 I-Chung Chen
I-Chung Chen Bidyadhar Sethy
Bidyadhar Sethy Jing-Ping Liou
Jing-Ping Liou- School of Pharmacy, College of Pharmacy, Taipei Medical University, Taipei, Taiwan
Modulating epigenetic modification has been recognized for over a decade as an effective therapeutic approach to cancer and many studies of histone deacetylase (HDAC), one of the best known epigenetic modulators, have been published. HDAC modulates cell proliferation and angiogenesis and plays an essential role in cell growth. Research shows that up-regulated HDACs are present in many cancer types and synthetic or natural HDAC inhibitors have been used to silence overregulated HDACs. Inhibiting HDACs may cause arrest of cell proliferation, angiogenesis reduction and cell apoptosis. Recent studies indicate that HDAC inhibitors can provide a therapeutic effect in various cancers, such as B-cell lymphoma, leukemia, multiple myeloma and some virus-associated cancers. Some evidence has demonstrated that HDAC inhibitors can increase the expression of immune-related molecules leading to accumulation of CD8 + T cells and causing unresponsive tumor cells to be recognized by the immune system, reducing tumor immunity. This may be a solution for the blockade of PD-1. Here, we review the emerging development of HDAC inhibitors in various cancer treatments and reduction of tumor immunity.
Introduction
Epigenetic modification plays an important role in regulating gene expression without changing deoxyribonucleic acid (DNA) sequence (Yoo and Jones, 2006). Recently, much evidence has shown that histone function, modulated by various types of reversible modifications, such as methylation and acetylation, is crucial in heritable deliverance and cancer progression. Among these modifications, histone acetylation which is controlled by histone acetyl transferase (HAT) and especially, histone deacetylases (HDAC) are regarded as effective fields of cancer therapy (Jenuwein and Allis, 2001; Li and Seto, 2016; Sanaei and Kavoosi, 2019).
In general, histone acetylation is related to chromatin expression. HATs free chromatin through acetylation of histone lysine tails, producing HDACs which oppose this effect (Berger, 2007). Human HDACs have 18 highly conserved members. Based on their functions and analogies to yeast, HDACs can be divided into two families and four classes, a zinc-dependent family (Class I, Class IIa, Class IIb, and Class IV) and a nicotinamide adenine dinucleotide (NAD)-dependent family (Class III) (Seto and Yoshida, 2014). In addition to histone deacetylation, HDACs have also been found to regulate acetylation of a variety of non-histone proteins (Choudhary et al., 2009). The balance between acetylation and deacetylation is often upset in cancer, and expression of aberrant HDACs may lead to inactivation of tumor suppressing genes. On this basis, many compounds has been identified as HDAC inhibitors (HDACI). These include hydroxamic acids, benzamides, short-chain fatty acids and cyclic peptides, all of which modulate overexpression of HDACs in cancer (Noureen et al., 2010). These HDACIs have marked effects on cancer cells where they induce apoptosis, arrest cell cycles and even modulate the immune system (Hull et al., 2016). Here, we review usage of HDACIs in reduction of lymphomas and tumor immunity.
Lymphoma
Lymphoma and leukemia are blood cancers, and while they share some common symptoms, they have different origins. Leukemia typically begins in the bone marrow, and lymphoma generally develops in the lymphatic system. The lymphatic system, including marrow, spleen and lymph nodes, are part of the immune system, which helps to protect against infection.
Hodgkin’s (HL) and non-Hodgkin’s lymphoma (NHL) are the two main subtypes of lymphoma. HL, a relatively aggressive lymphoma, is characterized by the presence of very large cells known as Reed-Sternberg (RS) cells, which can be classified into two main types: classic Hodgkin lymphoma (cHL) and nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) (Table 1). On the other hand, in the view of the Leukemia and Lymphoma Society (LLS), NHL is broadly categorized into two groups: B-cell lymphomas and natural killer (NK)/T-cell lymphomas (Table 2). NHL is nine times more common than HL, and there are more than 60 subtypes of NHL, some “aggressive” (fast-growing) and others “indolent” (slow-growing). This classification determines the treatment options.

Table 1. Classification of Hodgkin lymphomas (cHL).
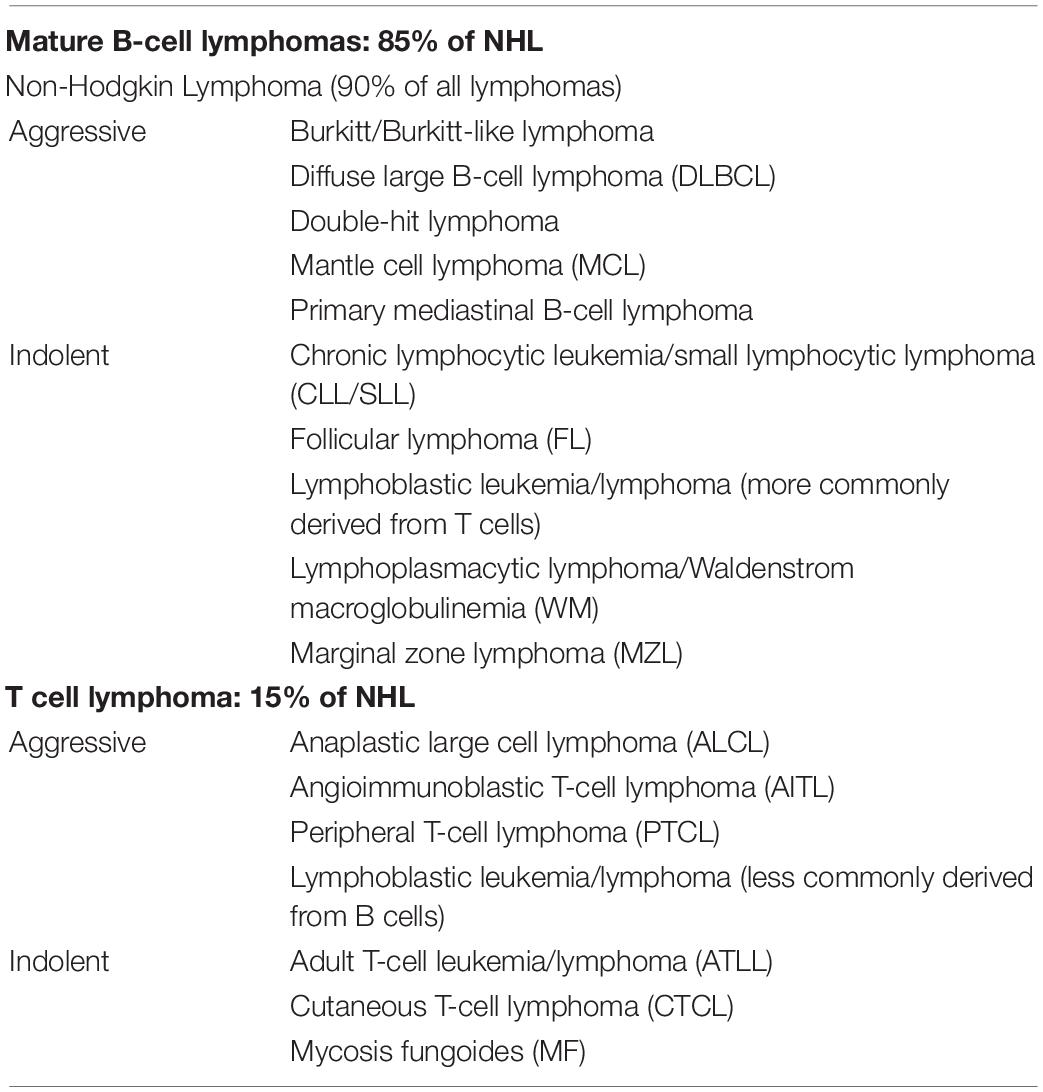
Table 2. Classification of Non-Hodgkin lymphomas.
The two principle therapies for lymphomas are radiation therapy and chemotherapy, and stem cell transplantation is another choice in some lymphoma types. Currently, increasing research efforts show that HDAC could be a therapeutic target in lymphomas (Table 3), and this has inspired us to try to understand its mechanism and development.
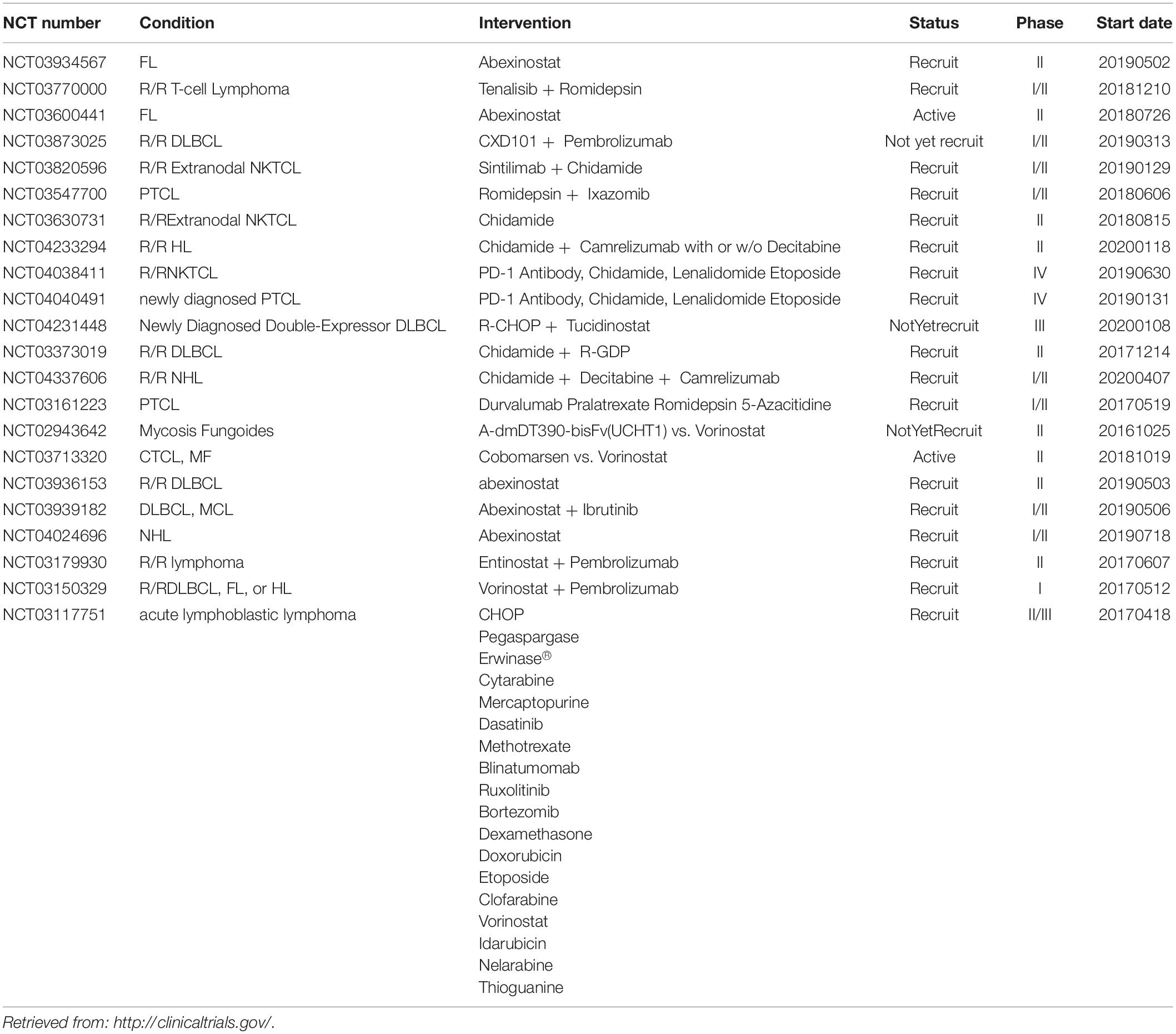
Table 3. HDAC related clinical trials started after 2017 for lymphoma treatment.
