 Yanping Li1
Yanping Li1 David Z. Qian
David Z. Qian Mu-Shui Dai
Mu-Shui Dai- 1Department of Molecular and Medical Genetics, School of Medicine, Portland, OR, United States
- 2The OHSU Knight Cancer Institute, Oregon Health and Science University, Portland, OR, United States
The transcription factor c-MYC (MYC thereafter) is a global regulator of gene expression. It is overexpressed or deregulated in human cancers of diverse origins and plays a key role in the development of cancers. Hypoxia-inducible factors (HIFs), a central regulator for cells to adapt to low cellular oxygen levels, is also often overexpressed and activated in many human cancers. HIF mediates the primary transcriptional response of a wide range of genes in response to hypoxia. Earlier studies focused on the inhibition of MYC by HIF during hypoxia, when MYC is expressed at physiological level, to help cells survive under low oxygen conditions. Emerging evidence suggests that MYC and HIF also cooperate to promote cancer cell growth and progression. This review will summarize the current understanding of the complex molecular interplay between MYC and HIF.
Introduction
Cancer cells undergo significant metabolic changes to sustain the rapid cell proliferation, adapt to environmental challenges such as hypoxia, and promote invasion and metastasis. While HIF signaling is essential for normal cell adaptation to oxygen homeostasis, it also plays a key role in the growth of solid tumors, which inevitably contain poorly vascularized regions due to rapid tumor cell proliferation (Brahimi-Horn and Pouyssegur, 2009; Rankin and Giaccia, 2016; Lee et al., 2020). Tumor hypoxia is typically correlated with more aggressive phenotype and poor prognosis partly due to its contribution to therapeutic resistance and tumor cell invasion and metastasis via activating various cell survival pathways. MYC signaling also plays pivotal roles in regulating cancer cell metabolism and vasculogenesis (Baudino et al., 2002; Dang, 2012b; Stine et al., 2015). In this review, we focus on the current understanding of the molecular interplay between MYC and HIF in cancer cell metabolism, growth and progression as well as its potential implication in cancer therapy.
The MYC oncoprotein is a master regulator of transcription that activates or represses gene expression to coordinate diverse cellular processes, including cellular division, differentiation, apoptosis, angiogenesis, DNA replication, RNA processing, metabolism, and ribosome biogenesis (Bretones et al., 2015; Kress et al., 2015; Baluapuri et al., 2020). MYC heterodimerizes with the MAX protein, via its C-terminal basic helix-loop-helix-leucine zipper (bHLH-LZ) domain, to bind to the consensus CACGTG (E-box) elements on chromatin. At the N-terminus of MYC lies its transactivation domain (TAD) that contains the conserved Myc box I (MBI) and MBII essential for both transcriptional activation and repression (Farrell and Sears, 2014; Tu et al., 2015; Baluapuri et al., 2020). Two important phosphorylation sites, Threonine 58 (T58) and Serine 62 (S62), within MBI are critical for the regulation of MYC stability and activity in response to cell growth signals (Farrell and Sears, 2014; Chen Y. et al., 2019). The central region of MYC contains the MBIIIa, MBIIIb, and MBIV which are important for transcriptional activity and pro-apoptotic activity (Cowling et al., 2006; Kurland and Tansey, 2008; Thomas et al., 2015, 2016). For example, MBIIIa interacts with the histone deacetylase HDAC3 to suppress transcription and MBIIIb associates with WDR5 to facilitate H3K4 methylation and MYC recruitment to chromatin (Thomas et al., 2015), whereas MBIV association with transcriptional coregulator host cell factor-1 (HCF-1) is critical for MYC-driven tumorigenesis (Thomas et al., 2016; Figure 1A). Recent genome-wide and gene specific studies have revealed several emerging models for MYC function, including specific-gene regulation, global gene activation, and gene-specific affinity models (Kress et al., 2015; Baluapuri et al., 2020). Together, MYC controls the expression of genes involved in almost all aspects of tumor hallmarks. Consistent with its roles in oncogenesis, MYC is frequently overexpressed in human cancers via various mechanisms, including gene amplification, chromosomal translocation, increased MYC translation, deregulated MYC protein stabilization, or constitutive activation of upstream pathways such as Wnt, Notch, Hedgehog signaling (Nesbit et al., 1999; Dang, 2012b; Kress et al., 2015). High levels of MYC expression is associated with poor patient outcomes. Transgenic overexpression of MYC induces tumorigenesis in mice and inactivation of MYC in MYC-driven tumors causes tumor regression in multiple tumor models (Gabay et al., 2014), further demonstrating the key role for MYC in tumorigenesis.
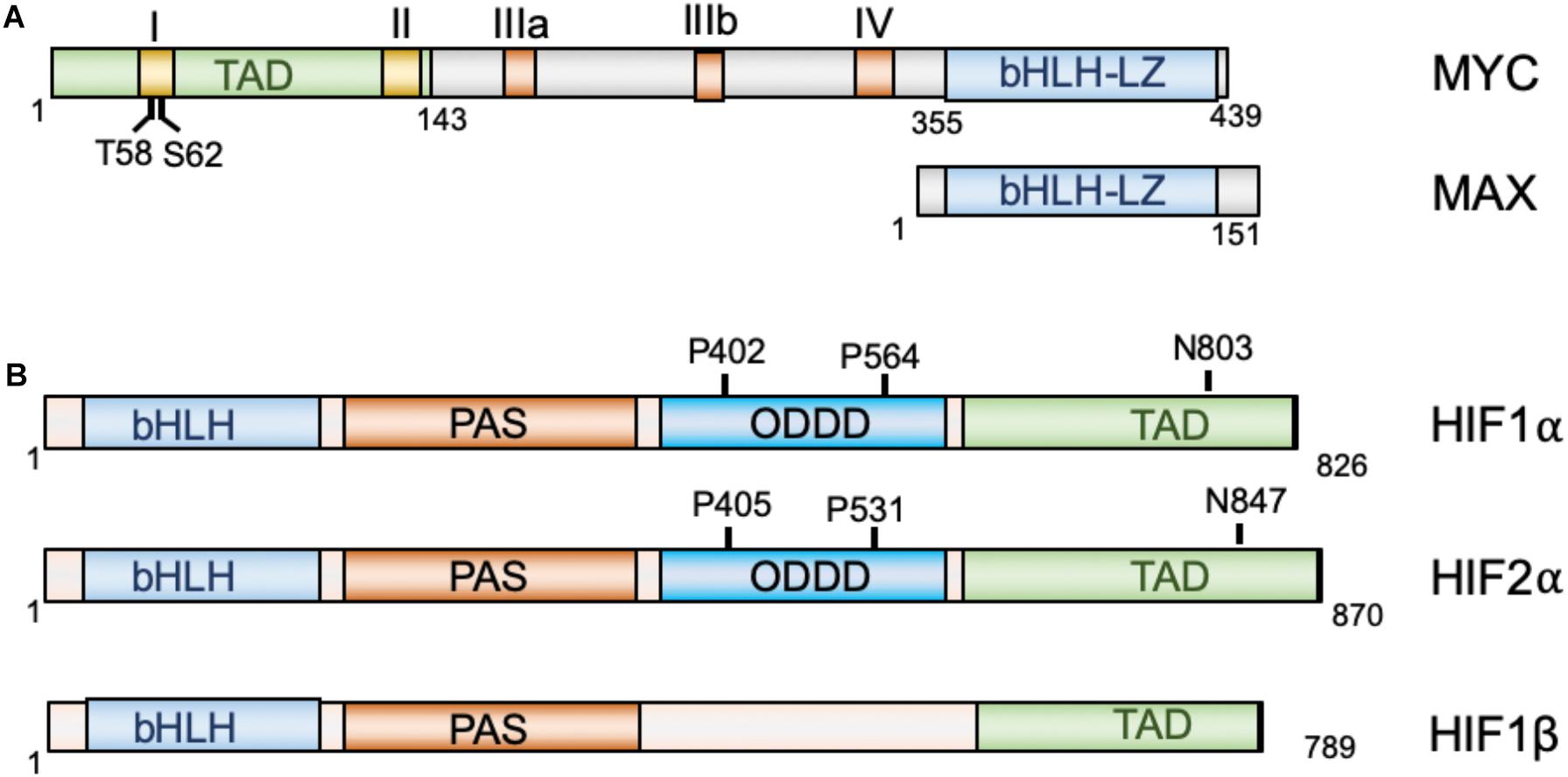
Figure 1. Diagrams of MYC and MAX (A) and HIF proteins (B). MYC and Max dimerize via their bHLH-LZ domains, whereas HIF1α and HIF2α dimerize with HIF1β via their N-terminal bHLH domains. TAD, transactivation domain; bHLH, basic helix-loop-helix; LZ, leucine zipper; PAS, Per-Arnt-Sim homology; ODDD, oxygen-dependent degradation domain.
