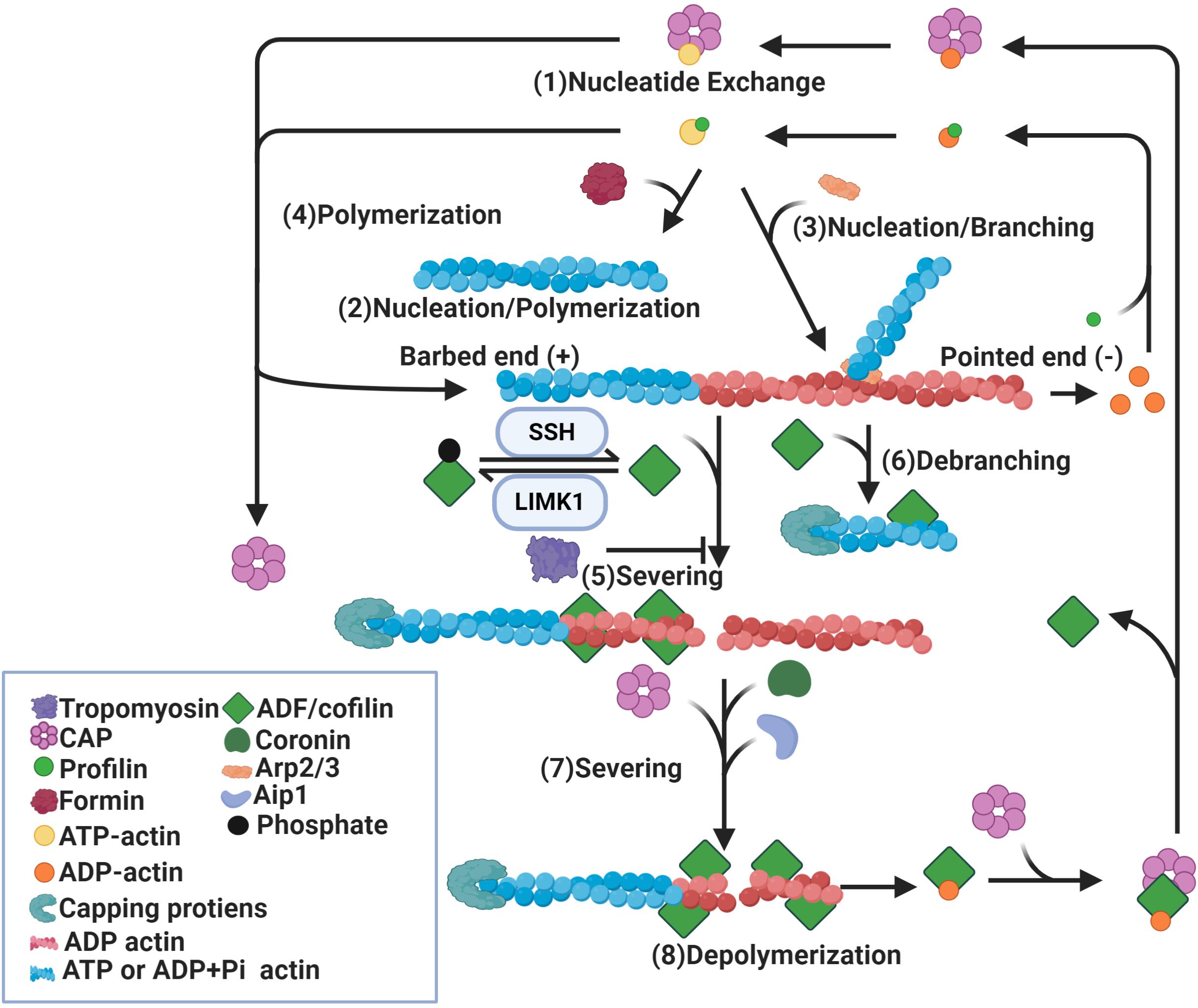
Figure 1. Regulation of actin dynamics by ADF/cofilin. Binding of profilin on ADP-actin monomers induces nucleotide exchange (1). Formin (2) and Arp2/3 (3) induce nucleation of actin monomers and formation of actin filaments. While formin induces parallel actin filament (2), Arp2/3 promotes branching of the original filament (3). In addition, actin filaments can be polymerized by the addition of ATP-actin monomers at the barbed ends (4). Binding of dephosphorylated/active ADF/cofilin to ADP-actin subunits of actin filaments causes severing of these filaments (5) and depolymerization at pointed ends (8). ADF/cofilin activity is mediated by phosphorylation and dephosphorylation by LIMK1 and SSH respectively. ADF/cofilin also debranch Arp2/3 nucleated actin filaments (6). The severing activity of ADF/cofilin is enhanced by Aip1, coronin and CAP (7) and diminished by tropomyosin (5). Binding of capping proteins at barbed ends blocks the growth of newly formed actin segments. CAP also dissociates ADF/cofilin from ADP-actin monomers and promotes nucleotide exchange on these monomers (1).
Multiple studies show a significant role for ADF/cofilin in actin filament assembly and disassembly (Lappalainen and Drubin, 1997; Rosenblatt et al., 1997; Loisel et al., 1999; Pollard and Borisy, 2003; Bernstein and Bamburg, 2010; Kanellos and Frame, 2016). Two models have been proposed to explain the disassembly function of ADF/cofilin. Actin-depolymerization factor/cofilin can increase the rate of depolymerization at pointed ends or sever existing actin filaments into smaller fragments (Blanchoin and Pollard, 1999; Pavlov et al., 2007). The best evidence for increased actin subunit dissociation comes from the “bulk sample of actin” experiment that measured the exchange of fluorescent or radiolabeled ADP-bound actin subunits incubated with ADF/cofilin for ATP in the medium (Carlier et al., 1997). Exchange of ADP into ATP occurs only on free actin monomers not on actin subunits in filaments, therefore nucleotide exchange can only happen after the dissociation of ADP-bound subunits from filaments. The observed increase in the rate of nucleotide exchange in this experiment can be interpreted to arise from the dissociation of actin subunits in the presence of ADF/cofilin (Carlier et al., 1997). Though this study suggests that depolymerization at the pointed end increases in the presence of ADF/cofilin, it does not provide direct evidence to support this conclusion. As neither the number of ends nor filament lengths were known, it was not possible to measure subunit dissociation from individual filament ends (Carlier et al., 1997). The filament disassembly severing model is supported by real-time microscopy assays which analyzed single actin filaments. Binding of ADF/cofilin to actin filaments was found to induce a conformational twist in these filaments resulting in fragmentation or severing of filaments (Andrianantoandro and Pollard, 2006). After binding of ADF/cofilin, an increase in actin depolymerization at pointed ends was observed in the presence of vitamin D binding proteins, which sequester free actin monomers. However, the detected rate of depolymerization was too slow to account for the observed rates of nucleotide exchange in bulk assays (Andrianantoandro and Pollard, 2006). Therefore, it could be concluded that severing of actin filaments by ADF/cofilin can produce many filament ends which may account for the nucleotide exchange rate (Ichetovkin et al., 2000; Pavlov et al., 2007). Recently, a study using single-filament approach based on microfluidics suggests that ADF/cofilin-induced actin disassembly is mediated by both severing and depolymerization activity (Wioland et al., 2017). Consistent with Andrianantoandro and Pollard (2006), pointed end depolymerization was enhanced by ADF/cofilin, though not to the extent predicted by bulk assays (Andrianantoandro and Pollard, 2006; Wioland et al., 2017). Actin-depolymerization factor/cofilin favor barbed-end depolymerization through either directly targeting the barbed ends of bare filaments, which is avoided when ATP-actin is present, or preventing the barbed ends of ADF/cofilin-saturated filaments from elongating and promoting barbed-end depolymerization, contrary to the general consensus (Wioland et al., 2017). As there is debate on how ADF/cofilin promote filament disassembly, they also promote assembly in multiple ways. Actin-depolymerization factor/cofilin could increase the rate of polymerization as detected in bulk assays (Carlier et al., 1997). However, single filament studies show that ADF/cofilin slow barbed-end polymerization (Andrianantoandro and Pollard, 2006). Another mechanism for cofilin-induced filament assembly is through severing, which may create more filament ends (Andrianantoandro and Pollard, 2006). Moreover, cofilin could stimulate nucleation by stabilizing long-pitch actin dimers, the first intermediate in spontaneous assembly and nucleation may be the main contribution of ADF to the increased rate of actin filament assembly (Andrianantoandro and Pollard, 2006). In summary, ADF/cofilin mediate both filament assembly and disassembly through multiple mechanisms, including depolymerization, severing, polymerization and nucleation (Figure 1).
The effect of ADF/cofilin on actin filaments depends on the relative concentration of ADF/cofilin to actin and interactions with other proteins (Ono, 2003; Winder and Ayscough, 2005; Pavlov et al., 2007). At a lower ADF/cofilin concentration, severing of actin filaments by ADF/cofilin is highest (Andrianantoandro and Pollard, 2006). When few ADF/cofilin molecules are bound to actin filaments, the number of strained interfaces between twisted and non-twisted region is the highest, resulting in frequent breakage (Bobkov et al., 2006). At a higher ADF/cofilin concentration, when actin filaments are largely covered with ADF/cofilin, severing is no longer observed, though there is still dissociation from the pointed ends (Pavlov et al., 2007). When ADF/cofilin levels are higher, they can nucleate filaments (Yeoh et al., 2002; Andrianantoandro and Pollard, 2006; Kudryashov et al., 2006). However, abnormally high levels of active ADF/cofilin can drive the formation of ADF/cofilin-actin rods that sequester a large fraction of the total ADF/cofilin, rendering ADF/cofilin incapable of promoting actin disassembly (Minamide et al., 2000). Other actin-binding proteins may alter ADF/cofilin’s ability to act on the actin cytoskeleton (Winder and Ayscough, 2005). These proteins include actin-interacting protein 1 (AIP1), tropomyosins (TPM), cortactin, actin-related proteins-2/3 (Arp2/3) and coronins (Ichetovkin et al., 2002; Brieher et al., 2006; Kueh et al., 2008; Ostrowska-Podhorodecka et al., 2020).
The reported rates of ADF/cofilin-mediated actin filament disassembly in vitro are lower than those observed in in vivo experiments, which could be due to a difference in cytosolic versus in vitro conditions (Lappalainen and Drubin, 1997). These results also suggest that additional cellular factors may be involved in regulating the activity of ADF/cofilin under physiological conditions. Many studies have demonstrated that other actin-binding proteins can potently modulate ADF/cofilin’s ability to act on the actin cytoskeleton (Winder and Ayscough, 2005). These actin-binding proteins include: (1) proteins structurally or functionally similar to ADF/Cofilin (e.g., AIP1), cyclase associated protein (CAP), and coronin; (2) proteins involved in F-actin filament assembly (e.g., Arp 2/3, profilin, and cortactin); (3) proteins generally antagonistic toward ADF/Cofilin activity (e.g., TPM) (Winder and Ayscough, 2005). Actin-interacting protein 1, coronin, and CAP are functionally similar to ADF/cofilin as they each promote F-actin disassembly. Both AIP1 and coronin facilitate the cofilin-mediated disassembly of Listeria comet tail and purified actin filaments even with a physiological concentration of actin monomers, a condition promoting actin assembly (Brieher et al., 2006; Kueh et al., 2008). This conclusion is supported further using internal reflection fluorescence microscopy to directly visualize the integrated actions of coronin and AIP in enhancing cofilin-mediated actin filaments disassembly (Jansen et al., 2015). Although AIP1 itself moderately enhances cofilin-mediated actin severing, the presence of coronin alone appears to inhibit severing by cofilin (Jansen et al., 2015). The inhibitory effect of coronin was also previously shown in bulk assay studies (Cai et al., 2007; Gandhi et al., 2009). This disparity is likely to arise due to differences in the nucleotides state of actin, as studies have shown that coronin may interfere with the binding of cofilin to ATP-actin, but not ADP-actin (Ge et al., 2014). In addition to the synergistic effect of coronin and AIP1 on cofilin-mediated actin severing, AIP1 has been shown to be able to bind to the newly generated barbed ends and block growth of the newly formed actin segments, enabling actin filament disassembly under cellular conditions which generally enables filament assembly (Jansen et al., 2015). Actin-interacting protein 1 is known to be regulated by STK16, a constitutive kinase. RNAi knockdown of STK16 in cultured cells resulted in significantly decreased F-actin levels and increased actin polymerization, demonstrating a potential link between AIP1 activity and actin dynamics (Liu et al., 2017). Interestingly, the mixture of cofilin, coronin and AIP1 failed to disassemble actin filaments with a physiological concentration of actin filaments until the addition of CAP (Normoyle and Brieher, 2012). As such, CAP was identified as a factor that promotes disassembly of cofilin-actin filaments. Cyclase associated protein was shown to associate with both actin monomers and filaments and is expressed in the hippocampus, striatum and cortex (Freeman et al., 1995; Bertling et al., 2004; Normoyle and Brieher, 2012). CAP1 knockdown in cultured cells results in abnormal cytoplasmic aggregates of cofilin and diminished actin depolymerization, suggesting a role of CAP in regulating the localization and function of cofilin-1 in mammalian cells (Bertling et al., 2004). Interestingly, CAP forms a hexameric structure that binds to actin filaments though its N-terminal segment and enhances cofilin-mediated actin severing (Jansen et al., 2014). The severing efficiency of CAP is directly proportional to the stoichiometry of their oligomerization, that is to say CAP tetramers and trimers show increased CAP-cofilin interaction compared to CAP monomers (Purde et al., 2019). Despite these studies suggesting a role of CAP in cofilin-mediated filament severing, recent studies report the inability of CAP to increase cofilin-mediated actin severing using single-filament microfluidics approach (Shekhar et al., 2019). In the same line, CAP accelerates actin depolymerization at the pointed end suggesting that CAP enhance cofilin-mediated disassembly through depolymerization not severing (Kotila et al., 2019; Shekhar et al., 2019). Cyclase associated protein can also bind to actin monomers through its C-terminal domain and catalyze nucleotide exchange on cofilin-bound ADP actin monomers (Jansen et al., 2014; Kotila et al., 2018). Proteins involved in F-actin assembly/nucleation shown to interact with ADF/cofilin include Arp2/3, profilin and formin (Weaver et al., 2001; Sagot et al., 2002; Bleicher et al., 2020). Early in vitro studies suggest a synergistic relationship between the Arp2/3 complex and cofilin in regulating filament assembly. The total number of newly polymerized filaments is increased in the presence of both the Arp2/3 complex and cofilin (Ichetovkin et al., 2002). In addition, the frequency of Arp2/3-nucleated branching, in newly formed actin filaments from cofilin-mediated severing, is higher than old pre-existing filaments. Other studies suggest cofilin promotes debranching and has an antagonistic relationship with Arp2/3 as cofilin promotes dissociation of actin filament branches induced by Arp 2/3 (Blanchoin et al., 2000). Moreover, binding of cofilin promotes structural changes in actin filaments, which decreases the affinity of Arp2/3 complex for actin resulting in dissociation of Arp2/3 complex from actin filaments and promoting dissociation of actin filament branches induced by Arp 2/3 complex (Chan et al., 2009). Therefore, the debranching activity of cofilin occurs via cofilin’s effect on actin filaments and not the Arp2/3 complex itself. However, some studies suggest that actin depolymerizing factor homology protein, known as glia maturation factor, functions more specifically as a debranching factor through direct interaction with the Arp2/3 complex (Ydenberg et al., 2013; Poukkula et al., 2014). Much like the process of disassembly, actin filament nucleation is synergistic and reliant on the concentration of both actin monomers and proteins required for polymerization. There is a certain degree of cooperativity between actin filament assembly and disassembly, as it has been shown that formin activity can preclude cofilin-mediated severing, although cofilin activity is required to produce the actin monomers needed to maintain network stability (Bleicher et al., 2020). Tropomyosin is a well characterized regulator of actin filament dynamics known to dampen ADF/cofilin activity (Gunning et al., 2015). Specifically, Tpm competes with ADF/cofilin-mediated actin disassembly by spatially restricting binding sites at the pointed ends of filaments (Kuhn and Bamburg, 2008; Gateva et al., 2017; Jansen and Goode, 2019). Notably, different Tpm isoforms have varying effects on ADF/cofilin-mediated actin dynamics; particularly, fast off-rate Tpm isoforms permit a relative increase in ADF/cofilin binding, allowing for greater F-actin turnover (Ostrowska-Podhorodecka et al., 2020). In summary, ADF/cofilin activity is intricately regulated by its interactions with diverse actin-binding proteins. Some of these proteins have been shown to play an important role in spine and synaptic plasticity and will be further discussed in later sections.
