
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 29 September 2020
Sec. Molecular and Cellular Pathology
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.528742
This article is part of the Research Topic Contribution of Ion Channels to Neuropathologies View all 5 articles
 Rebecca Martinez-Moreno1,2
Rebecca Martinez-Moreno1,2 Elisabet Selga1,2,3,4
Elisabet Selga1,2,3,4 Helena Riuró2David Carreras2Mered Parnes5Chandra Srinivasan6Michael F. Wangler7,8Guillermo J. Pérez1,4*†
Helena Riuró2David Carreras2Mered Parnes5Chandra Srinivasan6Michael F. Wangler7,8Guillermo J. Pérez1,4*† Fabiana S. Scornik1,4*†Ramon Brugada1,2,4,9*†
Fabiana S. Scornik1,4*†Ramon Brugada1,2,4,9*†Voltage-gated sodium (NaV) channels are transmembrane proteins that initiate and propagate neuronal and cardiac action potentials. NaV channel β subunits have been widely studied due to their modulatory role. Mice null for Scn1b, which encodes NaV β1 and β1b subunits, have defects in neuronal development and excitability, spontaneous generalized seizures, cardiac arrhythmias, and early mortality. A mutation in exon 3 of SCN1B, c.308A>T leading to β1_p.D103V and β1b_p.D103V, was previously found in a patient with a history of proarrhythmic conditions with progressive atrial standstill as well as cognitive and motor deficits accompanying structural brain abnormalities. We investigated whether β1 or β1b subunits carrying this mutation affect NaV1.5 and/or NaV1.1 currents using a whole cell patch-clamp technique in tsA201 cells. We observed a decrease in sodium current density in cells co-expressing NaV1.5 or NaV1.1 and β1D103V compared to β1WT. Interestingly, β1bD103V did not affect NaV1.1 sodium current density but induced a positive shift in the voltage dependence of inactivation and a faster recovery from inactivation compared to β1bWT. The β1bD103V isoform did not affect NaV1.5 current properties. Although the SCN1B_c.308A>T mutation may not be the sole cause of the patient’s symptoms, we observed a clear loss of function in both cardiac and brain sodium channels. Our results suggest that the mutant β1 and β1b subunits play a fundamental role in the observed electrical dysfunction.
Voltage-gated sodium (NaV) channels are transmembrane proteins that initiate and propagate neuronal and cardiac action potentials (Catterall, 2000; Catterall et al., 2005). These channels have an α subunit composed of four domains (DI–DIV), each with six transmembrane segments (S1–S6). NaVα subunits are associated with two or more auxiliary β subunits (Kaplan et al., 2016). NaVβ subunits modulate sodium channel biophysical properties in excitable and non-excitable tissues and function as cell adhesion molecules, which are critical for extracellular/intracellular communication (Chen, 2004; Watanabe et al., 2008; Patino et al., 2011).
Five NaVβ subunits have been identified in mammals: β1, β2, β3, β4 (encoded by SCN1B, SCN2B, SCN3B, and SCN4B, respectively), and β1b, a β1 splice variant. All β subunits with the exception of β1b are composed of an extracellular N-terminal immunoglobulin-like (Ig) domain (ECD), a transmembrane region, and an intracellular C-terminal domain (Brackenbury and Isom, 2011). Similar to the other β subunits, β1b is composed of an N-terminal region encoded by exons 1–3 of SCN1B. However, instead of a transmembrane domain, β1b has a different C-terminal region caused by partial retention of intron 3, which contains a stop codon (Hu et al., 2012). Apart from its structural differences, β1b is the only extracellularly secreted β subunit (Patino et al., 2011; O’Malley and Isom, 2016). However, it is predicted to have the same cellular interactions as the β1 subunit because it shares the same ECD (O’Malley and Isom, 2016).
In a series of experiments using NaVβ subunit chimeras, McCormick et al. (1999) demonstrated that the ECD is necessary and sufficient for sodium channel modulation (McCormick et al., 1999). Mutations in the ECD of the β1 subunit have been associated with brain diseases, such as epileptic encephalopathy and genetic (or generalized) epilepsy with febrile seizures plus (Wallace et al., 1998; Audenaert et al., 2003; Scheffer et al., 2007; Brackenbury and Isom, 2011; Darras et al., 2019). Some of these mutations have been proven to disrupt the interaction between NaV1.1 and the β1 subunit, impairing excitability or reducing cell-cell interactions (Meadows et al., 2002; Chen, 2004; Spampanato, 2004; Patino and Isom, 2010). Although NaVα subunit alone is sufficient to form a conducting channel, β1 subunits enhance protein expression and modulate gating and voltage dependency (McCormick et al., 1999; Meadows et al., 2002; Yu and Catterall, 2003; Olesen et al., 2012). Scn1b-null mice have defective neuronal development and excitability, present spontaneous generalized seizures and cardiac arrhythmias, and die by postnatal day 21 (Chen, 2004; Patino and Isom, 2010; Brackenbury and Isom, 2011; Lin et al., 2015).
The regulatory effect of the β1WT subunit on NaV1.5 has been widely studied. SCN1B mutations are reported to cause diseases such as Brugada syndrome (Watanabe et al., 2008; Peeters et al., 2015), long QT syndrome (Riuró et al., 2014), and atrial fibrillation (Olesen et al., 2012), demonstrating the critical effect of β1 on cardiac excitability. In addition, Lopez-Santiago et al. (2007) showed that Scn1b-null mice display prolonged QT and RR intervals. In addition, mutations in β1b have been associated with cardiac arrhythmias, such as Brugada syndrome, long QT syndrome, cardiac conduction disease, and SIDS (Watanabe et al., 2008; Hu et al., 2012; Riuró et al., 2014).
We investigated a mutation located in exon 3 of SCN1B [Chr19:35524503 A>T (hg19): NM_001037.4 c.308A>T] identified in a patient with a history of proarrhythmic conditions with progressive atrial standstill, cognitive and motor deficits, and structural brain abnormalities. The patient was part of a sequencing study by Eldomery et al. (2017) that proposed a dual diagnosis for this case. As a result, this β1 variant (β1D103V) (dbSNP ID: 1057519457) is indexed as pathogenic in the ClinVar database1. However, this variant has not been functionally characterized.
The mutation leads to substitution of an aspartic acid for a valine at position 103 of the protein (p.D103V). Exon 3 is part of the Ig-like loop of the ECD, which is common between β1 and β1b. Therefore, this substitution is present in both β1 and β1b subunit isoforms at a highly conserved position. Thus, this mutation likely affects the function of both β1 and β1b proteins, and could play a role in the patient’s pathophysiologic phenotype. This work characterizes the effects of mutant β1 and β1b subunits on the biophysical properties of both cardiac (NaV1.5) and neuronal (NaV1.1) sodium channels.
The pCMV vector harboring SCN1A was a generous gift from Dr. Alfred George Jr. (Vanderbilt University Division of Genetic Medicine, Nashville, TN, United States). The pcDNA3 vector harboring the complementary DNA (cDNA) of human SCN5A was a generous gift from Dr. Matteo Vatta (Baylor College of Medicine, Houston, TX, United States).
Commercially available human SCN1B cDNA (pCMV6-XL4-SCN1B OriGene Technologies Inc., Rockland, MD, United States) was subcloned into a bicistronic vector encoding enhanced green fluorescent protein (GFP) (pIRES-GFP, Clontech Laboratories Inc., Mountain View, CA, United States). We constructed a bicistronic vector encoding SCN1Bb-GFP (pIRES-GFP-SCN1Bb). Human SCN1Bb cDNA was cloned from the human right ventricle (Riuró et al., 2014).
pIRES-GFP-SCN1B and pIRES-GFP-SCN1Bb were used as templates to engineer the p.D103V mutation using a QuikChange Lightning site-directed mutagenesis kit (Stratagene, La Jolla, CA, United States) and the following primers (mutation underlined):
Forward: 5′ CACCAAAGACCTGCAGGTTCTGTCTATCTT CATCA 3′.
Reverse: 5′ TGATGAAGATAGACAGAACCTGCAGGTCTT TGGTG 3′.
The inserts were sequenced to verify presence of the desired mutation and absence of unwanted variations.
Human embryonic kidney (HEK)-293 tsA201 cells (Health Protection Agency Culture Collections, Salisbury, United Kingdom) were used to express the sodium channel subunits. Cells were maintained at 37°C and 5% CO2 in Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 1% GlutaMAX (all from Gibco, Thermo Fisher Scientific Inc., Waltham, MA, United States). HEK cells were plated on 35 mm dishes coated with poly-L-lysine (Sigma-Aldrich Co., St. Louis, MO, United States). Cells were transiently transfected 24 h after plating using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific Inc.) and Opti-MEM (Gibco, Thermo Fisher Scientific Inc.) with 2 μg of total DNA encoding NaV1.5 alone, NaV1.5+β1WT, NaV1.5+β1D103V, NaV1.1 alone, NaV1.1+β1WT, NaV1.1+β1D103V, NaV1.1+β1bWT, or NaV1.1+β1bD103V at a 1:2 (α:β) molar ratio. The effect of the β1b subunit on NaV1.5 current was studied by transfecting NaV1.5 alone, NaV1.5+β1bWT, or NaV1.5+β1bD103V at a 1:2 molar ratio using a Genecellin transfection kit (BioCellChallenge, Toulon, France). HEK cells were split and re-plated 24 h after transfection to obtain single cells. Electrophysiological studies were performed 48 h after transfection.
Sodium currents from cells displaying green fluorescence were studied at room temperature using the patch clamp whole cell configuration. The bath solution contained (in mmol/l): 140 NaCl, 3 KCl, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 1.8 CaCl2 and 1.2 MgCl2, pH 7.4 (NaOH). The pipette solution contained (in mmol/l): 130 CsCl, 1 ethylene glycol-bis(2-amino-ethylether)-N,N,N′,N′-tetra-acetic acid (EGTA), 10 HEPES, 10 NaCl and 2 ATP-Mg2+, pH 7.2 (CsOH). Osmolality of bath and pipette solutions was adjusted with glucose to 325 and 308 mOsm, respectively. Pipettes were pulled from borosilicate glass capillaries (Sutter Instrument, Novato, CA, United States) with a Narishige PC-10 puller (Narishige International LTD, London, United Kingdom), and their resistance ranged 2–3.5 MΩ when filled with pipette solution. Voltage clamp experiments were conducted with an Axopatch 200B amplifier and pClamp10.2/Digidata 1440A acquisition system (Axon Instruments, Molecular Devices, Sunnyvale, CA, United States) at a sampling rate of 20 kHz. Leak subtraction was not used. OriginPro8 software was used for data analysis and statistics (OriginLab Corporation, Northampton, MA, United States). Data was filtered at 5 kHz. Series resistance compensation of 80–90% was used when necessary. To permit current stabilization, recordings were performed at least 5 min after entering into the whole cell configuration. Membrane potentials were not corrected for junction potentials that arose between the bath and pipette solution.
