 Sanne van der Rijt
Sanne van der Rijt Marte Molenaars
Marte Molenaars Rebecca L. McIntyre
Rebecca L. McIntyre Georges E. Janssens
Georges E. Janssens Riekelt H. Houtkooper
Riekelt H. Houtkooper
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 26 November 2020
Sec. Signaling
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.594416
This article is part of the Research TopicMechanisms and Pathways Contributing to the Diversity of Aging across the Tree of LifeView all 15 articles
Since the identification and definition of the hallmarks of aging, these aspects of molecular and cellular decline have been most often described as isolated or distinct mechanisms. However, there is significant evidence demonstrating interplay between most of these hallmarks and that they have the capacity to influence and regulate one another. These interactions are demonstrable across the tree of life, yet not all aspects are conserved. Here, we describe an integrative view on the hallmarks of aging by using the hallmark “mitochondrial dysfunction” as a focus point, and illustrate its capacity to both influence and be influenced by the other hallmarks of aging. We discuss the effects of mitochondrial pathways involved in aging, such as oxidative phosphorylation, mitochondrial dynamics, mitochondrial protein synthesis, mitophagy, reactive oxygen species and mitochondrial DNA damage in relation to each of the primary, antagonistic and integrative hallmarks. We discuss the similarities and differences in these interactions throughout the tree of life, and speculate how speciation may play a role in the variation in these mechanisms. We propose that the hallmarks are critically intertwined, and that mapping the full extent of these interactions would be of significant benefit to the aging research community.
Aging is a multifactorial and complex process, affecting organisms at the molecular, cellular, tissue, and system levels (Gems and Partridge, 2013; Cohen, 2018). This progressive deterioration throughout life leads to age-related diseases such as cardiovascular diseases, cancer, osteoarthritis, diabetes mellitus type II and neurodegenerative diseases such as Alzheimer’s Disease (AD) and Parkinson’s Diseases (PD) (López-Otín et al., 2013; Franceschi et al., 2018). While lifespan has steadily increased in humans (Oeppen and Vaupel, 2002), many of these diseases remain prevalent and possess no cures. With the aim of uncovering mechanisms of aging and discovering interventions that promote healthier aging, scientists have utilized model organisms from across the phylogenetic tree. The most common model organisms are short lived, with well annotated genomes and molecular toolkits to intervene genetically in aging, such as S. cerevisiae, C. elegans, D. melanogaster, and M. musculus (Mitchell et al., 2015). Outside these traditional model organisms, aging researchers have studied extremely long-lived species, such as Hydra and the naked mole rat (Buffenstein, 2005; Boehm et al., 2012), as well as emerging models for aging research such as the killifish (Harel and Brunet, 2016).
Geroprotective interventions, either environmental or genetic, can extend the lifespan of model organisms (Mitchell et al., 2015). Through such experiments, nine hallmarks of aging have been defined, with indications that these are also conserved in humans (López-Otín et al., 2013). While more hallmarks are likely to be described in the future, as was the case for the hallmarks of cancer (Hanahan and Weinberg, 2011), we provide evidence of the interplay of between these current nine hallmarks of aging. The nine cellular and molecular hallmarks of aging are divided into three groups; (a) the primary hallmarks, (b) antagonistic hallmarks, and (c) integrative hallmarks (López-Otín et al., 2013). The primary hallmarks, which are unequivocally deleterious to the cell, include genomic instability, telomere attrition, epigenetic alterations, and loss of proteostasis. The antagonistic hallmarks, which are beneficial at low levels but at high levels become deleterious, are deregulated nutrient sensing, cellular senescence, and mitochondrial dysfunction. Finally, the integrative hallmarks, affecting tissue homeostasis and function, are stem cell exhaustion and altered intercellular communication. These hallmarks of aging have been presented throughout various research disciplines as nine separate hallmarks, and, while possessing crosstalk, are still considered largely independent. In this review, we provide evidence of the interplay between hallmarks, by highlighting one, mitochondrial dysfunction, and how it interacts with all others. In doing so, we aim to provide a broader view of the mitochondria’s role in the aging process throughout the tree of life and suggest reflection on the integrative nature of complex biological processes such as aging.
The role of mitochondrial dysfunction in aging is well documented and for comprehensive reviews on mitochondrial function in aging we refer the reader to Sun et al. (2016), Zhang et al. (2018), Bar-Ziv et al. (2020), Rath (2020). Here we summarize the various aspects of mitochondrial biology and aging across the prominent model organisms that are relevant to this review and the hallmark of mitochondrial dysfunction.
Mitochondria generate energy in the cell, via the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS). In general, mitochondrial function is impaired with age, marked by altered mitochondrial respiration, decreased energy production in the form of adenosine triphosphate (ATP), as well as widespread changes in metabolites associated with mitochondrial function (Lesnefsky and Hoppel, 2006; Houtkooper et al., 2011). Moreover, as a consequence of energy production, mitochondria also produce reactive oxygen species (ROS), which are formed during OXPHOS, causing oxidative damage to the mitochondria (Lesnefsky and Hoppel, 2006). ROS can contribute to damage to mitochondrial DNA (mtDNA), oxidation of mitochondrial proteins, less effective electron transport chain and impaired quality control in mitophagy (Bratic and Larsson, 2013; López-Otín et al., 2013). Yet, antioxidants do not provide lifespan benefits in model organisms through ROS scavenging, suggesting that ROS generation and oxidative stress do not themselves cause aging (Schulz et al., 2007). Instead, ROS have even been described as important signaling molecules in the cell, eliciting protective gene expression beneficial to longevity (Schulz et al., 2007). In line with this, a systematic review showed that there was no beneficial effect of antioxidant supplementation on the mortality in patients with various diseases (Bjelakovic et al., 2012). Recently, comparative analysis between mammalian species ranging in longevity identified a correlation between low levels of specific OXPHOS complex I proteins, ROS production, and lifespan in a species-specific manner (Mota-Martorell et al., 2020). Therefore, speciation may affect the role of ROS in aging and vice versa. In other words, ROS might have evolved different roles and physiologically normal levels depending on the environment of the species. These different selective pressures result in differences in the role of ROS.
While mitochondrial dysfunction shows detrimental effects at old age, it can also provide protection via hormetic stresses that promote longevity, especially when targeted early in life. For instance, reduction of nuclear encoded OXPHOS proteins via RNAi leads to extended life span in when the treatment is initiated in the larval stages of C. elegans (Dillin et al., 2002; Houtkooper et al., 2013). The proteins of the OXPHOS complexes are encoded by both nuclear DNA (nDNA) and mtDNA. Inhibiting mitochondrial translation and thus lowering synthesis of mtDNA-encoded OXPHOS proteins extends lifespan in C. elegans, and the link between mitochondrial translation and lifespan is conserved in natural populations of mice (Houtkooper et al., 2013). Inhibiting mitochondrial translation causes a mitonuclear protein imbalance that activates the protective mitochondrial unfolded protein response (UPRmt). The UPRmt is a stress response and one example of how dysfunctional mitochondria communicate to the nucleus where it activates mitochondrial chaperones and proteases, ROS detoxification enzymes, and mitochondrial protein import components (Nargund et al., 2015). In addition to UPRmt, several other pathways exist to protect mitochondria from intrinsic or extrinsic damage, including mitoCPR, MAGIC, UPRam, and mito-cytosolic translational balance (Molenaars et al., 2020a,b).
It is not only mitochondrial function that plays a role in aging, but also mitochondrial form. Mitochondria are constantly dynamically rearranging and degrading via the processes of fission, fusion and mitophagy. Mitochondria use these processes as quality control systems, degrading poorly-functional mitochondria on the go, and while specific proteins vary between model organisms, these processes are highly conserved (Liu et al., 2020a). Mitophagy involves the elimination of non-functional mitochondria, for instance upon their loss of membrane potential. Mitophagy is reduced throughout the aging process, and conversely, inducing mitophagy through genetic or pharmacological means leads to a longer lifespan (Wu et al., 2013; Ryu et al., 2016). Aging and age-related diseases are often accompanied by a lack of mitochondrial fusion, which leads to fragmented mitochondria (Chen et al., 2007; Houtkooper et al., 2013; Sharma et al., 2019), and altering mitochondrial dynamics of fission and fusion can extend lifespan in worms (Weir et al., 2017; Liu et al., 2020b).
Each of these aspects of mitochondrial dysfunction plays a significant role in aging. We next look beyond mitochondrial dysfunction as an individual hallmark and explore how these mitochondrial processes and age-related deteriorations interrelate and communicate with the other hallmarks of aging.
The primary hallmarks of aging are defined as unequivocally deleterious to the cell. This means that proper functioning of these processes is important for the viability of the cell and the dysfunction that occurs with age leads to cellular damage. Mitochondrial dysfunction interacts with each of these primary hallmarks, thus leading to progression of the aging process.
