 Yizhe Zhang
Yizhe Zhang Xiao Yang
Xiao Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 18 September 2020
Sec. Molecular and Cellular Pathology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.567682
This article is part of the Research TopicTGF-β in Human Disease: Friend or Foe?View all 11 articles
Cerebrovascular diseases are one of the leading causes of death worldwide, however, little progress has been made in preventing or treating these diseases to date. The transforming growth factor-β (TGF-β) signaling pathway plays crucial and highly complicated roles in cerebrovascular development and homeostasis, and dysregulated TGF-β signaling contributes to cerebrovascular diseases. In this review, we provide an updated overview of the functional role of TGF-β signaling in the cerebrovascular system under physiological and pathological conditions. We discuss the current understanding of TGF-β signaling in cerebral angiogenesis and the maintenance of brain vessel homeostasis. We also review the mechanisms by which disruption of TGF-β signaling triggers or promotes the progression of cerebrovascular diseases. Finally, we briefly discuss the potential of targeting TGF-β signaling to treat cerebrovascular diseases.
According to the latest Global Burden of Disease Study, cerebrovascular disease is the second leading cause of mortality worldwide (Naghavi et al., 2017). Emerging clinical research data show that cerebrovascular disease is also the cause of many central nervous system diseases (Turner et al., 2016; Iadecola and Gottesman, 2018; Kummer et al., 2019). However, due to the lack of techniques to study cerebrovascular development and its regulatory mechanisms at the whole-animal level, our understanding of cerebrovascular diseases is still very limited. Emerging studies have begun to uncover the molecular mechanisms of cerebrovascular development and homeostasis, providing new basis and treatment strategies for the prevention and treatment of cerebrovascular and central nervous system diseases (Vanlandewijck et al., 2018; Munji et al., 2019).
Blood vessels of the brain form a highly specialized vascular network, which have complex interactions with the central nervous system, and has important physiological functions in the development and maintenance of the central nervous system (Zhao et al., 2015; Iadecola, 2017; Paredes et al., 2018). The development of cerebrovasculature begins with the angiogenic sprouting of perineural vascular plexus (PNVP) blood vessels, which forms a delicate hierarchical vascular structure through continued sprouting and remodeling (Tata et al., 2015). Blood vessels of the brain are mainly composed of highly specialized vascular endothelial cells (ECs), which have an arteriovenous differentiation pattern similar to that of peripheral vascular ECs. Brain ECs have obvious heterogeneity, complex tight junctions, more pericyte coverage, and form the neurovascular unit (NVU) together with pericytes, smooth muscle cells (SMCs), astrocytes, and neurons. The brain ECs, pericytes and the endfeet of astrocytes together form the unique blood–brain barrier (BBB) to restrict potentially harmful substances and molecules from entering the brain. The nutrients, energy metabolites, metabolic waste and other essential molecules cross the brain endothelium via various substrate-specific transporters to ensure physiological functioning of the brain. The primitive BBB is formed at embryonic day 15 (E15) in mice and varies in other species (Zhao et al., 2015). BBB continues to mature under the influence of neural environment over a brief period after birth.
Cerebrovascular development is a highly conserved and complex process involving multiple signaling pathways. Using various model organisms, researchers have successively identified many genes and signaling pathways that regulate the formation and homeostasis of blood vessels of the brain, including vascular endothelial growth factor (VEGF), sonic hedgehog/Patched (Shh/PTC-1), platelet-derived growth factor B/platelet-derived growth factor receptor β (PDGFB/PDGFRβ), Wnt/β-catenin, orphan G protein coupled receptor 124 (GPR124), as well as transforming growth factor β/SMAD (TGF-β/SMAD) signaling (Stenman et al., 2008; Ferrari et al., 2009; James et al., 2009; Alvarez et al., 2011; Cullen et al., 2011; Posokhova et al., 2015; Sweeney et al., 2016). As one of the most important and complex signaling pathways in vascular development, TGF-β/SMAD signaling plays diverse functions during the development and homeostasis of the brain vessel, and dysfunction in this signaling pathway has been linked to various cerebrovascular diseases (Park et al., 2009; Nguyen et al., 2011; Maddaluno et al., 2013). In this review, we discuss the latest research progress on the physiological function of TGF-β signaling in cerebrovascular development, and the mechanisms by which disruption of TGF-β signaling causes cerebrovascular diseases.
The TGF-β signaling pathway is highly conserved in evolution, and plays multiple and complex physiological functions in the regulation of embryonic development and tissue homeostasis in a highly context-dependent manner (Morikawa et al., 2016; David and Massague, 2018; Zinski et al., 2018).
The TGF-β signaling pathway comprises of more than 30 kinds of ligands, mainly divided into subfamilies such as TGF-βs, bone morphogenetic proteins (BMPs), activins, inhibin, Nodal, anti-Müllerian hormone, and growth and differentiation factors (GDFs) (Figure 1). Most TGF-β ligands function as paracrine factors on adjacent cells. The TGF-β ligands are expressed in latent forms with latency-associated peptide (LAP) shadowing the active domains of TGF-βs in the latent complex, and mature TGF-β ligands are activated through cleavage by extracellular protease from the LAP or physical tension by integrins. Several milieu molecules interact specifically with latent TGF-β and are essential for the bioavailability of TGF-β ligands. It is widely accepted that αVβ6 and αVβ8 integrins convert the cytoskeletal tension into a mechanical force to dissociate LAP from the TGF-β active domain, thereby releasing the activated TGF-β molecule and initiating the signaling cascade (Aluwihare et al., 2009). Very recently, researchers used cryo-electron microscopy to analyze the intermediate conformation of the interaction between αVβ8 integrin and latent TGF-β, and found that latent TGF-β binding with αVβ8 can expose the active domain and directly activate the TGF-β signaling pathway without release of the mature conformation (Campbell et al., 2020).
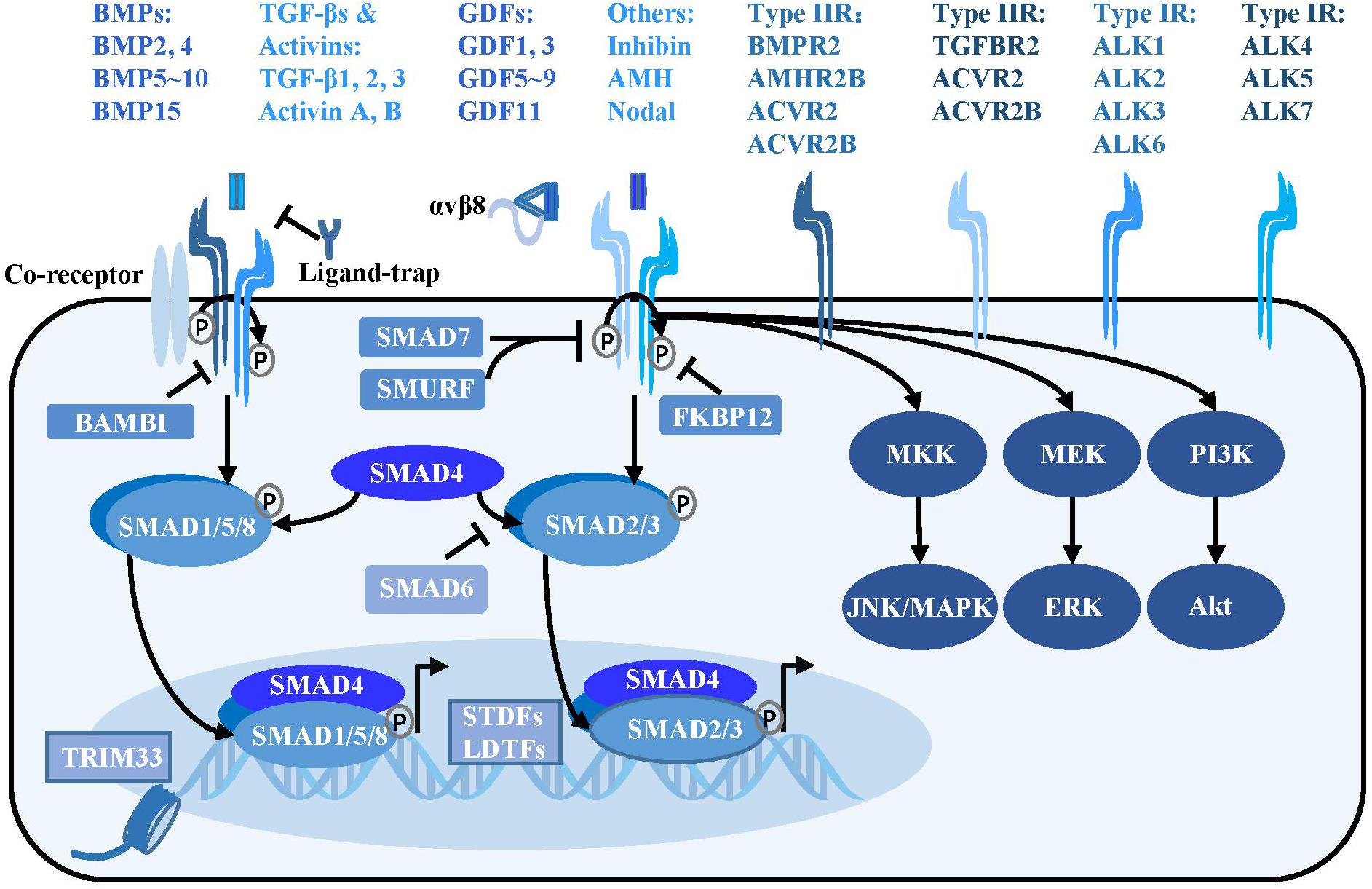
Figure 1. TGF-β signaling pathway. Activated TGF-β ligands with or without αVβ8 integrins bind at serine/threonine protein kinase type II receptors, sometimes with the assistance of the co-receptors such as endoglin. The type II receptors subsequently phosphorylate type I receptors to form a tetrameric receptor complex, which subsequently phosphorylates the SMAD2, SMAD3 or SMAD1, SMAD5, and SMAD8 to form a trimeric complex with SMAD4 in cytoplasm. The SMAD complex then translocates into nucleus and binds at special loci under the guidance of the SDTFs and LDTFs to initiate the transcriptional response. Besides, TRIM33, a modulator of TGF-β signaling, is able to regulate chromatin accessibility and remodeling. In addition to the canonical TGF-β signaling, there are SMAD-independent pathways, such as PI3K/Akt, MEK/ERK and MKK/JNK/MAPK downstream of the TGF-β receptors. TGF-β signaling is negatively regulated at multiple levels. Various ligand traps (noggin, chordin, follistatin, gremlin, coco, and cerberus) can prevent TGF-β ligands from binding to receptors, while FKBP12 and BAMBI can dock at cytoplasmic domain of TGF-β type I receptors to inhibit TGF-β signaling. In addition, inhibitory SMADs including SMAD6 and SMAD7 play a critical function in suppressing the SMAD-mediated signaling.
Activated TGF-β ligands, which are usually disulfide-linked homodimers, directly bind to the serine/threonine protein kinase type II receptors on the cell membrane surface, sometimes with the assistance of co-receptors such as endoglin and β-glycan (Goumans and Ten Dijke, 2018). Various proteins including noggin, chordin, follistatin, gremlin, coco, and cerberus act as ligand-traps to prevent TGF-β ligands from binding to receptors (David and Massague, 2018). Regulatory molecules such as FKBP12 and BAMBI inhibit the signaling pathway by docking at the cytoplasmic domain of TGF-β type I receptors (Wang et al., 1996; Onichtchouk et al., 1999).