The phosphorylated form of ADF/cofilin is considered the inactive form and is not involved in actin cytoskeleton regulation (Bernstein and Bamburg, 2010; Ridley, 2011; Kanellos and Frame, 2016). Few studies have shown a role of phosphorylated ADF/cofilin in muscarinic receptor−mediated stimulation of phospholipase D1 (PLD1), which is independent of actin regulation (Schmidt et al., 1999; Han et al., 2007). Phosphorylated ADF/cofilin can bind and activate PLD1, leading to the hydrolysis of phosphatidylcholine to phosphatidic acid by PLD1 in the cell membrane, and is considered to be involved in a large variety of early and late cellular responses. These responses include calcium mobilization, secretion, superoxide production, endocytosis, exocytosis, vesicle trafficking, glucose transport, mitogenesis and apoptosis (Exton, 2002). In HEK-293 and neuroblastoma cells, factors known to increase ADF/cofilin phosphorylation, such as LIM domain containing kinase (LIMK) 1 and inactive slingshot phosphatase (SSH) enhance the activity of PLD1, whereas expression of wild-type SSH, which abolishes ADF/cofilin phosphorylation, and constitutively active unphosphorylatable (S3A) cofilin compromise PLD stimulation (Han et al., 2007). Phospholipase D1 activity has been linked to neurite outgrowth and LTD, suggesting its involvement in synaptic plasticity but further characterization is required (Cai et al., 2006; Santa-Marinha et al., 2020). Thus, even in its phosphorylated, presumed inactive form, ADF/cofilin is likely to fulfil important biological roles.
Actin-depolymerization factor/cofilin phosphorylation/dephosphorylation at serine 3 (Ser 3) serves as a key convergence point for many signaling pathways to regulate ADF/cofilin activity in response to various intrinsic and external signals (Bamburg, 1999; Bamburg and Bernstein, 2008). Actin-depolymerization factor/cofilin phosphorylation at Ser 3 inhibits actin binding, whereas dephosphorylation activates actin binding (Bernstein and Bamburg, 2010; Kanellos and Frame, 2016). Actin-depolymerization factor/cofilin Ser 3 phosphorylation is mediated by LIMK and testicular protein kinase (TESK), which are serine/threonine kinases (Arber et al., 1998; Yang et al., 1998; Toshima et al., 2001). LIMKs are extensively studied and contain two family members; LIMK1 is predominantly expressed in the nervous system and LIMK2 is widespread throughout the body (Scott and Olson, 2007; Cuberos et al., 2015). LIMK1/2 have high specificity for Ser3 of ADF/cofilin, due to the interaction between the LIMK catalytic domain and the actin binding helix of ADF/cofilin. Targeted mutations at the phosphorylation site inhibit functional inactivation of cofilin-1 by LIMK1 in yeast and mammalian cells (Hamill et al., 2016). LIMK1/2 can be phosphorylated and activated by the Rho-associated protein kinases (ROCKs) and p21-activated kinases (PAKs; Scott and Olson, 2007; Arber et al., 1998; Yang et al., 1998; Cuberos et al., 2015). Phosphorylation of LIMK1 by PAK1 and LIMK2 by PAK4 occurs at Thr 508 and 505, respectively (Maekawa et al., 1999; Ohashi et al., 2000; Sumi et al., 2001). Both PAKs and ROCKs are protein kinases associated with and activated by the Rho family of small GTPases, the central mediators of actin reorganization in response to diverse signaling processes (Govek et al., 2005). The importance of LIMK1 for ADF/cofilin phosphorylation and actin regulation is shown by reduced ADF/cofilin phosphorylation and altered F-actin in LIMK1 knockout (KO) mice (Meng et al., 2002). Cofilin dephosphorylation at Ser 3 is mediated by two protein phosphatases; chronophin, which is highly specific for cofilin, and SSH, which can also dephosphorylate and inactivate LIMK1 (Niwa et al., 2002; Bernstein and Bamburg, 2010). SSH can be phosphorylated and inactivated by PAK4 and protein kinase D1 (Soosairajah et al., 2005; Eiseler et al., 2009). Both chronophin and SSH regulate ADF/cofilin in a spatially precise manner, proximal to the membrane, this can allow for the formation of membrane protrusions (Nagata-Ohashi et al., 2004; Gohla et al., 2005; Nishita et al., 2005). For example, during lamellipodium formation, dephosphorylation of SSH induces its release from scaffolding protein 14-3-3 in the cytoplasm and its translocation on growing actin filaments to induce dephosphorylation/activation of cofilin within lamellipodium (Nagata-Ohashi et al., 2004).
In addition to Ser 3, phosphorylation at tyrosine (Tyr 68) has been shown to be important for cofilin-1 regulation (Yoo et al., 2010). This regulation is unique to cofilin-1, since ADF does not have Tyr 68 (Yoo et al., 2010). In HEK cells, phosphorylation at Tyr 68 does not directly affect the actin-depolymerizing activity, however it increases ubiquitination and proteasome degradation of cofilin-1 sufficiently to reduce cofilin-1 levels and cellular distribution (Yoo et al., 2010). Oxidation has also been introduced as a mechanism for ADF/cofilin regulation (Bernstein and Bamburg, 2010; Kanellos and Frame, 2016). Under oxidative stress conditions in T cells, ADF/cofilin can undergo oxidative modification. Oxidation of the thiol groups of cysteine residues in ADF/cofilin molecules leads to the formation of both intra and intermolecular disulfide bonds which causes oxidized ADF/cofilin to interact weakly with LIMKs and this results in an increase in unphosphorylated/active ADF/cofilin (Klemke et al., 2008). Another mode of ADF/cofilin regulation is through binding to phosphatidylinositol 4,5-bisphosphate (PIP2; Bernstein and Bamburg, 2010; Kanellos and Frame, 2016). In vitro studies show that PIP2 directly binds to ADF/cofilin and inhibits their actin-depolymerizing activities (Yonezawa et al., 1990). This was confirmed using biochemical and spectroscopic studies showing that ADF/cofilin cluster PIP2 molecules at the membrane through their interaction with multiple PIP2 headgroups and that a small decrease in PIP2 density efficiently activated ADF/cofilin in carcinoma cells (Zhao et al., 2010). pH in vitro and in vivo is also shown to modulate mammalian ADF/cofilin activity (Bernstein et al., 2000; Pavlov et al., 2006). The in vivo mechanism is highlighted by the ability of cofilin to act as a cellular pH sensor, with increased activity at higher pH and that this ability involves the inhibition of cofilin activity by binding PIP2, as discussed earlier (Frantz et al., 2008). Studies in neurons have shown other regulatory mechanisms in addition to those introduced above and these include mRNA availability and translation (Feuge et al., 2019), and temporal and spatial regulation of subcellular distribution (e.g., Zhou et al., 2011; Pontrello et al., 2012; Bosch et al., 2014). These mechanisms are particularly important for the regulation of ADF/cofilin activity during spine and synaptic plasticity, which will be discussed further in later sections.
One of the most important features of neuronal synapses is their ability to change the strength of synaptic transmission in response to external stimuli, which is referred to as synaptic plasticity. In the mammalian central nervous system, most excitatory synapses are located on small dendritic protrusions called dendritic spines (Carlisle and Kennedy, 2005; Alvarez and Sabatini, 2007; Tønnesen and Nägerl, 2016; Gipson and Olive, 2017). Synaptic plasticity, including LTP and LTD, is closely associated with changes in the number and morphology of dendritic spines and these changes are typically referred to as structural plasticity (Lamprecht and LeDoux, 2004; Carlisle and Kennedy, 2005; Alvarez and Sabatini, 2007; Bernardinelli et al., 2014; Borovac et al., 2018; Lai and Ip, 2013; Sheppard et al., 2019). For example, using glutamate uncaging, an enlargement of dendritic spines during the induction of LTP at single spines of hippocampal CA1 pyramidal neurons is observed (Matsuzaki et al., 2004). On the other hand, the induction of LTD using low frequency stimulation is accompanied by shrinkage of dendric spines in acute hippocampal slices from neonatal rats (Zhou et al., 2004). As actin is the main cytoskeletal component of the dendritic spine, it is not surprising that actin dynamics play a key role in the regulation of spine morphology (Matus et al., 1982; Hotulainen and Hoogenraad, 2010; Miermans et al., 2017; Basu and Lamprecht, 2018). Using two-photon imaging, a dynamic pool of actin filaments is seen at the tips of spines from CA1 pyramidal neuron in rat hippocampal slices (Honkura et al., 2008). These actin filaments can be quickly treadmilled to generate an expansive force to mediate changes in spines (Honkura et al., 2008). Two-photon Forster resonance energy transfer (FRET) imaging shows that activity-dependent actin polymerization and depolymerization in dendritic spines during LTP and LTD (Okamoto et al., 2004). During synaptic plasticity, the actin cytoskeleton is highly regulated and goes through phases of polymerization and depolymerization (Bosch et al., 2014; Kim et al., 2015; Borovac et al., 2018). For example, the reorganization of actin during LTP appears to have two distinct but overlapping phases (Bosch et al., 2014). Within the first 5 min after LTP induction, there is remodeling of the actin cytoskeleton through rapid periods of actin filaments disassembly followed by periods of actin filament assembly, which result in enlargement of dendritic spines and LTP. After 5 min of LTP induction, there is a net increase in actin and newly polymerized actin filaments in the spine which result in long-term stabilization and consolidation of early synaptic changes (Bosch et al., 2014).
One of the earliest indications that ADF/cofilin is important for spine and synaptic regulation comes from studies on LIMK1/2 KO mice, which show altered spine morphology and impaired synaptic function and that these alterations are associated with a dramatic reduction in cofilin phosphorylation (Meng et al., 2002, 2004). In addition, bidirectional changes in ADF/cofilin phosphorylation and dephosphorylation can be induced rapidly by activation of glutamate receptors and signaling molecules at the synapse (Meng et al., 2002). A subsequent study using immunoelectron microscopy shows that cofilin-1 is concentrated in the shell of spines rich in dynamic actin and within postsynaptic density in the stratum radiatum of the rat hippocampus (Racz and Weinberg, 2006). More direct evidence to support cofilin function at the synapse comes from molecular and genetic manipulations of ADF/cofilin and their upstream regulators. Overexpression of constitutively active unphosphorylatable cofilin (S3A) in neurons leads to reduced spine size and immature morphology (Shi et al., 2009). The expression of constitutively inactive phosphomimetic cofilin (S3D) restores mature spine morphology (Shi et al., 2009). Longer dendritic protrusions and slower actin turnover are also observed when cofilin-1 expression is reduced using siRNA in primary hippocampal neurons (Hotulainen et al., 2009). In cofilin-1 conditional KO mice where cofilin-1 is selectively deleted in the excitatory neurons of the postnatal forebrain, increased synapse density and enlargement of dendritic spines are found in the hippocampus and these structural changes are associated with impaired late phase LTP and LTD (Rust et al., 2010). Although ADF KO mice show no deficits in spine properties or synaptic function (Görlich et al., 2011), ADF and cofilin-1 double KO mice exhibit greater changes in spine enlargement than cofilin-1 conditional KO mice, suggesting that ADF also plays a role in spine regulation (Wolf et al., 2015). Other evidence supporting the role of ADF/cofilin in basal spine properties comes from manipulations of their upstream regulators in addition to LIMK1/2 (Meng et al., 2002, 2004; Todorovski et al., 2015). These include PAK1/3 (Meng et al., 2005; Asrar et al., 2009; Huang et al., 2011), ROCK2 (Zhou et al., 2009), chronophin (Kim et al., 2016) and Rho GTPases (Nakayama et al., 2000; Li et al., 2002; Martino et al., 2013), all of which affect either spine morphology or density. For example, both PAK1/3 double and ROCK2 KO mice have reduced spine density and immature morphology and these spine changes are associated with a significant reduction in cofilin phosphorylation (Zhou et al., 2009; Huang et al., 2011). Overexpression of chronophin in mice results in the shrinkage of dendritic spines and knocking out chronophin causes dendritic spine enlargement in hippocampal neurons (Kim et al., 2016).
Actin-depolymerization factor/cofilin are not only important for basal spine morphology and density but also required for spine changes during synaptic plasticity for which the temporal and spatial regulation of ADF/cofilin appears to be particularly important (Bernstein and Bamburg, 2010; Lai and Ip, 2013; Noguchi et al., 2016; Borovac et al., 2018). In general, it has been shown that ADF/cofilin inactivation is associated with and required for actin assembly and spine enlargement and stabilization during LTP, whereas ADF/cofilin activation can drive actin filament disassembly and spine shrinkage during LTD. However, single spine imaging studies have revealed that changes in ADF/cofilin during LTP are much more complex and dynamic, exhibiting multiple phases of regulation (Bosch et al., 2014). During the initial phase (<5 min) of dendritic spine enlargement induced by glutamate uncaging at single spines, the amount of ADF/cofilin in the spine increases and this is accompanied by an increase in the amount of actin and spine enlargement (Bosch et al., 2014). This is consistent with the observation that cofilin undergoes translocation in its unphosphorylated/active form following glutamate uncaging (Noguchi et al., 2016). During a later phase (>5 min), there is sustained accumulation of phosphorylated/inactive ADF/cofilin at the base of the spine head where cofilin forms a stable complex with actin filaments (Bosch et al., 2014). These results suggest that ADF/cofilin phosphorylation is needed to retain its accumulation within spines. It is shown that wild type and constitutively inactive cofilin (S3E) accumulate in the stimulated spines for 30?min after spine enlargement following glutamate uncaging, whereas constitutively active cofilin (S3A) more rapidly diffuses away from the enlarged spine (Noguchi et al., 2016). These single spine imaging results are consistent with earlier studies using protocols to induce LTP at the global level. PAK and ADF/cofilin phosphorylation in rat hippocampal slices are found to be increased 2–7 min after theta-burst stimulation, a time window consistent with the transition from the initial to later phase (Chen et al., 2007). In cultured neurons, the levels of phosphorylated cofilin declines 5 min after the onset of chemically induced LTP, but significantly increases by 30 min (Gu et al., 2010). These studies suggest that dynamic regulation of ADF/cofilin phosphorylation and spine accumulation contributes to different phases of spine plasticity during LTP. During spine shrinkage and LTD, although the precise time course of ADF/cofilin involvement has not be investigated at single spines, ADF/cofilin dephosphorylation and spine accumulation are consistently found to be associated with and required for LTD in both cultured neurons and brain slices (Zhou et al., 2011; Pontrello et al., 2012). The mechanisms by which ADF/cofilin is regulated during LTP and LTD will be discussed further in later sections.
How changes in ADF/cofilin activity, either through spine accumulation or phosphorylation, regulate spine morphology remain unclear. However, a recent study using a fluorescent reporter to monitor membrane-proximal actin filaments (MPA) may provide new insight (Bisaria et al., 2020). In this study, it is shown that amount of MPA is lower at the front compared to the back during membrane protrusion and cell migration, and that increased cofilin activity is required for this MPA gradient and the initiation of new membrane protrusions. These results are consistent with earlier studies showing SSH can regulate ADF/cofilin in a spatially precise manner proximal to the membrane (Nagata-Ohashi et al., 2004; Soosairajah et al., 2005) and suggest that cofilin activity driven by SSH at the front is essential. The major isoform of slingshot is SSH-1L, which is only active in its cofilin dephosphorylation activity when it is bound to F-actin (Nagata-Ohashi et al., 2004; Soosairajah et al., 2005). In contrast to the accumulation of SSH1L, LIMK1 diffusely distributes in the cytoplasm (Nagata-Ohashi et al., 2004; Nishita et al., 2005). These findings suggest that spatially distinct localization of LIMK1 and SSH1L during protrusions formations may also play a role in spine formation and morphological changes.
