 Yangchun Xie1*
Yangchun Xie1* Jiao Liu
Jiao Liu Daolin Tang
Daolin Tang- 1Department of Oncology, The Second Xiangya Hospital, Central South University, Changsha, China
- 2The Third Affiliated Hospital, Guangzhou Medical University, Guangzhou, China
- 3Department of Surgery, University of Texas Southwestern Medical Center, Dallas, TX, United States
Mitochondria are multifunctional organelles that regulate cancer biology by synthesizing macromolecules, producing energy, and regulating cell death. The understanding of mitochondrial morphology, function, biogenesis, fission and fusion kinetics, and degradation is important for the development of new anticancer strategies. Mitophagy is a type of selective autophagy that can degrade damaged mitochondria under various environmental stresses, especially oxidative damage and hypoxia. The key regulator of mitophagy is the autophagy receptor, which recognizes damaged mitochondria and allows them to enter autophagosomes by binding to MAP1LC3 or GABARAP, and then undergo lysosomal-dependent degradation. Many components of mitochondria, including mitochondrial membrane proteins (e.g., PINK1, BNIP3L, BNIP3, FUNDC1, NIPSNAP1, NIPSNAP2, BCL2L13, PHB2, and FKBP8) and lipids (e.g., cardiolipin and ceramides), act as mitophagy receptors in a context-dependent manner. Dysfunctional mitophagy not only inhibits, but also promotes, tumorigenesis. Similarly, mitophagy plays a dual role in chemotherapy, radiotherapy, and immunotherapy. In this review, we summarize the latest advances in the mechanisms of mitophagy and highlight the pathological role of mitophagy receptors in tumorigenesis and treatment.
Introduction
Autophagy, which was first observed under an electron microscope by Belgian scientist Christian de Duve in the 1950s, is a cellular phenomenon of “self-eating” by lysosomes (Yang and Klionsky, 2010). At present, based on the transport mode of cytosolic cargoes to lysosomes, autophagy is mainly divided into three types: macroautophagy (hereafter referred to as autophagy), microautophagy, and chaperone-mediated autophagy (Dikic and Elazar, 2018). As an important degradation mechanism, the process of autophagy involves the formation of lipid-related autophagosomes by wrapping various cargoes (e.g., damaged organelles, unused proteins, and invading pathogens), and then fusing them with lysosomes to form autophagosomes and degrading their contents (Klionsky and Emr, 2000; Xie et al., 2020b). At the molecular level, autophagy-related (ATG) genes and proteins play a vital role in regulating the dynamic formation of autophagic membrane structures, mainly through protein-protein interactions (Kang et al., 2011; Dikic and Elazar, 2018; Figure 1). These ATG protein interactions are further modulated by various factors, especially kinase-mediated protein posttranslational modification (McEwan and Dikic, 2011; Xie et al., 2015). Generally, the activation of autophagy is an important defense mechanism that promotes cell survival and recovery under harmful stresses, such as starvation and hypoxia (Kroemer et al., 2010). The autophagic degradation products can be reused for protein synthesis and energy production, although the underlying mechanism of this process is unclear. In contrast, an excessive activation of autophagy may lead to cell death, which is called autophagy-dependent cell death (Bialik et al., 2018; Galluzzi et al., 2018; Tang et al., 2019). In particular, recent studies indicate that ferroptosis is a type of autophagy-dependent cell death (Hou et al., 2016; Bai et al., 2019; Li et al., 2020; Xie et al., 2020a), highlighting the importance of autophagy in the degradation of proteins involved in iron and lipid metabolism (Zhou et al., 2020; Liu et al., 2020). It is also worth noting that the term “autophagic cell death” is used to describe the phenotype of increased autophagy during the induction of cell death, regardless of the effect of autophagic response on cell fate (Kroemer and Levine, 2008). Therefore, autophagy plays a dual role in cell survival and cell death, which is related to human disease, especially cancer and neurodegenerative diseases (Levine and Kroemer, 2019).
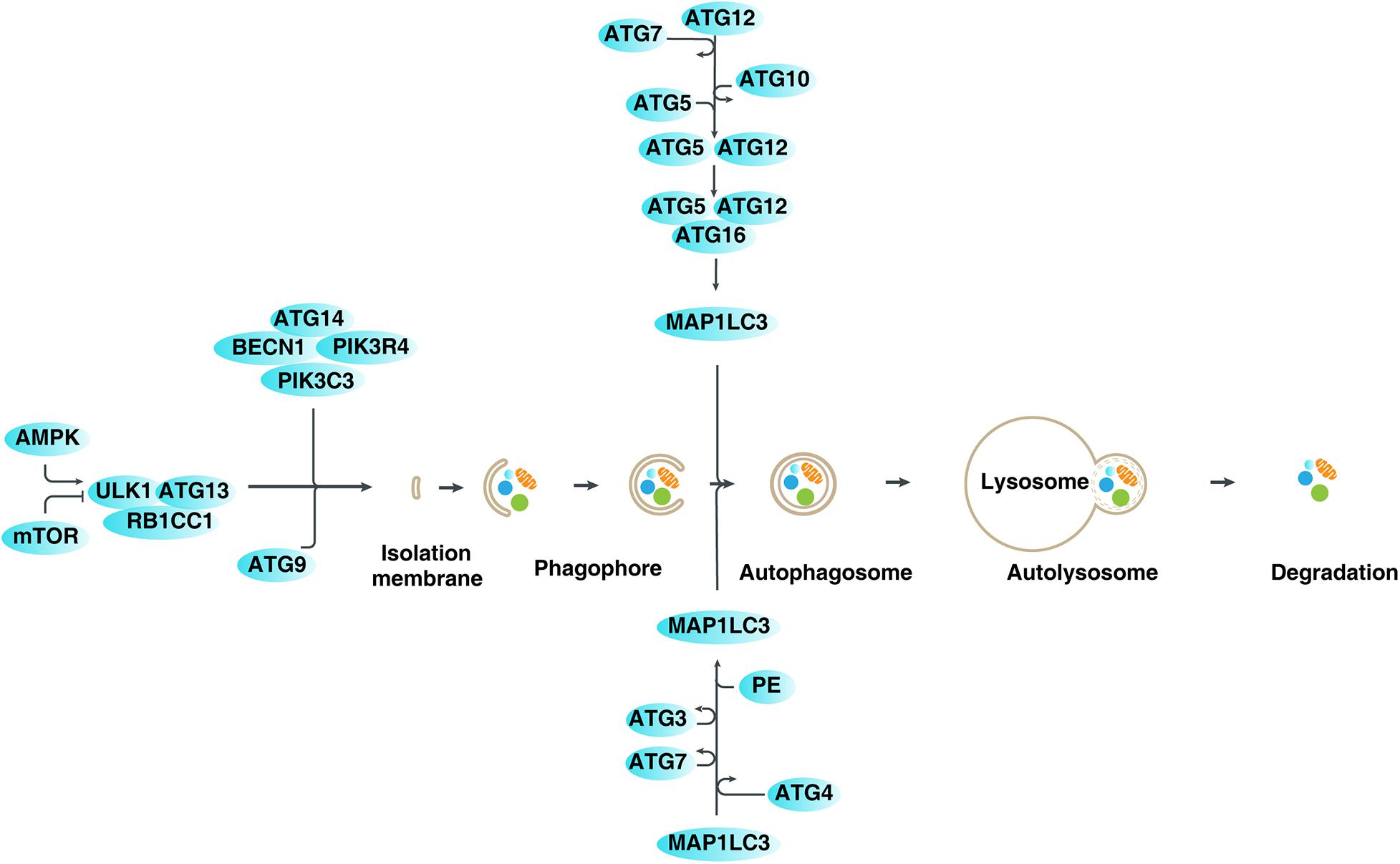
Figure 1. Mechanisms and regulation of autophagy in mammalian cells. Autophagy is a dynamic process involving the formation of several specific membrane structures, such as phagophores, autophagosomes, and autolysosomes. ATG proteins, in association with various regulators, are involved in regulating the dynamic process of membrane structure formation, leading to the degradation of various cargoes in lysosomes.
Depending on whether specific autophagic receptors (also known as autophagic adaptor proteins) are needed to degrade specific substrates, autophagy can be non-selective or selective (Zaffagnini and Martens, 2016). In recent years, a large number of types of selective autophagy have been found to regulate cell homeostasis, such as mitophagy (Harper et al., 2018), pexophagy (Cho et al., 2018), lipophagy (Kounakis et al., 2019), ferritinophagy (Mancias et al., 2014), and clockophagy (Yang et al., 2019). Among them, mitophagy is the most-studied selective autophagy, which eliminates damaged or aging mitochondria by recognizing different components of mitochondrial structure via various autophagy receptors (Lemasters, 2005). Dysregulated mitophagy is closely related to many physiological and pathological processes, such as aging, neurodegenerative diseases, and cancer (Palikaras et al., 2018). In this review, we first introduce the structure and function of mitochondria, and then focus on the molecular mechanisms of mitophagy. Finally, we describe the pathologic role of mitophagy regulators in tumor development and therapy, and will discuss new directions for cancer treatment.