During aging, DNA damage accumulates, for instance due to ROS or environmental sources such as radiation (Fakouri et al., 2019). When DNA damage occurs, the DNA damage repair (DDR) is triggered (Niedernhofer et al., 2018; Fakouri et al., 2019). DDR activates several pathways, such as apoptosis, repair, or senescence. Every day, over 104 mutations occur in the genome (Niedernhofer et al., 2018). These mutations can be repaired by several mechanisms such as base excision repair, nucleotide excision repair, interstrand crosslink repair, non-homologous end joining and homologous recombination (Niedernhofer et al., 2018). The capacity for DNA repair decreases with aging, which results in accumulations of mutations with age and genomic instability (Niedernhofer et al., 2018).
The increasing amounts of DNA damage occurring during aging lead to activation of poly (ADP-ribose) polymerase 1 (PARP1), which serves as a DNA damage sensor and activator of various target proteins involved in the DNA damage response (Gupte et al., 2017). PARP1 uses nicotinamide adenine dinucleotide (NAD+) as a substrate in this process, and increased amounts of DNA damage therefore require large amounts of NAD+, resulting in a depletion of the NAD+ pool (Berger, 1985). This depletion of the NAD+ pool contributes to mitochondrial dysfunction considering that NAD+ acts as a co-factor to activate sirtuins. Sirtuins play important roles for mitochondrial function and longevity, including the activation the transcription factor peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1α), which stimulates mitochondrial biogenesis and function, and NAD+ depletion reduces this activation (Houtkooper et al., 2012). In line with this observation, preserving NAD+ by inhibiting PARP activity leads to healthier cells. Possible detrimental effects. Indeed, mice with mutations in PARP1 or PARP2 or those treated with pharmacological PARP inhibitors showed improved mitochondrial function and organismal fitness (Bai et al., 2011; Pirinen et al., 2014), and worms with PARP inhibition had extended lifespan (Mouchiroud et al., 2013). However, it should also be taken into account that PARP is important for DNA repair and PARP inhibition can possibly lead to genomic instability and cancer. Therefore, it should be investigated what are the long-term side effects of PARP inhibition and how these can be prevented (Bai et al., 2011).
Another avenue in which mitochondrial dysfunction and genomic instability directly meet is through the mutation of mtDNA genes. Mutations in mtDNA is generally 10-fold higher than in nuclear DNA (Wang et al., 2005). mtDNA damage can have various downstream consequences related to aging, such as damage in neurons, increasing the risk on neurodegenerative diseases such as AD (Wang et al., 2005). Another way by which mitochondria might contribute to DNA maintenance is through their role in nucleotide synthesis. Indeed, mitochondrial dysfunction leads to impaired synthesis of nucleotides (Tufi et al., 2014), and although not formally demonstrated one might expect downstream consequences on telomere attrition and genome stability. To summarize, genome instability, for instance caused by endogenous or exogenous DNA damage sources can damage mtDNA or indirectly impair mitochondrial function through NAD+.
All species must maintain the integrity of the ends of their chromosomes. How this is achieved varies greatly per species, for instance through (a) retrotransposons in Drosophila melanogaster, (b) having circular DNA and therefore no chromosome ends, such as with bacteria, or (c) possessing “telomeres” at the ends of chromosomes, such as in humans. When cells divide, telomeres become shorter, and when telomeres become too short, cells enter the state of senescence, which is an irreversible growth arrest. Cellular senescence is itself a hallmark of aging that contributes to age-related dysfunction and disease (López-Otín et al., 2013). Rapid telomere shortening can therefore coincide with an acceleration of aging and, indeed, studies in humans demonstrated that individuals with shorter telomeres have mortality rates twice as high as those with longer telomeres. Additionally, individuals with shorter telomeres have a significantly higher chance of having heart disease and age-related diseases compared to individuals with longer telomeres (Cawthon et al., 2003).
Mitochondria can directly contribute to an acceleration of telomere shorting, due to their production of ROS, which lead to DNA damage and telomere shortening (Liu et al., 2002). For example, cells treated with the protonophore carbonyl p-trifluoromethoxyphenylhydrazone (FCCP), a compound which depolarize the mitochondrial membrane potential and disrupts ATP synthesis, have increased ROS production (Liu et al., 2002) as well as shorter telomeres compared to untreated cells. Both ROS levels and shorter telomeres were rescued by treating the cells with ROS scavenger molecules, indicating that ROS leads to the shortening of telomeres (Liu et al., 2002). Similarly, irradiating cells with UV-A caused an increased level of 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG), a marker of DNA oxidation, as well as accelerated telomere shortening (Kawanishi and Oikawa, 2004).
The presence of critically short telomeres leads to the activation of the tumor suppressor gene p53 in mice (Sahin et al., 2011). P53 itself is known to inhibit PGC-1α/β, leading to the inhibition of mitochondrial biogenesis, OXPHOS, decreased mitochondrial mass, less ATP production and increased ROS production (Sahin et al., 2011). Therefore, initial damage to telomeres by mitochondrial ROS can cause a cycle of increasing damage, whereby PGC-1α inhibition further impairs mitochondrial function, creating additional ROS, which then contributes to further telomere damage. This is in line with the observations of telomere shortening, oxidative stress and aging in PGC-1α knock-out mice (Xiong et al., 2015). Generally, mitochondrial dysfunction can cause telomere attrition, which inhibits PGC-1α/β, resulting in further mitochondrial dysfunction. Moreover, future work might reveal more crosstalk links.
Reversible modifications of DNA and chromosomes, called epigenetics, are another hallmark of aging (López-Otín et al., 2013). Out of all the epigenetic changes that occur, most is known regarding DNA methylation and histone modifications. DNA methylation occurs through the activity of DNA methyltransferases (DNMTs), on regions of cytosines followed by guanines (termed CpG islands). During aging, hypermethylation of these CpG regions occurs, while hypomethylation occurs outside of these CpG regions (Christensen et al., 2009). Histone modifications are also affected during aging. Trimethylation on the lysine 4 residue of histone 3 (H3K4me3) leads to transcriptional activation, while trimethylation of lysine 27 of histone 3 (H3K27me3) is a repressive mark. Knockdown of one of the complexes of the H3K4me3 methyltransferase results in a longer lifespan in C. elegans (Greer et al., 2010). Another epigenetic modification is through histone deacetylases (HDACs), removing acetyl groups on histones. HDACs regulate the chromatin structure and play a critical role in transcription regulation. HDAC inhibitor compounds and their effects on each of the hallmarks of aging have previously been reviewed (McIntyre et al., 2019). HDAC inhibitors can increase lifespan in yeast, worms and flies (Lee et al., 2012; McIntyre et al., 2019).
Mitochondrial dysfunction plays a role in determining epigenetics changes during aging. Increased ROS leads to changes in the methylome, inducing aspects of the epigenetic aging process (Kietzmann et al., 2017). ROS causes DNA lesions and the most common base lesion is 8-hydroxyguanine (8-OHdG), which is in fact used as a measure for the level of oxidative stress (Turk et al., 1995). 8-OHdG lesions in cells caused by ROS inhibit DNA methylation, in line with the hypothesis that mitochondrial dysfunction and ROS production may be a driver of altered DNA methylation observed during aging (Turk et al., 1995).
HDAC inhibitors such as suberoylanilide hydroxamic acid (SAHA) and trapoxin A result in mitochondrial elongation, leading to healthy aging (Lee et al., 2012). In C. elegans, mitochondrial stress can lead to changes in chromatin structures such as H3K9 di-methylation (Tian et al., 2016). Normally these marks lead to silenced chromatin, however, some parts open up which lead to the expression of the mitochondrial unfolded protein response (UPRmt), including activation of chaperones and quality control proteases (Tian et al., 2016). H3K27 demethylases, jmjd-1.2 and jmjd-1.3, are activated when there is a perturbation in the ETC, which leads to the demethylation of H3K27 and UPRmt activation, inducing longevity in C. elegans (Merkwirth et al., 2016).
Another study looked at a genome wide blood methylome profile, or DNA methylation age clock, in 656 individuals with the age between 19 and 101. Three methylation sites that change with age were found (Hannum et al., 2013). For instance, a missense mutation was found in the locus of GTP binding protein 10 (GTPBP10), located close to the gene STEAP2. The methylation state of GTPBP10 also lowers the gene expression of STEAP2 and influences the synthesis of iron and copper (Hannum et al., 2013), elements that are required for proper OXPHOS (Ohgami et al., 2006).