HDAC and T Cell Lymphoma
Role of HDACs in T Cell Development
HDACs have been reported to be necessary for t cell development. CD4 lineage integrity is regulated by HDAC1 and HDAC2 members through downregulation of Runx3/CBFβ complexes, which induce CD8 lineage programs in CD4 + t cells (Boucheron et al., 2014; Ellmeier, 2015). An HDAC1 and HDAC2 knockout test of t lymphocytes also has been found to result in cell cycle arrest and reduction of thymocytes. These events will eventually lead to decrease of the peripheral t cells and appearance of CD4 + and CD8 + t cells (Preglej et al., 2020). Another experiment with knockout HDAC1 and HDAC2 in mice showed the HDAC1/2-Sin3A-NuRD complex is disrupted. This may block double-negative (DN) to double-positive (DP) transition, and failure of proliferation. Moreover, insufficiency of HDAC1 or HDAC2 may lead to overacetylation of histones and chromosomal instability, finally causing t cell lymphoma (Dovey et al., 2013). Above all, this research indicates that HDAC1 and HDAC2 are essential for t cell development.
HDAC3 has also been found to be indispensable in steps of t cell progression, including commitment of CD4 and CD8, positive selection, and peripheral t cell maturation (Wang P. et al., 2020). HDAC3 deficiency in DP thymocytes terminates CD4-lineage program and redirects the MHC class II-restricted thymocytes toward the CD8-lineage program, due to the acetylation of histone expressing CD8-lineage genes, such as Runx3 and Patz1 (Philips et al., 2019). In a CD2-icre HDAC3 conditional knockout (HDAC3-cKO) mice, t cell development is blocked at the positive selection step, resulting in fewer CD4 and CD8 T cells, and cannot be rescued by TCR-transgene. The absence of HDAC3 renders RORγt unable to down-regulate although positively selected and fails to upregulate Bcl-2, which may lead to apoptosis (Philips et al., 2016). For t cell maturation, HDAC3 forms complexes with NF-kappaB-activating-protein (NKAP), which is necessary for recent thymic emigrants (RTE) to gain functional competency and transfer into a long-lived naive t cell pool. Lack of HDAC3 may cause CD50 downregulation which leads to t cell immaturation. In peripheral T cells, HDAC3 deficiency creates a defect in TNF licensing after TCR/CD28 stimulation (Hsu et al., 2015). Another distinct subset of t cells, nature killer t (NKT) cells, are also HDAC3 dependent during development. As was previously mentioned, NKAP activated through formation of complexes with HDAC3, also participates in invariant NKT (iNKT) cells lineage. Furthermore, HDAC3-deficient iNKT cells show low expression of nutrient receptors GLUT1, CD71 and CD98, and this results in incremental autophagy (Thapa et al., 2016, 2017).
Class IIa HDACs are also involved in t cell development. HDAC5 is implicated in t-regulatory (treg) cells homeostasis. In an HDAC5–/– mice model, Treg cells show reduced suppressive function, CD4 + t cells convert poorly into treg cells, and increasing acetylation of Foxo1 causes treg cells to experience difficulty in maintenance of the phenotype. CD8 + t cells have found to be less able to produce IFN-γ in HDAC5–/– mice (Xiao et al., 2016). HDAC7, a thymus-specific HDAC, acts as a regulator of t cell apoptosis and endothelial cell functions, is highly expressed in DP thymocytes, and inhibits Nur77 that is involved in apoptosis and negative selection. During TCR activation, HDAC7 is exported from the nucleus, leading to Nur77 expression and mediating TCR-mediated apoptosis (Dequiedt et al., 2003; Martin et al., 2008). HDAC6, a class IIb member of the HDACs, controls the production of immunosuppressive cytokine IL-10, and induction of antigen-presenting cells (APCs) that activate antigen-specific naïve t cells through formation of a complex with STAT3 (Cheng et al., 2014). HDAC10, another class IIb HDAC, mediates the inactivation of Foxp3. Foxp3 + treg cells are known to suppress immune responses, and HDAC10 dysfunction may cause some inflammatory disorders (Dahiya et al., 2020). HDAC11, which is the only member of class IV, negatively regulates the expression of IL-10. Overexpression of IL-10 will induce inflammatory APCs, priming naïve t cells and restoring the responsiveness of tolerant CD4 + t cells. Its adjustment with HDAC6 determines t cell activation (Villagra et al., 2009). HDAC11 acts as a positive controller of Foxp3 + tregs, and lack of HDAC11 will increase Foxp3 and TGF-β expression, which may lead to inflammation. The dynamic interaction between HDAC10 and HDAC11 serves to balance the immune response (Huang et al., 2017).
Evidence of HDACs in T Cell Lymphomas
Investigating the elaboration of HDACs in t cell lymphoma would assist an understanding of their pathogenesis, prognosis and role as a therapeutic target. Although the precise mechanism underlying this behavior has not been elucidated, it can be investigated through HDACIs.
Gene expression that mediates a balance between HAT and HDAC histone modification is important because it is also marks initiation and progression of cancer cells. HDACs intervene in carcinogenesis through the deacetylation of histone and non-histone proteins (Jenuwein and Allis, 2001). Recent research has shown that HDACs are involved in the expression of numerous oncogenes such as Bcl- xL-, BCL2-, c-Myc, TCRβ and Notch3 (Palermo et al., 2012; Kunami et al., 2014; Loosveld et al., 2014; Stengel et al., 2015). HDACs also participate in cytokine regulation. In the study of cutaneous t-cell lymphoma (CTCL) patients, 30% demonstrated the high affinity of the IL-2 receptor, which can be perturbed by HDACIs (Shao et al., 2002). Furthermore, HDAC1 and HDAC6 were also found to be upregulated in CTCL. This causes excessive secretion of IL-15, which mediates the inflammation that is crucial in CTCL, suggesting this oncogenic loop can be controlled by modulation of HDAC1 and HDAC6 (Mishra et al., 2016).
In t cell malignancies, HDACs act as negative controllers of apoptosis, and upregulation of HDACs will silence the pro-apoptotic gene and Bcl2 family expression. During the signaling pathway, HDACs can acetylate the chaperone heat shock protein 90 (HSP90), which stabilizes the client proteins RASGRP1 and c-RAF. These client proteins activate the mitogen-activated pathway, leading to the down regulation of the Bcl2 family (Ding et al., 2017). Upon treatment of peripheral t cell lymphoma with HDACi, chidamide induces cell apoptosis by downregulating Bcl2 and upregulating Caspase3 and Bax protein (Lu et al., 2016). The expression of a tumor suppressor gene was also found to be modulated by HDACs in CTCL cell lines. A combination of an HDACI, romidepsin and a demethylating agent, azacitidine leads to the induction of RHoB, the tumor suppressor gene, and to cell apoptosis (Rozati et al., 2016). The downstream apoptotic pathway regulated by HDACs may serve as a potent therapeutic target for apoptosis induction in a treatment for t cell malignancies (Figure 1).
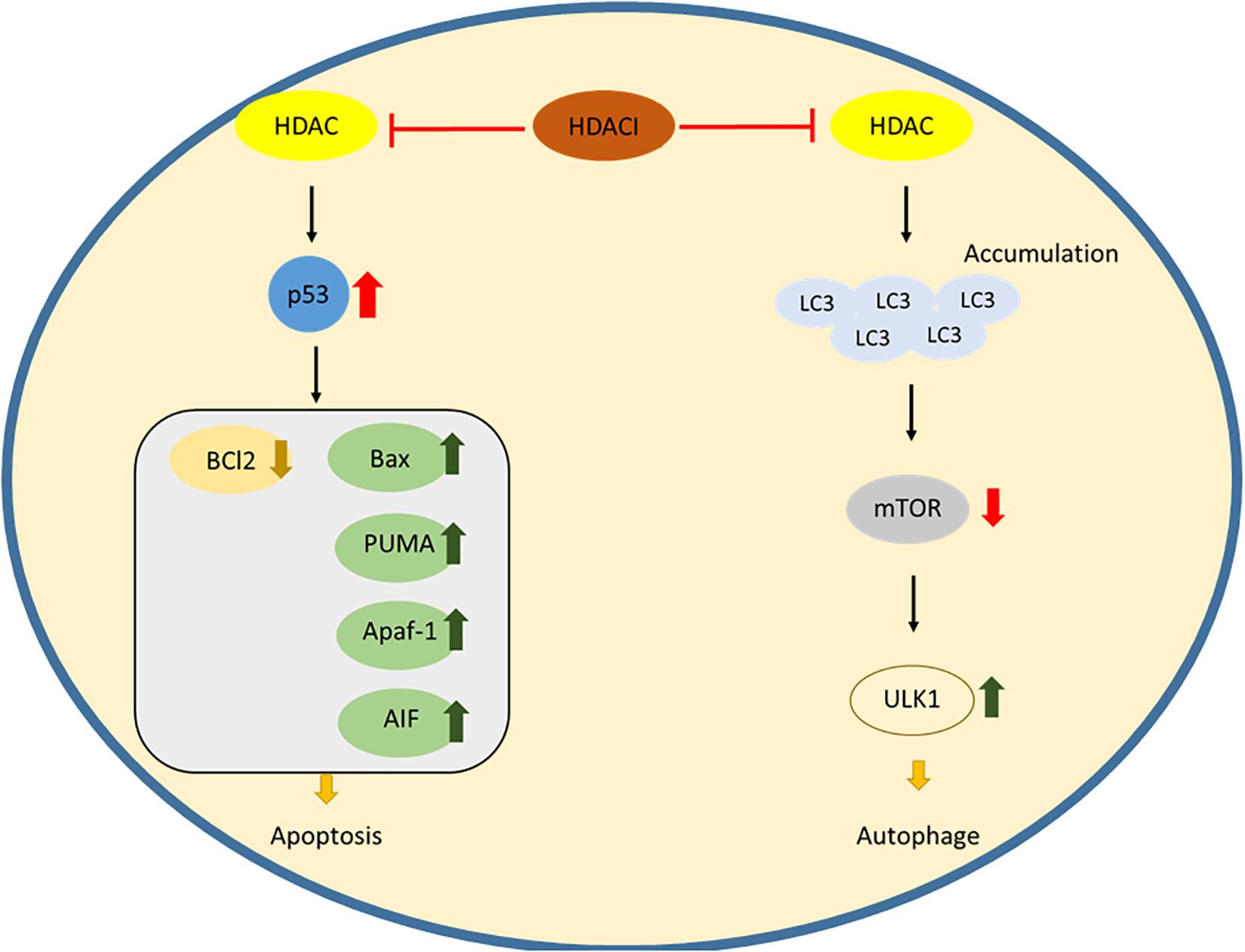
Figure 1. The action of HDAC inhibitors inducing apoptosis and autophagy.