Structure and Function of Mitochondria
Mitochondria are double-membrane organelles present in most eukaryotic cells, and their size, number, and appearance are different on different cells (Herst et al., 2017; Pfanner et al., 2019). Like chloroplasts in plants and algae, mitochondria may have evolved from primitive bacteria (Gray, 2012). The main chemical components of mitochondria include water, protein, and lipids. In addition, mitochondria have a small amount of small molecules, such as coenzymes and nucleic acids. Proteins, including soluble and insoluble proteins, account for 65 to 70% of the dry weight of mitochondria. Soluble proteins are mainly the enzymes located in the mitochondrial matrix and the periphery of the membrane, whereas insoluble proteins constitute the main body of the membrane, part of which is composed of mosaic proteins or enzymes (Pfanner et al., 2019). Lipids in mitochondria are mainly distributed in two layers of membranes, accounting for 20 to 30% of the dry weight. Phospholipids in mitochondria account for more than 75% of total lipids. The amount of phospholipids in the mitochondrial membrane of different tissues of the same organism is relatively stable. Abundant cardiolipin and less cholesterol are the obvious differences between the structure of mitochondria and other cell membranes (Montero et al., 2010).
From the outside to the inside, the mitochondria can be divided into four functional areas: the outer mitochondrial membrane (OMM), the intermembrane space (the space between the outer and inner membranes), the inner mitochondrial membrane (IMM), and the matrix (space within the inner membrane) (Pfanner et al., 2019). The OMM is smoother and acts as the boundary membrane of organelles, while the IMM folds inward to form mitochondrial cristae (e.g., lamellar cristae, tubular cristae, and vesicular cristae), which complicate biochemical reactions. Mitochondria are the main sites for oxidative phosphorylation and synthesis of adenosine triphosphate (ATP) in cells, and provide chemical energy for cellular activities as the “powerhouse of the cell.” In addition to supplying energy for cells, mitochondria are also involved in various processes, such as cell differentiation, signal transduction, cell growth, the cell cycle, and cell death (Herst et al., 2017; Bock and Tait, 2020). Dysfunctional mitochondria are unable to execute oxidative phosphorylation and consequently accumulate reactive oxygen species (ROS) in the cells. Mitochondrial oxidative stress is associated with a variety of pathologies, especially age-related diseases (e.g., cancer). In order to avoid mitochondrial dysfunction, some conservative mechanisms have evolved to control the quality of mitochondria. Among them, mitophagy plays a central role in preventing mitochondrial damage by promoting mitochondrial turnover. Understanding the signal transduction and molecular modification of mitophagy is important for improving the homeostasis of mitochondria (Palikaras et al., 2018).
Molecular Mechanisms of Mitophagy
Mitochondrial depolarization refers to the process in which the membrane potential of the mitochondria changes from negative to positive in the direction of depolarization (Zorova et al., 2018). During the electron transport process, as electrons flow down the chain of the redox complex located in the IMM, protons flow into the space between the IMM and the OMM. Therefore, the intermembrane space becomes positive, and the IMM becomes electrochemically polarized. The backflow of protons is related to the production of ATP. In this state, the mitochondria are polarized. When proton flow is independent of ATP production, mitochondria are considered to be depolarized (Zorova et al., 2018). Fission-induced mitochondrial depolarization is an important factor that triggers mitophagy to reduce oxidative stress (Twig and Shirihai, 2011). The molecular mechanisms involved in mitophagy are complex, and recognition of depolarized mitochondria requires a variety of cargo receptors and regulators. In general, mitophagy can be mediated through ubiquitin (Ub)-dependent and Ub-independent receptor pathways, as described below (Harper et al., 2018; Figure 2).
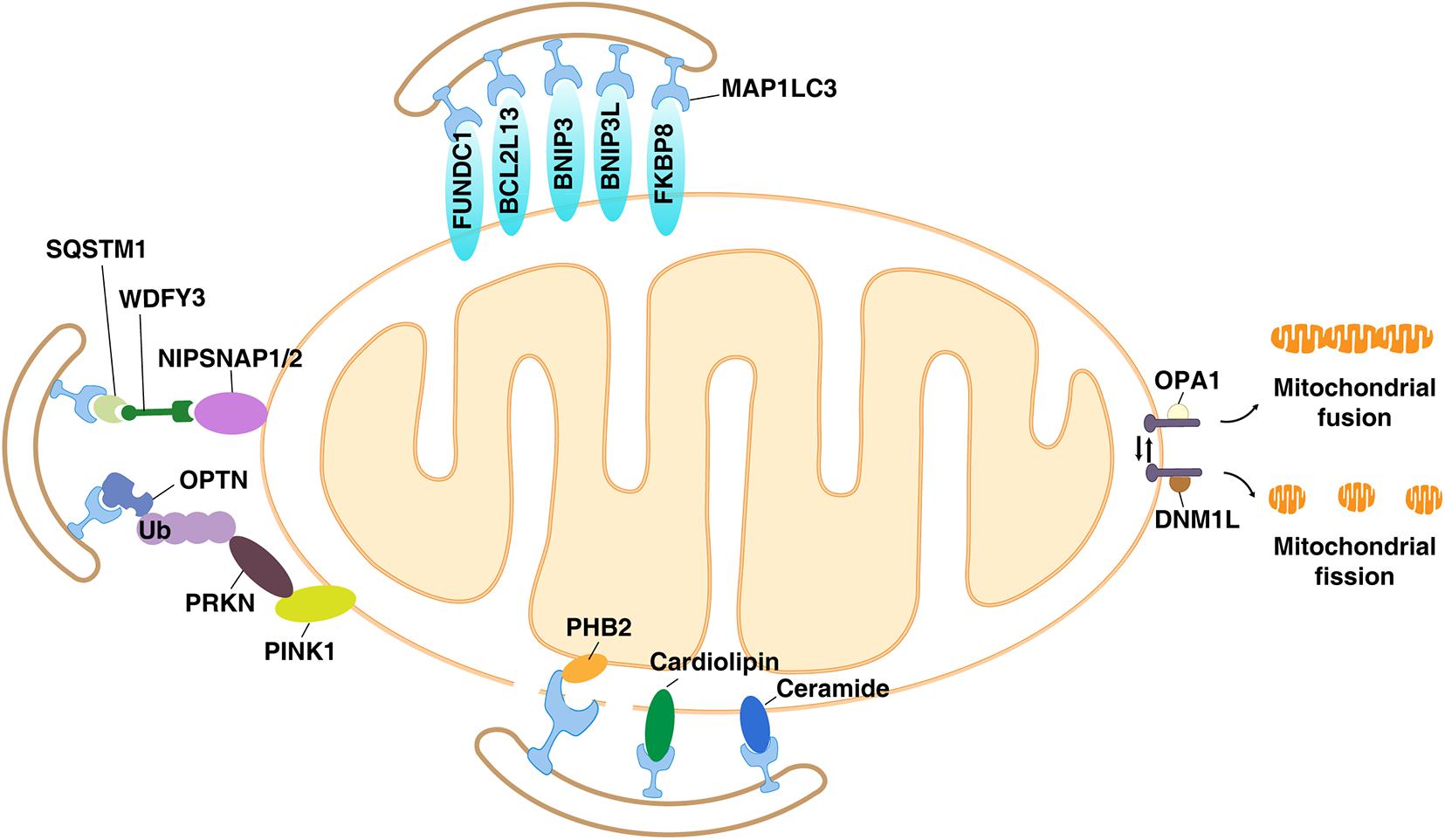
Figure 2. The Ub-dependent and Ub-independent receptor pathways of mitophagy. Many components of the mitochondria, including mitochondrial membrane proteins (e.g., PINK1, BNIP3L, BNIP3, FUNDC1, NIPSNAP1, NIPSNAP2, BCL2L13, PHB2, and FKBP8) and lipids (e.g., cardiolipin and ceramides), act as mitophagy receptors in a context-dependent manner.
Ub-Dependent Receptors
PTEN-induced kinase 1 (PINK1) is a serine/threonine protein kinase that localizes to mitochondria (Valente et al., 2004). Parkin RBR E3 ubiquitin protein ligase (PRKN/PARKIN/PARK2) is a component of the multiprotein E3 ubiquitin ligase complex, which can catalyze the covalent attachment of the ubiquitin part to the substrate protein (Shimura et al., 2000). Mutations in PINK1 and PRKN are implicated in Parkinson’s disease, an aging-related disease associated with mitochondrial abnormalities and motor nerve damage (Pickrell and Youle, 2015). Importantly, the activation of the PINK1-PRKN pathway is the first and most studied regulatory mechanism of mitophagy. When mitochondria are damaged or depolarized, PINK1 stabilizes on the OMM, where it recruits and activates PRKN (Vives-Bauza et al., 2010). After being transported from the cytoplasm to the mitochondria, PRKN ubiquitinates many OMM proteins [e.g., translocase of outer mitochondrial membrane 20 (TOMM20/TOM20), mitofusin 1 (MFN1), and mitofusin 2 (MFN2)], resulting in the recruitment of autophagy receptors such as sequestosome 1 (SQSTM1/p62) and optineurin (OPTN) (Geisler et al., 2010; Wong and Holzbaur, 2014). The ubiquitinated proteins with autophagy receptors are then bound to the autophagosome-associated proteins [microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3/Atg8) or GABA type A receptor-associated protein (GABARAP)] via their LC3 interacting domain (LIR) to direct the isolation membrane/phagophore of growing autophagosomes to surround damaged mitochondria (Wild et al., 2014). Finally, engulfed mitochondria are degraded and eliminated in autolysosomes (Geisler et al., 2010; Wong and Holzbaur, 2014). The activity and function of PINK1 or PRKN in mitophagy is further regulated by various binding partners and changes in mitochondrial dynamics (e.g., mitochondrial fission). Of note, some mammalian cells (Eiyama and Okamoto, 2015) do not express PINK1 or PRKN, indicating that PINK1- and PRKN-mediated mitophagy may have tissue and cell-specific effects in preventing mitochondrial dysfunction.