The type II receptors phosphorylate the type I receptors to form a receptor complex, which then phosphorylates the receptor regulated SMADs (R-SMADs) intracellularly. Type I receptors for the TGFβ subfamily (ALK4, ALK5, and ALK7) mainly phosphorylate SMAD2 and SMAD3, whereas type I receptors for the BMP subfamily (ALK1, ALK2, ALK3, and ALK6) mainly phosphorylate SMAD1, SMAD5, and SMAD8. The activated R-SMADs form a complex with the central mediator SMAD4 and translocate into the nucleus, where it binds to specific gene loci under the guidance of signal-driven transcription factors (SDTFs) and lineage-determined transcription factors (LDTFs) as well as tripartite motif 33 (TRIM33) to regulate chromatin accessibility and gene transcription (Xi et al., 2011; David and Massague, 2018). A negative feedback loop of TGF-β signaling is mediated by the inhibitory SMADs: SMAD6 and SMAD7. SMAD7 can recruit E3 ubiquitin protein ligase SMURF2 to degrade the TGF-β receptor (Kavsak et al., 2000). SMAD6 not only interferes with the activation of SMAD2 phosphorylation by the receptor, but binds to R-SMAD and inhibits its binding to SMAD4 (Imamura et al., 1997; Hata et al., 1998). The TGF-β ligands can also signal through SMAD-independent pathways including the mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways (Derynck and Zhang, 2003; Figure 1).
It is intriguing that this seemingly simple “two-step” signal transduction of the TGF-β pathway has various and even opposite biological effects on a wide range of physiological processes, thereby reflecting the high spatiotemporal specificity of TGF-β signaling. The complexity of TGF-β signaling is manifested in the abundance and different combinations of its ligands, receptors and intracellular co-factors collaborating with SMADs. A single ligand can trigger multiple receptors [one of the 5 type II receptors (TGFBR2, BMPR2, ACVR2, ACVR2B, and AMHR2) in combination with one of the 7 type I receptors (ALK1-7)], and single receptor can interact with different ligands as well. The diversity of TGF-β ligand-receptor combinations leads to superimposed, synergistic or antagonistic effects on cells harboring different transcription factors and co-factors interacting with SMADs, resulting in complex biological effects (Massague, 2012).
The cerebrovascular network is developed via sprouting angiogenesis. The primary vessels of PNVP penetrate the CNS parenchyma and undergo remodeling to form a hierarchical vascular system composing of branched arteries and veins as well as capillaries, which is regulated by various signaling pathways including TGF-β signaling (Paredes et al., 2018).
Endothelial TGF-β signaling has been shown to be essential for cerebral angiogenesis, since Tgfbr2 or Alk5 gene knockout blood vessels fail to invade into the neuroepithelial layers and exhibit intracerebral hemorrhage (Nguyen et al., 2011). Genetic disruption of Smad4 in brain ECs leads to increased EC proliferation, impaired endothelial-pericyte interaction and intracerebral hemorrhage, providing a strong evidence that brain endothelial canonical TGF-β signaling plays essential roles in regulating brain angiogenesis and maintaining cerebrovascular integrity (Li et al., 2011).
A previous study has revealed the anti-angiogenic effect of TGF-β signaling in CNS vascular development (Arnold et al., 2014). Activated TGF-β signaling, by αVβ8 integrin, distributes as highest concentration in ventral brain regions and decreases in a gradient toward the dorsal brain regions, which is accompanied with stabilized vessels in ventral brain regions and greater vascular density, branch points and filopodia in dorsal brain region, suggesting that TGF-β signaling may play an anti-angiogenic role in cerebral angiogenesis. Consistently, loss of β8 integrin (Itgb8) or TGF-β1 or knockout of Alk5 or Tgfbr2 in ECs causes excessive vascular sprouting, branching and proliferation, which eventually leads to vascular dysplasia and cerebral hemorrhage (Arnold et al., 2014; Hirota et al., 2015). It has been further verified that neuroepithelial Itgb8 and endothelial neuropilin 1 (Nrp1) cooperatively promote cerebral angiogenesis by balancing TGF-β signaling. Endothelial Nrp1 inhibits β8 integrin activated TGF-β signaling to promote brain sprouting angiogenesis, and EC specific ablation of Nrp1 leads to increased levels of phosphorylated SMADs and embryonic lethality associated with defective sprouting angiogenesis and cerebral hemorrhage (Hirota et al., 2015).
Transforming growth factor-β signaling has also been shown to promote angiogenesis. TGF-β1 derived from radial glial cells promotes murine microcapillary brain EC migration and tube formation in vitro and stimulates cerebrovascular branching angiogenesis in the cerebral cortex, and this effect may be mediated by the balanced expression of pro-angiogenic gene GPR124 or anti-angiogenic gene, brain-specific angiogenesis inhibitor-1 (BAI-1) (Siqueira et al., 2018).
Bone morphogenetic protein-ALK1 signaling has been shown to limit EC number and maintain the quiescence of nascent vessels. BMP9 and BMP10 are physiological ligands of ALK1 during vascular development (Chen et al., 2013). In zebrafish, ALK1 functions in transducing hemodynamic forces into a biochemical signal which limits nascent vessel caliber (Corti et al., 2011). In mouse, ALK1 has been shown to mediate fluid shear stress by inducing BMP9 to inhibit endothelial proliferation and promote the recruitment of mural cells, thus maintaining vascular quiescence (Baeyens et al., 2016). Circulating BMP10 acts through endothelial ALK1 to activate pSMAD1/5/8 which decreases pro-angiogenic chemokine receptor cxcr4a expression and induces vasoconstrictive peptide endothelin 1 (Edn1), thereby limiting EC number and stabilizing nascent arterial caliber (Laux et al., 2013).
The mechanisms by which BMP-ALK1 regulates cerebrovascular development are quite limited; therefore, certain studies on the developmental mechanisms of mouse retinal vasculature can help us understand the related processes. The study using heterozygous Acvrl1+/- mice revealed that BMP9-ALK1 signaling inhibits EC proliferation and migration by activating PTEN to inhibit PI3K/Akt and MEK/ERK cascades, thereby maintaining retinal vascular quiescence (Alsina-Sanchis et al., 2018). Consistently, simultaneously silencing Bmp10 and Bmp9 in developing mice increases the retinal vascular density by promoting angiogenesis (Ricard et al., 2012).
Transforming growth factor-β signaling has been implicated in BBB formation and permeability by regulating tight and adherens junctions. The BBB is mainly composed of ECs which are characterized by the presence of tight and adherens junctions, and pericytes play an important role in the formation and maintenance of the BBB (Armulik et al., 2010; Bell et al., 2010; Daneman et al., 2010; Li et al., 2011). Endothelial TGF-β/SMAD4 signaling upregulates the adhesion molecule N-cadherin to facilitate the EC-pericyte interaction and BBB formation, in collaboration with Notch signal transduction (Li et al., 2011). Knockout of Smad4 in the brain ECs causes decreased expression of N-cadherin and pericyte detachment, leading to intraventricular hemorrhage and BBB breakdown during the perinatal period (Li et al., 2011). Besides, TGF-β1 derived from pericytes upregulates the expression of claudin-5 and promotes BBB maturation via decreasing endothelial CD146 expression (Chen et al., 2017).
Bone morphogenetic protein signaling has also been demonstrated to participate in the maintenance of BBB function. In zebrafish, BMP3 has been shown to regulate BBB integrity by promoting pericyte coverage (Lei et al., 2017). In rat cerebral vessel, BMP9/ALK1 signaling increases expression of endothelial transporters such as organic anion transporting polypeptide 1a4 at the BBB (Abdullahi et al., 2017). And BMP9/Alk1 is required for BBB stability, since ALK1 haploinsufficiency worsens the vascular leakage in diabetic mice. Mechanistically, ALK1 signaling inhibits VEGF-induced VE-cadherin phosphorylation and induces occludin expression, thereby enhancing the BBB function (Akla et al., 2018).
Cerebral angiogenesis is not only programmed in ECs, but also orchestrated by dynamic TGF-β signaling in other cell types within or outside the NVU, including pericytes, astrocytes, oligodendrocyte precursor cells, neural progenitors, preosteoblasts and periosteal dura cells.
Brain pericytes have been shown to induce and upregulate the functions of BBB through continuous TGF-β production (Dohgu et al., 2005). Pericyte ALK5 upregulates tissue inhibitor of matrix metalloproteinase 3 (TIMP3) to control endothelial morphogenesis in the germinal matrix. Specific knockout of Alk5 in embryonic mouse pericytes causes degradation of the basement membrane by upregulated matrix metalloproteinases (MMPs), resulting in severe germinal matrix hemorrhage-intraventricular hemorrhage (GMH-IVH) (Dave et al., 2018).
Astrocytes, whose endfeet interact with ECs of the neural capillaries, play a critical role in cerebral angiogenesis and BBB formation though BMP signaling. Targeted disruption of BMP type IA receptor (BMPR1A) in telencephalic neural stem cells leads to upregulated expression of VEGF in mutant astrocytes, impaired EC-astrocyte interaction, and cerebrovascular malformation, demonstrating that BMP signaling in astrocytes is essential for a functional BBB (Araya et al., 2008). A very recent study showed that BBB breakdown in aging humans and rodents is associated with hyperactivation of TGF-β signaling in astrocytes. Conditional genetic knockdown of astrocytic TGF-β receptor-coding genes or pharmacological inhibition of TGF-β signaling rescues the phenotypes in aged mice (Senatorov et al., 2019).
Oligodendrocyte precursor cells have also been shown to maintain BBB integrity through TGF-β signaling. TGF-β1 derived from oligodendrocyte progenitor cells can activate the MEK/ERK signaling pathway in ECs to promote tight junction protein expression and improve BBB integrity, and knockout of Tgfbr1 in oligodendrocyte progenitor cells leads to cerebral hemorrhage and disruptive BBB in mice (Seo et al., 2014).
Several neural progenitors have also been shown to play important roles in brain region-specific angiogenesis via TGF-β signaling. Tgfbr2 silencing in forebrain-derived neural progenitors and neural cells impedes EC migration and sprouting, decreases vessel density and branching via altered secretion of pro- and anti-angiogenic factors, thereby leading to intracerebral hemorrhage in the telencephalon (Hellbach et al., 2014). Neural progenitor S1P signaling regulates integrin β8 gene expression, thereby activating local TGF-β signaling that promotes germinal matrix vasculature development. Disruption of S1P signaling in neural progenitors results in defective angiogenesis and hemorrhage, as well as phenotypes mimicking the germinal matrix hemorrhage in humans (Ma et al., 2017).
In addition, BMP2 and BMP4 derived from preosteoblasts and periosteal dura are essential for dural cerebral vein formation. Loss of Twist1 or BMP2/4 signaling in skull progenitor cells and dura leads to cerebral vein malformations, similar to that in humans with craniosynostosis (Tischfield et al., 2017).
Endothelial-to-Mesenchymal Transition is a complex biological process and mainly refers to the trans-differentiation of ECs into mesenchymal stem cells, fibroblasts, SMCs or pericytes (Dejana and Lampugnani, 2018). During the process of EndMT, ECs lose the expression of endothelial markers (such as CD31 and VE-cadherin), and exhibit increased expression of mesenchymal transcription factors and molecular markers [such as Snail1, Slug (Snail2), Twist, ZEBs, vimentin, α-SMA, fibroblast-specific protein-1 (FSP-1; also known as S100A4 protein), fibroblast activating protein (FAP), and fibrillary collagens type I and type III] to obtain a mesenchymal morphology. Mesenchymal cells derived from EndMT gain enhanced ability of cell migration and invasion via disturbing the paracellular connection and polarity of ECs.
Activation of the TGF-β signaling pathway is the most important onset of EndMT (Ma et al., 2020). All three TGF-βs (TGF-β1, TGF-β2, and TGF-β3) have been shown to induce EndMT, while TGF-β2 seems to be more effective than TGF-β1 or TGF-β3 (Sabbineni et al., 2018). TGF-β ligands activate the TGFBR2 and ALK2 or ALK5 in ECs, and induce the pSMAD2/3/4 complex to translocate into the nucleus, where they interact with other transcription factors required for EndMT including Snail1, Snail2, Zeb1, Zeb2, KLF4, TCF3, and Twist and subsequently trigger the expression of mesenchymal transcription factors and molecular markers. TGF-β ligands also trigger EndMT through the non-canonical TGF-β pathways including MAPK, PI3K, and RhoA pathways (Piera-Velazquez and Jimenez, 2019).
Emerging studies have revealed that BMP signaling serves as a gatekeeper by antagonizing TGF-β-induced EndMT in ECs. BMP7 has been shown to inhibit hypoxia-induced EndMT and gremlin-1-mediated EndMT (Zhang et al., 2018, 2020). Loss of Bmpr2 in ECs leads to EndMT characterized by conversion of VE-cadherin to junctional N-cadherin, Slug and Twist upregulation, as well as increased expression of extracellular matrix (ECM) proteins (Hiepen et al., 2019). BMPR2-JNK signaling axis has also been shown to antagonize inflammation-induced EndMT (Sanchez-Duffhues et al., 2019).
