 Xiuli Wang1†
Xiuli Wang1† Hanlin Peng
Hanlin Peng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 05 November 2020
Sec. Signaling
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.574706
This article is part of the Research Topic Inflammatory Signaling in Cardiovascular Diseases: Pathogenesis and Anti-inflammation Therapy View all 12 articles
The nuclear factor-kappa B (NF-κB) signaling pathway regulates a variety of biological functions in the body, and its abnormal activation contributes to the pathogenesis of many diseases, such as cardiovascular and respiratory diseases and cancers. Therefore, to ensure physiological homeostasis of body systems, this pathway is strictly regulated by IκBα transcription, IκBα synthesis, and the IκBα-dependent nuclear transport of NF-κB. Particularly, the post-translational modifications of IκBα including phosphorylation, ubiquitination, SUMOylation, glutathionylation and hydroxylation are crucial in the abovementioned regulatory process. Because of the importance of the NF-κB pathway in maintaining body homeostasis, understanding the post-translational modifications of IκBα can not only provide deeper insights into the regulation of NF-κB pathway but also contribute to the development of new drug targets and biomarkers for the diseases.
The nuclear factor-kappa B (NF-κB) is a critical class of transcriptional regulators belonging to the NF-κB/Rel protein family and is found in almost all cell types. In mammals, the NF-κB family has five members, namely RelA (p65), c-Rel, RelB, NF-κB1 (p50) and NF-κB2 (p52), which form homodimers and heterodimers, and the heterodimer of p50 and p65 is the most common form (Giridharan and Srinivasan, 2018). NF-κB plays a pivotal part in the immune response, inflammatory reaction, cell proliferation and apoptosis (Wu et al., 1996; Elewaut et al., 1999; Ellis et al., 2000; Jo et al., 2000). The overactivation of the NF-κB pathway can result in the inflammatory diseases, cardiovascular diseases, autoimmune diseases and cancers; therefore, the NF-κB pathway is tightly regulated to ensure the physiological homeostasis of body systems (Barnes and Karin, 1997; Wong et al., 1999; Kis et al., 2003; DiDonato et al., 2012). The inhibitors of NF-κB (IκBs), including classical IκB proteins (IκBα, IκBβ and IκBε), non-classical IκB proteins (Bcl-3, IκBζ, IκBNS, IκBη and IκBL) and precursor IκB proteins (p105 and p100), are important molecules that regulate its activation. Among these proteins, IκBα is the first in the IκB family to be cloned (Hinz et al., 2012; Annemann et al., 2016) and is one of the members widely studied (Hinz et al., 2012; Annemann et al., 2016). Following exposure to inflammatory cytokines or microbial products, the IκBα protein is degraded, resulting in a decreased inhibitory effect on NF-κB; NF-κB is then translocates from the cytoplasm to the nucleus, where it regulates the transcription of NF-κB target genes (Hoffmann et al., 2006). The regulation of IκBα mainly occurs at post-translational level, including phosphorylation, ubiquitination and SUMOylation (Perkins, 2006). This review focuses on the advances in the molecular structure, function, post-translational modifications and the regulatory mechanisms of IκBα.
IκB is an important protein regulating the activity of NF-κB. It forms a trimer with a homologous or heterodimer of NF-κB and retains inactive NF-κB in the cytoplasm (Mothes et al., 2015). IκBα is the most common member of the IκB protein family and can shuttle between the cytoplasm and nucleus. Therefore, it is located in both the cytoplasm and nucleus, and its intracellular distribution is dynamic (Huang et al., 2000).
The molecular mechanism by which IκBα inhibits NF-κB activity is closely related to its protein structure. The human IκBα protein has 317 amino acids (aa) with a molecular weight of 36 kDa. It mainly consists of the N-terminal signal-receiving domain (SRD; 1–72 aa), the intermediate ankyrin repeat domain (ARD; 73–280 aa), the C-terminal proline-glutamic-serine-threonine domain (PEST domain; 281–317 aa) and two nuclear export sequences (NESs; 45–54 aa and 265–277 aa) (Lin et al., 1996; Arenzana-Seisdedos et al., 1997; Johnson et al., 1999; Truhlar et al., 2006; Yazdi et al., 2015).
The large amount of amide exchange and 8-anilino-1-napthalenesulphonic acid-binding suggest that free IκBα has molten globule structure (Croy et al., 2004). The details are as follows (Figure 1): (1) N-terminal SRD is critical for receiving phosphorylation, ubiquitination and SUMOylation signals as it contains the phosphorylation sites Ser32, Ser36, and Tyr42, ubiquitination sites Lys21 and Lys22, and SUMOylation sites Lys21 and Lys38 (Mabb and Miyamoto, 2007; Hendriks et al., 2017). Additionally, SRD is significant to the activation of NF-κB. Furthermore, it consists of three stable α-helices to lie between residues 8–15 aa, 22–30 aa and 44–50 aa, which complement the six ankyrin repeats (ARs) present in crystallized IκBα. This finding lays a foundation for further studies on the signal transduction of SRD in the IκBα degron (Yazdi et al., 2015). (2) The AR is the characteristic structure of IκB and one of the most common protein sequence motifs that mediates protein–protein interactions (Sedgwick and Smerdon, 1999; Li et al., 2006). Particularly, human IκBα contains six ARs, each consisting of approximately 33 aa. An AR is folded into a β-hairpin, followed by two antiparallel α-helices and a variable loop, which connects it to the next. The α-helical stacks that are thought to form the small hydrophobic cores, whereas the β-hairpin “fingers” form the main protein–protein interaction sites (Sedgwick and Smerdon, 1999; Hendriks et al., 2017). The thermal stability of the ankyrin folding has been suggested to be mainly due to the local interaction between adjacent structural units and needs to be repeated many times to form a stable folding ARD. Amide exchange experiments have revealed that AR3 is the most compact; AR2 and AR4 are less compact; and AR1 and AR6 were solvent-exposed (Croy et al., 2004). ARD contains glutathionylation modification site Cys186 and hydroxylation modification sites Asn210 and Asn244 (Cockman et al., 2006; Kil et al., 2008). (3) The C-terminal PEST domain is highly solvent accessible (Croy et al., 2004); it is not necessary for the binding of IκBα with NF-κB but plays a pivotal role in inhibiting the competitive binding of NF-κB and DNA (Ernst et al., 1995; Sue and Dyson, 2009). The PEST domain contains phosphorylation modification sites Ser283, Ser289, Ser293, Thr291 and Thr299 (McElhinny et al., 1996; Schwarz et al., 1996). (4) Lastly, the two NES suquences are located in the N-terminal SRD domain (45–54 aa) and the C-terminal (265–277 aa). The N-terminal NES is necessary for the shuttling of IκBα between the nucleus and cytoplasm (Huang and Miyamoto, 2001; Lee and Hannink, 2001). Similarly, the C-terminal NES is involved in IκBα-mediated nuclear export of the retroviral oncoprotein v-Rel, which has structural homology with the NF-κB protein family members. However, the substitution of alanine in the C-terminal NES of IκBα did not affect the cytoplasmic relocalization of the mutant IκBα and c-Rel induced by the leptomycin B (Lee and Hannink, 2001). The abovementioned detailed protein structure can be found in references (Croy et al., 2004; Sue and Dyson, 2009; Truhlar et al., 2006; Yazdi et al., 2015).
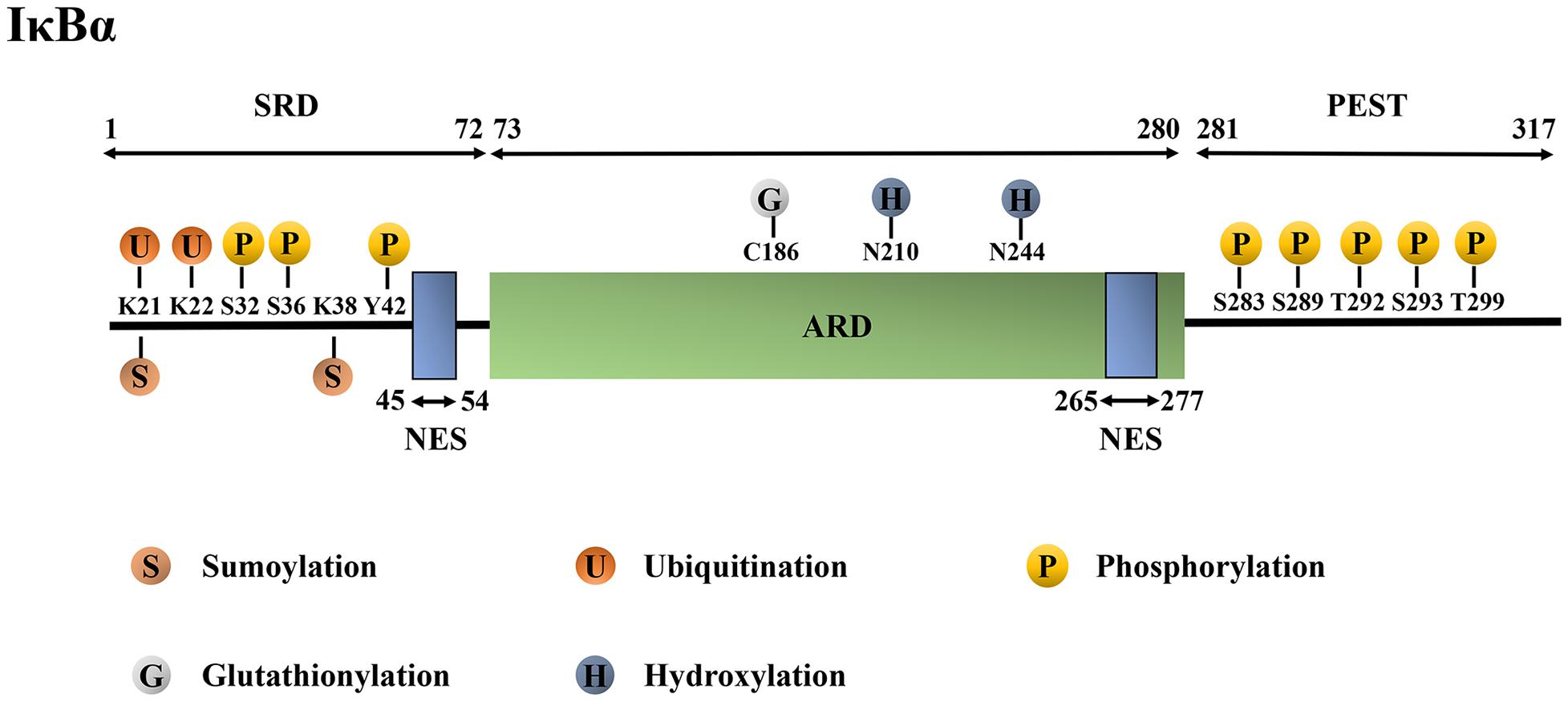
Figure 1. Structure and post-translational modification sites of the human IκBα protein. IκBα, nuclear factor-kappa B inhibitor alpha; SRD, signal-receiving domain; ARD, ankyrin repeat domain; PEST domain, proline–glutamic–serine–threonine domain; NES, nuclear export sequence; K, Lys, lysine; S, Ser, serine; Y, Tyr, tyrosine; C, Cys, cysteine; N, Asn, asparagine; T, Thr, threonine.
IκBα and NF-κB form a broad non-continuous binding surface, and all six repeat sequences are involved in the formation of the non-continuous contact surface (Croy et al., 2004). NF-κB family members, such as p65, contain three main domains: the N-terminal domain (NTD), dimerization domain, and transactivation domain (Komives, 2012). AR1–3 contacts the nuclear localization signal (NLS) polypeptide in the N-terminal Rel-homology domain (RHD) of p65, whereas AR4–6 contacts the dimerization domain of p50. Meanwhile, the inner helices of AR5 and AR6 contact the dimerization domain of p65, and the PEST region interacts with the NTD of p65 (Croy et al., 2004). Compared with IκBα in the IκBα–NF-κB complex, free IκBα has higher flexibility (Yazdi et al., 2015). The abovementioned detailed protein structure can be found in references (Croy et al., 2004; Komives, 2012; Yazdi et al., 2015).