Ub-Independent Receptors
BCL2 Interacting Protein 3-Like
The BCL2 apoptosis regulator (BCL2) family includes pro-apoptotic proteins [e.g., BCL2-associated X, apoptosis regulator (BAX), BCL2 antagonist/killer 1 (BAK1/BAK), and BH3 interacting domain death agonist (BID)] and anti-apoptotic proteins [e.g., BCL2 and BCL2-like 1 (BCL2L1/BCLXL)]. Members of the anti-apoptotic BCL2 protein family are overexpressed in many malignant tumors and become targets for tumor treatment (Adams and Cory, 2007). In addition to regulating mitochondrial apoptosis by controlling mitochondrial outer membrane permeabilization, the OMM protein BCL2 interacting protein 3-like (BNIP3L/NIX, a pro-apoptotic BCL2 family member) is involved in mediating mitophagy (Schweers et al., 2007; Sandoval et al., 2008). Unlike binding BCL2 during apoptosis (Zhang and Ney, 2009), BNIP3L directly binds to MAP1LC3 or GABARAP during mitophagy (Schwarten et al., 2009). Moreover, BNIP3L-mediated mitophagy may not be associated with the ubiquitination of BNIP3L, indicating that it is an Ub-independent receptor for mitophagy (Ney, 2015).
Functionally, the activation of BNIP3L-dependent mitophagy is essential for the programmed mitochondrial elimination during erythroid maturation, and BNIP3L-depleted mice show anemia (Schweers et al., 2007; Sandoval et al., 2008). Furthermore, BNIP3L instead of PRKN is responsible for mitophagy induction in HeLa cells (a cell line derived from patients with cervical cancer) (Ding et al., 2010). In addition, transcriptional factor hypoxia-inducible factor 1 subunit alpha (HIF1A/HIF1α)-mediated BNIP3L upregulation is required for hypoxia-induced mitophagy (Sowter et al., 2001), indicating a potential role of BNIP3L-mediated mitophagy in hypoxic tumor microenvironments (TMEs). Structurally, BNIP3L positioning on OMM requires the transmembrane domain, and BNIP3L dimerization is responsible for MAP1LC3 recruitment (Marinkovic et al., 2020). BNIP3L further binds to MAP1LC3 at the amino terminus of BNIP3L through the LIR motif (Schwarten et al., 2009). These structural studies provide information for the development of BNIP3L-targeted drugs.
BCL2 Interacting Protein 3
BCL2 interacting protein 3 (BNIP3) is a BH3-only protein and acts as a pro-apoptotic BCL2 family member (Vande Velde et al., 2000). It interacts with the anti-apoptotic BCL2, thereby overcoming the inhibitory effect of BCL2 on apoptosis (Zhang and Ney, 2009). BNIP3 at the OMM regulates the opening of the pores in the mitochondrial double membrane to mediate the transport of lysosomal proteins from the cytoplasm to the mitochondrial matrix, thereby leading to the degradation of damaged proteins in the mitochondria in response to oxidative damage (Zhang and Ney, 2009). BNIP3 also has a LIR domain through which it interacts with MAP1LC3 and mediates mitochondrial degradation through mitophagy (Novak et al., 2010; Park et al., 2013). Like its homolog BNIP3L, BNIP3 forms a dimer during mitophagy and its expression is regulated by HIF1A during hypoxia (Sowter et al., 2001; O’Sullivan et al., 2015). BNIP3 is also highly expressed in the hypoxic environment of solid tumors. Although both BNIP3 and BNIP3L mediate hypoxia-induced mitophagy (Sowter et al., 2001), the functional complementarity and differences of these two proteins in cancer-related mitophagy remain largely unclear. In addition, BNIP3 may affect the fission or fusion of mitochondria by binding to OPA1 mitochondrial dynamin-like GTPase (OPA1) or dynamin 1-like (DNM1L/DRP1), thereby promoting mitophagy (Lee et al., 2011). These findings highlight the role of mitochondrial dynamics in regulating mitophagy.
FUN14 Domain Containing 1
In addition to BNIP3L and BNIP3, FUN14 domain containing 1 (FUNDC1) was also found to be expressed in OMM as an autophagy receptor for mitophagy during hypoxia (Liu et al., 2012). The activity of FUNDC1 in hypoxia-induced mitophagy is regulated by phosphorylation and dephosphorylation events. Unc-51–like autophagy-activating kinase 1 (ULK1/ATG1) is the only kinase of the ATG family and a component of the ULK1-ATG13 RB1-inducible coiled-coil 1 (RB1CC1) complex, which initiates the formation of autophagosomes in mammalian cells (Hosokawa et al., 2009; Jung et al., 2009). ULK1 has many phosphorylation sites, and these phosphorylation sites have different functions in regulating autophagy (Xie et al., 2015). Phosphorylation of ULK1 at serine17 mediates ULK1 translocation to mitochondria and subsequently binds to FUNDC1 during hypoxia (Wu et al., 2014). In contrast, the dephosphorylation of FUNDC1 by PGAM family member 5, mitochondrial serine/threonine protein phosphatase (PGAM5) under hypoxia increases its binding to MAP1LC3 through LIR, and recruits the isolation membrane that binds to MAP1LC3, further forming autophagosomes to engulf damaged mitochondria in mammalian cells (Liu et al., 2012; Chen et al., 2014). Certain proteins [e.g., mitochondrial E3 ligase membrane-associated ring CH-type finger 5 (MARCHF5/MARCH5) and cytosolic molecular chaperone heat shock protein family A (hsp70) member 8 (HSPA8/HSC70)] bind to FUNDC1, which further regulate the protein stability of FUNDC1 to fine-tune mitophagy during hypoxia (Chen et al., 2017; Li et al., 2019b). Interestingly, the PRKN-mediated ubiquitination may promote the transport of MARCHF5 from mitochondria to peroxisomes, resulting in a decrease in mitophagy (Koyano et al., 2019). FUNDC1 also acts as a platform for regulating mitochondrial dynamics (e.g., fission and fusion) and mitophagy by interacting with DNM1L and OPA1 (Chen et al., 2016). In particular, the dissociation of FUNDC1 from DNML1 to form a complex with OPA1 inhibits mitochondrial fission and mitophagy (Chen et al., 2016). These findings further support the idea that mitochondrial dynamics and quality control are inseparably intertwined.
The 4-Nitrophenylphosphatase Domain and Non-neuronal SNAP25-Like Protein Homolog 1
Both 4-nitrophenylphosphatase domain and non-neuronal SNAP25-like protein homolog 1 (NIPSNAP1) and NIPSNAP2 play a major role in vesicular transport (Seroussi et al., 1998). Under normal conditions, they are located in the IMM and act as modulators of calcium channels (Brittain et al., 2012). However, they also localize to the OMM during mitochondrial depolarization to recruit autophagy receptors, MAP1LC3 homologs, and other proteins, and effectively serve as an “eat me” signal for triggering PRKN-dependent mitophagy (Princely Abudu et al., 2019). For example, the recruitment of autophagy receptors, such as calcium binding and coiled-coil domain 2 (CALCOCO2/NDP52), SQSTM1, NBR1 autophagy cargo receptor (NBR1), tax1-binding protein 1 (TAX1BP1), and WD repeat and FYVE domain containing 3 (WDFY3/ALFY), to depolarized mitochondria is mediated by NIPSNAP1 and NIPSNAP2 during mitophagy (Princely Abudu et al., 2019). Accordingly, NIPSNAP1 and NIPSNAP2, which require OMM localization, interact with MAP1LC3 or GABARAP as preferred interaction partners (Princely Abudu et al., 2019). Although zebrafish lacking Nipsnap1 show a decrease in mitochondria in the brain, which is coupled with the production of ROS, the loss of dopaminergic neurons, and a strong decrease in movement (Princely Abudu et al., 2019), the impact of NIPSNAP1 or NIPSNAP2-mediated mitophagy in neurodegenerative disease in mice or humans remains unknown.
