
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 22 October 2020
Sec. Stem Cell Research
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.592893
This article is part of the Research Topic Stem Cells and Cardiovascular Diseases View all 29 articles
 Wener Li1†
Wener Li1† Michael Stauske2,3†
Michael Stauske2,3† Xiaojing Luo1
Xiaojing Luo1 Stefan Wagner2,4Meike Vollrath1Carola S. Mehnert1
Stefan Wagner2,4Meike Vollrath1Carola S. Mehnert1 Mario Schubert1
Mario Schubert1 Lukas Cyganek2,3Simin Chen3
Lukas Cyganek2,3Simin Chen3 Sayed-Mohammad Hasheminasab5,6
Sayed-Mohammad Hasheminasab5,6 Gerald Wulf4Ali El-Armouche1Lars S. Maier2,7
Gerald Wulf4Ali El-Armouche1Lars S. Maier2,7 Gerd Hasenfuss2,3
Gerd Hasenfuss2,3 Kaomei Guan1,2,3*
Kaomei Guan1,2,3*Brugada syndrome (BrS) is one of the major causes of sudden cardiac death in young people, while the underlying mechanisms are not completely understood. Here, we investigated the pathophysiological phenotypes and mechanisms using induced pluripotent stem cell (iPSC)-derived cardiomyocytes (CMs) from two BrS patients (BrS-CMs) carrying a heterozygous SCN5A mutation p.S1812X. Compared to CMs derived from healthy controls (Ctrl-CMs), BrS-CMs displayed a 50% reduction of INa density, a 69.5% reduction of NaV1.5 expression, and the impaired localization of NaV1.5 and connexin 43 (Cx43) at the cell surface. BrS-CMs exhibited reduced action potential (AP) upstroke velocity and conduction slowing. The Ito in BrS-CMs was significantly augmented, and the ICaL window current probability was increased. Our data indicate that the electrophysiological mechanisms underlying arrhythmia in BrS-CMs may involve both depolarization and repolarization disorders. Cilostazol and milrinone showed dramatic inhibitions of Ito in BrS-CMs and alleviated the arrhythmic activity, suggesting their therapeutic potential for BrS patients.
Brugada syndrome (BrS), a genetic heart disease, is one of the major causes of sudden cardiac death in young people. The diagnosis of BrS is based on the changes in the electrocardiogram (ECG) with a transient or persistent ST-segment elevation in the right precordial leads and the right bundle branch block (Antzelevitch and Fish, 2006). In the past two decades, extensive research on BrS has revealed parts of its genetic basis. To date, 293 different mutations in SCN5A encoding the α-subunit of the cardiac sodium channel (NaV1.5) have been implicated as possible causes of BrS (Kapplinger et al., 2010). The implantation of an automatic implantable cardiac defibrillator (ICD) or the ablation of the arrhythmogenic substrate in the right ventricular outflow tract area have been proven effective in treating ventricular tachycardia and fibrillation and preventing sudden cardiac death in BrS patients. In addition, drug therapy with low-dose quinidine has been shown to prevent arrhythmic events in BrS patients after arrhythmic storm and should be considered according to current guidelines (class IIa indication). However, prophylactic use in asymptomatic patients is controversially discussed and worldwide commercial availability is limited. Thus, a detailed understanding of arrhythmic mechanisms in BrS is required to develop novel drug therapies that are urgently needed.
For more than a decade, the underlying pathophysiological mechanism of BrS has been a matter of debate between the depolarization versus repolarization hypotheses (Wilde et al., 2010). The depolarization theory relies on right ventricular conduction slowing and involvement of structural abnormalities including increased epicardial and interstitial fibrosis, and the reduced gap junction expression (Nademanee et al., 2015). The repolarization theory is mainly supported by the late fractionated low-voltage potentials found in right ventricle epicardium but not in endocardium by using canine heart wedge model, which are associated with repolarization defects (Szel and Antzelevitch, 2014; Patocskai et al., 2017). Nonetheless, there is a general consensus that SCN5A mutations resulting in NaV1.5 loss-of-function either by the decreased expression of functional NaV1.5 at the sarcolemma and/or by the altered channel gating properties lead to a diminution of INa and contribute to the development of arrhythmias in BrS (Patocskai et al., 2017). Recently, single cardiomyocytes (CMs) derived from patient-specific and genome-edited induced pluripotent stem cells (iPSCs) were used to recapitulate the phenotypes of patients with BrS and to study the underlying mechanism (Liang et al., 2016; Veerman et al., 2016; Ma et al., 2018). iPSC-CMs from BrS patients who tested negative for mutations in the known BrS-associated genes do not exhibit clear cellular electrophysiological abnormalities (Veerman et al., 2016). Patient-specific iPSC-CMs with a double missense mutation (p.R620H and p.R811H) or with a deletion (Δ1397) in SCN5A could recapitulate single-cell phenotype features of BrS, including the blunted INa, an increased triggered activity, and an abnormal Ca2+ handling (Liang et al., 2016).
Interestingly, a more recent study reported a remarked lower INa density and an increased phase-1 repolarization at a slow pacing frequency (0.1 Hz) in BrS patient-specific iPSC-CMs with a compound SCN5A mutation (p.A226V and p.R1629X), leading to pro-arrhythmic action potential (AP) morphology (Ma et al., 2018). The dominant repolarizing currents in human CMs are the transient-outward potassium current (Ito) conducted by KV4.2 and KV4.3 channels which contribute to Ito,fast, and by KV1.4 which accounts for Ito,slow and the rapidly and slowly activating delayed rectifier potassium currents (IKr and IKs) conducted by HERG and KVLQT1/mink channels, respectively (Nerbonne and Kass, 2005). Previous study identified gain-of-function mutations in KCND3 encoding KV4.3 in patients with BrS as a result of the increased Ito (Giudicessi et al., 2011). These studies suggest that the underlying mechanism for BrS might be different in BrS patients with different mutations and both depolarization and repolarization mechanisms might coexist.
In the present study, we utilized iPSC-CMs from two BrS patients (BrS1-CMs and BrS2-CMs) with a heterozygous SCN5A point mutation p.S1812X causing a premature termination codon. We show that BrS-CMs from both patients reveal a reduced INa and a delayed sodium channel activation, which result in a slowed AP upstroke velocity and a reduced field potential (FP) conduction velocity (CV), similar to the phenotype observed in BrS patients with SCN5A loss-of-function mutations. Importantly, BrS-CMs also exhibited the enhanced Ito and an augmented ICaL (L-type calcium current) window current, which may contribute to the arrhythmic phenotype. Exposure of BrS-CMs to cilostazol and milrinone, two clinically used phosphodiesterase (PDE) blockers, lowered Ito and alleviated arrhythmic activity, without affecting on the CV. These data indicate that the electrophysiological mechanisms underlying arrhythmia in BrS-CMs may involve an impaired coordination of INa, Ito, and ICaL and cilostazol and milrinone can reduce the arrhythmia but have no effect on the CV.
The study was approved by the Institutional Ethics Committee of University Medical Center Göttingen (approval number 21/1/11) and of Technical University of Dresden (approval number EK 422092019) and carried out in accordance with the approved guidelines. Bone marrow aspirate (10 mL) and peripheral blood (15 mL) were taken from the BrS1 and BrS2 patients, respectively, after obtaining written informed consent. For the Ctrl1, bone marrow aspirate of a 45-year-old female (without known cardiac disease) left over from diagnostic purposes was used. For the Ctrl2, skin biopsy was taken. Bone marrow-derived mesenchymal stem cells, peripheral blood mononuclear cells, and skin fibroblasts were cultured and reprogrammed into iPSCs using the STEMCCA lentivirus (BrS1-iPSCs, Ctrl1-iPSCs, Ctrl2-iPSCs) or Sendai virus system (BrS2-iPSCs), as described previously (Streckfuss-Bomeke et al., 2013; Cyganek et al., 2018). The generated iPSCs from the BrS patients and the healthy control were adapted to feeder-free culture conditions and cultivated on tissue culture plates pre-coated with Geltrex® (Thermo Fisher Scientific) in Essential 8TM medium (Thermo Fisher Scientific).
Karyotyping of BrS- and Ctrl-iPSCs was performed using standard methodology. Prior to karyotyping, the iPSCs were cultured on feeder-free conditions. The cells were treated with 100 ng/mL colcemid (Thermo Fisher Scientific) for 16 h. The metaphases were prepared according to standard methodology and subjected to Giemsa staining before analysis with a light microscope (Zeiss Axio Imager.M2). The karyotype was analyzed and documented by using the software Case Data Manager 6.0 (Applied Spectral Imaging).
The genomic DNA of Ctrl- and BrS-iPSCs was isolated and purified using the automated Maxwell® 16 cell DNA purification kit (Promega) according to the manufacturer’s instructions. DNA sequence comprising the SCN5A point mutation site in the generated Ctrl- and BrS-iPSCs was amplified by PCR. The primer set used is given in Supplementary Table 3. The PCR products were purified using the QIAquick® gel extraction kit (Qiagen) according to the manufacturer’s instructions and sequenced by a commercial sequencing facility (Seqlab, Göttingen).
For spontaneous in vitro differentiation experiments, the cells were digested with 200 U/mL collagenase type 4 (Worthington Biochemicals) into big clusters and cultivated in suspension to form multicellular aggregates, known as embryoid bodies (EBs) for 8 days. The differentiation medium contained IMDM (Thermo Fisher Scientific), 20% fetal bovine serum, 1x non-essential amino acid (Thermo Fisher Scientific), and monothioglycerol (450 μM, Sigma–Aldrich). EBs were thereafter plated onto 0.1% gelatin-coated tissue culture dishes, cultivated up to 1 month and used for further analyses.
BrS1- and BrS2-iPSCs and Ctrl1-iPSCs were characterized for the expression of pluripotency genes NANOG, LIN28, GDF3, and FOXD3 using reverse transcriptase-PCR (RT-PCR). Expression of tissue-specific genes ALB (albumin), α-MHC (myosin heavy chain), TNNT2 [cardiac troponin T (cTnT)], and TH (thyroxine hydroxylase) was analyzed during EB differentiation. To assess the expression of KCND2/3 and KCNA4 genes, 3-month-old CMs derived from Ctrl- and BrS-iPSCs were collected as cell pellets for further use.
Total mRNA was isolated as described in the SV Total RNA Isolation System with on-column DNase digestion (Promega). DNase-treated RNA (200 ng) was used for the first-strand cDNA synthesis by using Murine Leukemia Virus Reverse Transcriptase and Oligo d(T)16 (Thermo Fisher Scientific). One-tenth of the cDNA reaction was taken as PCR template and amplified for 25–35 cycles, denaturation at 95°C for 15 s, annealing at 52–64°C for 15–30 s depending on the primer melting temperatures, and elongation at 72°C for 30 s. The primer sequences (forward and reverse), annealing temperatures, and the cycles used for RT-PCR analyses are listed in Supplementary Table 3.
Quantitative real-time PCR were carried out using SsoAdvanced Universal SYBR Green PCR super mix, Hard-Shell Optical 96-well plates, and the BIO-RAD CFX96 Real-time PCR system (Bio-Rad) according to the manufacturer’s instructions. Initial denaturation was performed at 95°C for 30 s, followed by 45 amplification cycles of 15 s at 95°C and 1 min elongation at 60°C. A melting curve was obtained for 65–95°C. PCR products were confirmed by the melting temperature and agarose gel electrophoresis. Expression was quantified using ΔΔCt method with RPL32 as a reference. Sequences for RPL32, TNNT2, KCND2/3, and KCNA4 real-time PCR analyses are listed in Supplementary Table 3.
To investigate their differentiation potential, the generated iPSCs were differentiated into derivatives of the three germ layers in vivo as previously described (Streckfuss-Bomeke et al., 2013). Briefly, the undifferentiated iPSCs were subcutaneously injected into 8-week-old recombination activating gene 2 and gamma C deficient (RAGC) mice. The mice were sacrificed 8–12 weeks after injection and the resulting teratomas were examined with hematoxylin and eosin staining.