A series of conformational changes occur when IκBα binds to NF-κB. First, the ARD of IκBα is slightly twisted, and the distance between residues 166 and 262 increases (Trelle et al., 2016). Thereafter, its AR5 and AR6, whose β-hairpins are highly dynamic in free IκBα, are folded. Amide hydrogen/deuterium (H/D) exchange is useful to identify protein regions that fold upon binding (Truhlar et al., 2006; Trelle et al., 2016). The amide H/D exchange experiments show that the β-hairpins of AR2 and AR3 are significantly resistant to the exchange, whereas those of AR5 and AR6 are completely exchanged in free IκBα within the first minute. When bound with NF-κB, this exchange is significantly reduced. The difference in amide H/D exchange between free and NF-κB-bound IκBα suggests that the β-hairpins in AR5 and AR6 undergo a folding transition upon binding (Truhlar et al., 2006). This folding involves multiple aspects of NF-κB regulation, such as regulating IκBα degradation, mediating its binding to different NF-κB dimers, and possibly promoting the dissociation of NF-κB from DNA (Truhlar et al., 2006). Notably, the AR5 and AR6 of IκBα in the free state are neither random coils nor completely unfolded. In fact, even if the amides in these two repeats are completely exchanged within one minute, when IκBα binds to NF-κB, no new secondary structure is formed (Ferreiro and Komives, 2010). Therefore, the AR5–6 area must be partially folded and may exhibit the molten globular structure in the free state (Sue and Dyson, 2009). Third, the NLS polypeptide of p65 is folded. NMR data revealed that free-form NSL is in a disordered, highly dynamic configuration. After binding to IκBα, its helices 3 and 4 can be folded, with helix 4 being essential for the interaction, particularly the single residue Phe309 of helix 4. The Phe309 ring acts as a “button” fixing the helix 4 to the “buttonhole” formed by Phe77 and Leu80 on the top surface of the ARD of IκBα. This highly specific interaction effectively covers the ARD, resulting in a significant beneficial binding energy (Cervantes et al., 2011). Lastly, the orientation of the p65 NTD is changed. Based on the structure, IκBα uses the bottom of AR6 and C-terminal PEST regions to interact with the p65 NTD. The main interaction occurs between the highly acidic patch of the PEST sequence (i.e., Glu282, Glu284, Asp285, Glu286 and Glu287) and the basic region of the NTD of p65 (Lys28, Arg30, Lys79, Arg158 and His181). Other residues (Trp258, Gln278, Met279 and Pro281) can also stabilize the interface through polar and van der Waals contact. These interactions are suggested to promote the allosteric regulation of DNA binding by NF-κB, i.e., the binding of IκBα changes the direction of the NTD of p65 and locks the NTD into a closed conformation, thereby interfering with the DNA binding of NF-κB (Sue and Dyson, 2009). The abovementioned detailed structure can be found in references (Truhlar et al., 2006; Sue and Dyson, 2009; Ferreiro and Komives, 2010; Cervantes et al., 2011; Trelle et al., 2016).
The ARD of IκBα in unstimulated cells binds to the RHD region of the NF-κB dimer, forming a trimer complex in the cytoplasm. When the cells are stimulated, IκBα undergoes post-translational modifications, such as phosphorylation and ubiquitination, and then is degraded by the proteasomes. The NLS in the RHD region of NF-κB is subsequently exposed and recognized by the nuclear transport receptor importin α/β on the cytoplasm. Their binding forms a nuclear complex that is transferred to the nucleus through the nuclear pore complex (NPC) in an energy-dependent manner, thereby initiating the transcription of the target gene (Turpin et al., 1999; Lee and Hannink, 2002) (Figure 2). Because IκBα is also a target gene of NF-κB, IκBα is rapidly replenished when the NF-κB pathway is activated. The newly synthesized IκBα enters the nucleus and translocates NF-κB dimer to the cytoplasm (Turpin et al., 1999; Chen and Greene, 2004) (Figure 2).
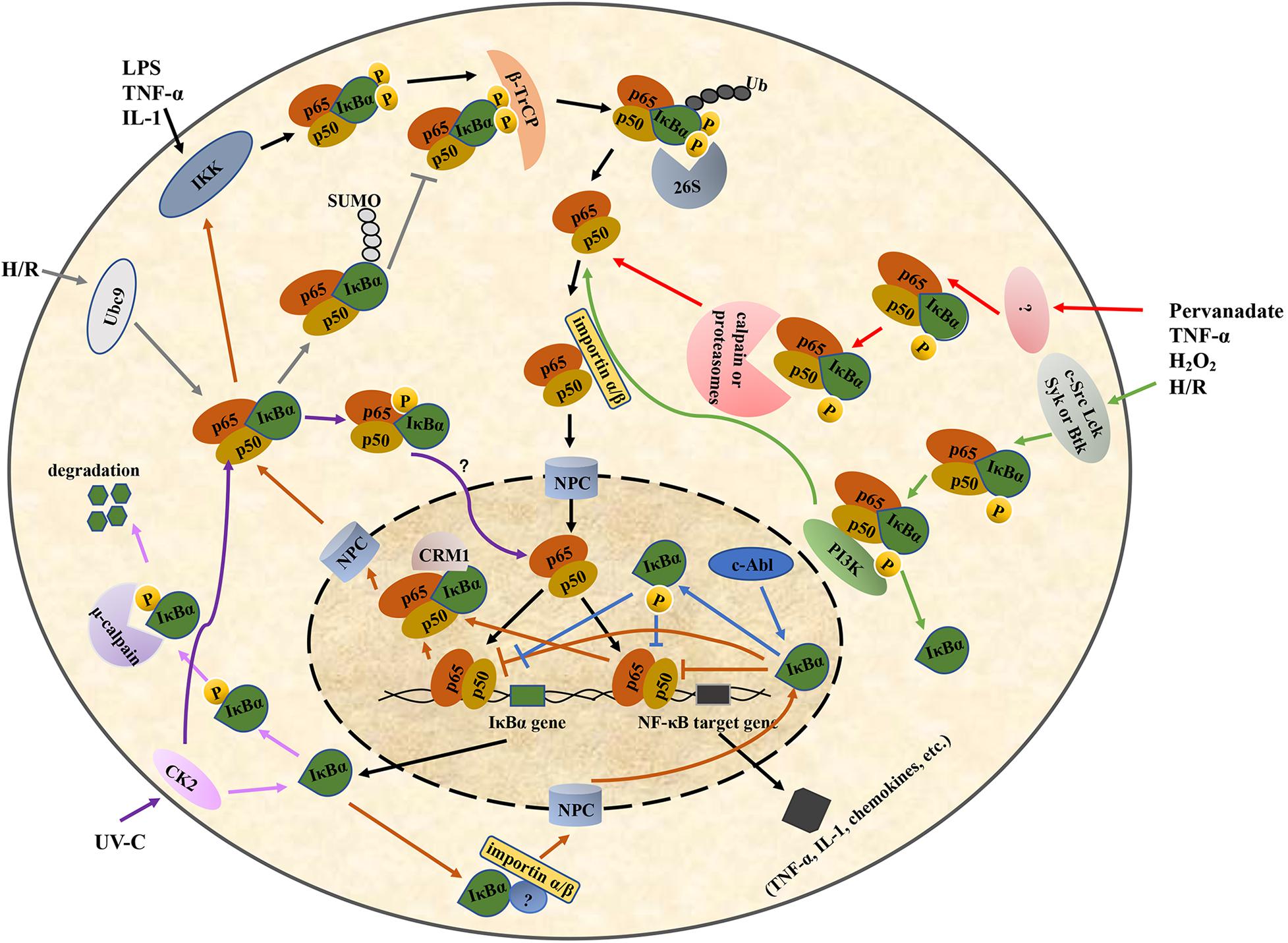
Figure 2. Regulation of the IκBα/NF-κB pathway. IκBα, nuclear factor-kappa B inhibitor alpha; NF-κB, nuclear factor-kappa B. LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α; IL-1, interleukin-1; H/R, hypoxia/reoxygenation; UV-C, ultraviolet-C; IKK, IκB kinase; CK2, casein kinase II; 26S, 26S proteasome; PI3K, phosphoinositide 3-kinase; Ubc9, ubiquitin-conjugating enzyme 9; β-TrCP, β-transducin repeat-containing protein; NPC, nuclear pore complex; CRM1, chromosome region maintenance 1.
IκBα lacks a classical basic NLS, which is a basic amino acid sequence required for nuclear translocation and is originally found in nucleoplasmin and SV40 T antigen. Thus, it cannot enter the nucleus in a classical pathway similar to NF-κB (Turpin et al., 1999). Sachdev et al. found that the AR2 deletion or the mutation in its hydrophobic residues markedly relocalizes IκBα to the cytoplasm (Sachdev et al., 1998). Additional experiments demonstrated that there was a functional discrete NLS in the AR2 region of IκBα, consisting of an N-terminal β-hairpin, an N-terminal α-helix, a C-terminal α-helix, and an adjacent C-terminal β-hairpin (Sachdev et al., 1998). In addition to ARs, some studies have suggested that the nuclear translocation of IκBα requires some unidentified proteins containing a basic NLS and can interact with the ARs of IκBα (Turpin et al., 1999). The nuclear-transport receptor importin α/β heterodimer in the cytoplasm subsequently recognizes the NLS and transports the nascent IκBα into the nucleus (Turpin et al., 1999) (Figure 2).
In the export NF-κB dimer to the cytoplasm, the C-terminal PEST domain and NES of IκBα play an important role. Negatively charged residues in the PEST domain of nuclear IκBα can interact with positively charged residues in the NTD of NF-κB p50, resulting in a conformational change in the NF-κB–DNA complex and facilitating the dissociation of DNA from NF-κB. Thereafter, the PEST domain of IκBα occupies the DNA-binding cavity of NF-κB, with the Glu287 residue interacting with a positively charged residue (typically Arg246) in the p65 NTD, thereby further hindering the binding between DNA and NF-κB (Potoyan et al., 2016). The NF-κB–IκBα complex is specifically recognized by the nuclear export receptor chromosome region maintenance (CRM1)/exportin-1 under the mediation of the NES at the N-terminus of IκBα. It is then transferred to the cytoplasm in the inactive NF-κB–IκBα form via the NPC, thus forming a spontaneous negative-feedback inhibition loop (Sachdev et al., 2000; Figure 2).
Furthermore, studies investigating the regulation of IκBα in the nucleus–cytoplasm shuttle have shown that the masking of the p50 NLS by IκBα in the cytoplasm is transient or incomplete. This leads to the partial exposure of the p50 NLS and causes the NF-κB–IκBα complex to enter the nucleus under the mediation of the NLS. An NES is specifically identified by the nuclear export receptor CRM1, thereby transporting the complex out of the nucleus. Therefore, the subcellular localization of the inactive NF-κB–IκBα complex is not static because of its dynamic shuttling between the nucleus and cytoplasm (Huang et al., 2000).
Protein phosphorylation refers to the process of transferring the γ-phosphate group of ATP or GTP to the amino acid residues of serine, threonine or tyrosine of the substrate protein and is catalyzed by protein kinases. “p-” designates that the protein is phosphorylated. A negatively charged phosphate group is added to the amino acid side chain of the protein, resulting in esterification. Esterification changes the protein configuration, activity and its ability to interact with other molecules, thereby greatly affecting cell signal transduction (Hunter, 2000; Kim et al., 2004). Meanwhile, protein dephosphorylation is the reverse process of protein phosphorylation, and is catalyzed by phosphatases (Domingo-Sananes et al., 2011). Therefore, the phosphorylation level of IκBα is regulated by both protein kinase-catalyzed phosphorylation and phosphatase-catalyzed dephosphorylation. Previous studies have shown that three main kinases promote IκBα protein phosphorylation, namely IκB kinase (IKK), casein kinase II (CK2) and tyrosine kinase (Perkins, 2006). The phosphatases that regulate IκBα dephosphorylation include calcineurin and protein tyrosine phosphatase L1 (Pons and Torres-Aleman, 2000; Wang et al., 2018).
The IKK complex can be activated by lipopolysaccharides (LPSs), viral proteins, oxygen free radicals, cytokines and other stimuli; it then phosphorylates the Ser36 and Ser32 residues in the SRD region of the N-terminus of IκBα (Traenckner et al., 1995; Hayden and Ghosh, 2004; Kato et al., 2012; Ko et al., 2017; Liu et al., 2018; Safi et al., 2018), leading to ubiquitination and the subsequent degradation of the IκBα in proteasome (Figure 2). IKK-dependent IκBα phosphorylation at the serine residues occurring in the cytosol is a key step in the release of active NF-κB and its nuclear translocation (Yazdi et al., 2017). A molecular dynamics simulation experiment showed that IKK first phosphorylates IκBα at Ser36, changing the local conformation of the N-terminal region and increasing the relative solvent-accessible surface area of Ser32. This enables Ser32 to interact with IKK via phosphorylation (Yazdi et al., 2017).
The subsequent changes in Ser32/Ser36 double-phosphorylated IκBα are as follows. (1) Conformational changes. First, the curved region between p-Ser32 and p-Ser36 is elongated, and the distance between them is increased. Then, the Met1 and Gln3 residues in the N-terminal move away from p-Ser32 to p-Ser36. Finally, the amides in the main chain of Met1 interact with the phosphate group of p-Ser36, whereas those in the main and side chains of Gln3 combine with the backbone carbonyl group of p-Ser36 to form hydrogen bonds (Yazdi et al., 2017). These changes result in the stabilization of the N-terminal tail. (2) Exposure of binding sites. The conformational change caused by double phosphorylation leads to the exposure of Leu34 and Lys21. Leu34 can interact with the β-transducin repeat-containing protein (β-TrCP), the main component of the IκBα-specific E3 ligase, whereas Lys21 is the ubiquitination site of IκBα. (3) Electrostatic interactions. In the non-phosphorylated state, strongly acidic residues (i.e., Asp27, Asp28, and Asp31 upstream of Ser32; and Asp39, Glu40, Glu41, and Glu43 downstream of Ser36) promote the formation of negatively charged regions on or near the protein surface around Ser32/Ser36. In contrast, the negative charge of the phosphate introduced during the double phosphorylation further changes the distribution of the surface charge and forms a more negatively charged protein surface, which is essential for the recognition of IκBα by the β-TrCP–S phase kinase-associated protein 1 (SKP1)–cullin 1 (CUL1)–F-box protein (SCF) complex and the ubiquitination of IκBα (Yazdi et al., 2017). More details can be found in reference (Yazdi et al., 2017).
Moreover, IKK-dependent IκBα phosphorylation might occur in the nucleus and play a transcriptional suppressive effect, together with transcriptional corepressors (Aguilera et al., 2004; Espinosa et al., 2011). Aguilera et al. have found that the nuclear IκBα bound to the promoter of the Notch target gene hes1 to facilitate the recruitment of histone acetyltransferases and deacetylases, correlating with transcriptional repression in unstimulated cells. In addition, IKK-α and -β were recruited to the hes1 promoter following TNF-α treatment, correlating with the IκBα phosphorylation, the release of chromatin-associated IκBα and gene transcriptional activation (Aguilera et al., 2004).