BCL2-Like 13
BCL2-like 13 (BCL2L13/Bcl-rambo) is an OMM protein, a member of the pro-apoptotic BCL2 family with four conserved BH domains (Murakawa et al., 2015). The overexpression of BCL2L13 induces caspase-3–dependent apoptosis, which can be blocked by co-expression of inhibitor of apoptotic proteins (IAPs) (Kataoka et al., 2001). However, unlike other BCL2 members, BCL2L13 does not require the BH domains to induce apoptosis, but instead relies on mitochondrial localization by the transmembrane domain (Kataoka et al., 2001). In addition to promoting apoptosis, BCL2L13 also acts as a homolog of Atg32 in mammalian cells, mediating mitochondrial fragmentation and subsequent mitophagy (Murakawa et al., 2015). The OMM protein Atg32 is an autophagy receptor for mitophagy in yeast, and interacts with Atg8 and Atg11 (Liu et al., 2012). BCL2L13 interacts with MAP1LC3 through a conserved LIR sequence, leading to autophagosome engulfment of damaged mitochondria (Otsu et al., 2015). BCL2L13-mediated mitochondria also require fission mechanisms to drive mitochondrial fragmentation (Murakawa et al., 2015). The BCL2L13 gene is involved in a wide range of cancers, but whether BCL2L13-mediated mitophagy affects tumor development is still poorly understood.
Prohibitin 2
Prohibitin 2 (PHB2) is a conserved protein found in the mitochondria and the nucleus of eukaryotic cells, and plays a role in development, lifespan regulation, and various cellular processes (including mitochondrial dynamics) (Wei et al., 2017; Zhou et al., 2018). Notably, PHB2 was initially identified as a specific repressor of estrogen receptor in the nucleus by competitively inhibiting the binding between nuclear receptor coactivator 1 (NCOA1/SRC-1) and estrogen receptors (Montano et al., 1999; Kasashima et al., 2006). PHB2 can combine with PHB1 to form a large ring complex on the mitochondrial membrane and act as a molecular chaperone to stabilize mitochondrial proteins, thereby supporting mitochondrial morphogenesis and preventing cell death (Tatsuta et al., 2005). Moreover, mitochondrial PHB2 acts as an autophagy receptor for the clearance of damaged mitochondria in mammalian cells and C. elegans (Wei et al., 2017). In many cases, the IMM protein requires the rupture of the OMM to recruit the mitophagy molecular machinery (including mitophagy receptors and MAP1LC3) (Wei et al., 2017). However, in some cases, this dynamic positional change of the IMM protein is not necessary for mitophagy. Alternatively, PHB2 may act as a direct autophagy receptor in the IMM and binds to MAP1LC3 through the classical LIR motif, thereby degrading mitochondria (Wei et al., 2017). However, PHB2 promotes PINK1-PRKN–mediated mitophagy in a MAP1LC3-independent manner via the presenilin-associated rhomboid-like (PARL)-PGAM5 axis (Yan et al., 2020). Thus, both OMM receptors and IMM receptors participate in mitophagy-mediated mitochondrial removal.
FKBP Prolyl Isomerase 8
FKBP prolyl isomerase 8 (FKBP8/FKBP38) is a member of the immunophilin family, which has a conserved peptidyl prolyl cis/trans-isomerase domain. FKBP8 not only plays a role in immune regulation, but also participates in protein quality control (e.g., protein folding and trafficking) (Okamoto et al., 2006; Janssens et al., 2014; Xu et al., 2019). When combined with calmodulin and calcium, FKBP8 becomes active (Edlich et al., 2005). FKBP8 is anchored by the transmembrane domain and is mainly distributed in mitochondria (Shirane-Kitsuji and Nakayama, 2014). Mitochondrial FKBP8 acts as a molecular chaperone of BCL2 or heat shock proteins to inhibit apoptosis (Chen et al., 2008; Misaka et al., 2018). In addition to its anti-apoptotic function in response to various mitochondrial stresses, FKBP8 is also an autophagy receptor for damaged mitochondria (Bhujabal et al., 2017). FKBP8 has a typical LIR motif, and can strongly recruit MAP1LC3 to damaged mitochondria in HeLa cells during mitophagy (Bhujabal et al., 2017). Consequently, the overexpression of FKBP8 promotes mitochondrial fission, leading to mitophagy (Bhujabal et al., 2017). Unlike other autophagy receptors that usually degrade with cargo, FKBP8 escapes autophagosome degradation during mitophagy and instead relocates to the endoplasmic reticulum to bind BCL2 (Bhujabal et al., 2017). Thus, FKBP8 partially protects mitochondria from damage through mitophagy activation.
Mitochondrial Membrane Lipids
Cardiolipin is a diphosphatidylglycerol lipid, first found in animal hearts. It is an important component of the IMM, accounting for 20% of its total lipid composition (Paradies et al., 2014). In addition to mitochondria, cardiolipin can also be found in the membranes of most bacteria (Carranza et al., 2017). Cardiolipin homeostasis plays a key role in regulating mitochondrial function, and is involved in metabolism, cell death, and mitochondrial quality control (Dudek, 2017). For example, cardiolipin is necessary for the enzymatic activity of the respiratory chain complex and acts as a proton trap for oxidative phosphorylation (Dudek, 2017). The distribution of cardiolipin on the OMM not only triggers apoptosis, but also induces mitophagy to clear damaged mitochondria by interacting with MAP1LC3 (Chu et al., 2013), indicating that cardiolipin is an important eat me signal that regulates cell death and survival after mitochondrial injury.
Other lipids that contribute to mitophagy come from ceramides, which are composed of sphingosine and fatty acids. For example, C18-ceramide synthesized by ceramide synthase 1 (CERS1) induces mitophagy and tumor suppression in head and neck squamous cell carcinoma cells (Sentelle et al., 2012) and acute myeloid leukemia cells (Dany et al., 2016) in vitro and in vivo. Mechanistically, ceramide can bind to MAP1LC3 on the mitochondrial membrane to trigger mitophagy after DNM1L-mediated mitochondrial fission (Dany and Ogretmen, 2015). These findings provide another strategy for removing damaged mitochondria through the phospholipid components of the mitochondrial membrane.
Mitophagy in Cancer
The role of autophagy in tumor biology is complex, which depends not only on the type of tumor, but also on the stage and context of the tumor (Levy et al., 2017). In general, autophagy plays a role in blocking the initiation of tumorigenesis because it inhibits genome instability and inflammation. In contrast, in established tumors, cancer cells may use autophagy to meet their metabolic requirements and enhance the resistance to cell death, leading to increased growth and invasiveness. Similarly, dysfunctional mitophagy is a characteristic phenomenon of cancer. Most mitophagy receptors or regulators are involved in cancer; however, whether they act as tumor promoters or tumor suppressors seems to be highly dependent on tumor type and TME (Table 1), which is described below (Panigrahi et al., 2019; Praharaj et al., 2019; Vara-Perez et al., 2019; Ferro et al., 2020).
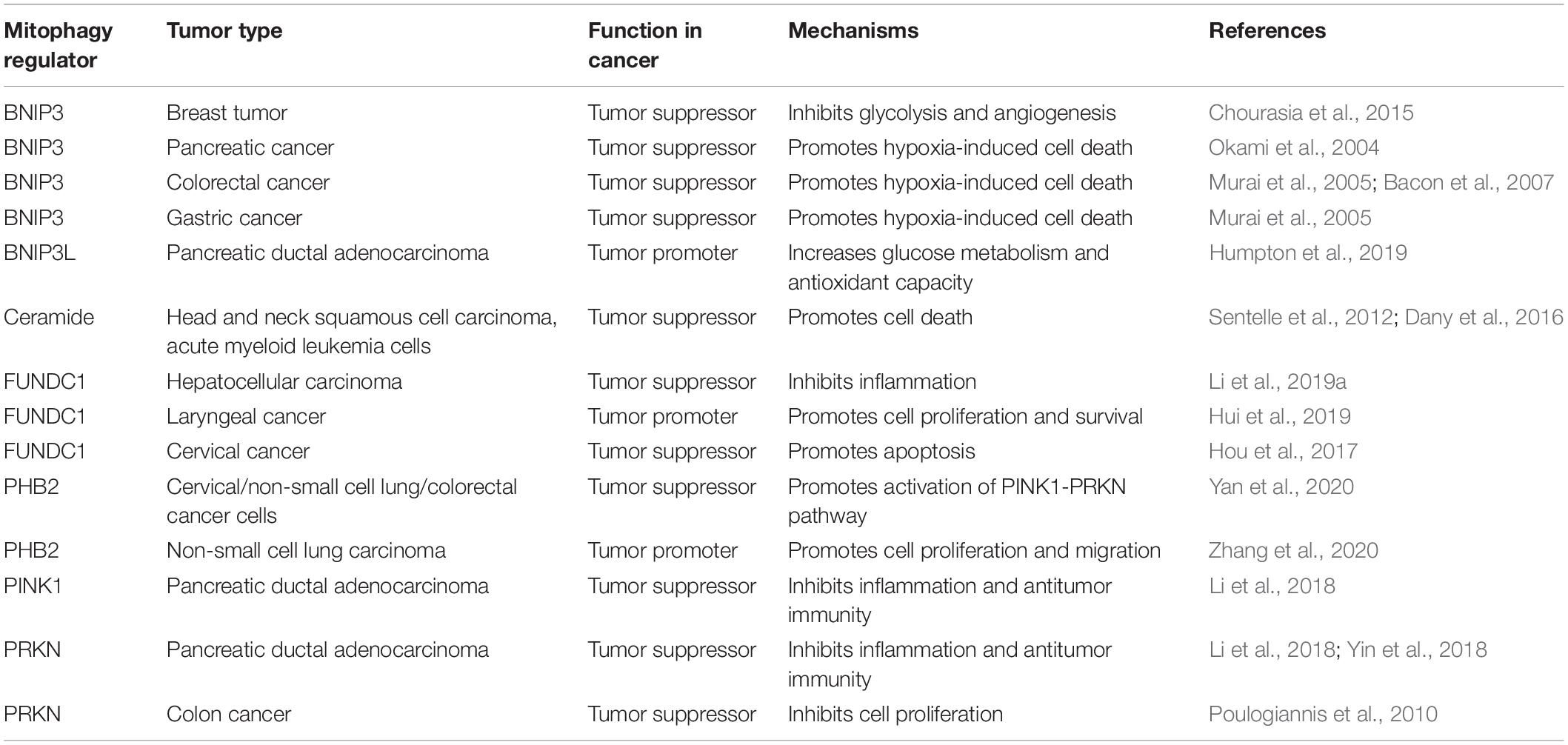
Table 1. Role of mitophagy regulators in tumorigenesis.