All iPSCs were cultured on Geltrex® in Essential 8TM medium as a monolayer with 80–100% confluence before the initiation of differentiation. At day 0, cells were cultured in RPMI1640 medium (Thermo Fisher Scientific) with Glutamax and HEPES (Thermo Fisher Scientific), 0.5 mg/mL human recombinant albumin, and 0.2 mg/mL L-ascorbic acid 2-phosphate and treated with 4 μM CHIR99021 (Merck Millipore), a highly selective inhibitor of GSK3β. After 48 h, CHIR99021 was removed and the cells were treated with 5 μM Wnt antagonist II (IWP2, Merck Millipore). Another 48 h later, the medium was replaced without IWP2. From day 8 on, the medium was replaced by RPMI1640 with Glutamax and HEPES containing 1x B27 supplement with insulin (Thermo Fisher Scientific). First beating CMs appeared around day 8. At day 25, the CMs were first treated with collagenase 2 (200 U/mL, Worthington) for 30 min so that beating clusters could be detached from the culture dish. These clusters were collected and digested with 0.25% trypsin/EDTA solution (Thermo Fisher Scientific) into single cells, which were replated onto Geltrex® -coated culture dishes as a monolayer. CMs were kept in culture for 3 months for maturation and further electrophysiological and gene and protein expression analyses.
For the allele-specific expression analysis of SCN5A in BrS-CMs, the mRNA was sequenced with primers containing specific barcodes using the Ion Torrent semiconductor sequencing system (Thermo Fisher Scientific). The genomic DNA was used as control.
Total mRNA was isolated and reverse transcribed into cDNA. The primer set (forward: 5’-GAG AGC ACC GAG CCC CTG AGT GAG G-3’ and reverse: 5’-CAC CAT GGG CAG GTC CAT GTT GAT G-3’) was used to amplify the region containing the mutation site. The PCR product was diluted 1/200 and used for a second PCR run with 12 cycles using the same forward primer and different custom-designed reverse primers (Sigma–Aldrich), which contained a unique barcode to distinguish individual samples. The DNA concentration of each sample was measured with Qubit 2.0 Fluorometer (Thermo Fisher Scientific) and 250 ng of each sample was pooled and electrophoretically separated on a 2% agar gel. The specific product was extracted from the gel in a QIAcube system using the QIAquick® gel extraction kit (Qiagen) and subsequently purified with Agencourt AMPure XP PCR purification kit (Beckman Coulter) according to the manufacturer’s instructions and eluted in low TE buffer (Thermo Fisher Scientific). The DNA quantity was determined by real-time PCR analysis in a 384-well plate using the GeneRead Library Quant kit (Qiagen). 400 μL of 10 pmol PCR product was used for clonal amplification onto Ion Sphere particles accomplished by an emulsion PCR in the Ion OneTouch (Thermo Fisher Scientific) system according to the manufacturer’s instructions. The Ion Sphere particles coated with the amplified template DNA were applied to an Ion Torrent sequencing chip and placed on the Ion Personal Genome Machine (Thermo Fisher Scientific) for sequencing.
Undifferentiated iPSCs or EB cultures at different differentiation stages as well as 3-month-old CMs were fixed with 4% paraformaldehyde (Sigma–Aldrich) in PBS at room temperature for 20 min and then blocked with 1% bovine serum albumin (Thermo Fisher Scientific) at 4°C overnight. The primary and respective secondary antibodies used are listed in Supplementary Table 4. Nuclei were counter-stained with 4-6-diamino-2-phenylindole (DAPI, 0.4 μg/mL, Sigma–Aldrich). Samples were mounted with Fluoromount-G mounting medium (eBioscience). Antibodies staining against non-transmembrane proteins were additionally treated with 0.1% Triton X-100 (Sigma–Aldrich) in PBS to permeabilize the cell membrane. Fluorescent images were captured with a fluorescence microscope (Zeiss Observer.Z1 or Axio Imager.M2). For some images, the ApoTome modus (Zeiss) was used.
For analysis of protein levels, Ctrl- and BrS-CMs were solubilized in 0.5% digitonin, 1.5% NP-40, and 1.0% Triton X 100 solubilization buffer for 35 min at 4°C. Protein concentrations were determined with a BCA assay kit. Equal amounts of protein were loaded on biphasic 6 and 10% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) and run at low voltage. After blotting to PVDF membranes, membranes were incubated with NaV1.5, Cx43, CaV1.2, KV4.3, and cTnT antibodies overnight. The primary and respective secondary antibodies used for Western blots are listed in Supplementary Table 4. Western blots were developed with a peqlab Fusion FX Vilber Lourmat camera and analyzed using the FusionCapt Advance 16.12 software.
Whole-cell clamp was used to measure membrane potential (current-clamp configuration), INa, late INa, Ito, and ICaL (voltage-clamp configuration), as previously described (Wagner et al., 2006; Streckfuss-Bomeke et al., 2013). All experiments were conducted with 3-month-old Ctrl- and BrS-CMs at room temperature. The pipette and extracellular solutions for various kinds of voltage and current patch-clamp recordings are listed in Supplementary Table 5.
Spontaneous APs were recorded immediately after rupture with the EPC10 amplifier (HEKA Elektronik) using the Patchmaster software (HEKA Elektronik). No current was injected into the cells. Signals were filtered with 2.9 and 10 kHz Bessel filters. At least 20 spontaneous APs in a row were analyzed and averaged using LabChart Pro software (ADInstruments) to determine Vmax, APA, and RMP.
For current measurements, all recordings started at least 1 min after rupture with the EPC10 amplifier using the Patchmaster software. Signals were filtered with 2.9 and 10 kHz Bessel filters and recorded with an EPC10 amplifier. Membrane capacitance (Cm) and series resistance (Rs) were compensated automatically after rupture, and currents were normalized to Cm.
The pulse stimulation of sodium current recording was picturized in inset of Figures 3B,D–F. The current–voltage (I–V) relationship was determined by increasing the voltage stepwise from −95 mV to +5 mV in 5 mV steps at a holding potential of −100 mV. Each pulse lasted 50 ms. Steady-state inactivation was measured by a two-pulse protocol at a holding potential of −100 mV. The voltage of the first conditioning pulse (500 ms) was increased stepwise from −120 to −20 mV in 5 mV steps. The second pulse to −20 mV was used to measure INa and normalized to the maximum INa (which was usually after the first pulse). For the measurement of the recovery from inactivation of sodium channels, a two-pulse protocol was used, with an increasing delay between the two pulses ranging from 1 to 165 ms. The first conditioning pulse (1000 ms) is used to induce sodium channel inactivation and the second pulse is used to measure INa generated by sodium channels that recover from inactivation. The INa from the second pulse was normalized to the maximal INa initiated by the conditioning pulse.
To study the effect of milrinone on INa in Ctrl- and BrS-CMs, an automated patch-clamp system (Patchliner Quattro, Nanion Technologies GmbH) was used with low resistance NPC-16 chips and INa was recorded at room temperature. iPSC-CMs were dissociated into single cells for automated patch-clamp analysis, as previously described (Li et al., 2019). The liquid junction potentials and series resistance were not compensated. The holding potential was −100 mV. INa was recorded using voltage steps from −90 to +70 mV for 20 ms in 5 mV steps at an interval of 2000 ms. Currents were sampled at 25 kHz and low-pass-filtered at 2.9 kHz.
The pulse protocols for Ito current were shown in inset of Figures 4B,C,F. The current–voltage (I–V) relationship was determined by increasing the voltage stepwise from −40 to +60 mV in 10 mV steps at a holding potential of −90 mV with a 20 ms prepulse at −35 mV to inactivate sodium channel. Each pulse lasted 600 ms. Steady-state inactivation was measured by using a two-pulse protocol at a holding potential of −90 mV. The voltage of the first conditioning pulse (600 ms) was increased stepwise from −90 to +40 mV in 10 mV steps after a 20 ms prepulse at −35 mV to inactivate sodium channels. The second pulse of 600 ms at +60 mV was used to measure Ito and normalized to the maximum Ito. For the measurement of the recovery from inactivation of Ito, a two-pulse protocol was used, with an increasing delay between the two pulses ranging from 0 to 10,240 ms. The pulse was depolarized from −90 to +40 mV and last for 600 ms. The Ito from the second pulse was normalized to the maximal Ito initiated by the conditioning pulse. The pulse intervals were 15 s for all the stimulations.
The impulse protocol for ICaL was shown in inset of Figures 4H,I,K, as previously described (Luo et al., 2020). The current–voltage (I–V) impulse depolarized from −90 to −80 mV then every 10 mV as step to +70 mV with a duration of 600 ms. Steady-state inactivation was measured by using a two-pulse protocol at a holding potential of −90 mV. The voltage of the first conditioning pulse (600 ms) was increased stepwise from −90 to +10 mV in 10 mV steps. The second pulse of 600 ms at 0 mV was used to measure ICaL and normalized to the maximum ICaL. For the measurement of the recovery from inactivation of ICaL, a two-pulse protocol was used, with an increasing delay between the two pulses ranging from 0 to 1460 ms. The pulse was depolarized from −90 to 0 mV and last for 600 ms. The ICaL from the second pulse was normalized to the maximal ICaL initiated by the conditioning pulse. The pulse intervals were 3 s for all the stimulations.
For the conductance (G/Gmax) calculation, a custom built-in formula and add-in modular in excel were used. The same fit functions were used for analyzing the kinetics of INa and ICaL. Steady-state activation and inactivation curves were fitted with a standard Boltzmann function: Y = 1/[1 + exp((V1/2 – V)/k∞)], where V1/2 is the half-maximal voltage of steady-state (in)activation and k∞ is the slope factor of the voltage dependence of (in)activation. The development of the recovery from inactivation was fitted with single exponential function: Y(t) = Y0 + A(1 – exp(−t/τrec)), where A is the amplitude and τrec the time constant of recovery from inactivation. Persistent INa was measured by integrating the current between 50 and 450 ms of a 1000 ms pulse from a holding potential of −100 to −20 mV. The probability of ICaL window current was calculated from steady-state (in)activation parameters through the following equation as previously described: Probability = 1/{1 + exp[(V1/2,act − V)/kact]} ∗ (1/{1 + exp[(V − V1/2,inact)/kinact]}) (Huang et al., 2011).
The data were analyzed using Clampfit (Axon Instruments), Fitmaster (HEKA Elektronik), and GraphPad Prism (GraphPad Software, Inc.) software.
The digested iPSC-CMs were seeded on Geltrex® -coated MEA chips. The final concentration was approximately 200,000 iPSC-CMs in 50 μL RPMI1640 medium supplemented with 1x B27. The medium was replenished to 1 mL after seeding for around 30 min. In this study, 60MEA200/30iR-Ti-gr chips were used (Multichannel Systems). The medium was changed one day after digestion and thereafter every two days until day 6, which performed the recording. All the recordings were carried out at 35–37°C by using Cardio 2D software (Multichannel Systems). The sample rate was 10,000 Hz. The spontaneous beating frequency and inter-beat interval were automatically generated by Cardio 2D. At least five stable continuous CV values were averaged from spontaneous beatings. For FP metrics [peak-to-peak amplitude (PPA), peak-to-peak duration (PPD), and peak-to-peak slope (PPS)] analysis, the averaged measurements (3 min) of the electrodes (Pin numbers: 22, 33, 44, 55, 66, and 77) were analyzed by Cardio 2D+ software. The rate of arrhythmia was quantified as standard deviation (SD) of the inter-beat interval, which was normalized to averaged inter-beat intervals.
Data are presented as the mean ± standard error of the mean (SEM). Statistical analysis was performed with GraphPad Prism 7 using the two-tailed unpaired Student’s t-test to compare differences between two independent groups, one-way ANOVA followed by Tukey’s post hoc test to compare more than two groups, and the paired Student’s t-test to compare differences between two dependent groups or the two-way ANOVA test for comparison of more groups and conditions. Results were considered statistically significant when the P value was <0.05 (∗P < 0.05; #P < 0.01; §P < 0.001).
We recruited two BrS patients, a 50-year-old male (BrS1) and his biological sister (BrS2) in the study. The ECG recordings of both patients at rest displayed the BrS-typical pattern, a coved-type ST-segment elevation in the right precordial leads V1 and V2 and a more saddleback pattern in V3 followed by a negative T wave (Supplementary Figure 1A). In addition, both patients revealed first-degree AV block. In long-term ECG recordings of patient BrS1, multiple episodes of polymorphic ventricular tachycardia appeared during stair climbing (Supplementary Figure 1B). Both patients received an ICD for the treatment. Genetic screening for possible mutations in several cardiac-specific genes (KCND3, KCNQ1, HERG, KCNE1, and SCN5A) revealed the heterozygous SCN5A point mutation C > A at position c.5435 in both patients (Schulze-Bahr et al., 2003). This non-sense mutation results in a premature termination codon causing the protein truncation at amino acid 1812 (p.S1812X). Genetic tests of their family members identified the mother as an asymptomatic carrier of the mutation, showing first-degree AV block, but no BrS-specific ECG pattern at rest. A brother died from sudden cardiac death at the age of 15. The father and another brother do not carry the mutation and show no BrS phenotype (Supplementary Figure 1C).