Heterozygous gain-of-function mutations associated with Ser32/Ser36 in IκBα can cause autosomal dominant form of anhidrotic ectodermal dysplasia with immunodeficiency. The mutations reported to date include missense mutations of Ser32, Ser36, or adjacent residues and nonsense mutations of upstream residues from Ser32, related to the reinitiation of truncated IκBα translation (Boisson et al., 2017). These mutations inhibit the phosphorylation of both Ser32 and Ser36, as well as the degradation of IκBα, thus enhancing the inhibitory activity of IκBα (Boisson et al., 2017). Fibroblasts, monocytes, and B and T cells of patients with these heterozygous mutations showed abnormal NF-κB regulatory responses to various surface receptor stimuli (Boisson et al., 2017). In addition, all patients suffered from severe B-cell deficiency, whereas some showed specific immunological characteristics, including increased lymphocytes and lack of peripheral lymph nodes. Often, purulent, mycobacterial, fungal and viral infections were also observed (Boisson et al., 2017). In patients with activated B-cell type (ABC) subgroup of diffuse large B-cell lymphoma, p-IκBα level in tissue microarrays of those with poor five-year survival rate was higher than those with good five-year survival rate, suggesting that p-IκBα is an independent prognostic marker of poor survival in ABC patients (Hussain et al., 2013).
CK2 is a ubiquitous serine/threonine kinase, and it can phosphorylate multiple serine and threonine residues in the PEST domain of the IκBα protein, including Ser283, Ser289, Ser293, Thr291 and Thr299, among which Ser293 is the most important (McElhinny et al., 1996; Schwarz et al., 1996). When CK2 binds to atypical protein kinase C-zeta (PKC-ζ), Ser293 will be preferentially phosphorylated (Bren et al., 2000).
When IκBα binds to NF-κB, its PEST domain is masked by NF-κB. Therefore, the CK2-dependent phosphorylation of the PEST domain mainly occurs in free IκBα protein, which are degraded directly by the proteasomes in a non-ubiquitin-dependent manner (Mathes et al., 2008). The PEST domain can bind to the μ-calmodulin-like domain of the large subunit of μ-calpain to trigger the degradation of IκBα associated with μ-calpain. The CK2-dependent phosphorylation of the serine/threonine residues in the PEST domain enhances IκBα degradation by μ-calpain (Shumway et al., 1999; Figure 2). CK2 phosphorylation of the free IκBα protein does not depend on stimulation but mainly regulates the basic turnover rate of IκBα in resting cells (Shumway et al., 1999; Mathes et al., 2008). However, some studies have shown that IκBα bound to NF-κB could also undergo CK2-dependent phosphorylation. Particularly, when epithelial HeLa cells are stimulated using short-wavelength ultraviolet radiation, CK2 is phosphorylated and activated by p38 MAP kinase through a phosphorylation- dependent allosteric mechanism, and the activated CK2 then phosphorylates the PEST domain of IκBα, leading to IκBα degradation and NF-κB activation, thus protecting cells from ultraviolet-induced cell death (Sayed et al., 2000; Kato et al., 2003) (Figure 2).
In addition to IKK and CK2, many types of tyrosine kinases are involved in the regulation of IκBα phosphorylation. Here, the modification sites of IκBα depend on the type of tyrosine kinases, stimulation factors and cell types, resulting in similar or opposite effects. The detailed regulation of tyrosine kinase-dependent phosphorylation is as follows.
In the pervanadate-stimulated human myeloid U937 cell, IκBα phosphorylation is induced at the Tyr42 residue, and IκBα is subsequently polyubiquitinated and degraded by the proteasome (Figure 2). This releases the NF-κB and translocates it to the nucleus, activating specific gene expression, including gene expression of IκBα (Mulero et al., 2013a). IκBα degradation and NF-κB activation are observed. However, the detailed regulation of tyrosine kinases in this process is unclear (Mukhopadhyay et al., 2000). U937 is a human leukemic monocyte-lymphoma cell line that is not homogeneous. In HIV-1 permissive clone 10 (plus), NF-κB p65 was constitutively phosphorylated, whereas in non-permissive clone 17 (minus), NF-κB p65 was not constitutively phosphorylated. In addition, both the α1 proteinase inhibitor and LPS induced the phosphorylation of NF-κB p65 Ser536 in the two clones; however, only the dephosphorylation of Ser529 was observed in the plus clone. These results indicated that the phosphorylation/dephosphorylation of NF-κB differs depending on the clone used, suggesting that the IκBα activity is also different (Bristow et al., 2008).
Second, the promotion of IκBα dissociation from the NF-κB complex occurs without its degradation (Figure 2). The tyrosine kinases c-Src, Lck, Syk or Btk are activated by many stimuli, such as TNF-α, pervanadate, hypoxia/reoxygenation, and H2O2, and phosphorylate IκBα at Tyr42, Tyr289 or Tyr305 residues. For example, when mouse bone marrow macrophages are stimulated by TNF-α, c-Src kinase is activated, and it then phosphorylates IκBα at Tyr42, resulting in the constant c-Src–IκBα association without IκBα degradation. This might be related to the release of the active RelA–p50 from IκBα–RelA–p50 complex and the subsequent of RelA–p50 nuclear translocation because the IκBα/RelA complex remains in the cytosol of c-Src-/- cell treated with TNF-α (Abu-Amer et al., 1998). Meanwhile, when epithelial HeLa cells were stimulated by pervanadate or hypoxia/reoxygenation, c-Src kinase promoted the transcriptional activity of NF-κB by phosphorylating IκBα at Tyr42 (Fan et al., 2003). Pervanadate and hypoxia/reoxygenation can also cause the phosphorylation of IκBα at Tyr42 in Jurkat cells, and this may be mediated by Lck. This activates NF-κB and is associated with the dissociation of IκBα–NF–κB complex (Imbert et al., 1996). Furthermore, H2O2 can induce the IκBα phosphorylation at tyrosine residues through Syk in human myeloid KBM-5 cells. This promoted the dissociation of the IκBα–NF–κB complex and the phosphorylation and nuclear translocation of p65 but did not affect IκBα degradation (Takada et al., 2003). Similarly, in anti-IgM-stimulated B cell, Btk is rapidly activated; thereafter, it then phosphorylates IκBα at Tyr305 and Tyr289 residues in the cytosol and associates with phosphorylated IκBα, which correlates with the nuclear translocation of p65 and mediated the early transcriptional activation of NF-κB-responsive genes activated via B cell receptor triggering (Pontoriero et al., 2019). Btk and NF-κB are upregulated in acute and chronic lymphocytic leukemia (Pal Singh et al., 2018). In addition, an inactivated mutation in Btk can lead to B-cell immunodeficiency, such as X-linked agammaglobulinemia, in humans (Pal Singh et al., 2018).
Regarding the mechanism by which IκBα phosphorylation at certain residues, such as Tyr42, activates the NF-κB pathway without accompanying IκBα degradation, the dissociation of IκBα from the NF-κB complex is speculated to be induced by other interacting proteins. Béraud et al. found that phosphoinositide 3-kinase (PI3K) is involved in NF-κB activation induced by IκBα phosphorylation at Tyr42, which is located in a consensus sequence of IκBα that binds to the SH2 domain of the PI3K p85 regulatory subunit. When Jurkat T cells were stimulated by pervanadate, the SH2 domain of PI3K p85 regulatory subunit could specifically bind to the phosphorylated Tyr42 of IκBα, causing the dissociation of IκBα from NF-κB/IκBα complex (Béraud et al., 1999).
Lastly, in human embryonic kidney 293T cells and human osteosarcoma U2OS cells, the nuclear non-receptor tyrosine kinase c-Abl can enhance the stability of nuclear IκBα through Tyr305 phosphorylation, allowing IκBα to accumulate in the nucleus and thereby inhibiting NF-κB activation caused by TNF-α stimulation (Kawai et al., 2002).
IκBα phosphorylation at serine and tyrosine residues plays distinct roles in different pathophysiological processes. For instance, in cardiomyopathic mice with cardiac-specific expression of TNF-α (TNF-1.6 mice), NF-κB activation was completely blocked after crossing them with IκBα (S32A, S36A, Y42F) transgenic mice; however, it only partially blocked after crossing with IκBα (S32A, S36A) transgenic mice. In addition, NF-κB activation following acute injury, including TNF-α and ischemia/reperfusion was completely blocked in both IκBα (S32A, S36A, Y42F) and IκBα (S32A, S36A) transgenic mice. These findings suggested that IκBα phosphorylation at tyrosine serves as the second signal for NF-κB activation in the pathogenesis of certain disease, although IκBα phosphorylation at Ser32 and Ser36 is dominant in the regulation of NF-κB activation, thus providing guidance for exploring the multiple targets of IκBα phosphorylation inhibitors in drug design and discovery (Brown et al., 2005).
As described in the abovementioned studies, almost all of IκBα phosphorylation modifications promote NF-κB activation. Similarly, the dephosphorylation of IκBα is critical for inhibiting the activation of NF-κB. Studies have revealed that calcineurin in astrocytes can be activated by insulin-like growth factor-I, which leads to site-specific dephosphorylation of p-Ser32, inhibition of IκBα degradation, and subsequent nuclear translocation of NF-κB p65 stimulated by TNF-α (Pons and Torres-Aleman, 2000). In addition, the protein tyrosine phosphatase L1 inhibits the development of high-grade serous ovarian cancer through dephosphorylating p-Tyr42 to stabilize IκBα and attenuate the nuclear translocation of NF-κB (Wang et al., 2018). Thyme quinone (a natural compound isolated from Nigella sativa) can induce the release of superoxide anion and hydrogen peroxide in ABC cells which promoted IκBα dephosphorylation, arrested NF-κB p65 nuclear translocation, and finally inhibited the NF-κB pathway-mediated ABC cell survival pathway (Hussain et al., 2013).
Ubiquitination is a cascade reaction involving the ubiquitin-proteasome-pathway (UPP), which is important for protein degradation in eukaryotes and is related to many biological processes including the cell cycle, apoptosis and inflammation. IκBα ubiquitination occurs following IκBα phosphorylation at Ser32/Ser36 and is recognized by the proteasome complex. The ubiquitinated IκBα is degraded in the proteasome, accompanied by the release of active NF-κB and the activation of the NF-κB pathway (Kanarek and Ben-Neriah, 2012).
The ubiquitination of IκBα is regulated by E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase (Kanarek and Ben-Neriah, 2012; Skaar et al., 2013). First, E1 ubiquitin-activating enzyme catalyzes the formation of a thioester bond between the C-terminal glycine residue of ubiquitin and the cysteine residue of E1. This activates ubiquitin in an ATP dependent manner and initiates ubiquitination modification. Thereafter, E1-ubiquitin intermediates transfer ubiquitin to E2, forming an E2-ubiquitin intermediate (Olsen et al., 2010; Singh et al., 2017). Finally, IκBα ubiquitination requires a specific E3 ubiquitin ligase, SCFβ-TrCP, comprising four subunits: Skp1, Cullin1, Rbx1 and β-TrCP. The crystal structure of SCFβ−TrCP shows that Cullin1, as a scaffold protein, can bind to the N-terminus of Skp1 and the C-terminus of cyclin Rbx1. Rbx1 can bind to E2 and promote the switch of ubiquitin from E2 to the substrate (Kanarek et al., 2010) (Figure 3). Meanwhile, the β-TrCP protein consists of two characteristic domains, the N-terminal F-box domain (190–228 aa) and the C-terminal tryptophan-aspartic acid (WD) repeat domain (301–590 aa) (Bai et al., 1996; Deshaies, 1999). The F-box domain was defined because it was first found in cyclin F. Particularly, the F-box domain of β-TrCP is mainly responsible for binding to Skp1, in which Ile143 and Leu152 residues play an important role in maintaining the hydrophobic surface of the β-TrCP–Skp1 interaction (Herter and Fuchs, 2002). The WD repeat domain is critical for substrate recognition (Hu et al., 2017) (Figure 3) and is a typical seven-blade propeller domain with an overall doughnut shaped. Each blade in WD repeat domain contains a conserved WD motif comprising approximately 40 residues and folded into four anti-parallel β chains. The central hole of the WD repeat domain usually mediates interactions with other proteins (Hu et al., 2017). In human IκBα, a degradation sequence, DSGLDS (31–36 aa), has been identified. Upon treatment with stimulating factors (such as TNFα), its Ser32 and Ser36 residues were phosphorylated to induce a conformational change in IκBα that is recognized by the WD repeat domain of the β-TrCP protein (Yaron et al., 1997).