CpG methylation of 12 other genes were later associated with aging, and two of these genes encode proteins that affect mitochondrial function (D’Aquila et al., 2019). The genes ras-related protein 32 (RAB32) and mitochondrial rho GTPase 2 (RHOT2), were hyper- and hypomethylated with age, respectively. Furthermore, RAB32 mRNA expression was decreased with age and RHOT2 expression was increased. RAB32 encodes for a GTPase and is important for mitochondrial fission and fusion, mitophagy and apoptosis (Alto et al., 2002). RHOT2 encodes for a protein in the outer mitochondrial membrane (OMM) and is important for the transport of molecules into the intermembrane space in the mitochondria, and also for mitochondrial fission and fusion (Fransson et al., 2006). These provide direct examples of how methylation state may influence mitochondrial biology during aging (D’Aquila et al., 2019).
To summarize, changes in mitochondrial biology with aging may result in increased ROS, which concomitantly alter epigenetic state at the DNA methylation level. Furthermore, DNA methylation as well as histone acetylation change during aging and impart alterations in gene expression of mitochondrial genes, creating a feedback loop of declining mitochondrial function.
Mitochondrial proteostasis has been extensively linked to aging (Jensen and Jasper, 2014; Moehle et al., 2019). Several pathways of mitochondrial proteostasis exist that restore mitochondrial function, including UPRam, which is activated by mistargeted mitochondrial precursor proteins, and mitoCPR which is activated upon mitochondrial import stress and (Wrobel et al., 2015; Weidberg and Amon, 2018). While these proteostasis pathways were predominantly described in yeast, the most extensively described pathway, UPRmt, was implicated in lifespan extension in worms, flies, and mice, suggesting a conserved role in the long-term maintenance of cellular homeostasis (Jovaisaite et al., 2014).
While the UPRmt ensures proteostasis specifically for mitochondrial proteins as a relatively independent protein quality control system, cytosolic proteins are simultaneously synthesized, folded, maintained and degraded in the cytosol. Stress can cause the unfolding and aggregation of proteins, and cells have different mechanisms to maintain proteostasis: specifically molecular chaperones, stress-response transcription factors and protein degradation via autophagy (Hartl et al., 2011). Another study showed that in C. elegans the mitochondrial ETC is a central regulator of the age-related decline of cytosolic proteostasis (Labbadia et al., 2017). Many more of these cytosolic proteostasis mechanisms linked to longevity are highly interconnected with mitochondria (reviewed in D’Amico et al., 2017; Molenaars et al., 2020a).
For instance, slowing down the synthesis of new proteins, by knockdown of cytosolic ribosomal proteins, extends lifespan in the model organism C. elegans (Hansen et al., 2007; Pan et al., 2007). Linking this to mitochondria, slowing down cytosolic protein synthesis is one of the consequences of mitochondrial disturbances that extend lifespan in C. elegans. For instance, ROS generated from dysfunctional mitochondria activates GCN-2-dependent eIF2α phosphorylation, leading to reduced cytosolic protein synthesis (Baker et al., 2012). Moreover, when looking at translational efficiencies in C. elegans with dysfunctional mitochondria, mRNAs coding for elements of the translation machinery were decreased and those coding for the OXPHOS and autophagy pathways were increased (Molenaars et al., 2018). Additionally, in C. elegans, mitochondrial translation and dynamics can synergistically regulate lifespan. This lifespan extension was dependent on the autophagy and lysosome biogenesis regulator, hlh-30 (TFEB in mammals) (Liu et al., 2020b), demonstrating a clear connection and communication between mitochondrial form and function with global cellular proteostasis.
Mitochondria are not only linked to cytosolic protein synthesis; accumulating evidence has shown that cytosolic proteins and aggregation-prone misfolded proteins can be translocated into the mitochondria. For instance, in yeast, cytosolic proteins prone to aggregation are imported into mitochondria for degradation which can be degraded via mitophagy (Ruan et al., 2017; Eldeeb and Fahlman, 2018).
In conclusion, both mitochondrial and cytosolic proteostasis, which can be linked via mito-cytosolic translational balance (Suhm et al., 2018; Molenaars et al., 2020b), are important for healthy aging. There is significant crosstalk between mitochondria and various aspects of proteostasis, both mitochondrial and cytosolic. At the same time there is a lot more to be discovered, especially with regard to the type of proteostatic response that is triggered depending on the context of the cellular or mitochondrial stress.
The antagonistic hallmarks of aging are hallmarks that can have beneficial or deleterious effects on the cell, depending on the level of intensity (López-Otín et al., 2013). When regulated properly, these hallmarks are beneficial or protective, but can be deleterious when levels are too high, or unregulated.
Nutrient sensing pathways involve the detection and cellular adaptation to nutritional challenges, either scarcity or excess. The activity of nutrient sensing pathways changes with age. Some of the best described nutrient sensing pathways involved in aging are the insulin/IGF1 pathway, the mechanistic target of rapamycin (mTOR), the sirtuin pathway, and AMP-activated protein kinase (AMPK) pathway (Houtkooper et al., 2010). Insulin/IGF1 and mTOR are typically activated under conditions of nutrient excess, leading to the activation of anabolic responses including glucose uptake, protein synthesis and inhibition of autophagy. Inhibition of these pathways, for instance inhibition of mTOR complex 1 (mTORC1) with rapamycin leads to marked lifespan extension in model organisms, including mice (Harrison et al., 2009). AMPK and sirtuins are activated upon caloric restriction and lead to activation of various catabolic pathways as well as stress defense systems. Activation of AMPK and sirtuins leads to enhanced autophagy, mitochondrial biogenesis and stress response, culminating in prolonged lifespan (Pearson et al., 2008). Importantly, the anabolic and catabolic nutrient sensing pathways function in intricate networks with bidirectional molecular communication (Houtkooper et al., 2010).
It is evident that mitochondrial (dys)function and nutrient sensing are connected. Through translational and transcriptional mechanisms, mTOR can regulate mitochondrial biogenesis, functions and dynamics (Cunningham et al., 2007; Morita et al., 2013, 2017; Gandin et al., 2016). Translation of nDNA-encoded proteins of OXPHOS complex I and V and fission protein mitochondrial fission process 1 (MTFP1) are stimulated by mTORC1 (Morita et al., 2013, 2017). When mTORC1 and thus MTFP1 is inhibited, this will lead to fusion of mitochondria and survival of the cell. In addition, mTORC1 can stimulate transcription of mitochondrial genes of the ETC leading to sustain high ATP production in the cell (Morita et al., 2013). Another mechanism connecting mTOR with mitochondrial function involves mitophagy. Mitophagy is reduced during aging. In TSC2-null cells, which have high mTORC1 activity, the activity of mitophagy was reduced (Bartolomé et al., 2017). This resulted an accumulation of defective mitochondria, leading to a shorter lifespan (Palikaras et al., 2015; Bartolomé et al., 2017).
Mitochondrial biogenesis regulates intracellular energy metabolism, in response to decreased energy. One of the regulators of mitochondrial biogenesis is AMPK. Activation of AMPK leads to initiation of mitophagy, for instance by activating the autophagy activating kinase 1 (Ulk1) (Egan et al., 2011; Laker et al., 2017). Furthermore, AMPK is important in fission of mitochondria (Toyama et al., 2016). The induction of AMPK can increase lifespan of model organisms such as C. elegans by maintaining mitochondrial homeostasis through the regulation of mitochondrial dynamics (Weir et al., 2017). Besides this, excess in nutrients, for instance in glucose or amino acids, can also lead to mitochondrial dysfunction (Houtkooper et al., 2010). Collectively, there is a tight-knit network of interacting nutrient sensing pathways that govern the cellular metabolic state, in part through modulating mitochondrial function, and thereby influence aging.
Cellular senescence is the transition to quiescence where cells cease dividing, and is characterized by the secretion of inflammatory signaling factors. This can be triggered by different mechanisms such as genomic instability or telomere attrition (McHugh and Gil, 2018). Senescent cells accumulate during aging in all tissues (Hudgins et al., 2018). This accumulation has an effect on cellular homeostasis. It increases inflammation, decreases tissue function and causes stem cell exhaustion, which all contribute to aging (López-Otín et al., 2013; McHugh and Gil, 2018).
One factor that can induce cellular senescence is oxidative stress. Indeed, when complex I, II and III are inhibited, by specific complex inhibitors, this will induce senescence (Yoon et al., 2003; Moiseeva et al., 2009). Similarly, when cells are treated with FCCP to depolarize the mitochondrial membrane potential, senescence is induced (Stöckl et al., 2006). All these findings indicate that defective OXPHOS leads to cellular senescence. Mitochondrial dynamics also play a role in cellular senescence. By knocking out a protein important for fission in mammalian cells, Fis1, mitochondria were elongated, which was accompanied by lower OXPHOS and higher ROS production, and this led to a significantly higher level of senescence (Lee et al., 2007). Another key aspect of senescence is the senescence-associated secretory phenotype (SASP), secreting pro-inflammatory cytokines, proteases and growth factors (Wiley et al., 2016). This secretory phenotype is activated upon mitochondrial dysfunction and depends on AMPK activation (Correia-Melo et al., 2016; Wiley et al., 2016). AMPK will phosphorylate the tumor suppressor gene p53 and this induces senescence (Wiley et al., 2016). These senescent cells will release the SASP, which amplifies the progression of senescence by inducing senescence in neighboring cells (Correia-Melo et al., 2016).