Sodium currents were studied with voltage-clamp step protocols. To determine the current-voltage relationship and voltage dependence of activation, cells were held at –120 mV and currents were elicited with depolarizing pulses of 50 ms from –80 to 80 mV in 5 mV increments. The voltage dependence of inactivation was determined by applying 50-ms prepulses from –140 to 5 mV in 5 mV increments, followed by a –20 mV test pulse. Recovery from inactivation was studied with a two-pulse protocol consisting of a first –20 mV pulse of 50 ms from a holding potential of –120 mV, followed by a –120 mV interpulse of varying duration (1–40 ms), and a second –20 mV pulse of 50 ms. Current density was obtained by normalizing the current at each potential by cell capacitance. Activation and steady-state inactivation data were fitted to a Boltzmann equation of the form G = Gmax/(1+exp(V1/2–V)/k) and I = Imax/(1+exp(V–V1/2)/k), respectively, where G is conductance, Gmax is maximum conductance, V1/2 is voltage at which half of channels are activated or inactivated, V is membrane potential, k is slope factor, I is peak current amplitude, and Imax is maximum current amplitude. Recovery from inactivation data were fitted to a mono-exponential function to obtain the time constant.
Cells were plated and transfected as above. Forty-eight hours after transfection, cells were washed three times with Dulbecco’s phosphate-buffered saline (DPBS) and scraped in Triton X-100 lysis buffer containing 1% Triton X-100, 50 mM Tris/HCl pH 7.4, 150 mM NaCl, 1 mM EDTA and cOmplete protease inhibitor cocktail (Roche, Madrid, Spain). Lysates were obtained after 1 h rotating at 4°C, and insoluble materials were removed by centrifugation. Proteins were quantified using a Pierce BCA protein assay kit (Thermo Scientific, Rockford, IL, United States) and resolved along with a protein marker (PageRuler Plus prestained protein ladder, Thermo Scientific) in 4–15% Mini-PROTEAN TGX Stain-Free precast gels (Bio-Rad Laboratories, Hercules, CA, United States). These gels include a trihalo compound that react with tryptophan residues in proteins, and allow rapid fluorescent detection of proteins in gels or on membranes without staining. Proteins were transferred to PVDF membranes (GE Healthcare Life Sciences, Chicago, IL, United States) overnight at 4°C. Gels were developed by exposure to UV light before and after protein transfer to the membrane. Membranes were probed with either a rabbit anti-human NaV1.5 antibody (Alomone Labs) at a 1:1000 dilution for 1 h at room temperature, or a rabbit anti-human NaV1.1 antibody (Alomone Labs, Jerusalem, Israel) at a 1:100 dilution overnight at 4°C. Membranes were further incubated with a secondary horseradish peroxidase-conjugated anti-rabbit antibody (Thermo Scientific) at a 1:10000 dilution for 1 h at room temperature. Chemiluminescent signal was obtained with Pierce ECL western blotting substrate (Thermo Scientific) and detected using standard X-ray films. Expression of NaV1.1 and NaV1.5 was quantified from digitized X-ray film images using ImageJ software (National Institutes of Health,2). Membrane intensity values for each sample were normalized by total lane density before protein transfer (Taylor et al., 2013).
Cell surface protein biotinilation was performed following the protocol from Tarradas et al. (2013). Briefly, cells were plated and transfected as described above. Twenty-four hours after transfection, cells were washed twice with DPBS supplemented with 0.9 mM CaCl2 and 0.49 mM MgCl2 (DPBS+). Membrane proteins were biotinylated by incubating cells with 1 mg/ml of EZ-Link Sulfo-NHS-SS-Biotin (Thermo Scientific) in DPBS+ for 30 min at 4°C. Cells were then washed three times in DPBS+ with 100 mM glycine, and scrapped in Triton X-100 lysis buffer [1% Triton X-100, 50 mM Tris/HCl pH 7.4, 150 mM NaCl, 1 mM EDTA and Complete Protease Inhibitor Cocktail (Roche, Madrid, Spain)]. Lysates were obtained after 1 h rotating at 4°C. Insoluble materials were removed by centrifugation. 10% of the supernatants were kept at −80°C (input samples), the rest (pull-down samples) were incubated with Ultralink Immobilized NeutrAvidin resin (Pierce, Thermo Scientific) overnight at 4°C. The resin was precipitated and washed with Triton X-100 lysis buffer, then in saline solution (5 mM EDTA, 350 mM NaCl and 0.1% TX-100 in DPBS+) and finally in 50 mM Tris/HCl pH 7.4, 150 mM NaCl and 1 mM EDTA. Input and pull-down samples were resuspended in SDS-PAGE loading buffer and heated for 5 min at 95°C. Proteins were resolved in 7.5% acrylamide gels using TGX Stain-Free FastCast Acrylamide Kit (Bio-Rad Laboratories) and transferred overnight at 4°C to PVDF membranes (Millipore, Billerica, MA, United States). Protein bands using Stain-Free gels were visualized by exposure to UV light before electroblotting to PVDF membranes. Membranes were probed with a rabbit anti-human NaV1.5 antibody (Alomone Labs) at a 1:1000 dilution either for 1 h at room temperature or overnight at 4°C. After washing, the membrane was further incubated with a secondary horseradish peroxidase-conjugated antibody (Thermo Scientific, Rockford, IL, United States) at a dilution of 1:10000 for 1 h at room temperature. Chemiluminescent signal was obtained with Clarity Western ECL Substrate (Bio-Rad Laboratories) and detected using the ChemiDoc Imaging System (Bio-Rad Laboratories). Expression of NaV1.5 was quantified using ImageJ software (National Institutes of Health,3). Membrane intensity values for each sample were normalized by total lane density before protein transfer (Taylor et al., 2013).
OriginPro 2019 software was used for statistical analysis. Results are presented as means ± standard error. Statistical comparisons were performed using one-way ANOVA with a post hoc Tukey test. Differences were considered statistically significant at p < 0.05.
The proband, currently an 8-year-old male, was part of a whole exome sequencing study by Eldomery et al. (2017). This study revealed that the proband was heterozygous for the SCN1B missense mutation c.308A>T. This mutation results in an amino acid change from a hydrophilic aspartic acid to a hydrophobic valine at position 103 (p.D103V) of the sodium channel auxiliary β subunit. D103 is probably important to channel function, as it is highly conserved among all sodium channel β subunits, except for β4, and is also conserved among different species (Figures 1A,B). Eldomery et al. indexed the β1 subunit variant D103V as pathogenic (Eldomery et al., 2017).

Figure 1. The protein region that includes the mutation is highly conserved. Sequence alignments of the region shared by β1 and β1b proteins containing the D103V mutation were performed using Uniprot. The position of the last amino acid of each sequence, and the reference for each protein according to Uniprot are indicated on the left side of each sequence. (A) Sequence alignment of human voltage-gated sodium channel β-subunit family members. (B) Sequence alignment of human voltage-gated sodium channel β1-subunit of different species.
In addition to the SCN1B variant, inherited from the child’s father, genetic analysis revealed that the proband was heterozygous for two pathogenic variants in POLR1C: c.614delG (p.G205Afs∗49), for which his mother was heterozygous, and c.88C>T (p.P30S), for which his father was heterozygous.
The clinical picture of the patient is complex and includes cardiac and neurological impairment. Arrhythmogenic features presented early in the neonatal stage. These included bradycardia from variable atrioventricular (AV) conduction disturbance along with evidence of a borderline prolongation of QTc interval, which appeared to be secondary to intraventricular conduction delay. This was treated with propranolol. Ventricular dysfunction was observed during the neonatal period but normalized at 18 months of age with medical management. Follow-up evaluations showed poorly discernible atrial activity and intraventricular conduction delays on surface electrocardiograms (ECG). The child developed recalcitrant tachycardia at 7 years of age, identified as cavo-tricuspid isthmus (CTI)-dependent macro-reentrant atrial tachycardia (cycle length ∼420 ms) with 1:1 AV conduction as well as 2:1 AV conduction during intracardiac electrophysiology study. At baseline, he was variably in junctional rhythm with brief periods of atrially mediated rhythm. Normal AV conduction was seen at baseline, with atrial pacing that worsened to 2:1 AV conduction on isoproterenol. Three-dimensional voltage mapping at baseline rhythm revealed extensive scarring of the right atrium and coronary sinus. Resting surface electrocardiogram from index patient (Figure 2A) shows poorly discernible atrial activity with low amplitude P wave, intraventricular conduction delay (QRS duration 114 ms) as well as prolonged QTc secondary to QRS abnormality (uncorrected QTc 477 ms). Intracardiac three-dimensional voltage map from index patient is contrasted against a healthy age-matched peer (Figure 2B). The patient underwent placement of a transvenous dual-chamber pacemaker. He was programmed in the VVI pacing mode due to progressive atrial undersensing and unreliable atrial capture.