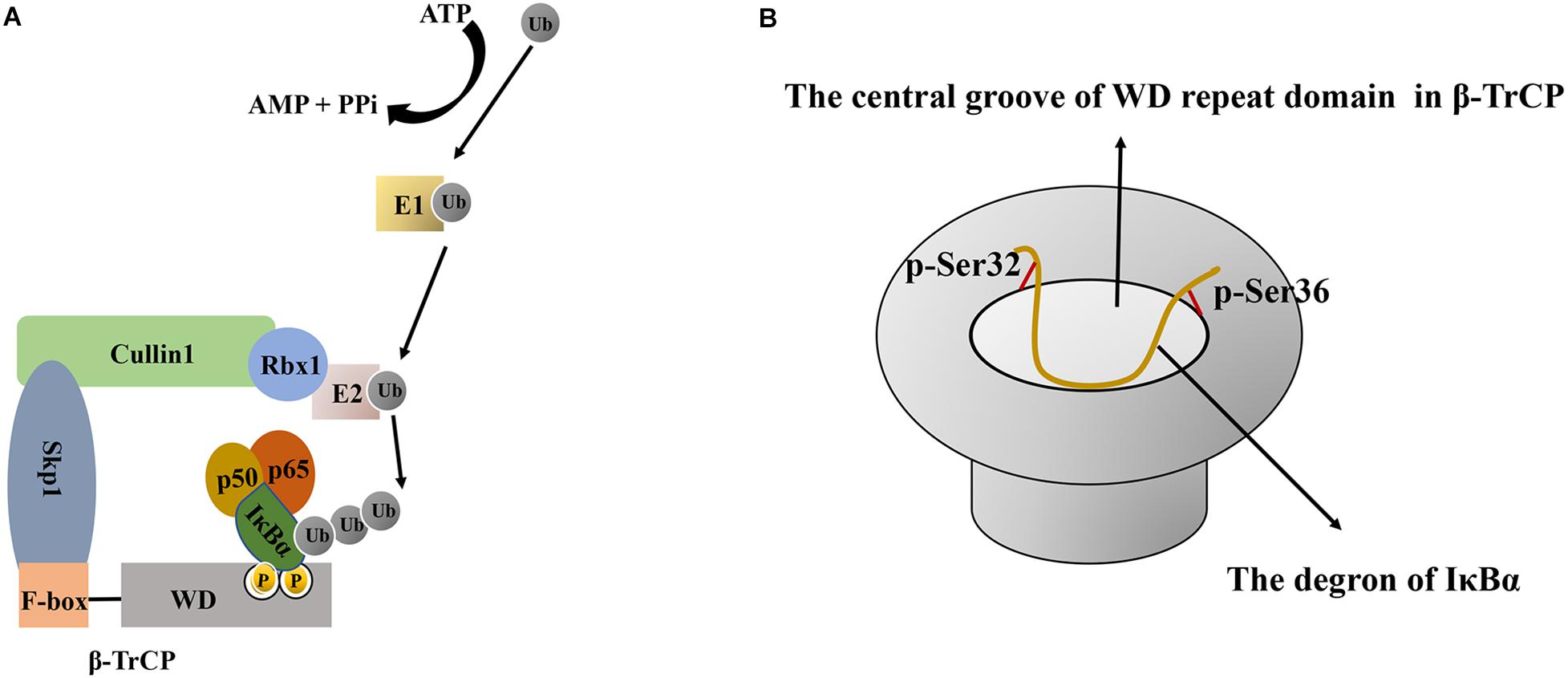
Figure 3. The schematic diagram of IκBα ubiquitination and the interaction between β-TrCP and IκBα. (A) Schematic diagram of IκBα ubiquitination. First, E1- activating enzyme activates ubiquitin in an ATP-dependent manner, and then, the E1-ubiquitin intermediate transfers ubiquitin to E2 to form an E2-ubiquitin intermediate. Finally, IκBα ubiquitination requires a specific E3 ubiquitin ligase comprising Skp1, Cullin1, Rbx1, and β-TrCP. As a scaffold protein, Cullin1 can bind to the N-terminus of Skp1 and the C-terminus of Rbx1. Rbx1 binds to E2 and promotes the conversion of ubiquitin from E2 to IκBα, whereas the β-TrCP protein has two characteristic domains, namely the N-terminal F-box domain and the C-terminal WD repeat domain. The F-box domain of β-TrCP is mainly responsible for binding to Skp1, whereas the WD repeat domain is essential for IκBα recognition. (B) Schematic diagram of the interaction between the central groove of the WD repeat domain in β-TrCP and the degron of IκBα. All six residues in the IκBα degron are in contact with β-TrCP, and the phosphate groups of p-Ser32 and p-Ser36 bind at the edge of the groove. Ub, Ubiquitin; ATP, adenosine triphosphate; AMP, adenosine monophosphate; PPi, pyrophosphoric acid; IκBα, nuclear factor-kappa B inhibitor alpha; E1, E1 ubiquitin-activating enzyme; E2, E2 ubiquitin-conjugating enzyme; Skp1, S phase kinase-associated protein 1; β-TrCP, β–transducin repeat–containing protein; WD, tryptophan-aspartic acid.
The following changes occur when IκBα combines with β-TrCP: 1) Conformational changes. The 24-residue phosphorylated peptide (24p-IκBα, 21–44 aa) in the free state exhibits a curvature around the 31DpSGLDpS36 residue. Upon binding with β-TrCP, in addition to the curve corresponding to the 31DpSGLDpS36 motif, the peptide has two bending regions (Lys22–Asp31 and Met37–Glu43) on both sides (Pons et al., 2007). NMR and crystal structure indicated that the interaction between IκBα and β-TrCP is due to the center curvature. The central bending of the DpSGLDpS motif region is responsible for high-affinity binding, and the N-terminal (25LLDDRH30) and C-terminal (35DpSMKDE40) turning regions can enhance the interaction between IκBα and the β-TrCP protein (Pons et al., 2007). 2) Interaction based on charge. In the DDR29HDpS32GLDpS36MKDE40E fragment, except for Gly33, Leu34 and Met37, all chain residues form a charged surface, which provides a reasonable binding area with the surface of the charged protein β-TrCP (Pons et al., 2007). 3) Interaction between WD40 and the degradation sequence of IκBα. The central groove through the middle of the WD propeller structure can accommodate the degradation sequence of IκBα. All six residues in the IκBα degradation sequence are in contact with β-TrCP. Particularly, the aspartic acid side chain, the skeleton of the hydrophobic residue glycine and a spacer residue are inserted into the farthest recess, enabling intermolecular contact in mostly buried environments. Meanwhile, the phosphate groups of p-Ser32 and p-Ser36 bind at the edge of the groove and, along with aspartic acid, form the maximum number of contacts through hydrogen bonding and electrostatic interactions with the β-TrCP residues around the groove (Kanarek et al., 2010) (Figure 3). All seven WD repeats of β-TrCP facilitate contact with the substrate, and p-Ser32 may be the major site of interaction with β-TrCP (Yazdi et al., 2017). The abovementioned detailed protein structural information can be found in references (Pons et al., 2007; Kanarek et al., 2010). The recognition of IκBα by β-TrCP prompts the transfer of ubiquitin from the thioester intermediate formed with E2 to Lys21 and Lys22 of IκBα. When the first ubiquitin molecule is attached to the target protein, the other ubiquitin molecules are successively linked to the Lys48 residue of the ubiquitin molecule linked to the substrate in the presence of SCFβ-TrCP E3, thereby forming a polyubiquitin chain, which is a target signal for the recognition and degradation of IκBα by the proteasome (Figure 2). Ubiquitinated IκBα enters the 26S proteasome and is degraded in the 20S catalytic center. Ubiquitins can be hydrolyzed from IκBα by deubiquitinating enzymes, such as ubiquitin-specific protease for reutilization, which is the deubiquitination of IκBα (Rape et al., 2006). In summary, the affinity of SCFβ-TrCP and the activity of deubiquitinating enzymes co-regulate IκBα ubiquitination.
In addition to β-TrCP, the muscular atrophy F-box protein has been shown to ubiquitinate IκBα in cardiomyocytes (Usui et al., 2011). Additionally, a novel E3 ligase, F-box and WD repeat domain-containing protein 7 (FBW7), targeting IκBα has been identified; its upregulation promotes the ubiquitin-dependent IκBα degradation, NF-κB activation, and the subsequent intestinal inflammation caused by intestinal epithelial cells, whereas its inhibition has the opposite effects (Meng et al., 2020).
Small ubiquitin-related modifier (SUMO) is a class of peptides comprising 98 aa and has low sequence identity with ubiquitin but forms a 3D structure similar to that of ubiquitin. To date, four members of the SUMO family (SUMO1–4) have been identified in mammalian cells (Carbia-Nagashima et al., 2007). SUMO proteins are expressed in a precursor or inactive form with a short C-terminus. Sentrin/SUMO-specific proteases (SENPs) possess hydrolase activity and cleave the inactive precursor of SUMO at the C-terminus to generate the active or mature SUMO. The human SENP family is composed of SENP1, SENP2, SENP3, SENP5, SENP6, SENP7, and SENP8. However, SENP8 had no effect on SUMO, and only SENP1, SENP2 and SENP5 can proteolyze the precursor SUMO proteins (Yeh, 2009; Kumar and Zhang, 2015; Kunz et al., 2018). SUMOylation is the process of protein coupling with SUMO through three enzymatic steps similar to the ubiquitin-binding cascade reaction.
The SUMOylation of IκBα can be activated by hypoxia/reoxygenation, adenosine receptor agonists (Liu et al., 2009). Particularly, in mammalian cells, the SUMO-specific E1-activating enzyme is the heterodimer SAE1/SAE2, and the E2-binding enzyme is the ubiquitin-conjugating enzyme 9 (Ubc9). In the presence of ATP, the IκBα SUMOylation in vitro only requires SAE1/SAE2 and Ubc9 and not E3 protein ligase. However, whether the E3 protein ligase is necessary for the SUMOylation of IκBα in vivo remains unclear (Desterro et al., 1999).
IκBα SUMOylation based on in vitro experiments is described as follows (Figure 4). First, the active site Cys173 in SAE2 forms a thioester bond with the glycine residue at the C-terminus of SUMO in an ATP-dependent mannerr. This transfers SUMO to the catalytic cysteine residue Cys93 of Ubc9 (Olsen et al., 2010). Thereafter, Ubc9 directly interacts with IκBα by recognizing the consensus sequence ψKxE (ψ represents a hydrophobic amino acid residue, whereas x represents any amino acid residue) and transfers SUMO to IκBα. The glycine residue in the C-terminus of SUMO forms a covalent isopeptide bond with the side chain of Lys21 in IκBα (Rodriguez et al., 2001). In addition, the results of high-throughput screening for SUMOylated proteins recently identified Lys38 as another SUMOylation site of IκBα (Hendriks et al., 2017).
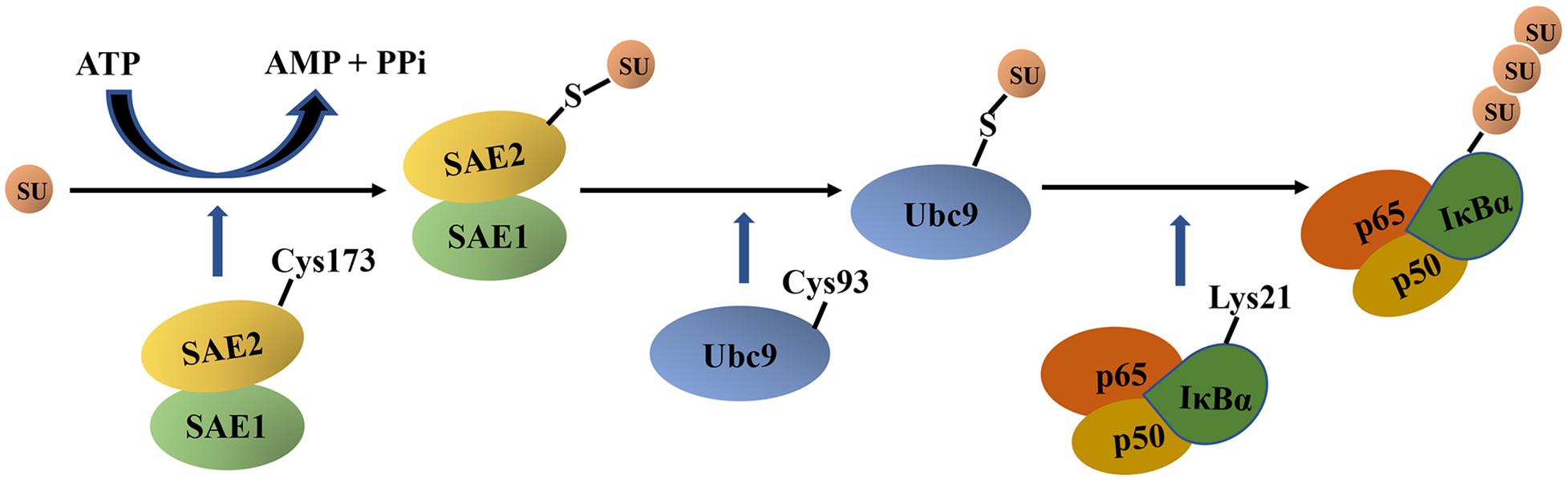
Figure 4. The schematic diagram of IκBα SUMOylation. First, a C-terminal glycine residue of mature SUMO forms a thioester bond with the active site Cys173 in SAE2 in an ATP-dependent manner. Thereafter, SUMO is transferred to the catalytic cysteine residue Cys93 of Ubc9, and Ubc9 directly interacts with IκBα and transfers SUMO to IκBα. Next, the glycine residue at the C terminal of mature SUMO forms a covalent isopeptide bond with the Lys21 side chain in IκBα. IκBα, nuclear factor-kappa B inhibitor alpha; SU, SUMO, small ubiquitin-like modifier; SAE, SUMO-1–activating enzyme; Ubc9, ubiquitin-conjugating enzyme 9; ATP, adenosine triphosphate; AMP, adenosine monophosphate; PPi, pyrophosphoric acid.