Together these results indicate that defects in OXPHOS, fission/fusion, high ROS production and altered mitochondrial biogenesis induce senescent cells and therefore the aging phenotype.
Integrative hallmarks of aging are hallmarks that have a more direct effect on the tissue homeostasis and function.
Stem cells can renew themselves and specific stem cells can give rise to a specific kind of tissue. However, aging reduces the renewal capability of stem cells (Ermolaeva et al., 2018). During aging, telomeres shorten and mutations occur in stem cells, which give rise to senescence of these stem cells. This stem cell exhaustion can, for instance, lead to neurodegenerative diseases or a decline in hematopoiesis which in turn leads to less production of adaptive immune responses (López-Otín et al., 2013; Ermolaeva et al., 2018). Though still a long way from application, it may eventually be possible to reprogram aged somatic stem cells into pluripotent stem cells as a way to prevent or reverse aspects of aging (Ahlqvist et al., 2015).
Stem cell exhaustion can also be caused by mitochondrial dysfunction. Increased ROS production in stem cells reduces the renewal of the stem cells in mice, which was rescued by antioxidant treatment (Jang and Sharkis, 2007). In line with that observation, mutator mice, which express proofreading defects in mitochondrial DNA polymerase gamma and thereby accumulate random mutations in mtDNA, present with an accelerated aging phenotype, demonstrated by graying fur, significant weight loss, osteoporosis and less ATP production in the heart when compared to wild type mice of the same age (Trifunovic et al., 2004). Moreover, these mice had both neural and hematopoietic stem cell dysfunction during their development. The neural stem cells in the mutator mice had decreased self-renewal capacity that was rescued by treating the cells with a ROS inhibitor (Ahlqvist et al., 2012), demonstrating that rescuing the mitochondrial dysfunction could rescue the stem cell dysfunction. Furthermore, lowering the mitochondrial activity in hematopoietic stem cell in mice by using carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), which chemically uncouples the electron transport chain, resulted in rapid stem cell differentiation (Vannini et al., 2016). These data suggest that mtDNA maintenance is important for healthy stem cells renewal.
Thus, mitochondrial dysfunction can cause stem cell exhaustion via several mechanisms. At the same time, this creates avenues for intervention since methods to improve mitochondrial function might improve stem cell renewal capacity and hence prevent its contribution to aging.
One example of altered intercellular communication is “inflamm-aging,” which is the development of chronic inflammation in aged people (Ferrucci and Fabbri, 2018). This chronic inflammation includes high levels of pro-inflammatory cytokines such as IL-1, IL-6, IFNα, transforming growth factor beta (TGFβ), and tumor necrosis factor (TNF) secreted by T and B cells (Ferrucci and Fabbri, 2018). Inflamm-aging is a risk factor for cancer, dementia and poor health status (Borrello et al., 2005; Gorelick, 2010; Leonardi et al., 2018). For instance, in many it was shown that the RET/PTC oncogene, a thyroid tumor, induces genes which are involved in inflammation, such as chemokines and cytokines (Borrello et al., 2005).
Mitochondria play an important role in altered intercellular communication. Upon inflammation, cells secrete signals to trigger an immune response. Damage associated molecular patterns (DAMPs) are danger signals released by cells if there is stress, apoptosis or necrosis. Mitochondria also secrete DAMPs, including molecules such as ATP, mtDNA and ROS (McGuire, 2019). For instance, it was reported that there is a high increase in mtDNA levels in plasma of people of 50+ years old with concomitant high levels of inflammatory cytokines (Pinti et al., 2014). Mitochondria can also trigger inflammatory responses via the mitochondrial antiviral signaling proteins (MAVS) (Koshiba et al., 2011). These proteins localize on the outer mitochondrial membrane, and phosphorylate interferon-regulatory factors 1/5/7 (IRF1/5/7) in an OXPHOS-dependent manner (Koshiba et al., 2011; Lazear et al., 2013). IRFs activate antiviral responses and this will lead to the production of type I interferon (IFN). Aged monocytes, however, have mitochondrial dysfunction including lower OXPHOS, but also lower IFR3/7. This results in lower IFN synthesis, and lower anti-viral response (Molony et al., 2017; Pence and Yarbro, 2018).
Alongside increased inflammation, the adaptive immune response declines with age. T-cells are important for the adaptive immune response to prevent infections (McGuire, 2019), but in the elderly the T-cell activity is reduced, making them more susceptible for diseases (Pieren et al., 2019). Interestingly, mice with T cells that were specifically deficient in a mitochondrial DNA–stabilizing protein exhibited multiple features associated with aging, including neurological, metabolic, muscular, and cardiovascular impairments (Desdín-Micó et al., 2020). The defective T cells initiated an early inflammatory program that induced premature senescence (Desdín-Micó et al., 2020). Another study showed that T cells from older subjects also had defects in autophagy and mitochondrial bioenergetics when compared to those of younger subjects, and that metformin alleviate aging-associated inflammation enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation (Bharath et al., 2020). T-cells with a cytochrome C oxidase 10 (Cox10), which is a part of OXPHOS complex IV, were infected with influenza (Tarasenko et al., 2017). These T-cells were not activated and were immunodeficient (Tarasenko et al., 2017), demonstrating that OXPHOS is important for T-cell activation.
To conclude, altered intercellular communication can cause mitochondrial dysfunction and vice versa, which contributes to age-related inflammation.
Since their classification, the nine hallmarks of aging have been used to describe and clarify the cellular and molecular processes involved in aging as a phenotype. However, these hallmarks are often thought of as discrete processes, as opposed to an intertwined system. In this review, we highlight the interrelation between the hallmarks of aging, with a focus on the mitochondria. Unifying mitochondrial factors such as decreased OXPHOS, increased ROS, reduced mitophagy, dysregulated fission or fusion, and mitochondrial proteostasis connect all of the hallmarks (Figure 1).