We generated iPSCs from these two patients (BrS1 and BrS2) by overexpression of the four Yamanaka factors SOX2, KLF4, OCT4, and c-MYC in somatic cells (Supplementary Figure 2). Three clones per individual were selected for further characterization (BrS1.1-3 and BrS2.1-3). In addition, we included control iPSC lines (Ctrl1.1-3 also known as MSC3-iPS1-3; and Ctrl2.1 and 2 also named as FB2-iPS1 and 2) from two independent healthy donors Ctrl1 and Ctrl2, which were described in our previous studies (Streckfuss-Bomeke et al., 2013; Cyganek et al., 2018). All generated BrS-iPSCs showed the typical morphology similar to Ctrl-iPSCs, were positive for pluripotency markers (Supplementary Figure 2A), showed expression of endogenous pluripotency genes (Supplementary Figure 3A), and revealed normal chromosome numbers in more than 90% of cells (Supplementary Figure 2C). Genomic sequencing confirmed the presence of the SCN5A mutation c.C5435A (p.S1812X) in BrS1- and BrS2-iPSCs and its absence in both Ctrl1- and Ctrl2-iPSCs (Supplementary Figure 2B). Furthermore, Ctrl- and BrS-iPSCs were differentiated into derivatives of all three embryonic germ layers in vitro (Supplementary Figures 3B,C) and developed teratomas in immune-deficient mice (Supplementary Figure 2D). To reduce the phenotypic variability among different cell lines, all three lines of every individual were used for the following studies.
We differentiated Ctrl- and BrS-iPSCs into ventricular CMs using the directed differentiation method, as described previously (Cyganek et al., 2018). This method gave rise to the differentiation efficiency with more than 90% of cells positive for cTnT at day 25 after the initiation of differentiation. No significant difference in cardiac differentiation efficiency was observed between Ctrl- and BrS-iPSCs (data not shown). Immunofluorescence analyses using antibodies against the cardiac-specific proteins cTnT and α-actinin revealed well-organized cross-striations in both 3-month-old BrS- and Ctrl-CMs (Figures 1A–C). To study whether the p.S1812X mutation affected the cellular localization of NaV1.5 in BrS-CMs, we applied an antibody that detects both full-length and truncated proteins of NaV1.5. We found a different distribution of NaV1.5 in Ctrl- and BrS-CMs (Figure 1B). Whereas equal distribution of NaV1.5 along cell membrane was clearly observed in Ctrl-CMs, NaV1.5 appeared irregular and intermittent on the cell surface. Similar to the findings of others (Malan et al., 2011; Davis et al., 2012), NaV1.5 was also expressed in a diffused or a fine granular-like pattern in the cytosol of both Ctrl- and BrS-CMs. In some Ctrl- and BrS-CMs, we observed that NaV1.5 was localized in a striated pattern, mostly overlapped with α-actinin (Figure 1B), as described in adult CMs (Mohler et al., 2004; Xi et al., 2009; Shy et al., 2014). The expression of connexin 43 (Cx43) was detected at the intercellular gap junctions in both Ctrl- and BrS-CMs (Figures 1C,D), indicating a cell-to-cell coupling. However, the co-staining of Cx43 with NaV1.5 revealed the strong overlap and interaction of Cx43 with NaV1.5 in Ctrl-CMs, but less in BrS-CMs (Figure 1D).
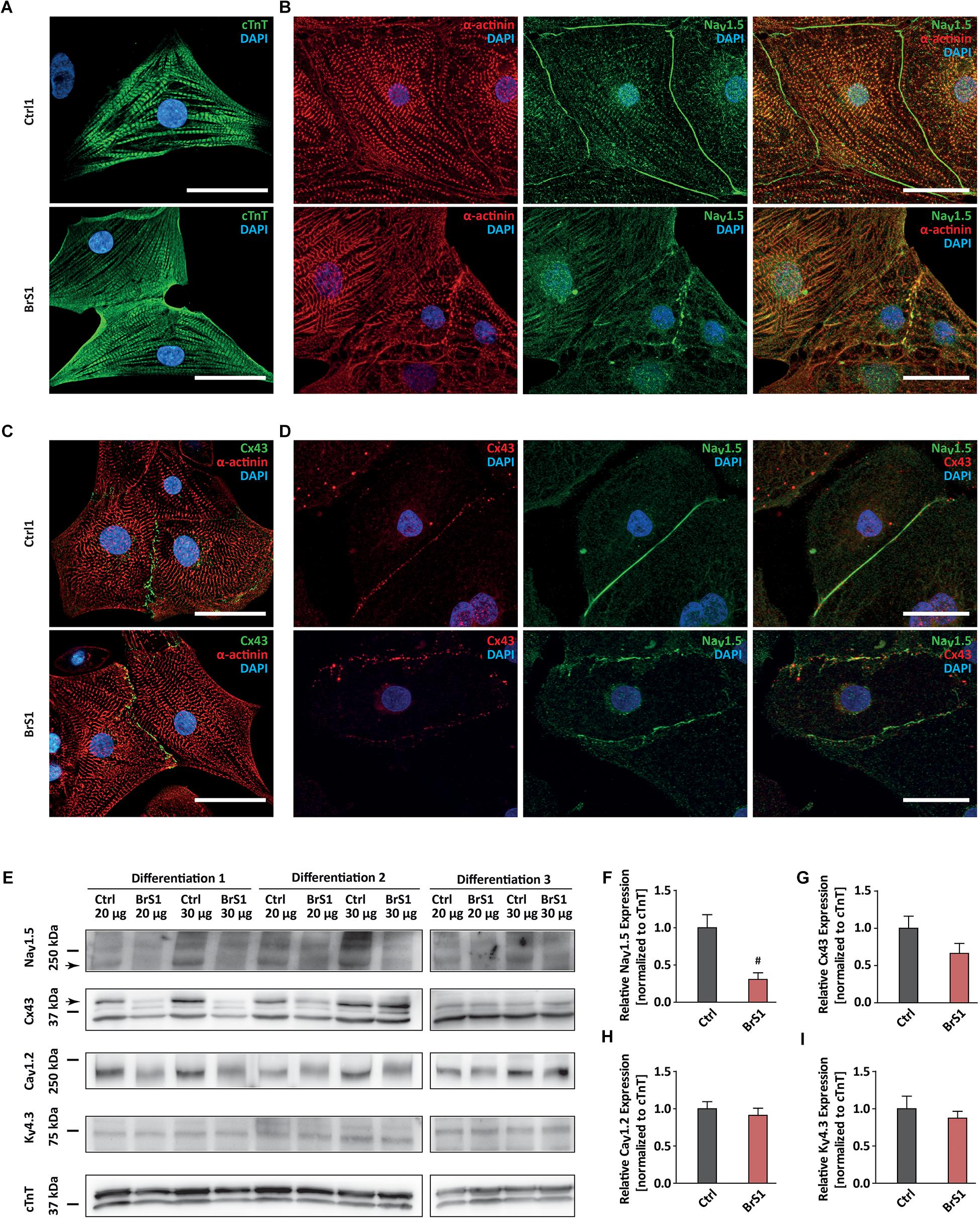
Figure 1. The expression of cardiac-specific proteins in Ctrl- and BrS-CMs. (A) Immunostaining of Ctrl- and BrS-CMs for cardiac markers cTnT. Scale bar, 20 μm. (B) Double-immunostaining of α-actinin and NaV1.5. Scale bar, 20 μm. (C) Double-immunostaining of Cx43 and α-actinin. Scale bar, 20 μm. (D) Double-immunostaining of Ctrl- and BrS-CMs with antibodies against Cx43 and NaV1.5. Scale bar, 20 μm. (E) Shown are original western blots for the decoration of NaV1.5 α subunit, Cx43, CaV1.2, KV4.3, and cTnT. The arrows represent the target bands used for quantification. Quantitative analysis of NaV1.5 α subunit (F), Cx43 (G), CaV1.2 (H), and KV4.3 (I) expression (normalized to cTnT). Data are presented as mean ± SEM. Two-tailed unpaired Student’s t-test was used for statistical analysis: #P < 0.01.
Western blot analyses using an antibody that only detects the full-length protein of NaV1.5 showed a significantly reduced expression of NaV1.5 in BrS-CMs by 69.5% compared to Ctrl-CMs (P = 0.0093; Figures 1E,F). Interestingly, Cx43 expression in BrS-CMs was also reduced by 34%, but not significantly, compared to Ctrl-CMs (P = 0.1443; Figures 1E,G). Furthermore, we assessed the expression of CaV1.2 and KV4.3 in Ctrl- and BrS-CMs and did not observe significant differences between Ctrl- and BrS-CMs (Figures 1E,H,I). Analysis of the allele-specific SCN5A expression in BrS1- and BrS2-CMs revealed the balanced expression of SCN5A between the wild-type and mutated alleles (Supplementary Figure 3D).
Clinically, both BrS patients experience severe arrhythmias and reveal first-degree AV block in the ECGs. To investigate whether the arrhythmic predisposition is exhibited in the monolayer culture of iPSC-CMs, we applied multi-electrode array (MEA) to compare the conduction properties of spontaneous electrical excitations in 3-month-old Ctrl- and BrS-CMs (Figure 2). The differences between Ctrl- and BrS-CMs in the FP metrics PPD (ms), PPA (mV), and PPS (V/s) were distinct after enlargement (Figure 2A). The beating frequency of BrS1-CMs (0.46 ± 0.07 Hz, n = 23) and BrS2-CMs (0.44 ± 0.02 Hz, n = 19) was comparable with Ctrl-CMs (0.45 ± 0.03 Hz, n = 37, Figure 2C). However, BrS-CMs revealed a smaller PPA and PPS but a prolonged PPD when compared to Ctrl-CMs (Figures 2D–F). In Figure 2B, typical excitation patterns and local activation times in reference to the trigger point were shown for both Ctrl2- and BrS1-CMs. The excitation of Ctrl2-CMs spread from one corner (the lower left corner as the trigger point) of the MEA to the other in around 10 ms (Figure 2B) whereas BrS1-CMs needed around 45 ms for the same distance (Figure 2B), indicating pronounced conduction slowing in BrS-CMs when compared to Ctrl-CMs. Quantitative analysis revealed that the CV of Ctrl-CMs was 15.47 ± 1.05 cm/s (n = 37), whereas 4.7 ± 0.44 and 4.4 ± 0.49 cm/s were observed in BrS1-CMs (n = 23, P < 0.001) and BrS2-CMs (n = 19, P < 0.001), respectively (Figure 2G). This dramatic difference can be vividly observed in our recorded movies (Supplementary Videos 1, 2). Moreover, BrS-CMs exhibited increased inter-beat interval variability compared to Ctrl-CMs, as demonstrated by the SD of the inter-beat interval normalized to the mean of the inter-beat intervals (Figure 2H).