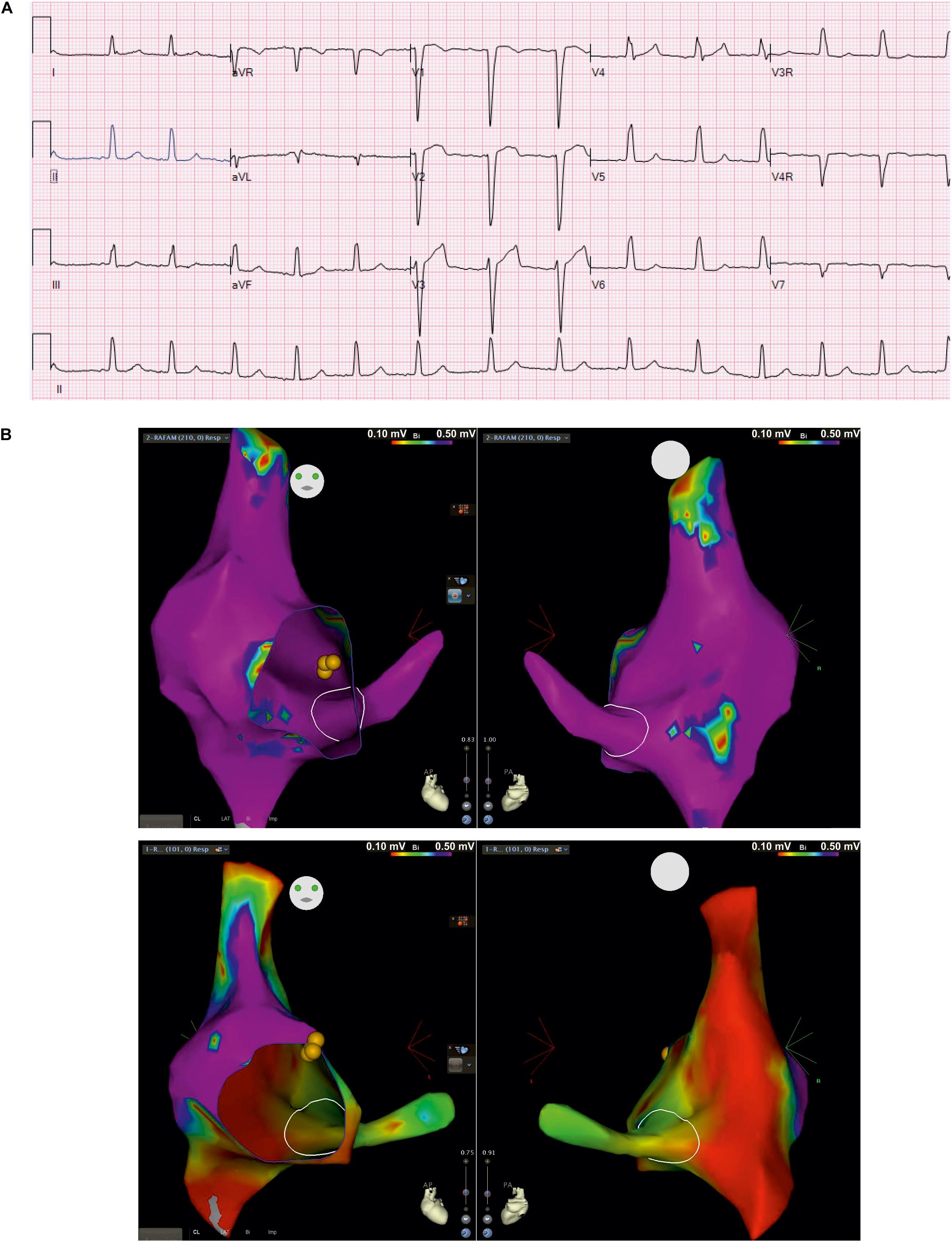
Figure 2. Surface electrocardiogram and intracardiac three-dimensional voltage mapping. (A) Resting surface electrocardiograms (ECG) at 5 years of age in index patient at 25 mm/second, 10 mm/mV; (B) Antero-posterior (left) and postero-anterior (right) projections of voltage map of right atrium and coronary sinus in baseline rhythm from index patient (top) and, healthy age-matched peer (bottom) for comparison (voltage color scale range: 0.1 – 0.5 mV, “purple” indicates voltage >0.5 mV)
By the 2 year of life, the patient also had global developmental delay and increased tone in the lower extremities. Neurological examination at age five demonstrated evidence of significant cognitive deficits (smiled socially, followed simple commands, gave one-word answers to questions), persistent large-amplitude horizontal nystagmus with primary gaze, normal tone in the upper extremities with mixed spasticity and dystonia in the lower extremities (spastic catches and intermittent dystonic extension and inward rotation of the lower extremities when excited). He demonstrated symmetrically brisk deep tendon reflexes with Babinski and Rossolimo signs, and significant dysmetria on the finger-to-nose test as well as axial ataxia affecting the neck and trunk. He sat with support and bore weight with assistance, with scissoring of the legs, and did not ambulate. Brain MRI revealed extensive polymicrogyria involving the bilateral cerebral hemispheres, extensive T2 hyperintense signal throughout the supratentorial white matter with less in the cerebellar white matter and dorsal brainstem, and markedly delayed myelination.
To date, no functional studies of the SCN1B mutation D103V have been published. Because this SCN1B variant was flagged as the best candidate for electrical dysfunction observed in the patient, we comprehensively analyzed the effects of this variant on heart and brain sodium currents.
Modulation of sodium channels by β subunits has been reported by different groups (Brackenbury and Isom, 2011; O’Malley and Isom, 2016). Our goal was to determine whether mutant β1 (β1D103V) and β1b (β1bD103V) subunits affected NaV1.5 and NaV1.1 sodium currents (INa). We first performed functional characterization of the sodium current in HEK-293T cells expressing NaV1.5 alone or co-expressed with either β1WT (NaV1.5+β1WT) or β1bWT (NaV1.5+β1bWT) subunits (Figure 3A). Co-expression of NaV1.5 with either β1WT or β1bWT increased peak INa density (41.2 and 42.8%, respectively) compared to NaV1.5 alone (Figure 3B and Tables 1, 2). Also, the β1WT subunit induced a negative shift of 5 mV in the voltage dependence of activation compared to NaV1.5 alone (Figure 3C left and Table 1). No changes were observed in voltage dependence of activation upon expression of NaV1.5+β1bWT (Figure 3C right and Table 2). Voltage dependence of inactivation was not altered by either β1WT or β1bWT subunits. Both NaV1.5+β1WT and NaV1.5+β1bWT currents displayed faster recovery from inactivation compared to NaV1.5 alone (Figure 3D and Tables 1, 2).
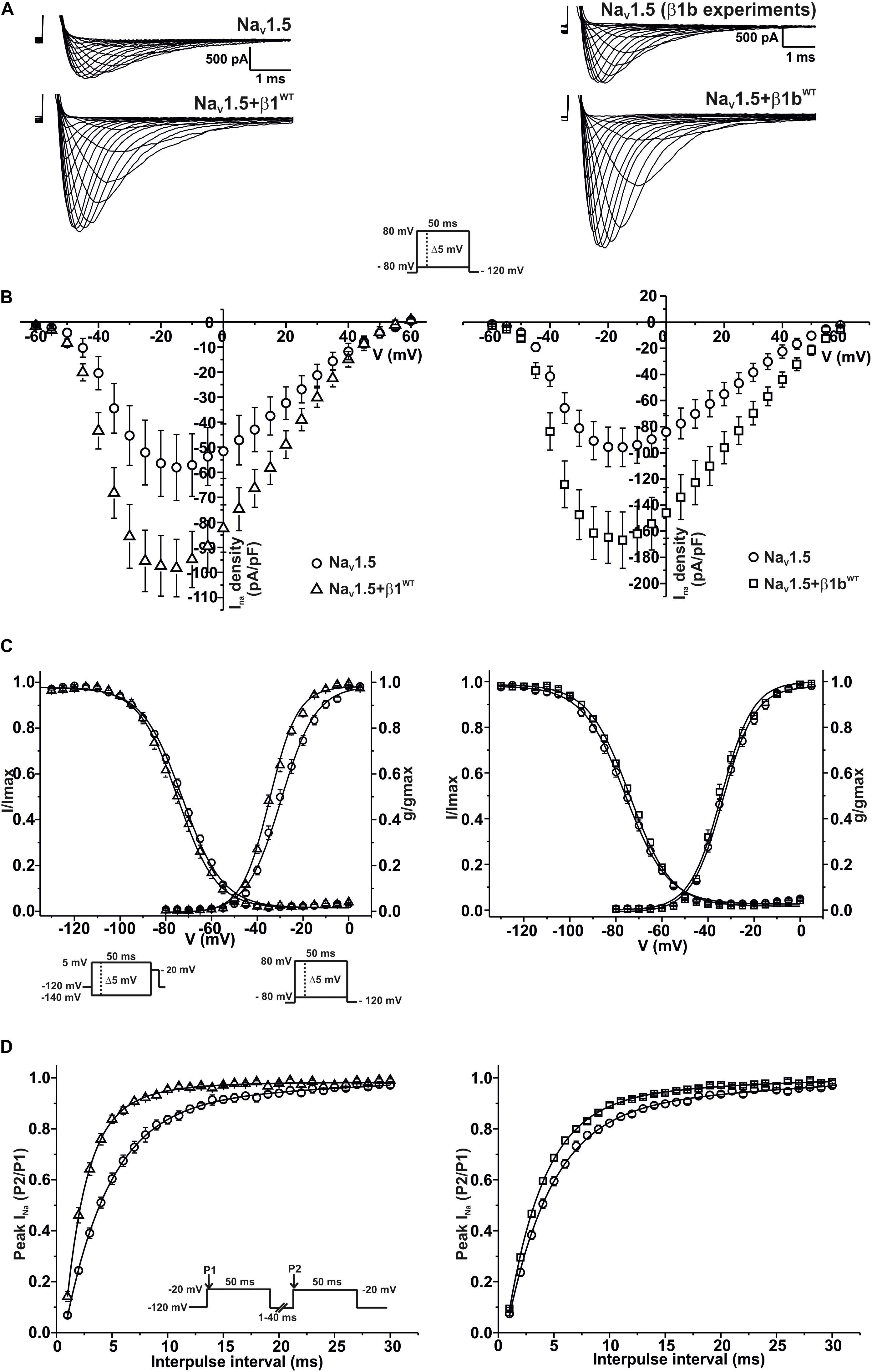
Figure 3. β1WT and β1bWT modulate NaV1.5 current. Biophysical properties of sodium currents measured from HEK-293T cells expressing NaV1.5 alone, or co-expressed with either β1WT or β1bWT. Whole cell currents were elicited by depolarizing potentials as shown in the insets. Circles are used to depict data for NaV1.5 alone, triangles for NaV1.5+β1WT, and squares for NaV1.5+β1bWT. Solid lines in panels (C) and (D) represent the fitted curves. Values are expressed as mean ± SEM. The studies with the β1 subunit are represented on the left side of the figure, and the studies with the β1b subunit on the right side. (A) Representative whole-cell Na+ current traces from HEK-293T cells expressing NaV1.5 alone (top left and top right), NaV1.5+β1WT (bottom left), and NaV1.5+β1bWT (bottom right). (B) Mean current-voltage relationship. INa amplitude was normalized by the cell capacitance to obtain INa density values. (C) INa steady-state voltage dependence of activation and inactivation plots. (D) Recovery from inactivation curves.
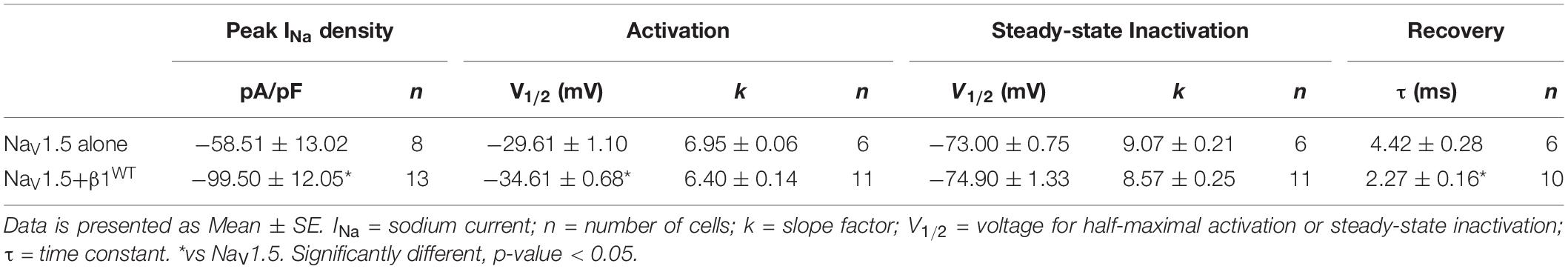
Table 1. Biophysical parameters of HEK cells expressing NaV1.5 alone or together with β1WT.