Physiologically, EndMT plays essential roles during cardiovascular development, such as angiogenic sprouting and cardiac valve formation (Kruithof et al., 2012; Welch-Reardon et al., 2014). Dysregulation of EndMT has been associated with pathological situations, such as malignant diseases, fibrotic disorders and vascular diseases (Piera-Velazquez and Jimenez, 2019).
Emerging evidence indicates that dysregulated EndMT contributes to certain cerebrovascular diseases (Piera-Velazquez and Jimenez, 2019). The first evidence that EndMT is involved in the pathological process of cerebrovascular diseases was from the study of cerebral cavernous malformation (CCM). TGF-β signaling mediated EndMT is a direct cellular mechanism leading to CCMs in either mouse models or human patients (Maddaluno et al., 2013; Cuttano et al., 2016). Shortly after, another study reported that a meningeal pathogen Group B Streptococcus infection induces Snail1 expression and endothelial dedifferentiation, leading to BBB disruption, suggesting that EndMT might also contribute to BBB deficiency (Kim et al., 2015). Very recently, several studies have revealed that EndMT occurs in multiple sclerosis (MS), ischemic stroke, as well as brain arteriovenous malformations (AVMs) in humans (Derada Troletti et al., 2019; Chen et al., 2020; Shoemaker et al., 2020). All these results indicate that dysregulated EndMT might be an important pathological process involved in a variety of cerebrovascular disorders. However, the causal link between EndMT and various cerebrovascular diseases needs to be further established.
Recent studies have shown that defects in TGF-β signaling are associated with human cerebrovascular diseases. Pathogenic mutations in TGF-β signaling, such as ENG, ALK1 gene mutations, are associated with type 1 and type 2 hereditary hemorrhagic telangiectasia (HHT), as well as Loeys-dietz syndrome with cerebrovascular events (McAllister et al., 1994; Cunha et al., 2017; Laterza et al., 2019). Some genome-wide association studies (GWAS) or whole exome trio sequencing have uncovered various pathogenic gene variants in the TGF-β pathway, which are associated with small vessel ischemic strokes, intracerebral hemorrhages and sporadic brain AVMs (Weinsheimer et al., 2016; Yilmaz et al., 2017; Wang et al., 2018; Chung et al., 2019). Increased expression of TGF-β1 has been found in the brain tissue after ischemic stroke, as well as in hereditary cerebral hemorrhage with amyloidosis-Dutch type (Krupinski et al., 1996; Grand Moursel et al., 2018), while a recent transcriptome-wide RNA sequencing study revealed that TGF-β signaling was downregulated in patients with brain AVMs (Hauer et al., 2020). All these evidences suggest that dysregulation of TGF-β signaling may contribute to the onset and progression of cerebrovascular diseases. While there are not many studies on the mechanisms of cerebrovascular diseases related to TGF-β dysfunction, we discuss the three most studied cerebrovascular diseases caused by dysregulation of TGF-β signaling.
Cerebral cavernous malformation is a cerebrovascular disease causing recurrent cerebral hemorrhage, headaches, seizures and stroke, which is histologically characterized by clusters of dilated vascular sacs with ECs lacking tight junctions and mural cell coverage (Goldstein and Solomon, 2017; Stapleton and Barker, 2018). Genetically, CCMs can be categorized into familial and sporadic types. Approximately, 20% of all CCMs are Familial CCMs which present autosomal dominant inheritance with loss-of-function germline mutations in any one of the following three genes: CCM1/KRIT1, CCM2/malcavernin, or CCM3/PDCD10 (Zafar et al., 2019). The sporadic CCMs are non-hereditary and are probably caused due to somatic mutations of CCM genes (McDonald et al., 2014).
In both familial and sporadic CCM patients, TGF-β signaling is activated during pathological progression, as indicated by nuclear accumulation of endothelial pSMAD3 accompanied by expression of EndMT markers in lesions of familial and sporadic cavernomas (Maddaluno et al., 2013; Bravi et al., 2016). Besides, Kruppel-like factor 2 (KLF2) and KLF4, the activators of the BMP signaling, are significantly upregulated in ECs of familial and sporadic CCM lesions (Cuttano et al., 2016; Zhou et al., 2016). Activated TGF-β/BMP signaling has also been observed in cultured cells wherein all three Ccm genes were knocked down in ECs (Maddaluno et al., 2013; Cuttano et al., 2016), especially under low fluid shear stress conditions (Li et al., 2019).
The activation of TGF-β/BMP signaling has been confirmed in endothelial specific Ccm1 and Ccm3 knockout mice. Ablation of Ccm1 in ECs activates the expression of endogenous Bmp6 which induces the upregulation of pSMAD1 and pSMAD3 and triggers EndMT resulting in cerebral vascular malformations (Maddaluno et al., 2013). The upregulation of Bmp6 caused by mutant Ccm1 could be mediated by KLF4 which directly binds to the promoters of Bmp6 and some EndMT markers to induce their expression (Cuttano et al., 2016; Figure 2). Moreover, increased levels of pSMAD1 and pSMAD3 were observed in ECs of endothelial Ccm3 knockout mice (Bravi et al., 2015). Small-molecule inhibitors of TGFBR, pSMAD or BMP signaling could prevent EndMT and reduce the size and number of cerebral malformations, demonstrating that dysregulation of TGF-β/BMP signaling directly contributes to the onset and pathological process of CCMs (Maddaluno et al., 2013).
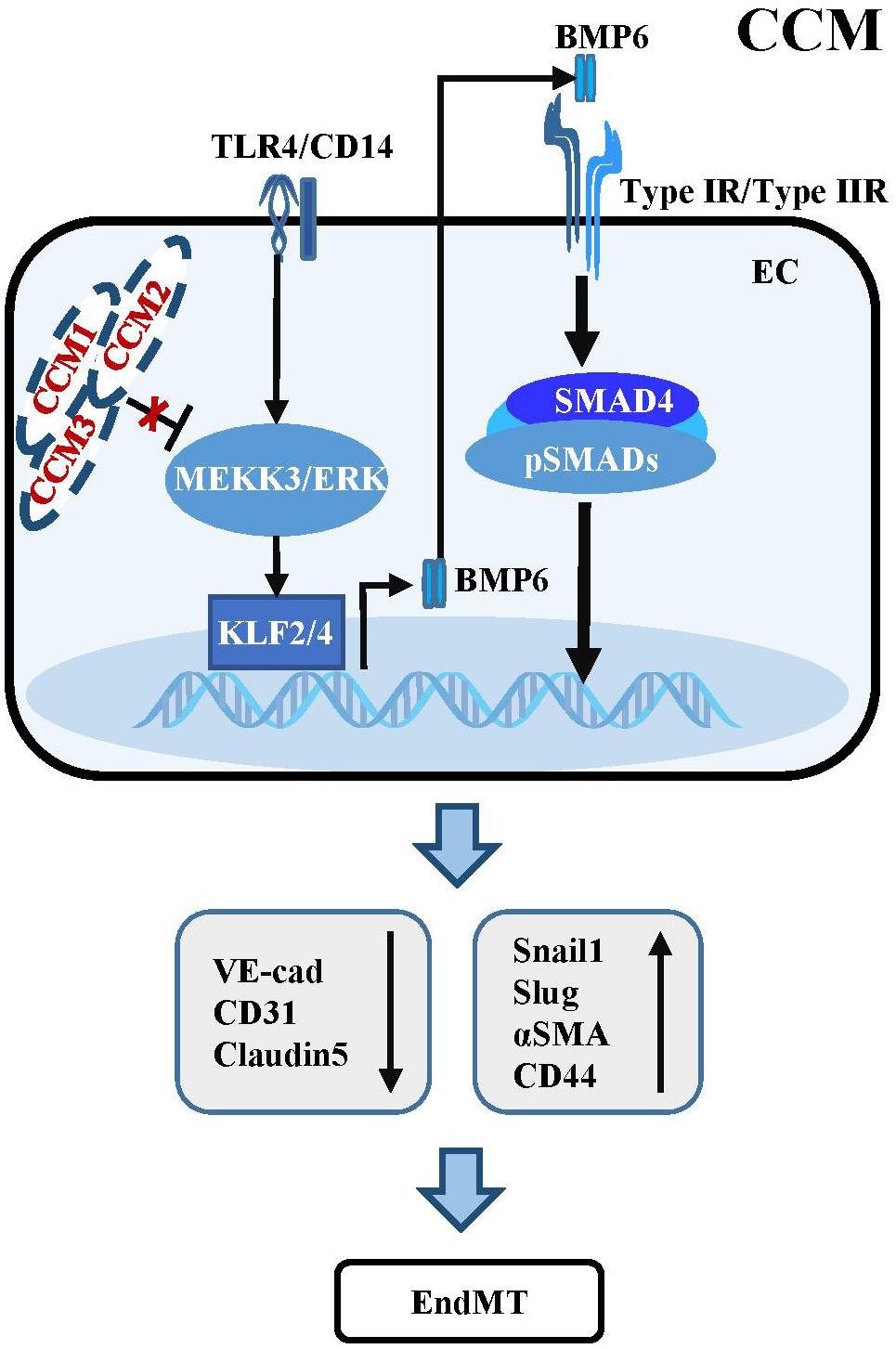
Figure 2. Dysregulation of TGF-β/BMP signaling in CCM. Mutant CCM release the inhibition of MEEK3/ERK pathway, which trigger the expression of KLF2/4. KLF2/4 subsequently suppress the expression of endothelial markers and induce the expression of EndMT-related molecules. KLF4 could also transcriptionally upregulate BMP6 to active TGF-β/BMP cascades in ECs. Besides, activation of TLR4 associated with CD14 by Gram-negative bacteria and lipopolysaccharide injection could increase the expression of Klf2/4 and promote CCM formation.
Some studies have uncovered the causal function of mitogen-activated protein kinase kinase Kinase 3 (MEKK3) and KLF2/4 in CCM pathogenesis, which is independent of TGF-β/SMAD signaling (Zhou et al., 2016). Endothelial-specific disruption of Mekk3, Klf2 or Klf4 significantly suppresses CCM and rescues the lethal phenotype in Ccm2 mutant mice. Consistently, the levels of KLF2 and KLF4 are increased in ECs of lesions in familial and sporadic CCM patients (Cuttano et al., 2016; Zhou et al., 2016). Supportively, ponatinib, a small-molecule compound inhibits MEKK3 activity to increase expression of the downstream Klf gene, suppresses CCM in neonatal Ccm1 deficient mouse models (Choi et al., 2018). In addition, activation of TLR4 by Gram-negative bacteria and lipopolysaccharide injection could increase the expression of Klf2/4 and promote CCM formation in Ccm1 and Ccm2 knockout mice (Tang et al., 2017; Figure 2). These inconsistencies with respect to the role of TGF-β signaling in the development and progression of CCM might be largely due to the different genetic backgrounds of the mouse models used, and the different stages of CCM pathogenesis analyzed in different experiments. Additional genetic rescue experiments might be helpful to further demonstrate the causal link between dysregulation of TGF-β signaling and the development and progression of CCM.
Hereditary hemorrhagic telangiectasia, also known as Osler-Weber-Rendu syndrome, is an autosomal dominant genetic disorder characterized as telangiectasia and AVMs affecting vessels in multiple organs and tissues including the brain (Brinjikji et al., 2015; Kritharis et al., 2018). Five types of HHT have been described, and HHT1 and HHT2 contribute to the disease in more than 80% of patients with definite HHT (Brinjikji et al., 2015). Some HHT patients display brain AVMs, often accompanied by cerebral hemorrhage, seizure, headache, or focal neurologic symptoms (Brinjikji et al., 2017a, b). Genetic screening of HHT patients has identified four mutated genetic loci, all of which are involved in the TGF-β signaling pathway, including BMP9 ligand encoding gene GDF2 (HHT5 or HHT like), type I receptor ALK1 encoding gene ACVRL1 (HHT2), co-receptor endoglin encoding gene ENG (HHT1) and intracellular mediator SMAD4 encoding gene MADH4 (JP-HHT) (McAllister et al., 1994; Johnson et al., 1996; Gallione et al., 2004; Wooderchak-Donahue et al., 2013; Figure 3).