Mitophagy Inhibits Tumorigenesis
The PINK1-PRKN pathway is considered to be the main pathway of mitophagy in cancer cells (Bernardini et al., 2017). A loss of PINK1 or PRKN function impairs mitochondrial quality control, which further leads to the accumulation of ROS, thereby affecting cell function. The mutation or depletion of PINK1 or PRKN is often detected in a variety of tumors, such as lung cancer, glioma, and colon cancer (Bernardini et al., 2017). For example, the PRKN gene and human colorectal cancer are obviously associated with adenomatous polyps, and the expression of PRKN can inhibit the proliferation of colon cancer cells (Poulogiannis et al., 2010). The hybridization of PRKN knockout mice with colorectal adenomatous polyposis mice significantly accelerates the development of intestinal adenomas in newborn mice, and the diversity of polyps is also significantly increased, indicating that PRKN is a tumor suppressor gene in colon cancer (Poulogiannis et al., 2010). In addition, in a KRAS-driven tumor model, the depletion of PINK1 or PRKN promotes pancreatic tumorigenesis in mice (Li et al., 2018). Mechanistically, PINK1- and PRKN-mediated autophagy degradation of mitochondrial iron importers [e.g., solute carrier family 25 member 37 (SLC25A37) and solute carrier family 25 member 28 (SLC25A28)] suppresses pancreatic tumors by attenuating mitochondrial iron accumulation, inflammasome activation, high-mobility group box 1 (HMGB1) release, and subsequent immune checkpoint expression (Li et al., 2018). Therefore, the pharmacological or genetic inhibition of mitochondrial iron-dependent signaling prolongs the survival of animals and reverses the phenotype of mitophagy deficient-mediated pancreatic tumors in vivo (Li et al., 2018). These findings suggest that PINK1-PRKN pathway-mediated mitophagy links iron metabolism to tumor immunity during tumor formation (Kang et al., 2019). Unlike extracellular HMGB1 that promotes tumor growth, intracellular HMGB1 can regulate autophagy and mitophagy to inhibit the development of pancreatic cancer (Tang et al., 2010, 2011; Kang et al., 2017; Kang and Tang, 2018).
As discussed above, the activation of HIF1A increases the expression of BNIP3 and subsequent mitophagy. In turn, the expression of BNIP3 may affect HIF1A stability. This HIF1A-BNIP3–mediated mitophagy pathway is also implicated in controlling tumorgenesis in some cancers, such as triple-negative breast cancer (TNBC) (Chourasia et al., 2015). In fact, during the metastasis of TNBC, HIF1A-dependent BNIP3 expression is often suppressed or absent (Chourasia et al., 2015). The combination of BNIP3 deletion and high HIF1A expression predicts poor metastasis-free survival for TNBC (Chourasia et al., 2015). The increased aggressiveness of breast tumors in BNIP3-depleted mice is related to a decrease in mitophagy and the increased stability of HIF1A, indicating that BNIP3 can inhibit HIF1A and mitochondrial dysfunction (Chourasia et al., 2015). In addition, BNIP3 has a tumor suppressor effect in pancreatic cancer (Okami et al., 2004), colorectal cancer (Murai et al., 2005; Bacon et al., 2007), and gastric cancer (Murai et al., 2005), which is related to hypermethylation of the BNIP3 promoter. Whether the epigenetic silencing of BNIP3 can help reduce mitophagy and subsequent tumorigenesis remains unanswered.
FUNDC1 is another player in hypoxia-mediated mitophagy through its dephosphorylation (Liu et al., 2012). In cervical cancer, the expression of FUNDC1 was higher in tumors than in adjacent normal tissues (Hou et al., 2017). This high FUNDC1 expression is negatively correlated with tumor progression and patient prognosis, indicating a potential role of FUNDC1 in the suppression of tumor growth of cervical cancer (Hou et al., 2017). In addition, FUNDC1-mediated mitophagy protects laryngeal cancer cells against oxidative stress (Hui et al., 2019), which correlates with tumorigenic potential. Conditional knockout of FUNDC1 in the liver also initiates liver cancer by activating inflammation (Li et al., 2019a).
Mitophagy Promotes Tumor Progression
In some cases, the activation of a specific mitophagy pathway may promote tumor growth and development. Although both BNIP3 and BNIP3L are similar modulators of mitophagy and apoptosis, BNIP3L, unlike BNIP3 which inhibits tumorgenesis, plays an opposite role in promoting tumorigenesis. For example, the loss of BNIP3L in the KPC (LSL-KrasG12D; Tp53R172H; Pdx1-Cre) model of pancreatic ductal adenocarcinoma (PDAC) delays tumor occurrence, which is associated with reduced mitophagy and attenuated progression from the pancreatic intraepithelial neoplasia stage to PDAC (Humpton et al., 2019). These findings raise an unsolved question about the role of BNIP3L-dependent mitophagy in mutated KRAS and mutated TP53-driven tumorigenesis. One possibility is that different types of mitophagy may produce different TMEs, which further affects inflammation response and tumor immunity. It is also a challenge to distinguish the mitophagy-dependent and -independent role of BNIP3L in tumor biology.
Mitophagy and Tumor Therapies
The main reason for treatment failure in cancer is the resistance of cancer cells to drugs, which leads to tumor recurrence and metastasis. Dysfunctional autophagy and mitophagy lead to drug resistance through multiple mechanisms, including inhibiting cell death, especially apoptosis (Levy et al., 2017). Cancer stem cells (CSCs) are self-renewing cell types that contribute to tumor onset, expansion, drug resistance, recurrence, and metastasis after treatment (Reya et al., 2001; Ward and Dirks, 2007). Mitochondria are an important source of ROS within most cells, including cancer cells. Elevated ROS production is a powerful inducer of apoptosis during chemotherapy. Mitophagy-mediated mitochondria degradation limits the production of ROS, thereby exerting a cytoprotective effect during chemotherapy and helping CSCs resist apoptosis (Ianniciello et al., 2018; Levy et al., 2020). Reversing mitophagy-mediated protective mechanisms may be one of the ways to reverse CSC chemotherapy resistance (Wang et al., 2020). For example, leukemia stem cells (LSCs) are resistant to traditional chemotherapy drugs because LSCs can attain a lower rate of energy metabolism and ROS production through fission-dependent mitophagy (Pei et al., 2018). LSCs increase the expression of fission, mitochondrial 1 (FIS1) through the adenosine 5′-monophosphate-activated protein kinase (AMPK) pathway (Pei et al., 2018). Blocking FIS1 gene expression blocks the mitophagy pathway by inhibiting glycogen synthase kinase 3 (GSK3) activity (Pei et al., 2018). The use of GSK3 inhibitors to target the AMPK-FIS1-GSK3-mediated mitophagy pathway may become a radical cure for acute myeloid leukemia (Pei et al., 2018). Doxorubicin (brand name: adriamycin) is used to treat different types of cancers. The inhibition of mitophagy enhances the anticancer activity of doxorubicin in colorectal cancer cells (Yan et al., 2017). Higher mitophagic levels are also found in CSCs in cisplatin-resistant oral squamous cell carcinoma and oxaliplatin-resistant human colorectal cancer (Naik et al., 2018; Takeda et al., 2019), supporting a widely pro-survival role of mitophagy in various chemo-resistant cancer cells.
In radiotherapy, increased mitophagy can also promote survival, which is mediated by the PINK1-PRKN pathway. Therefore, the inhibition of PINK1-PRKN–mediated mitophagy restores the radiosensitivity of tumor cells (Zheng et al., 2015). Temozolomide-perillyl alcohol (TMZ-POH) conjugate induces lysosomal dysfunction and subsequent impaired mitochondrial flux in non-small cell lung cancer cells and makes them sensitive to radiation, thereby showing TMZ-POH as a potential radiosensitizer (Chang et al., 2018). Ionizing radiation can trigger a series of cellular DNA damage responses, and the dynamic interaction between these responses and mitophagy remains to be revealed.