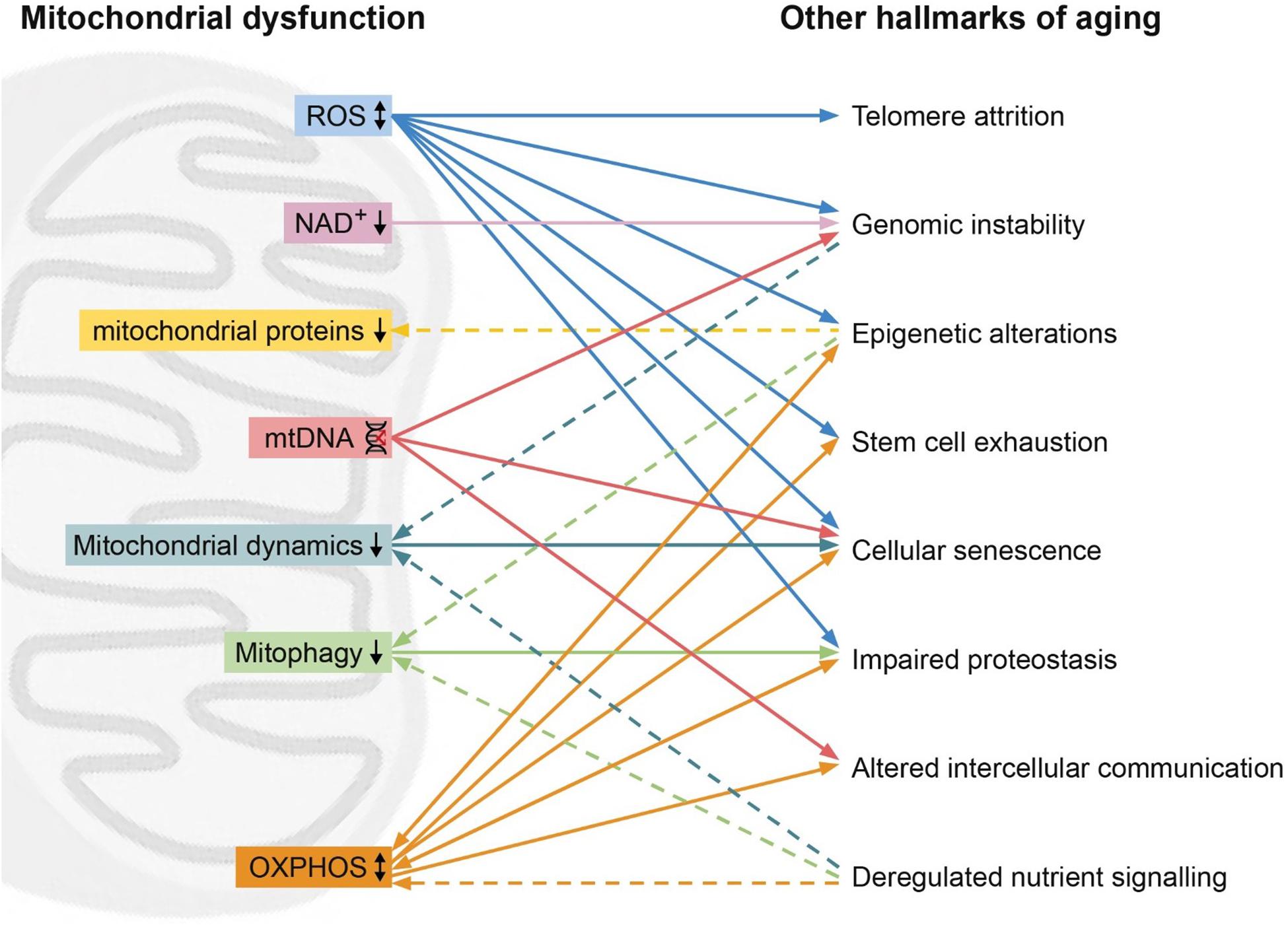
Figure 1. Mitochondrial dysfunction (left) and its relation with other hallmarks (right). High levels of reactive oxygen species (ROS, blue) can lead to telomere attrition, genomic instability, epigenetic alternations, stem cell exhaustion, cellular senescence. On the other hand, ROS can also improve proteostasis. Low levels of NAD+ (pink) can lead to genomic instability. Epigenetic alterations can lead to reduced mitochondrial proteins (yellow) such as STEAP2, RHOT2, and RAB32. Mitochondrial DNA (mtDNA) damage (red) can result in genomic instability, cellular senescence and altered intercellular communication. Hallmarks of aging genomic instability and deregulated nutrient sensing can lead to reduced mitochondrial dynamics (turquoise), while reduced mitochondrial dynamics induces cellular senescence. Reduced mitophagy (green) leads to impaired proteostasis, while epigenetic alterations and deregulated nutrient sensing induce reduced mitophagy. Oxidative phosphorylation (OXPHOS, orange) affects the hallmarks epigenetic alterations, stem cell exhaustion, cellular senescence, impaired proteostasis, altered intercellular communication and is in turn affected by deregulated nutrient sensing, epigenetic alterations and impaired proteostasis. Dashed line represents an effect of a hallmarks of aging on the mitochondrial dysfunction.
The majority of mechanistic aging research occurs in model organisms such as C. elegans, mice and D. melanogaster, and the interactions we describe are based on such traditional experimental models. However, resources such as the AnAge database demonstrate the dramatic variation in organism aging and longevity, which all can contribute to our understanding of these processes (Tacutu et al., 2018). The emergence of new model organisms provides further insight into aging mechanisms. The killifish (Nothobranchius furzeri) is a vertebrate, allowing for confirmation of findings in invertebrate organisms before moving to experiments in mammals. For example, the manipulation of the mitonuclear balance in assembly of the respiratory chain was shown to extend lifespan in C. elegans (Houtkooper et al., 2013). Recently, using comparative analysis of the genomes of various species within the killifish family, these genes were linked to evolution of lifespans in vertebrates as well (Sahm et al., 2017). These findings demonstrate not only the benefit of studying a wide variety of model organisms, but that comparative studies within the tree of life can also provide important insights into the mechanisms of aging.
However, the same reasons these organisms are often chosen as models can also make them less than suitable representations of the entirety of the phylogenetic tree. For instance, killifish, along with yeast, worms, flies, and mice, are all short-lived and fast-aging (Cohen, 2018). Extremely long-lived species, such as the naked mole rat and turtles, or seemingly immortal species, such as Hydra, can also provide valuable insights into molecular aging, such as the role of mitochondrial dysfunction plays.
The naked mole rat (Heterocephalus glaber) is well-known in the aging field as it boasts the most extreme longevity relative to its body size (Gorbunova et al., 2008). This extraordinary lifespan can potentially be connected to mitochondrial function. Naked mole rats exhibit remarkably less age-related changes in mitochondrial mass and efficiency, as well as lipid peroxidation (Buffenstein, 2008; Stoll et al., 2016). Particularly, OXPHOS complex IV expression and activity remains stable throughout lifespan (Stoll et al., 2016). When comparing the skeletal and heart muscle of naked mole rats to mice, one study found that the two species generated essentially equal quantities of mitochondrial H2O2 as a proxy of overall ROS metabolism. However, naked mole rats had a significantly greater capacity to consume ROS compared to mice (Munro et al., 2019). The naked mole rat therefore demonstrates a clear model for the aging benefits of maintaining mitochondrial function. Another example is birds. Birds have a relatively long lifespan considering their high metabolic activity and small body size, and it is shown that birds have less ROS production and longer telomeres (Hickey et al., 2012). Similarly, it has been noted that hypoxia in the freshwater turtle Trachemys scripta elegans inhibits mitochondrial respiration and ROS production, preventing oxidative damage (Bundgaard et al., 2019). These examples illustrate that different species have different ways to maintain mitochondrial function in the face of physiological challenges, and thereby sustain a relatively long and healthy lifespan.
The cnidarian Hydra (hydra vulgaris) is capable of continuous self-renewal and is therefore considered immortal (Schaible et al., 2015). This renewal is mainly due to the robust activity of three stem cell populations (Tomczyk et al., 2015). However, Hydra also have a well-described stress response system, including antioxidant processes (Dash et al., 2007). The FoxO transcription factor is expressed in stem cells, and reduction in FoxO levels negatively affect the proliferation of stem cells (Boehm et al., 2012). While there are few mitochondria-specific studies in Hydra, the interaction of these stem cells and FoxO regulation provide an example of interacting hallmarks in regulation of Hydra immortality. Therefore, interactions between hallmarks are not only present outside the classical aging model organisms, but also that all hallmarks connect together, not just within the lens of mitochondrial dysfunction.