Complex interactions between the SUMOylated IκBα and other IκBα modifications, such as ubiquitination and phosphorylation, affect the functional effect of IκBα SUMOylation (Scherer et al., 1995; Desterro et al., 1998; Hay et al., 1999; Liu et al., 2009; Aillet et al., 2012; Mulero et al., 2013b; Marruecos et al., 2020). The effects of the complex interactions is described as follows. (1) The SUMO1 modification of IκBα inhibits the ubiquitination of Lys21 and Lys22 in IκBα and blocks IκBα degradation (Figure 2). Studies have shown that the SUMO1 overexpression blocks the ubiquitination and degradation of IκBα caused by TNF-α and IL-1 in African green monkey kidney fibroblast cell line COS7 (Desterro et al., 1998). The following mechanisms may be involved in the inhibition of IκBα ubiquitination by SUMO1 modification. First, because Lys21 is the site of both SUMO and ubiquitin modifications, SUMO1 can competitively bind to it. Many SUMO1 modifications can stabilize IκBα and protect it from degradation by the proteasome system, thus inhibiting NF-κB activation (Desterro et al., 1998). Moreover, the NTD of IκBα is flexible, and the SUMOylation of Lys21 may cause the conformational change of IκBα to prevent it from being recognized by the SCF complex. Second, ubiquitination can occur solely at Lys22 to promote the proteasome-mediated degradation of IκBα without modification at Lys21. However, the SUMOylation of Lys21 having a spherical structure may prevent Lys22 ubiquitination by the β-TrCP SCF complex (Scherer et al., 1995; Mabb and Miyamoto, 2007). (2) The SUMO2/3 modification of IκBα can promote the ubiquitin-dependent degradation of IκBα (Aillet et al., 2012). Studies have shown that the SUMO2/3 modification of IκBα can be detected under physiological conditions. Upon TNF-α stimulation, the heterologous chains of SUMO2/3 and ubiquitin can promote IκBα degradation by 26S proteasome more effectively than SUMO2/3 or ubiquitin alone (Aillet et al., 2012). (3) The SUMOylation of IκBα is blocked by its phosphorylation. Phosphorylated IκBα at Ser32/Ser36 cannot undergo SUMOylation, suggesting that the SUMO modification is only effective before IKK phosphorylates IκBα (Hay et al., 1999). (4) The phosphorylation and SUMOylation of IκBα (PS-IκBα) coexist and exert a NF-κB independent transcription repressive effect (Mulero et al., 2013b; Marruecos et al., 2020). In unstimulated basal keratinocytes, IκBα undergoes IKK-independent phosphorylation modification and is then modified by SUMO2/3 to form PS-IκBα, which is retained in the nucleus. Nuclear PS-IκBα binds to the chromatin of the regulatory regions of target genes, such as HOX and IRX, and inhibits their transcription by recruiting the polycomb repressive complex 2 (PRC2), which is involved in the regulation of skin homeostasis and development. When keratinocytes are exposed to inflammatory stimuli or differentiation inducer, PS-IκBα dissociates from the chromatin, resulting in the inability to recruit PRC2 to the promoters of the target genes and subsequently upregulating gene expression. This ultimately establishes a link between inflammatory signals and skin homeostasis (Mulero et al., 2013b). Similarly, in intestinal crypt cells, phosphorylated and SUMOylated nuclear IκBα bind the PRC2 subunit SUZ12 and suppress the expression of fetal intestinal stem cell genes, which participated in the regulation of intestinal stem cell homeostasis and the intestinal response to inflammation, damage and repair (Marruecos et al., 2020). The abovementioned mechanism underlying nuclear PS-IκBα deepens the understanding of the IκBα function (Mulero et al., 2013b; Marruecos et al., 2020).
In addition to acting as a hydrolase to promote SUMO maturation, SENP possesses isopeptidase activity and cleaves the isopeptide bond between the glycine at the C-terminal of SUMO and lysine of the substrate protein. This leads to the release of the SUMO protein from its substrate, thereby arresting SUMOylation (Kumar and Zhang, 2015). SENP1, SENP2, SENP3, SENP5, SENP6, and SENP7 have been revealed to possess isopeptidase activities. SENP1 and SENP2 have broad specificity for SUMO-1/2/3, whereas the other SENPs preferentially dissociate SUMO-2/3 from its substrate (Kumar and Zhang, 2015).
Glutathione is a tripeptide composed of glutamic acid, cysteine and glycine. It exists in an oxidized (GSSG) form and a reduced (GSH) form, which can protect cells from damage by reactive oxygen species and heavy metals (Lv et al., 2019). Glutathionylation is a reversible post-translational modification involving formation of a disulfide bond between glutathione and protein cysteine thiol and can be reversed by glutathionase (thiol transferase) in a process called for de-glutathionylation.
In IκBα, Cys186 is the target of glutathionylation (Kil et al., 2008). The stimulation of HeLα cells with an oxidant (such as diamide) that induces GSH oxidation can activate IκBα glutathionylation (Kil et al., 2008). Compared with the native protein, glutathionylated IκBα displayed increased quantum yield of the emission spectra and a red shift of the maximum emission wavelength at 337 nm. Moreover, the accessibility of the hydrophobic regions in glutathionylated IκBα is higher than that in the native protein (Kil et al., 2008). Regarding the functional effect of glutathionylated IκBα, Kil et al. found that the glutathionylation of IκBα suppressed IKK- and CK2-dependment IκBα phosphorylation and IκBα ubiquitination in vitro. The mechanism may be related to the conformational change of IκBα induced by glutathionylation (Kil et al., 2008). In addition, Seidel et al. found that IκBα glutathionylation inhibited IκBα degradation, NF-κB p65 nuclear entry, and NF-κB/DNA binding, therefore downregulating the expression of NF-κB target genes in airway smooth muscle cells (Seidel et al., 2011).
Both in vitro and in vivo experiments have demonstrated that human hypoxia-inducible factor hydroxylase can effectively hydroxylate Asn244 and Asn210 in IκBα, with Asn244 being more effectively hydroxylated than Asn210. Both these residues are located in the hairpin loops that connect the ARs (Cockman et al., 2006). However, the regulatory effect of IκBα hydroxylation modification on the NF-κB signaling pathway remains unclear. Considering that the hydroxylation of collagen prolyl and lysyl residues can promote the stability of the extracellular matrix structure, asparagine hydroxylation in IκBα is speculated to play a similar role in stabilizing the protein structure (Myllyharju and Kivirikko, 2004).
Abnormal NF-κB activation is related to various diseases, such as cardiovascular, neurodegenerative and autoimmune diseases and cancer. For example, the increase in p-IκBα and the activation of NF-κB p65 were detected in monocrotaline-induced pulmonary hypertensive rats, mice with Alzheimer’s disease, rats with autoimmune myocarditis, and gastric cancer caused by Helicobacter pylori (Nozaki et al., 2005; Feng et al., 2017; Zhang et al., 2017; Fang et al., 2019). As a key regulator of the NF-κB signaling pathway, IκBα has attracted increasing research attention and its post-translational modification provides new therapeutic opportunities for diseases related to abnormal NF-κB activation (Vrábel et al., 2019).
For instance, proteasome inhibitors have been clinically used in the treatment of multiple myeloma (MM) (Tundo et al., 2020). Previous studies have demonstrated that the dysregulation of NF-κB pathway contribute to the development and clinical manifestations of MM via NF-κB target genes, including the growth factor interleukin-6 and insulin-like growth factor-1, cell cycle regulators cyclin D and c-Myc, and pro-angiogensis factors vascular endothelial growth factor-C and placental growth factor (Vrábel et al., 2019). Because the ubiquitination and subsequent degradation of IκBα in the proteasome are predominant mechanisms of the NF-κB pathway activation (Feng et al., 2017; Tundo et al., 2020), bortezomib, a reversible proteasome inhibitor, was used in the treatment of MM. Bortezomib binds directly to the β5-subunit of the proteasome and inhibits the enzymatic activity of the proteasome complex. As reported by many clinical trials, bortezomib alone or in combination with other drugs has dramatically improved the outcomes for patients with relapsed MM (Richardson et al., 2003; Lonial et al., 2011), which was in accordance with experimental results showing that bortezomib inhibited NF-κB activation in MM cells by blocking IκBα degradation in the proteasome (Hideshima et al., 2002). In addition, carfilzomib, a second-generation proteasome inhibitor, has been shown to significantly reduce mortality compared with bortezomib. Carfilzomib is the first drug to increase the overall survival rate of MM patients (Dimopoulos et al., 2017). Thereafter, the first oral proteasome inhibitor, ixazomib, was developed for the treatment of relapsed or refractory MM (Narayanan et al., 2020). Compared with bortezomib, ixazomib inhibited tumor proteasome activity for a longer duration and exhibited greater antitumor effect in OCI-Ly10 and PHTX22L lymphoma mouse models (Narayanan et al., 2020). In addition to carfilzomib and ixazomib, second-generation proteasome inhibitors include oprzomib, dilanzomib and malizomib, and their clinical trials are underway (Tundo et al., 2020).
It should be emphasized that in addition to the NF-κB pathway, proteasome inhibitors regulate many other important cellular pathways that depend on proteasome function and affect these signal transduction pathways in both normal and cancer cells, resulting in some limitations to their clinical use, including dose-limiting side effects and the rapid onset of secondary drug resistance (Begalli et al., 2017). For example, bortezomib usually causes peripheral neuropathy, whereas carfilzomib can cause cardiotoxicity, acute renal failure, pulmonary toxicity and other adverse reactions (Narayanan et al., 2020). Therefore, it seems reasonable that the inhibition of IκBα ubiquitination may represent a safer alternative to proteasomal inhibition for blocking NF-κB activation.
Furthermore, because of their central role in regulating IκBα ubiquitination, β-TrCP and its interaction with IκBα are attractive targets (Kanarek and Ben-Neriah, 2012). Studies have found that brain-specific TRIpartite motif protein 9, A49, and GS143 can inhibit IκBα ubiquitination and NF-κB activation by binding to β-TrCP (Nakajima et al., 2008; Mansur et al., 2013; Shi et al., 2014). The IκBα-super repressor (IκBα-SR), a mutant form of IκBα, which can neither be phosphorylated nor ubiquitinated, is found to inhibit NF-κB activation. Furthermore, the intraperitoneal injection of purified IκBα-SR-loaded exosomes attenuates mortality and systemic inflammation in septic mouse models (Choi et al., 2020). IκBα-SR can also increase the sensitivity of pancreatic cancer and pancreatic ductal adenocarcinoma to chemotherapeutics and inhibit tumor development in hepatitis-related cancer murine models (Sato et al., 2003; Pikarsky et al., 2004; Waters et al., 2015). Moreover, the microinjection of IκBα phosphopeptides into TNF-α-stimulated cells blocked NF-κB activation by competing with endogenous IκBα for binding to β-TrCP (Yaron et al., 1997). Cyclic IκBα phosphopeptides, the backbone of which was cyclized to improve stability and selectivity, were designed to effectively block IκBα ubiquitination (Qvit et al., 2009). However, because the E3 ubiquitin ligase SCFβ-TrCP has many substrates other than IκBα, potential adverse effects of β-TrCP inhibitors due to the accumulation of SCFβ-TrCP may occur, thus partly limiting their clinical usage. Therefore, further in-depth studies are required to develop clinical drugs targeting IκBα post-translational modification.
Moreover, a new method, fragment-based virtual E-pharmacophore screening, has been developed to promote the development of drug design targeting on the NF-κB/IκBα complex (Kanan et al., 2019). An excellent study conducted by Kanan et al. investigated the structure of the binding pocket of NF-κB p65/p50 heterodimer complex with IκBα in detail, and therefore constructed the e-pharmacophore models to discover potential ligands with strong binding affinity as candidate NF-κB/IκBα inhibitors. Furthermore, the screening small molecules and known inhibitors were deeply analyzed on the MetaCore/MetaDrug platform and their therapeutic activity, pharmacokinetic and toxicity profile were predicted. The abovementioned computational biological study might provide a novel approach for exploring the potent and low toxicity NF-κB/IκBα inhibitors and give a new perspective of clinical treatment of diseases with abnormal NF-κB activation.
In this review, we summarized the recent literature on post-translational modification of IκBα, including the associated structural changes and regulatory processes and their functional effect on NF-κB activation. These studies provide a theoretical basis for therapeutic interventions targeting IκBα post-translational modification for NF-κB-related disease. Many clinical trials on drugs targeting the IκBα post-translational modification achieved significant clinical benefit, thus confirming the therapeutic significance of these post-translational modifications.
However, there are many notable issues in the drug development based on the post-translational modification of IκBα to be addressed for their future clinical usage. First, considering the unavoidable adverse effects of the IκBα-related NF-κB inhibitors (Pancheri et al., 2020), the pathophysiological mechanisms and the significance of the IκBα–NF-κB pathway in the development of diseases should be further clarified to provide the best ratio of benefit to risk. Second, the drug release controllability to ensure the controllable release rate under certain specific pathophysiological conditions should be taken into account because of the vast regulatory role of NF-κB pathway (Chen and Stark, 2019). For example, endogenous sulfur dioxide (SO2) is found to play extensive regulatory roles in the cardiovascular system as a novel gasotransmitter (Huang et al., 2016). Along with the increasing studies on the pathophysiological significance of endogenous SO2 in the cardiovascular diseases, SO2 donors and prodrugs with different triggering mechanisms, such as thiol-activated SO2 prodrugs, thermally activated SO2 prodrugs and hydrolysis-based SO2 prodrugs, were designed in recent years (Day et al., 2016; Wang and Wang, 2018). The abovementioned studies might provide useful evidence for the drug design targeting the inhibition of NF-κB pathway. Third, novel post-translational modifications, such as persulfidation/sulfhydration (Mustafa et al., 2009; Fu et al., 2019), are being discovered. As reported in the previous studies, persulfidation of target protein characterized by a chemical modification of protein cysteinyl thiols to persulfides could change the protein function (Du et al., 2014). Therefore, whether this new kind of post-translational modification occurs in the IκBα protein should be investigated. Fourth, the interaction among different post-translational modifications of IκBα is important and should be stressed. For example, the discovery of PS-IκBα in the nucleus provides a new target for the treatment of intestinal inflammatory diseases and skin cancers such as squamous cell carcinoma (Mulero et al., 2013b; Colomer et al., 2017; Marruecos et al., 2020).