The hypoxia-inducible factors (HIFs) are transcription factors that regulate the transcription of a large array of genes involved in metabolism, cell survival, proliferation, migration, invasion, angiogenesis, immune evasion and resistance to therapies in response to hypoxia (Brahimi-Horn and Pouyssegur, 2009; Rankin and Giaccia, 2016; Lee et al., 2020) and are recognized as the master regulators of oxygen homeostasis (Semenza, 2014). HIFs are heterodimers consisting of an oxygen-regulated α-subunit and a non-oxygen-regulated stable β subunit. HIFα consists of three isoforms: HIF1α, HIF2α, and HIF3α (Brahimi-Horn and Pouyssegur, 2009; Semenza, 2014). Both HIF α and β subunits contain a basic helix-loop-helix (bHLH) domain and the Per-Arnt-Sim homology (PAS) domain, which accounts for the dimerization between the α and β subunits (Wang et al., 1995). HIF1α and HIF2α dimerizes with HIF1β to bind to the 5′-RCGTG-3′ (R = A or G) core sequence of the hypoxia-response elements (HREs) on DNA to regulate gene expression (Semenza et al., 1996). The C-terminal half of HIFα contains the TAD that is critical for the recruitment of the p300/CREB-binding protein (CBP) coactivators (Arany et al., 1996). In the middle region of HIFα lies the oxygen-dependent degradation domain (ODDD), which regulates HIFα protein stability (Figure 1B). Under normoxic conditions, two proline residues at the ODDD undergo hydroxylation by prolyl-hydroxylase domain enzymes (PHD1-3) encoded by the EGLN1-3 genes (Semenza, 2014; Ivan and Kaelin, 2017; Semenza, 2020). Hydroxylation of proline residues enables recognition of HIFα by the E3 ligase, von Hippel-Lindau protein (pVHL), thereby promoting ubiquitination and proteasome-mediated degradation of HIFα (Ivan et al., 2001; Jaakkola et al., 2001; Yu et al., 2001). In addition, factor inhibiting HIF1 (FIH1) inhibits HIFα activity by hydroxylating an asparaginyl residue at the C-terminal TAD of the HIF1α and HIF2α to block the binding of p300/CBP coactivators (Lando et al., 2002a,b). PHD utilizes O2, ferrous iron, and α-ketoglutarate (2-OG) as substrates (Markolovic et al., 2015). In response to hypoxia, the activity of PHD is first inhibited due to lack of sufficient oxygen, resulting in HIFα escaping the hydroxylation and consequent stabilization. The enzymatic activity of FIH1 is also dependent on substrates oxygen and 2-OG and ferrous iron, and thus its activity is also inhibited in response to hypoxia. Consequently, the stabilized HIFα translocates into the nucleus, dimerizes with HIFβ and activates the transcription of hundred genes involved in cellular adaptation and survival under hypoxic conditions, including genes involved in glycolysis, erythropoietin (Epo) and VEGF, etc. While HIF1α and HIF2α cooperatively regulate genes involved in angiogenesis and metastasis (Gao et al., 2009), HIF1α preferentially induces genes involved in glycolysis (Hu et al., 2003; Wang et al., 2005) and HIF2α stimulates genes important for tumor growth and metastasis, amino acid and lipid metabolism, cell cycle progression and maintaining stem cell pluripotency (Hu et al., 2003; Covello et al., 2006; Gruber et al., 2007; Xia et al., 2009). Consistent with their role in metabolic reprogramming, angiogenesis, metastasis, therapeutic resistance (Rankin and Giaccia, 2016; Lv et al., 2017), HIFs are frequently overexpressed in various cancer cells (Zhong et al., 1999; Semenza, 2003; Soni and Padwad, 2017).
The Regulation of MYC by HIF
In physiological normoxic conditions, MYC and HIF are expressed at low levels due to rapid protein degradation. Yet, both are essential for normal cell homeostasis and animal development. Homozygous deletion of c-MYC is embryonic lethal at E10.5 as it is essential for normal cell growth and proliferation (Davis et al., 1993). Homozygous deletion of HIF1α is also embryonic lethal at E10.5 due to defects in circulatory system and hematopoiesis (Iyer et al., 1998; Compernolle et al., 2003; Yoon et al., 2006). HIF2α null mice mostly die by E13.5 but sometimes survive only until birth due to impaired lung maturation, bradycardia, vascular defects and mitochondrial dysfunction (Tian et al., 1998; Peng et al., 2000; Compernolle et al., 2002). It is not clear whether MYC functionally interacts with HIFs in these development events.
Under hypoxia, MYC activity is inhibited by HIF1α as an adaptive response that promotes cell survival under low oxygen conditions. Evidence suggests several mechanisms underlying this inhibition (Figure 2A). First, HIF1α can antagonize MYC transcriptional activity at MYC target genes by interfering with MYC binding to protein partners. HIF1α binds to MAX and disrupts MYC/MAX complexes, leading to reduced cyclin D2 expression, induction of p21, and G1-phase arrest (Gordan et al., 2007). The MAD family of proteins MAD and MXI1 compete with MYC for binding to MAX, thus inhibiting MYC activity (Conacci-Sorrell et al., 2014). HIF1-dependent induction of MXI1 under hypoxia directly represses MYC target genes that are involved in mitochondrial biogenesis, such as PGC1β (Zhang et al., 2007), or apoptosis, for example, ornithine decarboxylase (ODC) (Corn et al., 2005). Second, HIF1α was also shown to directly inhibit MYC transcriptional activity by DNA-binding site competition. It was shown that HIF1α displaces MYC binding from the p21 promoter and upregulates the expression of p21 (Koshiji et al., 2004). HIF1α also competes against MYC for binding to SP1, a known coactivator of MYC, at the promoters of MYC target genes, such as MSH2, MSH6, and NBS1, which encode DNA repair proteins (Koshiji et al., 2005; To et al., 2006), and the recently reported E−type prostanoid (EP4) receptors (Seira et al., 2018). Third, several studies showed that HIF promotes proteasomal degradation of MYC under chronic hypoxia condition (Zhang et al., 2007; Wong et al., 2013). HIF is required for the hypoxia induced degradation of MYC depending on the cell type and system used (Zhang et al., 2007; Li et al., 2009; Wong et al., 2013; Zarrabi et al., 2017). In addition, an early study also showed that HIF1α physically interacts with MYC through its N-terminus containing bHLA/PAS domains, which is sufficient to induce p21 expression and G1 arrest (Koshiji et al., 2004). It remains to be determined whether such physical interaction directly inhibits MYC activity.
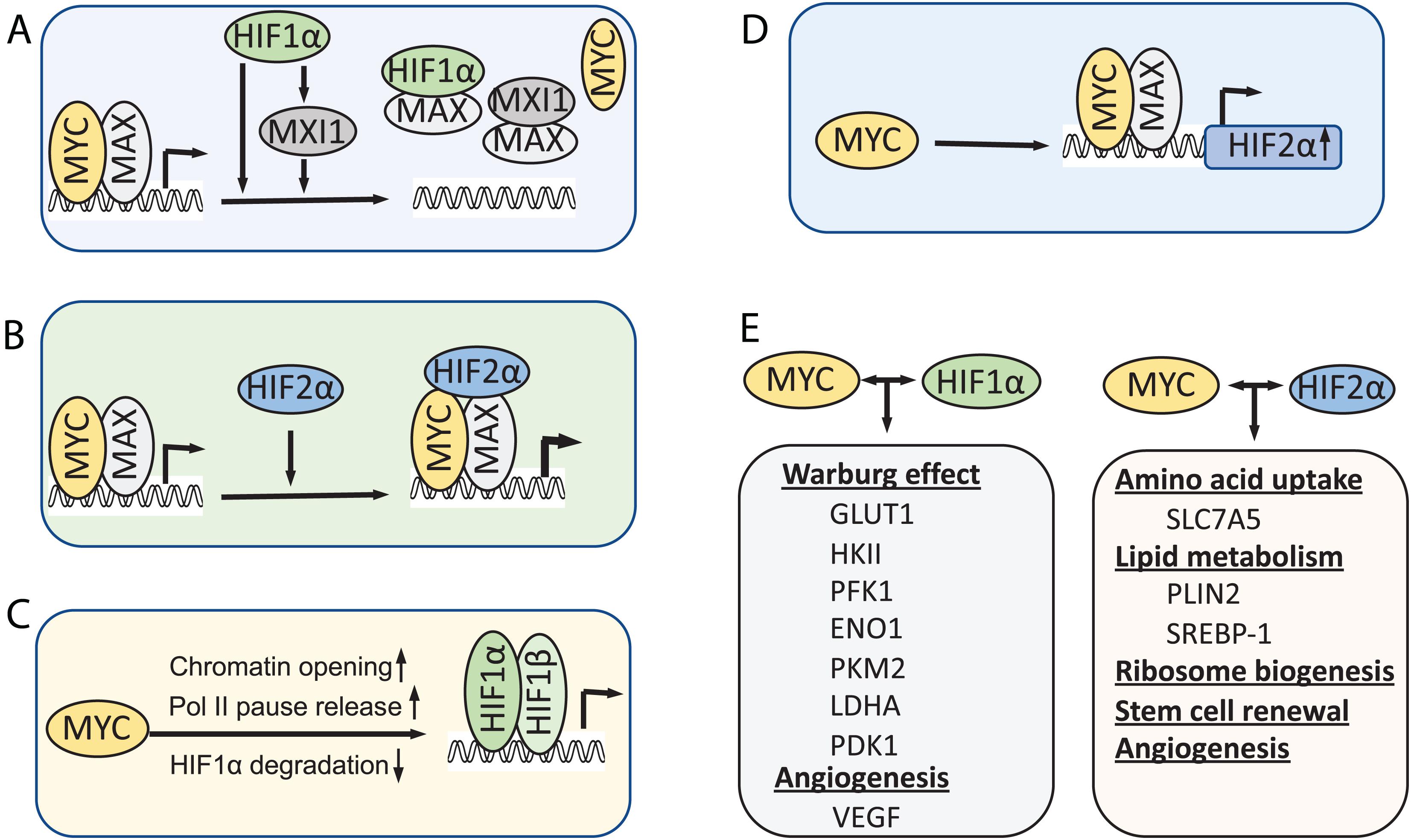
Figure 2. Models for the interplay between MYC and HIF. (A) HIF1α inhibits MYC activity under hypoxia. (B) HIF2α promotes MYC activity by stabilizing MYC-MAX complex. (C) MYC, when overexpressed, increases HIF levels and activity. (D) MYC also directly promotes HIF2α expression. (E) In cancer cells when MYC levels are high, MYC collaborates with HIFs to induce metabolic rewiring, tumor angiogenesis and CSC renewal, thereby promoting tumor cell growth and progression.