While the induction of LTP and LTD at many synapses requires the activation of N-Methyl-D-aspartic acid (NMDA) glutamate receptors, their expression involves modification of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors, the principal mediator of fast excitatory synaptic transmission (Bliss and Collingridge, 1993; Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Collingridge et al., 2004, 2010; Segal, 2005; Derkach et al., 2007; Rebola et al., 2010; Ho et al., 2011; Huganir and Nicoll, 2013; Henley and Wilkinson, 2016; Diering and Huganir, 2018). These modifications include channel properties and receptor abundance at the synapses (Lau and Zukin, 2007; Huganir and Nicoll, 2013; Henley and Wilkinson, 2016; Diering and Huganir, 2018). In particular, receptor trafficking at the synapse attracts the most attention due to its potent effects on synaptic strength (Collingridge et al., 2004; Henley and Wilkinson, 2016; Diering and Huganir, 2018; Park, 2018). Several studies have shown that ADF/cofilin play an important role in the regulating trafficking and accumulation of AMPA receptors within synapses during LTP and this process appears to be distinct from its role in spine morphological plasticity (Gu et al., 2010; Rust et al., 2010). Elevated ADF/cofilin activity markedly enhances addition of AMPARs to the surface after chemical induction of LTP in cultured neurons, whereas the inhibition of ADF/cofilin activity suppresses the addition of AMPA receptors (Gu et al., 2010). The role of ADF/cofilin in AMPA receptor trafficking has also been demonstrated in animal models. Lateral diffusion of the AMPA receptors subunit GluA2 was shown to be compromised in the extrasynaptic compartment of hippocampal neurons from cofilin-1 mutant mice (Rust et al., 2010). The exchange of AMPA receptors between synaptic and extrasynaptic domains by lateral diffusion is thought to represent a key mechanism to control the level of synaptic AMPA receptors during synaptic plasticity (Choquet and Hosy, 2020; Groc and Choquet, 2020). The stabilization of actin filament by jasplakinolide reduces the mobility of the extrasynaptic AMPA receptor subunit GluA2, whereas destabilization of actin filament by latrunculin A results in increased movement of GluA2 subunits (Rust et al., 2010). This suggests that the effect of ADF/cofilin on AMPA receptors mobility and surface expression is mediated by actin-dependent mechanisms. These results are consistent with studies where direct manipulations of the actin cytoskeleton affects LTP and AMPA receptor trafficking (Allison et al., 1998; Kim and Lisman, 1999; Zhou et al., 2001; Cingolani and Goda, 2008; Yang et al., 2008; Hanley, 2014; Basu and Lamprecht, 2018). ADF/cofilin have also been shown to play a role in AMPA receptor internalization during LTD (Zhou et al., 2011). The induction of metabotropic glutamate receptor-dependent LTD (mGluR-LTD) induces ADF/cofilin dephosphorylation, spine shrinkage and a decrease in synaptic AMPA receptors. Actin-depolymerization factor/cofilin-dependent regulation of AMPA receptor trafficking is also seen following learning. Extinction of conditioned taste aversion leads to temporally enhanced ADF/cofilin activity in the infralimbic cortex of the rats and manipulations of ADF/cofilin activity accelerates or inhibits memory extinction by regulating the recruitment of AMPA receptors at the synaptic surface (Wang et al., 2013). These studies support that ADF/cofilin regulates synaptic transmission through AMPA receptor trafficking in addition to spine morphological changes.
At many central synapses, the induction of LTP and LTD requires Ca2+-dependent signaling pathways, including protein kinases [e.g., activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) during LTP] and phosphatases (e.g., calcineurin) during LTD (Malinow and Malenka, 2002; Collingridge et al., 2004; Malenka and Bear, 2004; Derkach et al., 2007; Citri and Malenka, 2008; Lüscher and Malenka, 2012; Bliss and Collingridge, 2013; Huganir and Nicoll, 2013; Henley and Wilkinson, 2016; Sanderson et al., 2016; Diering and Huganir, 2018). Accumulating evidence indicates that multiple mechanisms exist at the synapse to link these Ca2+-dependent pathways to regulate ADF/cofilin (Meng et al., 2004; Jia et al., 2009; Rex et al., 2009; Martinez and Tejada-Simon, 2011; Yasuda, 2017; Nakahata and Yasuda, 2018) and these are summarized in Figure 2.
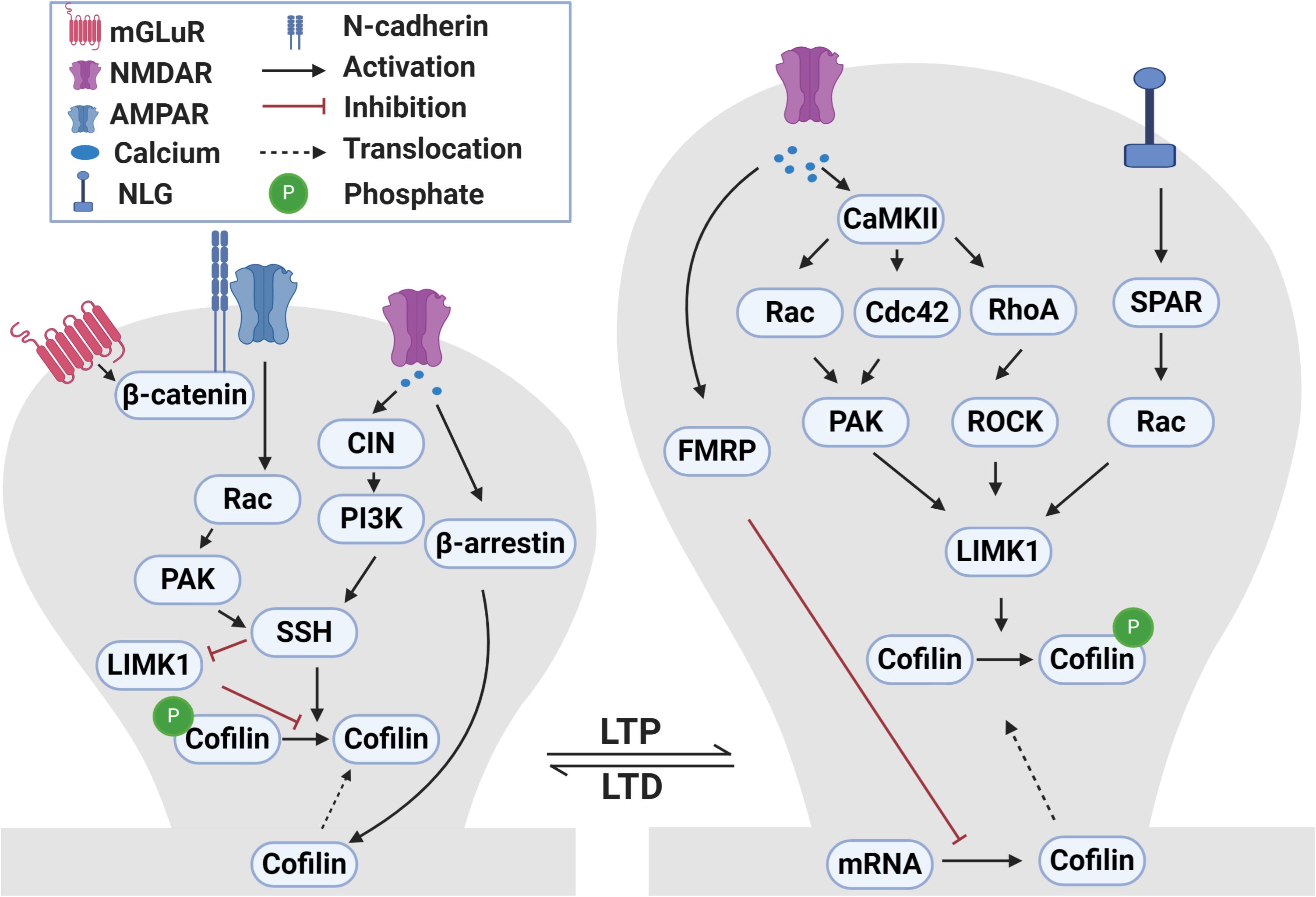
Figure 2. Signaling pathways that regulate ADF/cofilin phosphorylation and dephosphorylation during LTP and LTD. During LTP, activation of NMDA receptors causes calcium influx into dendritic spines. The increased intracellular calcium activates CaMKII which in turn activates small Rho GTPases, including Rac, Cdc42 and RhoA. These small GTPases bind to and activate PAKs and ROCKs that can directly phosphorylate and activate LIMK1. LIMK1 can also be activated following the activation of neuroligin 1 receptors during LTP through SPAR-Rac signaling pathway. Activated LIMK1 phosphorylates and inactivates cofilin resulting in the enlargement of dendritic spines. In addition, the LTP-induced calcium influx diminishes local translation of cofilin mRNA in dendrites through a FRMP 1-dependent manner. Translocation of cofilin into spines during LTP occurs through yet to be discovered mechanisms. During LTD, the activation of NMDA receptors and influx of calcium activates CIN which activates SSH through the PI3K-dependent pathway. Activated SSH dephosphorylates and activates cofilin which results in dendritic spine shrinkage. In addition, LTD-induced calcium influx mediates translocation of cofilin into spines in a β-arrestin 2-dependent manner. During mGLuR-LTD, GluA2 interaction with cadherin/β-catenin activates Rac-PAK which then activate SSH. Additionally, SSH can also dephosphorylate and inactivate LIMK1.
Many studies have shown that ADF/cofilin is a downstream effector of Rho GTPases and their effector protein kinases such as PAKs, ROCKs and LIMKs during LTP and spine enlargement (Borovac et al., 2018; Nakahata and Yasuda, 2018; Kovaleva et al., 2019). Rho proteins, including RhoA, Rac and Cdc42, are activated during LTP (Govek et al., 2005; Rex et al., 2009; Martinez and Tejada-Simon, 2011). For example, stimulation of NMDA receptors leads to activation of Rac1 and rapid enlargement of dendritic spines (Xie et al., 2007). Overexpression of either Rac1 or Rac3 causes an increase in spine density (Wiens et al., 2005; Pennucci et al., 2019). Double knockouts of Rac1 and Rac3 inhibit the formation of dendritic spines and induce an increase in filopodia-like spines (Pennucci et al., 2019). PAK1 and PAK3 double KO mice show decreased actin filaments and phosphorylated ADF/cofilin which are associated with immature spines and LTP impairments (Huang et al., 2011). Similarly, ROCK2 KO mice are altered in spine morphology accompanied by reduced phosphorylated ADF/cofilin (Zhou et al., 2009). LIMK1 KO mice exhibit significant abnormalities in the actin cytoskeleton, reduced phosphorylated ADF/cofilin and impaired late phase LTP (Meng et al., 2004; Todorovski et al., 2015). These genetic studies are consistent with results from manipulations of the Rho GTPases and their effectors in cultured neurons and slices (Luo et al., 1996; Nakayama et al., 2000; Tashiro et al., 2000; Rex et al., 2009; Shi et al., 2009). Therefore, ADF/cofilin phosphorylation mediated by the activation of the Rho GTPase-PAK/ROCK-LIMK pathway is a key mechanism that is responsible for ADF/cofilin inactivation, actin assembly and spine enlargement during LTP. Actin-depolymerization factor/cofilin dephosphorylation through activation of chronophin might also be important spine enlargement during LTP as chronophin KO mice are impaired in late-phase LTP (Kim et al., 2016). The effect of chronophin could be mediated through regulating the coupling of GluN2A subunits with postsynaptic proteins (Kim et al., 2016).
In addition to ADF/cofilin phosphorylation, reduced protein translation of ADF/cofilin has also been reported to be associated with chemical LTP and this translational regulation is mediated by fragile X mental retardation protein 1 (FMRP1; Feuge et al., 2019). In cultured hippocampal neurons, glycine induced LTP is accompanied by reduced ADF/cofilin mRNA availability and translation, and these changes are impaired in FMRP1 KO mice. How FMRP1-mediated suppression of ADF/cofilin translation is achieved remains unknown, but it is known that this mRNA-binding protein is a potent regulator of activity-dependent local protein synthesis involving the mTOR and ERK1/2 pathways (Brown et al., 2001), and therefore it is possible these pathways are also important for downregulating ADF/cofilin protein level during this form of LTP. Interestingly, the FMR1 KO mice also show elevated activation of the Rac-PAK-LIMK pathway, resulting in increased ADF/cofilin phosphorylation, under basal conditions, and overexpression of active ADF/cofilin rescues some of the behavior defects in FMR1 KO mice (Pyronneau et al., 2017). These results indicate that in addition to translational regulation of ADF/cofilin, FMRP1 also acts as a negative regulator of ADF/cofilin phosphorylation through the Rac-PAK/LIMK signaling process.
During LTD and spine shrinkage, Ca2+-dependent phosphatases are important for ADF/cofilin dephosphorylation and activation. Inhibition of calcineurin in CA1 pyramidal neurons blocks cofilin-dependent spine reduction during LTD induced by low frequency stimulation (Zhou et al., 2004). Chemical LTD induced by application of NMDA is associated with dendritic spine shrinkage and loss of synaptic proteins, and these changes require ADF/cofilin dephosphorylation and spine accumulation (Pontrello et al., 2012). Although calcineurin dependent activation of phosphatidylinositol 3-kinase (PI3K) is important for ADF/cofilin dephosphorylation, the intermediates between PI3K and ADF/cofilin are not yet identified. In non-neuronal cells, PI3K regulates cofilin dephosphorylation through activation of SSH (Nishita et al., 2004). Furthermore, calcineurin has been shown to mediate ADF/cofilin dephosphorylation by SSH in response to calcium influx (Wang Y. et al., 2005). In neuronal cells, ephrin-induced dendritic spine retraction and ADF/cofilin dephosphorylation requires activation of calcineurin and subsequent activation of SSH (Zhou et al., 2012). These studies suggest that the calcineurin-P13K-SSH pathway may mediate ADF/cofilin dephosphorylation and activation during LTD. The spine accumulation of ADF/cofilin during chemical LTD requires β-arrestin 2 as NMDA-induced spine remodeling and cofilin translocation are impaired in β-arrestin 2 KO neurons (Pontrello et al., 2012). The NMDA-induced cofilin dephosphorylation appears to be independent of spine accumulation as blocking the PI3K pathway does not prevent cofilin translocation to the spine. These results suggest that there are two distinct pathways respectively regulating cofilin dephosphorylation and spine accumulation that are activated during NMDA-induced spine shrinkage and LTD.
During mGluR-LTD, the mechanisms governing ADF/cofilin regulation are also distinct from those involved in NMDA receptor dependent LTD (Zhou et al., 2011). mGluR-LTD induces ADF/cofilin dephosphorylation and spine accumulation and these changes are required for both mGluR-dependent spine shrinkage and synaptic depression. Interestingly, ADF/cofilin dephosphorylation is dependent on the AMPA receptor subunit GluA2 and its interaction with the cell adhesion molecule N-cadherin/β-catenin and subsequent activation of Rac1. How the activation of Rac1 leads to dephosphorylation and activation of ADF/cofilin is not known, but it could involve inactivation of the PAK-LIMK pathway or activation of the SSH pathway.
In addition to glutamate receptors, other neuronal surface proteins and receptors have also been shown to regulate ADF/cofilin activity through similar mechanisms discussed above but may involve additional processes. For example, the c-terminal domain of the cell adhesion molecule neuroligin 1 induces spine enlargement and cofilin phosphorylation that are mediated by neuroligin 1’s interaction with spine-associated Rap GTPase-activating protein (SPAR) and subsequent activation of the Rac1-LIMK pathway (Liu et al., 2016). Neurotrophic factors and their receptors regulate spine growth and LTP which are dependent on changes in ADF/cofilin phosphorylation and dephosphorylation mediated by Rac1 and RhoA (Gehler et al., 2004; Soulé et al., 2006; Wong et al., 2019). Glucocorticoid hormone promotes learning-induced spine formation mediated by activation of LIMK1 and ADF/cofilin phosphorylation (Liston and Gan, 2011; Liston et al., 2013). These studies together indicate the complexity of the regulatory mechanisms governing ADF/cofilin activity at the synapse.
Actin is also abundantly expressed in presynaptic terminals (Matus et al., 1982; Gotow et al., 1991). Pharmacological studies have identified a role for actin in regulating synaptic vesicle mobilization and exocytosis (Morales et al., 2000; Gorovoy et al., 2005). Like actin, ADF/cofilin are also expressed in presynaptic terminals (Rust, 2015), suggesting a presynaptic function. This is initially supported by alterations in presynaptic properties in LIMK1 KO mice where the frequency of neurotransmitter release and synaptic depression in response to sustained neuronal activity are both increased and (Meng et al., 2002). However, neurotransmitter release and presynaptic short-term plasticity are not affected in cofilin-1 KO mice (Rust et al., 2010). In addition, the recruitment and exocytosis of synaptic vesicles are unchanged in ADF KO mice (Görlich et al., 2011). The lack of presynaptic defects in ADF KO mice may be explained by the elevated cofilin-1 levels observed in these mice (Görlich et al., 2011). Indeed, ADF and cofilin-1 double KO mice have more severely impaired actin dynamics as well as altered distribution and exocytosis of synaptic vesicles (Wolf et al., 2015; Zimmermann et al., 2015). Electron microscopy and biochemical data from these double KO mice show a shift in the distribution from the active zone to the reserve pool as well increased docking of synaptic vesicles at CA1 synapse (Wolf et al., 2015). In addition, electron microscopy data from the double KO mice show an increase in the presynaptic bouton area and an increased number of docked vesicles at the active zone of striatal synapses, resulting in increased overall glutamate release at the striatal synapses (Zimmermann et al., 2015). Interestingly, a decrease in glutamate release is detected within the hippocampus and this decrease could be caused by defective vesicle recruitment as shown by reduced glutamate release during sustained synaptic stimulation (Wolf et al., 2015). Therefore, although cofilin-1 is a limiting factor in postsynaptic plasticity and cannot be substituted by ADF, the presence of either ADF or cofilin-1 appears to be sufficient to regulate actin remodeling during presynaptic vesicle release, suggesting an overlapping functions presynaptically (Rust et al., 2010; Wolf et al., 2015; Zimmermann et al., 2015). In line with the role of ADF/cofilin in presynaptic function, the disruption of upstream regulators, including RhoA, ROCK2, PAK1/3, LIMK1, and SSH, all impair some aspects of vesicle exocytosis and neurotransmitter release (Meng et al., 2002; Wang H. G. et al., 2005; Asrar et al., 2009; Yuen et al., 2010; Huang et al., 2011).
long-term potentiation and depression are regarded as key mechanisms for learning and memory (Bliss and Collingridge, 1993; Citri and Malenka, 2008; Neves et al., 2008; Kandel et al., 2014). The demonstrated role of ADF/cofilin in these forms of synaptic plasticity, as discussed earlier, suggests that they are important in memory formation and this is supported by several studies. For example, the conditional deletion of cofilin-1 in postnatal principal neurons results in severe impairments in associative learning, but not exploratory or latent learning (Rust et al., 2010). Activation and inhibition of ADF/cofilin activities using peptides facilitated or impeded contextual fear memory extinction in rats, respectively (Wang et al., 2013). Increased phosphorylated, inactive, ADF/cofilin is observed in the hippocampal CA1 region of rats after learning in an enriched environment (Fedulov et al., 2007). Neonatal social isolation inactivates ADF/cofilin and leads to an increase in stable actin fractions at the dendritic spines in the juvenile medial prefrontal cortex (Tada et al., 2016) and barrel cortex of rats (Tada et al., 2017). Other evidence supporting the importance of ADF/cofilin in memory comes from memory abnormalities observed in the absence of ADF/cofilin upstream regulators. Impaired learning has been documented for mice lacking LIMK1, PAK1/3 and Rho GTPases (Meng et al., 2002, 2005; van Galen and Ramakers, 2005; Huang et al., 2011; Todorovski et al., 2015). For example, LIMK1 KO mice are drastically impaired in long-term but not short-term memory during fear conditioning and the Morris water maze (Meng et al., 2002; Todorovski et al., 2015). Expression of dominant-negative PAK3 alters cofilin phosphorylation and impairs social recognition memory (Leung et al., 2018). Actin-depolymerization factor/cofilin also play a role in other behaviors including reward learning (Toda et al., 2006; Rothenfluh and Cowan, 2013) and anxiety (Goodson et al., 2012). Conditional cofilin-1 KO mice show impaired novel object recognition, but normal social behavior including social recognition (Sungur et al., 2018). Moreover, ADF/cofilin conditional double KO mice also demonstrate abnormal nesting behavior, increased activity and impulsive behavior, as well as reduced non-associative learning and working memory (Zimmermann et al., 2015).