Finally, the localization of NF-κB and IκBα in the mitochondrion and their regulation of mitochondrial DNA activities should be investigated. NF-κB p65 and IκBα have been detected in the mitochondrial (Bottero et al., 2001; Zamora et al., 2004; Pazarentzos et al., 2014). In addition, TNF-α stimulation induces mitochondrial IκBα phosphorylation and proteasome-independent IκBα degradation and promotes the accumulation of mitochondrial NF-κB p65, thus leading to decreased expression of mitochondrial DNA cytochrome c oxidase III and cytochrome b in U937 cells (Cogswell et al., 2003). Further, in the retina of dark-adapted rats exposed to bright light, the mitochondrial translocation of NF-κB p65 was associated with decreased cytochrome c oxidase III expression (Tomita et al., 2016). Studies on the mitochondrial IκBα–NF-κB may provide an alternative mechanism by which IκBα–NF-κB pathway regulates mitochondrial homeostasis, thus serving as a novel target for the treatment of mitochondria-related diseases.
XW sorted out, reviewed, and analyzed the literatures, drew the diagrams, and wrote the manuscript. HJ devised the concept. HP sorted out, reviewed, and analyzed the literatures and revised the manuscript. HJ, WK, QC, JD, and YH supervised the writing. All the authors revised and approved the final version of the manuscript.
This research was funded by the National Natural Science Foundation of China (81970424 and 81921001), Beijing Natural Science Foundation (7171010, 7191012, and 7182168), Peking University Clinical Medicine Plus X – Young Scholars Project (PKU2019LCXQ011) and the Fundamental Research Funds for the Central Universities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
β -TrCP, β -transducin repeat-containing protein; ABC, activated B-cell type; AR, ankyrin repeat; ARD, ankyrin repeat domain; CK2, casein kinase II; Cox III, cytochrome c oxidase III; CRM1, chromosome region maintenance 1; FBW7, F-box and WD repeat domain-containing protein 7; GSH, glutathione; GSSG, glutathione disulfide; HKII, hexokinase II; I κ B α-SR, I κ B α-super repressor; I κ B, inhibitor of NF- κ B; IKK, I κ B kinase; LPS, lipopolysaccharide; MM, multiple myeloma; NES, nuclear export sequence; NF- κ B, nuclear factor-kappa B; NLS, nuclear localization signal; NPC, nuclear pore complex; NTD, N-terminal domain; PEST, proline–glutamic–serine–threonine; PI3K, phosphoinositide 3-kinase; PKC- ζ, protein kinase C-zeta; PRC2, polycomb repressive complex 2; PS-I κ B α, phosphorylation and SUMOylation of I κ B α; RHD, Rel-homology domain; SAE, SUMO-1-activating enzyme; SCF, S phase kinase-associated protein 1 (SKP1)–cullin 1 (CUL1)–F-box protein; SENP, sentrin/SUMO-specific protease; SO2, sulfur dioxide; SRD, signal-receiving domain; SUMO, small ubiquitin-like modifier; Ub, ubiquitin; Ubc9, ubiquitin-conjugating enzyme 9; UPP, ubiquitin-proteasome-pathway; WD, tryptophan-aspartic acid.
Abu-Amer, Y., Ross, F. P., McHugh, K. P., Livolsi, A., Peyron, J. F., and Teitelbaum, S. L. (1998). Tumor necrosis factor-alpha activation of nuclear transcription factor-kappaB in marrow macrophages is mediated by c-Src tyrosine phosphorylation of Ikappa Balpha. J. Biol. Chem. 273, 29417–29423. doi: 10.1074/jbc.273.45.29417
Aguilera, C., Hoya-Arias, R., Haegeman, G., Espinosa, L., and Bigas, A. (2004). Recruitment of IkappaBalpha to the hes1 promoter is associated with transcriptional repression. Proc. Natl. Acad. Sci. U.S.A. 101, 16537–16542. doi: 10.1073/pnas.0404429101
Aillet, F., Lopitz-Otsoa, F., Egaña, I., Hjerpe, R., Fraser, P., Hay, R. T., et al. (2012). Heterologous SUMO-2/3-ubiquitin chains optimize IκBα degradation and NF-κB activity. PLoS One 7:e51672. doi: 10.1371/journal.pone.0051672
Annemann, M., Plaza-Sirvent, C., Schuster, M., Katsoulis-Dimitriou, K., Kliche, S., Schraven, B., et al. (2016). Atypical IκB proteins in immune cell differentiation and function. Immunol. Lett. 171, 26–35. doi: 10.1016/j.imlet.2016.01.006
Arenzana-Seisdedos, F., Turpin, P., Rodriguez, M., Thomas, D., Hay, R. T., Virelizier, J. L., et al. (1997). Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J. Cell Sci. 110(Pt 3), 369–378.
Bai, C., Sen, P., Hofmann, K., Ma, L., Goebl, M., Harper, J. W., et al. (1996). SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell 86, 263–274. doi: 10.1016/s0092-8674(00)80098-7
Barnes, P. J., and Karin, M. (1997). Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336, 1066–1071. doi: 10.1056/NEJM199704103361506
Begalli, F., Bennett, J., Capece, D., Verzella, D., D’Andrea, D., Tornatore, L., et al. (2017). Unlocking the NF-κB conundrum: embracing complexity to achieve specificity. Biomedicines 5:50. doi: 10.3390/biomedicines5030050
Béraud, C., Henzel, W. J., and Baeuerle, P. A. (1999). Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-kappaB activation. Proc. Natl. Acad. Sci. U.S.A. 96, 429–434. doi: 10.1073/pnas.96.2.429
Boisson, B., Puel, A., Picard, C., and Casanova, J. (2017). Human IκBα gain of function: a severe and syndromic immunodeficiency. J. Clin. Immunol. 37, 397–412. doi: 10.1007/s10875-017-0400-z
Bottero, V., Rossi, F., Samson, M., Mari, M., Hofman, P., and Peyron, J. F. (2001). Ikappa b-alpha, the NF-kappa B inhibitory subunit, interacts with ANT, the mitochondrial ATP/ADP translocator. J. Biol. Chem. 276, 21317–21324. doi: 10.1074/jbc.M005850200
Bren, G. D., Pennington, K. N., and Paya, C. V. (2000). PKC-zeta-associated CK2 participates in the turnover of free IkappaBalpha. J. Mol. Biol. 297, 1245–1258. doi: 10.1006/jmbi.2000.3630
Bristow, C. L., Wolkowicz, R., Trucy, M., Franklin, A., Di, M. F., Kozlowski, M. T., et al. (2008). NF-kappaB signaling, elastase localization, and phagocytosis differ in HIV-1 permissive and nonpermissive U937 clones. J. Immunol. 180, 492–499. doi: 10.4049/jimmunol.180.1.492
Brown, M., McGuinness, M., Wright, T., Ren, X., Wang, Y., Boivin, G. P., et al. (2005). Cardiac-specific blockade of NF-kappaB in cardiac pathophysiology: differences between acute and chronic stimuli in vivo. Am. J. Physiol. Heart Circ. Physiol. 289, H466–H476. doi: 10.1152/ajpheart.00170.2004
Carbia-Nagashima, A., Gerez, J., Perez-Castro, C., Paez-Pereda, M., Silberstein, S., Stalla, G. K., et al. (2007). RSUME, a small RWD-containing protein, enhances SUMO conjugation and stabilizes HIF-1alpha during hypoxia. Cell 131, 309–323. doi: 10.1016/j.cell.2007.07.044
Cervantes, C. F., Bergqvist, S., Kjaergaard, M., Kroon, G., Sue, S.-C., Dyson, H. J., et al. (2011). The RelA nuclear localization signal folds upon binding to IκBα. J. Mol. Biol. 405, 754–764. doi: 10.1016/j.jmb.2010.10.055
Chen, J., and Stark, L. A. (2019). Insights into the relationship between nucleolar stress and the NF-κB pathway. Trends Genet. 35, 768–780. doi: 10.1016/j.tig.2019.07.009
Chen, L. F., and Greene, W. C. (2004). Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 5, 392–401. doi: 10.1038/nrm1368
Choi, H., Kim, Y., Mirzaaghasi, A., Heo, J., Kim, Y. N., Shin, J. H., et al. (2020). Exosome-based delivery of super-repressor IκBα relieves sepsis-associated organ damage and mortality. Sci. Adv. 6:eaaz6980. doi: 10.1126/sciadv.aaz6980
Cockman, M. E., Lancaster, D. E., Stolze, I. P., Hewitson, K. S., McDonough, M. A., Coleman, M. L., et al. (2006). Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc. Natl. Acad. Sci. U.S.A. 103, 14767–14772. doi: 10.1073/pnas.0606877103
Cogswell, P. C., Kashatus, D. F., Keifer, J. A., Guttridge, D. C., Reuther, J. Y., Bristow, C., et al. (2003). NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J. Biol. Chem. 278, 2963–2968. doi: 10.1074/jbc.M209995200
Colomer, C., Marruecos, L., Vert, A., Bigas, A., and Espinosa, L. (2017). NF-κB members left home: NF-κB-independent roles in cancer. Biomedicines 5:26. doi: 10.3390/biomedicines5020026
Croy, C. H., Bergqvist, S., Huxford, T., Ghosh, G., and Komives, E. A. (2004). Biophysical characterization of the free IkappaBalpha ankyrin repeat domain in solution. Protein Sci. 13, 1767–1777. doi: 10.1110/ps.04731004
Day, J. J., Yang, Z., Chen, W., Pacheco, A., and Xian, M. (2016). Benzothiazole sulfinate: a water-soluble and slow-releasing sulfur dioxide donor. ACS Chem. Biol. 11, 1647–1651. doi: 10.1021/acschembio.6b00106
Deshaies, R. J. (1999). SCF and cullin/ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 15, 435–467. doi: 10.1146/annurev.cellbio.15.1.435
Desterro, J. M., Rodriguez, M. S., and Hay, R. T. (1998). SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 2, 233–239. doi: 10.1016/s1097-2765(00)80133-1
Desterro, J. M., Rodriguez, M. S., Kemp, G. D., and Hay, R. T. (1999). Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J. Biol. Chem. 274, 10618–10624. doi: 10.1074/jbc.274.15.10618
DiDonato, J. A., Mercurio, F., and Karin, M. (2012). NF-κB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400. doi: 10.1111/j.1600-065X.2012.01099.x
Dimopoulos, M. A., Goldschmidt, H., Niesvizky, R., Joshua, D., Chng, W., Oriol, A., et al. (2017). Carfilzomib or bortezomib in relapsed or refractory multiple myeloma (ENDEAVOR): an interim overall survival analysis of an open-label, randomised, phase 3 trial. Lancet Oncol. 18, 1327–1337. doi: 10.1016/S1470-2045(17)30578-8
Domingo-Sananes, M. R., Kapuy, O., Hunt, T., and Novak, B. (2011). Switches and latches: a biochemical tug-of-war between the kinases and phosphatases that control mitosis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 3584–3594. doi: 10.1098/rstb.2011.0087
Du, J., Huang, Y., Yan, H., Zhang, Q., Zhao, M., Zhu, M., et al. (2014). Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor κB (NF-κB) pathway. J. Biol. Chem. 289, 9741–9753. doi: 10.1074/jbc.M113.517995
Elewaut, D., DiDonato, J. A., Kim, J. M., Truong, F., Eckmann, L., and Kagnoff, M. F. (1999). NF-kappa B is a central regulator of the intestinal epithelial cell innate immune response induced by infection with enteroinvasive bacteria. J. Immunol. 163, 1457–1466.
Ellis, L., Gilston, V., Soo, C. C., Morris, C. J., Kidd, B. L., and Winyard, P. G. (2000). Activation of the transcription factor NF-kappaB in the rat air pouch model of inflammation. Ann. Rheum. Dis. 59, 303–307. doi: 10.1136/ard.59.4.303
Ernst, M. K., Dunn, L. L., and Rice, N. R. (1995). The PEST-like sequence of I kappa B alpha is responsible for inhibition of DNA binding but not for cytoplasmic retention of c-Rel or RelA homodimers. Mol. Cell. Biol. 15, 872–882. doi: 10.1128/mcb.15.2.872
Espinosa, L., Bigas, A., and Mulero, M. C. (2011). Alternative nuclear functions for NF-κB family members. Am. J. Cancer Res. 1, 446–459.
Fan, C., Li, Q., Ross, D., and Engelhardt, J. F. (2003). Tyrosine phosphorylation of I kappa B alpha activates NF kappa B through a redox-regulated and c-Src-dependent mechanism following hypoxia/reoxygenation. J. Biol. Chem. 278, 2072–2080. doi: 10.1074/jbc.M206718200
Fang, W., Liao, W., Zheng, Y., Huang, X., Weng, X., Fan, S., et al. (2019). Neurotropin reduces memory impairment and neuroinflammation via BDNF/NF-κB in a transgenic mouse model of Alzheimer’s disease. Am. J. Transl. Res. 11, 1541–1554.