In contrast to HIF1α, HIF2α has been shown to promote MYC activity. Phosphorylation of HIF2α at T324 by protein kinase D1 prevents HIF2α from competing with MYC for SP1 binding (To et al., 2006). Instead, overexpression of HIF2α enhances SP1 activity and promote MYC-driven IL-8 expression in human microvascular endothelial cells (Florczyk et al., 2011). HIF2α also enhances MYC activity in VHL-deficient clear cell renal carcinoma cells (ccRCCs) and primary mouse embryo fibroblasts (Gordan et al., 2007, 2008). Consistently, HIF2α deletion reduced MYC target transcriptome in mouse ccRCC models (Hoefflin et al., 2020). It was shown that HIF2α promotes MYC activity by stabilizing the MYC/MAX complex (Zhang et al., 2007; Xue et al., 2015; Figure 2B). Interestingly, HIF2α-induced stabilization of MYC/MAX heterodimer is much stronger than HIF1α-induced degradation of MYC in cancer cells, leading to MYC activation under hypoxia (Xue et al., 2015).
The Regulation of HIF by MYC
Emerging evidence has indicated that MYC regulates the levels and activity of HIF1α (Figure 2C). Transient knockdown of MYC down-regulates HIF1α protein levels in multiple myeloma (MM) cells (Zhang et al., 2009). Overexpression of MYC in colon cancer and esophageal cancer cells promoted the expression of HIF1α at post-transcriptional level (Chen et al., 2013; Weili et al., 2019). Overexpression of MYC significantly stabilizes HIF1α and enhances HIF1α accumulation under both normoxic and hypoxic conditions in normal immortalized mammary epithelial cells and breast cancer cells (Doe et al., 2012). Accumulation of HIF1α by MYC leads to the induction of HIF1α targets and is required for MYC-induced anchorage-independent cell growth and proliferation (Doe et al., 2012). Mechanistically, It was shown that MYC prevents HIF1α degradation via reducing HIF1α binding to the pVHL complex, although it increases the level of pVHL complex components (Doe et al., 2012). Further, a recent study showed that MYC promotes pVHL SUMOylation while repressing its ubiquitination, thereby inhibiting HIF1α ubiquitination and proteasomal degradation (Fu et al., 2016). Besides hypoxia, HIF1α expression can be increased via oxygen-independent mechanisms under certain normoxic conditions such as reactive oxygen and nitrogen species. MYC increases mitochondrial oxidative phosphorylation (mtOXPHOS) and the generation of reactive oxygen species (ROS) (Lee et al., 2017). Increased levels of mitochondrial-generated ROS lead to the stabilization and accumulation of HIF1α by inhibiting PHDs in non-hypoxic conditions (Patten et al., 2010; Lee et al., 2017). Supporting this notion, MYC inhibits brusatol-induced HIF1α degradation by increasing mitochondrial ROS production and subsequent ROS-mediated transition of ferrous iron to ferric iron (Oh et al., 2017).
MYC may also increase HIF1α activity at the chromatin levels. HIF1α preferentially binds to transcriptionally active loci marked by the presence of histone H3 lysine 4 methylation and RNA polymerase II (Xia and Kung, 2009). MYC may promote chromatin opening by recruiting histone acetyl-transferase (HAT) and chromatin remodeling complexes (Tu et al., 2015) and facilitate RNA polymerase II pausing release (Rahl et al., 2010). MYC binding may be required for recruitment of transcriptional activators or repressor to the promoter of HIF1 target genes (Mongiardi et al., 2016). It will be interesting to examine whether MYC dynamically interplays with HIF1α at the promoter of genes that are both MYC and HIF targets, via physical interaction or sequential events at the promoters.
Recently, MYC has been shown to regulate the HIF2α expression (Figure 2D). MYC binds to HIF2α gene promoter preferentially in Sca1+ cancer stem cells (CSCs) in a MYC-driven mouse T-cell leukemia model and the equivalent ABCG2+ CSC population in human acute lymphoblastic lymphomas and activates HIF2α expression (Das et al., 2019). HIF2α is known to regulate stem cell function by inducing the expression of Oct4 (Covello et al., 2006) and ALKBH5, an m6A demethylase that demethylates Nanog mRNA and increases Nanog expression (Soleymani Abyaneh et al., 2018). Indeed, the stem cell factors Nanog and Sox2 facilitate MYC regulation of HIF2α, playing a critical role in stem cell renewal and tumor stemness (Das et al., 2019).
MYC and HIF Cooperate to Reprogram Cancer Cell Metabolism and Promote Tumor Cell Growth and Progression
Cancer cells undergo metabolic switch from oxidative phosphorylation to aerobic glycolysis even under aerobic conditions, called Warburg effect (Vander Heiden et al., 2009). This metabolic rewiring is under direct management by various oncogenes, such as MYC and HIF1 (Cantor and Sabatini, 2012; Dang, 2012a,b). MYC regulates multiple stages of cell metabolism and plays a key role in cancer cell metabolic reprogramming. MYC promotes glucose flux by inducing the expression of glucose transporter1 (GLUT1) and lactate dehydrogenase A (LDHA) (Shim et al., 1997; Osthus et al., 2000); It also upregulates almost all glycolytic enzymes, including hexokinase II (HK II), phosphofructokinase (PFKM), enolase 1 (ENO1), pyruvate dehydrogenase kinase1 (PDK1), and pyruvate kinase (PKM2) (Shim et al., 1997; Osthus et al., 2000; Li et al., 2003; Kim et al., 2004; Stine et al., 2015). MYC upregulates hnRNP proteins to regulate the pyruvate kinase alternative splicing that favors aerobic glycolysis (David et al., 2010). Furthermore, MYC promotes mitochondrial biogenesis by stimulating the expression of mitochondrial transcription factor A (TFAM), a key mitochondria transcription and DNA replication factor, and many genes involved in mitochondrial structure and function (Li et al., 2005). Overall, mitochondrial function is promoted by MYC, allowing cancer cells to proliferate in oxygen and nutrient sufficient conditions. Moreover, MYC also regulates glutaminolysis (Dang et al., 2009; Stine et al., 2015), which converts glutamine to glutamate by glutaminase and then to α-ketoglutarate (α-KG) by glutamate dehydrogenase. α-KG can enter TCA cycle for producing ATP and precursors for lipid, nucleotide and amino acid biosynthesis. MYC enhances glutamine uptake and metabolism by directly stimulating the transcription of glutamine metabolism genes, such as the glutamine transporter SLC1A5/ASCT2 (Wise et al., 2008) and glutaminase 2 (GLS2) (Xiao et al., 2015). MYC also increases the expression of glutaminase 1 (GLS1) indirectly by transcriptionally repressing miR-23a/b, which targets the gene 5′-UTR (Gao et al., 2009). Consequently, deregulated MYC makes cancer cells addicted to glutamine and deprivation of glutamine causes cell death (Wise et al., 2008; Cairns et al., 2011).
Similar to MYC, HIF1 is also a driver for metabolic switch from oxidative to glycolysis by upregulating glucose transporters and most glycolytic enzymes such as HK II, phosphofructokinase 1 (PFK1), fructose-bisphosphate aldolase A (ALDOA), ENO1, PKM2, and LDHA, as well as pentose phosphate pathway (PPP) enzymes (Iyer et al., 1998; Mathupala et al., 2001; Kim et al., 2006; Papandreou et al., 2006; Moldogazieva et al., 2020). In contrast to MYC, both HIF1 and HIF2 inhibit mitochondrial biogenesis in response to hypoxia. Under hypoxia, HIF1 promotes the transcription and activity of FOXO3a (Bakker et al., 2007), which represses a group of nuclear-encoded mitochondrial genes by directly antagonizing MYC on gene promoters (Jensen et al., 2011). Transcription of TFAM is facilitated by nuclear respiratory factor (NRF) interactions with peroxisome proliferator-activated receptor γ coactivator family members (PGC1α, PGC1β, and PRC1) in mitochondria biogenesis (Li et al., 2017). HIF1 also inhibits the transcription of PGC1β by antagonizing MYC (Zhang et al., 2007). A recent study shows that both HIF1α and HIF2α contribute to hypoxia-mediated inhibition of PGC1β and TFAM in human pulmonary endothelial cells (Zarrabi et al., 2017).
HIF1 directly transcriptionally activates PDK1 which inactivates pyruvate dehydrogenase (PDH), an enzyme converting mitochondrial pyruvate into acetyl coenzyme A, thus blocking pyruvate metabolism via the Krebs cycle (Kim et al., 2006; Papandreou et al., 2006). PDK1 not only reduces the mitochondrial oxygen consumption rate but also suppresses reactive oxygen species (ROS) production (Papandreou et al., 2006). HIF1 also fine-tunes hypoxic cell respiration by mediating switches in the subunit composition of cytochrome c oxidase from COX4-1 to COX4-2 (Fukuda et al., 2007). Furthermore, HIF1 reduces the overall mitochondrial mass by inducing mitochondrial autophagy. When cells are subjected to prolonged hypoxia, HIF1-dependent expression of BNIP3 protein triggers mitochondrial degradation through autophagy and stops the excessive production of mitochondrial ROS (Zhang et al., 2008). Such adaptive metabolic response may reduce energy consuming anabolic synthesis and prevent increased levels of ROS and cell death to increase cell survival during hypoxia.