Another self-devouring process similar to apoptosis is the source of dysfunction in t cell malignancies. Besides deacetylation of lysine residues in the histone, HDACs also have functions in regulation of cytosolic proteins which have a variety of cellular functions, including autophagy. SAHA (vorinostat), a pan-HDAC inhibitor, upregulates the expression of an autophagic factor LC3, inhibiting mTOR, the mammalian target of rapamycin and leading to activation of the autophagic protein kinase ULK1 (Figure 1; Gammoh et al., 2006). HDACs are inseparable by autophagy in cellular survival, and targeting autophagy by inhibition of HDACs could offer an effective treatment for t cell lymphomas.
The main function of HDACIs might be interference with histone and chromatin modification, but acetylation of histone and non-histone proteins may cause DNA damage, expression of suppressing genes in oncogenesis, and either lowering of the apoptotic threshold, or triggering autophagy response. These physiological processes have proved to be indispensable for HDACs in cancer pathogenesis and prognosis, and this makes them a prospective target for t cell malignancies.
Application of HDACIs in T Cell Lymphoma
Vorinostat (SAHA)
Vorinostat Figure 2, also been known as SAHA, is a hydroxamic acid HDAC inhibitor. It shows inhibitory activity in both class I and class II HDACIs with an IC50 less than 86 nM (Molecule of the month, 2006). To date, usage of SAHA has mostly been restricted to the treatment of CTCL. In cellular studies, SAHA shows a surprising anti-proliferative effect on human mantle lymphoma cells, CTCL cells, freshly isolated ATL cells and circulating malignant CTCL cells from patients by upregulating p21, decreasing the phosphorylation level of STAT6, increasing NF-κB in cytoplasm, and arresting the cell cycle (Nishioka et al., 2008; Zhang et al., 2005). In clinical trials, SAHA was tested in patients with refractory and relapsed CTCL with an ORR of 24 or 30% and duration of the response for 4 months or more in two phase II studies. SAHA was approved by FDA in 2006 as a treatment for refractory or relapsed CTCL (Duvic and Vu, 2007; Duvic et al., 2007; Olsen et al., 2007).
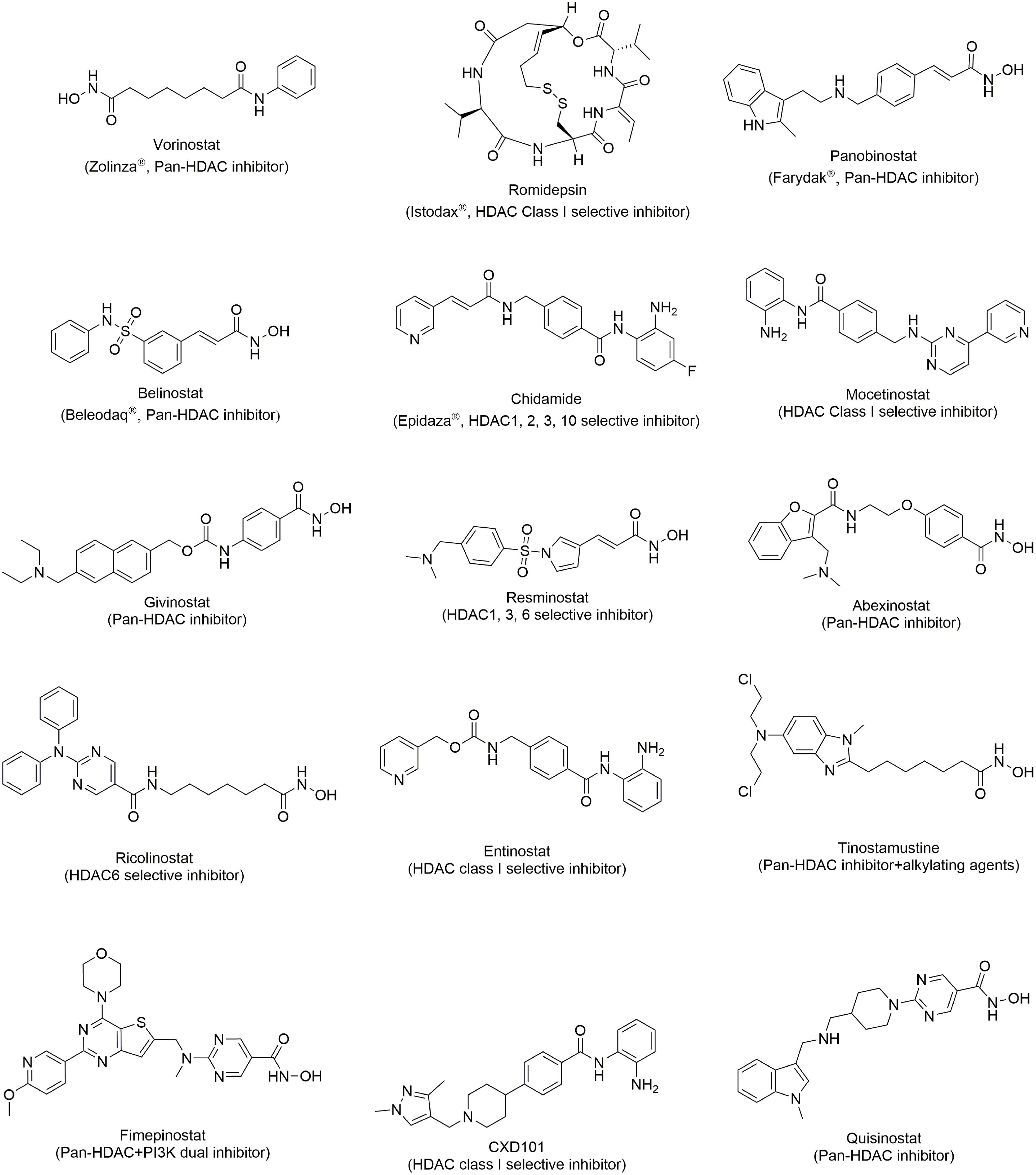
Figure 2. Structure of HDAC inhibitors.
In combination therapy, vorinostat combined with azacitidine has been tested. This resulted in 88% event-free and overall survival rates in t cell lymphoma patients (Nieto et al., 2016). In another clinical trial, vorinostat been used in a combination with gemcitabine, busulfan, and melphalan, and demonstrated high efficacy in refractory or poor-risk relapsed t cell lymphomas (Nieto et al., 2015). Vorinostat was also found to increase the effect of rituximab in a phase II study in newly diagnosed or relapsed/refractory NHL patients (Chen et al., 2015). Using a standard cyclophosphamide, hydroxydaunorpubicin, oncovin, and prednisone (CHOP) treatment with newly diagnosed peripheral t cell lymphoma patients in a phase I clinical trial, vorinostat also obtained a good therapeutic effect (Oki et al., 2013b). Combination of vorinostat with PI3K inhibitors or HSP90 inhibitors resulted in cytotoxic antagonism in CTCL cells, and investigation of this could be useful (Wozniak et al., 2010; Hutt et al., 2014). The proteasome inhibitor, bortezomib also causes a synergetic effect inducing apoptosis in CTCL patients (Heider et al., 2009). However, combination therapies do not always give positive results. For example, a study of a combination of lenalidomide, vorinostat, and dexamethasone used to treat patients with relapsed/refractory peripheral t cell lymphoma (PTCL), resulted in median-progression free survival and low overall survival (Hopfinger et al., 2014). Thus, vorinostat is still significant in lymphoma therapy.
Romidepsin (FR901228)
Romidepsin Figure 2 is a natural depsipeptide isolated from bacteria. It displays selective inhibition toward class I HDACs but is weak in HDAC IIB (Ueda et al., 1994). The single use of romidepsin, exhibits effectiveness in relapsed/refractory CTCL patients and was approved by FDA in 2009 for the treatment of CTCL (Imam et al., 2013; Reddy, 2016). Romidepsin demonstrates outstanding clinical response in relapsed/refractory PTCL patients and it was also approved in 2011 for PTCL treatment (Barbarotta and Hurley, 2015; Iyer and Foss, 2015; Irlé and Weintraub, 2016; Foss et al., 2017).
In combination therapies, romidepsin has been combined with conventional drugs for hematological malignances, such as methotrexate, vincristine, imatinib, cytarabine, carboplatin, doxorubicin, 4-hydroperoxy-cyclophosphamide, etoposide, 6-mercaptopurine, and SN-38. All of these showed an additive result, indicating that combination therapy with romidepsin is promising. Combining CHOP with romidepsin in newly diagnosed PTCL patients exhibited a surprising therapeutic effect with an overall survival of 71% at the median follow-up of 30 months (Dupuis et al., 2015). Unlike vorinostat, romidepsin shows synergistic effects with lenalidomide in relapsed/refractory lymphomas (Cosenza et al., 2016). Aurora kinase inhibitors are a promising agents for treatment of TCL, and combined with romidepsin their therapeutic effect is highly synergized (Zullo et al., 2014). Clinical trials of this combination are in progress. Combinations of romidepsin with other drugs, including pralatrexate, gemcitabine, and ICE (Wozniak et al., 2010; Sung et al., 2014; Foss et al., 2017) are being studied. Romidepsin seems to be useful in combination therapies, although more investigation is necessary.
Belinostat (PXD101)
Belinostat Figure 2 is pan-HDAC inhibitor with a sulfonamide-hydroxamic acid structure. It exhibited nanomolar inhibition against HDACI, II, and IV (Chowdhury et al., 2011). The clinical data from relapsed/refractory PTCL patients shows that belinostat, with high efficacy and low toxicity is an ideal drug for cancer treatment (Campbell and Thomas, 2017). With promising results, belinostat was approved for sale in 2014 for the treatment of relapsed or refractory PTCL (Poole, 2014). Because of its safety, belinostat is a first-line drug for relapsed or refractory PTCL or various drug combination therapies.
For the combination therapies, belinostat has been used with CHOP. In spite of its use as a first-line treatment for relapsed or refractory PTCL, its combination with CHOP delivered a poor prognosis with relapse within 5 years (Johnston et al., 2015). Other usage in combination with bortezomib, volasertib, zidovudine, or carfilzomib, has already been published. Most of these show a potential therapeutic effect, and this provides more alternative options for treatment of lymphoma patients.
Panobinostat (LBH-589)
Panobinostat Figure 2 is a cinnamic hydroxamic acid HDAC inhibitor which inhibits HDACI, II and IV and is 10-fold more potent than SAHA (Atadja, 2009). In clinical trials, panobinostat was demonstrated to be effective in patients with advanced CTCL (Duvic et al., 2013). In a clinical trial, panobinostat was acceptably tolerable and led to a modest overall response. However, it failed in the phase II trial due to its low response and short time to progression in refractory CTCL patients (Ellis et al., 2008; McCann and Story, 2013). Panobinostat is now undergoing a clinical trial with PTCL (Tan et al., 2015).