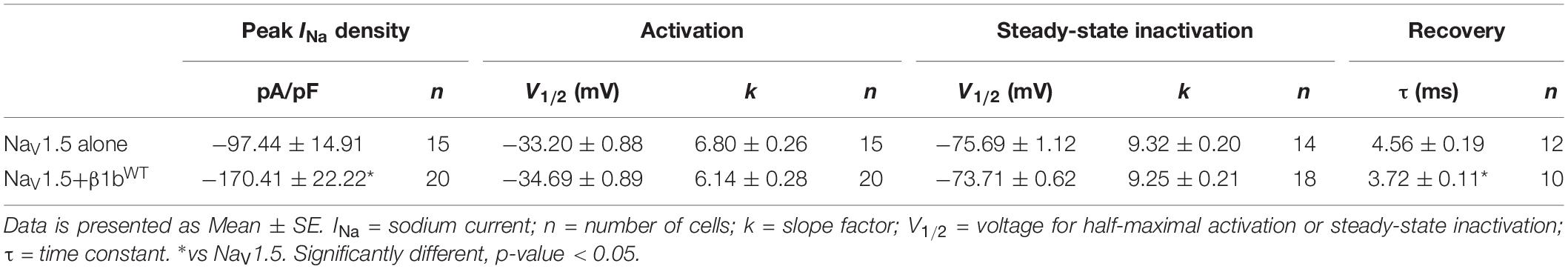
Table 2. Biophysical parameters of HEK cells expressing NaV1.5 alone or together with β1bWT.
To determine whether β1D103V affected the cardiac NaV1.5 sodium current, we co-expressed NaV1.5 with either the β1WT or β1D103V subunit (Figure 4A). NaV1.5+β1D103V decreased sodium current density by 45.7% compared to NaV1.5+β1WT (Figure 4B). β1D103V induced a 3.37-mV positive shift of the voltage dependence of activation compared to β1WT. The voltage dependence of steady state inactivation was similar between NaV1.5+β1WT and NaV1.5+β1D103V (Figure 4C). Likewise, the recovery from inactivation time constants were similar when INa was measured in both experimental conditions (Figure 4D and Table 3).
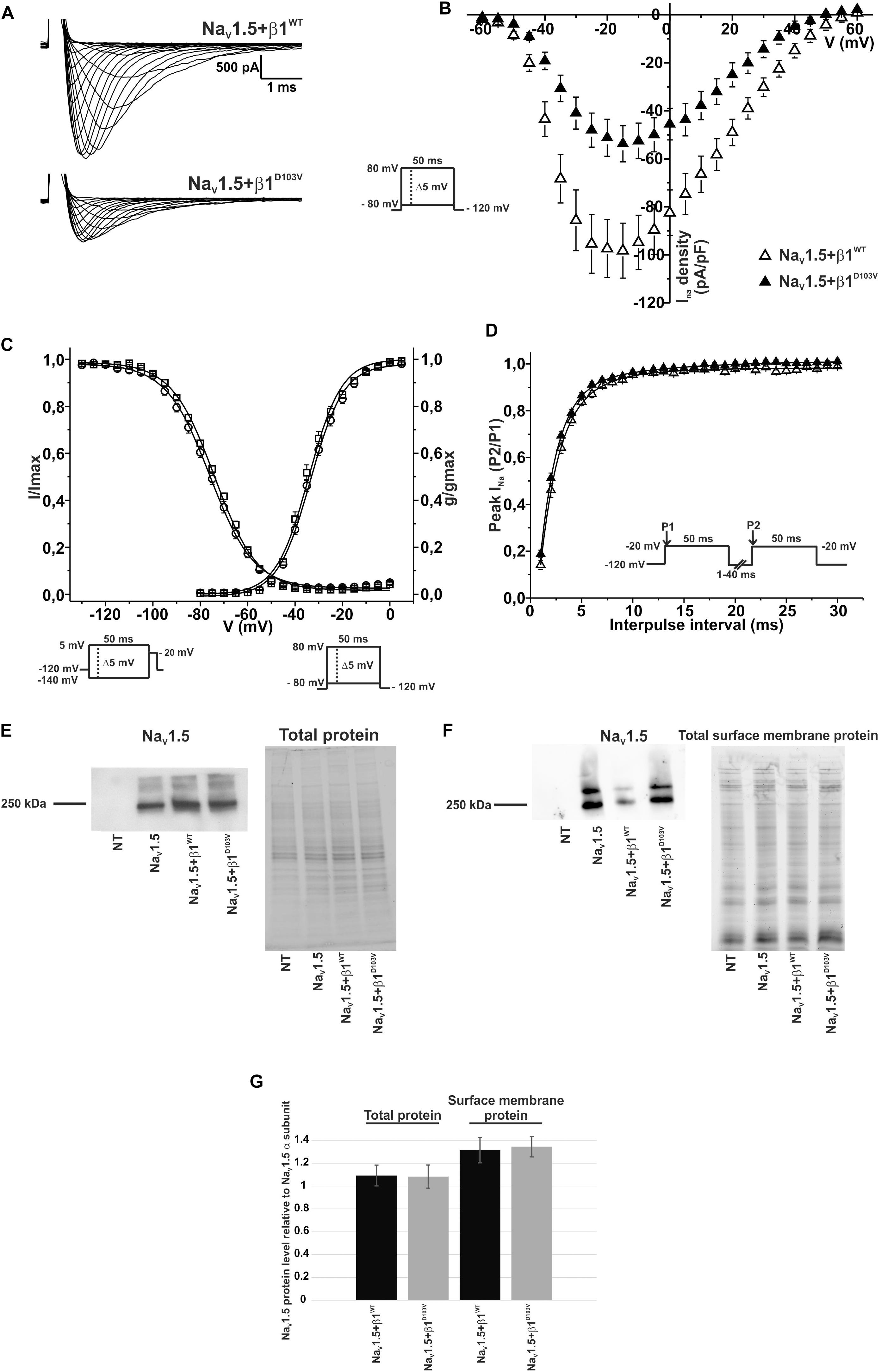
Figure 4. β1D103V modifies the gating properties of NaV1.5 channel. Biophysical properties of sodium currents measured from HEK-293T cells expressing NaV1.5 alone, or co-expressed with either β1WT or β1D103V. Whole cell currents were elicited by depolarizing potentials as shown in the insets. Open triangles are used to depict data for NaV1.5+β1WT and filled triangles for NaV1.5+β1D103V. Solid lines in panels (C) and (D) represent the fitted curves. Values are expressed as mean ± SEM. (A) Representative whole-cell Na+ current traces from HEK-293T cells expressing NaV1.5+β1WT (top), and NaV1.5+β1D103V (bottom). (B) Mean current-voltage relationship. INa amplitude was normalized by the cell capacitance to obtain INa density values. (C) INa steady-state voltage dependence of activation and inactivation plots. (D) Recovery from inactivation curves. (E) Western blot detection of NaV1.5 (left) and corresponding total protein stain-free gel (right), from HEK-293T cells transfected with either NaV1.5, NaV1.5+β1WT, or NaV1.5+β1D103V or non- transfected cells (NT) (n = 2). (F) Western blot detection of NaV1.5 after cell surface biotinylation (left) and corresponding total protein stain-free gel (right), from HEK-293T cells transfected with either NaV1.5, NaV1.5+β1WT, or NaV1.5+β1D103V or non-transfected cells (NT). (G) Bar graph depicts the relative protein expression normalized by the NaV1.5 α subunit of total protein (left, n = 2) and biotinylated protein (right, n = 4) from HEK-293T cells transfected with either NaV1.5+β1WT or NaV1.5+β1D103V. Both visible bands from the biotinylated samples were used for quantification, and the ratio was obtained by normalizing each condition with its respective input.
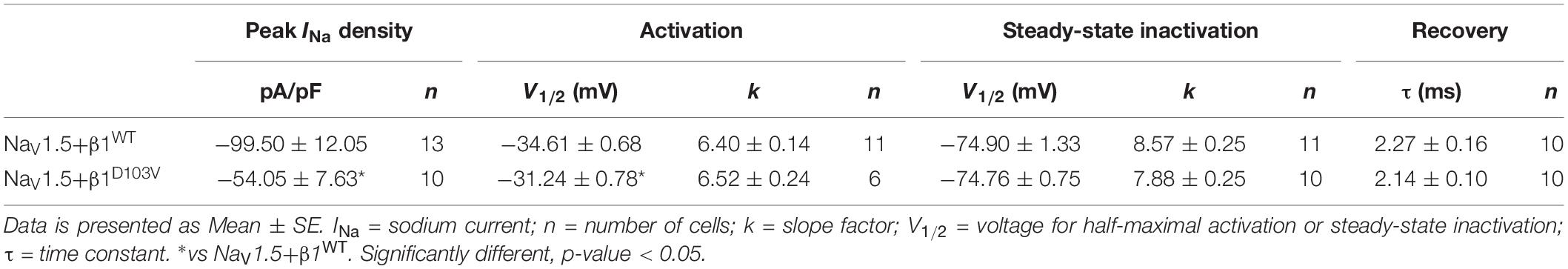
Table 3. Biophysical parameters of HEK cells cotransfected with NaV1.5 and β1WT or β1D103V.
Notably, we did not detect any sodium current in 34 of 75 cells transfected with NaV1.5+β1D103V. However, only 11 of 81 cells expressing NaV1.5+β1WT had no detectable INa. Nevertheless, β1 D103V mutation did not affect protein expression. We observed approximately the same amount of total NaV1.5 in all three conditions (NaV1.5 alone, NaV1.5+β1WT, and NaV1.5+β1D103V; Figures 4E,G). Moreover, the amount of NaV1.5 in the plasma membrane was unaffected by the β1 mutation. To determine this we performed immunoblotting analysis of biotinylated surface membrane proteins. Figures 4F,G show that the relative amount of plasma membrane NaV1.5+β1WT and NaV1.5+β1D103V channels was similar.
We finally evaluated the effects of β1bD103V on NaV1.5 sodium current properties. The β1bD103V mutation did not modify INa density compared to NaV1.5+β1bWT. Likewise, voltage dependence of activation, steady state inactivation, and recovery from inactivation were not altered by mutation compared to the wildtype subunit (Figure 5 and Table 4).