Feng, S., Chen, S., Yu, W., Zhang, D., Zhang, C., Tang, C., et al. (2017). HS inhibits pulmonary arterial endothelial cell inflammation in rats with monocrotaline-induced pulmonary hypertension. Lab. Invest. 97, 268–278. doi: 10.1038/labinvest.2016.129
Ferreiro, D. U., and Komives, E. A. (2010). Molecular mechanisms of system control of NF-kappaB signaling by IkappaBalpha. Biochemistry 49, 1560–1567. doi: 10.1021/bi901948j
Fu, L., Liu, K., He, J., Tian, C., Yu, X., and Yang, J. (2019). Direct proteomic mapping of cysteine persulfidation. Antioxid. Redox Signal. doi: 10.1089/ars.2019.7777 [Epub ahead of print].
Giridharan, S., and Srinivasan, M. (2018). Mechanisms of NF-κB p65 and. strategies for therapeutic manipulation. J. Inflamm. Res. 11, 407–419. doi: 10.2147/JIR.S140188
Hay, R. T., Vuillard, L., Desterro, J. M., and Rodriguez, M. S. (1999). Control of NF-kappa B transcriptional activation by signal induced proteolysis of I kappa B alpha. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1601–1609. doi: 10.1098/rstb.1999.0504
Hayden, M. S., and Ghosh, S. (2004). Signaling to NF-kappaB. Genes Dev. 18, 2195–2224. doi: 10.1101/gad.1228704
Hendriks, I. A., Lyon, D., Young, C., Jensen, L. J., Vertegaal, A. C. O., and Nielsen, M. L. (2017). Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 24, 325–336. doi: 10.1038/nsmb.3366
Herter, J. R., and Fuchs, S. Y. (2002). Recognition of substrate and Skp1 by the homologue of Slimb (HOS) ubiquitin ligase receptor D role of the F-box. Med. Sci. Monit. 8, BR283–BR288.
Hideshima, T., Chauhan, D., Richardson, P., Mitsiades, C., Mitsiades, N., Hayashi, T., et al. (2002). NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 277, 16639–16647. doi: 10.1074/jbc.M200360200
Hinz, M., Arslan, S. Ç., and Scheidereit, C. (2012). It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol. Rev. 246, 59–76. doi: 10.1111/j.1600-065X.2012.01102.x
Hoffmann, A., Natoli, G., and Ghosh, G. (2006). Transcriptional regulation via the NF-kappaB signaling module. Oncogene 25, 6706–6716. doi: 10.1038/sj.onc.1209933
Hu, X., Li, T., Wang, Y., Xiong, Y., Wu, X., Zhang, D., et al. (2017). Prokaryotic and highly-repetitive WD40 proteins: a systematic study. Sci. Rep. 7:10585. doi: 10.1038/s41598-017-11115-1
Huang, T. T., Kudo, N., Yoshida, M., and Miyamoto, S. (2000). A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc. Natl. Acad. Sci. U.S.A. 97, 1014–1019. doi: 10.1073/pnas.97.3.1014
Huang, T. T., and Miyamoto, S. (2001). Postrepression activation of NF-kappaB requires the amino-terminal nuclear export signal specific to IkappaBalpha. Mol. Cell. Biol. 21, 4737–4747. doi: 10.1128/MCB.21.14.4737-4747.2001
Huang, Y., Tang, C., Du, J., and Jin, H. (2016). Endogenous sulfur dioxide: a new member of gasotransmitter family in the cardiovascular system. Oxid. Med. Cell. Longev. 2016:8961951. doi: 10.1155/2016/8961951
Hussain, A. R., Uddin, S., Ahmed, M., Al-Dayel, F., Bavi, P. P., and Al-Kuraya, K. S. (2013). Phosphorylated IκBα predicts poor prognosis in activated B-cell lymphoma and its inhibition with thymoquinone induces apoptosis via ROS release. PLoS One 8:e60540. doi: 10.1371/journal.pone.0060540
Imbert, V., Rupec, R. A., Livolsi, A., Pahl, H. L., Traenckner, E. B., Mueller-Dieckmann, C., et al. (1996). Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha. Cell 86, 787–798. doi: 10.1016/s0092-8674(00)80153-1
Jo, H., Zhang, R., Zhang, H., McKinsey, T. A., Shao, J., Beauchamp, R. D., et al. (2000). NF-kappa B is required for H-ras oncogene induced abnormal cell proliferation and tumorigenesis. Oncogene 19, 841–849. doi: 10.1038/sj.onc.1203392
Johnson, C., Van Antwerp, D., and Hope, T. J. (1999). An N-terminal nuclear export signal is required for the nucleocytoplasmic shuttling of IkappaBalpha. EMBO J. 18, 6682–6693. doi: 10.1093/emboj/18.23.6682
Kanan, T., Kanan, D., Erol, I., Yazdi, S., Stein, M., and Durdagi, S. (2019). Targeting the NF-κB/IκBα complex via fragment-based E-pharmacophore virtual screening and binary QSAR models. J. Mol. Graph. Model. 86, 264–277. doi: 10.1016/j.jmgm.2018.09.014
Kanarek, N., and Ben-Neriah, Y. (2012). Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol. Rev. 246, 77–94. doi: 10.1111/j.1600-065X.2012.01098.x
Kanarek, N., London, N., Schueler-Furman, O., and Ben-Neriah, Y. (2010). Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2:a000166. doi: 10.1101/cshperspect.a000166
Kato, K., Tokuda, H., Matsushima-Nishiwaki, R., Natsume, H., Kondo, A., Ito, Y., et al. (2012). AMPK limits IL-1-stimulated IL-6 synthesis in osteoblasts: involvement of IκB/NF-κB pathway. Cell. Signal. 24, 1706–1712. doi: 10.1016/j.cellsig.2012.04.012
Kato, T., Delhase, M., Hoffmann, A., and Karin, M. (2003). CK2 is a C-terminal. IkappaB kinase responsible for NF-kappaB activation during the UV response. Mol. Cell 12, 829–839. doi: 10.1016/s1097-2765(03)00358-7
Kawai, H., Nie, L., and Yuan, Z. (2002). Inactivation of NF-kappaB-dependent cell survival, a novel mechanism for the proapoptotic function of c-Abl. Mol. Cell. Biol. 22, 6079–6088. doi: 10.1128/mcb.22.17.6079-6088.2002
Kil, I. S., Kim, S. Y., and Park, J. (2008). Glutathionylation regulates IkappaB. Biochem. Biophys. Res. Commun. 373, 169–173. doi: 10.1016/j.bbrc.2008.06.007
Kim, J. H., Lee, J., Oh, B., Kimm, K., and Koh, I. (2004). Prediction of phosphorylation sites using SVMs. Bioinformatics 20, 3179–3184. doi: 10.1093/bioinformatics/bth382
Kis, A., Yellon, D. M., and Baxter, G. F. (2003). Role of nuclear factor-kappa B activation in acute ischaemia-reperfusion injury in myocardium. Br. J. Pharmacol. 138, 894–900. doi: 10.1038/sj.bjp.0705108
Ko, E., Cho, S., Kwon, S., Eom, C., Jeong, M., Lee, W., et al. (2017). The roles of NF-κB and ROS in regulation of pro-inflammatory mediators of inflammation induction in LPS-stimulated zebrafish embryos. Fish Shellfish Immunol. 68, 525–529. doi: 10.1016/j.fsi.2017.07.041
Komives, E. A. (2012). Consequences of fuzziness in the NFκB/IκBα interaction. Adv. Exp. Med. Biol. 725, 74–85. doi: 10.1007/978-1-4614-0659-4_5
Kumar, A., and Zhang, K. Y. J. (2015). Advances in the development of SUMO specific protease (SENP) inhibitors. Comput. Struct. Biotechnol. J. 13, 204–211. doi: 10.1016/j.csbj.2015.03.001
Kunz, K., Piller, T., and Müller, S. (2018). SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 131:jcs211904. doi: 10.1242/jcs.211904
Lee, S., and Hannink, M. (2002). Characterization of the nuclear import and export functions of Ikappa B(epsilon). J. Biol. Chem. 277, 23358–23366. doi: 10.1074/jbc.M111559200
Lee, S. H., and Hannink, M. (2001). The N-terminal nuclear export sequence of IkappaBalpha is required for RanGTP-dependent binding to CRM1. J. Biol. Chem. 276, 23599–23606. doi: 10.1074/jbc.M011197200
Li, J., Mahajan, A., and Tsai, M. (2006). Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45, 15168–15178. doi: 10.1021/bi062188q
Lin, R., Beauparlant, P., Makris, C., Meloche, S., and Hiscott, J. (1996). Phosphorylation of IkappaBalpha in the C-terminal PEST domain by casein kinase II affects intrinsic protein stability. Mol. Cell. Biol. 16, 1401–1409. doi: 10.1128/mcb.16.4.1401
Liu, J., Yuan, Y., Xu, J., Xiao, K., Xu, Y., Guo, T., et al. (2018). β-TrCP restricts lipopolysaccharide (LPS)-induced activation of TRAF6-IKK pathway upstream of IκBα signaling. Front. Immunol. 9:2930. doi: 10.3389/fimmu.2018.02930
Liu, Q., Li, J., Khoury, J., Colgan, S. P., and Ibla, J. C. (2009). Adenosine signaling mediates SUMO-1 modification of IkappaBalpha during hypoxia and reoxygenation. J. Biol. Chem. 284, 13686–13695. doi: 10.1074/jbc.M809275200
Lonial, S., Mitsiades, C. S., and Richardson, P. G. (2011). Treatment options for relapsed and refractory multiple myeloma. Clin. Cancer Res. 17, 1264–1277. doi: 10.1158/1078-0432.CCR-10-1805
Lv, H., Zhen, C., Liu, J., Yang, P., Hu, L., and Shang, P. (2019). Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid. Med. Cell. Longev. 2019:3150145. doi: 10.1155/2019/3150145
Mabb, A. M., and Miyamoto, S. (2007). SUMO and NF-kappaB ties. Cell. Mol. Life Sci. 64, 1979–1996. doi: 10.1007/s00018-007-7005-2
Mansur, D. S., Maluquer de Motes, C., Unterholzner, L., Sumner, R. P., Ferguson, B. J., Ren, H., et al. (2013). Poxvirus targeting of E3 ligase β-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog. 9:e1003183. doi: 10.1371/journal.ppat.1003183
Marruecos, L., Bertran, J., Guillén, Y., González, J., Batlle, R., López-Arribillaga, E., et al. (2020). IκBα deficiency imposes a fetal phenotype to intestinal stem cells. EMBO Rep. 21:e49708. doi: 10.15252/embr.201949708
Mathes, E., O’Dea, E. L., Hoffmann, A., and Ghosh, G. (2008). NF-kappaB dictates the degradation pathway of IkappaBalpha. EMBO J. 27, 1357–1367. doi: 10.1038/emboj.2008.73
McElhinny, J. A., Trushin, S. A., Bren, G. D., Chester, N., and Paya, C. V. (1996). Casein kinase II phosphorylates I kappa B alpha at S-283, S-289, S-293, and T-291 and is required for its degradation. Mol. Cell. Biol. 16, 899–906. doi: 10.1128/mcb.16.3.899
Meng, Q., Wu, W., Pei, T., Xue, J., Xiao, P., Sun, L., et al. (2020). miRNA-129/FBW7/NF-κB, a novel regulatory pathway in inflammatory bowel disease. Mol. Ther. Nucleic Acids 19, 731–740. doi: 10.1016/j.omtn.2019.10.048
Mothes, J., Busse, D., Kofahl, B., and Wolf, J. (2015). Sources of dynamic variability in NF-κB signal transduction: a mechanistic model. Bioessays 37, 452–462. doi: 10.1002/bies.201400113
Mukhopadhyay, A., Manna, S. K., and Aggarwal, B. B. (2000). Pervanadate-induced nuclear factor-kappaB activation requires tyrosine phosphorylation and degradation of IkappaBalpha. Comparison with tumor necrosis factor-alpha. J. Biol. Chem. 275, 8549–8555. doi: 10.1074/jbc.275.12.8549
Mulero, M. C., Bigas, A., and Espinosa, L. (2013a). IκBα beyond the NF-kB dogma. Oncotarget 4, 1550–1551. doi: 10.18632/oncotarget.1325
Mulero, M. C., Ferres-Marco, D., Islam, A., Margalef, P., Pecoraro, M., Toll, A., et al. (2013b). Chromatin-bound IκBα regulates a subset of polycomb target genes in differentiation and cancer. Cancer Cell 24, 151–166. doi: 10.1016/j.ccr.2013.06.003
Mustafa, A. K., Gadalla, M. M., Sen, N., Kim, S., Mu, W., Gazi, S. K., et al. (2009). H2S signals through protein S-sulfhydration. Sci. Signal. 2:ra72. doi: 10.1126/scisignal.2000464
Myllyharju, J., and Kivirikko, K. I. (2004). Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 20, 33–43. doi: 10.1016/j.tig.2003.11.004
Nakajima, H., Fujiwara, H., Furuichi, Y., Tanaka, K., and Shimbara, N. (2008). A novel small-molecule inhibitor of NF-kappaB signaling. Biochem. Biophys. Res. Commun. 368, 1007–1013. doi: 10.1016/j.bbrc.2008.01.166
Narayanan, S., Cai, C., Assaraf, Y. G., Guo, H., Cui, Q., Wei, L., et al. (2020). Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updat. 48:100663. doi: 10.1016/j.drup.2019.100663
Nozaki, K., Tanaka, H., Ikehara, Y., Cao, X., Nakanishi, H., Azuma, T., et al. (2005). Helicobacter pylori-dependent NF-kappa B activation in newly established Mongolian gerbil gastric cancer cell lines. Cancer Sci. 96, 170–175. doi: 10.1111/j.1349-7006.2005.00030.x