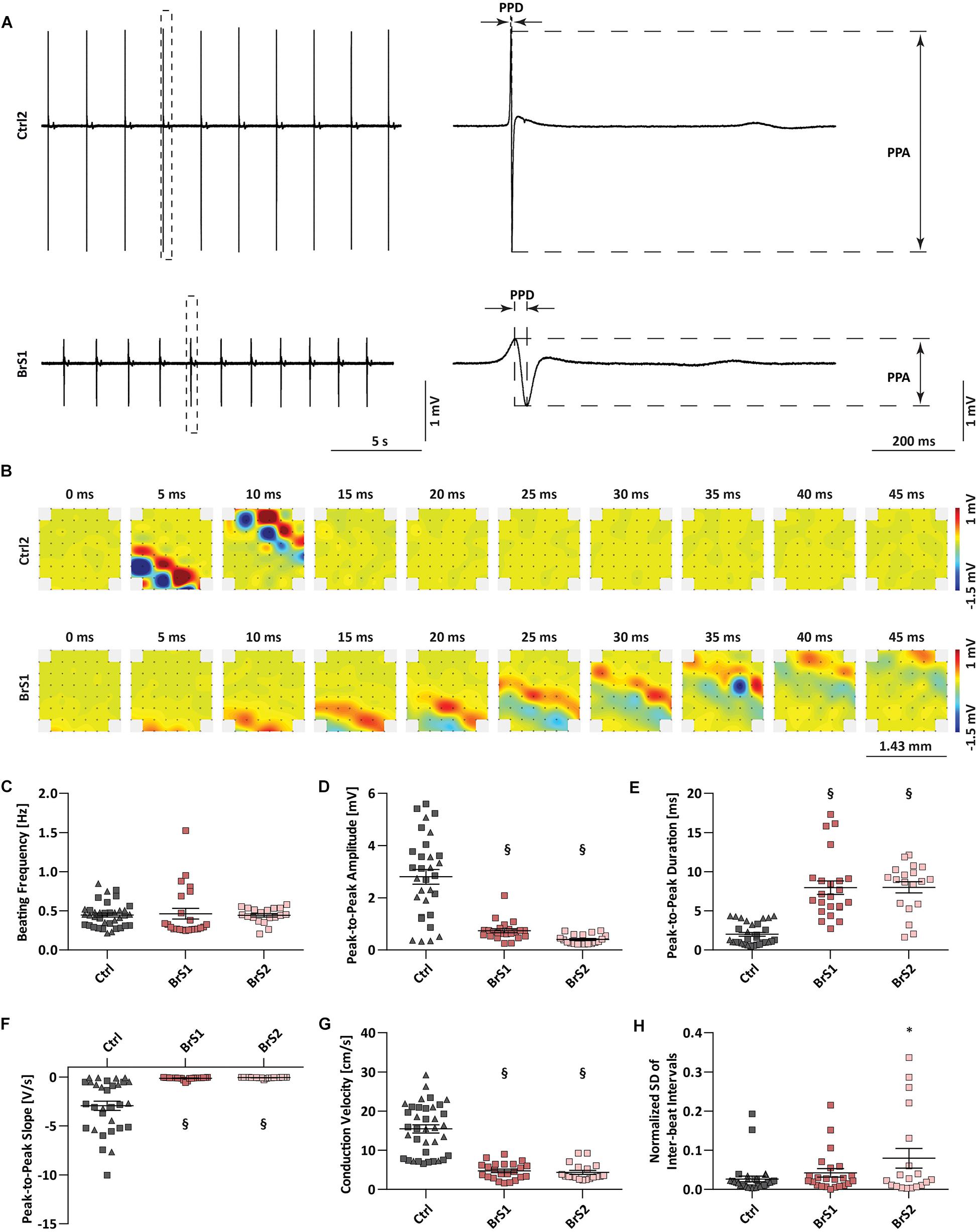
Figure 2. Field potential and conduction velocity of Ctrl- and BrS-CMs. (A) Represented original field potential traces of Ctrl2- and BrS1-CMs were enlarged to show PPA, PPD, and PPS. (B) Represented beats of Ctrl2- and BrS1-CMs were dissected to 10 frames (0, 5, 10, 15, 20, 25, 30, 35, 40, 45 ms), respectively. The width and length of the electrodes distributed area were 1.43 mm. The metrics of MEA measurements including beating frequency (C), PPA (D), PPD (E), PPS (F), CV (G), and normalized SD of inter-beat interval (H) in Ctrl-CMs, BrS1-CMs, and BrS2-CMs. Gray triangle represents Ctrl1 and gray rectangle represents Ctrl2. Data are mean ± SEM. One-way ANOVA followed by Tukey’s post hoc test was used for statistical analysis: ∗P < 0.05, §P < 0.001. PPA, peak-to-peak amplitude; PPD, peak-to-peak duration; PPS, peak-to-peak slope; CV, conduction velocity; SD, standard deviation.
To assess the effect of the SCN5A mutation p.S1812X on the electrophysiological properties of BrS-CMs, INa density and gating properties of sodium channels were compared in 3-month-old BrS-CMs versus Ctrl-CMs using the whole-cell voltage-clamp technique (Figure 3). We observed no differences in the current density, voltage dependence, and gating properties of sodium channels between Ctrl1- and Ctrl2-CMs (Supplementary Figure 4). For a better overview and comparison of the data, we pooled the data of Ctrl1- and Ctrl2-CMs, as shown in Figure 3. Representative examples of INa recordings were shown in Figure 3A. Compared to the Ctrl-CMs, the INa densities were significantly reduced in BrS1- and BrS2-CMs (Figure 3B). The maximal peak INa in Ctrl-CMs (n = 60) at −35 mV showing −38.3 ± 1.8 A/F was reduced by more than 50% in BrS-CMs showing −18.1 ± 1.5 A/F in BrS1-CMs (n = 37, P < 0.001) and −16.7 ± 1.4 A/F in BrS2-CMs (n = 19, P < 0.001). There was no significant difference in the current–voltage relationship and the INa densities between BrS1- and BrS2-CMs (Figure 3B). However, current–voltage relationships normalized to the maximum peak INa showed a shift of peak current toward the positive direction in BrS-CMs compared to Ctrl-CMs (Figure 3C).
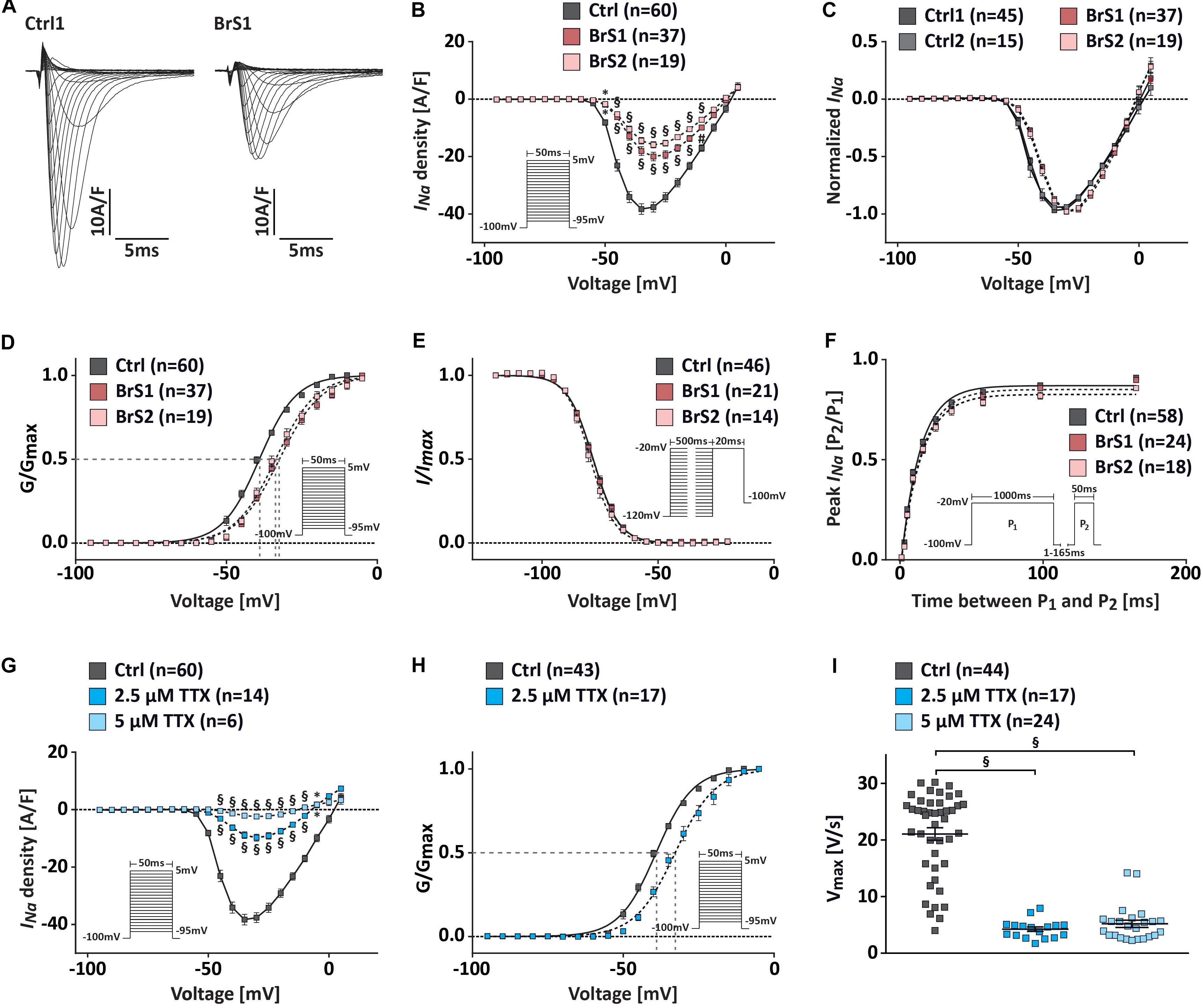
Figure 3. INa characterization in Ctrl- and BrS-CMs. (A) Examples of original INa traces elicited by 5 mV-step depolarization from −95 to 5 mV at a holding potential of −100 mV. Left, Na+ current in one Ctrl1-CM. Right, Na+ current in one BrS1-CM. (B) Average I–V relationships of the INa in Ctrl- and BrS-CMs. The protocol is shown as inset. (C) Current–voltage relationships normalized to the maximum peak INa show a positive shift of the peak INa in BrS1- and BrS2-CMs compared to Ctrl1- and Ctrl2-CMs. No differences were observed between two controls or two BrS. (D) Average of the steady-state voltage dependence of activation of the INa in Ctrl- and BrS-CMs. The protocol is shown as inset. (E) Average of the steady-state voltage dependence of inactivation of INa in Ctrl- and BrS-CMs. The protocol is shown as inset. (F) Average of the recovery from inactivation of INa in Ctrl- and BrS-CMs. The protocol is shown as inset. (G) Average current–voltage relationships of the INa in Ctrl-CMs with and without 2.5 or 5 μM TTX treatment. Protocol is shown as inset. (H) Average steady-state voltage dependence of activation in Ctrl-CMs treated with and without 2.5 μM TTX. Protocol is shown as inset. (I) Vmax in Ctrl-CMs with and without 2.5 or 5 μM TTX treatment. Data are presented as mean ± SEM. Two-way repeated measures ANOVA (B–H) and one-way ANOVA followed by Tukey’s post hoc test were used for statistical analysis: ∗P < 0.05, #P < 0.01, §P < 0.001.
To study the changes in channel kinetics, we compared the steady-state (in)activation and the recovery from inactivation of the sodium channel in BrS-CMs versus Ctrl-CMs. The steady-state activation curve was significantly shifted in a depolarizing direction in BrS-CMs compared to Ctrl-CMs (Figure 3D). V1/2 was −39.1 ± 0.2 mV for Ctrl-CMs (n = 60), −32.4 ± 0.2 mV for BrS1-CMs (n = 37, P < 0.001), and −33.7 ± 0.3 mV for BrS2-CMs (n = 25, P < 0.001). Slope factor k∞ was 6.1 ± 0.2 mV for Ctrl-CMs, significantly smaller than 6.9 ± 0.2 mV for BrS1-CMs (P < 0.001) and 6.7 ± 0.2 mV for BrS2-CMs (P < 0.05) (Supplementary Table 1). The steady-state inactivation of sodium channels in BrS-CMs did not differ significantly from Ctrl-CMs (Figure 3E and Supplementary Table 1). Next, we determined the fraction (A) of sodium channels recovered from an inactivation pulse (1000 ms at −20 mV) and the time constant (τrec) for this recovery. We found a slight but not significant slowing of the recovery from the inactivation in BrS-CMs versus Ctrl-CMs (Figure 3F and Supplementary Table 1). However, no difference in persistent INa was detected in our study (Supplementary Figures 4F,G).