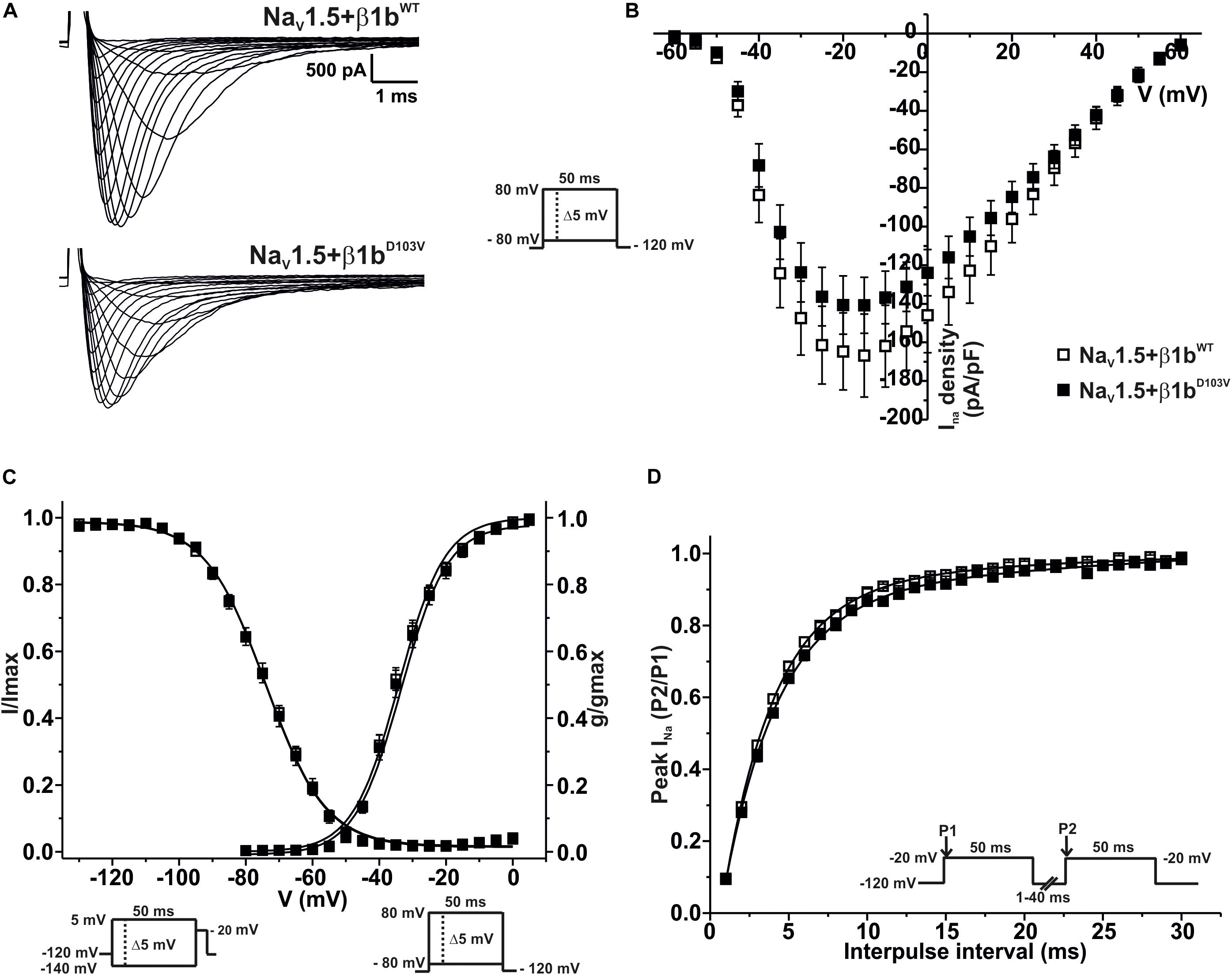
Figure 5. NaV1.5 current properties are not modified by β1bD103V. Biophysical properties of sodium currents measured from HEK-293T cells expressing NaV1.5 co-expressed with either β1bWT or β1bD103V. Whole cell currents were elicited by depolarizing potentials as shown in the insets. Open squares are used to depict data for NaV1.5+β1WT and filled squares for NaV1.5+β1D103V. Solid lines in panels (C) and (D) represent the fitted curves. Values are expressed as mean ± SEM. (A) Representative whole-cell Na+ current traces from HEK-293T cells expressing NaV1.5+β1bWT (top), and NaV1.5+β1bD103V (bottom). (B) Mean current-voltage relationship. INa amplitude was normalized to the cell capacitance to obtain INa density values. (C) INa steady-state voltage dependence of activation and inactivation plots. (D) Recovery from inactivation curves.
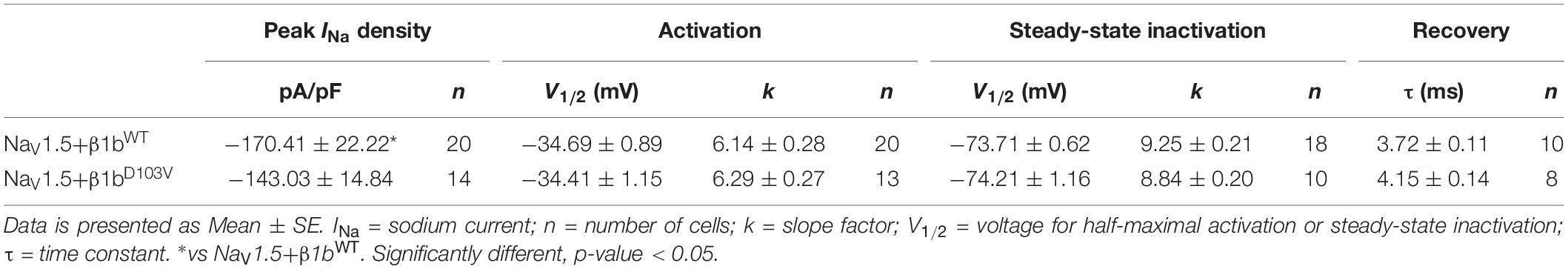
Table 4. Biophysical parameters of HEK cells cotransfected with NaV1.5 and β1bWT or β1bD103V.
Because the proband also experienced brain pathologies, we investigated whether the D130V mutation in β1 and β1b isoforms also affected the most predominant brain-type sodium current: NaV1.1. We performed functional characterization of the sodium current in HEK-293T cells expressing NaV1.1 alone or co-expressed with β1WT or β1bWT subunits.
We measured macroscopic sodium currents (INa) at varying potentials from these transfected cells (Figure 6A). There was a 45.9% increase of peak INa density when NaV1.1 was co-expressed with β1WT compared to NaV1.1 alone. However, co-expression of NaV1.1 with β1bWT did not significantly increase INa density compared to NaV1.1 alone (Figure 6B and Table 5). Neither β1WT nor β1bWT caused any significant changes in the voltage dependence of activation (Table 5). Steady-state inactivation of the NaV1.1 current was not altered by β1WT. On the contrary, co-expression of the β1bWT isoform caused a 7.21 mV negative shift of the voltage dependence of inactivation compared to NaV1.1 alone (Figure 6C and Table 5). β1WT markedly decreased the recovery from inactivation time in comparison to NaV1.1 alone. However, this decrease was not observed when NaV1.1 was co-expressed with β1bWT (Figure 6D and Table 5).
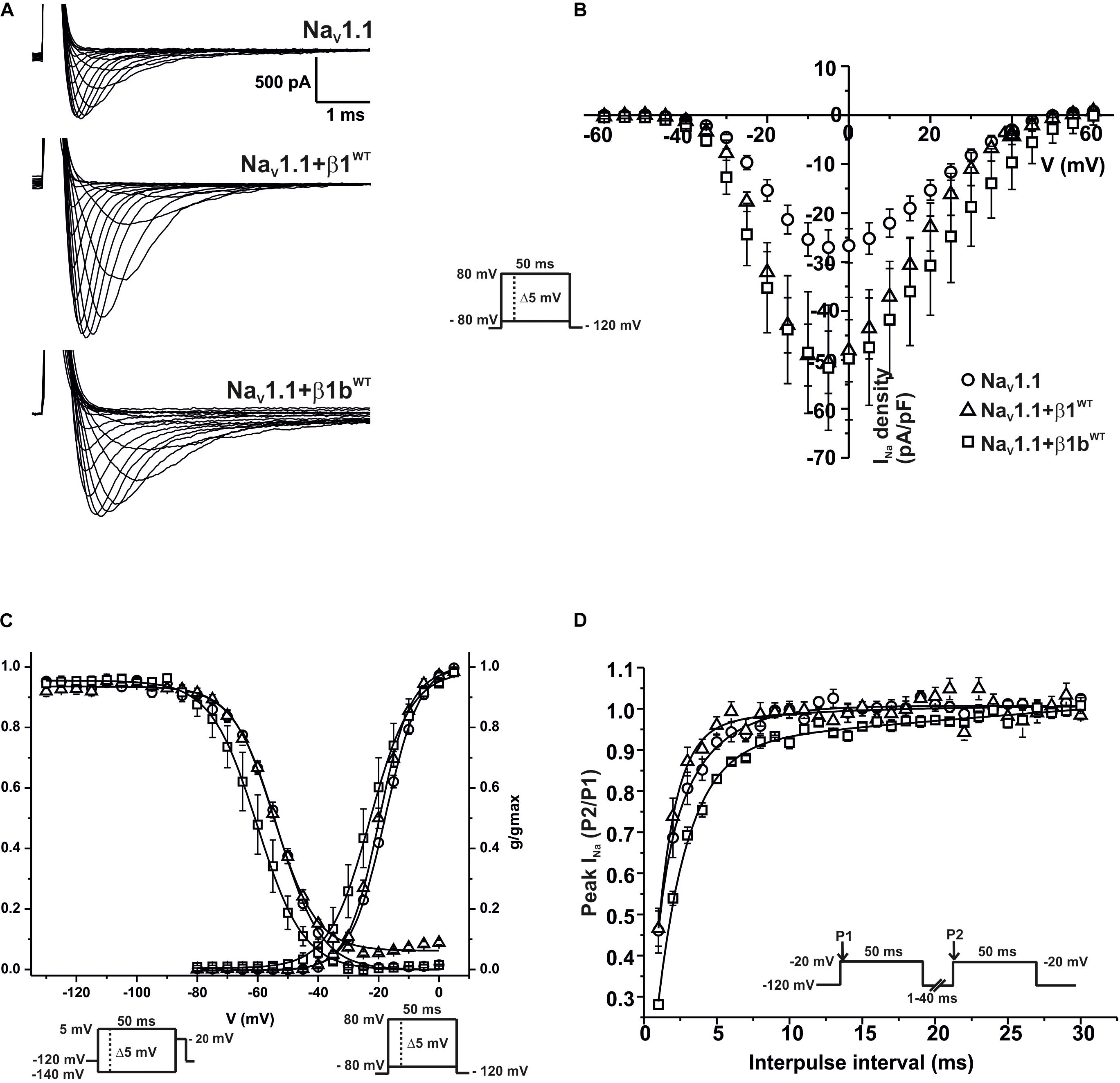
Figure 6. β1WT and β1bWT modulate NaV1.1 current. Biophysical properties of sodium currents measured from HEK-293T cells expressing NaV1.1 alone, or co-expressed with either β1WT or β1bWT. Whole cell currents were elicited by depolarizing potentials as shown in the insets. Circles are used to depict data for NaV1.1 alone, triangles for NaV1.1+β1WT, and squares for NaV1.1+β1bWT. Solid lines in panels (C) and (D) represent the fitted curves. Values are expressed as mean ± SEM. (A) Representative whole-cell Na+ current traces from HEK-293T cells expressing NaV1.1 alone (top), NaV1.1+β1WT (middle), and NaV1.1+β1bWT (bottom). (B) Mean current-voltage relationship. INa amplitude was normalized by the cell capacitance to obtain INa density values. (C) INa steady-state voltage dependence of activation and inactivation plots. (D) Recovery from inactivation curves.