Across the phylogenetic tree, the hallmarks of aging connect to and influence one another. Although we chose to focus our review on the hallmark of mitochondrial dysfunction, a clear pattern emerges that all hallmarks of aging influence one another. For instance, cellular senescence can be induced by genomic instability or telomere attrition (Lidzbarsky et al., 2018) and epigenetic alternations can lead to genomic instability (Pal and Tyler, 2016). It is hence evident that the hallmarks of aging are not discrete entities as how are often presented, but instead operate in a large and tightly connected network. Targeting one factor of this network can result in affecting other hallmarks and thus influence the whole network of aging. Although this complicates our interpretation of anti-aging interventions and requires a more holistic approach, it also opens opportunities for treatment options that not only target one hallmark but in fact act on the entire, or at least a large section of the network. In relation to the phylogenetic tree of life, while the exact details of the hallmarks of aging may differ, the main commonality that unifies aging across all species is the fact that all their hallmarks interconnect. Taking the entirety of this network into account will benefit the aging research community, and ultimately allow for a greater understanding of the aging processes and the progression of age-related disease.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Work in the Houtkooper group was financially supported by an ERC Starting grant (No. 638290), a VIDI grant from ZonMw (No. 91715305), and a grant from the Velux Stiftung (No. 1063). GJ was supported by a VENI grant from ZonMw (No. 09150161810014, https://www.zonmw.nl).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ahlqvist, K. J., Hämäläinen, R. H., Yatsuga, S., Uutela, M., Terzioglu, M., Götz, A., et al. (2012). Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in polg mutator mice. Cell Metab. 15, 100–109. doi: 10.1016/j.cmet.2011.11.012
Ahlqvist, K. J., Suomalainen, A., and Hämäläinen, R. H. (2015). Stem cells, mitochondria and aging. Biochim. Biophys. Acta Bioenerg. 1847, 1380–1386. doi: 10.1016/j.bbabio.2015.05.014
Alto, N. M., Soderling, J., and Scott, J. D. (2002). Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J. Cell Biol. 158, 659–668. doi: 10.1083/jcb.200204081
Bai, P., Cantó, C., Oudart, H., Brunyánszki, A., Cen, Y., Thomas, C., et al. (2011). PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468. doi: 10.1016/j.cmet.2011.03.004
Baker, B. M., Nargund, A. M., Sun, T., and Haynes, C. M. (2012). Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 8:e1002760. doi: 10.1371/journal.pgen.1002760
Bartolomé, A., García-Aguilar, A., Asahara, S.-I., Kido, Y., Guillén, C., Pajvani, U. B., et al. (2017). MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol. Cell. Biol. 37:e00441-17. doi: 10.1128/mcb.00441-17
Bar-Ziv, R., Bolas, T., and Dillin, A. (2020). Systemic effects of mitochondrial stress. EMBO Rep. 21:e50094. doi: 10.15252/embr.202050094
Berger, N. A. (1985). Poly(ADP-Ribose) in the cellular response to DNA damage. Radiat. Res. 101:4. doi: 10.2307/3576299
Bharath, L. P., Agrawal, M., McCambridge, G., Nicholas, D. A., Hasturk, H., Liu, J., et al. (2020). Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55.e6. doi: 10.1016/j.cmet.2020.04.015
Bjelakovic, G., Nikolova, D., Gluud, L. L., Simonetti, R. G., and Gluud, C. (2012). Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 3:CD007176. doi: 10.1002/14651858.cd007176.pub2
Boehm, A. M., Khalturin, K., Anton-Erxleben, F., Hemmrich, G., Klostermeier, U. C., Lopez-Quintero, J. A., et al. (2012). FoxO is a critical regulator of stem cell maintenance in immortal Hydra. Proc. Natl. Acad. Sci. U.S.A. 109, 19697–19702. doi: 10.1073/pnas.1209714109
Borrello, M. G., Alberti, L., Fischer, A., Degl’Innocenti, D., Ferrario, C., Gariboldi, M., et al. (2005). Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc. Natl. Acad. Sci. U.S.A. 102, 14825–14830. doi: 10.1073/pnas.0503039102
Bratic, A., and Larsson, N.-G. (2013). The role of mitochondria in aging. J. Clin. Invest. 123, 951–957. doi: 10.1172/JCI64125
Buffenstein, R. (2005). The naked mole-rat: a new long-living model. J. Gerontol. A Biol. Sci. Med. Sci. 60, 1369–1377. doi: 10.1093/gerona/60.11.1369
Buffenstein, R. (2008). Negligible senescence in the longest living rodent, the naked mole-rat: insights from a successfully aging species. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 178, 439–445. doi: 10.1007/s00360-007-0237-5
Bundgaard, A., Qvortrup, K., Rasmussen, L. J., and Fago, A. (2019). Turtles maintain mitochondrial integrity but reduce mitochondrial respiratory capacity in the heart after cold acclimation and anoxia. J. Exp. Biol. 222:jeb200410. doi: 10.1242/jeb.200410
Cawthon, R. M., Smith, K. R., O’Brien, E., Sivatchenko, A., and Kerber, R. A. (2003). Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 361, 393–395. doi: 10.1016/S0140-6736(03)12384-7
Chen, H., McCaffery, J. M., and Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562. doi: 10.1016/j.cell.2007.06.026
Christensen, B. C., Houseman, E. A., Marsit, C. J., Zheng, S., Wrensch, M. R., Wiemels, J. L., et al. (2009). Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 5:e1000602. doi: 10.1371/journal.pgen.1000602
Cohen, A. A. (2018). Aging across the tree of life: the importance of a comparative perspective for the use of animal models in aging. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 2680–2689. doi: 10.1016/J.BBADIS.2017.05.028
Correia-Melo, C., Marques, F. D., Anderson, R., Hewitt, G., Hewitt, R., Cole, J., et al. (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 35, 724–742. doi: 10.15252/embj.201592862
Cunningham, J. T., Rodgers, J. T., Arlow, D. H., Vazquez, F., Mootha, V. K., and Puigserver, P. (2007). mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 450, 736–740. doi: 10.1038/nature06322
D’Amico, D., Sorrentino, V., and Auwerx, J. (2017). Cytosolic proteostasis networks of the mitochondrial stress response. Trends Biochem. Sci. 42, 712–725. doi: 10.1016/j.tibs.2017.05.002
D’Aquila, P., Montesanto, A., De Rango, F., Guarasci, F., Passarino, G., and Bellizzi, D. (2019). Epigenetic signature: implications for mitochondrial quality control in human aging. Aging 11, 1240–1251. doi: 10.18632/aging.101832
Dash, B., Metz, R., Huebner, H. J., Porter, W., and Phillips, T. D. (2007). Molecular characterization of two superoxide dismutases from Hydra vulgaris. Gene 387, 93–108. doi: 10.1016/j.gene.2006.08.020
Desdín-Micó, G., Soto-Heredero, G., Aranda, J. F., Oller, J., Carrasco, E., Gabandé-Rodríguez, E., et al. (2020). T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376. doi: 10.1126/science.aax0860
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., et al. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398–2401. doi: 10.1126/science.1077780
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Eldeeb, M. A., and Fahlman, R. P. (2018). Does too much MAGIC lead to mitophagy? Trends Biochem. Sci. 43, 485–487. doi: 10.1016/j.tibs.2018.04.008
Ermolaeva, M., Neri, F., Ori, A., and Rudolph, K. L. (2018). Cellular and epigenetic drivers of stem cell ageing. Nat. Rev. Mol. Cell Biol. 19, 594–610. doi: 10.1038/s41580-018-0020-3
Fakouri, N. B., Hou, Y., Demarest, T. G., Christiansen, L. S., Okur, M. N., Mohanty, J. G., et al. (2019). Toward understanding genomic instability, mitochondrial dysfunction and aging. FEBS J. 286, 1058–1073. doi: 10.1111/febs.14663
Ferrucci, L., and Fabbri, E. (2018). Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522. doi: 10.1038/s41569-018-0064-2
Franceschi, C., Garagnani, P., Morsiani, C., Conte, M., Santoro, A., Grignolio, A., et al. (2018). The continuum of aging and age-related diseases: common mechanisms but different rates. Front. Med. 5:61. doi: 10.3389/fmed.2018.00061
Fransson, Å, Ruusala, A., and Aspenström, P. (2006). The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 344, 500–510. doi: 10.1016/j.bbrc.2006.03.163
Gandin, V., Masvidal, L., Hulea, L., Gravel, S. P., Cargnello, M., McLaughlan, S., et al. (2016). NanoCAGE reveals 5’ UTR features that define specific modes of translation of functionally related MTOR-sensitive mRNAs. Genome Res. 26, 636–648. doi: 10.1101/gr.197566.115
Gems, D., and Partridge, L. (2013). Genetics of longevity in model organisms: debates and paradigm shifts. Annu. Rev. Physiol. 75, 621–644. doi: 10.1146/annurev-physiol-030212-183712
Gorbunova, V., Bozzella, M. J., and Seluanov, A. (2008). Rodents for comparative aging studies: from mice to beavers. Age 30, 111–119. doi: 10.1007/s11357-008-9053-4