Panobinostat also guided some combination therapies. A combination of panobinostat and bortezomib in PTCL highly synergized the ubiquitination ability in preclinical studies (Samimi et al., 2013). In further clinical studies, a combination of bortezomib displayed promising efficacy, improving the outcome following a single dosage. This Phase III clinical trial for PTCL treatment (Tan et al., 2015) has been completed. In other clinical studies, conspicuously, administration of everolimus and panobinostat to TCL patients decreased serum cytokine levels (Oki et al., 2013a). The severe adverse effects of panbinostat makes its development difficult, but it still offers a new approach for lymphoma therapies. These results in combination with other agents may a sign of a new era of PTCL therapies.
Quisinostat
Quisinostat Figure 2 is a broad spectrum HDAC inhibitor, which has strong inhibition activity in HDACs, except for HDAC6, 7, and 9. Its clinical trial for CTCL treatment has been completed in 2016 (NCT01486277). However, the result was failed to be superior comparing to other HDACIs, such as vorinostat or romidepsin while treating with CTCL patients. Furthermore, after dosing quisinostat 5 of 26 patients have grade 3 drug related adverse effect, including, hypertension, lethargy, chills, pyrexia, pruritus and hyperkalemia, even though the safety and tolerability profile is similar to other pan-HDACIs, the overall outcome limited its development (Child et al., 2016). Recently, quisinostat has been combined with bortezomib and dexamethasone for multiple myeloma (NCT01464112), but no any clinical trials in lymphomas.
Perspective
Recently, HDACIs has been approved for TCL treatment, and most of them belongs to pan-inhibitor, such as vorinostat and belinostat. Indeed, these pan-HDAC inhibitors brought effectiveness in PTCL patients, but not in CTCL patients. Interestingly, romidepsin, a class I HDACs selective inhibitor, showed promising result in CTCL patients. Above all, inhibition of other HDAC subtypes would decrease the efficacy in CTCL, which inspired us targeting class I HDACs might increase the application of HDACI in TCL therapies. Moreover, romidepsin also showed broader availability comparing to other pan-HDACIs in combination with other target therapies or chemotherapies. Thus, further pre-clinical studies are necessary to understand the precise mechanism.
HDAC and B Cell Lymphoma
Role of HDACs in B Cell Development
In B cell differentiation, HDAC1 and HDAC2 promote the development in the pre-B cell stage that progresses the cell cycle from G1 to S. Knockout of HDAC1 and HDAC2 leads to cell cycle arrest and expression of p21 and p57, which may cause apoptosis (Yamaguchi et al., 2010). At the terminal stage of B cell development, Blimp-1 restrains c-myc through the aggregation of HDAC1 and HDAC2 (Yu et al., 2000). In HDAC3 knockdown mice, the progenitor B cells cause impaired B cell maturation, and defects in VDJ (varies, diversity, joining) recombination (Stengel et al., 2017). HDACs participate in complex formation in different stages of B cell development. HDACs have been considered to be a component of the STAT5a-LSD1 complex, which demonstrates the possible activation of STAT5a in the early stages of B cell development (Nanou et al., 2017). In mature B cells, Bach2 recruits the HDAC3-NCoR1/NCoR2-Rif1 complex to repress Pdm1 transcription thus blocking the differentiation between B cells and plasma cells (Tanaka et al., 2016).
BCL6, a sequence-specific repressor of transcription, requires formation of complexes with specific HDACs. HDAC3, HDAC4, and HDAC9 have been found to be a corepressor of BCL6 (Lemercier et al., 2002; Pasqualucci et al., 2011; Gil et al., 2016). In the HDAC7 conditional deletion mice experiment, HDAC7 was deemed to control the pro-B cell to pre-B cell transition. In pro-B cells, the transcription factor ME2FC is complexed with HDAC7, which silences the lineage-inappropriate genes, ensuring the correct B cell differentiation (Barneda-Zahonero et al., 2015; Azagra et al., 2016). HDAC6 was also shown to be a controller of PD-L1 in B cells, regulating the immunogenicity (Powers et al., 2014). Selective HDAC6 inhibitors are considered to be a new target for immunotherapy, but the precise mechanism is needed for further investigation.
Evidence of HDACs in B Cell Lymphoma
In the pathogenesis of lymphoma, HDAC-BCL6 complexes are often aberrant in the transcription step. For instance, the germinal centers (GC) of B cells in CREBBP-regulated/active enhancers are negatively regulated through H3K27 deacetylation by the BCL6-SMRT-HDAC3 complex. In folicullar lymphoma (FL) and diffuse large B cell lymphoma (DLBCL), however, CREBBP mutations lead to unopposed deacetylation by BCL6-SMRT-HDAC3 at an enhancer of B cell signal transduction and expression of immune response genes, which results in lymphomagenesis (Pasqualucci et al., 2011; Jiang et al., 2017). In B-NHL cells, the abnormal expression of HDAC9-BCL6 complex may cause B-lymphoproliferative disorders. Overexpression of HDAC9 contributes to alter pathways involved in growth and survival, as well as modulation of BCL6 activity and p53 tumor suppressor function (Gil et al., 2016). HDAC4 plays a key role in suppressing oncogenes. Dysfunction of HDAC4 disrupts the complex with BCL6 and this may lead to induce uncontrolled proliferation, clonogenic potential, and decreased apoptosis (Lemercier et al., 2002; Sandhu et al., 2012). Targeting these HDACs might therefore have promising effect in B cell lymphoma therapies.
Application of HDACIs in B Cell Lymphoma
Vorinostat (SAHA)
Vorinostat, the first approved HDAC inhibitor, has been used in a phase II clinical trial for relapsed DLBCL therapies. But the overall response rate (ORR) was only 5.6%, which indicates that in single usage, vorinostat is limited (Crump et al., 2008). However, other clinical trials conducted using vorinostat as an FL treatment showed 8-times better ORRs of 47 and 49% (Kirschbaum et al., 2011; Ogura et al., 2014).
In the combination therapies vorinostat, combined with rituximab or R-CHOP in NHL patients, also showed enhanced effects, especially in DLBCL patients with an 81% ORR (Chen et al., 2015; Straus et al., 2015; Persky et al., 2018). A combination of R-ICE and vorinostat for relapsed or refractory NHL patients also had 70% ORR (Budde et al., 2013). Pre-clinical experiments showed that SAHA and topoisomerase inhibitors surprisingly defeated lymphoma cells, and this might be a new aspect for NHL therapies (Seo, 2015).
Belinostat (PXD101)
Similar to vorinostat, belinostat behaves poorly in monotherapies. A Phase II clinical trial showed that administration of belinostat to relapsed or refractory aggressive B-NHL patients resulted in only 10.5% ORR (Puvvada et al., 2016). This result terminated the research on monotherapies of belinostat in B cell lymphoma patients. Several combination therapies, however, are still in clinical trials, for example a trial in combination with carfilzomib, a proteasome inhibitor, is still ongoing (NCT02142530).
Chidamide
Chidamide Figure 2 is a selective HDAC class I inhibitor, and is now approved only in China (Ning et al., 2012). Its therapeutic effect in relapsed or refractory B-NHL is still being evaluated in clinical trials (NCT03245905 and NCT03410004).
Combination of chidamide with other chemotherapies have also been investigated in clinical trials. Such combinations include R-GDP (NCT03373019), vinorelbine, liposomal doxorubicin, dexamethasone and thalidomide (VDDT) (NCT02733380), dexamethasone and ICE (DICE) (NCT03105596), and R-CHOP (NCT03201471) in relapsed or refractory B-NHL. A clinical trial of R-CHOP combined with chidamide (NCT02753647) in untreated elderly DLBCL patients is progressing (Wang L. et al., 2020).
Mocetinostat
Mocetinostat Figure 2 is a selective HDAC I and IV inhibitor, which has been approved by FDA for use in cases of relapsed or refractory CTCL (Boumber et al., 2011). The effect of mocetinostat in a Phase II clinical trial showed low ORR in both DLBCL (18.9%) and FL (11.5%) (Batlevi et al., 2017). These results are similar to those from other HDAC inhibitors, whether selective or not, and show low ORR in B cell lymphoma patients. Therefore, HDACIs may significantly increase the therapeutic effects in combination with other chemotherapies. However, mocetinostat only been used with azacitidine (NCT00543582) in a clinical trial, and further research is necessary.
CXD101
CXD101 Figure 2 is a selective class I HDACs inhibitor, which is now undergoing phase I clinical trial to assess its tolerability, safety, pharmacokinetics and pharmacodynamics in advanced malignancies (NCT01977638). It has been hypothesized that selectively inhibiting class I HDACs could reduce the toxicity, which is brought by off target inhibition on class II HDACs. Preliminary result shows that, PXD101 has lower toxicity and higher tolerability than other non-selective inhibitor (Eyre et al., 2019).
Besides, CXD101 also test with the combination of pembrolizumab for R/R DLBCL treatment (NCT03873025), but no any other result has been reported.
Ricolinostat (ACY-1215)
Ricolinostat Figure 2 is the only HDAC6 selective inhibitor, which entered clinical trial for NHL therapies (NCT02091063). However, there is no any further development in single agent therapies for NHL.
Surprisingly, ricolinostat has synergizing effect combining with other drug or regimen. Such as ibrutinib (Amengual et al., 2017), carfilzomib (Dasmahapatra et al., 2014), bendamustine (Cosenza et al., 2017), and crizotinib (Liu et al., 2018) in DLBCL or MCL models. These combination therapies showed great potency toward DLBCL and MCL, but its efficacy in human is still under investigation.
Fimepinostat (CUDC-907)
Fimepinostat Figure 2, targeting HDAC and PI3K, is the first dual-target inhibitor that has been approved for R/R DLBCL treatment in clinical trials. Its phase I study shows fimepinostat has better tolerability, and lower toxicity than other FDA approved single target HDAC or PI3K inhibitor (Younes et al., 2016). Now the efficacy is evaluating in phase II clinical trial (NCT02909777).
Perspective
Single agents of HDACIs has low response in all types of BCL. However, the significantly synergizing effect with other drugs makes it worth to be developed, especially selective HDAC inhibitors. Class I HDAC inhibitor improved the tolerability and reduced the toxicity and HDAC6 inhibitor also showed promising effect in combination with other drugs even overcome the drug resistance. These results may encourage us to fully understand the mechanism and develop more specific selective HDACIs.
Furthermore, fimepinostat, the first-in-class dual target inhibitor for DLBCL therapies, successfully decrease the toxicity comparing to dose single target agent. This may also give us an inspiration to develop other dual-target inhibitor.
Hodgkin Lymphomas
Introduction
One of the most curable cancer types, Hodgkin lymphoma (HL) is a type of B cell lymphoma, which has specific and unique characteristics. Hodgkin lymphoma was first been identified by Hodgkin in 1832 (Hodgkin, 1832). Subsequently, this lymphoma was named after him in 1865 by Wilks (Wilks, 1856). According to World Health organization (WHO), HL can be classified into two main types, classical Hodgkin lymphoma (cHL) and nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL). cHL accounts for 95% of all HL patients and the remainder are NLPHL. In this review we will mainly focus on cHL (Stathis and Younes, 2015).