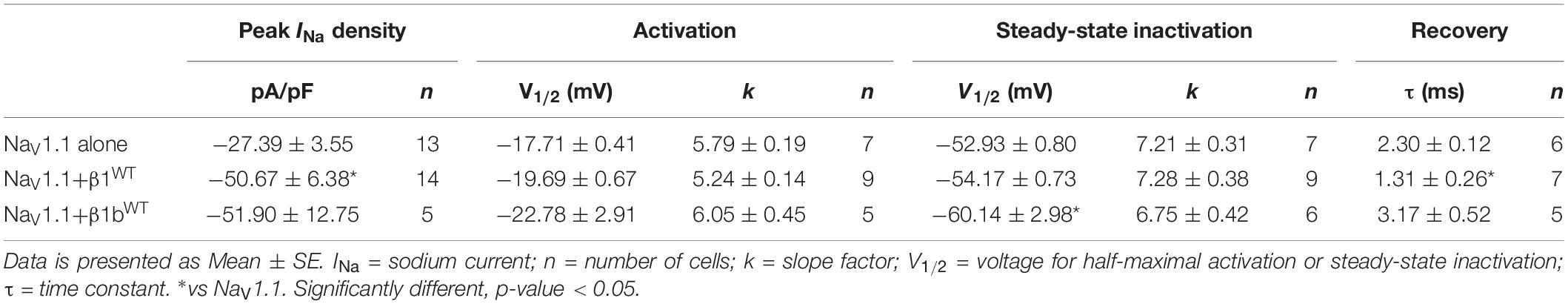
Table 5. Biophysical parameters of NaV1.1 channels alone or cotransfected with β1WT or β1bWT.
To determine whether the β1 mutation identified in the patient had an effect on the neuronal NaV1.1 sodium current, we performed functional characterization in HEK-293T cells expressing NaV1.1+β1D103V compared to those expressing NaV1.1+β1WT. Peak current density measured from cells expressing NaV1.1+β1D103V was 66.9% smaller than that in NaV1.1+β1WT-expressing cells (Figures 7A,B and Table 6). Voltage dependence of activation and steady-state inactivation were not altered by β1D103V (Figure 7C and Table 6). Recovery from inactivation time constants were similar in both experimental conditions (Figure 7D and Table 6).
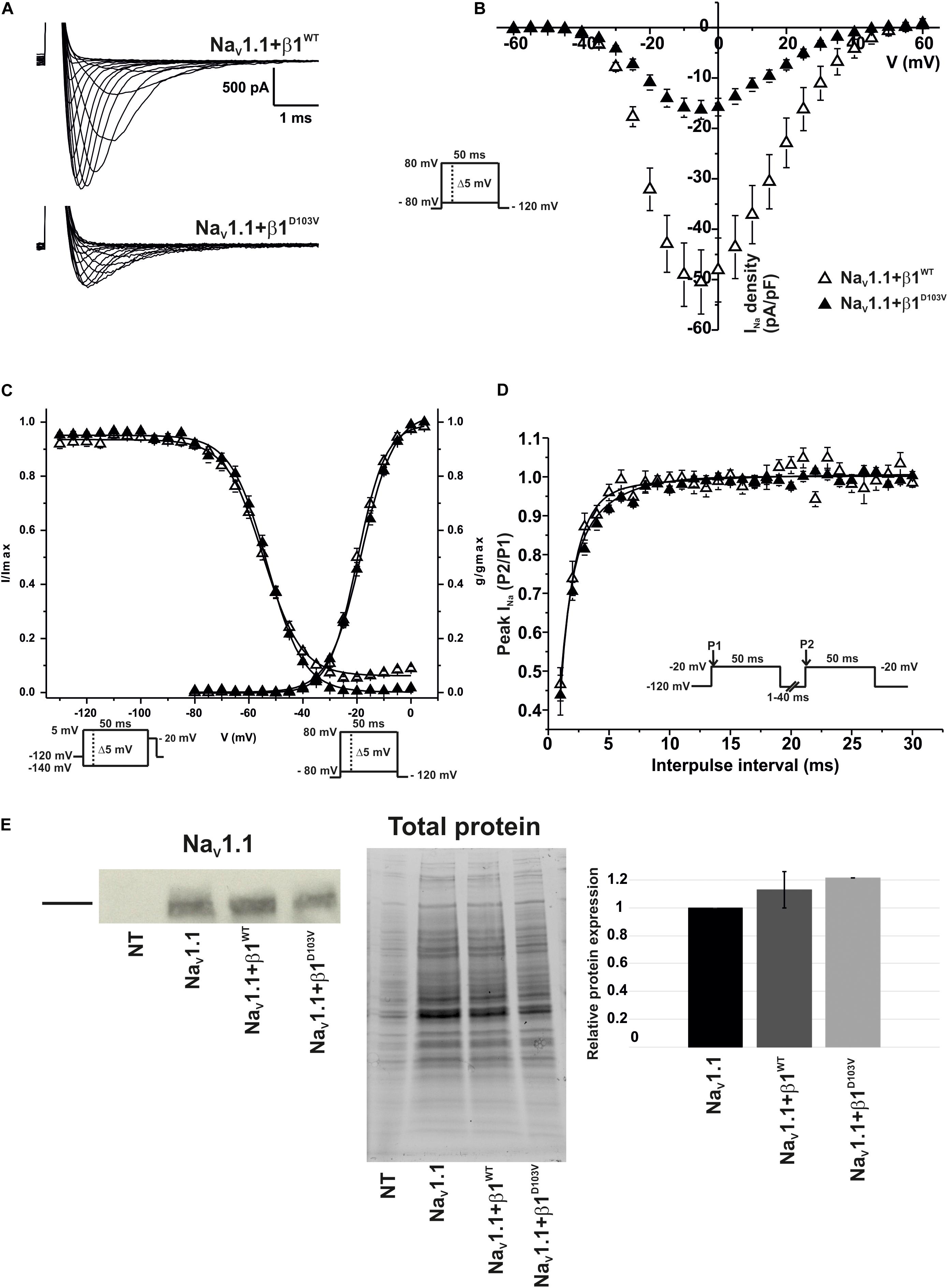
Figure 7. β1D103V significantly decreases peak INa on NaV1.1. Biophysical properties of sodium currents measured from HEK-293T cells expressing NaV1.1 co-expressed with either β1WT or β1D103V. Whole cell currents were elicited by depolarizing potentials as shown in the insets. Open triangles are used to depict data for NaV1.1+β1WT and filled triangles for NaV1.1+β1D103V. Solid lines in panels (C) and (D) represent the fitted curves. Values are expressed as mean ± SEM. (A) Representative whole-cell Na+ current traces from HEK-293T cells expressing NaV1.1+β1WT (top), and NaV1.1+β1D103V (bottom). (B) Mean current-voltage relationship. INa amplitude was normalized by the cell capacitance to obtain INa density values. (C) INa steady-state voltage dependence of activation and inactivation plots. (D) Recovery from inactivation curves. (E) Western blot detection of NaV1.1 (left) and corresponding total protein stain-free gel (middle), from HEK-293T cells transfected with either NaV1.1, NaV1.1+β1WT, or NaV1.1+β1D103V or non- transfected cells (NT). Bar graph on the right depicts the relative protein expression normalized by the NaV1.1 alone (n = 2).
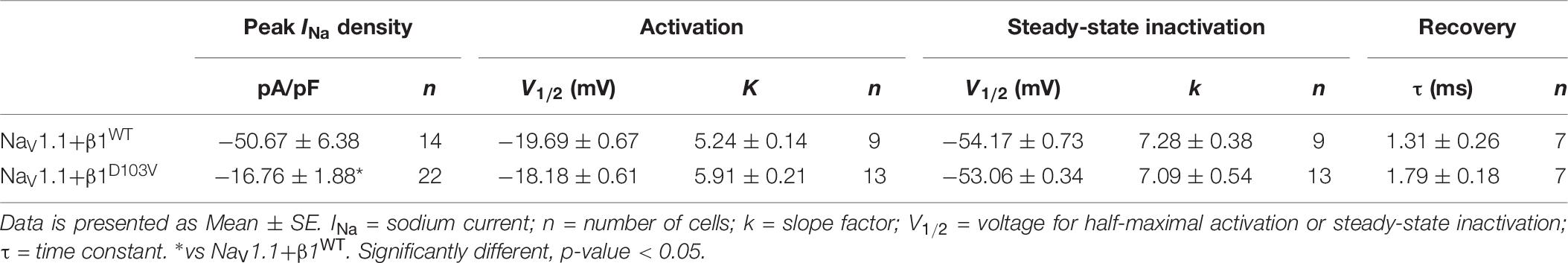
Table 6. Biophysical parameters of NaV1.1 channels cotransfected with β1WT or β1D103V.
Similar to NaV1.5, we did not detect any current in 23 out of 59 cells expressing NaV1.1+β1D103V. However, when cells were transfected with NaV1.1+β1WT, only 4 of 56 cells showed no current. Western blot analysis showed no significant differences in NaV1.1 expression between the three conditions (Figure 7E).
Because the D103V mutation is located in a region shared by both β1 and β1b subunits, we assessed the effect of β1bD103V on NaV1.1 current properties. We co-expressed NaV1.1 with either β1bWT or β1bD103V subunits (Figure 8A). Co-expression of β1bD103V with NaV1.1 did not change INa density compared to NaV1.1+β1bWT (Figure 8B and Table 7). Likewise, the β1bD103V subunit did not significantly change the voltage dependence of activation compared to β1bWT. However, β1bD103V caused a 6.04-mV right shift of the voltage dependence of steady-state inactivation when compared to the wildtype subunit (Figure 8C). This change resulted in a positive shift of the window current of NaV1.1+β1bD103V compared to NaV1.1+β1bWT (Figure 8D and Table 7). β1bD103V significantly reduced the recovery from inactivation time constant compared to NaV1.1+β1bWT (Figure 8E and Table 7).