Gorelick, P. B. (2010). Role of inflammation in cognitive impairment: results of observational epidemiological studies and clinical trials. Ann. N. Y. Acad. Sci. 1207, 155–162. doi: 10.1111/j.1749-6632.2010.05726.x
Greer, E. L., Maures, T. J., Hauswirth, A. G., Green, E. M., Leeman, D. S., Maro, G. S., et al. (2010). Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383–387. doi: 10.1038/nature09195
Gupte, R., Liu, Z., and Kraus, W. L. (2017). Parps and adp-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126. doi: 10.1101/gad.291518.116
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Hannum, G., Guinney, J., Zhao, L., Zhang, L., Hughes, G., Sadda, S., et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367. doi: 10.1016/j.molcel.2012.10.016
Hansen, M., Taubert, S., Crawford, D., Libina, N., Lee, S. J., and Kenyon, C. (2007). Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6, 95–110. doi: 10.1111/j.1474-9726.2006.00267.x
Harel, I., and Brunet, A. (2016). The African turquoise killifish: a model for exploring vertebrate aging and diseases in the fast lane. Cold Spring Harb. Symp. Quant. Biol. 80, 275–279. doi: 10.1101/sqb.2015.80.027524
Harrison, D. E., Strong, R., Sharp, Z. D., Nelson, J. F., Astle, C. M., Flurkey, K., et al. (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395. doi: 10.1038/nature08221
Hartl, F. U., Bracher, A., and Hayer-Hartl, M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. doi: 10.1038/nature10317
Hickey, A. J. R., Jüllig, M., Aitken, J., Loomes, K., Hauber, M. E., and Phillips, A. R. J. (2012). Birds and longevity: does flight driven aerobicity provide an oxidative sink? Ageing Res. Rev. 11, 242–253. doi: 10.1016/j.arr.2011.12.002
Houtkooper, R. H., Argmann, C., Houten, S. M., Cantìo, C., Jeninga, E. H., Andreux, ṔA., et al. (2011). The metabolic footprint of aging in mice. Sci. Rep. 1:134. doi: 10.1038/srep00134
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. doi: 10.1038/nature12188
Houtkooper, R. H., Pirinen, E., and Auwerx, J. (2012). Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. doi: 10.1038/nrm3293
Houtkooper, R. H., Williams, R. W., and Auwerx, J. (2010). Metabolic networks of longevity. Cell 142, 9–14. doi: 10.1016/j.cell.2010.06.029
Hudgins, A. D., Tazearslan, C., Tare, A., Zhu, Y., Huffman, D., and Suh, Y. (2018). Age- and tissue-specific expression of senescence biomarkers in mice. Front. Genet. 9:59. doi: 10.3389/fgene.2018.00059
Jang, Y.-Y., and Sharkis, S. J. (2007). A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 110, 3056–3063. doi: 10.1182/blood-2007-05-087759
Jensen, M. B., and Jasper, H. (2014). Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 20, 214–225. doi: 10.1016/j.cmet.2014.05.006
Jovaisaite, V., Mouchiroud, L., and Auwerx, J. (2014). The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J. Exp. Biol. 217, 137–143. doi: 10.1242/jeb.090738
Kawanishi, S., and Oikawa, S. (2004). Mechanism of telomere shortening by oxidative stress. Ann. N. Y. Acad. Sci. 1019, 278–284. doi: 10.1196/annals.1297.047
Kietzmann, T., Petry, A., Shvetsova, A., Gerhold, J. M., and Görlach, A. (2017). The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 174, 1533–1554. doi: 10.1111/bph.13792
Koshiba, T., Yasukawa, K., Yanagi, Y., and Kawabata, S. I. (2011). Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal. 4:ra7. doi: 10.1126/scisignal.2001147
Labbadia, J., Brielmann, R. M., Neto, M. F., Lin, Y. F., Haynes, C. M., and Morimoto, R. I. (2017). Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep. 21, 1481–1494. doi: 10.1016/j.celrep.2017.10.038
Laker, R. C., Drake, J. C., Wilson, R. J., Lira, V. A., Lewellen, B. M., Ryall, K. A., et al. (2017). Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 8, 1–13. doi: 10.1038/s41467-017-00520-9
Lazear, H. M., Lancaster, A., Wilkins, C., Suthar, M. S., Huang, A., Vick, S. C., et al. (2013). IRF-3, IRF-5, and IRF-7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog. 9:e1003118. doi: 10.1371/journal.ppat.1003118
Lee, J. S., Yoon, Y. G., Yoo, S. H., Jeong, N. Y., Jeong, S. H., Lee, S. Y., et al. (2012). Histone deacetylase inhibitors induce mitochondrial elongation. J. Cell. Physiol. 227, 2856–2869. doi: 10.1002/jcp.23027
Lee, S., Jeong, S.-Y., Lim, W.-C., Kim, S., Park, Y.-Y., Sun, X., et al. (2007). Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 282, 22977–22983. doi: 10.1074/jbc.M700679200
Leonardi, G. C., Accardi, G., Monastero, R., Nicoletti, F., and Libra, M. (2018). Ageing: from inflammation to cancer. Immun. Ageing 15:1. doi: 10.1186/s12979-017-0112-5
Lesnefsky, E. J., and Hoppel, C. L. (2006). Oxidative phosphorylation and aging. Ageing Res. Rev. 5, 402–433. doi: 10.1016/j.arr.2006.04.001
Lidzbarsky, G., Gutman, D., Shekhidem, H. A., Sharvit, L., and Atzmon, G. (2018). Genomic instabilities, cellular senescence, and aging: In vitro, in vivo and aging-like human syndromes. Front. Med. 5:104. doi: 10.3389/fmed.2018.00104
Liu, L., Trimarchi, J. R., Smith, P. J. S., and Keefe, D. L. (2002). Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 1, 40–46. doi: 10.1046/j.1474-9728.2002.00004.x
Liu, Y. J., McIntyre, R. L., Janssens, G. E., and Houtkooper, R. H. (2020a). Mitochondrial fission and fusion: a dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 186:111212. doi: 10.1016/j.mad.2020.111212
Liu, Y. J., McIntyre, R. L., Janssens, G. E., Williams, E. G., Lan, J., van Weeghel, M., et al. (2020b). Mitochondrial translation and dynamics synergistically extend lifespan in C. elegans through HLH-30. J. Cell Biol. 219:e201907067. doi: 10.1083/jcb.201907067
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The hallmarks of aging. Cell 153, 1194–1217. doi: 10.1016/j.cell.2013.05.039
McGuire, P. J. (2019). Mitochondrial dysfunction and the aging immune system. Biology 8:26. doi: 10.3390/biology8020026
McHugh, D., and Gil, J. (2018). Senescence and aging: causes, consequences, and therapeutic avenues. J. Cell Biol. 217, 65–77. doi: 10.1083/jcb.201708092
McIntyre, R. L., Daniels, E. G., Molenaars, M., Houtkooper, R. H., and Janssens, G. E. (2019). From molecular promise to preclinical results: HDAC inhibitors in the race for healthy aging drugs. EMBO Mol. Med. 11:e9854. doi: 10.15252/emmm.201809854
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223. doi: 10.1016/j.cell.2016.04.012
Mitchell, S. J., Scheibye-Knudsen, M., Longo, D. L., and de Cabo, R. (2015). Animal models of aging research: implications for human aging and age-related diseases. Annu. Rev. Anim. Biosci. 3, 283–303. doi: 10.1146/annurev-animal-022114-110829
Moehle, E. A., Shen, K., and Dillin, A. (2019). Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 294, 5396–5407. doi: 10.1074/jbc.TM117.000893
Moiseeva, O., Bourdeau, V., Roux, A., Deschênes-Simard, X., and Ferbeyre, G. (2009). Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 29, 4495–4507. doi: 10.1128/MCB.01868-08
Molenaars, M., Daniels, E. G., Meurs, A., Janssens, G. E., and Houtkooper, R. H. (2020a). Mitochondrial cross-compartmental signalling to maintain proteostasis and longevity. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 375:20190414. doi: 10.1098/rstb.2019.0414
Molenaars, M., Janssens, G. E., Santermans, T., Lezzerini, M., Jelier, R., MacInnes, A. W., et al. (2018). Mitochondrial ubiquinone–mediated longevity is marked by reduced cytoplasmic mRNA translation. Life Sci. Alliance 1:e201800082. doi: 10.26508/lsa.201800082
Molenaars, M., Janssens, G. E., Williams, E. G., Jongejan, A., Lan, J., Rabot, S., et al. (2020b). A conserved mito-cytosolic translational balance links two longevity pathways. Cell Metab. 31, 549–563.e7. doi: 10.1016/j.cmet.2020.01.011
Molony, R. D., Nguyen, J. T., Kong, Y., Montgomery, R. R., Shaw, A. C., and Iwasaki, A. (2017). Aging impairs both primary and secondary RIG-I signaling for interferon induction in human monocytes. Sci. Signal. 10:eaan2392. doi: 10.1126/scisignal.aan2392
Morita, M., Gravel, S. P., Chénard, V., Sikström, K., Zheng, L., Alain, T., et al. (2013). MTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 18, 698–711. doi: 10.1016/j.cmet.2013.10.001
Morita, M., Prudent, J., Basu, K., Goyon, V., Katsumura, S., Hulea, L., et al. (2017). mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell 67, 922–935.e5. doi: 10.1016/j.molcel.2017.08.013
Mota-Martorell, N., Jove, M., Pradas, I., Sanchez, I., Gómez, J., Naudi, A., et al. (2020). Low abundance of NDUFV2 and NDUFS4 subunits of the hydrophilic complex I domain and VDAC1 predicts mammalian longevity. Redox Biol. 34:101539. doi: 10.1016/j.redox.2020.101539