Compared with chemotherapy and radiotherapy, immunotherapy (e.g., using cytokines, antibodies, or immune checkpoint inhibitors) has shown emerging and great potential in inhibiting tumor growth. Accordingly, more research has focused on the dual roles of mitophagy in immunotherapy. On the one hand, inhibition of the mitophagic axis enhances tumor necrosis factor-based immunotherapy to control the survival and progression of cervical and gastric cancer cells (Yan et al., 2018; Zhao et al., 2019). On the other hand, enhanced mitophagy may induce immunogenic cell death, thereby inhibiting tumor growth through the activation of cytotoxic T lymphocytes in liver cancer cells (Yu et al., 2020). These findings further support that mitophagy may be an effective target for modified tumor immunotherapy.
Conclusion and Perspectives
Mitochondria are multifunctional organelles that play an important role in cancer through the synthesis of macromolecules, energy production, and cell death regulation. Understanding the regulation of mitochondrial morphology, function, biogenesis, fission and fusion dynamics, and degradation is important for the development of new anticancer strategies. Dysfunctional mitophagy is a feature of the TME in many cancers and plays multiple roles in regulating tumor metabolism. On the one hand, mitophagy prevents the accumulation of damaged mitochondria, thereby maintaining energy production for tumor growth. On the other hand, mitophagy may suppress tumors by limiting the production of ROS, which is a well-known factor in causing gene mutation and chromosomal instability. Therefore, it is not surprising that mitophagy is a regulator of tumor biology, acting as either a suppressor or a facilitator of tumorigenesis.
The identification of various mitophagy-related autophagic receptors (including mitochondrial OMM, IMM, or lipid components) has accelerated our knowledge of the complexity of mitophagy in tumor biology. Therefore, understanding the molecular mechanism and function of mitophagy during different types of mitochondrial stress and damage may be critical for developing the next generation of cancer treatment methods. It is also important to develop convenient and reliable methods or biomarkers to assess the activity of mitophagy in humans. In addition, distinguishing the function of mitophagy between normal cells and cancer cells may be important for improving the targeting of tumor therapy and reducing its toxicity.
Author Contributions
YX and DT conceived of the topic for this review. All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
YX was supported by the National Natural Science Foundation of China (No. 81802476).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript.
References
Adams, J. M., and Cory, S. (2007). The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337. doi: 10.1038/sj.onc.1210220
Bacon, A. L., Fox, S., Turley, H., and Harris, A. L. (2007). Selective silencing of the hypoxia-inducible factor 1 target gene BNIP3 by histone deacetylation and methylation in colorectal cancer. Oncogene 26, 132–141. doi: 10.1038/sj.onc.1209761
Bai, Y., Meng, L., Han, L., Jia, Y., Zhao, Y., Gao, H., et al. (2019). Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003. doi: 10.1016/j.bbrc.2018.12.039
Bernardini, J. P., Lazarou, M., and Dewson, G. (2017). Parkin and mitophagy in cancer. Oncogene 36, 1315–1327. doi: 10.1038/onc.2016.302
Bhujabal, Z., Birgisdottir, A. B., Sjottem, E., Brenne, H. B., Overvatn, A., Habisov, S., et al. (2017). FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep 18, 947–961. doi: 10.15252/embr.201643147
Bialik, S., Dasari, S. K., and Kimchi, A. (2018). Autophagy-dependent cell death - where, how and why a cell eats itself to death. J. Cell Sci. 131:jcs215152. doi: 10.1242/jcs.215152
Bock, F. J., and Tait, S. W. G. (2020). Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100. doi: 10.1038/s41580-019-0173-8
Brittain, J. M., Wang, Y., Wilson, S. M., and Khanna, R. (2012). Regulation of CREB signaling through L-type Ca2+ channels by Nipsnap-2. Channels 6, 94–102. doi: 10.4161/chan.19415
Carranza, G., Angius, F., Ilioaia, O., Solgadi, A., Miroux, B., and Arechaga, I. (2017). Cardiolipin plays an essential role in the formation of intracellular membranes in Escherichia coli. Biochim. Biophys. Acta Biomembr. 1859, 1124–1132. doi: 10.1016/j.bbamem.2017.03.006
Chang, M., Song, X., Geng, X., Wang, X., Wang, W., Chen, T. C., et al. (2018). Temozolomide-Perillyl alcohol conjugate impairs Mitophagy flux by inducing lysosomal dysfunction in non-small cell lung Cancer cells and sensitizes them to irradiation. J. Exp. Clin. Cancer Res. 37:250.
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell. 54, 362–377. doi: 10.1016/j.molcel.2014.02.034
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, Y., Sternberg, P., and Cai, J. (2008). Characterization of a Bcl-XL-interacting protein FKBP8 and its splice variant in human RPE cells. Invest. Ophthalmol. Vis. Sci. 49, 1721–1727. doi: 10.1167/iovs.07-1121
Chen, Z., Liu, L., Cheng, Q., Li, Y., Wu, H., Zhang, W., et al. (2017). Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep. 18, 495–509. doi: 10.15252/embr.201643309
Cho, D. H., Kim, Y. S., Jo, D. S., Choe, S. K., and Jo, E. K. (2018). Pexophagy: molecular mechanisms and implications for health and diseases. Mol. Cells 41, 55–64.
Chourasia, A. H., Tracy, K., Frankenberger, C., Boland, M. L., Sharifi, M. N., Drake, L. E., et al. (2015). Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 16, 1145–1163. doi: 10.15252/embr.201540759
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Dany, M., Gencer, S., Nganga, R., Thomas, R. J., Oleinik, N., Baron, K. D., et al. (2016). Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 128, 1944–1958. doi: 10.1182/blood-2016-04-708750
Dany, M., and Ogretmen, B. (2015). Ceramide induced mitophagy and tumor suppression. Biochim. Biophys. Acta 1853, 2834–2845. doi: 10.1016/j.bbamcr.2014.12.039
Dikic, I., and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364. doi: 10.1038/s41580-018-0003-4
Ding, W. X., Ni, H. M., Li, M., Liao, Y., Chen, X., Stolz, D. B., et al. (2010). Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J. Biol. Chem. 285, 27879–27890. doi: 10.1074/jbc.m110.119537
Dudek, J. (2017). Role of cardiolipin in mitochondrial signaling pathways. Front. Cell Dev. Biol. 5:90. doi: 10.3389/fcell.2017.00090
Edlich, F., Weiwad, M., Erdmann, F., Fanghanel, J., Jarczowski, F., Rahfeld, J. U., et al. (2005). Bcl-2 regulator FKBP38 is activated by Ca2+/calmodulin. EMBO J. 24, 2688–2699. doi: 10.1038/sj.emboj.7600739
Eiyama, A., and Okamoto, K. (2015). PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol 33, 95–101. doi: 10.1016/j.ceb.2015.01.002
Ferro, F., Servais, S., Besson, P., Roger, S., Dumas, J. F., and Brisson, L. (2020). Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 98, 129–138. doi: 10.1016/j.semcdb.2019.05.029
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death. Differ 25, 486–541.
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Harper, J. W., Ordureau, A., and Heo, J. M. (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108. doi: 10.1038/nrm.2017.129
Herst, P. M., Rowe, M. R., Carson, G. M., and Berridge, M. V. (2017). Functional mitochondria in health and disease. Front. Endocrinol. 8:296. doi: 10.3389/fendo.2017.00296
Hosokawa, N., Hara, T., Kaizuka, T., Kishi, C., Takamura, A., Miura, Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 20, 1981–1991. doi: 10.1091/mbc.e08-12-1248
Hou, H., Er, P., Cheng, J., Chen, X., Ding, X., Wang, Y., et al. (2017). High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med 6, 1871–1881. doi: 10.1002/cam4.1112
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. doi: 10.1080/15548627.2016.1187366
Hui, L., Wu, H., Wang, T. W., Yang, N., Guo, X., and Jang, X. J. (2019). Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 21, 596–606. doi: 10.1007/s12094-018-1958-5
Humpton, T. J., Alagesan, B., DeNicola, G. M., Lu, D., Yordanov, G. N., Leonhardt, C. S., et al. (2019). Oncogenic KRAS induces NIX-mediated mitophagy to promote pancreatic cancer. Cancer Discov. 9, 1268–1287. doi: 10.1158/2159-8290.cd-18-1409
Ianniciello, A., Rattigan, K. M., and Helgason, G. V. (2018). The ins and outs of autophagy and metabolism in hematopoietic and leukemic stem cells: food for thought. Front. Cell Dev. Biol. 6:120. doi: 10.3389/fcell.2018.00120
Janssens, S., Pulendran, B., and Lambrecht, B. N. (2014). Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 15, 910–919. doi: 10.1038/ni.2991
Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., et al. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. doi: 10.1091/mbc.e08-12-1249
Kang, R., and Tang, D. (2018). The dual role of HMGB1 in pancreatic cancer. J. Pancreatol. 1, 19–24.