To assess whether the reduced INa density in BrS-CMs is related to the positive shift of the steady-state activation, we treated Ctrl-CMs with the Na+ channel blocker tetrodotoxin (TTX; 2.5 and 5 μM, Figures 3G–I). The treatment with 5 μM TTX resulted in a robustly reduced INa, decreasing maximal INa to 5% of the basal level (Figure 3G). The peak INa density at −35 mV in Ctrl-CMs treated with 2.5 μM TTX (−9.7 ± 1.2 A/F, n = 14) showed a significant reduction compared to non-treated Ctrl-CMs (−38.3 A/F, n = 60) (Figure 3G). We also observed a significant positive shift in the steady-state activation, as demonstrated by the change in V1/2 in Ctrl-CMs treated with 2.5 μM TTX (−32.7 ± 0.3 mV, n = 14) compared to non-treated Ctrl-CMs (−39.1 ± 0.2 mV, n = 60, P < 0.001) (Figure 3H). These data suggest that the reduced INa density in BrS-CMs is associated with the positive shift of the steady-state activation.
To study whether the blockage of Na+ channel by TTX can mimic the FP properties observed in BrS-CMs, we treated Ctrl-CMs with 2.5 μM TTX for 10 min. We found that the application of TTX in Ctrl-CMs led to a significantly reduced PPA, PPS, and CV, but a prolonged PPD (Supplementary Figure 5). In addition, TTX reduced beating frequency (Supplementary Figure 5). Taken together, these data indicate that BrS-CMs recapitulate the conduction abnormalities seen in patients with loss-of-function sodium channel mutations, and the conduction slowing can be evoked in Ctrl-CMs by TTX and is associated with the diminution of INa.
Previous studies reported that in the presence of weak INa, the unopposed outward K+ current Ito cause accentuation of the AP notch in the right ventricular epicardium, resulting in accentuated J wave and ST-segment elevation associated with the Brugada pattern (Szel and Antzelevitch, 2014). To test whether Ito is pivotal in generating the disease phenotype in BrS, we recorded Ito from single iPSC-CMs (Figure 4). The representative Ito traces of Ctrl1-CMs and BrS1-CMs were shown in Figure 4A. Surprisingly, the BrS1-CMs and BrS2-CMs exhibited significantly larger Ito in comparison to Ctrl-CMs (Figure 4B). The maximum peak currents at +60 mV in BrS1-CMs (14.91 ± 2.27 A/F, n = 27, P < 0.001) and BrS2-CMs (12.14 ± 1.42 A/F, n = 38, P < 0.001) were 2.4 and 1.9 times bigger than those in Ctrl-CMs (6.14 ± 0.85 A/F, n = 73) (Figure 4B). When the cells were treated with 4-aminopyridine (4-AP), an Ito inhibitor, 60.9% reduction of the currents was observed in Ctrl-CMs (from 5.29 ± 0.67 to 2.07 ± 0.24 A/F, paired, n = 29; Figure 4D), which is comparable to 65.9% reduction in BrS-CMs (from 13.97 ± 1.8 to 4.8 ± 0.84 A/F, paired, n = 34; Figure 4E), indicating the measured currents are the 4-AP-sensitive Ito. Nevertheless, we did not detect any significant differences in V1/2 for the steady-state inactivation between Ctrl-CMs (−30.41 ± 1.6 mV, n = 14) and BrS-CMs (−30.42 ± 0.72 mV, n = 24) (Figure 4C and Supplementary Table 1). Recovery from the inactivation of Ito in Ctrl-CMs showed fast (fast τrec, 279.7 ± 121.1 ms) and slow phases (slow τrec, 2854 ± 581.7 ms), which were comparable to the fast (fast τrec, 211.9 ± 135.8 ms) and slow phases (slow τrec, 2773 ± 462.6 ms) in BrS-CMs, respectively (Figure 4F and Supplementary Table 1). Using real-time PCR technique, we assessed the expression of Ito-related genes KCND2/3 and KCNA4. It turned out the expressions of KCND2 and KCND3 were significantly higher in BrS1- and BrS2-CMs when compared to Ctrl-CMs (Figures 4G,H). However, no difference regarding the expression of KCNA4 was found between Ctrl- and BrS-CMs (Figure 4I). The TNNT2 expression was normalized to reference gene RPL32 like the other three genes (Figure 4J).
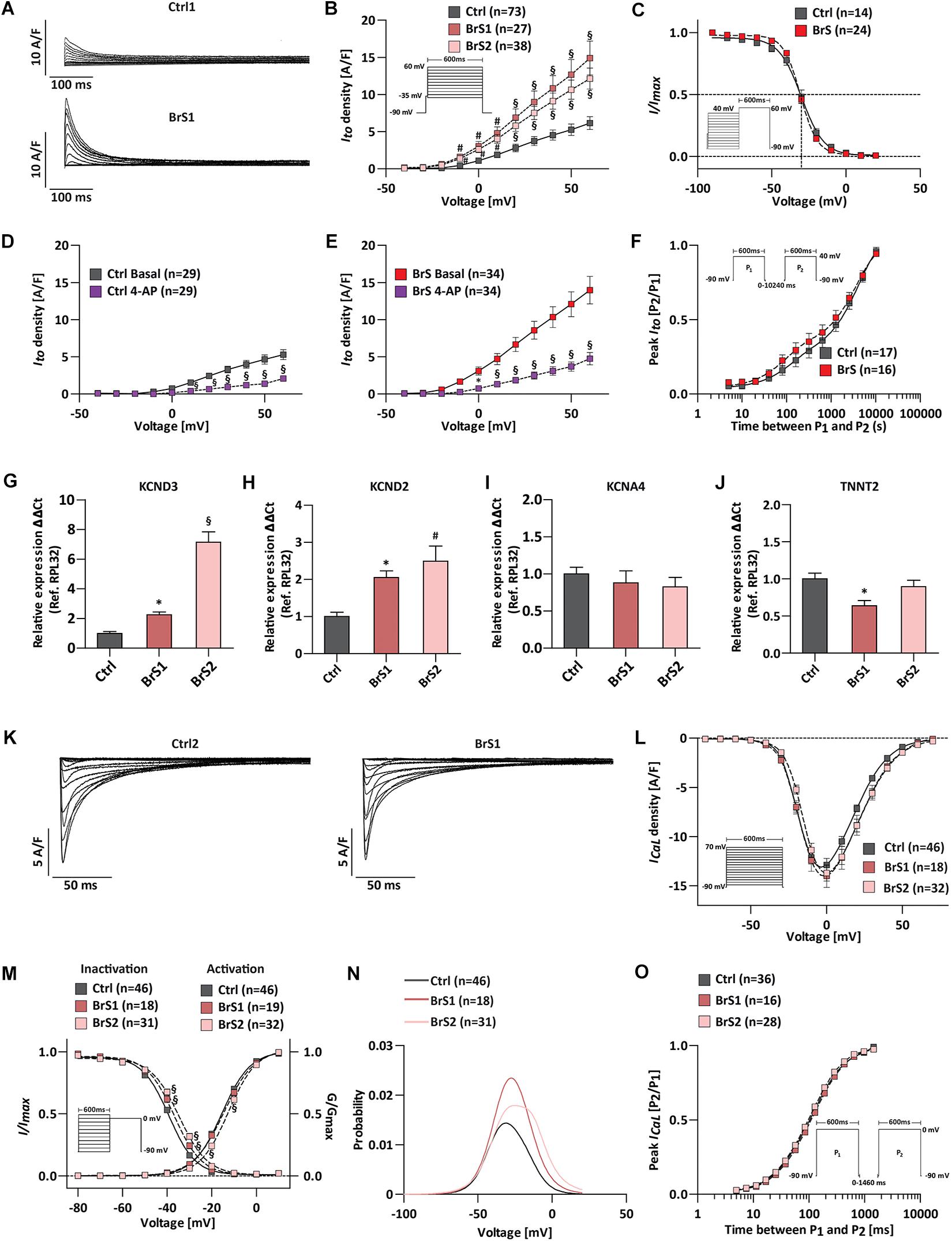
Figure 4. ICaL and Ito characterization of Ctrl- and BrS-CMs. (A) Examples of original Ito traces elicited by 10 mV-step depolarization from −40 to 60 mV at a holding potential of −90 mV. Top, Ito in one Ctrl1-CM. Bottom, Ito in one BrS1-CM. (B) Average I–V relationships of the Ito current in Ctrl- and BrS-CMs. Protocol is shown as inset. (C) Average of the steady-state voltage dependence of inactivation Ito current in Ctrl- and BrS-CMs. The pulse protocol is shown as inset. No significant differences were observed. The Ito current density in Ctrl-CMs (D) and BrS-CMs (E) with and without 4-AP (1 mM) treatment for 1 min. (F) Average of recovery from inactivation of Ito current in Ctrl- and BrS-CMs. The pulse protocol is shown as inset. No significant differences were observed. Expression profile of Ito-related genes: KCND3 (G), KCND2 (H), KCNA4 (I), and TNNT2 (J). Data were presented relative to RPL32 expression (Ctrl: n = 6, BrS1: n = 6, BrS2: n = 6 independent differentiation experiments). (K) Examples of original ICaL traces of a Ctrl2-CM and a BrS1-CM. (L) Average I–V relationships of the ICaL in Ctrl-, BrS1-, and BrS2-CMs. The protocol is shown as inset. (M) The steady-state of activation (G/Gmax) and steady-state of inactivation (I/Imax) are overlaid to show the window current of ICaL. Window currents are the areas under the intersecting current–voltage curves. The impulse for inactivation is shown as inset. (N) The probability being within ICaL window current is plotted. (O) Average of recovery from inactivation of ICaL in Ctrl-, BrS1-, and BrS2-CMs. The pulse protocol is shown as inset. Data are presented as mean ± SEM. Two-way repeated measures ANOVA was used for Ito and ICaL statistical analysis. One-way ANOVA was used for gene expression analyses. ∗P < 0.05, #P < 0.01, §P < 0.001.
We further checked the ICaL and did not detect significant differences in the peak ICaL at 0 mV in BrS1-CMs (−13.99 ± 1.18 A/F, n = 18) or BrS2-CMs (−13.71 ± 0.8 A/F, n = 32) compared to Ctrl-CMs (−12.88 ± 0.66 A/F, n = 46) (Figures 4K,L). The curve of the steady-state activation was shifted to more depolarizing voltage in BrS2-CMs (V1/2 = −12.51 ± 0.50 mV, n = 32, P < 0.001) but not in BrS1-CMs (V1/2 = −14.45 ± 0.84 mV, n = 19, P > 0.05) when compared to Ctrl-CMs (V1/2 = −15.54 ± 0.38 mV, n = 46) (Figure 4M and Supplementary Table 1). The steady-state inactivation curves were also significantly shifted toward more depolarizing voltage for BrS1-CMs (V1/2 = −36.83 ± 0.38 mV, n = 18, P < 0.001) and BrS2-CMs (V1/2 = −34.61 ± 0.40 mV, n = 31, P < 0.001) when compared to Ctrl-CMs (V1/2 = −39.18 ± 0.22 mV, n = 46) (Figure 4M and Supplementary Table 1). Notably, the shifts in the voltages for half-(in)activation V1/2 and a slight increase in slopes of (in)activation k∞ resulted in the significant increases in the window current (area under the curves in Figure 4N). After calculating with a window current probability formula, we found the probabilities of the ICaL window current increased 1.59 times for BrS1-CMs and 1.56 times for BrS2-CMs when compared to Ctrl-CMs (Figure 4N). As window currents are steady-state currents, bigger window currents indicate the increased Ca2+ influx that may contribute to the formation of afterdepolarization and arrhythmia in BrS-CMs. The maximum ratio of peak ICaL [P2/P1] of the recovery from the inactivation for Ctrl-CMs is comparable to those for BrS1-CMs and BrS2-CMs (Figure 4O and Supplementary Table 1). Taken together, our results demonstrate that BrS-CMs exhibit distinct anomalies not only in Na+ channel function but also in K+ and Ca2+ channel functions compared to Ctrl-CMs, which may result in the abnormal AP phenotype.