Although HIF1α and MYC have opposing effects on mitochondrial function and biogenesis, they share common target genes on regulating glycolysis (Figure 2E) such as HK-II, PFK1, ENO1, LDHA. Most genes of glycolytic enzymes have MYC and HIF-1α DNA binding consensus sequences (Marbaniang and Kma, 2018). The relative expression levels of MYC and HIF1α proteins determine how they will interplay with each other during this process. It appears that when MYC is overexpressed, it overrides the inhibitory effects by HIF1α. HIF1α effects on MYC via binding to Max may be stoichiometrically diminished by increased MYC-Max complex formation. In Burkitt lymphoma, in which both MYC and HIF1α are highly expressed, HIF1α can actually collaborate with MYC to induce the expression of specific target genes, such as HK2, PDK1, and vascular endothelial growth factor (VEGF) (Kim et al., 2007) under hypoxia and confer resistance to cisplatin treatment (Nakajima et al., 2019). Similarly, high levels of N-MYC in N-MYC amplified neuroblastoma cells override HIF1α inhibition of cell cycle progression under hypoxia and cooperates with HIF1α to promote the expression of phosphoglycerate kinase 1 (PGK1), HK2, and LDHA (Qing et al., 2010). Ectopic expression of a stable form of HIF1 increases MYC-mediated tumorigenesis, while knockdown of HIF1α in B-cell lymphoma P493 cells suppresses its tumorigenesis (Gao et al., 2007). MYC overexpression can override the need for HIF1 for cell survival and propagation in response to hypoxia by inducing glutaminolysis and de novo lipogenesis (Munksgaard Thoren et al., 2017). For example, MYC and L-MYC amplified small cell lung carcinoma (SCLC) cells are dependent on glutamine, but not on glucose, for growth and survival (Munksgaard Thoren et al., 2017). Knockdown of HIF1α in these cells does not affect cell growth and cell survival at hypoxic conditions (Munksgaard Thoren et al., 2017). In addition to common targets regulated by both HIF-1α and HIF-2α, HIF-2α also stimulates the expression of specific genes involved in cell cycle progression, amino acid and lipid metabolism, angiogenesis and vasculature remodeling, tumor growth, metastasis, stemness as well as regulating tumor microenvironment (Covello et al., 2006; Keith et al., 2011; Elorza et al., 2012; Rankin and Giaccia, 2016; Hoefflin et al., 2020). Both MYC (Yue et al., 2017) and HIF-2α (Elorza et al., 2012) activate the expression of the amino acid transporter SLC7A5 to promote essential amino acid (EAA) uptake. Both MYC (Gouw et al., 2019; Casciano et al., 2020) and HIF-2α (Rankin et al., 2009; Qiu et al., 2015) regulate lipid metabolism.
MYC and HIFs also cooperate to promote tumor angiogenesis (Figure 2E). Under hypoxic conditions, HIF-1α can stimulate the expression of various pro-angiogenic factors, including VEGF, VEGF receptors FLT-1 and FLK-1, placental growth factor (PlGF), platelet-derived growth factor B (PDGF-B), plasminogen activator inhibitor-1 (PAI-1), the TIE-2 receptor, matrix metalloproteinases (MMP-2 and MMP-9) and angiopoietins (ANG-1 and ANG -2) (Hickey and Simon, 2006; Zimna and Kurpisz, 2015). Among all of these pro-angiogenic factors, VEGF is one of the most potent mediators of physiologic and pathological angiogenesis. Tumor angiogenesis can also be stimulated by MYC (Baudino et al., 2002). It was shown that oncogenic MYC cooperates with HIF1α to trigger VEGF production and secretion (Kim et al., 2007; Zhang et al., 2009). Although no conserved canonical MYC binding E box exists on VEGF promoter, MYC binds to the same genomic region where HIF1 binds (Kim et al., 2007). MYC also cooperates with HIF2α to promote tumor angiogenesis and hematogenous metastasis by transcriptional repression of miR-15-16 in hypoxia (Xue et al., 2015). miR-15-16 is an important negative regulator of fibroblast growth factor-2 (FGF2) which was proved to promote angiogenesis and metastasis (Cao et al., 2011).
The interplay of HIF family proteins with MYC plays an important role in tumorigenesis and progression. It has been shown that antioxidants such as N-acetylcycteine and vitamin C inhibit MYC-driven lymphoma xenograft growth in vivo by targeting HIF1, which can be rescued by the expression of oxygen-independent HIF1 mutant (Gao et al., 2007), supporting a key role of HIF1 in MYC-driven tumors. MYC activation in combination with Vhl and Ink4a/Arf deletion results in tumors in mice resembling human ccRCCs (Bailey et al., 2017). Interestingly, HIF-1α expression is frequently lost in ccRCCs correlating with poor patient survival (Monzon et al., 2011; Shen et al., 2011). Knockdown of HIF-1α promotes ccRCC cell proliferation and xenograft tumor growth (Shen et al., 2011). In VHL deficient ccRCCs with the expression of HIF-2α, but not HIF-1α, MYC activity is elevated together with the stimulation of cell cycle targets, cell proliferation and resistance to replication stress (Gordan et al., 2008). VHL–/HIF2α+ tumors that show more aggressiveness and proliferative capacity than VHL–/HIF1α+ tumors (Gordan et al., 2008). These data suggest that HIF-2α plays a major role in ccRCC initiation while HIF1a seems to play a role in inhibiting aggressive tumor behaviors. However, a recent study using Vhl/p53/Rb1 deletion mouse model showed that HIF-1α is actually essential for ccRCC formation, whereas deletion of HIF-2α has moderate effects on tumor onset and growth but leads to increased intra-tumoral immune activation. Yet, deletion of HIF-1α in immortalized Vhl/p53 null MEF cells increased cell proliferation, highlighting the role of HIF-1α in inhibiting proliferation of mouse Vhl deletion cells and tumor onset in the autochthonous setting. These seemingly contradicted findings by comparing the role of HIF-1α and HIF-2α in cell culture to that in xenograft models highlights the oncogenic role of HIF-1α in ccRCC initiation, the altered HIF-1α and HIF-2α balance in tumor development, contextual genetic background, and the role of HIF-2α in regulating tumor microenvironment. It would be interesting to further study the direct crosstalk of MYC with HIF-1α and HIF-2α in vivo in MYC activation in combination with Vhl/Ink4a null ccRCC model. Given the heterogeneity of tumor hypoxia, the expression of HIF-1α and HIF-2α may differ in different tumor areas. For example, compared to the cells in the tumor core, cells in the edge of tumors may exhibit chronic hypoxia, more stemness features, close interaction with tumor microenvironment and thus higher expression of HIF-2α (Mortezaee, 2020). HIF-1α can inhibit MYC to slow cell cycle progression under severe hypoxia condition whereas in mild hypoxia, HIF-2α may promote MYC activity by facilitating MYC-MAX dimerization.
Given the importance of the MYC-HIF interplay in cancer cells, it would be critical to further understand how MYC interplays with HIF in jointly regulating the expression of metabolic genes, mitochondria biogenesis, and the production of mitochondria intermediates for nucleotides, fatty acids, and bioamine biosynthesis. Does MYC set up the basal mitochondria activity while HIF shift the balance to glycolysis? The interplay may also heavily rely on the degree of oxygen deprivation and may differ at mild vs. severe hypoxia or acute vs. chronic hypoxia. There may also be different interplay in chromatin and mitochondria.
Targeting MYC-HIF Crosstalk for Cancer Therapy
Given the essential roles of MYC and HIF in tumor progression and metastasis, there has been great clinical value in developing inhibitors targeting MYC and HIF as well as their regulators or downstream targets. Various approaches have been explored for targeting the MYC pathway (Whitfield et al., 2017). Although targeting MYC itself has often proven very challenging because of its nucleus localization and the absence of a deep surface-binding pocket, recent studies have shown that Omomyc, initially designed as a dominant-negative MYC peptide (Soucek et al., 1998, 2002) that competitively binds to E-box elements as heterodimer with MAX or homodimer and suppresses the binding of MYC to E-box (Beaulieu et al., 2019; Demma et al., 2019; Masso-Valles and Soucek, 2020), has cell-penetrating activity and the therapeutic potential in vivo in various cancer models with only mild and reversible side effects, demonstrating its potential in drug development for directly targeting MYC in cancer (Beaulieu et al., 2019; Wang et al., 2019; Masso-Valles and Soucek, 2020). Interestingly, Omomyc strongly inhibited the expression of a subset of genes directly regulated by HIF1α by reducing HIF1α binding to target promoters, thus inhibiting hypoxia-dependent glycolytic reprogramming and mitochondrial functionality in glioblastoma multiforme cells (Mongiardi et al., 2016). BET bromodomain inhibitors such as JQ1 downregulate MYC transcription by disrupting BRD4 binding at a distal MYC “super enhancer,” followed by genome-wide downregulation of Myc-dependent target genes (Delmore et al., 2011; Loven et al., 2013). Also, a number of MYC synthetic lethal pathways have been explored for targeting MYC-driven cancers (reviewed in Cermelli et al., 2014; Hsieh and Dang, 2016).