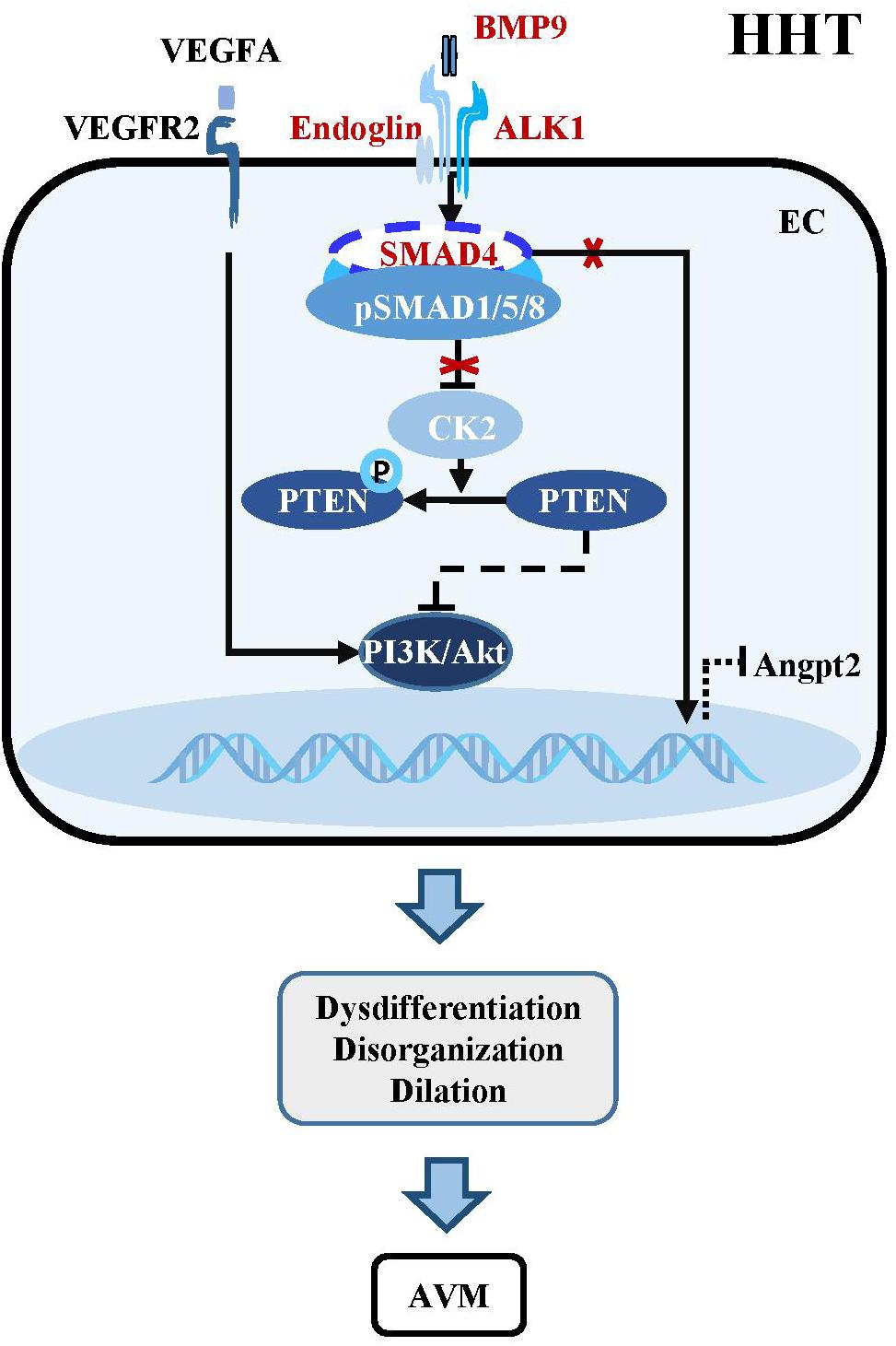
Figure 3. Dysregulation of BMP-ALK1-Smad4 signaling in HHTs. The mutant components within the BMP-ALK1 pathway including BMP9, ALK1, Endoglin, and SMAD4 are unable to suppress the transcription of CK2. Therefore, PTEN is phosphorylated by CK2 and loss the capacity to inhibit the PI3K/Akt pathway, which result in dysdifferentiation, disorganization, dilation and subsequent AVMs in cerebrovasculatures. Besides, AVMs-caused hypoxia increases the level of VEGFA, which subsequently binds VEGFR2 to active downstream PI3K/Akt signaling to aggravate the pathological process of AVMs. In addition, mutant SMAD4 increases the expression of angiopoietin-2 (Angpt2), leading to AVMs.
BMP9/10-ALK1 signaling suppresses HHTs through SMAD-dependent or SMAD-independent pathways. Endothelial-specific knockout of Alk1 triggers cerebral AVMs mimicking the pathologic characteristics of HHT (Park et al., 2009). In adult mouse, combined with VEGF stimulation, knockout of Alk1 could alter cerebral arteriovenous molecule specificity and induce AVMs (Walker et al., 2011). Zebrafish harboring mutations in Bmp9 and duplicate Bmp10 paralogs, Bmp10 and Bmp10-like exhibit cranial AVMs mimicking Acvrl1 mutants (Capasso et al., 2020). In postnatal retina, BMP9/10 ligand blockade and endothelial-specific homozygous ALK1 inactivation induces excessive angiogenesis via activating VEGF and PI3K/Akt signaling (Ola et al., 2016; Ruiz et al., 2016; Alsina-Sanchis et al., 2018). Pharmacological or genetic inhibition of PI3K rather than VEGFR could abolish ALK1-induced vascular hyperplasia in vivo, confirming that PI3K/Akt is the core mechanism downstream of BMP9/10-ALK1 signaling in maintaining vascular quiescence (Alsina-Sanchis et al., 2018; Ola et al., 2018; Iriarte et al., 2019; Figure 3).
Mice with homozygous or heterozygous deletion of Eng with VEGF treatment exhibit brain AVMs (Choi et al., 2012, 2014), and endothelial-specific Eng knockout mice spontaneously develop AVMs in the retina or brain (Mahmoud et al., 2010; Choi et al., 2014). In cerebral and retinal vessels, the Eng-null ECs cannot migrate against blood flow toward the arteries, leading to the accumulation and proliferation of ECs thereby triggering AVMs. Increased VEGFA expression which activates PI3K/Akt signaling through VEGFR2 may be responsible for stimulating sprouting angiogenesis and promoting venous differentiation in Eng mutant mice (Jin et al., 2017). Consistently, a recent study showed that ECs lacking Eng exhibit increased VEGF sensitivity and abnormal proliferation resulting in the formation of peripheral AVM (Tual-Chalot et al., 2020).
The essential role of endothelial SMAD4 in the maintenance of cerebrovascular integrity has been demonstrated by the study using a brain endothelial specific Smad4 knockout mouse, which develops phenotypes partially simulating HHT patients, such as dilated vessels, increased EC proliferation, intracranial hemorrhage and BBB breakdown (Li et al., 2011). Postnatally inducible endothelial Smad4 knockout results in AVM in neonatal and adult mice, which is comparable with the phenotypes observed in inducible endothelial Alk1 and Eng knockout mice (Crist et al., 2018; Kim et al., 2018; Ola et al., 2018). Mechanistically, SMAD4-mediated BMP9/10-ALK1 signaling inhibits the transcription of casein kinase 2 (CK2) which limits PTEN phosphorylation and PI3K/Akt activation, thereby preventing AVMs in the brain, retina, and gastrointestinal tract (Ola et al., 2018). In addition, Smad4 knockout leads to increased angiopoietin-2 (Angpt2) expression in ECs, which might cause AVM by changing the size and shape of ECs in the retina of Smad4 mutant mice (Lan et al., 2007; Crist et al., 2019; Figure 3).
These studies based on mouse models that mimic human HHT patients have provided the causal link between dysregulated TGF-β signaling and the pathogenesis of HHT. Blood flow stimulates BMP9-ALK1-ENG-SMAD4 signaling to maintain EC quiescence by suppressing EC proliferation and inducing pericyte recruitment (Baeyens et al., 2016), which involves PI3K/Akt signaling, Angpt2 signaling and possibly other factors (Alsina-Sanchis et al., 2018; Ola et al., 2018; Crist et al., 2019). It is worth noting that AVMs develop due to a combination of gene mutations in TGF-β signaling with angiogenic induction (via VEGF stimulation or wounding) (Park et al., 2009; Garrido-Martin et al., 2014), supporting the “Two hit mechanism” in HHT. Consistently, the tissues that are most vulnerable to AVMs or telangiectasia are those-susceptible to repeated damage and repair, such as the face, lips, and fingers in HHT patients (Brinjikji et al., 2015). Further investigation is required to elucidate whether dysregulation of other signaling pathways which cross talk with the TGF-β signaling pathway could serve as the second hits in the pathogenesis of HHT.
Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy is a rare autosomal recessive cerebrovascular disease that mainly occurs in cerebral white matter and basal ganglia, causing early adult-onset dementia, gait disturbance, alopecia, and low back pain (Nozaki et al., 2014; Tikka et al., 2014). Histologically, CARASIL displays cerebral arteriopathy showing fibrous proliferation of intima, loss of vascular SMCs and thickening of meningeal and parenchymal arteries. Fibrous hyperplasia in arteries results in the impaired contraction, leading to subcortical lacunar infarcts and subsequent vascular dementia (Oide et al., 2008; Ito et al., 2016). Genetically, mutations in high-temperature requirement serine peptidase A1 (HTRA1) gene have been identified to be associated with CARASIL (Hara et al., 2009). In patients, TGF-β1-pSMAD2 activation was observed in cerebral small arteries (Hara et al., 2009; Shiga et al., 2011).
The mechanistic role of TGF-β signaling in the pathogenesis of CARASIL, is a debatable topic. HRTA1 is a serine protease which is strongly expressed in ECs, vessel SMCs and pericytes (De Luca et al., 2003). HtrA1 knockout mice display a significantly decreased retinal vascular density which coincides with patients presenting reduced cerebral small vessels (Zhang et al., 2012). Mechanistically, HTRA1 cleaves the pro-domain of proTGF-β1 and TGF-β receptors to antagonize TGF-β signaling (Oka et al., 2004; Shiga et al., 2011; Graham et al., 2013). Consistently, HtrA1 knockout either in vivo or in cultured cells induces the expression of TGF-β ligands and activates pSMAD2/3 signaling (Zhang et al., 2012; Klose et al., 2019). All these results indicate that abnormal activation of TGF-β signaling contributes to the pathogenesis of CARASIL. However, there are studies showing that impaired TGF-β signaling is involved in CARASIL pathogenesis (Beaufort et al., 2014; Fasano et al., 2020). Loss of HtrA1 leads to defective HTRA1-mediated LTBP-1 processing and reduced TGF-β signaling (Beaufort et al., 2014), and fibroblasts derived from HTRA1 mutation carriers exhibit no significant change in pSMAD2/3 expression (Fasano et al., 2020). The possible reasons for the discrepancy might partially be due to different HTRA1 mutations leading to different outcomes (Verdura et al., 2015; Lee et al., 2018). Future studies should identify the natural characteristics of CARASIL associated mutations and develop animal models that accurately mimic all pathological and molecular aspects of human CARASIL patients, which will help uncover the mechanisms of CARASIL and discover new therapeutic targets.
Current therapies for CCM, HHT, and CARASIL patients mainly rely on surgery or relieving complications (Tikka et al., 2014; Kritharis et al., 2018; Stapleton and Barker, 2018). Recent advances in understanding the mechanisms of dysfunctional TGF-β signaling which results in cerebrovascular diseases has provided hope to develop pharmacological and genetic therapies for these diseases.