Kang, R., Xie, Y., Zeh, H. J., Klionsky, D. J., and Tang, D. (2019). Mitochondrial quality control mediated by PINK1 and PRKN: links to iron metabolism and tumor immunity. Autophagy 15, 172–173. doi: 10.1080/15548627.2018.1526611
Kang, R., Xie, Y., Zhang, Q., Hou, W., Jiang, Q., Zhu, S., et al. (2017). Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Res. 27, 916–932. doi: 10.1038/cr.2017.51
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death. Differ. 18, 571–580. doi: 10.1038/cdd.2010.191
Kasashima, K., Ohta, E., Kagawa, Y., and Endo, H. (2006). Mitochondrial functions and estrogen receptor-dependent nuclear translocation of pleiotropic human prohibitin 2. J. Biol. Chem. 281, 36401–36410. doi: 10.1074/jbc.m605260200
Kataoka, T., Holler, N., Micheau, O., Martinon, F., Tinel, A., Hofmann, K., et al. (2001). Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J. Biol. Chem. 276, 19548–19554. doi: 10.1074/jbc.m010520200
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. doi: 10.1126/science.290.5497.1717
Kounakis, K., Chaniotakis, M., Markaki, M., and Tavernarakis, N. (2019). Emerging roles of lipophagy in health and disease. Front. Cell Dev. Biol. 7:185. doi: 10.3389/fcell.2019.00185
Koyano, F., Yamano, K., Kosako, H., Kimura, Y., Kimura, M., Fujiki, Y., et al. (2019). Parkin-mediated ubiquitylation redistributes MITOL/March5 from mitochondria to peroxisomes. EMBO Rep. 20:e47728.
Kroemer, G., and Levine, B. (2008). Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010. doi: 10.1038/nrm2529
Kroemer, G., Marino, G., and Levine, B. (2010). Autophagy and the integrated stress response. Mol. Cell. 40, 280–293. doi: 10.1016/j.molcel.2010.09.023
Lee, Y., Lee, H. Y., Hanna, R. A., and Gustafsson, A. B. (2011). Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 301, H1924–H1931.
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Levine, B., and Kroemer, G. (2019). Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. doi: 10.1016/j.cell.2018.09.048
Levy, A., Stedman, A., Deutsch, E., Donnadieu, F., Virgin, H. W., Sansonetti, P. J., et al. (2020). Innate immune receptor NOD2 mediates LGR5(+) intestinal stem cell protection against ROS cytotoxicity via mitophagy stimulation. Proc. Natl. Acad. Sci. U.S.A. 117, 1994–2003. doi: 10.1073/pnas.1902788117
Levy, J. M. M., Towers, C. G., and Thorburn, A. (2017). Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542.
Li, C., Zhang, Y., Cheng, X., Yuan, H., Zhu, S., Liu, J., et al. (2018). PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 46, 441–455. doi: 10.1016/j.devcel.2018.07.012
Li, C., Zhang, Y., Liu, J., Kang, R., Klionsky, D. J., and Tang, D. (2020). Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 18, 1–13. doi: 10.1080/15548627.2020.1739447
Li, W., Li, Y., Siraj, S., Jin, H., Fan, Y., Yang, X., et al. (2019a). FUN14 domain-containing 1-mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology 69, 604–621. doi: 10.1002/hep.30191
Li, Y., Xue, Y., Xu, X., Wang, G., Liu, Y., Wu, H., et al. (2019b). A mitochondrial FUNDC1/HSC70 interaction organizes the proteostatic stress response at the risk of cell morbidity. EMBO J. 38:e98786.
Liu, J., Kuang, F., Kroemer, G., Klionsky, D. J., Kang, R., and Tang, D. (2020). Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem. Biol. 27, 420–435. doi: 10.1016/j.chembiol.2020.02.005
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W., and Kimmelman, A. C. (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. doi: 10.1038/nature13148
Marinkovic, M., Sprung, M., and Novak, I. (2020). Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy. [Epub ahead of print]. doi: 10.1007/7651_2017_14
McEwan, D. G., and Dikic, I. (2011). The three Musketeers of autophagy: phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 21, 195–201. doi: 10.1016/j.tcb.2010.12.006
Misaka, T., Murakawa, T., Nishida, K., Omori, Y., Taneike, M., Omiya, S., et al. (2018). FKBP8 protects the heart from hemodynamic stress by preventing the accumulation of misfolded proteins and endoplasmic reticulum-associated apoptosis in mice. J. Mol. Cell Cardiol. 114, 93–104. doi: 10.1016/j.yjmcc.2017.11.004
Montano, M. M., Ekena, K., Delage-Mourroux, R., Chang, W., Martini, P., and Katzenellenbogen, B. S. (1999). An estrogen receptor-selective coregulator that potentiates the effectiveness of antiestrogens and represses the activity of estrogens. Proc. Natl. Acad. Sci. U.S.A. 96, 6947–6952. doi: 10.1073/pnas.96.12.6947
Montero, J., Mari, M., Colell, A., Morales, A., Basanez, G., Garcia-Ruiz, C., et al. (2010). Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim. Biophys. Acta 1797, 1217–1224. doi: 10.1016/j.bbabio.2010.02.010
Murai, M., Toyota, M., Suzuki, H., Satoh, A., Sasaki, Y., Akino, K., et al. (2005). Aberrant methylation and silencing of the BNIP3 gene in colorectal and gastric cancer. Clin. Cancer Res. 11, 1021–1027.
Murakawa, T., Yamaguchi, O., Hashimoto, A., Hikoso, S., Takeda, T., Oka, T., et al. (2015). Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 6:7527.
Naik, P. P., Mukhopadhyay, S., Panda, P. K., Sinha, N., Das, C. K., Mishra, R., et al. (2018). Autophagy regulates cisplatin-induced stemness and chemoresistance via the upregulation of CD44, ABCB1 and ADAM17 in oral squamous cell carcinoma. Cell Prolif. 51:e12411.
Ney, P. A. (2015). Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 1853, 2775–2783. doi: 10.1016/j.bbamcr.2015.02.022
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Okami, J., Simeone, D. M., and Logsdon, C. D. (2004). Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res. 64, 5338–5346. doi: 10.1158/0008-5472.can-04-0089
Okamoto, T., Nishimura, Y., Ichimura, T., Suzuki, K., Miyamura, T., Suzuki, T., et al. (2006). Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 25, 5015–5025. doi: 10.1038/sj.emboj.7601367
O’Sullivan, T. E., Johnson, L. R., Kang, H. H., and Sun, J. C. (2015). BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity 43, 331–342. doi: 10.1016/j.immuni.2015.07.012
Otsu, K., Murakawa, T., and Yamaguchi, O. (2015). BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy 11, 1932–1933. doi: 10.1080/15548627.2015.1084459
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Panigrahi, D. P., Praharaj, P. P., Bhol, C. S., Mahapatra, K. K., Patra, S., Behera, B. P., et al. (2019). The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin. Cancer Biol. 66, 45–58.
Paradies, G., Paradies, V., Ruggiero, F. M., and Petrosillo, G. (2014). Cardiolipin and mitochondrial function in health and disease. Antioxid. Redox. Signal. 20, 1925–1953. doi: 10.1089/ars.2013.5280
Park, C. W., Hong, S. M., Kim, E. S., Kwon, J. H., Kim, K. T., Nam, H. G., et al. (2013). BNIP3 is degraded by ULK1-dependent autophagy via MTORC1 and AMPK. Autophagy 9, 345–360. doi: 10.4161/auto.23072
Pei, S., Minhajuddin, M., Adane, B., Khan, N., Stevens, B. M., Mack, S. C., et al. (2018). AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell 23:e6.
Pfanner, N., Warscheid, B., and Wiedemann, N. (2019). Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20, 267–284. doi: 10.1038/s41580-018-0092-0
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Poulogiannis, G., McIntyre, R. E., Dimitriadi, M., Apps, J. R., Wilson, C. H., Ichimura, K., et al. (2010). PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. U.S.A. 107, 15145–15150. doi: 10.1073/pnas.1009941107
Praharaj, P. P., Naik, P. P., Panigrahi, D. P., Bhol, C. S., Mahapatra, K. K., Patra, S., et al. (2019). Intricate role of mitochondrial lipid in mitophagy and mitochondrial apoptosis: its implication in cancer therapeutics. Cell Mol. Life. Sci. 76, 1641–1652. doi: 10.1007/s00018-018-2990-x
Princely Abudu, Y., Pankiv, S., Mathai, B. J., Hakon Lystad, A., Bindesboll, C., Brenne, H. B., et al. (2019). NIPSNAP1 and NIPSNAP2 Act as “Eat Me” Signals for Mitophagy. Dev. Cell 49:e12.
Reya, T., Morrison, S. J., Clarke, M. F., and Weissman, I. L. (2001). Stem cells, cancer, and cancer stem cells. Nature 414, 105–111.