HIFs is also recognized as an attractive target for anticancer agents. But the complexity involved in the regulation of the HIF pathway has made developing specific HIF inhibitors very challenging too. Currently, there are no clinically approved HIF specific inhibitors. Yet, there are a number of molecules inhibiting HIF1 directly or indirectly, including targeting HIF1 transcription, translation, and protein degradation as well as targeting HIF-2α dimerization. Most of the reported HIF1 inhibitors were originally used for targeting other endogenous proteins and later they were found to inhibit HIF1 activity (Masoud and Li, 2015; Wigerup et al., 2016; Soni and Padwad, 2017). For example, EZN-2698, a synthetic antisense oligonucleotide, and Aminoflavone directly inhibit HIF1α mRNA expression, the PI3K/Akt/mTOR pathway inhibitors and topoisomerase I inhibitors suppress HIF1α translation, while HSP90 and HDAC inhibitors inhibit HIF1α degradation (Masoud and Li, 2015; Wigerup et al., 2016; Soni and Padwad, 2017). HIF1 inhibitor is thought to cooperate with anti-angiogenesis agents to overcome hypoxia-mediated therapy resistance. In addition, Acriflavine, a HIF1 inhibitor targeting HIF dimerization (Lee et al., 2009), inhibits chronic myelogenous leukemia cell growth in vitro and in vivo, which is partially associated with the reduction of MYC (Cheloni et al., 2017).
HIF-2α has recently emerged as a promising target in ccRCCs as it plays a major role in ccRCC tumorigenesis and progression. Following the discovery of the large internal cavity in the HIF-2α PAS-B domain that allows for ligand binding (Key et al., 2009; Scheuermann et al., 2009), several HIF-2α-specific antagonists have been discovered that disrupt the HIF-2α dimerization and show promising effects from pre-clinical to clinical trials, including PT2385, PT2399 and PT2977 (MK-6482) developed by Peloton Therapeutics. PT2399 was shown to effectively inhibit tumor growth in HIF-2α-high ccRCC cell lines and xenograft tumors (Chen et al., 2016; Cho et al., 2016). PT2385 also inhibited HIF-2α-driven gene expression and induced ccRCC tumor regression (Wallace et al., 2016). Recent result from a phase I clinic trial showed response, partial response or stable disease in ∼66% of patients with favorable safety profile and well tolerance for PT2385 (Courtney et al., 2018, 2020). Phase II clinic trials for PT2385 are ongoing in ccRCC (NCT03108066) and recurrent glioblastoma (NCT03216499). Preliminary results for phase II clinic trial for MK-6482 (PT2399) showed reduced size of target lesions in 86.9% (53/61) of pre-treated advanced ccRCC patients with 27.9% partial response rate (Eric Jonasch et al., 2020). A phase III clinic trial for MK-6482 in advanced ccRCC (NCT04195750) is currently in progress. If these clinic trials show promising efficacy, these HIF-2α inhibitors could be extended to other high grade and late stage solid tumors with high expression of HIF-2α.
As aerobic glycolysis and angiogenesis are common downstream effectors of MYC and HIF, which cooperate to drive the expression of many genes involved in both processes in cancer cells, targeting Warburg effect and pro-angiogenic factors have been of great interest. N-MYC amplified neuroblastoma cells are addicted to LDHA, which converts pyruvate to lactate. Knockdown of LDHA completely inhibits tumorigenesis in vivo and targeting LDHA could be a promising approach in treating neuroblastoma patients with N-MYC amplification (Qing et al., 2010). HKII is another potential therapeutic target to overcome cisplatin resistance in B-cell lymphoma (Nakajima et al., 2019). Inhibitors targeting metabolism or angiogenesis in combination with other chemotherapeutic drugs is an attractive strategy. For example, combination of apigenin and gefitinib, an epidermal growth factor receptor (EGFR) inhibitor, to treat EGFR-resistant mutant non-small cell lung cancers impairs energy utilization and suppresses cell growth and malignant behavior. They inhibit the activity of several oncogenic drivers such as MYC, HIF1α, and EGFR, reduce the protein expression of Gluts and MCT1, and inactivate the 5′ adenosine monophosphate-activated protein kinase (AMPK) signaling (Chen Z. et al., 2019). Together, targeting these downstream signaling pathways controlled by MYC-HIF crosstalk (Figure 2E) or directly co-targeting MYC and HIFs could emerge as effective therapeutics in advanced human cancers such as advanced ccRCCs.
Conclusion and Perspectives
High MYC level was significantly associated with stabilized HIF1α expression in various cancers, such as prostate cancer, triple-negative breast cancer (TNBC) and animal model of CNS primitive neuroectodermal tumors (Malchenko et al., 2017; Boldrini et al., 2019; Cui and Jiang, 2019). Deregulated MYC cooperates with HIFs to regulate cancer cell adaptation to hypoxia, rewire metabolism, and promote angiogenesis. Also, high MYC and HIF expression is associated with poor outcome in various cancers such as prostate and breast cancers and clear cell renal cell carcinomas (Maroto et al., 2017; Boldrini et al., 2019; Cui and Jiang, 2019). Therefore, MYC and HIFs are potential biomarkers for targeting both pathways. The HIF-MYC interplay also plays a critical role in adaptive and innate immunity by regulating T cell development, activation, differentiation, metabolism and thus anti-tumor immunity (reviewed in Rankin and Giaccia, 2016; Gnanaprakasam et al., 2017). While HIF-1α deletion results in CD8+ T-cell infiltration, HIF-2α deletion leads to both CD4+ and CD8+ T-cell infiltration and activation as well as increased antigen presentation and interferon signaling in mouse ccRCC models, suggesting a role for HIF-2α in suppressing T-cell inflammation and intra-tumoral immune activation (Hoefflin et al., 2020). Therefore, it is important to further understand the role of MYC-HIF interplay in shaping tumor immune microenvironment and metastasis. Also, the MYC-HIF crosstalk may be dynamic and divergent in different type of tumors, various tumor stages, and hypoxia heterogeneity. As in other targeted therapy, therapeutic resistance is of a concern. For example, a gatekeeper mutation G323E in HIF-2α was identified to be responsible for PT2385 resistance after prolonged treatment (Courtney et al., 2020). Thus, further insights into the understanding of the MYC-HIF interplay are warranted for developing novel targeted therapeutics. It is also conceivable that the HIF-MYC axis further interplays with other oncogenic pathways such as ERK/MAPK, Akt/mTOR, WNT, and Notch signaling to alter cell metabolism, cell cycle, ribosome biogenesis, and genomic stability in tumorigenesis. Thus, the HIF-MYC targeted therapy may be in combination with inhibitors targeting these pathways as well.
Author Contributions
YL, X-XS, DQ, and M-SD wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the NIH grants R01 CA186241 and R01 GM130604 to M-SD and R01 CA207377 to DQ.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Arany, Z., Huang, L. E., Eckner, R., Bhattacharya, S., Jiang, C., Goldberg, M. A., et al. (1996). An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. U.S.A. 93, 12969–12973. doi: 10.1073/pnas.93.23.12969
Bailey, S. T., Smith, A. M., Kardos, J., Wobker, S. E., Wilson, H. L., Krishnan, B., et al. (2017). MYC activation cooperates with Vhl and Ink4a/Arf loss to induce clear cell renal cell carcinoma. Nat. Commun. 8:15770.
Bakker, W. J., Harris, I. S., and Mak, T. W. (2007). FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol. Cell 28, 941–953. doi: 10.1016/j.molcel.2007.10.035
Baluapuri, A., Wolf, E., and Eilers, M. (2020). Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 21, 255–267. doi: 10.1038/s41580-020-0215-2
Baudino, T. A., McKay, C., Pendeville-Samain, H., Nilsson, J. A., Maclean, K. H., White, E. L., et al. (2002). c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 16, 2530–2543. doi: 10.1101/gad.1024602
Beaulieu, M. E., Jauset, T., Masso-Valles, D., Martinez-Martin, S., Rahl, P., Maltais, L., et al. (2019). Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 11:eaar5012, doi: 10.1126/scitranslmed.aar5012
Boldrini, L., Bartoletti, R., Giordano, M., Manassero, F., Selli, C., Panichi, M., et al. (2019). C-MYC, HIF-1alpha, ERG, TKT, and GSTP1: an Axis in Prostate Cancer? Pathol. Oncol. Res. 25, 1423–1429. doi: 10.1007/s12253-018-0479-4
Brahimi-Horn, M. C., and Pouyssegur, J. (2009). HIF at a glance. J. Cell Sci. 122, 1055–1057. doi: 10.1242/jcs.035022
Bretones, G., Delgado, M. D., and Leon, J. (2015). Myc and cell cycle control. Biochim. Biophys. Acta 1849, 506–516.
Cairns, R. A., Harris, I. S., and Mak, T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95.
Cantor, J. R., and Sabatini, D. M. (2012). Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898. doi: 10.1158/2159-8290.cd-12-0345
Cao, Y., Arbiser, J., D’Amato, R. J., D’Amore, P. A., Ingber, D. E., Kerbel, R., et al. (2011). Forty-year journey of angiogenesis translational research. Sci. Transl. Med. 3:114rv113.
Casciano, J. C., Perry, C., Cohen-Nowak, A. J., Miller, K. D., Vande Voorde, J., Zhang, Q., et al. (2020). MYC regulates fatty acid metabolism through a multigenic program in claudin-low triple negative breast cancer. Br. J. Cancer 122, 868–884. doi: 10.1038/s41416-019-0711-3
Cermelli, S., Jang, I. S., Bernard, B., and Grandori, C. (2014). Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harb. Perspect. Med. 4:a014209. doi: 10.1101/cshperspect.a014209
Cheloni, G., Tanturli, M., Tusa, I., Ho DeSouza, N., Shan, Y., Gozzini, A., et al. (2017). Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood 130, 655–665. doi: 10.1182/blood-2016-10-745588
Chen, C., Cai, S., Wang, G., Cao, X., Yang, X., Luo, X., et al. (2013). c-Myc enhances colon cancer cell-mediated angiogenesis through the regulation of HIF-1alpha. Biochem. Biophys. Res. Commun. 430, 505–511. doi: 10.1016/j.bbrc.2012.12.006
Chen, W., Hill, H., Christie, A., Kim, M. S., Holloman, E., Pavia-Jimenez, A., et al. (2016). Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 539, 112–117.