Olsen, S. K., Capili, A. D., Lu, X., Tan, D. S., and Lima, C. D. (2010). Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 463, 906–912. doi: 10.1038/nature08765
Pal Singh, S., Dammeijer, F., and Hendriks, R. W. (2018). Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 17:57. doi: 10.1186/s12943-018-0779-z
Pancheri, E., Guglielmi, V., Wilczynski, G. M., Malatesta, M., Tonin, P., Tomelleri, G., et al. (2020). Non-hematologic toxicity of bortezomib in multiple myeloma: the neuromuscular and cardiovascular adverse effects. Cancers 12:E2540. doi: 10.3390/cancers12092540
Pazarentzos, E., Mahul-Mellier, A., Datler, C., Chaisaklert, W., Hwang, M., Kroon, J., et al. (2014). IκBα inhibits apoptosis at the outer mitochondrial membrane independently of NF-κB retention. EMBO J. 33, 2814–2828. doi: 10.15252/embj.201488183
Perkins, N. D. (2006). Post-translational modifications regulating the activity and. function of the nuclear factor kappa B pathway. Oncogene 25, 6717–6730. doi: 10.1038/sj.onc.1209937
Pikarsky, E., Porat, R. M., Stein, I., Abramovitch, R., Amit, S., Kasem, S., et al. (2004). NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 431, 461–466. doi: 10.1038/nature02924
Pons, J., Evrard-Todeschi, N., Bertho, G., Gharbi-Benarous, J., Sonois, V., Benarous, R., et al. (2007). Structural studies on 24P-IkappaBalpha peptide derived from a human IkappaB-alpha protein related to the inhibition of the activity of the transcription factor NF-kappaB. Biochemistry 46, 2958–2972. doi: 10.1021/bi061401f
Pons, S., and Torres-Aleman, I. (2000). Insulin-like growth factor-I stimulates dephosphorylation of ikappa B through the serine phosphatase calcineurin (protein phosphatase 2B). J. Biol. Chem. 275, 38620–38625. doi: 10.1074/jbc.M004531200
Pontoriero, M., Fiume, G., Vecchio, E., de Laurentiis, A., Albano, F., Iaccino, E., et al. (2019). Activation of NF-κB in B cell receptor signaling through Bruton’s tyrosine kinase-dependent phosphorylation of IκB-α. J. Mol. Med. 97, 675–690. doi: 10.1007/s00109-019-01777-x
Potoyan, D. A., Zheng, W., Ferreiro, D. U., Wolynes, P. G., and Komives, E. A. (2016). PEST control of molecular stripping of NFκB from DNA transcription sites. J. Phys. Chem. B 120, 8532–8538. doi: 10.1021/acs.jpcb.6b02359
Qvit, N., Hatzubai, A., Shalev, D. E., Ben-Neriah, Y., and Gilon, C. (2009). Design and synthesis of backbone cyclic phosphopeptides: the IkappaB model. Biopolymers 91, 157–168. doi: 10.1002/bip.21098
Rape, M., Reddy, S. K., and Kirschner, M. W. (2006). The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell 124, 89–103. doi: 10.1016/j.cell.2005.10.032
Richardson, P. G., Barlogie, B., Berenson, J., Singhal, S., Jagannath, S., Irwin, D., et al. (2003). A phase 2 study of bortezomib in relapsed, refractory myeloma. N. Engl. J. Med. 348, 2609–2617. doi: 10.1056/NEJMoa030288
Rodriguez, M. S., Dargemont, C., and Hay, R. T. (2001). SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659. doi: 10.1074/jbc.M009476200
Sachdev, S., Bagchi, S., Zhang, D. D., Mings, A. C., and Hannink, M. (2000). Nuclear import of IkappaBalpha is accomplished by a ran-independent transport pathway. Mol. Cell. Biol. 20, 1571–1582. doi: 10.1128/mcb.20.5.1571-1582.2000
Sachdev, S., Hoffmann, A., and Hannink, M. (1998). Nuclear localization of IkappaB alpha is mediated by the second ankyrin repeat: the IkappaB alpha ankyrin repeats define a novel class of cis-acting nuclear import sequences. Mol. Cell. Biol. 18, 2524–2534. doi: 10.1128/mcb.18.5.2524
Safi, S. Z., Shah, H., Qvist, R., Bindal, P., Mansor, M., Yan, G. O. S., et al. (2018). Beta adrenergic receptors stimulation attenuates phosphorylation of NF-κB and IκBα in hyperglycemic endothelial cells. Cell. Physiol. Biochem. 51, 1429–1436. doi: 10.1159/000495591
Sato, T., Odagiri, H., Ikenaga, S., Maruyama, M., and Sasaki, M. (2003). Chemosensitivity of human pancreatic carcinoma cells is enhanced by IkappaBalpha super-repressor. Cancer Sci. 94, 467–472. doi: 10.1111/j.1349-7006.2003.tb01466.x
Sayed, M., Kim, S. O., Salh, B. S., Issinger, O. G., and Pelech, S. L. (2000). Stress-induced activation of protein kinase CK2 by direct interaction with p38 mitogen-activated protein kinase. J. Biol. Chem. 275, 16569–16573. doi: 10.1074/jbc.M000312200
Scherer, D. C., Brockman, J. A., Chen, Z., Maniatis, T., and Ballard, D. W. (1995). Signal-induced degradation of I kappa B alpha requires site-specific ubiquitination. Proc. Natl. Acad. Sci. U.S.A. 92, 11259–11263. doi: 10.1073/pnas.92.24.11259
Schwarz, E. M., Van Antwerp, D., and Verma, I. M. (1996). Constitutive phosphorylation of IkappaBalpha by casein kinase II occurs preferentially at serine 293: requirement for degradation of free IkappaBalpha. Mol. Cell. Biol. 16, 3554–3559. doi: 10.1128/mcb.16.7.3554
Sedgwick, S. G., and Smerdon, S. J. (1999). The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem. Sci. 24, 311–316. doi: 10.1016/s0968-0004(99)01426-7
Seidel, P., Roth, M., Ge, Q., Merfort, I., S’ng, C. T., and Ammit, A. J. (2011). IκBα glutathionylation and reduced histone H3 phosphorylation inhibit eotaxin and RANTES. Eur. Respir. J. 38, 1444–1452. doi: 10.1183/09031936.00129610
Shi, M., Cho, H., Inn, K., Yang, A., Zhao, Z., Liang, Q., et al. (2014). Negative regulation of NF-κB activity by brain-specific TRIpartite motif protein 9. Nat. Commun. 5:4820. doi: 10.1038/ncomms5820
Shumway, S. D., Maki, M., and Miyamoto, S. (1999). The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mu-calpain. J. Biol. Chem. 274, 30874–30881. doi: 10.1074/jbc.274.43.30874
Singh, R. K., Kazansky, Y., Wathieu, D., and Fushman, D. (2017). Hydrophobic patch of ubiquitin is important for its optimal activation by ubiquitin activating enzyme E1. Anal. Chem. 89, 7852–7860. doi: 10.1021/acs.analchem.6b04194
Skaar, J. R., Pagan, J. K., and Pagano, M. (2013). Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell Biol. 14, 369–381. doi: 10.1038/nrm3582
Sue, S.-C., and Dyson, H. J. (2009). Interaction of the IkappaBalpha C-terminal PEST sequence with NF-kappaB: insights into the inhibition of NF-kappaB DNA binding by IkappaBalpha. J. Mol. Biol. 388, 824–838. doi: 10.1016/j.jmb.2009.03.048
Takada, Y., Mukhopadhyay, A., Kundu, G. C., Mahabeleshwar, G. H., Singh, S., and Aggarwal, B. B. (2003). Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 278, 24233–24241. doi: 10.1074/jbc.M212389200
Tomita, H., Tabata, K., Takahashi, M., Nishiyama, F., and Sugano, E. (2016). Light induces translocation of NF-κB p65 to the mitochondria and suppresses expression of cytochrome c oxidase subunit III (COX III) in the rat retina. Biochem. Biophys. Res. Commun. 473, 1013–1018. doi: 10.1016/j.bbrc.2016.04.008
Traenckner, E. B., Pahl, H. L., Henkel, T., Schmidt, K. N., Wilk, S., and Baeuerle, P. A. (1995). Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. EMBO J. 14, 2876–2883. doi: 10.1002/j.1460-2075.1995.tb07287.x
Trelle, M. B., Ramsey, K. M., Lee, T. C., Zheng, W., Lamboy, J., Wolynes, P. G., et al. (2016). Binding of NFκB appears to twist the ankyrin repeat domain of IκBα. Biophys. J. 110, 887–895. doi: 10.1016/j.bpj.2016.01.001
Truhlar, S. M. E., Torpey, J. W., and Komives, E. A. (2006). Regions of IkappaBalpha that are critical for its inhibition of NF-kappaB.DNA interaction fold upon binding to NF-kappaB. Proc. Natl. Acad. Sci. U.S.A. 103, 18951–18956. doi: 10.1073/pnas.0605794103
Tundo, G. R., Sbardella, D., Santoro, A. M., Coletta, A., Oddone, F., Grasso, G., et al. (2020). The proteasome as a druggable target with multiple therapeutic potentialities: cutting and non-cutting edges. Pharmacol. Ther. 213:107579. doi: 10.1016/j.pharmthera.2020.107579
Turpin, P., Hay, R. T., and Dargemont, C. (1999). Characterization of IkappaBalpha nuclear import pathway. J. Biol. Chem. 274, 6804–6812. doi: 10.1074/jbc.274.10.6804
Usui, S., Maejima, Y., Pain, J., Hong, C., Cho, J., Park, J. Y., et al. (2011). Endogenous muscle atrophy F-box mediates pressure overload-induced cardiac hypertrophy through regulation of nuclear factor-kappaB. Circ. Res. 109, 161–171. doi: 10.1161/CIRCRESAHA.110.238717
Vrábel, D., Pour, L., and Ševèíková, S. (2019). The impact of NF-κB signaling on pathogenesis and current treatment strategies in multiple myeloma. Blood Rev. 34, 56–66. doi: 10.1016/j.blre.2018.11.003
Wang, W. Y., and Wang, B. H. (2018). SO2 donors and prodrugs, and their possible applications: a review. Front. Chem. 6:559. doi: 10.3389/fchem.2018.00559
Wang, Y., Li, M., Huang, T., and Li, J. (2018). Protein tyrosine phosphatase L1 inhibits high-grade serous ovarian carcinoma progression by targeting IκBα. Onco Targets Ther. 11, 7603–7612. doi: 10.2147/OTT.S167106
Waters, J. A., Matos, J., Yip-Schneider, M., Aguilar-Saavedra, J. R., Crean, C. D., Beane, J. D., et al. (2015). Targeted nuclear factor-kappaB suppression enhances gemcitabine response in human pancreatic tumor cell line murine xenografts. Surgery 158, 881–888; discussion 888–889. doi: 10.1016/j.surg.2015.04.043
Wong, H. K., Kammer, G. M., Dennis, G., and Tsokos, G. C. (1999). Abnormal NF-kappa B activity in T lymphocytes from patients with systemic lupus erythematosus is associated with decreased p65-RelA protein expression. J. Immunol. 163, 1682–1689.
Wu, M., Lee, H., Bellas, R. E., Schauer, S. L., Arsura, M., Katz, D., et al. (1996). Inhibition of NF-kappaB/Rel induces apoptosis of murine B cells. EMBO J. 15, 4682–4690. doi: 10.1002/j.1460-2075.1996.tb00845.x
Yaron, A., Gonen, H., Alkalay, I., Hatzubai, A., Jung, S., Beyth, S., et al. (1997). Inhibition of NF-kappa-B cellular function via specific targeting of the I-kappa-B-ubiquitin ligase. EMBO J. 16, 6486–6494. doi: 10.1093/emboj/16.21.6486
Yazdi, S., Durdagi, S., Naumann, M., and Stein, M. (2015). Structural modeling of the N-terminal signal-receiving domain of IκBα. Front. Mol. Biosci. 2:32. doi: 10.3389/fmolb.2015.00032
Yazdi, S., Naumann, M., and Stein, M. (2017). Double phosphorylation-induced structural changes in the signal-receiving domain of IκBα in complex with NF-κB. Proteins 85, 17–29. doi: 10.1002/prot.25181
Yeh, E. T. H. (2009). SUMOylation and De-SUMOylation: wrestling with life’s processes. J. Biol. Chem. 284, 8223–8227. doi: 10.1074/jbc.R800050200
Zamora, M., Meroño, C., Viñas, O., and Mampel, T. (2004). Recruitment of NF-kappaB into mitochondria is involved in adenine nucleotide translocase 1 (ANT1)-induced apoptosis. J. Biol. Chem. 279, 38415–38423. doi: 10.1074/jbc.M404928200
Keywords: IκBα, NF-κB, post-translational modifications, phosphorylation, ubiquitination, SUMOylation
Citation: Wang X, Peng H, Huang Y, Kong W, Cui Q, Du J and Jin H (2020) Post-translational Modifications of IκBα: The State of the Art. Front. Cell Dev. Biol. 8:574706. doi: 10.3389/fcell.2020.574706
Received: 21 June 2020; Accepted: 19 October 2020;
Published: 05 November 2020.
Edited by:
Hui-Hua Li, Dalian Medical University, ChinaReviewed by:
Matthias Stein, Max Planck Institute for Dynamics of Complex Technical Systems, Max Planck Society (MPG), GermanyCopyright © 2020 Wang, Peng, Huang, Kong, Cui, Du and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongfang Jin, amluaG9uZ2Zhbmc1MUAxMjYuY29t; amluaG9uZ2ZhbmdAYmptdS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.