According to the following stringent criteria, the majority of 3-month-old iPSC-CMs (81%) reveal ventricular-like APs by using the C-clamp mode of patch-clamp technique (Figure 5 and Supplementary Table 2). Ventricular-like APs exhibit a relatively negative resting membrane potential (RMP; <−60 mV), a rapid maximal upstroke velocity (Vmax), a prominent plateau phase, and an AP amplitude (APA) over 95 mV. Atrial-like APs reveal similar properties as ventricular-like APs but lack the plateau phase. Nodal-like APs exhibit a more positive RMP (≥−55 mV), a slow Vmax (≤5 V/s), and an APA < 85 mV. Overall, no significant differences in RMP and APA were observed in BrS-CMs compared to Ctrl-CMs (Supplementary Table 2). Since sodium channels are responsible for the fast upstroke of the atrial and ventricular APs, as expected, the Vmax was significantly slower in spontaneously beating ventricular- and atrial-like BrS1- and BrS2-CMs compared to Ctrl-CMs (Figure 5A). Similarly, Ctrl-CMs after the treatment with TTX showed a slower Vmax (Figure 3I). We also observed a significantly slower Vmax in BrS2-CMs than in BrS1-CMs (Figure 5B), which is consistent with the more pronounced INa reduction in BrS2-CMs than in BrS1-CMs. Additionally, we found a pronounced AP notch in BrS-CMs compared to Ctrl-CMs (Figure 5C), which is in line with the augmented Ito density in BrS-CMs. Notably, we detected prominent irregularities in spontaneous AP recordings in BrS-CMs, including early afterdepolarization (EAD), EAD-triggered activities, delayed afterdepolarization (DAD), and DAD-triggered activities (Figure 5C). We observed the irregularities in 40% of BrS1-CMs (n = 43; P < 0.001, Fisher’s exact test) and 42% of BrS2-CMs (n = 31; P < 0.001) compared to 11% of Ctrl-CMs (n = 74, Figure 5D). Taken together, these results demonstrate that BrS-CMs exhibit distinctly abnormal AP phenotype compared with Ctrl-CMs.
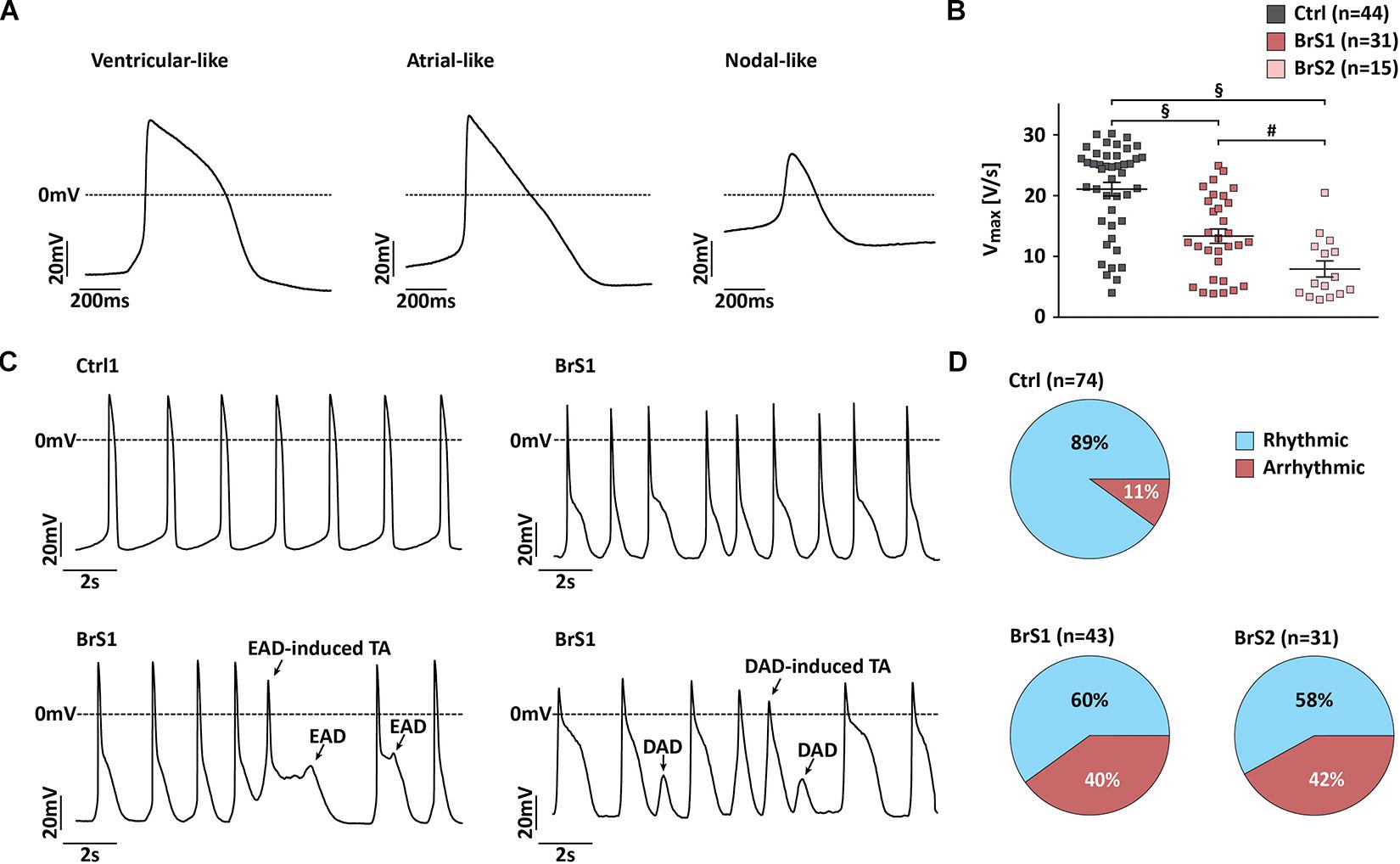
Figure 5. Action potential characterization of Ctrl- and BrS-CMs. (A) Representative traces of spontaneous action potentials (APs) measured in ventricular-, atrial-, and nodal-like CMs. (B) Vmax in ventricular- and atrial-like BrS- and Ctrl-CMs. Data are presented as mean ± SEM. One-way ANOVA followed by Tukey’s post hoc test was used for statistical analysis: #P < 0.01, §P < 0.001. (C) Representative traces of spontaneous AP recordings from Ctrl- and BrS-CMs. (D) Percentage of Ctrl- and BrS-CMs with spontaneously (ar)rhythmic beating. Two-tailed Fisher’s exact test was used for statistical analysis.
Given that two clinically used PDE inhibitors cilostazol and milrinone have been shown to suppress the hypothermia-induced ventricular tachycardia/ventricular fibrillation by reversing the repolarization abnormalities (Gurabi et al., 2014), we tested their effects on our iPSC-CMs at therapeutically relevant concentrations (Szel et al., 2013). After 10 μM cilostazol treatment of BrS-CMs for half an hour, we observed a 50.9% reduction of Ito in BrS1-CMs (from 14.91 to 7.3 A/F) and a 35.4% reduction in BrS2-CMs (from 12.14 to 7.8 A/F) at +60 mV pulse stimulation, whereas only a slight but not significant Ito reduction of 14.4% was observed for the Ctrl-CMs (from 6.14 to 5.25 A/F) (Figures 6A–C). The similar pattern was found when we tested the treatment with 2.5 μM milrinone at the same condition: Ito was significantly reduced from 14.91 to 9.5 A/F in BrS1-CMs (36.3% reduction) and from 12.14 to 7.3 A/F in BrS2-CMs (40% reduction), but only a 10.9% reduction in Ctrl-CMs (from 6.14 to 5.7 A/F) (Figures 6A–C).
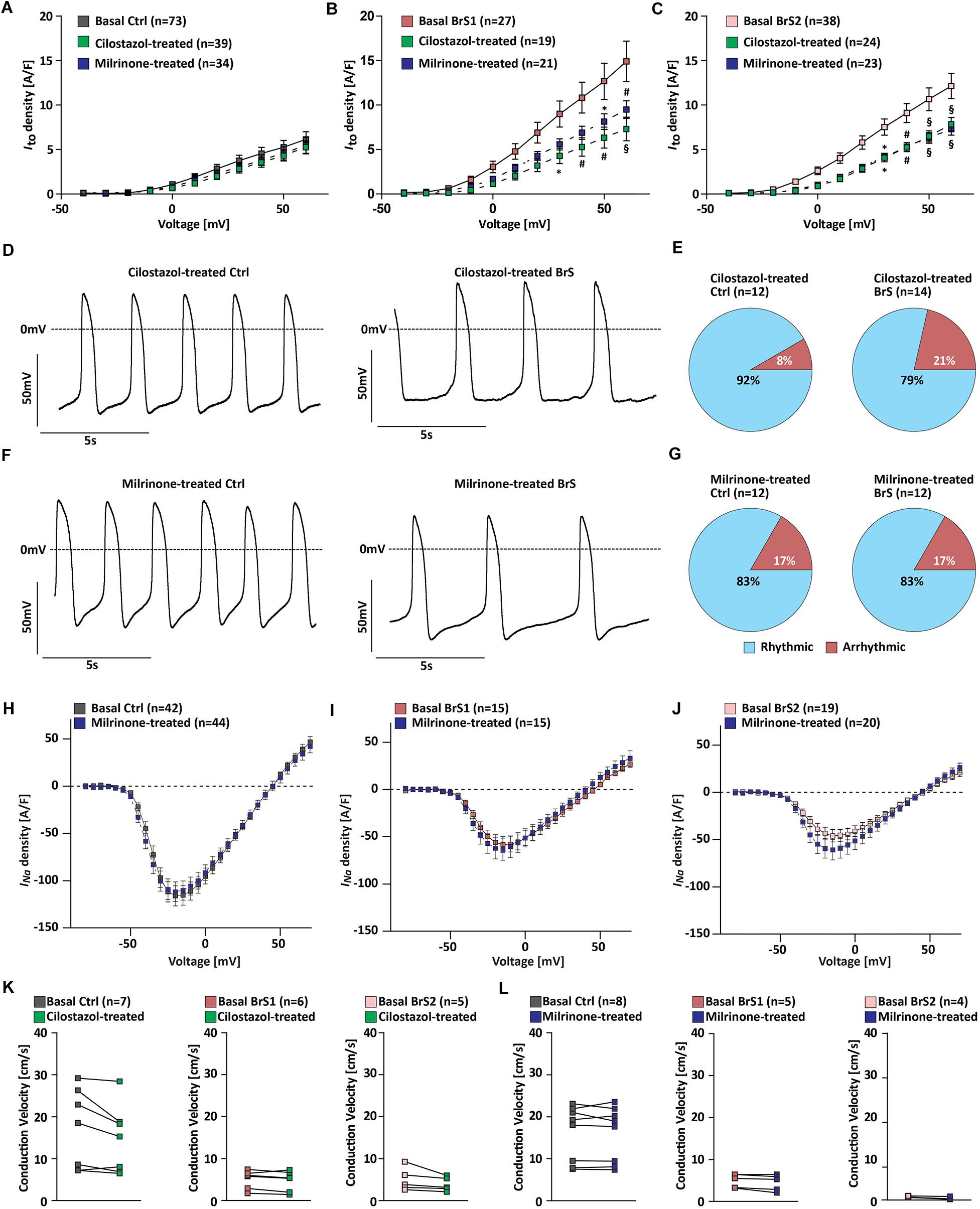
Figure 6. Effects of PDE inhibitor cilostazol or milrinone on electrophysiological properties of Ctrl- and BrS-CMs. Average current–voltage relationships of Ito in Ctrl-CMs (A), BrS1-CMs (B), and BrS2-CMs (C) with and without cilostazol (10 μM) or milrinone (2.5 μM) treatment for half an hour. Data are presented mean ± SEM. (D) Representative original action potential (AP) traces of Ctrl- and BrS-CMs after 10 μM cilostazol treatment. (E) Percentages of Ctrl- and BrS-CMs with rhythmic and arrhythmic APs after 10 μM cilostazol treatment. (F) Representative original AP traces of Ctrl- and BrS-CMs after 2.5 μM milrinone treatment. (G) Percentages of Ctrl- and BrS-CMs with rhythmic and arrhythmic APs after 2.5 μM milrinone treatment. Average I–V curves of INa (automated patch-clamp) in Ctrl-CMs (H), BrS1-CMs (I), and BrS2-CMs (J) with and without 2.5 μM milrinone treatment for half an hour. No statistically significant differences were observed. (K) Quantitative analysis of the conduction velocity in Ctrl-, BrS1-, and BrS2-CMs with and without cilostazol (10 μM) treatment. No statistically significant differences were observed. (L) Quantitative analysis of the conduction velocity in Ctrl-, BrS1, and BrS2-CMs with and without 2.5 μM milrinone treatment. No statistically significant differences were observed. Two-way repeated measures ANOVA (A–C,H–J) and two-tailed paired Student’s t-test (K,L) were used for statistical analysis: ∗P < 0.05, #P < 0.01, §P < 0.001.