Chen, Z., Tian, D., Liao, X., Zhang, Y., Xiao, J., Chen, W., et al. (2019). Apigenin combined with gefitinib blocks autophagy flux and induces apoptotic cell death through inhibition of HIF-1alpha, c-Myc, p-EGFR, and Glucose Metabolism in EGFR L858R+T790M-Mutated H1975 cells. Front. Pharmacol. 10:260.
Chen, Y., Sun, X. X., Sears, R. C., and Dai, M. S. (2019). Writing and erasing MYC ubiquitination and SUMOylation. Genes Dis. 6, 359–371. doi: 10.1016/j.gendis.2019.05.006
Cho, H., Du, X., Rizzi, J. P., Liberzon, E., Chakraborty, A. A., Gao, W., et al. (2016). On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 539, 107–111. doi: 10.1038/nature19795
Compernolle, V., Brusselmans, K., Acker, T., Hoet, P., Tjwa, M., Beck, H., et al. (2002). Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat. Med. 8, 702–710. doi: 10.1038/nm721
Compernolle, V., Brusselmans, K., Franco, D., Moorman, A., Dewerchin, M., Collen, D., et al. (2003). Cardia bifida, defective heart development and abnormal neural crest migration in embryos lacking hypoxia-inducible factor-1alpha. Cardiovasc. Res. 60, 569–579. doi: 10.1016/j.cardiores.2003.07.003
Conacci-Sorrell, M., McFerrin, L., and Eisenman, R. N. (2014). An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 4:a014357. doi: 10.1101/cshperspect.a014357
Corn, P. G., Ricci, M. S., Scata, K. A., Arsham, A. M., Simon, M. C., Dicker, D. T., et al. (2005). Mxi1 is induced by hypoxia in a HIF-1-dependent manner and protects cells from c-Myc-induced apoptosis. Cancer Biol. Ther. 4, 1285–1294. doi: 10.4161/cbt.4.11.2299
Courtney, K. D., Infante, J. R., Lam, E. T., Figlin, R. A., Rini, B. I., Brugarolas, J., et al. (2018). Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2alpha Antagonist in Patients With Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 36, 867–874. doi: 10.1200/jco.2017.74.2627
Courtney, K. D., Ma, Y., Diaz de Leon, A., Christie, A., Xie, Z., Woolford, L., et al. (2020). HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 26, 793–803. doi: 10.1158/1078-0432.ccr-19-1459
Covello, K. L., Kehler, J., Yu, H., Gordan, J. D., Arsham, A. M., Hu, C. J., et al. (2006). HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557–570. doi: 10.1101/gad.1399906
Cowling, V. H., Chandriani, S., Whitfield, M. L., and Cole, M. D. (2006). A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol. Cell Biol. 26, 4226–4239. doi: 10.1128/mcb.01959-05
Cui, J., and Jiang, H. (2019). Prediction of postoperative survival of triple-negative breast cancer based on nomogram model combined with expression of HIF-1alpha and c-myc. Medicine 98, e17370. doi: 10.1097/md.0000000000017370
Dang, C. V. (2012a). Links between metabolism and cancer. Genes Dev. 26, 877–890. doi: 10.1101/gad.189365.112
Dang, C. V., Le, A., and Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 15, 6479–6483. doi: 10.1158/1078-0432.ccr-09-0889
Das, B., Pal, B., Bhuyan, R., Li, H., Sarma, A., Gayan, S., et al. (2019). MYC Regulates the HIF2alpha Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells versus Non-Stem Cancer Cells. Cancer Res. 79, 4015–4025. doi: 10.1158/0008-5472.can-18-2847
David, C. J., Chen, M., Assanah, M., Canoll, P., and Manley, J. L. (2010). HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368. doi: 10.1038/nature08697
Davis, A. C., Wims, M., Spotts, G. D., Hann, S. R., and Bradley, A. (1993). A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 7, 671–682. doi: 10.1101/gad.7.4.671
Delmore, J. E., Issa, G. C., Lemieux, M. E., Rahl, P. B., Shi, J., Jacobs, H. M., et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917.
Demma, M. J., Mapelli, C., Sun, A., Bodea, S., Ruprecht, B., Javaid, S., et al. (2019). Omomyc reveals new mechanisms to inhibit the MYC Oncogene. Mol. Cell Biol. 39:e00248-19.
Doe, M. R., Ascano, J. M., Kaur, M., and Cole, M. D. (2012). Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res. 72, 949–957. doi: 10.1158/0008-5472.can-11-2371
Elorza, A., Soro-Arnaiz, I., Melendez-Rodriguez, F., Rodriguez-Vaello, V., Marsboom, G., de Carcer, G., et al. (2012). HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 48, 681–691. doi: 10.1016/j.molcel.2012.09.017
Eric Jonasch, F. D., Othon, I., Wendy, K. R., Vivek, N., Benjamin, L. M., and Stephane, O. (2020). Phase II study of the oral HIF-2α inhibitor MK-6482 for Von Hippel-Lindau disease–associated renal cell carcinoma. J. Clin. Oncol. 38:5003. doi: 10.1200/jco.2020.38.15_suppl.5003
Farrell, A. S., and Sears, R. C. (2014). MYC degradation. Cold Spring Harb. Perspect. Med. 4:a014365. doi: 10.1101/cshperspect.a014365
Florczyk, U., Czauderna, S., Stachurska, A., Tertil, M., Nowak, W., Kozakowska, M., et al. (2011). Opposite effects of HIF-1alpha and HIF-2alpha on the regulation of IL-8 expression in endothelial cells. Free Radic. Biol. Med. 51, 1882–1892. doi: 10.1016/j.freeradbiomed.2011.08.023
Fu, R., Chen, Y., Wang, X. P., An, T., Tao, L., Zhou, Y. X., et al. (2016). Wogonin inhibits multiple myeloma-stimulated angiogenesis via c-Myc/VHL/HIF-1alpha signaling axis. Oncotarget 7, 5715–5727. doi: 10.18632/oncotarget.6796
Fukuda, R., Zhang, H., Kim, J. W., Shimoda, L., Dang, C. V., and Semenza, G. L. (2007). HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122. doi: 10.1016/j.cell.2007.01.047
Gabay, M., Li, Y., and Felsher, D. W. (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 4:a014241. doi: 10.1101/cshperspect.a014241
Gao, P., Tchernyshyov, I., Chang, T. C., Lee, Y. S., Kita, K., Ochi, T., et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. doi: 10.1038/nature07823
Gao, P., Zhang, H., Dinavahi, R., Li, F., Xiang, Y., Raman, V., et al. (2007). HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12, 230–238. doi: 10.1016/j.ccr.2007.08.004
Gnanaprakasam, J. N. R., Sherman, J. W., and Wang, R. (2017). MYC and HIF in shaping immune response and immune metabolism. Cytokine Growth Factor Rev. 35, 63–70. doi: 10.1016/j.cytogfr.2017.03.004
Gordan, J. D., Bertout, J. A., Hu, C. J., Diehl, J. A., and Simon, M. C. (2007). HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 11, 335–347. doi: 10.1016/j.ccr.2007.02.006
Gordan, J. D., Lal, P., Dondeti, V. R., Letrero, R., Parekh, K. N., Oquendo, C. E., et al. (2008). HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 14, 435–446. doi: 10.1016/j.ccr.2008.10.016
Gouw, A. M., Margulis, K., Liu, N. S., Raman, S. J., Mancuso, A., Toal, G. G., et al. (2019). The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab. 30:e555.
Gruber, M., Hu, C. J., Johnson, R. S., Brown, E. J., Keith, B., and Simon, M. C. (2007). Acute postnatal ablation of Hif-2alpha results in anemia. Proc. Natl. Acad. Sci. U.S.A. 104, 2301–2306. doi: 10.1073/pnas.0608382104
Hickey, M. M., and Simon, M. C. (2006). Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr. Top. Dev. Biol. 76, 217–257. doi: 10.1016/s0070-2153(06)76007-0
Hoefflin, R., Harlander, S., Schafer, S., Metzger, P., Kuo, F., Schonenberger, D., et al. (2020). HIF-1alpha and HIF-2alpha differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 11:4111.
Hsieh, A. L., and Dang, C. V. (2016). MYC, metabolic synthetic lethality, and cancer. Recent Results Cancer Res. 207, 73–91. doi: 10.1007/978-3-319-42118-6_4
Hu, C. J., Wang, L. Y., Chodosh, L. A., Keith, B., and Simon, M. C. (2003). Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell Biol. 23, 9361–9374.