Translocation of ADF/cofilin to the mitochondria is important for induction of apoptosis in multiple cell types, including neurons, neutrophils, lymphoma, neuroblastomas, and prostate cancer (Chua et al., 2003; Zhu et al., 2006; Klamt et al., 2009). Actin-depolymerization factor/cofilin undergo oxidation during inflammatory stress (Klamt et al., 2009; Bernstein and Bamburg, 2010), and when oxidation is prevented, apoptosis is inhibited (Klamt et al., 2009). In mouse embryonic fibroblasts, oxidation of ADF/cofilin cause them to lose their affinity for actin and translocate to the mitochondria, where they induce swelling and cytochrome c release by mediating the opening of the permeability transition pore (Klamt et al., 2009). Knocking down endogenous ADF/cofilin using targeted small interfering (siRNA) inhibits apoptosis, which is restored by expression of wild type ADF/cofilin (Klamt et al., 2009). The apoptotic effect of ADF/cofilin is independent of ADF/cofilin’s role in actin cytoskeleton regulation (Bernstein and Bamburg, 2010).
Several studies have implicated ADF/cofilin in the regulation of neuronal apoptosis (Yang et al., 2004; Bernstein and Bamburg, 2010; Li et al., 2013). Knocking down ADF/cofilin from primary cortical neurons results in decreased excitotoxic neuronal death caused by excess glutamate (Posadas et al., 2012). During excitotoxic neuronal death, ADF/cofilin interacts with the proapoptotic protein Bax, carrying it to the mitochondria and contributing to the depolarization of the mitochondrial membrane, the release of apoptotic factors and neuronal death (Posadas et al., 2012). Actin-depolymerization factor/cofilin is also involved in ischemia-induced neuronal death (Madineni et al., 2016). The activation of ADF/cofilin occurs during ischemia in cortical neurons and knocking down ADFcofilin increases neuronal viability (Madineni et al., 2016). These results are consistent with work on LIMK1 and SSH (Yang et al., 2004; Posadas et al., 2012), showing that overexpression of LIMK1 and inhibition of SSH protects cells from apoptosis by inactivating ADF/cofilin.
In addition to direct involvement in neuronal apoptosis, recent studies suggest that ADF/cofilin contribute to neuronal apoptosis through other cell types like astrocytes (Alhadidi et al., 2016). Astrocytes express glutamate transporters which regulate the clearance of glutamate released from synapses (Anderson and Swanson, 2000). Dysfunction of astrocytic glutamate transporters trigger neuronal death by excessive glutamate and excitotoxicity (Rossi et al., 2000). In primary astrocyte cultures, the actin cytoskeleton has an important role in regulating the activity of glial glutamate transporters as inhibition of actin polymerization by cytochalasin-B reduces cell surface expression of these transporters (Adolph et al., 2007). Also, Rottlerin, a polyphenol natural product, decreases the activity of astrocyte glutamate transporters and disrupts actin filament dynamics (Sheean et al., 2013). Endocytosis of astrocyte glutamate transporters is also dependent on actin dynamics (Yan et al., 2014). These studies suggest that ADF/cofilin mediate actin changes and astrocytic glutamate transporters which in turn contribute to glutamate uptake and neuronal apoptosis.
ADF/cofilin have also been suggested to be involved in the regulation of neuroinflammation (Rasmussen et al., 2010; Gitik et al., 2014). Microglia and astrocytes are the first cells to be activated following brain injuries (Taylor and Sansing, 2013). In the Ra2 microglia cell line, ADF/cofilin knockdown inhibits nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase activity and reactive oxygen species (ROS) formation which result in decline in cells activity (Rasmussen et al., 2010). Microglial cell phagocytic activity is also highly influenced by ADF/cofilin activation (Gitik et al., 2014). Similarly, ADF/cofilin activation is required for the restoration of the myelin sheath and involve in control of phagocytosis of degenerated myelin by microglia and macrophages (Hadas et al., 2012). Actin depolymerization by ADF/cofilin has also been shown to be important for exosome formation, which plays an essential role in facilitating neuroinflammation (Gupta and Pulliam, 2014).
Given the involvement of ADF/cofilin in the regulation of dendritic spines, synaptic plasticity and learning and memory, it is not surprising that deficits in ADF/cofilin are implicated in a wide range of brain disorders (Bamburg and Wiggan, 2002). These conditions include autism spectrum disorders (Duffney et al., 2015; Sungur et al., 2018), Williams syndrome (Hoogenraad et al., 2004), intellectual disability (Newey et al., 2005; van Galen and Ramakers, 2005; Zamboni et al., 2018), drug addiction (Rothenfluh and Cowan, 2013), sleep deprivation (Havekes et al., 2016) and neurodegenerative diseases such as AD (Liu et al., 2019), which will be discussed briefly below.
Alzheimer’s disease is a neurodegenerative condition characterized by memory loss and cognitive decline, resulting in the loss of independence and a shorter life span (Hsiao et al., 1996; Hsia et al., 1999; Li et al., 2014). Pathologically, AD is characterized by neurofibrillary tangles and senile plaques, consisting mainly of extracellular amyloid β (Aβ) peptides (Hsiao et al., 1996; Hsia et al., 1999; Hardy and Selkoe, 2002; Butterfield, 2014). In the brain, Aβ results from the proteolytic processing of the amyloid precursor protein (APP) and it has been proposed that the accumulation of toxic Aβ42 plays a major role in the development of dementia (Hardy and Selkoe, 2002; Palop and Mucke, 2010; Mucke and Selkoe, 2012). The effect of Aβ on the synapse and synaptic function, including LTP and LTD, is of great interest due to their direct relevance to learning and memory (Varadarajan et al., 2000; Selkoe, 2002; Lacor, 2007; Mucke and Selkoe, 2012; Sheng et al., 2012). Given the function of ADF/cofilin in synaptic plasticity, learning and memory, several studies have described the role of ADF/cofilin in the pathophysiology of AD. Actin-depolymerization factor/cofilin were discovered to accumulate in senile plaques in AD tissue and AD mouse models (Bamburg and Bernstein, 2016; Sun et al., 2019). Several studies show that brain tissue from AD patients and AD mouse models such as the APP/PS1 model exhibit elevated levels of inactive phosphorylated cofilin-1 (Barone et al., 2014; Gu et al., 2014; Han et al., 2017; Kang and Woo, 2019). On the other hand, multiple studies show that active dephosphorylated cofilin-1 forms aberrant cofilin-actin rods, which blocks axonal trafficking and may contribute to deficits in synaptic plasticity (Davis et al., 2011; Mendoza-Naranjo et al., 2012; Barone et al., 2014; Kang and Woo, 2019). Recently, it was shown that knocking down CAP2 in hippocampal neurons results in abnormal dendritic spines and impaired synaptic plasticity. This effect of CAP2 is relevant to ADF/cofilin because the CAP protein family is known to form a complex with ADF/cofilin and promote actin disassembly as discussed earlier. Moreover, chemical induction of LTP triggers CAP2 translocation to the spines and increases the formation of dimers, promoting the association of CAP2 with cofilin. In the hippocampal synapses of AD patients and mouse models, there is an increase in cofilin levels accompanied by a reduction in CAP2 synaptic availability, leading to a decrease in CAP2 dimer formation at the synapse (Pelucchi et al., 2020). Studies have also shown that the protein level of cofilin-2 is elevated in the brain and blood of AD patient brains and mouse models (Sun et al., 2015, 2019), but the significance of these changes will need further studies.
The disturbance in cofilin activity in AD may contribute to the loss of dendritic spines and synapses (Kang and Woo, 2019; Pelucchi et al., 2020). Decreased dendritic spine density and active synapses are seen in rat hippocampal pyramidal neurons from organotypic slices after exposure to Aβ oligomers (Shankar et al., 2007). Aβ-induced spine loss can be blocked by prevention of ADF/cofilin activation by expression of constitutively inactive cofilin (S3D) (Shankar et al., 2007). In the same line, Aβ42 oligomers promote ADF/cofilin dephosphorylation and activation in the hippocampus derived HT22 cell line and primary cortical neurons (Woo et al., 2015). Genetic reduction in ADF/cofilin activity activation rescues Aβ42-induced synaptic protein loss as well as deficits in LTP and contextual memory in APP/PS1 mice (Woo et al., 2015). These studies suggest the involvement of active cofilin in AD synaptic dysfunction. In addition, cofilin may also contribute to accumulation of Aβ aggregate and development of AD. For example, Aβ deposition in APP/PS1 mice is significantly decreased by genetically reducing cofilin using small interfering RNA (Liu et al., 2019). The effect of cofilin in Aβ accumulation may be through dual and opposing endocytic mechanisms promoting Aβ production in neurons and inhibiting Aβ clearance in microglia (Liu et al., 2019).
Despite the strong evidence for a role of ADF/cofilin dephosphorylation and activation in AD pathogenesis, several studies show that ADF/cofilin phosphorylation and inactivation may also play a role in AD pathogenesis (Kang and Woo, 2019). Acute exposure to Aβ oligomers increases the level of phosphorylated cofilin-1 at the postsynaptic compartment, leading to a subsequent stabilization of spine actin filaments, as well as impairment of chemically induced LTP (Rush et al., 2018). Also, Aβ oligomers increase cofilin phosphorylation and actin polymerization selectively in cholinergic basal forebrain neurons via increasing PAK1 phosphorylation and activity (Gu et al., 2014). There is also an increase in level of phosphorylated cofilin in synaptic fractions from APP/PS1 mice and AD patients’ brains (Rush et al., 2018). In APP/PS1 mouse brains, level of phosphorylated/inactive cofilin-1 is reduced at 4 months of age and increases at 10 months of age (Barone et al., 2014). In addition, different Aβ species and conformations seem to act differently on ADF/cofilin, depending on the locality, age and neuronal type (Kang and Woo, 2019). Despite this, the genetic reduction of ADF/cofilin rescues neurodegeneration (Woo et al., 2012), as well as LTP and contextual memory deficits in APP/PS1 mice (Woo et al., 2015). In summary, dysregulation of ADF/cofilin activity through either phosphorylation or dephosphorylation may contribute to the neurotoxic effects induced by Aβ in AD.
Several studies show that ADF/cofilin changes and synaptic dysfunction induced by Aβ are caused by both LIMK1 and SSH pathways (Kang and Woo, 2019). Treatment of hippocampal neurons with fibrillar amyloid beta increases the phosphorylation and activity of LIMK1 and these are accompanied by abnormalities in actin cytoskeleton, neuritic dystrophy and neuronal cell death (Heredia et al., 2006). Hippocampal neurons treated with Aβ42 oligomer induced LIMK1 activation which is regulated by an increase in the activity of Rac1 and Cdc42 Rho-GTPases and subsequent activation of PAK1 (Mendoza-Naranjo et al., 2012). Despite increased LIMK1 activation, Aβ42 treatments induce dephosphorylation of ADF/cofilin, suggesting the involvement of SSH. This is supported by work showing that overexpression of SSH prevents actin cytoskeleton abnormalities induced by Aβ42 treatments (Mendoza-Naranjo et al., 2012). In addition, Aβ42-induced ADF/cofilin dephosphorylation in the hippocampus-derived HT22 cell line is mediated by β1−integrin, a cell receptor important in the maintenance of synapses (Lilja and Ivaska, 2018), and the subsequent activation of SSH (Woo et al., 2015). These studies suggest that ADF/cofilin activity is regulated by bifurcating pathways that stimulate PAK1 and LIMK1 as well as SSH. In contrast, deficits in both PAK1 and PAK3 levels are detected in AD patients’ brains, which lead to activation of ADF/cofilin (Zhao et al., 2006). The application of Aβ42 can directly result in abnormally low levels of PAK in primary neurons (Zhao et al., 2006). Despite low levels of total PAK in AD brains, phosphorylated active PAK is increased around Aβ deposits (Zhao et al., 2006). Arsenault et al. (2013) confirm the loss of PAK in cortex of AD patients’ brains and cortex of AD mouse model (3xTg) and show that expression of a dominant-negative form of PAK results in deficits in social recognition.
In addition to the dysregulation of ADF/cofilin activity, formation of ADF/cofilin-actin rods may contribute to the pathology of AD (Bamburg and Bernstein, 2016; Kang and Woo, 2019). For example, an increase in ADF/cofilin-actin rods/aggregates have been reported in AD patients and AD mouse models including APP/PS1 and 3xTg (Rahman et al., 2014; Bamburg and Bernstein, 2016; Kang and Woo, 2019). Multiple studies have also shown that Aβ dimers/trimers promote the formation of ADF/cofilin-actin rods in neurons, associated with the dephosphorylation, activation of cofilin (Maloney et al., 2005; Davis et al., 2011; Mendoza-Naranjo et al., 2012; Barone et al., 2014). Bernstein et al. (2012) show that intermolecular disulfide bonds between cofilin subunits form in vitro by cofilin oxidation and is critical for cofilin-actin rod formation in stressed neurons. These less dynamic ADF/cofilin actin rods consist of ADF/cofilin and actin in 1:1 ratio and are shown to disrupt the integrity of dendritic microtubules, block intracellular transport of mitochondria and induce significant loss of dendritic spines (Bamburg and Bernstein, 2016; Walsh et al., 2014).
Targeting ADF/cofilin regulation may provide therapeutic targets to improve synaptic function and reduce memory impairment in AD patients (Shaw and Bamburg, 2017). Inhibition of cofilin-1 stabilizes the function and activity of dendritic spines in LTD mouse model (Zhou et al., 2004). Cofilin-1 inhibition is achieved using a phosphorylated peptide containing the first 16 amino acids of cofilin-1 (p-Cofilin peptide), which inhibits cofilin-1 activation through competitive binding to phosphatases. The use of this phosphorylated peptide in an AD mouse model (5 × FAD) rescues the deficits in surface expression and function of AMPA and NMDA receptors (Deng et al., 2016). Cofilin-1 inhibition by the peptide also partially improves working memory and novel object recognition in the model (Deng et al., 2016). In summary, ADF/cofilin contribute to AD pathology through multiple mechanisms including phosphorylation, dephosphorylation and formation of less dynamic ADF/cofilin-actin rods. Therefore, targeting ADF/cofilin holds promise to mitigate the physiological and behavioral abnormality in AD.