Moreover, we assessed whether these two drugs can alleviate the arrhythmic beating degree of BrS-CMs. We found that the treatment of BrS-CMs with cilostazol led to the decrease of cells with arrhythmic beating from 40.5% (n = 74) to 21% (n = 14) (Figures 6D,E). We detected the similar effect of milrinone on BrS-CMs and the arrhythmic beating degree of BrS-CMs dropped to 17% (n = 12) (Figures 6F,G). Both cilostazol and milrinone treatment had no obvious effect on Ctrl-CMs (Figures 6D–G). The most interesting thing is that no EAD- or EAD-triggered activities were observed in these AP recordings.
We further studied the effect of milrinone on INa using the automated patch-clamp system and found no significant changes in INa in both Ctrl- and BrS-CMs after 2.5 μM milrinone treatment for half an hour (Figures 6H–J). In line with these data, there were no significant effects of cilostazol and milrinone on the CV in Ctrl-, BrS1-, and BrS2-CMs (Figures 6K,L), but inter-beat interval variability was slightly reduced (Supplementary Figures 4H–K).
In this study, we applied patient-specific iPSC-CMs carrying the non-sense mutation p.S1812X in SCN5A and investigated the pathophysiological phenotypes and possible pharmacological therapy. Our results demonstrate the ability of patient-specific BrS-CMs to recapitulate the loss-of-function of NaV1.5 resulting from the truncation of SCN5A as evidenced by the significant reduction of peak INa, the significantly slower of the steady-state activation and the 69% reduction of NaV1.5 α-subunit protein expression. In line with these findings, we also observed the decreased AP upstroke velocity and the conduction slowing in BrS-CMs. Notably, we also found the increased Ito and the augmented window current of ICaL in BrS-CMs when compared to Ctrl-CMs. We believe that the impaired interplay between INa, Ito, and ICaL may be involved in the generation of arrhythmia in BrS-CMs. Furthermore, we showed the positive effect of PDE inhibitors cilostazol and milrinone on the reduction of the arrhythmia in BrS-CMs.
In this study, we, for the first time, applied the MEA technology and demonstrated the conduction slowing in BrS-CMs. It is well known that the INa and the electrically coupling degree of gap junctions are the key determinants of cardiac conduction (Kucera et al., 2002). We demonstrated that the INa density was reduced in BrS-CMs, which is consistent with other observations in iPSC-CMs with BrS-related mutations in SCN5A (Liang et al., 2016; Ma et al., 2018). Sodium channels play an important role in phase 0 of the cardiac AP and determine the upstroke velocity (Satin et al., 2004). In BrS-CMs, Vmax was significantly lower compared to Ctrl-CMs, consistent with observed reduction in peak INa density. These findings were also demonstrated in patient-specific iPSC-CMs harboring the SCN5A1795insD mutation (Davis et al., 2012). Our findings that the INa activation curve shifted toward more positive potential with an increased slope factor are in accordance with the later activation of sodium channels resulted from loss-of-function mutations in SCN5A as previously reported for BrS patients (Wilde and Amin, 2018). Additionally, the diminution of INa and the altered gating properties are clearly associated with the expression reduction of full-length NaV1.5 protein and the disrupted localization at the cell surface due to the non-sense mutation p.S1812X in SCN5A in BrS-CMs. Our data are in line with the previous study demonstrating that in HEK293 cells overexpressing the wild-type and the BrS-associated mutant p.V2016M SCN5A together, the surface expression of NaV1.5 is reduced due to its disturbed interaction with SAP97 via a PDZ domain (the last three residues SIV of NaV1.5), with a subsequent decrease of INa (Shy et al., 2014).
In line with previous studies (Kucera et al., 2002; Edokobi and Isom, 2018), we found the co-localization of NaV1.5 and Cx43 at the sites of cell-to-cell appositions in both Ctrl- and BrS-CMs, however, in BrS-CMs the co-localization of NaV1.5 and Cx43 seems altered. Physical and functional interactions of sodium channel complex with gap junction and desmosomal components at intercalated discs in CMs are essential for cardiac excitability and conduction (Delmar, 2012; Cerrone and Delmar, 2014). Previous study demonstrated that loss of the gap junction protein Cx43 resulted in diminished NaV1.5 expression at intercalated discs (Jansen et al., 2012). In our study, we observed a reduced expression of Cx43 in BrS-CMs, this brings up an open question: how the reduced expression of NaV1.5 regulates the Cx43 expression, which should be clarified in future studies. Nonetheless, our data indicate that BrS-CMs with loss-of-function mutation p.S1812X in SCN5A reveal some pathophysiological phenotypes including the conduction slowing, supporting the “depolarization disorder” theory as the underlying mechanism of BrS.
Notably, we show that Ito is present in 3-month-old iPSC-CMs and, to our surprise, the Ito was significantly larger in BrS-CMs than in Ctrl-CMs. However, genetic analysis in both patients did not find mutations in KCND3, KCNQ1, HERG, and KCNE1 (Schulze-Bahr et al., 2003). The 4-AP sensitivity experiments confirmed that the currents we recorded were Ito. Moreover, we observed a prominent AP notch in BrS-CMs. Previous studies showed that Ito is pivotal in generating the disease phenotype in BrS (Di Diego et al., 2002; Giudicessi et al., 2011). The direct relationship between Ito and BrS was first reported by applying the Ito activator NS5806 in isolated canine ventricular wedge preparations. NS5806 could increase phase 1 and notch amplitude of the AP in the epicardium, but not in the endocardium, and accentuate J-wave in the ECG (Calloe et al., 2009). It is well accepted that the orderly sequence of repolarization in the heart is linked to heterogeneous distribution of the fast component of Ito in right ventricle epicardium and endocardium. The highest density of Ito is seen in epicardial CMs, whereas the lowest density is observed in endocardial CMs (Litovsky and Antzelevitch, 1988; Wettwer et al., 1994; Costantini et al., 2005). The changes in repolarization might lead to the so-called phase 2 re-entry and polymorphic ventricular tachycardia. One limitation of our study is that the in vitro BrS-CM model cannot recapitulate the in vivo heterogeneous distribution of Ito in right ventricular epicardium and endocardium. Nonetheless, our data demonstrate that the increased Ito in BrS-CMs is associated to the disease phenotype, and are in line with the previous studies (Delpon et al., 2008; Giudicessi et al., 2011; Perrin et al., 2014). Gain-of-function mutations in KCND3 encoding KV4.3 were found in patients with BrS, leading to the loss of the AP dome in the modified Luo-Rudy II AP model as a result of the increased Ito (Giudicessi et al., 2011). A mutation in KCNE3 encoding a β-subunit of the KV4.3 channel was found in a patient with BrS, which resulted in the reduction of its inhibitory effect on the KV4.3 channel, leading to an increase in Ito (Delpon et al., 2008). Additionally, the mutation p.D612N in KCND2 coding for KV4.2 was identified responsible for the increased J-wave manifestation (Perrin et al., 2014). On the contrary, two recent studies reported some different findings. Different with our increased mRNA expression of KCND2/3, a significant decrease in the gene expression of KCND3 was revealed in CMs derived from iPSCs generated from two BrS patients carrying the SCN5A R620H-R811H mutation and SCN5A Δ1397 mutation, respectively (Liang et al., 2016). Moreover, the Ito current density remained unaltered in BrS iPSC-CMs with SCN5A A226V-R1629X variant (Ma et al., 2018). Although our data showing the increased Ito in BrS-CMs support the “repolarization disorder” theory of BrS, further studies need to investigate how the SCN5A mutation p.S1812X affects the transient-outward potassium channels, especially when we did not observe changes in KV4.3 protein expression in BrS-CMs. We postulate that the augmented Ito may result from the post-translational modification of Kv4.3 channel and impaired protein-protein interaction. It is worth mentioning that there is an inverse relationship between the maximum velocities of depolarization and repolarization, as demonstrated in murine isolated ventricular CMs as well as in HEK293 cells overexpressing both KV4.3 and NaV1.5 (Portero et al., 2018). Therefore, it is of vital importance to further study the entire molecular complexity, in which NaV1.5 is naturally embedded in human CMs and to assess the functional interactions of different ion channels.
Moreover, in our study higher arrhythmic tendencies in BrS-CMs were observed. Spontaneous AP recordings revealed a higher percentage of BrS-CMs with arrhythmogenic events, and FP recordings displayed the increased SD of the inter-beat intervals. Unlike type 3 long QT syndrome, in which most of the SCN5A mutations cause a significantly enhanced persistent INa contributing to the life-threatening ventricular arrhythmias (Malan et al., 2011; Ma et al., 2013), the mutation p.S1812X in our study did not alter the persistent INa in BrS-CMs. To our surprise, the window current probability of ICaL was significantly increased in BrS-CMs compared to Ctrl-CMs, which is associated with the alterations in the steady-state activation and steady-state inactivation of the L-type calcium channels. Previous studies demonstrated that the increased ICaL window current contributed to the EAD formation and EAD-mediated arrhythmias (Madhvani et al., 2015). In iPSC-CMs derived from BrS patients without identified mutations, no significant changes in the steady-state activation and steady-state inactivation of the L-type calcium channels were observed, but the probability of ICaL window current was not calculated (Veerman et al., 2016). Future studies should investigate how the different channels in CMs, including the channels responsible for ICaL, Ito, and INa, co-operate and regulate the arrhythmia in BrS-CMs.
Based on the Ito augment in our BrS-CMs, we speculate that the alleviation of AP arrhythmia could be possibly achieved through the reduction of Ito currents. Cilostazol and milrinone are two oral PDE3 inhibitors, which might be potentially antiarrhythmic drugs for BrS patients (Tsuchiya et al., 2002). Previous study reported that the ventricular fibrillation of a 67-year-old patient with BrS was prevented by oral cilostazol administration (Tsuchiya et al., 2002). In the setting of an arterially perfused right ventricular wedge, cilostazol and milrinone could suppress arrhythmogenesis associated with BrS (Szel et al., 2013; Szel and Antzelevitch, 2014). The effects of cilostazol and milrinone may be related to an elevation of intracellular cyclic AMP concentration via inhibition of PDE activity, and consequently to suppression of Ito and to an increase in ICa, as previously discussed (Tsuchiya et al., 2002; Szel et al., 2013; Szel and Antzelevitch, 2014). Patocskai et al. (2016) demonstrated that both cilostazol and milrinone reduced Ito in canine single left ventricular CMs, and addition of cilostazol or milrinone to the coronary perfusate restored the AP dome at all epicardial sites, reduced epicardial and transmural dispersion of repolarization, decreased J point and ST segment elevation, and terminated all arrhythmic activity in an experimental model of early repolarization syndrome. In our study, inhibition effects of cilostazol and milrinone on Ito and arrhythmogenic events in BrS-CMs suggest a new therapeutic potential to alleviate BrS probably via Ito reduction. Nevertheless, cilostazol and milrinone do not have effects on INa or CV.
By using iPSC-CMs from two BrS patients harboring the SCN5A mutation p.S1812X, we demonstrate here that the mutation results in the reduced INa but augmented Ito and increased ICaL window current probability as well as conduction slowing, indicating that both repolarization and depolarization disorders coexist in BrS-CMs. Our findings indicate that the electrophysiological mechanisms underlying conduction slowing and arrhythmia in BrS-CMs involve not only the NaV1.5 loss-of-function but also an impaired coordination of INa, Ito, and ICaL. Moreover, pharmacological treatments with the PDE3 inhibitors cilostazol and milrinone reduce the Ito and proarrhythmic events in BrS, suggesting their therapeutic potential for BrS patients.
All datasets presented in this study are included in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by the Institutional Ethics Committee of University Medical Center Göttingen (approval number 21/1/11) and of Technical University of Dresden (approval number EK 422092019) and carried out in accordance with the approved guidelines. The patients/participants provided their written informed consent to participate in this study.