Ivan, M., and Kaelin, W. G. Jr. (2017). The EGLN-HIF O2-sensing system: multiple inputs and feedbacks. Mol. Cell 66, 772–779. doi: 10.1016/j.molcel.2017.06.002
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., et al. (2001). HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468. doi: 10.1126/science.1059817
Iyer, N. V., Kotch, L. E., Agani, F., Leung, S. W., Laughner, E., Wenger, R. H., et al. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 12, 149–162. doi: 10.1101/gad.12.2.149
Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., et al. (2001). Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472. doi: 10.1126/science.1059796
Jensen, K. S., Binderup, T., Jensen, K. T., Therkelsen, I., Borup, R., Nilsson, E., et al. (2011). FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J. 30, 4554–4570. doi: 10.1038/emboj.2011.323
Keith, B., Johnson, R. S., and Simon, M. C. (2011). HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 12, 9–22. doi: 10.1038/nrc3183
Key, J., Scheuermann, T. H., Anderson, P. C., Daggett, V., and Gardner, K. H. (2009). Principles of ligand binding within a completely buried cavity in HIF2alpha PAS-B. J. Am. Chem. Soc. 131, 17647–17654. doi: 10.1021/ja9073062
Kim, J. W., Gao, P., Liu, Y. C., Semenza, G. L., and Dang, C. V. (2007). Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 27, 7381–7393. doi: 10.1128/mcb.00440-07
Kim, J. W., Tchernyshyov, I., Semenza, G. L., and Dang, C. V. (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. doi: 10.1016/j.cmet.2006.02.002
Kim, J. W., Zeller, K. I., Wang, Y., Jegga, A. G., Aronow, B. J., O’Donnell, K. A., et al. (2004). Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell Biol. 24, 5923–5936. doi: 10.1128/mcb.24.13.5923-5936.2004
Koshiji, M., Kageyama, Y., Pete, E. A., Horikawa, I., Barrett, J. C., and Huang, L. E. (2004). HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. EMBO J. 23, 1949–1956. doi: 10.1038/sj.emboj.7600196
Koshiji, M., To, K. K., Hammer, S., Kumamoto, K., Harris, A. L., Modrich, P., et al. (2005). HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell 17, 793–803. doi: 10.1016/j.molcel.2005.02.015
Kress, T. R., Sabo, A., and Amati, B. (2015). MYC: connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 15, 593–607. doi: 10.1038/nrc3984
Kurland, J. F., and Tansey, W. P. (2008). Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 68, 3624–3629. doi: 10.1158/0008-5472.can-07-6552
Lando, D., Peet, D. J., Gorman, J. J., Whelan, D. A., Whitelaw, M. L., and Bruick, R. K. (2002a). FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 16, 1466–1471. doi: 10.1101/gad.991402
Lando, D., Peet, D. J., Whelan, D. A., Gorman, J. J., and Whitelaw, M. L. (2002b). Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858–861. doi: 10.1126/science.1068592
Lee, K., Zhang, H., Qian, D. Z., Rey, S., Liu, J. O., and Semenza, G. L. (2009). Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. U.S.A. 106, 17910–17915. doi: 10.1073/pnas.0909353106
Lee, K. M., Giltnane, J. M., Balko, J. M., Schwarz, L. J., Guerrero-Zotano, A. L., Hutchinson, K. E., et al. (2017). MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab. 26, 633–647 e637.
Lee, P., Chandel, N. S., and Simon, M. C. (2020). Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283. doi: 10.1038/s41580-020-0227-y
Li, F., Wang, Y., Zeller, K. I., Potter, J. J., Wonsey, D. R., O’Donnell, K. A., et al. (2005). Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell Biol. 25, 6225–6234. doi: 10.1128/mcb.25.14.6225-6234.2005
Li, P. A., Hou, X., and Hao, S. (2017). Mitochondrial biogenesis in neurodegeneration. J. Neurosci. Res. 95, 2025–2029. doi: 10.1002/jnr.24042
Li, Q., Kluz, T., Sun, H., and Costa, M. (2009). Mechanisms of c-myc degradation by nickel compounds and hypoxia. PLoS One 4:e8531. doi: 10.1371/journal.pone.0008531
Li, Z., Van Calcar, S., Qu, C., Cavenee, W. K., Zhang, M. Q., and Ren, B. (2003). A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. U.S.A. 100, 8164–8169. doi: 10.1073/pnas.1332764100
Loven, J., Hoke, H. A., Lin, C. Y., Lau, A., Orlando, D. A., Vakoc, C. R., et al. (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. doi: 10.1016/j.cell.2013.03.036
Lv, X., Li, J., Zhang, C., Hu, T., Li, S., He, S., et al. (2017). The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 4, 19–24. doi: 10.1016/j.gendis.2016.11.003
Malchenko, S., Sredni, S. T., Bi, Y., Margaryan, N. V., Boyineni, J., Mohanam, I., et al. (2017). Stabilization of HIF-1alpha and HIF-2alpha, up-regulation of MYCC and accumulation of stabilized p53 constitute hallmarks of CNS-PNET animal model. PLoS One 12:e0173106. doi: 10.1371/journal.pone.0173106
Marbaniang, C., and Kma, L. (2018). Dysregulation of glucose metabolism by oncogenes and tumor suppressors in cancer cells. Asian Pac. J. Cancer Prev. 19, 2377–2390.
Markolovic, S., Wilkins, S. E., and Schofield, C. J. (2015). Protein Hydroxylation Catalyzed by 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem. 290, 20712–20722. doi: 10.1074/jbc.r115.662627
Maroto, P., Esteban, E., Parra, E. F., Mendez-Vidal, M. J., Domenech, M., Perez-Valderrama, B., et al. (2017). HIF pathway and c-Myc as biomarkers for response to sunitinib in metastatic clear-cell renal cell carcinoma. Onco Targets Ther. 10, 4635–4643. doi: 10.2147/ott.s137677
Masoud, G. N., and Li, W. (2015). HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 5, 378–389. doi: 10.1016/j.apsb.2015.05.007
Masso-Valles, D., and Soucek, L. (2020). Blocking Myc to treat cancer: reflecting on two decades of omomyc. Cells 9:883. doi: 10.3390/cells9040883
Mathupala, S. P., Rempel, A., and Pedersen, P. L. (2001). Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J. Biol. Chem. 276, 43407–43412. doi: 10.1074/jbc.m108181200
Moldogazieva, N. T., Mokhosoev, I. M., and Terentiev, A. A. (2020). Metabolic heterogeneity of cancer cells: an interplay between HIF-1, GLUTs, and AMPK. Cancers 12:862. doi: 10.3390/cancers12040862
Mongiardi, M. P., Savino, M., Falchetti, M. L., Illi, B., Bozzo, F., Valle, C., et al. (2016). c-MYC inhibition impairs hypoxia response in glioblastoma multiforme. Oncotarget 7, 33257–33271. doi: 10.18632/oncotarget.8921
Monzon, F. A., Alvarez, K., Peterson, L., Truong, L., Amato, R. J., Hernandez-McClain, J., et al. (2011). Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. Mod. Pathol. 24, 1470–1479. doi: 10.1038/modpathol.2011.107
Mortezaee, K. (2020). Hypoxia induces core-to-edge transition of progressive tumoral cells: A critical review on differential yet corroborative roles for HIF-1alpha and HIF-2alpha. Life Sci. 242:117145. doi: 10.1016/j.lfs.2019.117145
Munksgaard Thoren, M., Vaapil, M., Staaf, J., Planck, M., Johansson, M. E., Mohlin, S., et al. (2017). Myc-induced glutaminolysis bypasses HIF-driven glycolysis in hypoxic small cell lung carcinoma cells. Oncotarget 8, 48983–48995. doi: 10.18632/oncotarget.16904
Nakajima, K., Kawashima, I., Koshiisi, M., Kumagai, T., Suzuki, M., Suzuki, J., et al. (2019). Glycolytic enzyme hexokinase II is a putative therapeutic target in B-cell malignant lymphoma. Exp. Hematol. 78:e43.
Nesbit, C. E., Tersak, J. M., and Prochownik, E. V. (1999). MYC oncogenes and human neoplastic disease. Oncogene 18, 3004–3016. doi: 10.1038/sj.onc.1202746
Oh, E. T., Kim, C. W., Kim, H. G., Lee, J. S., and Park, H. J. (2017). Brusatol-Mediated Inhibition of c-Myc Increases HIF-1alpha Degradation and Causes Cell Death in Colorectal Cancer under Hypoxia. Theranostics 7, 3415–3431. doi: 10.7150/thno.20861
Osthus, R. C., Shim, H., Kim, S., Li, Q., Reddy, R., Mukherjee, M., et al. (2000). Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 275, 21797–21800. doi: 10.1074/jbc.c000023200
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L., and Denko, N. C. (2006). HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197. doi: 10.1016/j.cmet.2006.01.012
Patten, D. A., Lafleur, V. N., Robitaille, G. A., Chan, D. A., Giaccia, A. J., and Richard, D. E. (2010). Hypoxia-inducible factor-1 activation in nonhypoxic conditions: the essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell 21, 3247–3257. doi: 10.1091/mbc.e10-01-0025
Peng, J., Zhang, L., Drysdale, L., and Fong, G. H. (2000). The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc. Natl. Acad. Sci. U.S.A. 97, 8386–8391. doi: 10.1073/pnas.140087397
Qing, G., Skuli, N., Mayes, P. A., Pawel, B., Martinez, D., Maris, J. M., et al. (2010). Combinatorial regulation of neuroblastoma tumor progression by N-Myc and hypoxia inducible factor HIF-1alpha. Cancer Res. 70, 10351–10361. doi: 10.1158/0008-5472.can-10-0740
Qiu, B., Ackerman, D., Sanchez, D. J., Li, B., Ochocki, J. D., Grazioli, A., et al. (2015). HIF2alpha-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 5, 652–667. doi: 10.1158/2159-8290.cd-14-1507
Rahl, P. B., Lin, C. Y., Seila, A. C., Flynn, R. A., McCuine, S., Burge, C. B., et al. (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. doi: 10.1016/j.cell.2010.03.030
Rankin, E. B., and Giaccia, A. J. (2016). Hypoxic control of metastasis. Science 352, 175–180. doi: 10.1126/science.aaf4405
Rankin, E. B., Rha, J., Selak, M. A., Unger, T. L., Keith, B., Liu, Q., et al. (2009). Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell Biol. 29, 4527–4538. doi: 10.1128/mcb.00200-09
Scheuermann, T. H., Tomchick, D. R., Machius, M., Guo, Y., Bruick, R. K., and Gardner, K. H. (2009). Artificial ligand binding within the HIF2alpha PAS-B domain of the HIF2 transcription factor. Proc. Natl. Acad. Sci.U.S.A. 106, 450–455. doi: 10.1073/pnas.0808092106
Seira, N., Yamagata, K., Fukushima, K., Araki, Y., Kurata, N., Yanagisawa, N., et al. (2018). Cellular density-dependent increases in HIF-1alpha compete with c-Myc to down-regulate human EP4 receptor promoter activity through Sp-1-binding region. Pharmacol. Res. Perspect. 6, e00441. doi: 10.1002/prp2.441
Semenza, G. L. (2003). Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732. doi: 10.1038/nrc1187
Semenza, G. L. (2014). Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 9, 47–71. doi: 10.1146/annurev-pathol-012513-104720
Semenza, G. L. (2020). The genomics and genetics of oxygen homeostasis. Annu. Rev. Genomics Hum. Genet. 21, 183–204. doi: 10.1146/annurev-genom-111119-073356
Semenza, G. L., Jiang, B. H., Leung, S. W., Passantino, R., Concordet, J. P., Maire, P., et al. (1996). Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 271, 32529–32537. doi: 10.1074/jbc.271.51.32529
Shen, C., Beroukhim, R., Schumacher, S. E., Zhou, J., Chang, M., Signoretti, S., et al. (2011). Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov. 1, 222–235. doi: 10.1158/2159-8290.cd-11-0098
Shim, H., Dolde, C., Lewis, B. C., Wu, C. S., Dang, G., Jungmann, R. A., et al. (1997). c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. U.S.A. 94, 6658–6663. doi: 10.1073/pnas.94.13.6658
Soleymani Abyaneh, H., Gupta, N., Alshareef, A., Gopal, K., Lavasanifar, A., and Lai, R. (2018). Hypoxia Induces the Acquisition of Cancer Stem-like Phenotype Via Upregulation and Activation of Signal Transducer and Activator of Transcription-3 (STAT3) in MDA-MB-231, a Triple Negative Breast Cancer Cell Line. Cancer Microenviron. 11, 141–152. doi: 10.1007/s12307-018-0218-0
Soni, S., and Padwad, Y. S. (2017). HIF-1 in cancer therapy: two decade long story of a transcription factor. Acta Oncol. 56, 503–515. doi: 10.1080/0284186x.2017.1301680
Soucek, L., Helmer-Citterich, M., Sacco, A., Jucker, R., Cesareni, G., and Nasi, S. (1998). Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 17, 2463–2472. doi: 10.1038/sj.onc.1202199
Soucek, L., Jucker, R., Panacchia, L., Ricordy, R., Tato, F., and Nasi, S. (2002). Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 62, 3507–3510.