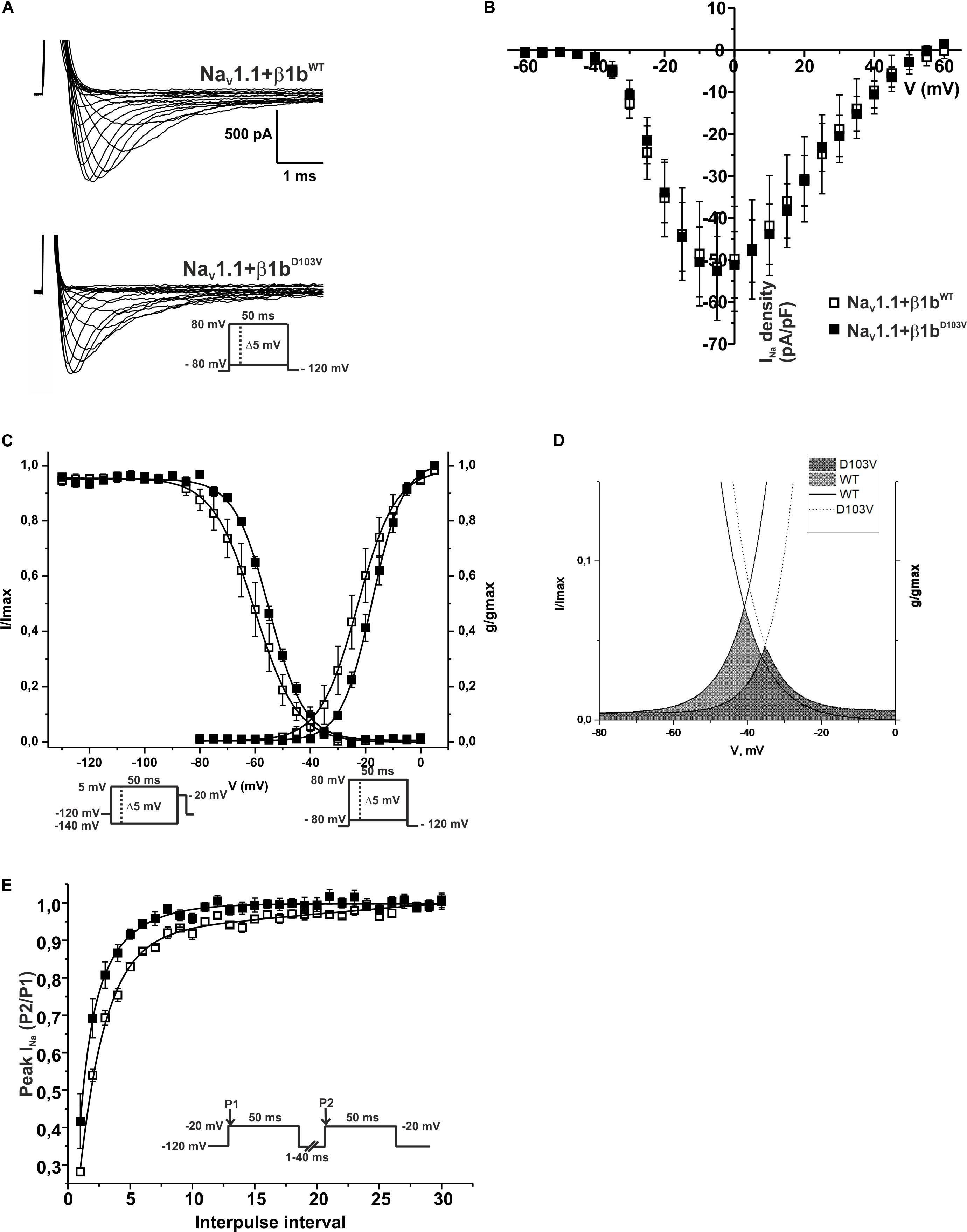
Figure 8. β1bD103V modifies the gating properties of NaV1.1 channel. Open squares are used to depict data for NaV1.1+β1bWT and filled squares for NaV1.1+β1bD103V. Solid lines represent the fitted curves. Values are expressed as mean ± SEM. Sodium currents were obtained with voltage clamp step protocols as shown in the insets. (A) Representative whole-cell Na+ current traces from HEK-293T cells expressing NaV1.1+β1bWT (top), and NaV1.1+β1bD103V (bottom). (B) Mean current-voltage relationship. INa amplitude was normalized by the cell capacitance to obtain INa density values. (C) INa steady-state voltage dependence of activation (right) and inactivation (left). (D) Window region bounded by the steady state activation and inactivation voltage dependent curves (NaV1.1+β1bWT solid gray and NaV1.1+β1bD103V diagonal pattern). (E) Recovery from inactivation curves.
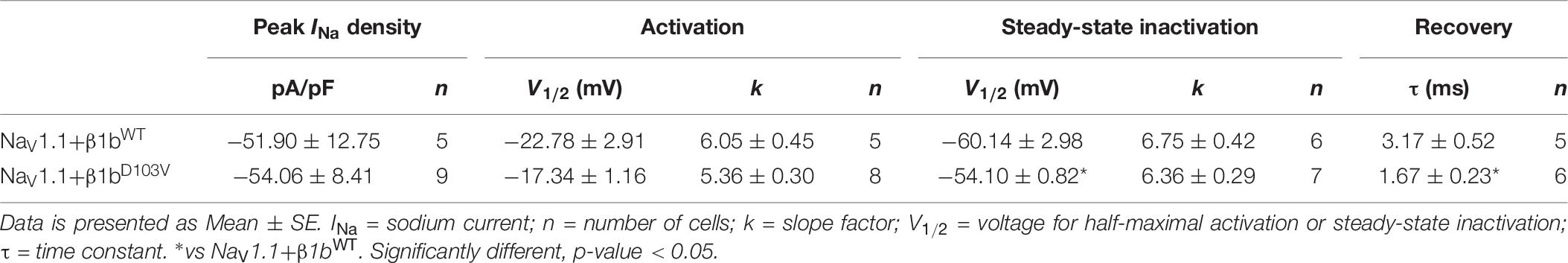
Table 7. Biophysical parameters of NaV1.1 cotransfected with β1bWT or β1bD103V.
We present functional characterization of an SCN1B missense variant (c.308A>T; p.D103V) affecting both β1 and β1b regulatory subunits. This mutation was found in a newborn with heart and brain pathologies. The mutation was previously reported as pathogenic by Eldomery et al. (2017), potentially causing cardiomyopathies and intellectual disability among other phenotypes. The goal of our study was to determine whether the mutant β1D103V and β1bD103V subunits affected NaV1.5 and NaV1.1 sodium currents.
HEK cells expressing NaV1.5+β1WT showed increased INa, consistent with Watanabe et al. (2008) and Qu et al. (1995). Also, we observed a shift of the voltage dependence of activation toward hyperpolarizing potentials in agreement with Ko et al. (2005) and Yuan et al. (2014). Lastly, we found a reduced recovery from inactivation time constant, consistent with Fahmi et al. (2001).
Mutations in the β1 subunit have been implicated in various cardiac arrhythmias, such as Brugada syndrome, atrial fibrillation, sudden infant death syndrome (SIDS), long QT syndrome, and cardiac conduction defect (Audenaert et al., 2003; Watanabe et al., 2008; Tan et al., 2010; Ricci et al., 2014; O’Malley and Isom, 2016). We show that the β1D103V subunit does not increase INa as β1WT does. In addition, the mutant subunit shifted the voltage dependence of activation toward more positive potentials, suggesting a physical interaction between β1D103V and the alpha subunit. This interaction, however, appears not to disrupt NaV1.5 trafficking to the plasma membrane. Our data does not support this interpretation as relative plasma membrane protein levels are similar in both β1WT and β1D103V conditions. Thus the mutant β1 is likely to exert its effect over NaV1.5 is directly on the channel electrical properties. Taking into account that the β1 subunit is normally present in cardiac cells, this difference between β1WT and β1D103V would be considered a loss-of-function of the channel, consistent with the progressive atrial standstill and cardiac conduction disorder observed in the patient. It is necessary to point out that the patient is heterozygous for the β1 mutation. Thus, whether the mutation has a dominant negative effect remains to be determined. Further experiments in either patient-specific iPS derived cardiomyocytes or heterocygous heterologous expression would be needed to further explore this possibility.
The effect of the β1bWT subunit on NaV1.5 current has not been well-studied. However, some groups have reported presence of the β1bWT subunit in human and rat adult hearts, and expression levels of β1b in the atria and ventricle are greater than those of β1 (Kazen-Gillespie et al., 2000; Yuan et al., 2014). We observed an increase in NaV1.5 current density in the presence of β1bWT, consistent with previous works, as well as a decrease in the recovery from inactivation time constant. Although NaV1.5 current was diminished by the presence of the β1bD103V subunit, acceleration in the recovery from inactivation was still observed with β1bD103V. This suggests that the interaction between NaV1.5 and the mutant β1b subunit is still present.
Our results for neuronal NaV1.1 current show that β1WT increases NaV1.1 INa density, in agreement with findings from Isom et al. (1992). In addition, we detected faster recovery from inactivation, as previously reported by Aman et al. (2009) and Barela (2006). Further, we show that the mutant β1 subunit strongly decreases NaV1.1 sodium current density compared to the wildtype β1 subunit, which represents a loss-of-function of the channel, in agreement with Meadows et al. (2002).
The modulatory effect of the β1b subunit on NaV1.1 is poorly studied and controversial. Some groups propose that β1b expression predominates in embryonic development and early life and is thus essential for brain development (Kazen-Gillespie et al., 2000; Patino et al., 2011; O’Malley and Isom, 2016). Patino et al. (2011) studied an epilepsy-related mutation that only affects the β1b subunit. Although their co-immunoprecipitation studies did not detect an association between β1b and NaV1.1 or NaV1.3 channels, they showed that β1b modulates the NaV1.3 sodium current in heterologous systems. Our study shows that the β1bWT subunit modulates voltage dependence of inactivation by shifting the curve to more negative potentials.
While β1bD103V does not have any effects on NaV1.1 current density, we found that it shifts the voltage dependence of inactivation in the depolarizing direction, thus causing the channel to be available for activation in a larger voltage range. In addition, β1bD103V strongly reduces the recovery from inactivation time constant compared to β1bWT. Thus, mutant channels are ready to be activated earlier than those without mutation. Assuming that β1b is normally present and interacts with NaV1.1 in native tissue, both effects would contribute to gain-of-function of the channel.
β1 subunit mutations that induce a gain-of-function of NaV1.1 have also been reported to cause brain dysfunction (Meadows et al., 2002; Kruger et al., 2016). Thus, it is possible that β1bD103V causes neuron hyperexcitability, destabilizing normal neuronal behavior of the brain. Also, given the importance of this subunit to brain development (O’Malley and Isom, 2016), this mutation may impair brain formation at embryonic stages, thus provoking the brain phenotype at an early age, and may be involved in the patient’s polymicrogyria.
Our results showed that the modulatory effects of β1D103V and β1bD103V on NaV1.1 INa are different, even opposing each other. Considering that β1b is predominantly expressed during embryonic development, we expect that the gain-of-function effect of β1bD103V is more important during that stage. A loss-of-function caused by β1D103V would be more important later in development. Since the p.D103V mutation affects both β1 and β1b isoforms, it could differentially compromise neuronal electrical activity during development.
In conclusion, our results strongly suggest that the SCN1B_c.308A>T mutation contributes to the patient’s phenotype. We show that the β1D103V mutant channels cause a loss-of-function of the cardiac-type sodium current, which could explain the clinical presentation of progressive atrial standstill, intra-atrial reentrant tachycardia, and cardiac conduction disorder in the child.
We surmise that the SCN1B variant could contribute to the patient’s brain phenotype at two different stages. During development and early life, when the β1b isoform is predominant, β1bD103V leads to a gain-of-function of the channel. During adulthood, when the β1 subunit is predominantly expressed, β1D103V produces loss-of-function of the sodium current. Both effects could contribute to the cognitive and motor deficits observed in the patient.
This family was part of a sequencing re-analysis project from which the SCN1B variant along with variants in POLR1C were flagged as the best candidates that potentially contribute to phenotypes manifested by both the proband and his elder sister. She had a fetal diagnosis of bradycardia and subsequent postnatal finding of complete AV block, leading to early neonatal death from multi-organ failure (Eldomery et al., 2017).