In cHL patients, mononuclear Hodgkin cells and multinucleated Reed-Stemberg (HRS) cells arise from monoclonal B lymphocytes in the germinal center of lymphoid tissue and effect the rearrangement of IgG genes (Diehl, 2007). According to statistics, around 40% of cHL patients are infected by Epstein-Barr virus (EBV) and 100% of patients are infected with the human immunodeficiency virus (HIV). The apoptosis of these abnormal cells was inhibited in a manner which correlates with the expression of NF-κB, Notch 1 and some other transcription factors (Re et al., 2005). Research shows that, CD30 surface receptors, a member of tumor necrosis factor (TNF) receptor superfamily, will be characteristically expressed in HRS cells. The expression of TNF receptors mediates various signaling pathways, including the activation of NF-κB (Dürkop et al., 1992; Duckett et al., 1997).
To date, around 80% of cHL patients can be cured after receiving radiation therapy and chemotherapy. Recently, early and advanced stages in cHL patients were treated with doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) in a first line and combined with bleomycin, etoposide, adriamycin, cyclophosphamide, vincristine, procarbazine, and prednisone (BEACOPP) in a second line chemotherapy regimen. Although BEACOPP showed better overall survival rate than ABVD, its high acute toxicity makes ABVD more acceptable (Carde et al., 2016). Targeting CD30 on the other hand, is another strategy for cHL. In a combination with brentuximab vedotin and ABVD, it showed a promising therapeutic effect, but resulted in high pulmonary toxicity. By omitting bleomycin the toxicity was dramatically reduced, and this type of B-AVD therapy has become popular (Younes et al., 2013). For relapsed and refractory cHL patients, platinum- or gemcitabine-based therapies were used in a first line followed by autologous stem-cell transplantation, which can cure 60% of R/R cHL patients (Clavio et al., 2005). Nowadays, cHL is almost a curable disease, but delayed treatment-related toxicity may lead to second malignancies and cardiovascular disease (Armitage, 2010), and this has inspired a search for new therapeutic strategies.
Evidence of HDAC in Classical Hodgkin Lymphoma
As was mentioned previously, expression of abnormal HDACs has been found in both t cell and B cell lymphomas, which made it a promising therapeutic target. Because cHL is a type of B cell lymphoma, HDACs also are overexpressed in cHL. Research from Tzankov et al. shows that HDAC1, 2, and 3 are highly expressed in HRS cells. Interestingly, after treatment with HDACIs, the inhibition of HDAC1 inhibition in HRS cells leads to a poorer prognostic effect, for reasons that are still under investigation. Notwithstanding this, HDAC is still deemed a potent therapeutic target for cHL (Adams et al., 2010).
Application of HDACIs in Classical Hodgkin Lymphomas
Recently, several HDACIs Figure 2 have been tested against R/R cHL in clinical trials, including panobinostat (Younes et al., 2012), vorinostat (NCT00132028), givinostat (NCT00496431), resminostat (NCT01037478), mocetinostat (NCT00358982), abexinostat (NCT00724984, NCT01149668), ricolinostat (NCT02091063), entinostat (NCT00866333), and tinostamustine (NCT02576496). These HDACIs were used as single treatment which brought patients positive results. Comparison, however, with other target therapies, such as PD-1 antibodies or some immunomodulatory antibodies, showed that HDACIs give relatively low overall response rates and comparable progression-free survivals (Adams et al., 2010). Above all, HDACIs might not be suitable for cHL treatment. On the other hand, HDACIs have been reported to have the ability to alter cytokines, which may enhance the immune response (Oki et al., 2014). Downregulation of PD1 on t cell and upregulation of OX40 ligand in HRS cells can exhibit antitumor immunity through HDAC11 inhibition (Buglio et al., 2011). This makes HDAC a favorable enhancer in numerous combination therapies and a number of clinical trials are now in progress. For instance, panobinostat has been used with everolimus (NCT00918333), lenalidomide (NCT01460940), cytarabine (NCT01321346), and ICE (NCT01169636). Other HDACIs are also being tested in combination therapies, such as combination of vorinostat with lenalidomide (NCT01116154), alisertib (NCT01567709), R-CHOP (NCT00667615), or a combination of mocetinostat with azacitidine (NCT00543582) and brentuximab vedotin (NCT02429375). Although some preliminary results showed high efficacy, further evaluation is necessary.
Perspective
Similar to the role of HDACIs in BCL therapies, HDACIs are more like an enhancer in cHL therapies. It is certainly single dosage of HDACIs provided some positive result. However, other target therapies exhibited more potent in cHL therapies. In spite of that, HDACIs displayed dramatically increase of efficacy, when combined with other cHL therapies. Though, the clinical studies haven’t been completed yet, still give us some inspiration of HDACIs‘ character in cHL therapies.
Tumor Immunity Reduction
Tumor immunity escape is an important issue in cancer therapy. As cancer cells are known to be abnormal proliferating cells, they have a unique microenvironment with which they can evade the immune system. The programmed death ligand-1/programmed death-1 (PD-L1/PD-1) pathway, is at the root of the cancer cells’ tolerance to the immune system. PD-1 overexpression in the tumor microenvironment causes an immunosuppressive effect (He et al., 2015; Jiang et al., 2019). Research on melanoma tumor cells has found that the tumor immunity is utilized by the host immune system, instead of by the tumor itself, which means that one can reduce the tumor immunity by modulating the immune system. B7 is a protein family that is found on antigen presenting cells (APC), and can bind to t cells. B7-H1, one of the B7 family members also known as PD-L1, has been found to be abundant in cancer cells. PD-L1 can be induced by cytokines such as IFN-γ or IL-8, and these cytokines are produced by CD8 + t cells, and the tumor microenvironment can be considered as a pro-inflammatory condition. Thus, targeting CD8 + t cell could be a solution to reduction of tumor immunity (Dong and Chen, 2003; Harlin et al., 2009; Spranger et al., 2013).
Application of HDACIs to Tumor Immunity Reduction
As mentioned previously, HDACs play a crucial role in t cell regulation. In CD8 + t cells, the expression of PD1/PD-L1 has been shown through an inhibition assay by ACY241, a selective HDAC6 inhibitor to be positively controlled by HDAC6. Notably, HDACs other than HDAC6 are important in the CD8 + t cell immune response pathway (Yu et al., 2002; Adcock, 2007). Therefore, several HDACIs have been examined for their ability to reduce the tumor immunity (Bae et al., 2018).
Vorinostat and panobinostat have been used with immune cell stimulating antibodies in renal and colon carcinomas, and showed a surprising effect in inhibition of tumor growth (Christiansen et al., 2011). The selective HDAC inhibitor, entinostat, combined with IL-2 is also effective in a renal cell carcinoma mice model (Kato et al., 2007). Besides conventional HDAC inhibitors, some new compounds were synthesized for this kind of HDAC mediated immunotherapy (Vo et al., 2009). A novel HDAC and HSP90 dual inhibitor also causes downregulation of PD-L1 expression (Mehndiratta et al., 2020). These results are still in pre-clinical stages, but they provide a new aspect in immunotherapies.
Conclusion
Lymphomas are a group of hematopoietic malignancies with complex pathogenesis and which easily relapse. Thus, new therapeutic targets are necessary. As HDAC being an important character in epigenetic modulation, HDACIs have been approved for the treatment of several cancers. These HDACIs also show high potency in treatment of lymphomas. In T cell lymphoma, single usage of HDACIs shows promising results and combination of HDACIs with conventional chemotherapies showed a synergistic effect comparable to that from a single dosage. However, in B cell lymphoma and Hodgkin lymphoma, single usage of HDACIs, shows low overall response rates, which means it may be unsuitable for both B cell lymphoma and Hodgkin lymphoma. Fortunately, when combined with chemotherapies or other targeting therapies, the therapeutic effect was surprisingly enhanced. In some of the cases, the overall response rate was increased to more than 80%. This result inspired us to focus on the character of HDACIs in combination therapies. We concluded that in studies of the role of HDACs in T cells and tumor immunity expression, HDACIs might act as an enhancer, which can reduce the tumor immunity, thereby increasing the drugs’ therapeutic effects. According to the information gained from B cell lymphoma and Hodgkin lymphoma treatments, we predict that HDACIs can not only arrest the cell cycle and trigger apoptosis, but modulate the t cell function in order to reduce tumor immunity. Indeed, there is some research in this area but still the precise mechanism should be clarified. Noticeably, HDACIs can both reduce the production of cytokines and lower the expression of PD-1. The modulation of HDACs by these two actions is already known, but their influence on the therapeutic effects remains unknown and further investigation is needed. HDACIs are still potent and prospectively useful either in immunotherapies or target therapies. We hope that HDACIs may lead us to cures for cancer in the future.
Author Contributions
J-PL: framework design and concept development. I-CC: article composition. BS: collection. All authors contributed to the article and approved the submitted version.
Funding
Ministry of Science and Technology, Taiwan (108-2113-M-038-005) has funded for the research and publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.576391/full#supplementary-material
References
Adams, H., Fritzsche, F. R., Dirnhofer, S., Kristiansen, G., and Tzankov, A. (2010). Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Ther. Targets 14, 577–584. doi: 10.1517/14728221003796609
Adcock, I. M. (2007). HDAC inhibitors as anti-inflammatory agents. Br. J. Pharmacol. 150, 829–831. doi: 10.1038/sj.bjp.0707166
Amengual, J. E., Prabhu, S. A., Lombardo, M., Zullo, K., Johannet, P. M., Gonzalez, Y., et al. (2017). Mechanisms of acquired drug resistance to the HDAC6 selective inhibitor ricolinostat reveals rational drug-drug combination with ibrutinib. Clin. Cancer Res. 23, 3084–3096. doi: 10.1158/1078-0432.CCR-16-2022
Armitage, J. O. (2010). Early-stage Hodgkin’s lymphoma. N. Engl. J. Med. 363, 653–662. doi: 10.1056/NEJMra1003733
Atadja, P. (2009). Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 280, 233–241. doi: 10.1016/j.canlet.2009.02.019
Azagra, A., Román-González, L., Collazo, O., Rodríguez-Ubreva, J., de Yébenes, V. G., Barneda-Zahonero, B., et al. (2016). In vivo conditional deletion of HDAC7 reveals its requirement to establish proper B lymphocyte identity and development. J. Exp. Med. 213, 2591–2601. doi: 10.1084/jem.20150821
Bae, J., Hideshima, T., Tai, Y. T., Song, Y., Richardson, P., Raje, N., et al. (2018). Histone deacetylase (HDAC) inhibitor ACY241 enhances anti-tumor activities of antigen-specific central memory cytotoxic t lymphocytes against multiple myeloma and solid tumors. Leukemia 32, 1932–1947. doi: 10.1038/s41375-018-0062-8
Barbarotta, L., and Hurley, K. (2015). Romidepsin for the treatment of peripheral T-cell lymphoma. J. Adv. Pract. Oncol. 6, 22–36.