Activated TGF-β/BMP signaling has been demonstrated to contribute to the onset and progression of CCMs in patients and mouse models (Maddaluno et al., 2013; Bravi et al., 2016; Cuttano et al., 2016). Therefore, it is expected that therapeutics targeting TGF-β/BMP signaling would be beneficial for CCMs. Indeed, TGFBR1/pSMAD inhibitors LY364947 and SB431542 as well as BMPR1 inhibitor dorsomorphin (DMH1) strikingly reduce the level of pSMAD1 and pSMAD3, prevent the expression of EndMT markers, and decrease the number and size of vascular malformation lesions in CCM1 mutant mice (Maddaluno et al., 2013). KLF4 has been shown to be a good therapeutic target for CCM. Ccm1 knockout results in MEKK3-MEK5-ERK5-MEF2 signaling dependent activation of KLF4 which promotes the expression of Bmp6. A specific MEK5 inhibitor BIX-02189 (Tatake et al., 2008) significantly decreases pERK5 and KLF4 expression, inhibits Bmp6 upregulation and EndMT in CCM1 deficient ECs (Cuttano et al., 2016), indicating that inhibitors of the MEKK3-MEK5-ERK5-MEF2 axis might be useful for suppressing BMP signaling and EndMT in the pathogenesis of CCM. There are several novel drugs targeting TGF-β signaling, developed through preclinical trials and further tested in clinical trials, including anti-ligand antisense oligonucleotides (ASOs), ligand-competitive peptides, antibodies targeting ligands, receptors or associated proteins, and inhibitors against TGF-β receptor kinases for various diseases (Akhurst and Hata, 2012; Graham et al., 2013; Kemaladewi et al., 2014; Aykul and Martinez-Hackert, 2016; Wu et al., 2017; Holmgaard et al., 2018). It is worth examining whether these candidate drugs that target TGF-β signaling could inhibit the progress of CCMs.
The majority of HHT patients have pathogenic loss of function mutations in TGF-β signaling. Although many studies have uncovered the molecular mechanisms underlying HHTs caused by dysfunctional TGF-β signaling, there is currently no efficient drug for HHT treatment. Current drug therapy regimens mainly focus on interfering with the downstream core signaling pathway such as activated VEGF and PI3K/Akt signaling (Alsina-Sanchis et al., 2018; Ola et al., 2018). Since haploinsufficiency of endoglin and ALK1 have been identified as the causes of HHT1 and HHT2, a better understanding of the regulation of their expression levels at the transcriptional level or post-transcriptional level will help developing therapeutic strategies targeting endoglin and ALK1 expression or function. Indeed, a very recent study shows that ALK1-overexpression could normalize SMAD and NOTCH target gene expression, restore the effect of BMP9 on suppression of p-Akt, and inhibit the development of AVMs in Alk1- and Eng-inducible knockout mice, suggesting that ALK1 overexpression or activation might be a potential therapeutic strategy for HHT patients (Kim et al., 2020).
Genome editing may serve as the final solution. CRISPR-based genome editing has been demonstrated as a powerful tool for treating genetic diseases (Pickar-Oliver and Gersbach, 2019). The CRISPR-Cas9 system has been demonstrated to efficiently correct gene mutations in various mouse models of human diseases, including cataracts, muscular dystrophy and many others (Wu et al., 2013; Long et al., 2014). Recent studies show that base editing can correct mutations in human cells and in a mouse model of genetic deafness, and a newly developed template-free Cas9 editing is able to precisely correct the pathogenic mutations in human cells (Gaudelli et al., 2017; Gao et al., 2018; Shen et al., 2018). A newly developed CRISPR-CasΦ system, with a molecular weight which is only half of Cas9 or Cas12a displays expanded target recognition capabilities and is functional in human cells as well (Pausch et al., 2020), providing a new genome editing tool for treating cerebrovascular diseases. Once the causal link between mutations in TGF-β signaling and cerebrovascular diseases has been established, genome editing will likely correct these mutant genes to heal the related cerebrovascular diseases.
Previous studies have demonstrated the crucial function of TGF-β signaling in cerebral vasculature development and integrity, and uncovered the causal link between the dysfunctional TGF-β signaling and the onset or progression of several cerebrovascular diseases such as CCM, HHT and CARASIL. However, the related mechanisms underlying the dysregulation of TGF-β signaling resulting in cerebrovascular diseases remains to be further elucidated. In recent years, using the rapidly developed single cell sequencing technology and advanced graphics algorithm, researchers have revealed the unappreciated heterogeneity and plasticity of human and mouse cerebral blood vessels, discovering not only new markers for different subtypes of ECs but also a new cell type adjacent to the blood vessel (Schaum et al., 2018; Vanlandewijck et al., 2018; Kalucka et al., 2020). Further investigation of the role and mechanism of TGF-β signaling in the regulation of cerebrovascular heterogeneity and plasticity will help to understand the function of TGF-β signaling in the occurrence and development of cerebrovascular diseases.
There are not many animal models that can accurately mimic the genetic and pathological characteristics of human cerebrovascular diseases. Rapid advances in genome editing technologies based on CRISPR-Cas systems provide powerful tools for generating animal models carrying genomic mutations precisely mimicking the ones in human patients (Pickar-Oliver and Gersbach, 2019). Studies using cell lineage tracing technology combined with single cell sequencing in animal models of human cerebrovascular diseases will help reveal the cellular and molecular mechanisms of cerebrovascular diseases and discover new therapeutic targets. In addition, human cortical organoids with functional cerebral vessels will provide valuable models for dissecting the roles of TGF-β signaling in the development and progression of human cerebrovascular diseases (Cakir et al., 2019).
Although recent advances have indicated that targeting TGF-β signaling will be a potential strategy for the treatment of cerebrovascular diseases, clinical transformation is still challenging. Considering the cell context-dependent pleiotropic roles of the TGF-β signaling pathway, the selectivity and dosage of targeted drugs may be crucial for the desired therapeutic effects. Previous research has identified various TGF-β inhibitory drugs involving almost every level in the TGF-β signaling cascade, some of which have been proved safe and effective for treating systemic sclerosis, cancers or idiopathic pulmonary fibrosis in clinical trials (Rice et al., 2015; Yingling et al., 2018; Joyce et al., 2019; Kelley et al., 2019; Papachristodoulou et al., 2019; Santini et al., 2019). These existing TGF-β inhibitory drugs provide potential therapeutic opportunities for treating cerebrovascular diseases with activated TGF-β signaling. For cerebrovascular diseases with loss-of-function mutations in TGF-β signaling, somatic genome editing may provide tools to correct the mutations or enhance TGF-β signaling.
Increased clinical research data shows that there is a close correlation between cerebrovascular and central nervous system diseases. Abnormal cerebrovascular structure and function are closely related to brain atrophy, dementia and various neurodegenerative disorders and cognitive impairment (Turner et al., 2016; Yang et al., 2017; Iadecola and Gottesman, 2018; Kummer et al., 2019). Dysregulated TGF-β signaling has been observed in neurodegenerative diseases accompanied by cerebrovascular abnormalities (von Bernhardi et al., 2015). Further studying the synergistic mechanisms by which TGF-β signaling maintains the homeostasis of the cerebrovascular and central nervous system might be very helpful in uncovering the direct causal link between cerebrovascular and central nervous system diseases, providing new theoretical basis and treatment strategies for joint preventing and treating cerebrovascular and central nervous system diseases.
YZ and XY wrote the review. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the National Key Research and Development Program of China (2016YFC1300600 to XY) and the National Natural Science Foundation of China (31430057 and 31630093 to XY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdullahi, W., Brzica, H., Ibbotson, K., Davis, T. P., and Ronaldson, P. T. (2017). Bone morphogenetic protein-9 increases the functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier via the activin receptor-like kinase-1 receptor. J. Cereb. Blood Flow Metab. 37, 2340–2345. doi: 10.1177/0271678X17702916
Akhurst, R. J., and Hata, A. (2012). Targeting the TGFbeta signalling pathway in disease. Nat. Rev. Drug Discov. 11, 790–811. doi: 10.1038/nrd3810
Akla, N., Viallard, C., Popovic, N., Lora Gil, C., Sapieha, P., and Larrivee, B. (2018). BMP9 (bone morphogenetic protein-9)/Alk1 (activin-like kinase receptor type I) signaling prevents hyperglycemia-induced vascular permeability. Arterioscler. Thromb. Vasc. Biol. 38, 1821–1836. doi: 10.1161/ATVBAHA.118.310733
Alsina-Sanchis, E., Garcia-Ibanez, Y., Figueiredo, A. M., Riera-Domingo, C., Figueras, A., Matias-Guiu, X., et al. (2018). ALK1 loss results in vascular hyperplasia in mice and humans through PI3K activation. Arterioscler. Thromb. Vasc. Biol. 38, 1216–1229. doi: 10.1161/ATVBAHA.118.310760
Aluwihare, P., Mu, Z., Zhao, Z., Yu, D., Weinreb, P. H., Horan, G. S., et al. (2009). Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J. Cell Sci. 122(Pt 2), 227–232. doi: 10.1242/jcs.035246
Alvarez, J. I., Dodelet-Devillers, A., Kebir, H., Ifergan, I., Fabre, P. J., Terouz, S., et al. (2011). The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334, 1727–1731. doi: 10.1126/science.1206936
Araya, R., Kudo, M., Kawano, M., Ishii, K., Hashikawa, T., Iwasato, T., et al. (2008). BMP signaling through BMPRIA in astrocytes is essential for proper cerebral angiogenesis and formation of the blood-brain-barrier. Mol. Cell Neurosci. 38, 417–430. doi: 10.1016/j.mcn.2008.04.003
Armulik, A., Genove, G., Mae, M., Nisancioglu, M. H., Wallgard, E., Niaudet, C., et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468, 557–561. doi: 10.1038/nature09522
Arnold, T. D., Niaudet, C., Pang, M. F., Siegenthaler, J., Gaengel, K., Jung, B., et al. (2014). Excessive vascular sprouting underlies cerebral hemorrhage in mice lacking alphaVbeta8-TGFbeta signaling in the brain. Development 141, 4489–4499. doi: 10.1242/dev.107193
Aykul, S., and Martinez-Hackert, E. (2016). Transforming growth factor-beta family ligands can function as antagonists by competing for type II receptor binding. J. Biol. Chem. 291, 10792–10804. doi: 10.1074/jbc.M115.713487
Baeyens, N., Larrivee, B., Ola, R., Hayward-Piatkowskyi, B., Dubrac, A., Huang, B., et al. (2016). Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 214, 807–816. doi: 10.1083/jcb.201603106