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A. L., and Dang, C. V. (2015). MYC, Metabolism, and Cancer. Cancer Discov. 5, 1024–1039.
Thomas, L. R., Foshage, A. M., Weissmiller, A. M., Popay, T. M., Grieb, B. C., Qualls, S. J., et al. (2016). Interaction of MYC with host cell factor-1 is mediated by the evolutionarily conserved Myc box IV motif. Oncogene 35, 3613–3618. doi: 10.1038/onc.2015.416
Thomas, L. R., Wang, Q., Grieb, B. C., Phan, J., Foshage, A. M., Sun, Q., et al. (2015). Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 58, 440–452. doi: 10.1016/j.molcel.2015.02.028
Tian, H., Hammer, R. E., Matsumoto, A. M., Russell, D. W., and McKnight, S. L. (1998). The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 12, 3320–3324. doi: 10.1101/gad.12.21.3320
To, K. K., Sedelnikova, O. A., Samons, M., Bonner, W. M., and Huang, L. E. (2006). The phosphorylation status of PAS-B distinguishes HIF-1alpha from HIF-2alpha in NBS1 repression. EMBO J. 25, 4784–4794. doi: 10.1038/sj.emboj.7601369
Tu, W. B., Helander, S., Pilstal, R., Hickman, K. A., Lourenco, C., Jurisica, I., et al. (2015). Myc and its interactors take shape. Biochim. Biophys. Acta 1849, 469–483. doi: 10.1016/j.bbagrm.2014.06.002
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. doi: 10.1126/science.1160809
Wallace, E. M., Rizzi, J. P., Han, G., Wehn, P. M., Cao, Z., Du, X., et al. (2016). A Small-Molecule Antagonist of HIF2alpha Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 76, 5491–5500. doi: 10.1158/0008-5472.can-16-0473
Wang, E., Sorolla, A., Cunningham, P. T., Bogdawa, H. M., Beck, S., Golden, E., et al. (2019). Tumor penetrating peptides inhibiting MYC as a potent targeted therapeutic strategy for triple-negative breast cancers. Oncogene 38, 140–150. doi: 10.1038/s41388-018-0421-y
Wang, G. L., Jiang, B. H., Rue, E. A., and Semenza, G. L. (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U.S.A. 92, 5510–5514. doi: 10.1073/pnas.92.12.5510
Wang, V., Davis, D. A., Haque, M., Huang, L. E., and Yarchoan, R. (2005). Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 65, 3299–3306. doi: 10.1158/0008-5472.can-04-4130
Weili, Z., Zhikun, L., Jianmin, W., and Qingbao, T. (2019). Knockdown of USP28 enhances the radiosensitivity of esophageal cancer cells via the c-Myc/hypoxia-inducible factor-1 alpha pathway. J. Cell Biochem. 120, 201–212. doi: 10.1002/jcb.27305
Whitfield, J. R., Beaulieu, M. E., and Soucek, L. (2017). Strategies to inhibit myc and their clinical applicability. Front. Cell Dev. Biol. 5:10.
Wigerup, C., Pahlman, S., and Bexell, D. (2016). Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 164, 152–169. doi: 10.1016/j.pharmthera.2016.04.009
Wise, D. R., DeBerardinis, R. J., Mancuso, A., Sayed, N., Zhang, X. Y., Pfeiffer, H. K., et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. U.S.A. 105, 18782–18787. doi: 10.1073/pnas.0810199105
Wong, W. J., Qiu, B., Nakazawa, M. S., Qing, G., and Simon, M. C. (2013). MYC degradation under low O2 tension promotes survival by evading hypoxia-induced cell death. Mol. Cell Biol. 33, 3494–3504. doi: 10.1128/mcb.00853-12
Xia, X., and Kung, A. L. (2009). Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 10:R113.
Xia, X., Lemieux, M. E., Li, W., Carroll, J. S., Brown, M., Liu, X. S., et al. (2009). Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc. Natl. Acad. Sci. U.S.A. 106, 4260–4265. doi: 10.1073/pnas.0810067106
Xiao, D., Ren, P., Su, H., Yue, M., Xiu, R., Hu, Y., et al. (2015). Myc promotes glutaminolysis in human neuroblastoma through direct activation of glutaminase 2. Oncotarget 6, 40655–40666. doi: 10.18632/oncotarget.5821
Xue, G., Yan, H. L., Zhang, Y., Hao, L. Q., Zhu, X. T., Mei, Q., et al. (2015). c-Myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2alpha and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene 34, 1393–1406. doi: 10.1038/onc.2014.82
Yoon, D., Pastore, Y. D., Divoky, V., Liu, E., Mlodnicka, A. E., Rainey, K., et al. (2006). Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J. Biol. Chem. 281, 25703–25711. doi: 10.1074/jbc.m602329200
Yu, F., White, S. B., Zhao, Q., and Lee, F. S. (2001). HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. U.S.A. 98, 9630–9635. doi: 10.1073/pnas.181341498
Yue, M., Jiang, J., Gao, P., Liu, H., and Qing, G. (2017). Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 21, 3819–3832. doi: 10.1016/j.celrep.2017.12.002
Zarrabi, A. J., Kao, D., Nguyen, D. T., Loscalzo, J., and Handy, D. E. (2017). Hypoxia-induced suppression of c-Myc by HIF-2alpha in human pulmonary endothelial cells attenuates TFAM expression. Cell Signal 38, 230–237. doi: 10.1016/j.cellsig.2017.07.008
Zhang, H., Bosch-Marce, M., Shimoda, L. A., Tan, Y. S., Baek, J. H., Wesley, J. B., et al. (2008). Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 283, 10892–10903. doi: 10.1074/jbc.m800102200
Zhang, H., Gao, P., Fukuda, R., Kumar, G., Krishnamachary, B., Zeller, K. I., et al. (2007). HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11, 407–420. doi: 10.1016/j.ccr.2007.04.001
Zhang, J., Sattler, M., Tonon, G., Grabher, C., Lababidi, S., Zimmerhackl, A., et al. (2009). Targeting angiogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res. 69, 5082–5090. doi: 10.1158/0008-5472.can-08-4603
Zhong, H., De Marzo, A. M., Laughner, E., Lim, M., Hilton, D. A., Zagzag, D., et al. (1999). Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 59, 5830–5835.
Keywords: MYC, HIF1α, HIF2α, transcription, protein stability, metabolism
Citation: Li Y, Sun X-X, Qian DZ and Dai M-S (2020) Molecular Crosstalk Between MYC and HIF in Cancer. Front. Cell Dev. Biol. 8:590576. doi: 10.3389/fcell.2020.590576
Received: 02 August 2020; Accepted: 21 October 2020;
Published: 05 November 2020.
Edited by:
Isabel Merida, Consejo Superior de Investigaciones Científicas (CSIC), SpainReviewed by:
Serge Roche, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceElisabetta Rovida, University of Florence, Italy
Silvia Martin-Puig, Spanish National Centre for Cardiovascular Research, Spain
Copyright © 2020 Li, Sun, Qian and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mu-Shui Dai, ZGFpbUBvaHN1LmVkdQ==