Barneda-Zahonero, B., Collazo, O., Azagra, A., Fernández-Duran, I., Serra-Musach, J., Islam, A. B., et al. (2015). The transcriptional repressor HDAC7 promotes apoptosis and c-Myc downregulation in particular types of leukemia and lymphoma. Cell Death Dis. 6:e1635. doi: 10.1038/cddis.2014.594
Batlevi, C. L., Crump, M., Andreadis, C., Rizzieri, D., Assouline, S. E., Fox, S., et al. (2017). A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 178, 434–441. doi: 10.1111/bjh.14698
Berger, S. (2007). The complex language of chromatin regulation during transcription. Nature 447, 407–412. doi: 10.1038/nature05915
Boucheron, N., Tschismarov, R., Goeschl, L., Moser, M. A., Lagger, S., Sakaguchi, S., et al. (2014). CD4+ t cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC2. Nat. Immunol. 15, 439–448. doi: 10.1038/ni.2864
Boumber, Y., Younes, A., and Garcia-Manero, G. (2011). Mocetinostat (MGCD0103): a review of an isotype-specific histone deacetylase inhibitor. Expert Opin. Investig. Drugs 20, 823–829. doi: 10.1517/13543784.2011.577737
Budde, L. E., Zhang, M. M., Shustov, A. R., Pagel, J. M., Gooley, T. A., Oliveira, G. R., et al. (2013). A phase I study of pulse high-dose vorinostat (V) plus rituximab (R), ifosphamide, carboplatin, and etoposide (ICE) in patients with relapsed lymphoma. Br. J. Haematol. 161, 183–191. doi: 10.1111/bjh.12230
Buglio, D., Khaskhely, N. M., Voo, K. S., Martinez-Valdez, H., Liu, Y. J., and Younes, A. (2011). HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood 117, 2910–2917. doi: 10.1182/blood-2010-08-303701
Campbell, P., and Thomas, C. M. (2017). Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J. Oncol. Pharm. Pract. 23, 143–147. doi: 10.1177/1078155216634178
Carde, P., Karrasch, M., Fortpied, C., Brice, P., Khaled, H., Casasnovas, O., et al. (2016). Eight cycles of ABVD versus four cycles of BEACOPPescalated plus four cycles of BEACOPPbaseline in stage III to IV, international prognostic score = 3, high-risk Hodgkin lymphoma: first results of the phase III EORTC 20012 intergroup trial. J. Clin. Oncol. 34, 2028–2036. doi: 10.1200/JCO.2015.64.5648
Chen, R., Frankel, P., Popplewell, L., Siddiqi, T., Ruel, N., Rotter, A., et al. (2015). A phase II study of vorinostat and rituximab for treatment of newly diagnosed and relapsed/refractory indolent non-Hodgkin lymphoma. Haematologica 100, 357–362. doi: 10.3324/haematol.2014.117473
Cheng, F., Lienlaf, M., Wang, H. W., Perez-Villarroel, P., Lee, C., Woan, K., et al. (2014). A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J. Immunol. 193, 2850–2862. doi: 10.4049/jimmunol.1302778
Child, F., Ortiz-Romero, P. L., Alvarez, R., Bagot, M., Stadler, R., Weichenthal, M., et al. (2016). Phase II multicentre trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA mycosis fungoides/Sézary syndrome. Br. J. Dermatol. 175, 80–88. doi: 10.1111/bjd.14427
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
Chowdhury, S., Howell, G. M., Teggart, C. A., Chowdhury, A., Person, J. J., Bowers, D. M., et al. (2011). Histone deacetylase inhibitor belinostat represses survivin expression through reactivation of transforming growth factor beta (TGFbeta) receptor II leading to cancer cell death. J. Biol. Chem. 286, 30937–30948. doi: 10.1074/jbc.M110.212035
Christiansen, A. J., West, A., Banks, K. M., Haynes, N. M., Teng, M. W., Smyth, M. J., et al. (2011). Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc. Natl. Acad. Sci. U.S.A. 108, 4141–4146. doi: 10.1073/pnas.1011037108
Clavio, M., Garrone, A., Pierri, I., Michelis, G. L., Balocco, M., Albarello, A., et al. (2005). Ifosfamide, epirubicin, etoposide (IEV) and autologous peripheral blood progenitor cell transplant: a feasible and effective salvage treatment for lymphoid malignancies. Oncol. Rep. 14, 933–940.
Cosenza, M., Civallero, M., Fiorcari, S., Pozzi, S., Marcheselli, L., Bari, A., et al. (2016). The histone deacetylase inhibitor romidepsin synergizes with lenalidomide and enhances tumor cell death in T-cell lymphoma cell lines. Cancer Biol. Ther. 17, 1094–1106. doi: 10.1080/15384047.2016.1219820
Cosenza, M., Civallero, M., Marcheselli, L., Sacchi, S., and Pozzi, S. (2017). Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 22, 827–840. doi: 10.1007/s10495-017-1364-4
Crump, M., Coiffier, B., Jacobsen, E. D., Sun, L., Ricker, J. L., Xie, H., et al. (2008). Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann. Oncol. 19, 964–969. doi: 10.1093/annonc/mdn031
Dahiya, S., Beier, U. H., Wang, L., Han, R., Jiao, J., Akimova, T., et al. (2020). HDAC10 deletion promotes Foxp3+ T-regulatory cell function. Sci. Rep. 10:424. doi: 10.1038/s41598-019-57294-x
Dasmahapatra, G., Patel, H., Friedberg, J., Quayle, S. N., Jones, S. S., and Grant, S. (2014). In vitro and in vivo interactions between the HDAC6 inhibitor ricolinostat (ACY1215) and the irreversible proteasome inhibitor carfilzomib in non-Hodgkin lymphoma cells. Mol. Cancer Ther. 13, 2886–2897. doi: 10.1158/1535-7163.MCT-14-0220
Dequiedt, F., Kasler, H., Fischle, W., Kiermer, V., Weinstein, M., Herndier, B. G., et al. (2003). HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 18, 687–698. doi: 10.1016/S1074-7613(03)00109-2
Diehl, V. (2007). Hodgkin’s disease–from pathology specimen to cure. N. Engl. J. Med. 357, 1968–1971. doi: 10.1056/NEJMe078173
Ding, H., Peterson, K. L., Correia, C., Koh, B., Schneider, P. A., Nowakowski, G. S., et al. (2017). Histone deacetylase inhibitors interrupt HSP90RASGRP1 and HSP90CRAF interactions to upregulate BIM and circumvent drug resistance in lymphoma cells. Leukemia 31, 1593–1602. doi: 10.1038/leu.2016.357
Dong, H., and Chen, L. (2003). B7-H1 pathway and its role in the evasion of tumor immunity. J. Mol. Med. 81, 281–287. doi: 10.1007/s00109-003-0430-2
Dovey, O. M., Foster, C. T., Conte, N., Edwards, S. A., Edwards, J. M., Singh, R., et al. (2013). Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 121, 1335–1344. doi: 10.1182/blood-2012-07-441949
Duckett, C. S., Gedrich, R. W., Gilfillan, M. C., and Thompson, C. B. (1997). Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol. Cell Biol. 17, 1535–1542. doi: 10.1128/mcb.17.3.1535
Dupuis, J., Morschhauser, F., Ghesquières, H., Tilly, H., Casasnovas, O., Thieblemont, C., et al. (2015). Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T-cell lymphoma: a non-randomised, phase 1b/2 study. Lancet Haematol. 2, e160–e165. doi: 10.1016/S2352-3026(15)00023-X
Dürkop, H., Latza, U., Hummel, M., Eitelbach, F., Seed, B., and Stein, H. (1992). Molecular cloning and expression of a new member of the nerve growth factor receptor family that is characteristic for Hodgkin’s disease. Cell 68, 421–427. doi: 10.1016/0092-8674(92)90180-k
Duvic, M., Dummer, R., Becker, J. C., Poulalhon, N., Romero, P. O., Bernengo, M. G., et al. (2013). Panobinostat activity in both bexarotene-exposed and -naïve patients with refractory cutaneous T-cell lymphoma: results of a phase II trial. Eur. J. Cancer 49, 386–394. doi: 10.1016/j.ejca.2012.08.017
Duvic, M., Talpur, R., Ni, X., Zhang, C., Hazarika, P., Kelly, C., et al. (2007). Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109, 31–39. doi: 10.1182/blood-2006-06-025999
Duvic, M., and Vu, J. (2007). Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 16, 1111–1120. doi: 10.1517/13543784.16.7.1111
Ellis, L., Pan, Y., Smyth, G. K., George, D. J., McCormack, C., Williams-Truax, R., et al. (2008). Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin. Cancer Res. 14, 4500–4510. doi: 10.1158/1078-0432.ccr-07-4262
Ellmeier, W. (2015). Molecular control of CD4(+) t cell lineage plasticity and integrity. Int. Immunopharmacol. 28, 813–817. doi: 10.1016/j.intimp.2015.03.050
Eyre, T. A., Collins, G. P., Gupta, A., Coupe, N., Sheikh, S., Whittaker, J., et al. (2019). A phase 1 study to assess the safety, tolerability, and pharmacokinetics of CXD101 in patients with advanced cancer. Cancer 125, 99–108. doi: 10.1002/cncr.31791
Foss, F., Pro, B., Miles Prince, H., Sokol, L., Caballero, D., Horwitz, S., et al. (2017). Responses to romidepsin by line of therapy in patients with relapsed or refractory peripheral T-cell lymphoma. Cancer Med. 6, 36–44. doi: 10.1002/cam4.939
Gammoh, N., Lam, D., Puente, C., Ganley, I., Marks, P. A., and Jiang, X. (2006). Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 109, 6561–6565. doi: 10.1073/pnas.1204429109
Gil, V. S., Bhagat, G., Howell, L., Zhang, J., Kim, C. H., Stengel, S., et al. (2016). Deregulated expression of HDAC9 in B cells promotes development of lymphoproliferative disease and lymphoma in mice. Dis. Model Mech. 9, 1483–1495. doi: 10.1242/dmm.023366
Harlin, H., Meng, Y., Peterson, A. C., Zha, Y., Tretiakova, M., Slingluff, C., et al. (2009). Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 69, 3077–3085. doi: 10.1158/0008-5472.CAN-08-2281
He, J., Hu, Y., Hu, M., and Li, B. (2015). Development of PD-1/PD-L1 pathway in tumor immune microenvironment and treatment for non-small cell lung cancer. Sci. Rep. 5:13110. doi: 10.1038/srep13110
Heider, U., Rademacher, J., Lamottke, B., Mieth, M., Moebs, M., von Metzler, I., et al. (2009). Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous t cell lymphoma. Eur. J. Haematol. 82, 440–449. doi: 10.1111/j.1600-0609.2009.01239.x
Hodgkin, T. (1832). On some morbid appearances of the absorbent glands and spleen. Med. Chir. Trans. 17, 68–114. doi: 10.1177/095952873201700106
Hopfinger, G., Nösslinger, T., Lang, A., Linkesch, W., Melchardt, T., Weiss, L., et al. (2014). Lenalidomide in combination with vorinostat and dexamethasone for the treatment of relapsed/refractory peripheral t cell lymphoma (PTCL): report of a phase I/II trial. Ann. Hematol. 93, 459–462. doi: 10.1007/s00277-014-2009-0
Hsu, F. C., Belmonte, P. J., Constans, M. M., Chen, M. W., McWilliams, D. C., Hiebert, S. W., et al. (2015). Histone deacetylase 3 is required for t cell maturation. J. Immunol. 195, 1578–1590. doi: 10.4049/jimmunol.1500435
Huang, J., Wang, L., Dahiya, S., Beier, U. H., Han, R., Samanta, A., et al. (2017). Histone/protein deacetylase 11 targeting promotes Foxp3+ Treg function. Sci. Rep. 7:8626.