Beaufort, N., Scharrer, E., Kremmer, E., Lux, V., Ehrmann, M., Huber, R., et al. (2014). Cerebral small vessel disease-related protease HtrA1 processes latent TGF-beta binding protein 1 and facilitates TGF-beta signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 16496–16501. doi: 10.1073/pnas.1418087111
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., LaRue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi: 10.1016/j.neuron.2010.09.043
Bravi, L., Malinverno, M., Pisati, F., Rudini, N., Cuttano, R., Pallini, R., et al. (2016). Endothelial cells lining sporadic cerebral cavernous malformation cavernomas undergo endothelial-to-mesenchymal transition. Stroke 47, 886–890. doi: 10.1161/STROKEAHA.115.011867
Bravi, L., Rudini, N., Cuttano, R., Giampietro, C., Maddaluno, L., Ferrarini, L., et al. (2015). Sulindac metabolites decrease cerebrovascular malformations in CCM3-knockout mice. Proc. Natl. Acad. Sci. U.S.A. 112, 8421–8426. doi: 10.1073/pnas.1501352112
Brinjikji, W., Iyer, V. N., Lanzino, G., Thielen, K. R., and Wood, C. P. (2017a). Natural history of brain capillary vascular malformations in hereditary hemorrhagic telangiectasia patients. J. Neurointerv. Surg. 9, 26–28. doi: 10.1136/neurintsurg-2015-012252
Brinjikji, W., Iyer, V. N., Sorenson, T., and Lanzino, G. (2015). Cerebrovascular manifestations of hereditary hemorrhagic telangiectasia. Stroke 46, 3329–3337. doi: 10.1161/STROKEAHA.115.010984
Brinjikji, W., Iyer, V. N., Wood, C. P., and Lanzino, G. (2017b). Prevalence and characteristics of brain arteriovenous malformations in hereditary hemorrhagic telangiectasia: a systematic review and meta-analysis. J. Neurosurg. 127, 302–310. doi: 10.3171/2016.7.JNS16847
Cakir, B., Xiang, Y., Tanaka, Y., Kural, M. H., Parent, M., Kang, Y. J., et al. (2019). Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175. doi: 10.1038/s41592-019-0586-5
Campbell, M. G., Cormier, A., Ito, S., Seed, R. I., Bondesson, A. J., Lou, J., et al. (2020). Cryo-EM reveals integrin-mediated TGF-beta activation without release from latent TGF-beta. Cell 180, 490–501e16. doi: 10.1016/j.cell.2019.12.030
Capasso, T. L., Li, B., Volek, H. J., Khalid, W., Rochon, E. R., Anbalagan, A., et al. (2020). BMP10-mediated ALK1 signaling is continuously required for vascular development and maintenance. Angiogenesis 23, 203–220. doi: 10.1007/s10456-019-09701-0
Chen, D., Li, L., Wang, Y., Xu, R., Peng, S., Zhou, L., et al. (2020). Ischemia-reperfusion injury of brain induces endothelial-mesenchymal transition and vascular fibrosis via activating let-7i/TGF-betaR1 double-negative feedback loop. FASEB J. 34, 7178–7191. doi: 10.1096/fj.202000201R
Chen, H., Brady Ridgway, J., Sai, T., Lai, J., Warming, S., Chen, H., et al. (2013). Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. U.S.A. 110, 11887–11892. doi: 10.1073/pnas.1306074110
Chen, J., Luo, Y., Hui, H., Cai, T., Huang, H., Yang, F., et al. (2017). CD146 coordinates brain endothelial cell-pericyte communication for blood-brain barrier development. Proc. Natl. Acad. Sci. U.S.A. 114, E7622–E7631. doi: 10.1073/pnas.1710848114
Choi, E. J., Chen, W., Jun, K., Arthur, H. M., Young, W. L., and Su, H. (2014). Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One 9:e88511. doi: 10.1371/journal.pone.0088511
Choi, E. J., Walker, E. J., Shen, F., Oh, S. P., Arthur, H. M., Young, W. L., et al. (2012). Minimal homozygous endothelial deletion of Eng with VEGF stimulation is sufficient to cause cerebrovascular dysplasia in the adult mouse. Cerebrovasc. Dis. 33, 540–547. doi: 10.1159/000337762
Choi, J. P., Wang, R., Yang, X., Wang, X., Wang, L., Ting, K. K., et al. (2018). Ponatinib (AP24534) inhibits MEKK3-KLF signaling and prevents formation and progression of cerebral cavernous malformations. Sci. Adv. 4:eaau0731. doi: 10.1126/sciadv.aau0731
Chung, J., Marini, S., Pera, J., Norrving, B., Jimenez-Conde, J., Roquer, J., et al. (2019). Genome-wide association study of cerebral small vessel disease reveals established and novel loci. Brain 142, 3176–3189. doi: 10.1093/brain/awz233
Corti, P., Young, S., Chen, C. Y., Patrick, M. J., Rochon, E. R., Pekkan, K., et al. (2011). Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development 138, 1573–1582. doi: 10.1242/dev.060467
Crist, A. M., Lee, A. R., Patel, N. R., Westhoff, D. E., and Meadows, S. M. (2018). Vascular deficiency of Smad4 causes arteriovenous malformations: a mouse model of Hereditary Hemorrhagic Telangiectasia. Angiogenesis 21, 363–380. doi: 10.1007/s10456-018-9602-0
Crist, A. M., Zhou, X., Garai, J., Lee, A. R., Thoele, J., Ullmer, C., et al. (2019). Angiopoietin-2 inhibition rescues arteriovenous malformation in a smad4 hereditary hemorrhagic telangiectasia mouse model. Circulation 139, 2049–2063. doi: 10.1161/CIRCULATIONAHA.118.036952
Cullen, M., Elzarrad, M. K., Seaman, S., Zudaire, E., Stevens, J., Yang, M. Y., et al. (2011). GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc. Natl. Acad. Sci. U.S.A. 108, 5759–5764. doi: 10.1073/pnas.1017192108
Cunha, S. I., Magnusson, P. U., Dejana, E., and Lampugnani, M. G. (2017). Deregulated TGF-beta/BMP signaling in vascular malformations. Circ. Res. 121, 981–999. doi: 10.1161/CIRCRESAHA.117.309930
Cuttano, R., Rudini, N., Bravi, L., Corada, M., Giampietro, C., Papa, E., et al. (2016). KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Mol. Med. 8, 6–24. doi: 10.15252/emmm.201505433
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Dave, J. M., Mirabella, T., Weatherbee, S. D., and Greif, D. M. (2018). Pericyte ALK5/TIMP3 axis contributes to endothelial morphogenesis in the developing brain. Dev. Cell 44, 665–678.e6. doi: 10.1016/j.devcel.2018.01.018
David, C. J., and Massague, J. (2018). Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 19, 419–435. doi: 10.1038/s41580-018-0007-0
De Luca, A., De Falco, M., Severino, A., Campioni, M., Santini, D., Baldi, F., et al. (2003). Distribution of the serine protease HtrA1 in normal human tissues. J. Histochem. Cytochem. 51, 1279–1284. doi: 10.1177/002215540305101004
Dejana, E., and Lampugnani, M. G. (2018). Endothelial cell transitions. Science 362, 746–747. doi: 10.1126/science.aas9432
Derada Troletti, C., Fontijn, R. D., Gowing, E., Charabati, M., van Het Hof, B., Didouh, I., et al. (2019). Inflammation-induced endothelial to mesenchymal transition promotes brain endothelial cell dysfunction and occurs during multiple sclerosis pathophysiology. Cell Death Dis. 10:45. doi: 10.1038/s41419-018-1294-2
Derynck, R., and Zhang, Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584. doi: 10.1038/nature02006
Dohgu, S., Takata, F., Yamauchi, A., Nakagawa, S., Egawa, T., Naito, M., et al. (2005). Brain pericytes contribute to the induction and up-regulation of blood-brain barrier functions through transforming growth factor-beta production. Brain Res. 1038, 208–215. doi: 10.1016/j.brainres.2005.01.027
Fasano, A., Formichi, P., Taglia, I., Bianchi, S., Di Donato, I., Battisti, C., et al. (2020). HTRA1 expression profile and activity on TGF-beta signaling in HTRA1 mutation carriers. J. Cell Physiol. 235, 7120–7127. doi: 10.1002/jcp.29609
Ferrari, G., Cook, B. D., Terushkin, V., Pintucci, G., and Mignatti, P. (2009). Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J. Cell Physiol. 219, 449–458. doi: 10.1002/jcp.21706
Gallione, C. J., Repetto, G. M., Legius, E., Rustgi, A. K., Schelley, S. L., Tejpar, S., et al. (2004). A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 363, 852–859. doi: 10.1016/S0140-6736(04)15732-2
Gao, X., Tao, Y., Lamas, V., Huang, M., Yeh, W. H., Pan, B., et al. (2018). Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221. doi: 10.1038/nature25164
Garrido-Martin, E. M., Nguyen, H. L., Cunningham, T. A., Choe, S. W., Jiang, Z., Arthur, H. M., et al. (2014). Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models–brief report. Arterioscler. Thromb. Vasc. Biol. 34, 2232–2236. doi: 10.1161/ATVBAHA.114.303984
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., et al. (2017). Programmable base editing of A∗T to G∗C in genomic DNA without DNA cleavage. Nature 551, 464–471. doi: 10.1038/nature24644
Goldstein, H. E., and Solomon, R. A. (2017). Epidemiology of cavernous malformations. Handb. Clin. Neurol. 143, 241–247. doi: 10.1016/B978-0-444-63640-9.00023-0
Goumans, M. J., and Ten Dijke, P. (2018). TGF-beta signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 10:a022210. doi: 10.1101/cshperspect.a022210
Graham, J. R., Chamberland, A., Lin, Q., Li, X. J., Dai, D., Zeng, W., et al. (2013). Serine protease HTRA1 antagonizes transforming growth factor-beta signaling by cleaving its receptors and loss of HTRA1 in vivo enhances bone formation. PLoS One 8:e74094. doi: 10.1371/journal.pone.0074094
Grand Moursel, L., Munting, L. P., van der Graaf, L. M., van Duinen, S. G., Goumans, M. T. H., Ueberham, U., et al. (2018). TGFbeta pathway deregulation and abnormal phospho-SMAD2/3 staining in hereditary cerebral hemorrhage with amyloidosis-Dutch type. Brain Pathol. 28, 495–506. doi: 10.1111/bpa.12533
Hara, K., Shiga, A., Fukutake, T., Nozaki, H., Miyashita, A., Yokoseki, A., et al. (2009). Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N. Engl. J. Med. 360, 1729–1739. doi: 10.1056/NEJMoa0801560
Hata, A., Lagna, G., Massague, J., and Hemmati-Brivanlou, A. (1998). Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 12, 186–197. doi: 10.1101/gad.12.2.186
Hauer, A. J., Kleinloog, R., Giuliani, F., Rinkel, G. J. E., de Kort, G. A., Berkelbach van der Sprenkel, J. W., et al. (2020). RNA-sequencing highlights inflammation and impaired integrity of the vascular wall in brain arteriovenous malformations. Stroke 51, 268–274. doi: 10.1161/STROKEAHA.119.025657
Hellbach, N., Weise, S. C., Vezzali, R., Wahane, S. D., Heidrich, S., Roidl, D., et al. (2014). Neural deletion of Tgfbr2 impairs angiogenesis through an altered secretome. Hum. Mol. Genet. 23, 6177–6190. doi: 10.1093/hmg/ddu338
Hiepen, C., Jatzlau, J., Hildebrandt, S., Kampfrath, B., Goktas, M., Murgai, A., et al. (2019). BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFbeta responses and altered cell mechanics. PLoS Biol. 17:e3000557. doi: 10.1371/journal.pbio.3000557
Hirota, S., Clements, T. P., Tang, L. K., Morales, J. E., Lee, H. S., Oh, S. P., et al. (2015). Neuropilin 1 balances beta8 integrin-activated TGFbeta signaling to control sprouting angiogenesis in the brain. Development 142, 4363–4373. doi: 10.1242/dev.113746
Holmgaard, R. B., Schaer, D. A., Li, Y., Castaneda, S. P., Murphy, M. Y., Xu, X., et al. (2018). Targeting the TGFbeta pathway with galunisertib, a TGFbetaRI small molecule inhibitor, promotes anti-tumor immunity leading to durable, complete responses, as monotherapy and in combination with checkpoint blockade. J. Immunother. Cancer 6:47. doi: 10.1186/s40425-018-0356-4