In summary, ADF/cofilin play multifaced roles in the regulation of synaptic structure and function in the brain. The temporal and spatial regulation of ADF/cofilin appears to be particularly important for the bi-directional effect on spine and synaptic plasticity. However, there are several key questions that need to be addressed further. First, although spine accumulation of ADF/cofilin is associated with both spine enlargement/LTP and LTD/spine shrinkage, how the increased ADF/cofilin in the spine leads to opposite changes in spine morphology remains unclear. Second, the relationship between spine accumulation and phosphorylation/dephosphorylation of ADF/cofilin needs further characterization. It remains unclear whether the translocation of the endogenous ADF/cofilin to the spine during LTP/spine enlargement or LTD/spine shrinkage requires ADF/cofilin dephosphorylation, although the exogenously expressed cofilin S3D appears unable to accumulate in the spine during LTP or LTD (Pontrello et al., 2012; Noguchi et al., 2016). Therefore, it is important to further elucidate how the translocation is regulated by protein phosphorylation and what protein kinases (e.g., LIMK1)/phosphatases (e.g., SSH) are involved. Third, the cooperation between ADF/cofilin and other actin-binding proteins (e.g., CAP2 and AIP) would provide another layer of regulation for ADF/cofilin activity at the synapse as some of these proteins also exhibit redistribution in the dendritic spine (Bosch et al., 2014; Pelucchi et al., 2020), but exactly when and how these interactions affect actin dynamics within the spine will require further studies. The use of photoactivatable ADF/cofilin (Noguchi et al., 2016; Senju et al., 2017; Borovac et al., 2018) or their upstream regulators (such as Rac1) (Wu et al., 2009; Fujii et al., 2013) in specific neuronal types and/or subcellular compartments within the spine should facilitate these investigations. Another emerging area is how ADF/cofilin-mediated actin dynamics are associated with and affect behavior in living animals, including different phases of learning and memory. As many brain disorders are associated with altered regulation of ADF/cofilin, a better understanding of this protein family could also aid in the understanding and treatment of these disorders.
All authors wrote and approved the manuscript.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR PJT155959 and CIHR PJT168922, ZJ), and Canadian Natural Science and Engineering Research Council (NSERC RGPIN341498 and RGPIN06295, ZJ) and the Hospital for Sick Children Foundation (ZJ). YB was supported by Libyan-North American Scholarship. NM was supported by University of Toronto Graduate Scholarship and the Dystonia Medical Research Foundation Canada.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Georgiana Forguson and Nina Huang for their technical assistance and comments on the manuscript.
Adolph, O., Köster, S., Räth, M., Georgieff, M., Weigt, H. U., Engele, J., et al. (2007). Rapid increase of glial glutamate uptake via blockade of the protein kinase A pathway. Glia 55, 1699–1707. doi: 10.1002/glia.20583
Agrawal, P. B., Joshi, M., Savic, T., Chen, Z., and Beggs, A. H. (2012). Normal myofibrillar development followed by progressive sarcomeric disruption with actin accumulations in a mouse Cfl2 knockout demonstrates requirement of cofilin-2 for muscle maintenance. Hum. Mol. Genet. 21, 2341–2356. doi: 10.1093/hmg/dds053
Alhadidi, Q., Bin Sayeed, M. S., and Shah, Z. A. (2016). Cofilin as a promising therapeutic target for ischemic and hemorrhagic stroke. Transl. Stroke Res. 7, 33–41. doi: 10.1007/s12975-015-0438-2
Allison, D. W., Gelfand, V. I., Spector, I., and Craig, A. M. (1998). Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J. Neurosci. 18, 2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998
Alvarez, V. A., and Sabatini, B. L. (2007). Anatomical and physiological plasticity of dendritic spines. Ann. Rev. Neurosci. 30, 79–97. doi: 10.1146/annurev.neuro.30.051606.094222
Anderson, C. M., and Swanson, R. A. (2000). Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32, 1–14. doi: 10.1002/1098-1136(200010)32:1<1::aid-glia10>3.0.co;2-w
Andrianantoandro, E., and Pollard, T. D. (2006). Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 24, 13–23. doi: 10.1016/j.molcel.2006.08.006
Arber, S., Barbayannis, F. A., Hanser, H., Schneider, C., Stanyon, C. A., Bernard, O., et al. (1998). Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393, 805–809. doi: 10.1038/31729
Arsenault, D., Dal-Pan, A., Tremblay, C., Bennett, D. A., Guitton, M. J., De Koninck, Y., et al. (2013). PAK inactivation impairs social recognition in 3xTg-AD Mice without increasing brain deposition of tau and Aβ. J. Neurosci. 33, 10729–10740. doi: 10.1523/JNEUROSCI.1501-13.2013
Asrar, S., Meng, Y., Zhou, Z., Todorovski, Z., Huang, W. W., and Jia, Z. (2009). Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1). Neuropharmacology 56, 73–80. doi: 10.1016/j.neuropharm.2008.06.055
Bamburg, J. R. (1999). Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Ann. Rev. Cell Dev. Biol. 15, 185–230. doi: 10.1146/annurev.cellbio.15.1.185
Bamburg, J. R., and Bernstein, B. W. (2008). ADF/cofilin. Curr. Biol. 18, R273–R275. doi: 10.1016/j.cub.2008.02.002
Bamburg, J. R., and Bernstein, B. W. (2016). Actin dynamics and cofilin-actin rods in alzheimer disease. Cytoskeleton (Hoboken N. J.) 73, 477–497. doi: 10.1002/cm.21282
Bamburg, J. R., and Wiggan, O. P. (2002). ADF/cofilin and actin dynamics in disease. Trends Cell Biol. 12, 598–605. doi: 10.1016/s0962-8924(02)02404-2
Barone, E., Mosser, S., and Fraering, P. C. (2014). Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and γ-secretase. Biochim. Biophys. Acta 1842(12 Pt A), 2500–2509. doi: 10.1016/j.bbadis.2014.10.004
Basu, S., and Lamprecht, R. (2018). The role of actin cytoskeleton in dendritic spines in the maintenance of long-term memory. Front. Mol. Neurosci. 11:143. doi: 10.3389/fnmol.2018.00143
Bernardinelli, Y., Nikonenko, I., and Muller, D. (2014). Structural plasticity: mechanisms and contribution to developmental psychiatric disorders. Front. Neuroanat. 8:123. doi: 10.3389/fnana.2014.00123
Bernstein, B. W., and Bamburg, J. R. (2010). ADF/cofilin: a functional node in cell biology. Trends Cell Biol. 20, 187–195. doi: 10.1016/j.tcb.2010.01.001
Bernstein, B. W., Painter, W. B., Chen, H., Minamide, L. S., Abe, H., and Bamburg, J. R. (2000). Intracellular pH modulation of ADF/cofilin proteins. Cell Motil. Cytosk. 47, 319–336. doi: 10.1002/1097-0169(200012)47:4<319::AID-CM6<3.0.CO;2-I
Bernstein, B. W., Shaw, A. E., Minamide, L. S., Pak, C. W., and Bamburg, J. R. (2012). Incorporation of cofilin into rods depends on disulfide intermolecular bonds: implications for actin regulation and neurodegenerative disease. J. Neurosci. 32, 6670–6681. doi: 10.1523/JNEUROSCI.6020-11.2012
Bertling, E., Hotulainen, P., Mattila, P. K., Matilainen, T., Salminen, M., and Lappalainen, P. (2004). Cyclase-associated protein 1 (CAP1) promotes cofilin-induced actin dynamics in mammalian nonmuscle cells. Mol. Biol. Cell 15, 2324–2334. doi: 10.1091/mbc.e04-01-0048
Bisaria, A., Hayer, A., Garbett, D., Cohen, D., and Meyer, T. (2020). Membrane-proximal F-actin restricts local membrane protrusions and directs cell migration. Science (New York N. Y.) 368, 1205–1210. doi: 10.1126/science.aay7794
Blanchoin, L., and Pollard, T. D. (1999). Mechanism of interaction of Acanthamoeba actophorin (ADF/Cofilin) with actin filaments. J. Biol. Chem. 274, 15538–15546. doi: 10.1074/jbc.274.22.15538
Blanchoin, L., and Pollard, T. D. (2002). Hydrolysis of ATP by polymerized actin depends on the bound divalent cation but not profilin. Biochemistry 41, 597–602. doi: 10.1021/bi011214b
Blanchoin, L., Boujemaa-Paterski, R., Sykes, C., and Plastino, J. (2014). Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 94, 235–263. doi: 10.1152/physrev.00018.2013
Blanchoin, L., Pollard, T. D., and Mullins, R. D. (2000). Interactions of ADF/cofilin, Arp2/3 complex, capping protein and profilin in remodeling of branched actin filament networks. Curr. Biol. 10, 1273–1282. doi: 10.1016/s0960-9822(00)00749-1
Bleicher, P., Sciortino, A., and Bausch, A. R. (2020). The dynamics of actin network turnover is self-organized by a growth-depletion feedback. Sci. Rep. 10, 6215. doi: 10.1038/s41598-020-62942-8
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Bliss, T. V., and Collingridge, G. L. (2013). Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol. Brain 6:5. doi: 10.1186/1756-6606-6-5
Bobkov, A. A., Muhlrad, A., Pavlov, D. A., Kokabi, K., Yilmaz, A., and Reisler, E. (2006). Cooperative effects of cofilin (ADF) on actin structure suggest allosteric mechanism of cofilin function. J. Mol. Biol. 356, 325–334. doi: 10.1016/j.jmb.2005.11.072
Borovac, J., Bosch, M., and Okamoto, K. (2018). Regulation of actin dynamics during structural plasticity of dendritic spines: signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 91, 122–130. doi: 10.1016/j.mcn.2018.07.001
Bosch, M., Castro, J., Saneyoshi, T., Matsuno, H., Sur, M., and Hayashi, Y. (2014). Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 82, 444–459. doi: 10.1016/j.neuron.2014.03.021
Bredt, D. S., and Nicoll, R. A. (2003). AMPA receptor trafficking at excitatory synapses. Neuron 40, 361–379. doi: 10.1016/s0896-6273(03)00640-8
Brieher, W. (2013). Mechanisms of actin disassembly. Mol. Biol. Cell 24, 2299–2302. doi: 10.1091/mbc.E12-09-0694
Brieher, W. M., Kueh, H. Y., Ballif, B. A., and Mitchison, T. J. (2006). Rapid actin monomer-insensitive depolymerization of Listeria actin comet tails by cofilin, coronin, and Aip1. J. Cell Biol. 175, 315–324. doi: 10.1083/jcb.200603149
Brown, V., Jin, P., Ceman, S., Darnell, J. C., O’Donnell, W. T., Tenenbaum, S. A., et al. (2001). Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107, 477–487. doi: 10.1016/s0092-8674(01)00568-2
Butterfield, D. A. (2014). The 2013 SFRBM discovery award: selected discoveries from the butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Radical Biol. Med. 74, 157–174. doi: 10.1016/j.freeradbiomed.2014.06.006
Cai, D., Zhong, M., Wang, R., Netzer, W. J., Shields, D., Zheng, H., et al. (2006). Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer’s disease-linked presenilin-1 mutant neurons. Proc. Natl. Acad. Sci. U.S.A. 103, 1936–1940. doi: 10.1073/pnas.0510710103
Cai, L., Makhov, A. M., and Bear, J. E. (2007). F-actin binding is essential for coronin 1B function in vivo. J. Cell Sci. 120(Pt 10), 1779–1790. doi: 10.1242/jcs.007641
Carlier, M. F., and Pantaloni, D. (1986). Direct evidence for ADP-Pi-F-actin as the major intermediate in ATP-actin polymerization. Rate of dissociation of Pi from actin filaments. Biochemistry 25, 7789–7792. doi: 10.1021/bi00372a001
Carlier, M. F., Laurent, V., Santolini, J., Melki, R., Didry, D., Xia, G. X., et al. (1997). Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J. Cell Biol. 136, 1307–1322. doi: 10.1083/jcb.136.6.1307
Carlisle, H. J., and Kennedy, M. B. (2005). Spine architecture and synaptic plasticity. Trends Neurosci. 28, 182–187. doi: 10.1016/j.tins.2005.01.008
Chan, C., Beltzner, C. C., and Pollard, T. D. (2009). Cofilin dissociates Arp2/3 complex and branches from actin filaments. Curr. Biol. 19, 537–545. doi: 10.1016/j.cub.2009.02.060
Chen, L. Y., Rex, C. S., Casale, M. S., Gall, C. M., and Lynch, G. (2007). Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci. 27, 5363–5372. doi: 10.1523/JNEUROSCI.0164-07.2007
Chin, S. M., Jansen, S., and Goode, B. L. (2016). TIRF microscopy analysis of human Cof1, Cof2, and ADF effects on actin filament severing and turnover. J. Mol. Biol. 428, 1604–1616. doi: 10.1016/j.jmb.2016.03.006
Choquet, D., and Hosy, E. (2020). AMPA receptor nanoscale dynamic organization and synaptic plasticities. Curr. Opin. Neurobiol. 63, 137–145. doi: 10.1016/j.conb.2020.04.003
Chua, B. T., Volbracht, C., Tan, K. O., Li, R., Yu, V. C., and Li, P. (2003). Mitochondrial translocation of cofilin is an early step in apoptosis induction. Nat. Cell Biol. 5, 1083–1089. doi: 10.1038/ncb1070
Cingolani, L. A., and Goda, Y. (2008). Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 9, 344–356. doi: 10.1038/nrn2373
Citri, A., and Malenka, R. C. (2008). Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33, 18–41. doi: 10.1038/sj.npp.1301559
Collingridge, G. L., Isaac, J. T., and Wang, Y. T. (2004). Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962. doi: 10.1038/nrn1556
Collingridge, G. L., Peineau, S., Howland, J. G., and Wang, Y. T. (2010). Long-term depression in the CNS. Nat. Rev. Neurosci. 11, 459–473. doi: 10.1038/nrn2867
Cuberos, H., Vallée, B., Vourc’h, P., Tastet, J., Andres, C. R., and Bénédetti, H. (2015). Roles of LIM kinases in central nervous system function and dysfunction. FEBS Lett. 589(24 Pt B), 3795–3806. doi: 10.1016/j.febslet.2015.10.032
Davis, R. C., Marsden, I. T., Maloney, M. T., Minamide, L. S., Podlisny, M., Selkoe, D. J., et al. (2011). Amyloid beta dimers/trimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin-phosphorylation. Mol. Neurodegenerat. 6:10. doi: 10.1186/1750-1326-6-10
Deng, Y., Wei, J., Cheng, J., Zhong, P., Xiong, Z., Liu, A., et al. (2016). Partial amelioration of synaptic and cognitive deficits by inhibiting cofilin dephosphorylation in an animal model of Alzheimer’s disease. J. Alzheimers Dis. 53, 1419–1432. doi: 10.3233/JAD-160167
Derkach, V. A., Oh, M. C., Guire, E. S., and Soderling, T. R. (2007). Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 8, 101–113. doi: 10.1038/nrn2055
Diering, G. H., and Huganir, R. L. (2018). The AMPA receptor code of synaptic plasticity. Neuron 100, 314–329. doi: 10.1016/j.neuron.2018.10.018
Duffney, L. J., Zhong, P., Wei, J., Matas, E., Cheng, J., Qin, L., et al. (2015). Autism-like deficits in Shank3-deficient Mice are rescued by targeting actin regulators. Cell Rep. 11, 1400–1413. doi: 10.1016/j.celrep.2015.04.064
Eiseler, T., Döppler, H., Yan, I. K., Kitatani, K., Mizuno, K., and Storz, P. (2009). Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat. Cell Biol. 11, 545–556. doi: 10.1038/ncb1861
Exton, J. H. (2002). Phospholipase D-structure, regulation and function. Rev. Physiol. Biochem. Pharmacol. 144, 1–94. doi: 10.1007/BFb0116585
Fedulov, V., Rex, C. S., Simmons, D. A., Palmer, L., Gall, C. M., and Lynch, G. (2007). Evidence that long-term potentiation occurs within individual hippocampal synapses during learning. J. Neurosci. 27, 8031–8039. doi: 10.1523/JNEUROSCI.2003-07.2007