Hull, E. E., Montgomery, M. R., and Leyva, K. J. (2016). HDAC inhibitors as epigenetic regulators of the immune system: impacts on cancer therapy and inflammatory diseases. Biomed Res. Int. 2016:8797206. doi: 10.1155/2016/8797206
Hutt, D. M., Roth, D. M., Vignaud, H., Cullin, C., and Bouchecareilh, M. (2014). The histone deacetylase inhibitor, Vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS One 9:e106224. doi: 10.1371/journal.pone.0106224
Imam, M. H., Shenoy, P. J., Flowers, C. R., Phillips, A., and Lechowicz, M. J. (2013). Incidence and survival patterns of cutaneous T-cell lymphomas in the United States. Leuk. Lymphoma 54, 752–759. doi: 10.3109/10428194.2012.729831
Irlé, C., and Weintraub, J. (2016). Long-term treatment with romidepsin in patients with peripheral T-cell lymphoma. Case Rep. Hematol. 2016:8175957. doi: 10.1155/2016/8175957
Iyer, S. P., and Foss, F. F. (2015). Romidepsin for the treatment of peripheral T-cell lymphoma. Oncologist 20, 1084–1091. doi: 10.1634/theoncologist.2015-0043
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jiang, X., Wang, J., Deng, X., Xiong, F., Ge, J., Xiang, B., et al. (2019). Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer. 18:10. doi: 10.1186/s12943-018-0928-4
Jiang, Y., Ortega-Molina, A., Geng, H., Ying, H. Y., Hatzi, K., Parsa, S., et al. (2017). CREBBP inactivation promotes the development of HDAC3-dependent lymphomas. Cancer Discov. 7, 38–53. doi: 10.1158/2159-8290.CD-16-0975
Johnston, P. B., Cashen, A. F., Nikolinakos, P. G., Beaven, A. W., Barta, S. K., Bhat, G., et al. (2015). Safe and effective treatment of patients with peripheral T-cell lymphoma (PTCL) with the novel HDAC inhibitor, belinostat, in combination with CHOP: results of the bel-CHOP phase 1 trial. Blood 126:253. doi: 10.1182/blood.V126.23.253.253
Kato, Y., Yoshimura, K., Shin, T., Verheul, H., Hammers, H., Sanni, T. B., et al. (2007). Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin. Cancer Res. 13, 4538–4546. doi: 10.1158/1078-0432.CCR-07-0014
Kirschbaum, M., Frankel, P., Popplewell, L., Zain, J., Delioukina, M., Pullarkat, V., et al. (2011). Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J. Clin. Oncol. 29, 1198–1203. doi: 10.1200/JCO.2010.32.1398
Kunami, N., Katsuya, H., Nogami, R., Ishitsuka, K., and Tamura, K. (2014). Promise of combining a Bcl-2 family inhibitor with bortezomib or SAHA for adult T-cell leukemia/lymphoma. Anticancer Res. 34, 5287–5294.
Lemercier, C., Brocard, M. P., Puvion-Dutilleul, F., Kao, H. Y., Albagli, O., and Khochbin, S. (2002). Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 277, 22045–22052. doi: 10.1074/jbc.M201736200
Li, Y., and Seto, E. (2016). HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 6:a026831. doi: 10.1101/cshperspect.a026831
Liu, Z., Cai, Y., Yang, Y., Li, A., Bi, R., Wang, L., et al. (2018). Activation of MET signaling by HDAC6 offers a rationale for a novel ricolinostat and crizotinib combinatorial therapeutic strategy in diffuse large B-cell lymphoma. J. Pathol. 246, 141–153. doi: 10.1002/path.5108
Loosveld, M., Castellano, R., Gon, S., Goubard, A., Crouzet, T., Pouyet, L., et al. (2014). Therapeutic targeting of c-Myc in T-cell acute lymphoblastic leukemia, T-ALL. Oncotarget 30, 3168–3172. doi: 10.18632/oncotarget.1873
Lu, X., Ning, Z., Li, Z., Cao, H., and Wang, X. (2016). Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis. Res. 5, 185–191. doi: 10.5582/irdr.2016.01024
Martin, M., Potente, M., Janssens, V., Vertommen, D., Twizere, J. C., Rider, M. H., et al. (2008). Protein phosphatase 2A controls the activity of histone deacetylase 7 during t cell apoptosis and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 4727–4732. doi: 10.1073/pnas.0708455105
McCann, S. A., and Story, S. K. (2013). Histone deacetylase inhibitors in cutaneous T-cell lymphoma. J. Dermatol. Nurs. Assoc. 5, 305–313. doi: 10.1097/JDN.0000000000000007
Mehndiratta, S., Lin, M. H., Wu, Y. W., Chen, C. H., Wu, T. Y., Chuang, K. H., et al. (2020). N-alkyl-hydroxybenzoyl anilide hydroxamates as dual inhibitors of HDAC and HSP90, downregulating IFN-γ induced PD-L1 expression. Eur. J. Med. Chem. 185:111725. doi: 10.1016/j.ejmech.2019.111725
Mishra, A., La Perle, K., Kwiatkowski, S., Sullivan, L. A., Sams, G. H., Johns, J., et al. (2016). Mechanism, consequences, and therapeutic targeting of abnormal IL15 signaling in cutaneous T-cell lymphoma. Cancer Discov. 6, 986–1005. doi: 10.1158/2159-8290
Nanou, A., Toumpeki, C., Lavigne, M. D., Lazou, V., Demmers, J., Paparountas, T., et al. (2017). The dual role of LSD1 and HDAC3 in STAT5-dependent transcription is determined by protein interactions, binding affinities, motifs and genomic positions. Nucleic Acids Res. 45, 142–154. doi: 10.1093/nar/gkw832
Nieto, Y., Valdez, B. C., Thall, P. F., Ahmed, S., Jones, R. B., Hosing, C., et al. (2015). Vorinostat combined with high-dose gemcitabine, busulfan, and melphalan with autologous stem cell transplantation in patients with refractory lymphomas. Biol. Blood Marrow Transplant. 21, 1914–1920. doi: 10.1016/j.bbmt.2015.06.003
Nieto, Y., Valdez, B. C., Thall, P. F., Jones, R. B., Wei, W., Myers, A., et al. (2016). Double epigenetic modulation of high-dose chemotherapy with azacitidine and vorinostat for patients with refractory or poor-risk relapsed lymphoma. Cancer 122, 2680–2688. doi: 10.1002/cncr.30100
Ning, Z. Q., Li, Z. B., Newman, M. J., Shan, S., Wang, X. H., Pan, D. S., et al. (2012). Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 69, 901–909. doi: 10.1007/s00280-011-1766-x
Nishioka, C., Ikezoe, T., Yang, J., Komatsu, N., Bandobashi, K., Taniguchi, A., et al. (2008). Histone deacetylase inhibitors induce growth arrest and apoptosis of HTLV-1-infected T-cells via blockade of signaling by nuclear factor κB. Leuk. Res. 32, 287–296. doi: 10.1016/j.leukres.2007.05.026
Noureen, N., Rashid, H., and Kalsoom, S. (2010). Identification of type-specific anticancer histone deacetylase inhibitors: road to success. Cancer Chemother. Pharmacol. 66, 625–633. doi: 10.1007/s00280-010-1324-y
Ogura, M., Ando, K., Suzuki, T., Ishizawa, K., Oh, S. Y., Itoh, K., et al. (2014). A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Br. J. Haematol. 165, 768–776. doi: 10.1111/bjh.12819
Oki, Y., Buglio, D., Fanale, M., Fayad, L., Copeland, A., Romaguera, J., et al. (2013a). Phase I study of panobinostat plus everolimus in patients with relapsed or refractory lymphoma. Clin. Cancer Res. 19, 6882–6890. doi: 10.1158/1078-0432.CCR-13-1906
Oki, Y., Younes, A., Copeland, A., Hagemeister, F., Fayad, L. E., McLaughlin, P., et al. (2013b). Phase I study of vorinostat in combination with standard CHOP in patients with newly diagnosed peripheral T-cell lymphoma. Br. J. Haematol. 162, 138–141. doi: 10.1111/bjh.12326
Oki, Y., Buglio, D., Zhang, J., Ying, Y., Zhou, S., Sureda, A., et al. (2014). Immune regulatory effects of panobinostat in patients with Hodgkin lymphoma through modulation of serum cytokine levels and T-cell PD1 expression. Blood Cancer J. 4:e236. doi: 10.1038/bcj.2014.58
Olsen, E. A., Kim, Y. H., Kuzel, T. M., Pacheco, T. R., Foss, F. M., Parker, S., et al. (2007). Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 25, 3109–3115. doi: 10.1200/JCO.2006.10.243
Palermo, R., Checquolo, S., Giovenco, A., Grazioli, P., Kumar, V., Campese, A. F., et al. (2012). Acetylation controls Notch3 stability and function in T-cell leukemia. Oncogene 31, 3807–3817. doi: 10.1038/onc.2011.533
Pasqualucci, L., Dominguez-Sola, D., Chiarenza, A., Fabbri, G., Grunn, A., Trifonov, V., et al. (2011). Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471, 189–195. doi: 10.1038/nature09730
Persky, D. O., Li, H., Rimsza, L. M., Barr, P. M., Popplewell, L. L., Bane, C. L., et al. (2018). A phase I/II trial of vorinostat (SAHA) in combination with rituximab-CHOP in patients with newly diagnosed advanced stage diffuse large B-cell lymphoma (DLBCL): SWOG S0806. Am. J. Hematol. 93, 486–493. doi: 10.1002/ajh.25010
Philips, R. L., Chen, M. W., McWilliams, D. C., Belmonte, P. J., Constans, M. M., and Shapiro, V. S. (2016). HDAC3 is required for the downregulation of RORγt during thymocyte positive selection. J. Immunol. 197, 541–554. doi: 10.4049/jimmunol.1502529
Philips, R. L., Lee, J. H., Gaonkar, K., Chanana, P., Chung, J. Y., Romero Arocha, S. R., et al. (2019). HDAC3 restrains CD8-lineage genes to maintain a bi-potential state in CD4+CD8+ thymocytes for CD4-lineage commitment. eLife 8:e43821. doi: 10.7554/eLife.43821
Poole, R. M. (2014). Belinostat: first global approval. Drugs 74, 1543–1554. doi: 10.1007/s40265-014-0275-8
Powers, J. J., Maharaj, K. K., Sahakian, E., Xing, L., PerezVillarroel, P., Knox, T., et al. (2014). Histone deacetylase 6 (HDAC6) as a regulator of immune check-point molecules in chronic lymphocytic leukemia (CLL). Blood 124, 3311–3311. doi: 10.1182/blood.V124.21.3311.3311
Preglej, T., Hamminger, P., Luu, M., Bulat, T., Andersen, L., Göschl, L., et al. (2020). Histone deacetylases 1 and 2 restrain CD4+ cytotoxic t lymphocyte differentiation. JCI Insight 5:e133393. doi: 10.1172/jci.insight.133393