Mouchiroud, L., Houtkooper, R. H., Moullan, N., Katsyuba, E., Ryu, D., Cantó, C., et al. (2013). The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. doi: 10.1016/j.cell.2013.06.016
Munro, D., Baldy, C., Pamenter, M. E., and Treberg, J. R. (2019). The exceptional longevity of the naked mole-rat may be explained by mitochondrial antioxidant defenses. Aging Cell 18, 1–13. doi: 10.1111/acel.12916
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P., and Haynes, C. M. (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPRmt. Mol. Cell 58, 123–133. doi: 10.1016/j.molcel.2015.02.008
Niedernhofer, L. J., Gurkar, A. U., Wang, Y., Vijg, J., Hoeijmakers, J. H. J., and Robbins, P. D. (2018). Nuclear genomic instability and aging. Annu. Rev. Biochem. 87, 295–322. doi: 10.1146/annurev-biochem-062917-012239
Oeppen, J., and Vaupel, J. W. (2002). Demography: broken limits to life expectancy. Science 296, 1029–1031. doi: 10.1126/science.1069675
Ohgami, R. S., Campagna, D. R., McDonald, A., and Fleming, M. D. (2006). The Steap proteins are metalloreductases. Blood 108, 1388–1394. doi: 10.1182/blood-2006-02-003681
Pal, S., and Tyler, J. K. (2016). Epigenetics and aging. Sci. Adv. 2:e1600584. doi: 10.1126/sciadv.1600584
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. doi: 10.1038/nature14300
Pan, K. Z., Palter, J. E., Rogers, A. N., Olsen, A., Chen, D., Lithgow, G. J., et al. (2007). Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 6, 111–119. doi: 10.1111/j.1474-9726.2006.00266.x
Pearson, K. J., Baur, J. A., Lewis, K. N., Peshkin, L., Price, N. L., Labinskyy, N., et al. (2008). Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 8, 157–168. doi: 10.1016/j.cmet.2008.06.011
Pence, B. D., and Yarbro, J. R. (2018). Aging impairs mitochondrial respiratory capacity in classical monocytes. Exp. Gerontol. 108, 112–117. doi: 10.1016/J.EXGER.2018.04.008
Pieren, D. K. J., Smits, N. A. M., van de Garde, M. D. B., and Guichelaar, T. (2019). Response kinetics reveal novel features of ageing in murine T cells. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-42120-1
Pinti, M., Cevenini, E., Nasi, M., De Biasi, S., Salvioli, S., Monti, D., et al. (2014). Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging.”. Eur. J. Immunol. 44, 1552–1562. doi: 10.1002/eji.201343921
Pirinen, E., Cantó, C., Jo, Y. S., Morato, L., Zhang, H., Menzies, K. J., et al. (2014). Pharmacological inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 19, 1034–1041. doi: 10.1016/j.cmet.2014.04.002
Rath, C. P. (2020). Models, Molecules and Mechanisms in Biogerontology. Cham: Springer. doi: 10.1007/978-981-32-9005-1
Ruan, L., Zhou, C., Jin, E., Kucharavy, A., Zhang, Y., Wen, Z., et al. (2017). Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543, 443–446. doi: 10.1038/nature21695
Ryu, D., Mouchiroud, L., Andreux, P. A., Katsyuba, E., Moullan, N., Nicolet-Dit-Félix, A. A., et al. (2016). Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 22, 879–888. doi: 10.1038/nm.4132
Sahin, E., Colla, S., Liesa, M., Moslehi, J., Müller, F. L., Guo, M., et al. (2011). Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 470, 359–365. doi: 10.1038/nature09787
Sahm, A., Bens, M., Platzer, M., and Cellerino, A. (2017). Parallel evolution of genes controlling mitonuclear balance in short-lived annual fishes. Aging Cell 16, 488–496. doi: 10.1111/acel.12577
Schaible, R., Scheuerlein, A., Dańko, M. J., Gampe, J., Martínez, D. E., and Vaupel, J. W. (2015). Constant mortality and fertility over age in Hydra. Proc. Natl. Acad. Sci. U.S.A. 112, 15701–15706. doi: 10.1073/pnas.1521002112
Schulz, T. J., Zarse, K., Voigt, A., Urban, N., Birringer, M., and Ristow, M. (2007). Glucose restriction extends caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 6, 280–293. doi: 10.1016/J.CMET.2007.08.011
Sharma, A., Smith, H. J., Yao, P., and Mair, W. B. (2019). Causal roles of mitochondrial dynamics in longevity and healthy aging. EMBO Rep. 20:e48395. doi: 10.15252/embr.201948395
Stöckl, P., Hütter, E., Zwerschke, W., and Jansen-Dürr, P. (2006). Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp. Gerontol. 41, 674–682. doi: 10.1016/j.exger.2006.04.009
Stoll, E. A., Karapavlovic, N., Rosa, H., Woodmass, M., Rygiel, K., White, K., et al. (2016). Naked mole-rats maintain healthy skeletal muscle and Complex IV mitochondrial enzyme function into old age. Aging 8, 3468–3485. doi: 10.18632/aging.101140
Suhm, T., Kaimal, J. M., Dawitz, H., Peselj, C., Masser, A. E., Hanzén, S., et al. (2018). Mitochondrial translation efficiency controls cytoplasmic protein homeostasis. Cell Metab. 27, 1309–1322.e6. doi: 10.1016/j.cmet.2018.04.011
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028
Tacutu, R., Thornton, D., Johnson, E., Budovsky, A., Barardo, D., Craig, T., et al. (2018). Human ageing genomic resources: new and updated databases. Nucleic Acids Res. 46, D1083–D1090. doi: 10.1093/nar/gkx1042
Tarasenko, T. N., Pacheco, S. E., Koenig, M. K., Gomez-Rodriguez, J., Kapnick, S. M., Diaz, F., et al. (2017). Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab. 25, 1254–1268.e7. doi: 10.1016/J.CMET.2017.05.007
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165, 1197–1208. doi: 10.1016/j.cell.2016.04.011
Tomczyk, S., Fischer, K., Austad, S., and Galliot, B. (2015). Hydra, a powerful model for aging studies. Invertebr. Reprod. Dev. 59, 11–16. doi: 10.1080/07924259.2014.927805
Toyama, E. Q., Herzig, S., Courchet, J., Lewis, T. L., Losón, O. C., Hellberg, K., et al. (2016). AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. doi: 10.1126/science.aab4138
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423. doi: 10.1038/nature02517
Tufi, R., Gandhi, S., De Castro, I. P., Lehmann, S., Angelova, P. R., Dinsdale, D., et al. (2014). Enhancing nucleotide metabolism protects against mitochondrial dysfunction and neurodegeneration in a PINK1 model of Parkinson’s disease. Nat. Cell Biol. 16, 157–166. doi: 10.1038/ncb2901
Turk, P. W., Laayoun, A., Smith, S. S., and Weitzman, S. A. (1995). DNA adduct 8-hydroxyl-2’-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 16, 1253–1255. doi: 10.1093/carcin/16.5.1253
Vannini, N., Girotra, M., Naveiras, O., Nikitin, G., Campos, V., Giger, S., et al. (2016). Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 7:13125. doi: 10.1038/ncomms13125
Wang, J., Xiong, S., Xie, C., Markesbery, W. R., and Lovell, M. A. (2005). Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J. Neurochem. 93, 953–962. doi: 10.1111/j.1471-4159.2005.03053.x
Weidberg, H., and Amon, A. (2018). MitoCPR–A surveillance pathway that protects mitochondria in response to protein import stress. Science 360:eaan4146. doi: 10.1126/science.aan4146
Weir, H. J., Yao, P., Huynh, F. K., Escoubas, C. C., Goncalves, R. L., Burkewitz, K., et al. (2017). Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. 26, 884–896.e5. doi: 10.1016/j.cmet.2017.09.024
Wiley, C. D., Velarde, M. C., Lecot, P., Liu, S., Sarnoski, E. A., Freund, A., et al. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 23, 303–314. doi: 10.1016/j.cmet.2015.11.011
Wrobel, L., Topf, U., Bragoszewski, P., Wiese, S., Sztolsztener, M. E., Oeljeklaus, S., et al. (2015). Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524, 485–488. doi: 10.1038/nature14951
Wu, J. J., Liu, J., Chen, E. B., Wang, J. J., Cao, L., Narayan, N., et al. (2013). Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of mTOR expression. Cell Rep. 4, 913–920. doi: 10.1016/j.celrep.2013.07.030
Xiong, S., Patrushev, N., Forouzandeh, F., Hilenski, L., and Alexander, R. W. (2015). PGC-1α modulates telomere function and DNA damage in protecting against aging-related chronic diseases. Cell Rep. 12, 1391–1399. doi: 10.1016/j.celrep.2015.07.047
Yoon, Y. S., Byun, H. O., Cho, H., Kim, B. K., and Yoon, G. (2003). Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induced Senescence-associated Growth Arrest. J. Biol. Chem. 278, 51577–51586. doi: 10.1074/jbc.M308489200
Keywords: aging, mitochondria, interplay, hallmarks of aging, tree of life
Citation: van der Rijt S, Molenaars M, McIntyre RL, Janssens GE and Houtkooper RH (2020) Integrating the Hallmarks of Aging Throughout the Tree of Life: A Focus on Mitochondrial Dysfunction. Front. Cell Dev. Biol. 8:594416. doi: 10.3389/fcell.2020.594416
Received: 13 August 2020; Accepted: 04 November 2020;
Published: 26 November 2020.
Edited by:
Owen Jones, University of Southern Denmark, DenmarkReviewed by:
Morten Scheibye-Knudsen, University of Copenhagen, DenmarkCopyright © 2020 van der Rijt, Molenaars, McIntyre, Janssens and Houtkooper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Riekelt H. Houtkooper, ci5oLmhvdXRrb29wZXJAYW1zdGVyZGFtdW1jLm5s
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.