WL, MiS, SW, and KG conceived the study and designed experiments. WL, MiS, XJL, MV, CSM, MaS, SC, GW, and S-MH performed experiments and acquired data. WL, MiS, XJL, SW, MV, MaS, and KG analyzed the data. WL, MiS, SW, LC, GH, LSM, AE-A, and KG contributed to the interpretation of the data. WL, MiS, and KG wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) grants to KG (SFB 1002 TP A04, TP S, GU 595/3-1, and project number 288034826–IRTG 2251), and by the Free State of Saxony and the European Union EFRE (SAB project “PhänoKard” and “PhenoCor”) to AE-A and KG, and by the Anschubsfinanzierung from University Medical Center Göttingen to KG and MiS. SW was funded by DFG grants WA 2539/4-1, 5-1, and 7-1. LSM was funded by DFG grants MA 1982/5-1 and 7-1. SW and LSM were also funded by the DFG SFB 1350 grant (project number 387509280, TPA6) and supported by the ReForM C program of the faculty. We acknowledge the support of the Open Access Funding by the Publication Fund of the TU Dresden.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Yvonne Wiegräfe, Yvonne Hintz, Anke Bode, Konstanze Fischer, Jessie Pöche, and Judith Müller for excellent technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.592893/full#supplementary-material
Antzelevitch, C., and Fish, J. M. (2006). Therapy for the Brugada syndrome. Handb. Exp. Pharmacol. 171, 305–330.
Calloe, K., Cordeiro, J. M., Di Diego, J. M., Hansen, R. S., Grunnet, M., Olesen, S. P., et al. (2009). A transient outward potassium current activator recapitulates the electrocardiographic manifestations of Brugada syndrome. Cardiovasc. Res. 81, 686–694. doi: 10.1093/cvr/cvn339
Cerrone, M., and Delmar, M. (2014). Desmosomes and the sodium channel complex: implications for arrhythmogenic cardiomyopathy and Brugada syndrome. Trends Cardiovasc. Med. 24, 184–190. doi: 10.1016/j.tcm.2014.02.001
Costantini, D. L., Arruda, E. P., Agarwal, P., Kim, K. H., Zhu, Y., Zhu, W., et al. (2005). The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell 123, 347–358. doi: 10.1016/j.cell.2005.08.004
Cyganek, L., Tiburcy, M., Sekeres, K., Gerstenberg, K., Bohnenberger, H., Lenz, C., et al. (2018). Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 3:e99941.
Davis, R. P., Casini, S., Van Den Berg, C. W., Hoekstra, M., Remme, C. A., Dambrot, C., et al. (2012). Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 125, 3079–3091. doi: 10.1161/circulationaha.111.066092
Delmar, M. (2012). Connexin43 regulates sodium current; ankyrin-G modulates gap junctions: the intercalated disc exchanger. Cardiovasc. Res. 93, 220–222. doi: 10.1093/cvr/cvr343
Delpon, E., Cordeiro, J. M., Nunez, L., Thomsen, P. E., Guerchicoff, A., Pollevick, G. D., et al. (2008). Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ. Arrhythm. Electrophysiol. 1, 209–218. doi: 10.1161/circep.107.748103
Di Diego, J. M., Cordeiro, J. M., Goodrow, R. J., Fish, J. M., Zygmunt, A. C., Perez, G. J., et al. (2002). Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation 106, 2004–2011. doi: 10.1161/01.cir.0000032002.22105.7a
Edokobi, N., and Isom, L. L. (2018). Voltage-gated sodium channel beta1/beta1B subunits regulate cardiac physiology and pathophysiology. Front. Physiol. 9:351. doi: 10.3389/fphys.2018.00351
Giudicessi, J. R., Ye, D., Tester, D. J., Crotti, L., Mugione, A., Nesterenko, V. V., et al. (2011). Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 8, 1024–1032. doi: 10.1016/j.hrthm.2011.02.021
Gurabi, Z., Koncz, I., Patocskai, B., Nesterenko, V. V., and Antzelevitch, C. (2014). Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ. Arrhythm. Electrophysiol. 7, 134–142. doi: 10.1161/circep.113.000919
Huang, H., Priori, S. G., Napolitano, C., O’leary, M. E., and Chahine, M. (2011). Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of Nav1.5 channels. Am. J. Physiol. Heart Circ. Physiol. 300, H288–H299.
Jansen, J. A., Noorman, M., Musa, H., Stein, M., De Jong, S., Van Der Nagel, R., et al. (2012). Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm 9, 600–607. doi: 10.1016/j.hrthm.2011.11.025
Kapplinger, J. D., Tester, D. J., Alders, M., Benito, B., Berthet, M., Brugada, J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 7, 33–46. doi: 10.1016/j.hrthm.2009.09.069
Kucera, J. P., Rohr, S., and Rudy, Y. (2002). Localization of sodium channels in intercalated disks modulates cardiac conduction. Circ. Res. 91, 1176–1182. doi: 10.1161/01.res.0000046237.54156.0a
Li, W., Luo, X., Ulbricht, Y., Wagner, M., Piorkowski, C., El-Armouche, A., et al. (2019). Establishment of an automated patch-clamp platform for electrophysiological and pharmacological evaluation of hiPSC-CMs. Stem Cell Res. 41:101662. doi: 10.1016/j.scr.2019.101662
Liang, P., Sallam, K., Wu, H., Li, Y., Itzhaki, I., Garg, P., et al. (2016). Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of Brugada syndrome. J. Am. Coll. Cardiol. 68, 2086–2096. doi: 10.1016/j.jacc.2016.07.779
Litovsky, S. H., and Antzelevitch, C. (1988). Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ. Res. 62, 116–126. doi: 10.1161/01.res.62.1.116
Luo, X., Li, W., Kunzel, K., Henze, S., Cyganek, L., Strano, A., et al. (2020). IP3R-mediated compensatory mechanism for calcium handling in human induced pluripotent stem cell-derived cardiomyocytes with cardiac ryanodine receptor deficiency. Front. Cell. Dev. Biol. 8:772. doi: 10.3389/fcell.2020.00772
Ma, D., Liu, Z., Loh, L. J., Zhao, Y., Li, G., Liew, R., et al. (2018). Identification of an INa-dependent and Ito-mediated proarrhythmic mechanism in cardiomyocytes derived from pluripotent stem cells of a Brugada syndrome patient. Sci. Rep. 8:11246.
Ma, D., Wei, H., Zhao, Y., Lu, J., Li, G., Sahib, N. B., et al. (2013). Modeling type 3 long QT syndrome with cardiomyocytes derived from patient-specific induced pluripotent stem cells. Int. J. Cardiol. 168, 5277–5286. doi: 10.1016/j.ijcard.2013.08.015
Madhvani, R. V., Angelini, M., Xie, Y., Pantazis, A., Suriany, S., Borgstrom, N. P., et al. (2015). Targeting the late component of the cardiac L-type Ca2+ current to suppress early afterdepolarizations. J. Gen. Physiol. 145, 395–404. doi: 10.1085/jgp.201411288
Malan, D., Friedrichs, S., Fleischmann, B. K., and Sasse, P. (2011). Cardiomyocytes obtained from induced pluripotent stem cells with long-QT syndrome 3 recapitulate typical disease-specific features in vitro. Circ. Res. 109, 841–847. doi: 10.1161/circresaha.111.243139
Mohler, P. J., Rivolta, I., Napolitano, C., Lemaillet, G., Lambert, S., Priori, S. G., et al. (2004). Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 101, 17533–17538. doi: 10.1073/pnas.0403711101
Nademanee, K., Raju, H., De Noronha, S. V., Papadakis, M., Robinson, L., Rothery, S., et al. (2015). Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J. Am. Coll. Cardiol. 66, 1976–1986. doi: 10.1016/j.jacc.2015.08.862
Nerbonne, J. M., and Kass, R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. doi: 10.1152/physrev.00002.2005
Patocskai, B., Barajas-Martinez, H., Hu, D., Gurabi, Z., Koncz, I., and Antzelevitch, C. (2016). Cellular and ionic mechanisms underlying the effects of cilostazol, milrinone, and isoproterenol to suppress arrhythmogenesis in an experimental model of early repolarization syndrome. Heart Rhythm 13, 1326–1334. doi: 10.1016/j.hrthm.2016.01.024
Patocskai, B., Yoon, N., and Antzelevitch, C. (2017). Mechanisms underlying epicardial radiofrequency ablation to suppress arrhythmogenesis in experimental models of Brugada syndrome. JACC Clin. Electrophysiol. 3, 353–363. doi: 10.1016/j.jacep.2016.10.011
Perrin, M. J., Adler, A., Green, S., Al-Zoughool, F., Doroshenko, P., Orr, N., et al. (2014). Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death. Circ. Cardiovasc. Genet. 7, 782–789. doi: 10.1161/circgenetics.114.000623
Portero, V., Wilders, R., Casini, S., Charpentier, F., Verkerk, A. O., and Remme, C. A. (2018). KV4.3 expression modulates NaV1.5 sodium current. Front. Physiol. 9:178. doi: 10.3389/fphys.2018.00178
Satin, J., Kehat, I., Caspi, O., Huber, I., Arbel, G., Itzhaki, I., et al. (2004). Mechanism of spontaneous excitability in human embryonic stem cell derived cardiomyocytes. J. Physiol. 559, 479–496. doi: 10.1113/jphysiol.2004.068213
Schulze-Bahr, E., Eckardt, L., Breithardt, G., Seidl, K., Wichter, T., Wolpert, C., et al. (2003). Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum. Mutat. 21, 651–652. doi: 10.1002/humu.9144
Shy, D., Gillet, L., Ogrodnik, J., Albesa, M., Verkerk, A. O., Wolswinkel, R., et al. (2014). PDZ domain-binding motif regulates cardiomyocyte compartment-specific NaV1.5 channel expression and function. Circulation 130, 147–160. doi: 10.1161/circulationaha.113.007852
Streckfuss-Bomeke, K., Wolf, F., Azizian, A., Stauske, M., Tiburcy, M., Wagner, S., et al. (2013). Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 34, 2618–2629. doi: 10.1093/eurheartj/ehs203
Szel, T., and Antzelevitch, C. (2014). Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J. Am. Coll. Cardiol. 63, 2037–2045. doi: 10.1016/j.jacc.2014.01.067
Szel, T., Koncz, I., and Antzelevitch, C. (2013). Cellular mechanisms underlying the effects of milrinone and cilostazol to suppress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm 10, 1720–1727. doi: 10.1016/j.hrthm.2013.07.047
Tsuchiya, T., Ashikaga, K., Honda, T., and Arita, M. (2002). Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. J. Cardiovasc. Electrophysiol. 13, 698–701. doi: 10.1046/j.1540-8167.2002.00698.x
Veerman, C. C., Mengarelli, I., Guan, K., Stauske, M., Barc, J., Tan, H. L., et al. (2016). hiPSC-derived cardiomyocytes from Brugada syndrome patients without identified mutations do not exhibit clear cellular electrophysiological abnormalities. Sci. Rep. 6:30967.
Wagner, S., Dybkova, N., Rasenack, E. C., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/jci26620
Wettwer, E., Amos, G. J., Posival, H., and Ravens, U. (1994). Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ. Res. 75, 473–482. doi: 10.1161/01.res.75.3.473
Wilde, A. A., Postema, P. G., Di Diego, J. M., Viskin, S., Morita, H., Fish, J. M., et al. (2010). The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J. Mol. Cell. Cardiol. 49, 543–553.
Wilde, A. A. M., and Amin, A. S. (2018). Clinical spectrum of SCN5A mutations: long qt syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 4, 569–579.
Keywords: Brugada syndrome, disease modeling, induced pluripotent stem cells, SCN5A mutation, depolarization, repolarization
Citation: Li W, Stauske M, Luo XJ, Wagner S, Vollrath M, Mehnert CS, Schubert M, Cyganek L, Chen S, Hasheminasab S-M, Wulf G, El-Armouche A, Maier LS, Hasenfuss G and Guan K (2020) Disease Phenotypes and Mechanisms of iPSC-Derived Cardiomyocytes From Brugada Syndrome Patients With a Loss-of-Function SCN5A Mutation. Front. Cell Dev. Biol. 8:592893. doi: 10.3389/fcell.2020.592893
Received: 08 August 2020; Accepted: 01 October 2020;
Published: 22 October 2020.
Edited by:
Mingtao Zhao, Nationwide Children’s Hospital, United StatesCopyright © 2020 Li, Stauske, Luo, Wagner, Vollrath, Mehnert, Schubert, Cyganek, Chen, Hasheminasab, Wulf, El-Armouche, Maier, Hasenfuss and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaomei Guan, a2FvbWVpLmd1YW5AdHUtZHJlc2Rlbi5kZQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.