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Schwarten, M., Mohrluder, J., Ma, P., Stoldt, M., Thielmann, Y., Stangler, T., et al. (2009). Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5, 690–698. doi: 10.4161/auto.5.5.8494
Schweers, R. L., Zhang, J., Randall, M. S., Loyd, M. R., Li, W., Dorsey, F. C., et al. (2007). NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. U.S.A. 104, 19500–19505. doi: 10.1073/pnas.0708818104
Sentelle, R. D., Senkal, C. E., Jiang, W., Ponnusamy, S., Gencer, S., Selvam, S. P., et al. (2012). Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 8, 831–838. doi: 10.1038/nchembio.1059
Seroussi, E., Pan, H. Q., Kedra, D., Roe, B. A., and Dumanski, J. P. (1998). Characterization of the human NIPSNAP1 gene from 22q12: a member of a novel gene family. Gene 212, 13–20. doi: 10.1016/s0378-1119(98)00098-5
Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., et al. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305. doi: 10.1038/77060
Shirane-Kitsuji, M., and Nakayama, K. I. (2014). Mitochondria: FKBP38 and mitochondrial degradation. Int. J. Biochem. Cell Biol. 51, 19–22. doi: 10.1016/j.biocel.2014.03.007
Sowter, H. M., Ratcliffe, P. J., Watson, P., Greenberg, A. H., and Harris, A. L. (2001). HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61, 6669–6673.
Takeda, M., Koseki, J., Takahashi, H., Miyoshi, N., Nishida, N., Nishimura, J., et al. (2019). Disruption of Endolysosomal RAB5/7 efficiently eliminates colorectal cancer stem cells. Cancer Res. 79, 1426–1437. doi: 10.1158/0008-5472.can-18-2192
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P., and Kroemer, G. (2019). The molecular machinery of regulated cell death. Cell Res. 29, 347–364. doi: 10.1038/s41422-019-0164-5
Tang, D., Kang, R., Livesey, K. M., Cheh, C. W., Farkas, A., Loughran, P., et al. (2010). Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190, 881–892. doi: 10.1083/jcb.200911078
Tang, D., Kang, R., Livesey, K. M., Kroemer, G., Billiar, T. R., Van Houten, B., et al. (2011). High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 13, 701–711. doi: 10.1016/j.cmet.2011.04.008
Tatsuta, T., Model, K., and Langer, T. (2005). Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol. Biol. Cell 16, 248–259. doi: 10.1091/mbc.e04-09-0807
Twig, G., and Shirihai, O. S. (2011). The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox. Signal. 14, 1939–1951. doi: 10.1089/ars.2010.3779
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Vande Velde, C., Cizeau, J., Dubik, D., Alimonti, J., Brown, T., Israels, S., et al. (2000). BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 20, 5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000
Vara-Perez, M., Felipe-Abrio, B., and Agostinis, P. (2019). Mitophagy in cancer: a tale of adaptation. Cells 8:493. doi: 10.3390/cells8050493
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., de Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383.
Wang, Y., Liu, H. H., Cao, Y. T., Zhang, L. L., Huang, F., and Yi, C. (2020). The role of mitochondrial dynamics and mitophagy in carcinogenesis. metastasis and therapy. Front Cell Dev Biol 8:413. doi: 10.3389/fcell.2020.00413
Ward, R. J., and Dirks, P. B. (2007). Cancer stem cells: at the headwaters of tumor development. Annu. Rev. Pathol. 2, 175–189. doi: 10.1146/annurev.pathol.2.010506.091847
Wei, Y., Chiang, W. C., Sumpter, R. Jr., Mishra, P., and Levine, B. (2017). Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168:e10.
Wild, P., McEwan, D. G., and Dikic, I. (2014). The LC3 interactome at a glance. J. Cell Sci. 127, 3–9. doi: 10.1242/jcs.140426
Wong, Y. C., and Holzbaur, E. L. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U.S.A. 111, E4439–E4448.
Wu, W., Tian, W., Hu, Z., Chen, G., Huang, L., Li, W., et al. (2014). ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15, 566–575. doi: 10.1002/embr.201438501
Xie, Y., Kang, R., Sun, X., Zhong, M., Huang, J., Klionsky, D. J., et al. (2015). Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 11, 28–45. doi: 10.4161/15548627.2014.984267
Xie, Y., Kuang, F., Liu, J., Tang, D., and Kang, R. (2020a). DUSP1 blocks autophagy-dependent ferroptosis in pancreatic cancer. J. Pancreatol. 3, 154–160. doi: 10.1097/JP9.0000000000000054
Xie, Y., Li, J., Kang, R., and Tang, D. (2020b). Interplay between lipid metabolism and autophagy. Front. Cell Dev. Biol. 8:431. doi: 10.3389/fcell.2020.00431
Xu, S. S., Xu, L. G., Yuan, C., Li, S. N., Chen, T., Wang, W., et al. (2019). FKBP8 inhibits virus-induced RLR-VISA signaling. J. Med. Virol. 91, 482–492. doi: 10.1002/jmv.25327
Yan, C., Gong, L., Chen, L., Xu, M., Abou-Hamdan, H., Tang, M., et al. (2020). PHB2 (prohibitin 2) promotes PINK1-PRKN/Parkin-dependent mitophagy by the PARL-PGAM5-PINK1 axis. Autophagy 16, 419–434. doi: 10.1080/15548627.2019.1628520
Yan, C., Luo, L., Guo, C. Y., Goto, S., Urata, Y., Shao, J. H., et al. (2017). Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 388, 34–42. doi: 10.1016/j.canlet.2016.11.018
Yan, H., Xiao, F., Zou, J., Qiu, C., Sun, W., Gu, M., et al. (2018). NR4A1-induced increase in the sensitivity of a human gastric cancer line to TNFalpha-mediated apoptosis is associated with the inhibition of JNK/Parkin-dependent mitophagy. Int. J. Oncol. 52, 367–378.
Yang, M., Chen, P., Liu, J., Zhu, S., Kroemer, G., Klionsky, D. J., et al. (2019). Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 5:eaaw2238. doi: 10.1126/sciadv.aaw2238
Yang, Z., and Klionsky, D. J. (2010). Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822. doi: 10.1038/ncb0910-814
Yin, J., Guo, J., Zhang, Q., Cui, L., Zhang, L., Zhang, T., et al. (2018). Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. Vitro 51, 1–10. doi: 10.1016/j.tiv.2018.05.001
Yu, Z., Guo, J., Hu, M., Gao, Y., and Huang, L. (2020). Icaritin exacerbates mitophagy and synergizes with doxorubicin to induce immunogenic cell death in hepatocellular carcinoma. ACS Nano 14, 4816–4828. doi: 10.1021/acsnano.0c00708
Zaffagnini, G., and Martens, S. (2016). Mechanisms of selective autophagy. J. Mol. Biol. 428, 1714–1724. doi: 10.1016/j.jmb.2016.02.004
Zhang, H., Yin, C., Liu, X., Bai, X., Wang, L., Xu, H., et al. (2020). Prohibitin 2/PHB2 in parkin-mediated mitophagy: a potential therapeutic target for non-small cell lung carcinoma. Med. Sci. Monit. 26:e923227.
Zhang, J., and Ney, P. A. (2009). Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death. Differ. 16, 939–946. doi: 10.1038/cdd.2009.16
Zhao, Q., Wang, W., and Cui, J. (2019). Melatonin enhances TNF-alpha-mediated cervical cancer HeLa cells death via suppressing CaMKII/Parkin/mitophagy axis. Cancer Cell Int. 19:58.
Zheng, R., Yao, Q., Xie, G., Du, S., Ren, C., Wang, Y., et al. (2015). TAT-ODD-p53 enhances the radiosensitivity of hypoxic breast cancer cells by inhibiting Parkin-mediated mitophagy. Oncotarget 6, 17417–17429. doi: 10.18632/oncotarget.4002
Zhou, B., Liu, J., Kang, R., Klionsky, D. J., Kroemer, G., and Tang, D. (2020). Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 66, 89–100. doi: 10.1016/j.semcancer.2019.03.002
Zhou, Z., Ai, H., Li, K., Yao, X., Zhu, W., Liu, L., et al. (2018). Prohibitin 2 localizes in nucleolus to regulate ribosomal RNA transcription and facilitate cell proliferation in RD cells. Sci. Rep. 8:1479.
Keywords: mitophagy, cancer, cell death, autophagy, mitochondria
Citation: Xie Y, Liu J, Kang R and Tang D (2020) Mitophagy Receptors in Tumor Biology. Front. Cell Dev. Biol. 8:594203. doi: 10.3389/fcell.2020.594203
Received: 12 August 2020; Accepted: 21 October 2020;
Published: 11 November 2020.
Edited by:
Nektarios Tavernarakis, Foundation for Research and Technology Hellas (FORTH), GreeceReviewed by:
Takao Kataoka, Kyoto Institute of Technology, JapanYong Liu, Xuzhou Medical University, China
Copyright © 2020 Xie, Liu, Kang and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yangchun Xie, eGlleWFuZ2NodW44OEBjc3UuZWR1LmNu; Daolin Tang, ZGFvbGluLnRhbmdAdXRzb3V0aHdlc3Rlcm4uZWR1