Iadecola, C. (2017). The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96, 17–42. doi: 10.1016/j.neuron.2017.07.030
Iadecola, C., and Gottesman, R. F. (2018). Cerebrovascular alterations in Alzheimer disease. Circ. Res. 123, 406–408. doi: 10.1161/CIRCRESAHA.118.313400
Imamura, T., Takase, M., Nishihara, A., Oeda, E., Hanai, J., Kawabata, M., et al. (1997). Smad6 inhibits signalling by the TGF-beta superfamily. Nature 389, 622–626. doi: 10.1038/39355
Iriarte, A., Figueras, A., Cerda, P., Mora, J. M., Jucgla, A., Penin, R., et al. (2019). PI3K (Phosphatidylinositol 3-Kinase) activation and endothelial cell proliferation in patients with hemorrhagic hereditary telangiectasia type 1. Cells 8:971. doi: 10.3390/cells8090971
Ito, S., Takao, M., Fukutake, T., Hatsuta, H., Funabe, S., Ito, N., et al. (2016). Histopathologic analysis of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL): a report of a new genetically confirmed case and comparison to 2 previous cases. J. Neuropathol. Exp. Neurol. 75, 1020–1030. doi: 10.1093/jnen/nlw078
James, J. M., Gewolb, C., and Bautch, V. L. (2009). Neurovascular development uses VEGF-A signaling to regulate blood vessel ingression into the neural tube. Development 136, 833–841. doi: 10.1242/dev.028845
Jin, Y., Muhl, L., Burmakin, M., Wang, Y., Duchez, A. C., Betsholtz, C., et al. (2017). Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat. Cell Biol. 19, 639–652. doi: 10.1038/ncb3534
Johnson, D. W., Berg, J. N., Baldwin, M. A., Gallione, C. J., Marondel, I., Yoon, S. J., et al. (1996). Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 13, 189–195. doi: 10.1038/ng0696-189
Joyce, C. E., Saadatpour, A., Ruiz-Gutierrez, M., Bolukbasi, O. V., Jiang, L., Thomas, D. D., et al. (2019). TGFbeta signaling underlies hematopoietic dysfunction and bone marrow failure in Shwachman-Diamond Syndrome. J. Clin. Invest. 129, 3821–3826. doi: 10.1172/JCI125375
Kalucka, J., de Rooij, L., Goveia, J., Rohlenova, K., Dumas, S. J., Meta, E., et al. (2020). Single-cell transcriptome atlas of murine endothelial cells. Cell 180, 764–779.e20. doi: 10.1016/j.cell.2020.01.015
Kavsak, P., Rasmussen, R. K., Causing, C. G., Bonni, S., Zhu, H., Thomsen, G. H., et al. (2000). Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 6, 1365–1375. doi: 10.1016/s1097-2765(00)00134-9
Kelley, R. K., Gane, E., Assenat, E., Siebler, J., Galle, P. R., Merle, P., et al. (2019). A phase 2 study of galunisertib (TGF-beta1 receptor type I inhibitor) and Sorafenib in patients with advanced hepatocellular carcinoma. Clin. Transl. Gastroenterol. 10:e00056. doi: 10.14309/ctg.0000000000000056
Kemaladewi, D. U., Pasteuning, S., van der Meulen, J. W., van Heiningen, S. H., van Ommen, G. J., Ten Dijke, P., et al. (2014). Targeting TGF-beta signaling by antisense oligonucleotide-mediated knockdown of TGF-beta type I receptor. Mol. Ther. Nucleic Acids 3:e156. doi: 10.1038/mtna.2014.7
Kim, B. J., Hancock, B. M., Bermudez, A., Del Cid, N., Reyes, E., van Sorge, N. M., et al. (2015). Bacterial induction of Snail1 contributes to blood-brain barrier disruption. J. Clin. Invest. 125, 2473–2483. doi: 10.1172/JCI74159
Kim, Y. H., Choe, S. W., Chae, M. Y., Hong, S., and Oh, S. P. (2018). SMAD4 deficiency leads to development of arteriovenous malformations in neonatal and adult mice. J. Am. Heart Assoc. 7:e009514. doi: 10.1161/JAHA.118.009514
Kim, Y. H., Phuong, N. V., Choe, S. W., Jeon, C. J., Arthur, H. M., Vary, C. P., et al. (2020). Overexpression of activin receptor-like kinase 1 in endothelial cells suppresses development of arteriovenous malformations in mouse models of hereditary hemorrhagic telangiectasia. Circ. Res. doi: 10.1161/CIRCRESAHA.119.316267 [Epub ahead of print],
Klose, R., Prinz, A., Tetzlaff, F., Weis, E. M., Moll, I., Rodriguez-Vita, J., et al. (2019). Loss of the serine protease HTRA1 impairs smooth muscle cells maturation. Sci. Rep. 9:18224. doi: 10.1038/s41598-019-54807-6
Kritharis, A., Al-Samkari, H., and Kuter, D. J. (2018). Hereditary hemorrhagic telangiectasia: diagnosis and management from the hematologist’s perspective. Haematologica 103, 1433–1443. doi: 10.3324/haematol.2018.193003
Kruithof, B. P., Duim, S. N., Moerkamp, A. T., and Goumans, M. J. (2012). TGFbeta and BMP signaling in cardiac cushion formation: lessons from mice and chicken. Differentiation 84, 89–102. doi: 10.1016/j.diff.2012.04.003
Krupinski, J., Kumar, P., Kumar, S., and Kaluza, J. (1996). Increased expression of TGF-beta 1 in brain tissue after ischemic stroke in humans. Stroke 27, 852–857. doi: 10.1161/01.str.27.5.852
Kummer, B. R., Diaz, I., Wu, X., Aaroe, A. E., Chen, M. L., Iadecola, C., et al. (2019). Associations between cerebrovascular risk factors and parkinson disease. Ann. Neurol. 86, 572–581. doi: 10.1002/ana.25564
Lan, Y., Liu, B., Yao, H., Li, F., Weng, T., Yang, G., et al. (2007). Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol. Cell Biol. 27, 7683–7692. doi: 10.1128/MCB.00577-07
Laterza, D., Ritelli, M., Zini, A., Colombi, M., Dell’Acqua, M. L., Vandelli, L., et al. (2019). Novel pathogenic TGFBR1 and SMAD3 variants identified after cerebrovascular events in adult patients with Loeys-dietz syndrome. Eur. J. Med. Genet. 62:103727. doi: 10.1016/j.ejmg.2019.103727
Laux, D. W., Young, S., Donovan, J. P., Mansfield, C. J., Upton, P. D., and Roman, B. L. (2013). Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development 140, 3403–3412. doi: 10.1242/dev.095307
Lee, Y. C., Chung, C. P., Chao, N. C., Fuh, J. L., Chang, F. C., Soong, B. W., et al. (2018). Characterization of heterozygous HTRA1 mutations in taiwanese patients with cerebral small vessel disease. Stroke 49, 1593–1601. doi: 10.1161/STROKEAHA.118.021283
Lei, D., Jin, X., Wen, L., Dai, H., Ye, Z., and Wang, G. (2017). bmp3 is required for integrity of blood brain barrier by promoting pericyte coverage in Zebrafish embryos. Curr. Mol. Med. 17, 298–303. doi: 10.2174/1566524017666171106114234
Li, F., Lan, Y., Wang, Y., Wang, J., Yang, G., Meng, F., et al. (2011). Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell 20, 291–302. doi: 10.1016/j.devcel.2011.01.011
Li, J., Zhao, Y., Coleman, P., Chen, J., Ting, K. K., Choi, J. P., et al. (2019). Low fluid shear stress conditions contribute to activation of cerebral cavernous malformation signalling pathways. Biochim. Biophys. Acta Mol. Basis Dis. 1865:165519. doi: 10.1016/j.bbadis.2019.07.013
Long, C., McAnally, J. R., Shelton, J. M., Mireault, A. A., Bassel-Duby, R., and Olson, E. N. (2014). Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 345, 1184–1188. doi: 10.1126/science.1254445
Ma, J., Sanchez-Duffhues, G., Goumans, M. J., and Ten Dijke, P. (2020). TGF-beta-induced endothelial to mesenchymal transition in disease and tissue engineering. Front. Cell Dev. Biol. 8:260. doi: 10.3389/fcell.2020.00260
Ma, S., Santhosh, D., Kumar, T. P., and Huang, Z. (2017). A brain-region-specific neural pathway regulating germinal matrix angiogenesis. Dev. Cell 41, 366–381.e4. doi: 10.1016/j.devcel.2017.04.014
Maddaluno, L., Rudini, N., Cuttano, R., Bravi, L., Giampietro, C., Corada, M., et al. (2013). EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498, 492–496. doi: 10.1038/nature12207
Mahmoud, M., Allinson, K. R., Zhai, Z., Oakenfull, R., Ghandi, P., Adams, R. H., et al. (2010). Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ. Res. 106, 1425–1433. doi: 10.1161/CIRCRESAHA.109.211037
Massague, J. (2012). TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630. doi: 10.1038/nrm3434
McAllister, K. A., Grogg, K. M., Johnson, D. W., Gallione, C. J., Baldwin, M. A., Jackson, C. E., et al. (1994). Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 8, 345–351. doi: 10.1038/ng1294-345
McDonald, D. A., Shi, C., Shenkar, R., Gallione, C. J., Akers, A. L., Li, S., et al. (2014). Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: evidence for a common biochemical pathway for CCM pathogenesis. Hum. Mol. Genet. 23, 4357–4370. doi: 10.1093/hmg/ddu153
Morikawa, M., Derynck, R., and Miyazono, K. (2016). TGF-beta and the TGF-beta family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 8:a021873. doi: 10.1101/cshperspect.a021873
Munji, R. N., Soung, A. L., Weiner, G. A., Sohet, F., Semple, B. D., Trivedi, A., et al. (2019). Profiling the mouse brain endothelial transcriptome in health and disease models reveals a core blood-brain barrier dysfunction module. Nat. Neurosci. 22, 1892–1902. doi: 10.1038/s41593-019-0497-x
Naghavi, M., Abajobir, A., Cristiana, A., Abbas, K. M., Abd-Allah, F., Abera, S. F., et al. (2017). Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390, 1151–1210. doi: 10.1016/S0140-6736(17)32152-9
Nguyen, H. L., Lee, Y. J., Shin, J., Lee, E., Park, S. O., McCarty, J. H., et al. (2011). TGF-beta signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab. Invest. 91, 1554–1563. doi: 10.1038/labinvest.2011.124
Nozaki, H., Nishizawa, M., and Onodera, O. (2014). Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 45, 3447–3453. doi: 10.1161/STROKEAHA.114.004236
Oide, T., Nakayama, H., Yanagawa, S., Ito, N., Ikeda, S., and Arima, K. (2008). Extensive loss of arterial medial smooth muscle cells and mural extracellular matrix in cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). Neuropathology 28, 132–142. doi: 10.1111/j.1440-1789.2007.00864.x
Oka, C., Tsujimoto, R., Kajikawa, M., Koshiba-Takeuchi, K., Ina, J., Yano, M., et al. (2004). HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development 131, 1041–1053. doi: 10.1242/dev.00999
Ola, R., Dubrac, A., Han, J., Zhang, F., Fang, J. S., Larrivee, B., et al. (2016). PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat. Commun. 7:13650. doi: 10.1038/ncomms13650
Ola, R., Kunzel, S. H., Zhang, F., Genet, G., Chakraborty, R., Pibouin-Fragner, L., et al. (2018). SMAD4 prevents flow induced arteriovenous malformations by inhibiting casein kinase 2. Circulation 138, 2379–2394. doi: 10.1161/CIRCULATIONAHA.118.033842
Onichtchouk, D., Chen, Y. G., Dosch, R., Gawantka, V., Delius, H., Massague, J., et al. (1999). Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 401, 480–485. doi: 10.1038/46794
Papachristodoulou, A., Silginer, M., Weller, M., Schneider, H., Hasenbach, K., Janicot, M., et al. (2019). Therapeutic targeting of TGFbeta ligands in glioblastoma using novel antisense oligonucleotides reduces the growth of experimental gliomas. Clin. Cancer Res. 25, 7189–7201. doi: 10.1158/1078-0432.CCR-17-3024
Paredes, I., Himmels, P., and Ruiz de Almodovar, C. (2018). Neurovascular communication during CNS development. Dev. Cell 45, 10–32. doi: 10.1016/j.devcel.2018.01.023
Park, S. O., Wankhede, M., Lee, Y. J., Choi, E. J., Fliess, N., Choe, S. W., et al. (2009). Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 119, 3487–3496. doi: 10.1172/JCI39482
Pausch, P., Al-Shayeb, B., Bisom-Rapp, E., Tsuchida, C. A., Li, Z., Cress, B. F., et al. (2020). CRISPR-CasPhi from huge phages is a hypercompact genome editor. Science 369, 333–337. doi: 10.1126/science.abb1400