Feuge, J., Scharkowski, F., Michaelsen-Preusse, K., and Korte, M. (2019). FMRP Modulates activity-dependent spine plasticity by binding cofilin1 mrna and regulating localization and local translation. Cerebral Cortex (New York N. Y. 1991) 29, 5204–5216. doi: 10.1093/cercor/bhz059
Frantz, C., Barreiro, G., Dominguez, L., Chen, X., Eddy, R., Condeelis, J., et al. (2008). Cofilin is a pH sensor for actin free barbed end formation: role of phosphoinositide binding. J. Cell Biol. 183, 865–879. doi: 10.1083/jcb.200804161
Freeman, N. L., Chen, Z., Horenstein, J., Weber, A., and Field, J. (1995). An actin monomer binding activity localizes to the carboxyl-terminal half of the Saccharomyces cerevisiae cyclase-associated protein. J. Biol. Chem. 270, 5680–5685. doi: 10.1074/jbc.270.10.5680
Fujii, M., Kawai, K., Egami, Y., and Araki, N. (2013). Dissecting the roles of Rac1 activation and deactivation in macropinocytosis using microscopic photo-manipulation. Sci. Rep. 3:2385. doi: 10.1038/srep02385
Gandhi, M., Achard, V., Blanchoin, L., and Goode, B. L. (2009). Coronin switches roles in actin disassembly depending on the nucleotide state of actin. Mol. Cell 34, 364–374. doi: 10.1016/j.molcel.2009.02.029
Gateva, G., Kremneva, E., Reindl, T., Kotila, T., Kogan, K., Gressin, L., et al. (2017). Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr. Biol. 27, 705–713. doi: 10.1016/j.cub.2017.01.018
Ge, P., Durer, Z. A., Kudryashov, D., Zhou, Z. H., and Reisler, E. (2014). Cryo-EM reveals different coronin binding modes for ADP- and ADP-BeFx actin filaments. Nat. Struct. Mol. Biol. 21, 1075–1081. doi: 10.1038/nsmb.2907
Gehler, S., Shaw, A. E., Sarmiere, P. D., Bamburg, J. R., and Letourneau, P. C. (2004). Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J. Neurosci. 24, 10741–10749. doi: 10.1523/JNEUROSCI.2836-04.2004
Gipson, C. D., and Olive, M. F. (2017). Structural and functional plasticity of dendritic spines – root or result of behavior? Genes Brain Behav. 16, 101–117. doi: 10.1111/gbb.12324
Gitik, M., Kleinhaus, R., Hadas, S., Reichert, F., and Rotshenker, S. (2014). Phagocytic receptors activate and immune inhibitory receptor SIRPα inhibits phagocytosis through paxillin and cofilin. Front. Cell. Neurosci. 8:104. doi: 10.3389/fncel.2014.00104
Gohla, A., Birkenfeld, J., and Bokoch, G. M. (2005). Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat. Cell Biol. 7, 21–29. doi: 10.1038/ncb1201
Goodson, M., Rust, M. B., Witke, W., Bannerman, D., Mott, R., Ponting, C. P., et al. (2012). Cofilin-1: a modulator of anxiety in mice. PLoS Genet. 8:e1002970. doi: 10.1371/journal.pgen.1002970
Görlich, A., Wolf, M., Zimmermann, A. M., Gurniak, C. B., Al Banchaabouchi, M., Sassoè-Pognetto, M., et al. (2011). N-cofilin can compensate for the loss of ADF in excitatory synapses. PLoS One 6:e26789. doi: 10.1371/journal.pone.0026789
Gorovoy, M., Niu, J., Bernard, O., Profirovic, J., Minshall, R., Neamu, R., et al. (2005). LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. J. Biol. Chem. 280, 26533–26542. doi: 10.1074/jbc.M502921200
Gotow, T., Miyaguchi, K., and Hashimoto, P. H. (1991). Cytoplasmic architecture of the axon terminal: filamentous strands specifically associated with synaptic vesicles. Neuroscience 40, 587–598. doi: 10.1016/0306-4522(91)90143-c
Govek, E. E., Newey, S. E., and Van Aelst, L. (2005). The role of the Rho GTPases in neuronal development. Genes Dev. 19, 1–49. doi: 10.1101/gad.1256405
Groc, L., and Choquet, D. (2020). Linking glutamate receptor movements and synapse function. Science (New York N. Y.) 368:eaay4631. doi: 10.1126/science.aay4631
Gu, J., Lee, C. W., Fan, Y., Komlos, D., Tang, X., Sun, C., et al. (2010). ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci. 13, 1208–1215. doi: 10.1038/nn.2634
Gu, J., Lee, C. W., Fan, Y., Komlos, D., Tang, X., Sun, C., et al. (2014). Aβ selectively impairs mGluR7 modulation of NMDA signaling in basal forebrain cholinergic neurons: implication in Alzheimer’s disease. J. Neurosci. 34, 13614–13628. doi: 10.1523/JNEUROSCI.1204-14.2014
Gunning, P. W., Hardeman, E. C., Lappalainen, P., and Mulvihill, D. P. (2015). Tropomyosin - master regulator of actin filament function in the cytoskeleton. J. Cell Sci. 128, 2965–2974. doi: 10.1242/jcs.172502
Gupta, A., and Pulliam, L. (2014). Exosomes as mediators of neuroinflammation. J. Neuroinflam. 11:68. doi: 10.1186/1742-2094-11-68
Gurniak, C. B., Chevessier, F., Jokwitz, M., Jönsson, F., Perlas, E., Richter, H., et al. (2014). Severe protein aggregate myopathy in a knockout mouse model points to an essential role of cofilin2 in sarcomeric actin exchange and muscle maintenance. Eur. J. Cell Biol. 93, 252–266. doi: 10.1016/j.ejcb.2014.01.007
Hadas, S., Spira, M., Hanisch, U. K., Reichert, F., and Rotshenker, S. (2012). Complement receptor-3 negatively regulates the phagocytosis of degenerated myelin through tyrosine kinase Syk and cofilin. J. Neuroinflam. 9:166. doi: 10.1186/1742-2094-9-166
Hamill, S., Lou, H. J., Turk, B. E., and Boggon, T. J. (2016). Structural Basis for noncanonical substrate recognition of cofilin/ADF proteins by LIM kinases. Mol. Cell 62, 397–408. doi: 10.1016/j.molcel.2016.04.001
Han, F., Zhuang, T. T., Chen, J. J., Zhu, X. L., Cai, Y. F., and Lu, Y. P. (2017). Novel derivative of Paeonol, Paeononlsilatie sodium, alleviates behavioral damage and hippocampal dendritic injury in Alzheimer’s disease concurrent with cofilin1/phosphorylated-cofilin1 and RAC1/CDC42 alterations in rats. PLoS One 12:e0185102. doi: 10.1371/journal.pone.0185102
Han, L., Stope, M. B., de Jesús, M. L., Oude Weernink, P. A., Urban, M., Wieland, T., et al. (2007). Direct stimulation of receptor-controlled phospholipase D1 by phospho-cofilin. EMBO J. 26, 4189–4202. doi: 10.1038/sj.emboj.7601852
Hanley, J. G. (2014). Actin-dependent mechanisms in AMPA receptor trafficking. Front. Cell. Neurosci. 8:381. doi: 10.3389/fncel.2014.00381
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science (New York N. Y) 297, 353–356. doi: 10.1126/science.1072994
Havekes, R., Park, A. J., Tudor, J. C., Luczak, V. G., Hansen, R. T., Ferri, S. L., et al. (2016). Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. eLife 5:e13424. doi: 10.7554/eLife.13424
Henley, J. M., and Wilkinson, K. A. (2016). Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 17, 337–350. doi: 10.1038/nrn.2016.37
Heredia, L., Helguera, P., de Olmos, S., Kedikian, G., Solá Vigo, F., LaFerla, F., et al. (2006). Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer’s disease. J. Neurosci. 26, 6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006
Ho, V. M., Lee, J. A., and Martin, K. C. (2011). The cell biology of synaptic plasticity. Science (New York N. Y.) 334, 623–628. doi: 10.1126/science.1209236
Honkura, N., Matsuzaki, M., Noguchi, J., Ellis-Davies, G. C., and Kasai, H. (2008). The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57, 719–729. doi: 10.1016/j.neuron.2008.01.013
Hoogenraad, C. C., Akhmanova, A., Galjart, N., and De Zeeuw, C. I. (2004). LIMK1 and CLIP-115: linking cytoskeletal defects to Williams syndrome. BioEssays 26, 141–150. doi: 10.1002/bies.10402
Hotulainen, P., and Hoogenraad, C. C. (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619–629. doi: 10.1083/jcb.201003008
Hotulainen, P., Llano, O., Smirnov, S., Tanhuanpää, K., Faix, J., Rivera, C., et al. (2009). Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J. Cell Biol. 185, 323–339. doi: 10.1083/jcb.200809046
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. U.S.A. 96, 3228–3233. doi: 10.1073/pnas.96.6.3228
Hsiao, K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., et al. (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science (New York N. Y.) 274, 99–102. doi: 10.1126/science.274.5284.99
Huang, W., Zhou, Z., Asrar, S., Henkelman, M., Xie, W., and Jia, Z. (2011). p21-Activated kinases 1 and 3 control brain size through coordinating neuronal complexity and synaptic properties. Mol. Cell. Biol. 31, 388–403. doi: 10.1128/MCB.00969-10
Huganir, R. L., and Nicoll, R. A. (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717. doi: 10.1016/j.neuron.2013.10.025
Ichetovkin, I., Grant, W., and Condeelis, J. (2002). Cofilin produces newly polymerized actin filaments that are preferred for dendritic nucleation by the Arp2/3 complex. Curr. Biol. 12, 79–84. doi: 10.1016/s0960-9822(01)00629-7
Ichetovkin, I., Han, J., Pang, K. M., Knecht, D. A., and Condeelis, J. S. (2000). Actin filaments are severed by both native and recombinant dictyostelium cofilin but to different extents. Cell Motil. Cytosk. 45, 293–306. doi: 10.1002/(SICI)1097-0169(200004)45:4<293::AID-CM5<3.0.CO;2-1
Jansen, S., and Goode, B. L. (2019). Tropomyosin isoforms differentially tune actin filament length and disassembly. Mol. Biol. Cell 30, 671–679. doi: 10.1091/mbc.E18-12-0815
Jansen, S., Collins, A., Chin, S. M., Ydenberg, C. A., Gelles, J., and Goode, B. L. (2015). Single-molecule imaging of a three-component ordered actin disassembly mechanism. Nat. Commun. 6:7202. doi: 10.1038/ncomms8202
Jansen, S., Collins, A., Golden, L., Sokolova, O., and Goode, B. L. (2014). Structure and mechanism of mouse cyclase-associated protein (CAP1) in regulating actin dynamics. J. Biol. Chem. 289, 30732–30742. doi: 10.1074/jbc.M114.601765
Jia, Z., Todorovski, Z., Meng, Y., Asrar, S., and Wang, L. Y. (2009). LIMK-1 and actin regulation of spine and synaptic function. New Encycl. Neurosci. 9, 467–472.
Kandel, E. R., Dudai, Y., and Mayford, M. R. (2014). The molecular and systems biology of memory. Cell 157, 163–186. doi: 10.1016/j.cell.2014.03.001
Kanellos, G., and Frame, M. C. (2016). Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 129, 3211–3218. doi: 10.1242/jcs.187849
Kang, D. E., and Woo, J. A. (2019). Cofilin, a master node regulating cytoskeletal pathogenesis in Alzheimer’s disease. J. Alzheimers Dis. 72, S131–S144. doi: 10.3233/JAD-190585
Kessels, H. W., and Malinow, R. (2009). Synaptic AMPA receptor plasticity and behavior. Neuron 61, 340–350. doi: 10.1016/j.neuron.2009.01.015
Kim, C. H., and Lisman, J. E. (1999). A role of actin filament in synaptic transmission and long-term potentiation. J. Neurosci. 19, 4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999
Kim, J. E., Kim, Y. J., Lee, D. S., Kim, J. Y., Ko, A. R., Hyun, H. W., et al. (2016). PLPP/CIN regulates bidirectional synaptic plasticity via GluN2A interaction with postsynaptic proteins. Sci. Rep. 6:26576. doi: 10.1038/srep26576
Kim, K., Lakhanpal, G., Lu, H. E., Khan, M., Suzuki, A., Hayashi, M. K., et al. (2015). A temporary gating of actin remodeling during synaptic plasticity consists of the interplay between the kinase and structural functions of CaMKII. Neuron 87, 813–826. doi: 10.1016/j.neuron.2015.07.023
Kiuchi, T., Ohashi, K., Kurita, S., and Mizuno, K. (2007). Cofilin promotes stimulus-induced lamellipodium formation by generating an abundant supply of actin monomers. J. Cell Biol. 177, 465–476. doi: 10.1083/jcb.200610005
Klamt, F., Zdanov, S., Levine, R. L., Pariser, A., Zhang, Y., Zhang, B., et al. (2009). Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol. 11, 1241–1246. doi: 10.1038/ncb1968
Klemke, M., Wabnitz, G. H., Funke, F., Funk, B., Kirchgessner, H., and Samstag, Y. (2008). Oxidation of cofilin mediates T cell hyporesponsiveness under oxidative stress conditions. Immunity 29, 404–413. doi: 10.1016/j.immuni.2008.06.016
Kotila, T., Kogan, K., Enkavi, G., Guo, S., Vattulainen, I., Goode, B. L., et al. (2018). Structural basis of actin monomer re-charging by cyclase-associated protein. Nat. Commun. 9:1892. doi: 10.1038/s41467-018-04231-7
Kotila, T., Wioland, H., Enkavi, G., Kogan, K., Vattulainen, I., Jégou, A., et al. (2019). Mechanism of synergistic actin filament pointed end depolymerization by cyclase-associated protein and cofilin. Nat. Commun. 10:5320. doi: 10.1038/s41467-019-13213-2
Kovaleva, T., Maksimova, N., Zhukov, I., Pershin, V., Mukhina, I., and Gainullin, M. (2019). Cofilin: molecular and cellular functions and its role in the functioning of the nervous system. Neurochem. J. 13, 11–19. doi: 10.1134/S1819712419010124
Kudryashov, D. S., Galkin, V. E., Orlova, A., Phan, M., Egelman, E. H., and Reisler, E. (2006). Cofilin cross-bridges adjacent actin protomers and replaces part of the longitudinal F-actin interface. J. Mol. Biol. 358, 785–797. doi: 10.1016/j.jmb.2006.02.029
Kueh, H. Y., Charras, G. T., Mitchison, T. J., and Brieher, W. M. (2008). Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 182, 341–353. doi: 10.1083/jcb.200801027
Kuhn, T. B., and Bamburg, J. R. (2008). Tropomyosin and ADF/cofilin as collaborators and competitors. Adv. Exp. Med. Biol. 644, 232–249. doi: 10.1007/978-0-387-85766-4_18
Lacor, P. N. (2007). Advances on the understanding of the origins of synaptic pathology in AD. Curr. Genom. 8, 486–508. doi: 10.2174/138920207783769530
Lai, K. O., and Ip, N. Y. (2013). Structural plasticity of dendritic spines: the underlying mechanisms and its dysregulation in brain disorders. Biochim. Biophys. Acta 1832, 2257–2263. doi: 10.1016/j.bbadis.2013.08.012
Lamprecht, R. (2011). The roles of the actin cytoskeleton in fear memory formation. Front. Behav. Neurosci. 5:39. doi: 10.3389/fnbeh.2011.00039
Lamprecht, R. (2016). The Role of actin cytoskeleton in memory formation in amygdala. Front. Mol. Neurosci. 9:23. doi: 10.3389/fnmol.2016.00023
Lamprecht, R., and LeDoux, J. (2004). Structural plasticity and memory. Nat. Rev. Neurosci. 5, 45–54. doi: 10.1038/nrn1301
Lappalainen, P., and Drubin, D. G. (1997). Cofilin promotes rapid actin filament turnover in vivo. Nature 388, 78–82. doi: 10.1038/40418
Lappalainen, P., Kessels, M. M., Cope, M. J., and Drubin, D. G. (1998). The ADF homology (ADF-H) domain: a highly exploited actin-binding module. Mol. Biol. Cell 9, 1951–1959. doi: 10.1091/mbc.9.8.1951
Lau, C. G., and Zukin, R. S. (2007). NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426. doi: 10.1038/nrn2153
Lee, S. H., and Dominguez, R. (2010). Regulation of actin cytoskeleton dynamics in cells. Mol. Cells 29, 311–325. doi: 10.1007/s10059-010-0053-8
Leung, C., Cao, F., Nguyen, R., Joshi, K., Aqrabawi, A. J., Xia, S., et al. (2018). Activation of entorhinal cortical projections to the dentate gyrus underlies social memory retrieval. Cell Rep. 23, 2379–2391. doi: 10.1016/j.celrep.2018.04.073
Li, G. B., Cheng, Q., Liu, L., Zhou, T., Shan, C. Y., Hu, X. Y., et al. (2013). Mitochondrial translocation of cofilin is required for allyl isothiocyanate-mediated cell death via ROCK1/PTEN/PI3K signaling pathway. Cell Commun. Signal. 11:50. doi: 10.1186/1478-811X-11-50