Puvvada, S. D., Li, H., Rimsza, L. M., Bernstein, S. H., Fisher, R. I., LeBlanc, M., et al. (2016). A phase II study of belinostat (PXD101) in relapsed and refractory aggressive B-cell lymphomas: SWOG S0520. Leuk. Lymphoma 57, 2359–2369. doi: 10.3109/10428194.2015.1135431
Re, D., Thomas, R. K., Behringer, K., and Diehl, V. (2005). From Hodgkin disease to Hodgkin lymphoma: biologic insights and therapeutic potential. Blood 105, 4553–4560. doi: 10.1182/blood-2004-12-4750
Reddy, S. A. (2016). Romidepsin for the treatment of relapsed/refractory cutaneous T-cell lymphoma (mycosis fungoides/Sézary syndrome): use in a community setting. Crit. Rev. Oncol. Hematol. 106, 99–107. doi: 10.1016/j.critrevonc.2016.07.001
Rozati, S., Cheng, P. F., Widmer, D. S., Fujii, K., Levesque, M. P., and Dummer, R. (2016). Romidepsin and azacitidine synergize in their epigenetic modulatory effects to induce apoptosis in CTCL. Clin. Cancer Res. 22, 2020–2031. doi: 10.1158/1078-0432.ccr-15-1435
Samimi, S., Morrissey, K., Anshelevich, S., Evans, K., Gardner, J., Musiek, A., et al. (2013). Romidepsin and interferon gamma: a novel combination for refractory cutaneous T-cell lymphoma. J. Am. Acad. Dermatol. 68, e5–e6. doi: 10.1016/j.jaad.2011.06.043
Sanaei, M., and Fraidoon, K. (2019). Histone deacetylases and histone deacetylase inhibitors: molecular mechanisms of action in various cancers. Adv. Biomed. Res. 8:63. doi: 10.4103/abr.abr_142_19
Sanaei, M., and Kavoosi, F. (2019). Histone deacetylases and histone deacetylase inhibitors: molecular mechanisms of action in various cancers. Adv. Biomed. Res. 8:63. doi: 10.4103/abr.abr_142_19
Sandhu, S. K., Volinia, S., Costinean, S., Galasso, M., Neinast, R., Santhanam, R., et al. (2012). miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Eμ-miR-155 transgenic mouse model. Proc. Natl. Acad. Sci. U.S.A. 109, 20047–20052. doi: 10.1073/pnas.1213764109
Seo, Y. H. (2015). Dual inhibitors against topoisomerases and histone deacetylases. J. Cancer Prev. 20, 85–91. doi: 10.15430/JCP.2015.20.2.85
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6:a018713. doi: 10.1101/cshperspect.a018713
Shao, R. H., Tian, X., Gorgun, G., Urbano, A. G., and Foss, F. M. (2002). Arginine butyrate increases the cytotoxicity of DAB389IL-2 in leukemia and lymphoma cells by upregulation of IL-2Rβ gene. Leuk. Res. 26, 1077–1083. doi: 10.1016/S0145-2126(02)00059-0
Spranger, S., Spaapen, R. M., Zha, Y., Williams, J., Meng, Y., Ha, T. T., et al. (2013). Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 5:200ra116. doi: 10.1126/scitranslmed.3006504
Stathis, A., and Younes, A. (2015). The new therapeutical scenario of Hodgkin lymphoma. Ann. Oncol. 26, 2026–2033. doi: 10.1093/annonc/mdv256
Stengel, K. R., Barnett, K. R., Wang, J., Liu, Q., Hodges, E., Hiebert, S. W., et al. (2017). Deacetylase activity of histone deacetylase 3 is required for productive VDJ recombination and B-cell development. Proc. Natl. Acad. Sci. U.S.A. 114, 8608–8613. doi: 10.1073/pnas.1701610114
Stengel, K. R., Zhao, Y., Klus, N. J., Kaiser, J. F., Gordy, L. E., Joyce, S., et al. (2015). Histone deacetylase 3 is required for efficient T cell development. Mol. Cell Biol. 35, 3854–3865. doi: 10.1128/mcb.00706-15
Straus, D. J., Hamlin, P. A., Matasar, M. J., Lia Palomba, M., Drullinsky, P. R., Zelenetz, A. D., et al. (2015). Phase I/II trial of vorinostat with rituximab, cyclophosphamide, etoposide and prednisone as palliative treatment for elderly patients with relapsed or refractory diffuse large B-cell lymphoma not eligible for autologous stem cell transplantation. Br. J. Haematol. 168, 663–670. doi: 10.1111/bjh.13195
Sung, J. J., Ververis, K., and Karagiannis, T. C. (2014). Histone deacetylase inhibitors potentiate photochemotherapy in cutaneous T-cell lymphoma MyLa cells. J. Photochem. Photobiol. B 131, 104–112. doi: 10.1016/j.jphotobiol.2014.01.009
Tan, D., Phipps, C., Hwang, W. Y., Tan, S. Y., Yeap, C. H., Chan, Y. H., et al. (2015). Panobinostat in combination with bortezomib in patients with relapsed or refractory peripheral T-cell lymphoma: an open-label, multicentre phase 2 trial. Lancet Haematol. 2, e326–e333. doi: 10.1016/S2352-3026(15)00097-6
Tanaka, H., Muto, A., Shima, H., Katoh, Y., Sax, N., Tajima, S., et al. (2016). Epigenetic regulation of the blimp-1 gene (Prdm1) in B cells involves Bach2 and histone deacetylase 3. J. Biol. Chem. 291, 6316–6330. doi: 10.1074/jbc.M116.713842
Thapa, P., Chen, M. W., McWilliams, D. C., Belmonte, P., Constans, M., Sant’Angelo, D. B., et al. (2016). NKAP regulates invariant NKT cell proliferation and differentiation into ROR-γt-expressing NKT17 cells. J. Immunol. 196, 4987–4998. doi: 10.4049/jimmunol.1501653
Thapa, P., Romero Arocha, S., Chung, J. Y., Sant’Angelo, D. B., and Shapiro, V. S. (2017). Histone deacetylase 3 is required for iNKT cell development. Sci. Rep. 7:5784. doi: 10.1038/s41598-017-06102-5
Ueda, H., Nakajima, H., Hori, Y., Goto, T., and Okuhara, M. (1994). Action of FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum no. 968, on Ha-ras transformed NIH3T3 cells. Biosci. Biotechnol. Biochem. 58, 1579–1583. doi: 10.1271/bbb.58.1579
Villagra, A., Cheng, F., Wang, H. W., Suarez, I., Glozak, M., Maurin, M., et al. (2009). The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 10, 92–100. doi: 10.1038/ni.1673
Vo, D. D., Prins, R. M., Begley, J. L., Donahue, T. R., Morris, L. F., Bruhn, K. W., et al. (2009). Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 69, 8693–8699. doi: 10.1158/0008-5472.CAN-09-1456
Wang, L., Qin, W., Huo, Y. J., Li, X., Shi, Q., Rasko, J., et al. (2020). Advances in targeted therapy for malignant lymphoma. Signal Transduct. Target. Ther. 5:15. doi: 10.1038/s41392-020-0113-2
Wang, P., Wang, Z., and Liu, J. (2020). Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 19:5. doi: 10.1186/s12943-019-1127-7
Wilks, S. (1856). Cases of enlargement of the lymphatic glands and spleen (or, Hodgkin’s disease) with remarks. Guys Hosp. Rep. 11, 56–67.
Wozniak, M. B., Villuendas, R., Bischoff, J. R., Aparicio, C. B., Martínez Leal, J. F., de La Cueva, P., et al. (2010). Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica 95, 613–621. doi: 10.3324/haematol.2009.013870
Xiao, H., Jiao, J., Wang, L., O’Brien, S., Newick, K., Wang, L. C., et al. (2016). HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. Int. J. Cancer 138, 2477–2486. doi: 10.1002/ijc.29979
Yamaguchi, T., Cubizolles, F., Zhang, Y., Reichert, N., Kohler, H., Seiser, C., et al. (2010). Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 24, 455–469. doi: 10.1101/gad.552310
Yoo, C. B., and Jones, P. A. (2006). Epigenetic therapy of cancer: past, present and future. Nat. Rev. Drug Discov. 5, 537–550. doi: 10.1038/nrd1930
Younes, A., Berdeja, J. G., Patel, M. R., Flinn, I., Gerecitano, J. F., Neelapu, S. S., et al. (2016). Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 17, 622–631. doi: 10.1016/S1470-2045(15)00584-7
Younes, A., Connors, J. M., Park, S. I., Fanale, M., O’Meara, M. M., Hunder, N. N., et al. (2013). Brentuximab vedotin combined with ABVD or AVD for patients with newly diagnosed Hodgkin’s lymphoma: a phase 1, open-label, dose-escalation study. Lancet Oncol. 14, 1348–1356. doi: 10.1016/S1470-2045(13)70501-1
Younes, A., Sureda, A., Ben-Yehuda, D., Zinzani, P. L., Ong, T. C., Prince, H. M., et al. (2012). Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II study. J. Clin. Oncol. 30, 2197–2203. doi: 10.1200/JCO.2011.38.1350
Yu, J., Angelin-Duclos, C., Greenwood, J., Liao, J., and Calame, K. (2000). Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol. Cell Biol. 20, 2592–2603. doi: 10.1128/mcb.20.7.2592-2603.2000
Yu, Z., Zhang, W., and Kone, B. C. (2002). Histone deacetylases augment cytokine induction of the iNOS gene. J. Am. Soc. Nephrol. 13, 2009–2017. doi: 10.1097/01.ASN.0000024253.59665.F1
Zhang, C., Richon, V., Ni, X., Talpur, R., and Duvic, M. (2005). Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J. Invest. Dermatol. 125, 1045–1052. doi: 10.1111/j.0022-202X.2005.23925.x
Zullo, K. M., Guo, Y., Cooke, L., Jirau-Serrano, X., Mangone, M., Scotto, L., et al. (2014). The investigational aurora A kinase inhibitor alisertib exhibits broad activity in preclinical models of T-cell lymphoma and is highly synergistic with romidepsin. Blood 124:4493. doi: 10.1182/blood.V124.21.4493.4493
Keywords: lymphomas, HDAC inhibitors, tumor immunity, chemotherapy regimen, clinical trials
Citation: Chen I-C, Sethy B and Liou J-P (2020) Recent Update of HDAC Inhibitors in Lymphoma. Front. Cell Dev. Biol. 8:576391. doi: 10.3389/fcell.2020.576391
Received: 26 June 2020; Accepted: 18 August 2020;
Published: 03 September 2020.
Edited by:
Ângela Sousa, University of Beira Interior, PortugalReviewed by:
Yingjie Zhang, Shandong University, ChinaEva Sahakian, Moffitt Cancer Center, United States
Copyright © 2020 Chen, Sethy and Liou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing-Ping Liou, anBsQHRtdS5lZHUudHc=