Pickar-Oliver, A., and Gersbach, C. A. (2019). The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 20, 490–507. doi: 10.1038/s41580-019-0131-5
Piera-Velazquez, S., and Jimenez, S. A. (2019). Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 99, 1281–1324. doi: 10.1152/physrev.00021.2018
Posokhova, E., Shukla, A., Seaman, S., Volate, S., Hilton, M. B., Wu, B., et al. (2015). GPR124 functions as a WNT7-specific coactivator of canonical beta-catenin signaling. Cell Rep. 10, 123–130. doi: 10.1016/j.celrep.2014.12.020
Ricard, N., Ciais, D., Levet, S., Subileau, M., Mallet, C., Zimmers, T. A., et al. (2012). BMP9 and BMP10 are critical for postnatal retinal vascular remodeling. Blood 119, 6162–6171. doi: 10.1182/blood-2012-01-407593
Rice, L. M., Padilla, C. M., McLaughlin, S. R., Mathes, A., Ziemek, J., Goummih, S., et al. (2015). Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Invest. 125, 2795–2807. doi: 10.1172/JCI77958
Ruiz, S., Zhao, H., Chandakkar, P., Chatterjee, P. K., Papoin, J., Blanc, L., et al. (2016). A mouse model of hereditary hemorrhagic telangiectasia generated by transmammary-delivered immunoblocking of BMP9 and BMP10. Sci. Rep. 5:37366. doi: 10.1038/srep37366
Sabbineni, H., Verma, A., and Somanath, P. R. (2018). Isoform-specific effects of transforming growth factor beta on endothelial-to-mesenchymal transition. J. Cell Physiol. 233, 8418–8428. doi: 10.1002/jcp.26801
Sanchez-Duffhues, G., Garcia de Vinuesa, A., van de Pol, V., Geerts, M. E., de Vries, M. R., Janson, S. G., et al. (2019). Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J. Pathol. 247, 333–346. doi: 10.1002/path.5193
Santini, V., Valcarcel, D., Platzbecker, U., Komrokji, R. S., Cleverly, A. L., Lahn, M. M., et al. (2019). Phase II study of the ALK5 inhibitor galunisertib in very low-, low-, and intermediate-risk myelodysplastic syndromes. Clin. Cancer Res. 25, 6976–6985. doi: 10.1158/1078-0432.CCR-19-1338
Schaum, N., Karkanias, J., Neff, N. F., May, A. P., Quake, S. R., Wyss-Coray, T., et al. (2018). Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372. doi: 10.1038/s41586-018-0590-4
Senatorov, V. V. Jr., Friedman, A. R., Milikovsky, D. Z., Ofer, J., Saar-Ashkenazy, R., Charbash, A., et al. (2019). Blood-brain barrier dysfunction in aging induces hyperactivation of TGFbeta signaling and chronic yet reversible neural dysfunction. Sci. Transl. Med. 11:eaaw8283. doi: 10.1126/scitranslmed.aaw8283
Seo, J. H., Maki, T., Maeda, M., Miyamoto, N., Liang, A. C., Hayakawa, K., et al. (2014). Oligodendrocyte precursor cells support blood-brain barrier integrity via TGF-beta signaling. PLoS One 9:e103174. doi: 10.1371/journal.pone.0103174
Shen, M. W., Arbab, M., Hsu, J. Y., Worstell, D., Culbertson, S. J., Krabbe, O., et al. (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature 563, 646–651. doi: 10.1038/s41586-018-0686-x
Shiga, A., Nozaki, H., Yokoseki, A., Nihonmatsu, M., Kawata, H., Kato, T., et al. (2011). Cerebral small-vessel disease protein HTRA1 controls the amount of TGF-beta1 via cleavage of proTGF-beta1. Hum. Mol. Genet. 20, 1800–1810. doi: 10.1093/hmg/ddr063
Shoemaker, L. D., McCormick, A. K., Allen, B. M., and Chang, S. D. (2020). Evidence for endothelial-to-mesenchymal transition in human brain arteriovenous malformations. Clin. Transl. Med. 10:e99. doi: 10.1002/ctm2.99
Siqueira, M., Francis, D., Gisbert, D., Gomes, F. C. A., and Stipursky, J. (2018). Radial glia cells control angiogenesis in the developing cerebral cortex through TGF-beta1 signaling. Mol. Neurobiol. 55, 3660–3675. doi: 10.1007/s12035-017-0557-8
Stapleton, C. J., and Barker, F. G. II (2018). Cranial cavernous malformations: natural history and treatment. Stroke 49, 1029–1035. doi: 10.1161/STROKEAHA.117.017074
Stenman, J. M., Rajagopal, J., Carroll, T. J., Ishibashi, M., McMahon, J., and McMahon, A. P. (2008). Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247–1250. doi: 10.1126/science.1164594
Sweeney, M. D., Ayyadurai, S., and Zlokovic, B. V. (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 19, 771–783. doi: 10.1038/nn.4288
Tang, A. T., Choi, J. P., Kotzin, J. J., Yang, Y., Hong, C. C., Hobson, N., et al. (2017). Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 545, 305–310. doi: 10.1038/nature22075
Tata, M., Ruhrberg, C., and Fantin, A. (2015). Vascularisation of the central nervous system. Mech. Dev. 138(Pt 1), 26–36. doi: 10.1016/j.mod.2015.07.001
Tatake, R. J., O’Neill, M. M., Kennedy, C. A., Wayne, A. L., Jakes, S., Wu, D., et al. (2008). Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem. Biophys. Res. Commun. 377, 120–125. doi: 10.1016/j.bbrc.2008.09.087
Tikka, S., Baumann, M., Siitonen, M., Pasanen, P., Poyhonen, M., Myllykangas, L., et al. (2014). CADASIL and CARASIL. Brain Pathol. 24, 525–544. doi: 10.1111/bpa.12181
Tischfield, M. A., Robson, C. D., Gilette, N. M., Chim, S. M., Sofela, F. A., DeLisle, M. M., et al. (2017). Cerebral vein malformations result from loss of twist1 expression and BMP signaling from skull progenitor cells and dura. Dev. Cell 42, 445–461.e5. doi: 10.1016/j.devcel.2017.07.027
Tual-Chalot, S., Garcia-Collado, M., Redgrave, R. E., Singh, E., Davison, B., Park, C., et al. (2020). Loss of endothelial endoglin promotes high-output heart failure through peripheral arteriovenous shunting driven by VEGF signaling. Circ. Res. 126, 243–257. doi: 10.1161/CIRCRESAHA.119.315974
Turner, M. R., Goldacre, R., Talbot, K., and Goldacre, M. J. (2016). Cerebrovascular injury as a risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 87, 244–246. doi: 10.1136/jnnp-2015-311157
Vanlandewijck, M., He, L., Mae, M. A., Andrae, J., Ando, K., Del Gaudio, F., et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. doi: 10.1038/nature25739
Verdura, E., Herve, D., Scharrer, E., Amador Mdel, M., Guyant-Marechal, L., Philippi, A., et al. (2015). Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain 138(Pt 8), 2347–2358. doi: 10.1093/brain/awv155
von Bernhardi, R., Cornejo, F., Parada, G. E., and Eugenin, J. (2015). Role of TGFbeta signaling in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 9:426. doi: 10.3389/fncel.2015.00426
Walker, E. J., Su, H., Shen, F., Choi, E. J., Oh, S. P., Chen, G., et al. (2011). Arteriovenous malformation in the adult mouse brain resembling the human disease. Ann. Neurol. 69, 954–962. doi: 10.1002/ana.22348
Wang, K., Zhao, S., Liu, B., Zhang, Q., Li, Y., Liu, J., et al. (2018). Perturbations of BMP/TGF-beta and VEGF/VEGFR signalling pathways in non-syndromic sporadic brain arteriovenous malformations (BAVM). J. Med. Genet. 55, 675–684. doi: 10.1136/jmedgenet-2017-105224
Wang, T., Li, B. Y., Danielson, P. D., Shah, P. C., Rockwell, S., Lechleider, R. J., et al. (1996). The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell 86, 435–444. doi: 10.1016/s0092-8674(00)80116-6
Weinsheimer, S., Bendjilali, N., Nelson, J., Guo, D. E., Zaroff, J. G., Sidney, S., et al. (2016). Genome-wide association study of sporadic brain arteriovenous malformations. J. Neurol. Neurosurg. Psychiatry 87, 916–923. doi: 10.1136/jnnp-2015-312272
Welch-Reardon, K. M., Ehsan, S. M., Wang, K., Wu, N., Newman, A. C., Romero-Lopez, M., et al. (2014). Angiogenic sprouting is regulated by endothelial cell expression of Slug. J. Cell Sci. 127(Pt 9), 2017–2028. doi: 10.1242/jcs.143420
Wooderchak-Donahue, W. L., McDonald, J., O’Fallon, B., Upton, P. D., Li, W., Roman, B. L., et al. (2013). BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am. J. Hum. Genet. 93, 530–537. doi: 10.1016/j.ajhg.2013.07.004
Wu, J., Jia, J., Liu, L., Yang, F., Fan, Y., Zhang, S., et al. (2017). Schisandrin B displays a protective role against primary pulmonary hypertension by targeting transforming growth factor beta1. J. Am. Soc. Hypertens 11, 148–157e1. doi: 10.1016/j.jash.2016.12.007
Wu, Y., Liang, D., Wang, Y., Bai, M., Tang, W., Bao, S., et al. (2013). Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13, 659–662. doi: 10.1016/j.stem.2013.10.016
Xi, Q., Wang, Z., Zaromytidou, A. I., Zhang, X. H., Chow-Tsang, L. F., Liu, J. X., et al. (2011). A poised chromatin platform for TGF-beta access to master regulators. Cell 147, 1511–1524. doi: 10.1016/j.cell.2011.11.032
Yang, T., Sun, Y., Lu, Z., Leak, R. K., and Zhang, F. (2017). The impact of cerebrovascular aging on vascular cognitive impairment and dementia. Ageing Res. Rev. 34, 15–29. doi: 10.1016/j.arr.2016.09.007
Yilmaz, B., Toktas, Z. O., Akakin, A., Isik, S., Bilguvar, K., Kilic, T., et al. (2017). Familial occurrence of brain arteriovenous malformation: a novel ACVRL1 mutation detected by whole exome sequencing. J. Neurosurg. 126, 1879–1883. doi: 10.3171/2016.6.JNS16665
Yingling, J. M., McMillen, W. T., Yan, L., Huang, H., Sawyer, J. S., Graff, J., et al. (2018). Preclinical assessment of galunisertib (LY2157299 monohydrate), a first-in-class transforming growth factor-beta receptor type I inhibitor. Oncotarget 9, 6659–6677. doi: 10.18632/oncotarget.23795
Zafar, A., Quadri, S. A., Farooqui, M., Ikram, A., Robinson, M., Hart, B. L., et al. (2019). Familial cerebral cavernous malformations. Stroke 50, 1294–1301. doi: 10.1161/STROKEAHA.118.022314
Zhang, H., Liu, Y., Yan, L., Du, W., Zhang, X., Zhang, M., et al. (2018). Bone morphogenetic protein-7 inhibits endothelial-mesenchymal transition in pulmonary artery endothelial cell under hypoxia. J. Cell Physiol. 233, 4077–4090. doi: 10.1002/jcp.26195
Zhang, L., Lim, S. L., Du, H., Zhang, M., Kozak, I., Hannum, G., et al. (2012). High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-beta family member growth differentiation factor 6. J. Biol. Chem. 287, 1520–1526. doi: 10.1074/jbc.M111.275990
Zhang, Y., Zhang, M., Xie, W., Wan, J., Tao, X., Liu, M., et al. (2020). Gremlin-1 is a key regulator of endothelial-to-mesenchymal transition in human pulmonary artery endothelial cells. Exp. Cell Res. 390, 111941. doi: 10.1016/j.yexcr.2020.111941
Zhao, Z., Nelson, A. R., Betsholtz, C., and Zlokovic, B. V. (2015). Establishment and dysfunction of the blood-brain barrier. Cell 163, 1064–1078. doi: 10.1016/j.cell.2015.10.067
Zhou, Z., Tang, A. T., Wong, W. Y., Bamezai, S., Goddard, L. M., Shenkar, R., et al. (2016). Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 532, 122–126. doi: 10.1038/nature17178
Keywords: cerebral cavernous malformation, hereditary hemorrhagic telangiectasia, cerebrovascular disease, endothelial-to-mesenchymal transition, cerebral angiogenesis, TGF-β signaling
Citation: Zhang Y and Yang X (2020) The Roles of TGF-β Signaling in Cerebrovascular Diseases. Front. Cell Dev. Biol. 8:567682. doi: 10.3389/fcell.2020.567682
Received: 03 June 2020; Accepted: 31 August 2020;
Published: 18 September 2020.
Edited by:
Xin-Ming Chen, Royal North Shore Hospital, AustraliaReviewed by:
Yue Liu, Xiyuan Hospital, ChinaCopyright © 2020 Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiao Yang, eWFuZ3hAYm1pLmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.