Li, X. L., Hu, N., Tan, M. S., Yu, J. T., and Tan, L. (2014). Behavioral and psychological symptoms in Alzheimer’s disease. BioMed Res. Int. 2014:927804. doi: 10.1155/2014/927804
Li, Z., Aizenman, C. D., and Cline, H. T. (2002). Regulation of rho GTPases by crosstalk and neuronal activity in vivo. Neuron 33, 741–750. doi: 10.1016/s0896-6273(02)00621-9
Lian, G., and Sheen, V. L. (2015). Cytoskeletal proteins in cortical development and disease: actin associated proteins in periventricular heterotopia. Front. Cell. Neurosci. 9:99. doi: 10.3389/fncel.2015.00099
Lilja, J., and Ivaska, J. (2018). Integrin activity in neuronal connectivity. J. Cell Sci. 131:jcs212803. doi: 10.1242/jcs.212803
Liston, C., and Gan, W. B. (2011). Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 16074–16079. doi: 10.1073/pnas.1110444108
Liston, C., Cichon, J. M., Jeanneteau, F., Jia, Z., Chao, M. V., and Gan, W. B. (2013). Circadian glucocorticoid oscillations promote learning-dependent synapse formation and maintenance. Nat. Neurosci. 16, 698–705. doi: 10.1038/nn.3387
Liu, A., Zhou, Z., Dang, R., Zhu, Y., Qi, J., He, G., et al. (2016). Neuroligin 1 regulates spines and synaptic plasticity via LIMK1/cofilin-mediated actin reorganization. J. Cell Biol. 212, 449–463. doi: 10.1083/jcb.201509023
Liu, J., Yang, X., Li, B., Wang, J., Wang, W., Liu, J., et al. (2017). STK16 regulates actin dynamics to control Golgi organization and cell cycle. Sci. Rep. 7:44607. doi: 10.1038/srep44607
Liu, T., Woo, J. A., Yan, Y., LePochat, P., Bukhari, M. Z., and Kang, D. E. (2019). Dual role of cofilin in APP trafficking and amyloid-β clearance. FASEB J. 33, 14234–14247. doi: 10.1096/fj.201901268R
Loisel, T. P., Boujemaa, R., Pantaloni, D., and Carlier, M. F. (1999). Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature 401, 613–616. doi: 10.1038/44183
Luo, L., Hensch, T. K., Ackerman, L., Barbel, S., Jan, L. Y., and Jan, Y. N. (1996). Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature 379, 837–840. doi: 10.1038/379837a0
Lüscher, C., and Malenka, R. C. (2012). NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harbor Perspect. Biol. 4:a005710. doi: 10.1101/cshperspect.a005710
Maciver, S. K., and Hussey, P. J. (2002). The ADF/cofilin family: actin-remodeling proteins. Genome Biol. 3:reviews3007. doi: 10.1186/gb-2002-3-5-reviews3007
Madineni, A., Alhadidi, Q., and Shah, Z. A. (2016). Cofilin inhibition restores neuronal cell death in oxygen-glucose deprivation model of ischemia. Mol. Neurobiol. 53, 867–878. doi: 10.1007/s12035-014-9056-3
Maekawa, M., Ishizaki, T., Boku, S., Watanabe, N., Fujita, A., Iwamatsu, A., et al. (1999). Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science (New York N. Y.) 285, 895–898. doi: 10.1126/science.285.5429.895
Malenka, R. C., and Bear, M. F. (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. doi: 10.1016/j.neuron.2004.09.012
Malinow, R., and Malenka, R. C. (2002). AMPA receptor trafficking and synaptic plasticity. Ann. Rev. Neurosci. 25, 103–126. doi: 10.1146/annurev.neuro.25.112701.142758
Maloney, M. T., Minamide, L. S., Kinley, A. W., Boyle, J. A., and Bamburg, J. R. (2005). Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: a feedforward mechanism for Alzheimer’s disease. J. Neurosci. 25, 11313–11321. doi: 10.1523/JNEUROSCI.3711-05.2005
Martinez, L. A., and Tejada-Simon, M. V. (2011). Pharmacological inactivation of the small GTPase Rac1 impairs long-term plasticity in the mouse hippocampus. Neuropharmacology 61, 305–312. doi: 10.1016/j.neuropharm.2011.04.017
Martino, A., Ettorre, M., Musilli, M., Lorenzetto, E., Buffelli, M., and Diana, G. (2013). Rho GTPase-dependent plasticity of dendritic spines in the adult brain. Front. Cell. Neurosci. 7:62. doi: 10.3389/fncel.2013.00062
Mateos-Aparicio, P., and Rodríguez-Moreno, A. (2019). The impact of studying brain plasticity. Front. Cell. Neurosci. 13:66. doi: 10.3389/fncel.2019.00066
Matsuzaki, M., Honkura, N., Ellis-Davies, G. C., and Kasai, H. (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. doi: 10.1038/nature02617
Matus, A., Ackermann, M., Pehling, G., Byers, H. R., and Fujiwara, K. (1982). High actin concentrations in brain dendritic spines and postsynaptic densities. Proc. Natl. Acad. Sci. U.S.A. 79, 7590–7594. doi: 10.1073/pnas.79.23.7590
Mendoza-Naranjo, A., Contreras-Vallejos, E., Henriquez, D. R., Otth, C., Bamburg, J. R., Maccioni, R. B., et al. (2012). Fibrillar amyloid-β1-42 modifies actin organization affecting the cofilin phosphorylation state: a role for Rac1/cdc42 effector proteins and the slingshot phosphatase. J. Alzheimers Dis. 29, 63–77. doi: 10.3233/JAD-2012-101575
Meng, J., Meng, Y., Hanna, A., Janus, C., and Jia, Z. (2005). Abnormal long-lasting synaptic plasticity and cognition in mice lacking the mental retardation gene Pak3. J. Neurosci. Off. J. Soc. Neurosci. 25, 6641–6650. doi: 10.1523/JNEUROSCI.0028-05.2005
Meng, Y., Takahashi, H., Meng, J., Zhang, Y., Lu, G., Asrar, S., et al. (2004). Regulation of ADF/cofilin phosphorylation and synaptic function by LIM-kinase. Neuropharmacology 47, 746–754. doi: 10.1016/j.neuropharm.2004.06.030
Meng, Y., Zhang, Y., Tregoubov, V., Janus, C., Cruz, L., Jackson, M., et al. (2002). Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35, 121–133. doi: 10.1016/s0896-6273(02)00758-4
Miermans, C. A., Kusters, R. P., Hoogenraad, C. C., and Storm, C. (2017). Biophysical model of the role of actin remodeling on dendritic spine morphology. PLoS One 12:e0170113. doi: 10.1371/journal.pone.0170113
Minamide, L. S., Striegl, A. M., Boyle, J. A., Meberg, P. J., and Bamburg, J. R. (2000). Neurodegenerative stimuli induce persistent ADF/cofilin-actin rods that disrupt distal neurite function. Nat. Cell Biol. 2, 628–636. doi: 10.1038/35023579
Moon, A. L., Janmey, P. A., Louie, K. A., and Drubin, D. G. (1993). Cofilin is an essential component of the yeast cortical cytoskeleton. J. Cell Biol. 120, 421–435. doi: 10.1083/jcb.120.2.421
Morales, M., Colicos, M. A., and Goda, Y. (2000). Actin-dependent regulation of neurotransmitter release at central synapses. Neuron 27, 539–550. doi: 10.1016/s0896-6273(00)00064-7
Mucke, L., and Selkoe, D. J. (2012). Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harbor Perspect. Med. 2:a006338. doi: 10.1101/cshperspect.a006338
Nagata-Ohashi, K., Ohta, Y., Goto, K., Chiba, S., Mori, R., Nishita, M., et al. (2004). A pathway of neuregulin-induced activation of cofilin-phosphatase Slingshot and cofilin in lamellipodia. J. Cell Biol. 165, 465–471. doi: 10.1083/jcb.200401136
Nakahata, Y., and Yasuda, R. (2018). Plasticity of spine structure: local signaling, translation and cytoskeletal reorganization. Front. Synap. Neurosci. 10:29. doi: 10.3389/fnsyn.2018.00029
Nakayama, A. Y., Harms, M. B., and Luo, L. (2000). Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 20, 5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000
Neves, G., Cooke, S. F., and Bliss, T. V. (2008). Bred. Nat. Rev. Neurosci. 9, 65–75. doi: 10.1038/nrn2303
Newey, S. E., Velamoor, V., Govek, E. E., and Van Aelst, L. (2005). Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 64, 58–74. doi: 10.1002/neu.20153
Nishita, M., Tomizawa, C., Yamamoto, M., Horita, Y., Ohashi, K., and Mizuno, K. (2005). Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J. Cell Biol. 171, 349–359. doi: 10.1083/jcb.200504029
Nishita, M., Wang, Y., Tomizawa, C., Suzuki, A., Niwa, R., Uemura, T., et al. (2004). Phosphoinositide 3-kinase-mediated activation of cofilin phosphatase Slingshot and its role for insulin-induced membrane protrusion. J. Biol. Chem. 279, 7193–7198. doi: 10.1074/jbc.M312591200
Niwa, R., Nagata-Ohashi, K., Takeichi, M., Mizuno, K., and Uemura, T. (2002). Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell 108, 233–246. doi: 10.1016/s0092-8674(01)00638-9
Noguchi, J., Hayama, T., Watanabe, S., Ucar, H., Yagishita, S., Takahashi, N., et al. (2016). State-dependent diffusion of actin-depolymerizing factor/cofilin underlies the enlargement and shrinkage of dendritic spines. Sci. Rep. 6:32897. doi: 10.1038/srep32897
Normoyle, K. P., and Brieher, W. M. (2012). Cyclase-associated protein (CAP) acts directly on F-actin to accelerate cofilin-mediated actin severing across the range of physiological pH. J. Biol. Chem. 287, 35722–35732. doi: 10.1074/jbc.M112.396051
Ohashi, K., Nagata, K., Maekawa, M., Ishizaki, T., Narumiya, S., and Mizuno, K. (2000). Rho-associated kinase ROCK activates LIM-kinase 1 by phosphorylation at threonine 508 within the activation loop. J. Biol. Chem. 275, 3577–3582. doi: 10.1074/jbc.275.5.3577
Okamoto, K., Nagai, T., Miyawaki, A., and Hayashi, Y. (2004). Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 7, 1104–1112. doi: 10.1038/nn1311
Ono, S. (2003). Regulation of actin filament dynamics by actin depolymerizing factor/cofilin and actin-interacting protein 1: new blades for twisted filaments. Biochemistry 42, 13363–13370. doi: 10.1021/bi034600x
Ostrowska-Podhorodecka, Z., śliwinska, M., Reisler, E., and Moraczewska, J. (2020). Tropomyosin isoforms regulate cofilin 1 activity by modulating actin filament conformation. Arch. Biochem. Biophys. 682:108280. doi: 10.1016/j.abb.2020.108280
Palop, J. J., and Mucke, L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci. 13, 812–818. doi: 10.1038/nn.2583
Park, M. (2018). AMPA Receptor trafficking for postsynaptic potentiation. Front. Cell. Neurosci. 12:361. doi: 10.3389/fncel.2018.00361
Pavlov, D., Muhlrad, A., Cooper, J., Wear, M., and Reisler, E. (2007). Actin filament severing by cofilin. J. Mol. Biol. 365, 1350–1358. doi: 10.1016/j.jmb.2006.10.102
Pavlov, D., Muhlrad, A., Cooper, J., Wear, M., and Reisler, E. (2006). Severing of F-actin by yeast cofilin is pH-independent. Cell Motil. Cytoskel. 63, 533–542. doi: 10.1002/cm.20142
Pelucchi, S., Stringhi, R., and Marcello, E. (2020). Dendritic spines in Alzheimer’s disease: how the actin cytoskeleton contributes to synaptic failure. Int. J. Mol. Sci. 21:908. doi: 10.3390/ijms21030908
Pennucci, R., Gucciardi, I., and de Curtis, I. (2019). Rac1 and Rac3 GTPases differently influence the morphological maturation of dendritic spines in hippocampal neurons. PLoS One 14:e0220496. doi: 10.1371/journal.pone.0220496
Pollard, T. D., and Borisy, G. G. (2003). Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465. doi: 10.1016/s0092-8674(03)00120-x
Pontrello, C. G., Sun, M. Y., Lin, A., Fiacco, T. A., DeFea, K. A., and Ethell, I. M. (2012). Cofilin under control of β-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc. Natl. Acad. Sci. U.S.A. 109, E442–E451. doi: 10.1073/pnas.1118803109
Posadas, I., Pérez-Martínez, F. C., Guerra, J., Sánchez-Verdú, P., and Ceña, V. (2012). Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J. Neurochem. 120, 515–527. doi: 10.1111/j.1471-4159.2011.07599.x
Poukkula, M., Hakala, M., Pentinmikko, N., Sweeney, M. O., Jansen, S., Mattila, J., et al. (2014). GMF promotes leading-edge dynamics and collective cell migration in vivo. Curr. Biol. 24, 2533–2540. doi: 10.1016/j.cub.2014.08.066
Purde, V., Busch, F., Kudryashova, E., Wysocki, V. H., and Kudryashov, D. S. (2019). Oligomerization affects the ability of human cyclase-associated proteins 1 and 2 to promote actin severing by cofilins. Int. J. Mol. Sci. 20:5647. doi: 10.3390/ijms20225647
Pyronneau, A., He, Q., Hwang, J. Y., Porch, M., Contractor, A., and Zukin, R. S. (2017). Aberrant Rac1-cofilin signaling mediates defects in dendritic spines, synaptic function, and sensory perception in fragile X syndrome. Sci. Signal. 10:eaan0852. doi: 10.1126/scisignal.aan0852
Racz, B., and Weinberg, R. J. (2006). Spatial organization of cofilin in dendritic spines. Neuroscience 138, 447–456. doi: 10.1016/j.neuroscience.2005.11.025
Rahman, T., Davies, D., Tannenberg, R., Fok, S., Shepherd, C., Dodd, P., et al. (2014). Cofilin rods and aggregates concur with tau pathology and the development of Alzheimer’s disease. J. Alzheimers Dis. 42, 1443–1460. doi: 10.3233/JAD-140393
Rasmussen, I., Pedersen, L. H., Byg, L., Suzuki, K., Sumimoto, H., and Vilhardt, F. (2010). Effects of F/G-actin ratio and actin turn-over rate on NADPH oxidase activity in microglia. BMC Immunol. 11:44. doi: 10.1186/1471-2172-11-44
Rebola, N., Srikumar, B. N., and Mulle, C. (2010). Activity-dependent synaptic plasticity of NMDA receptors. J. Physiol. 588(Pt 1), 93–99. doi: 10.1113/jphysiol.2009.179382
Rex, C. S., Chen, L. Y., Sharma, A., Liu, J., Babayan, A. H., Gall, C. M., et al. (2009). Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J. Cell Biol. 186, 85–97. doi: 10.1083/jcb.200901084
Ridley, A. J. (2011). Life at the leading edge. Cell 145, 1012–1022. doi: 10.1016/j.cell.2011.06.010
Rosenblatt, J., Agnew, B. J., Abe, H., Bamburg, J. R., and Mitchison, T. J. (1997). Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol. 136, 1323–1332. doi: 10.1083/jcb.136.6.1323
Rossi, D. J., Oshima, T., and Attwell, D. (2000). Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321. doi: 10.1038/35002090
Rothenfluh, A., and Cowan, C. W. (2013). Emerging roles of actin cytoskeleton regulating enzymes in drug addiction: actin or reactin’? Curr. Opin. Neurobiol. 23, 507–512. doi: 10.1016/j.conb.2013.01.027
Rush, T., Martinez-Hernandez, J., Dollmeyer, M., Frandemiche, M. L., Borel, E., Boisseau, S., et al. (2018). Synaptotoxicity in Alzheimer’s disease involved a dysregulation of actin cytoskeleton dynamics through cofilin 1 phosphorylation. J. Neurosci. 38, 10349–10361. doi: 10.1523/JNEUROSCI.1409-18.2018
Rust, M. B. (2015). Novel functions for ADF/cofilin in excitatory synapses – lessons from gene-targeted mice. Commun. Integrat. Biol. 8, e1114194. doi: 10.1080/19420889.2015.1114194
Rust, M. B., Gurniak, C. B., Renner, M., Vara, H., Morando, L., Görlich, A., et al. (2010). Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 29, 1889–1902. doi: 10.1038/emboj.2010.72
Sagot, I., Rodal, A. A., Moseley, J., Goode, B. L., and Pellman, D. (2002). An actin nucleation mechanism mediated by Bni1 and profilin. Nat. Cell Biol. 4, 626–631. doi: 10.1038/ncb834
Sanderson, J. L., Gorski, J. A., and Dell’Acqua, M. L. (2016). NMDA receptor-dependent LTD requires transient synaptic incorporation of Ca2+-permeable AMPARs mediated by AKAP150-anchored PKA and calcineurin. Neuron 89, 1000–1015. doi: 10.1016/j.neuron.2016.01.043
Santa-Marinha, L., Castanho, I., Silva, R. R., Bravo, F. V., Miranda, A. M., Meira, T., et al. (2020). Phospholipase D1 ablation disrupts mouse longitudinal hippocampal axis organization and functioning. Cell Rep. 30, 4197–4208. doi: 10.1016/j.celrep.2020.02.102
Schmidt, M., Voss, M., Weernink, P. A., Wetzel, J., Amano, M., Kaibuchi, K., et al. (1999). A role for rho-kinase in rho-controlled phospholipase D stimulation by the m3 muscarinic acetylcholine receptor. J. Biol. Chem. 274, 14648–14654. doi: 10.1074/jbc.274.21.14648
Scott, R. W., and Olson, M. F. (2007). LIM kinases: function, regulation and association with human disease. J. Mol. Med. (Berlin Germany) 85, 555–568. doi: 10.1007/s00109-007-0165-6
Segal, M. (2005). Dendritic spines and long-term plasticity. Nat. Rev. Neurosci. 6, 277–284. doi: 10.1038/nrn1649
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science (New York N. Y.) 298, 789–791. doi: 10.1126/science.1074069
Senju, Y., Kalimeri, M., Koskela, E. V., Somerharju, P., Zhao, H., Vattulainen, I., et al. (2017). Mechanistic principles underlying regulation of the actin cytoskeleton by phosphoinositides. Proc. Natl. Acad. Sci. U.S.A. 114, E8977–E8986. doi: 10.1073/pnas.1705032114
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007
Shaw, A. E., and Bamburg, J. R. (2017). Peptide regulation of cofilin activity in the CNS: a novel therapeutic approach for treatment of multiple neurological disorders. Pharmacol. Therap. 175, 17–27. doi: 10.1016/j.pharmthera.2017.02.031
Sheean, R. K., Lau, C. L., Shin, Y. S., O’Shea, R. D., and Beart, P. M. (2013). Links between L-glutamate transporters, Na+/K+-ATPase and cytoskeleton in astrocytes: evidence following inhibition with rottlerin. Neurosci. 254, 335–346. doi: 10.1016/j.neuroscience.2013.09.043
Shekhar, S., Chung, J., Kondev, J., Gelles, J., and Goode, B. L. (2019). Synergy between cyclase-associated protein and cofilin accelerates actin filament depolymerization by two orders of magnitude. Nat. Commun. 10:5319. doi: 10.1038/s41467-019-13268-1
Sheng, M., Sabatini, B. L., and Südhof, T. C. (2012). Synapses and Alzheimer’s disease. Cold Spring Harbor Perspect. Biol. 4:a005777. doi: 10.1101/cshperspect.a005777
Sheppard, P., Choleris, E., and Galea, L. (2019). Structural plasticity of the hippocampus in response to estrogens in female rodents. Mol. Brain 12:22. doi: 10.1186/s13041-019-0442-7
Shi, Y., Pontrello, C. G., DeFea, K. A., Reichardt, L. F., and Ethell, I. M. (2009). Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J. Neurosci. 29, 8129–8142. doi: 10.1523/JNEUROSCI.4681-08.2009
Shishkin, S., Eremina, L., Pashintseva, N., Kovalev, L., and Kovaleva, M. (2016). Cofilin-1 and Other ADF/cofilin superfamily members in human malignant cells. Int. J. Mol. Sci. 18:10. doi: 10.3390/ijms18010010
Soosairajah, J., Maiti, S., Wiggan, O., Sarmiere, P., Moussi, N., Sarcevic, B., et al. (2005). Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J. 24, 473–486. doi: 10.1038/sj.emboj.7600543
Soulé, J., Messaoudi, E., and Bramham, C. R. (2006). Brain-derived neurotrophic factor and control of synaptic consolidation in the adult brain. Biochem. Soc. Transact. 34(Pt 4), 600–604. doi: 10.1042/BST0340600
Spence, E. F., and Soderling, S. H. (2015). Actin out: regulation of the synaptic cytoskeleton. J. Biol. Chem. 290, 28613–28622. doi: 10.1074/jbc.R115.655118
Sumi, T., Matsumoto, K., and Nakamura, T. (2001). Specific activation of LIM kinase 2 via phosphorylation of threonine 505 by ROCK, a Rho-dependent protein kinase. J. Biol. Chem. 276, 670–676. doi: 10.1074/jbc.M007074200
Sun, Y., Liang, L., Dong, M., Li, C., Liu, Z., and Gao, H. (2019). Cofilin 2 in serum as a novel biomarker for Alzheimer’s disease in han Chinese. Front. Aging Neurosci. 11:214. doi: 10.3389/fnagi.2019.00214
Sun, Y., Rong, X., Lu, W., Peng, Y., Li, J., Xu, S., et al. (2015). Translational study of Alzheimer’s disease (AD) biomarkers from brain tissues in AβPP/PS1 mice and serum of AD patients. J. Alzheimers Dis. 45, 269–282. doi: 10.3233/JAD-142805
Sungur, A. Ö, Stemmler, L., Wöhr, M., and Rust, M. B. (2018). Impaired object recognition but normal social behavior and ultrasonic communication in cofilin1 mutant mice. Front. Behav. Neurosci. 12:25. doi: 10.3389/fnbeh.2018.00025
Tada, H., Miyazaki, T., Takemoto, K., Jitsuki, S., Nakajima, W., Koide, M., et al. (2017). Social isolation suppresses actin dynamics and synaptic plasticity through ADF/cofilin inactivation in the developing rat barrel cortex. Sci. Rep. 7:8471. doi: 10.1038/s41598-017-08849-3
Tada, H., Miyazaki, T., Takemoto, K., Takase, K., Jitsuki, S., Nakajima, W., et al. (2016). Neonatal isolation augments social dominance by altering actin dynamics in the medial prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 113, E7097–E7105. doi: 10.1073/pnas.1606351113
Tashiro, A., Minden, A., and Yuste, R. (2000). Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cerebral cortex (New York N. Y. 1991) 10, 927–938. doi: 10.1093/cercor/10.10.927
Taylor, R. A., and Sansing, L. H. (2013). Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin. Dev. Immunol. 2013:746068. doi: 10.1155/2013/746068
Thirion, C., Stucka, R., Mendel, B., Gruhler, A., Jaksch, M., Nowak, K. J., et al. (2001). Characterization of human muscle type cofilin (CFL2) in normal and regenerating muscle. Eur. J. Biochem. 268, 3473–3482. doi: 10.1046/j.1432-1327.2001.02247.x
Toda, S., Shen, H. W., Peters, J., Cagle, S., and Kalivas, P. W. (2006). Cocaine increases actin cycling: effects in the reinstatement model of drug seeking. J. Neurosci. 26, 1579–1587. doi: 10.1523/JNEUROSCI.4132-05.2006
Todorovski, Z., Asrar, S., Liu, J., Saw, N. M., Joshi, K., Cortez, M. A., et al. (2015). LIMK1 regulates long-term memory and synaptic plasticity via the transcriptional factor CREB. Mol. Cell. Biol. 35, 1316–1328. doi: 10.1128/MCB.01263-14
Tønnesen, J., and Nägerl, U. V. (2016). Dendritic spines as tunable regulators of synaptic signals. Front. Psychiatry 7:101. doi: 10.3389/fpsyt.2016.00101
Toshima, J., Toshima, J. Y., Takeuchi, K., Mori, R., and Mizuno, K. (2001). Cofilin phosphorylation and actin reorganization activities of testicular protein kinase 2 and its predominant expression in testicular Sertoli cells. J. Biol. Chem. 276, 31449–31458. doi: 10.1074/jbc.M102988200
van Galen, E. J., and Ramakers, G. J. (2005). Rho proteins, mental retardation and the neurobiological basis of intelligence. Prog. Brain Res. 147, 295–317. doi: 10.1016/S0079-6123(04)47022-8
Varadarajan, S., Yatin, S., Aksenova, M., and Butterfield, D. A. (2000). Review: Alzheimer’s amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 130, 184–208. doi: 10.1006/jsbi.2000.4274
Vartiainen, M. K., Mustonen, T., Mattila, P. K., Ojala, P. J., Thesleff, I., Partanen, J., et al. (2002). The three mouse actin-depolymerizing factor/cofilins evolved to fulfill cell-type-specific requirements for actin dynamics. Mol. Biol. Cell 13, 183–194. doi: 10.1091/mbc.01-07-0331
Walsh, K. P., Minamide, L. S., Kane, S. J., Shaw, A. E., Brown, D. R., Pulford, B., et al. (2014). Amyloid-β and proinflammatory cytokines utilize a prion protein-dependent pathway to activate NADPH oxidase and induce cofilin-actin rods in hippocampal neurons. PLoS One 9:e95995. doi: 10.1371/journal.pone.0095995
Wang, H. G., Lu, F. M., Jin, I., Udo, H., Kandel, E. R., de Vente, J., et al. (2005). Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron 45, 389–403. doi: 10.1016/j.neuron.2005.01.011
Wang, Y., Dong, Q., Xu, X. F., Feng, X., Xin, J., Wang, D. D., et al. (2013). Phosphorylation of cofilin regulates extinction of conditioned aversive memory via AMPAR trafficking. J. Neurosci. 33, 6423–6433. doi: 10.1523/JNEUROSCI.5107-12.2013
Wang, Y., Shibasaki, F., and Mizuno, K. (2005). Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J. Biol. Chem. 280, 12683–12689. doi: 10.1074/jbc.M411494200
Weaver, A. M., Karginov, A. V., Kinley, A. W., Weed, S. A., Li, Y., Parsons, J. T., et al. (2001). Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr. Biol. 11, 370–374. doi: 10.1016/s0960-9822(01)00098-7
Wegner, A. (1982). Treadmilling of actin at physiological salt concentrations. An analysis of the critical concentrations of actin filaments. J. Mol. Biol. 161, 607–615. doi: 10.1016/0022-2836(82)90411-9
Wiens, K. M., Lin, H., and Liao, D. (2005). Rac1 induces the clustering of AMPA receptors during spinogenesis. J. Neurosci. 25, 10627–10636. doi: 10.1523/JNEUROSCI.1947-05.2005
Winder, S. J., and Ayscough, K. R. (2005). Actin-binding proteins. J. Cell Sci. 118(Pt 4), 651–654. doi: 10.1242/jcs.01670
Wioland, H., Guichard, B., Senju, Y., Myram, S., Lappalainen, P., Jégou, A., et al. (2017). ADF/cofilin accelerates actin dynamics by severing filaments and promoting their depolymerization at both ends. Curr. Biol. 27, 1956–1967. doi: 10.1016/j.cub.2017.05.048
Wolf, M., Zimmermann, A. M., Görlich, A., Gurniak, C. B., Sassoè-Pognetto, M., Friauf, E., et al. (2015). ADF/cofilin controls synaptic actin dynamics and regulates synaptic vesicle mobilization and exocytosis. Cerebral Cortex (New York N. Y. 1991) 25, 2863–2875. doi: 10.1093/cercor/bhu081
Wong, L. W., Tann, J. Y., Ibanez, C. F., and Sajikumar, S. (2019). The p75 neurotrophin receptor is an essential mediator of impairments in hippocampal-dependent associative plasticity and memory induced by sleep deprivation. J. Neurosci. 39, 5452–5465. doi: 10.1523/JNEUROSCI.2876-18.2019
Woo, J. A., Boggess, T., Uhlar, C., Wang, X., Khan, H., Cappos, G., et al. (2015). RanBP9 at the intersection between cofilin and Aβ pathologies: rescue of neurodegenerative changes by RanBP9 reduction. Cell Death Dis. 6:1676. doi: 10.1038/cddis.2015.37
Woo, J. A., Jung, A. R., Lakshmana, M. K., Bedrossian, A., Lim, Y., Bu, J. H., et al. (2012). Pivotal role of the RanBP9-cofilin pathway in Aβ-induced apoptosis and neurodegeneration. Cell Death Different. 19, 1413–1423. doi: 10.1038/cdd.2012.14
Wu, Y. I., Frey, D., Lungu, O. I., Jaehrig, A., Schlichting, I., Kuhlman, B., et al. (2009). A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461, 104–108. doi: 10.1038/nature08241
Xie, Z., Srivastava, D. P., Photowala, H., Kai, L., Cahill, M. E., Woolfrey, K. M., et al. (2007). Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56, 640–656. doi: 10.1016/j.neuron.2007.10.005
Yan, X., Yadav, R., Gao, M., and Weng, H. R. (2014). Interleukin-1 beta enhances endocytosis of glial glutamate transporters in the spinal dorsal horn through activating protein kinase C. Glia 62, 1093–1109. doi: 10.1002/glia.22665
Yang, E., Kim, H., Lee, J., Shin, J. S., Yoon, H., Kim, S. J., et al. (2004). Overexpression of LIM kinase 1 renders resistance to apoptosis in PC12 cells by inhibition of caspase activation. Cell. Mol. Neurobiol. 24, 181–192. doi: 10.1023/b:cemn.0000018615.84358.33
Yang, N., Higuchi, O., Ohashi, K., Nagata, K., Wada, A., Kangawa, K., et al. (1998). Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812. doi: 10.1038/31735
Yang, Y., Wang, X. B., Frerking, M., and Zhou, Q. (2008). Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc. Natl. Acad. Sci. U.S.A. 105, 11388–11393. doi: 10.1073/pnas.0802978105
Yasuda, R. (2017). Biophysics of biochemical signaling in dendritic spines: implications in synaptic plasticity. Biophys. J. 113, 2152–2159. doi: 10.1016/j.bpj.2017.07.029
Ydenberg, C. A., Padrick, S. B., Sweeney, M. O., Gandhi, M., Sokolova, O., and Goode, B. L. (2013). GMF severs actin-Arp2/3 complex branch junctions by a cofilin-like mechanism. Curr. Biol. 23, 1037–1045. doi: 10.1016/j.cub.2013.04.058
Yeoh, S., Pope, B., Mannherz, H. G., and Weeds, A. (2002). Determining the differences in actin binding by human ADF and cofilin. J. Mol. Biol. 315, 911–925. doi: 10.1006/jmbi.2001.5280
Yonezawa, N., Nishida, E., Iida, K., Yahara, I., and Sakai, H. (1990). Inhibition of the interactions of cofilin, destrin, and deoxyribonuclease I with actin by phosphoinositides. J. Biol. Chem. 265, 8382–8386.
Yoo, Y., Ho, H. J., Wang, C., and Guan, J. L. (2010). Tyrosine phosphorylation of cofilin at Y68 by v-Src leads to its degradation through ubiquitin-proteasome pathway. Oncogene 29, 263–272. doi: 10.1038/onc.2009.319
Yuen, E. Y., Liu, W., Kafri, T., van Praag, H., and Yan, Z. (2010). Regulation of AMPA receptor channels and synaptic plasticity by cofilin phosphatase Slingshot in cortical neurons. J. Physiol. 588(Pt 13), 2361–2371. doi: 10.1113/jphysiol.2009.186353
Zamboni, V., Armentano, M., Berto, G., Ciraolo, E., Ghigo, A., Garzotto, D., et al. (2018). Hyperactivity of Rac1-GTPase pathway impairs neuritogenesis of cortical neurons by altering actin dynamics. Sci. Rep. 8:7254. doi: 10.1038/s41598-018-25354-3
Zhao, H., Hakala, M., and Lappalainen, P. (2010). ADF/cofilin binds phosphoinositides in a multivalent manner to act as a PIP(2)-density sensor. Biophys. J. 98, 2327–2336. doi: 10.1016/j.bpj.2010.01.046
Zhao, L., Ma, Q. L., Calon, F., Harris-White, M. E., Yang, F., Lim, G. P., et al. (2006). Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat. Neurosci. 9, 234–242. doi: 10.1038/nn1630
Zhou, L., Jones, E. V., and Murai, K. K. (2012). EphA signaling promotes actin-based dendritic spine remodeling through slingshot phosphatase. J. Biol. Chem. 287, 9346–9359. doi: 10.1074/jbc.M111.302802
Zhou, Q., Homma, K. J., and Poo, M. M. (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757. doi: 10.1016/j.neuron.2004.11.011
Zhou, Q., Xiao, M., and Nicoll, R. A. (2001). Contribution of cytoskeleton to the internalization of AMPA receptors. Proc. Natl. Acad. Sci. U.S.A. 98, 1261–1266. doi: 10.1073/pnas.031573798
Zhou, Z., Hu, J., Passafaro, M., Xie, W., and Jia, Z. (2011). GluA2 (GluR2) regulates metabotropic glutamate receptor-dependent long-term depression through N-cadherin-dependent and cofilin-mediated actin reorganization. J. Neurosci. 31, 819–833. doi: 10.1523/JNEUROSCI.3869-10.2011
Zhou, Z., Meng, Y., Asrar, S., Todorovski, Z., and Jia, Z. (2009). A critical role of Rho-kinase ROCK2 in the regulation of spine and synaptic function. Neuropharmacology 56, 81–89. doi: 10.1016/j.neuropharm.2008.07.031
Zhu, B., Fukada, K., Zhu, H., and Kyprianou, N. (2006). Prohibitin and cofilin are intracellular effectors of transforming growth factor beta signaling in human prostate cancer cells. Cancer Res. 66, 8640–8647. doi: 10.1158/0008-5472.CAN-06-1443
Keywords: ADF/cofilin, dendritic spine, LTP, LTD, AMPA glutamate receptor
Citation: Ben Zablah Y, Merovitch N and Jia Z (2020) The Role of ADF/Cofilin in Synaptic Physiology and Alzheimer’s Disease. Front. Cell Dev. Biol. 8:594998. doi: 10.3389/fcell.2020.594998
Received: 14 August 2020; Accepted: 23 October 2020;
Published: 12 November 2020.
Edited by:
Lei-Miao Yin, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Marco Rust, University of Marburg, GermanyCopyright © 2020 Ben Zablah, Merovitch and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengping Jia, emhlbmdwaW5nLmppYUBzaWNra2lkcy5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.