The proband’s father is heterozygous for the c.308A>T variant in SCN1B and for the c.88C>T variant in POLR1C, and his mother is heterozygous for the c.614delG variant in POLR1C. Both mother and father are asymptomatic. Incomplete penetrance is a common feature of channelopathies, and the idea of monogenetic disease has changed in recent years (Symonds and Zuberi, 2018). However, neither the SCN1B variant nor POLR1C variants found in the child could be the sole cause of his complex clinical picture.
POLR1C has been associated with autosomal recessive hypomyelinating leukodystrophy (Thiffault et al., 2015). This gene also has been described to cause recessive Treacher Collins syndrome 3 (OMIM #248390). Our proband did not show clinical manifestations of either of these conditions (Eldomery et al., 2017). However, the patient exhibited several neurological phenotypes consistent with POLR1C mutations, including hypomyelination, ataxia, and nystagmus, but the patient’s other clinical features are not described in POLR1C-related cases. Thus, it is possible that electrical disturbances caused by the SCN1B variant become more severe in the context of POLR1C mutations. Also, three-dimensional voltage mapping at baseline rhythm revealed extensive scarring of the right atrium and coronary sinus. Thus, electrical dysfunction potentially caused by the mutant β1 subunit could be aggravated by a damaged tissue substrate.
In summary, although the overall pathophysiology of the patient is complex, the NaV1.5 loss of function caused by the mutant β1 subunit D103V largely explains the clinical manifestations related to the patient’s heart dysfunction. In addition, loss of function of NaV1.1 caused by the β1 and β1b mutant subunits might aggravate a brain condition caused by the combination of the two POLR1C mutations.
The datasets generated for this study are available on request to the corresponding author.
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
RM-M performed most of the experiments and analysis, made the figures, drafted the manuscript and contributed to the final version. ES revised all versions of the manuscript. HR performed the initial experiments and participated in the planning of the project. DC performed the cell surface protein biotinylation experiments. MP provided and supervised the neurology clinical aspects of the manuscript. CS provided and supervised the clinical cardiological aspects of the manuscript. MW provided the genetic data and participated in the planning of the project. GP and FS directed the project, supervised the experiments, and revised the data analysis and all versions of the manuscript. RB participated in the initial planning of the project and revised the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Fundació La Marató de TV3 (FS: 20153910), Obra social “la Caixa,” Centro Nacional de Investigaciones Cardiovasculares (RB: CNIC-03-2008), Instituto de Salud Carlos III (RB: FIS-PI08/1800 and Fondo Europeo de Desarrollo Regional) and Universitat de Girona (GP: MPCUdG2016). ES, RB, GP, and FS are members of the CIBERCV, an initiative of the Instituto de Salud Carlos III, Spanish Ministry of Economy and Competitiveness.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aman, T. K., Grieco-Calub, T. M., Chen, C., Rusconi, R., Slat, E. A., Isom, L. L., et al. (2009). Regulation of persistent Na current by interactions between B subunits of voltage-gated na channels. J. Neurosci. 29, 2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009
Audenaert, D., Claes, L., Ceulemans, B., Löfgren, A., Van Broeckhoven, C., and De Jonghe, P. (2003). A deletion in SCN1B is associated with febrile seizures and early-onset absence epilepsy. Neurology 61, 854–856. doi: 10.1212/01.WNL.0000080362.55784.1C
Barela, A. J. (2006). An epilepsy mutation in the sodium channel SCN1A that decreases channel excitability. J. Neurosci. 26, 2714–2723. doi: 10.1523/JNEUROSCI.2977-05.2006
Brackenbury, W. J., and Isom, L. L. (2011). Na+ channel B subunits: overachievers of the ion channel family. Front. Pharmacol. 2:53. doi: 10.3389/fphar.2011.00053
Catterall, W. A. (2000). From ionic currents to molecular review mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25. doi: 10.1016/S0896-6273(00)81133-2
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409. doi: 10.1124/pr.57.4.4.and
Chen, C. (2004). Mice lacking sodium channel 1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J. Neurosci. 24, 4030–4042. doi: 10.1523/JNEUROSCI.4139-03.2004
Darras, N., Ha, T. K., Rego, S., Barroso, E., Slavotinek, A. M., and Cilio, M. R. (2019). Developmental and epileptic encephalopathy in two siblings with a novel, homozygous missense variant in SCN1B. Am. J. Med. Genet. 179A, 2190–2195. doi: 10.1002/ajmg.a.61344
Eldomery, M. K., Coban-Akdemir, Z., Harel, T., Rosenfeld, J. A., Gambin, T., Stray-Pedersen, A., et al. (2017). Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 9, 1–15. doi: 10.1186/s13073-017-0412-6
Fahmi, A. I., Patel, M., Stevens, E. B., Fowden, A. L., John, J. E., Lee, K., et al. (2001). The sodium channel β-subunits SCN3b modulates the kinetics of SCN5a and is expressed heterogeneously in sheep heart. J. Physiol. 537, 693–700. doi: 10.1113/jphysiol.2001.012691
Hu, D., Barajas-Martínez, H., Medeiros-Domingo, A., Crotti, L., Veltmann, C., Schimpf, R., et al. (2012). A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Nav1.5 and Kv4.3 channel currents. Heart Rhythm 9, 760–769. doi: 10.1016/j.hrthm.2011.12.006
Isom, L. L., De Jongh, K. S., Patton, D. E., Reber, B. F., Offord, J., Charbonneau, H., et al. (1992). Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256, 839–842. doi: 10.1126/science.256.5058.839
Kaplan, D. I., Isom, L. L., and Petrou, S. (2016). Role of sodium channels in epilepsy. Cold Spring Harb. Perspect. Med. 6, 1–18. doi: 10.1101/cshperspect.a022814
Kazen-Gillespie, K. A., Ragsdale, D. S., D’Andreall, M. R., Mattei, L. N., Rogers, K. E., and Isom, L. L. (2000). Cloning, localization, and functional expression of sodium channel β1A subunits. J. Biol. Chem. 275, 1079–1088. doi: 10.1074/jbc.275.2.1079
Ko, S. H., Lenkowski, P. W., Lee, H. C., Mounsey, J. P., and Patel, M. K. (2005). Modulation of Nav1.5 by β1- and β3-subunit co-expression in mammalian cells. Pflugers Arch. Eur. J. Physiol. 449, 403–412. doi: 10.1007/s00424-004-1348-4
Kruger, L. C., O’Malley, H. A., Hull, J. M., Kleeman, A., Patino, G. A., and Isom, L. L. (2016). B1-C121W is down but not out: epilepsy-associated Scn1b-C121W results in a deleterious gain-of-function. J. Neurosci. 36, 6213–6224. doi: 10.1523/JNEUROSCI.0405-16.2016
Lin, X., O’Malley, H., Chen, C., Auerbach, D., Foster, M., Shekhar, A., et al. (2015). Scn1b deletion leads to increased tetrodotoxin-sensitive sodium current, altered intracellular calcium homeostasis and arrhythmias in murine hearts. J. Physiol. 593, 1389–1407. doi: 10.1113/jphysiol.2014.277699
Lopez-Santiago, L. F., Meadows, L. S., Ernst, S. J., Chen, C., Malhotra, J. D., McEwen, D. P., et al. (2007). Sodium channel Scn1b null mice exhibit prolonged QT and RR intervals. J. Mol. Cell. Cardiol. 43, 636–647. doi: 10.1016/j.yjmcc.2007.07.062
McCormick, K. A., Srinivasan, J., White, K., and Scheuer, T. (1999). The extracellular domain of the b1 subunit is both necessary and sufficient for b1- like modulation of sodium channel gating. J. Biol. Chem 274, 32638–32646. doi: 10.1074/jbc.274.46.32638
Meadows, L. S., Malhotra, J., Loukas, A., Thyagarajan, V., Kazen-Gillespie, K. A., Koopman, M. C., et al. (2002). Functional and biochemical analysis of a sodium channel B1 subunit mutation responsible for generalized epilepsy with febrile seizures plus type 1. J. Neurosci. 22, 10699–10709. doi: 10.1523/jneurosci.22-24-10699.2002
Olesen, M. S., Holst, A. G., Svendsen, J. H., Haunsø, S., and Tfelt-Hansen, J. (2012). SCN1Bb R214Q found in 3 patients: 1 With Brugada syndrome and 2 with lone atrial fibrillation. Heart Rhythm 9, 770–773. doi: 10.1016/j.hrthm.2011.12.005
O’Malley, H. A., and Isom, L. L. (2016). Sodium channel β subunits: emerging targets in channelopathies. Annu. Rev. Physiol. 77, 481–504. doi: 10.1146/annurev-physiol-021014-071846.Sodium
Patino, G. A., Brackenbury, W. J., Bao, Y., Lopez-Santiago, L. F., O’Malley, H. A., Chen, C., et al. (2011). Voltage-gated Na+ Channel B1B: a secreted cell adhesion molecule involved in human epilepsy. J. Neurosci. 31, 14577–14591. doi: 10.1523/JNEUROSCI.0361-11.2011
Patino, G. A., and Isom, L. L. (2010). Electrophysiology and beyond: multiple roles of Na+channel β subunits in development and disease. Neurosci. Lett. 486, 53–59. doi: 10.1016/j.neulet.2010.06.050
Peeters, U., Scornik, F., Riuró, H., Pérez, G., Komurcu-Bayrak, E., Van Malderen, S., et al. (2015). Contribution of cardiac sodium channel β-subunit variants to brugada syndrome. Circ. J. 79, 2118–2129. doi: 10.1253/circj.CJ-15-0164
Qu, Y., Isom, L. L., Westenbroek, R. E., Rogers, J. C., Tanada, T. N., McCormick, K. A., et al. (1995). Modulation of cardiac Na+ channel expression in Xenopus oocytes by beta 1 subunits. J. Biol. Chem. 270, 25696–25701. doi: 10.1074/jbc.270.43.25696
Ricci, M. T., Menegon, S., Vatrano, S., Mandrile, G., Cerrato, N., Carvalho, P., et al. (2014). SCN1B gene variants in Brugada syndrome: a study of 145 SCN5A-negative patients. Sci. Rep. 4, 1–6. doi: 10.1038/srep06470
Riuró, H., Campuzano, O., Arbelo, E., Iglesias, A., Batlle, M., Pérez-Villa, F., et al. (2014). A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying long QT syndrome. Heart Rhythm 11, 1202–1209. doi: 10.1016/j.hrthm.2014.03.044
Scheffer, I. E., Harkin, L. A., Grinton, B. E., Dibbens, L. M., Turner, S. J., Zielinski, M. A., et al. (2007). Temporal lobe epilepsy and GEFS+ phenotypes associated with SCN1B mutations. Brain 130, 100–109. doi: 10.1093/brain/awl272
Spampanato, J. (2004). A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for subunit interaction. J. Neurosci. 24, 10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004
Symonds, J. D., and Zuberi, S. M. (2018). Genetics update: monogenetics, polygene disorders and the quest for modifying genes. Neuropharmacology 132, 3–19. doi: 10.1016/j.neuropharm.2017.10.013
Tan, B. H., Pundi, K. N., Van Norstrand, D. W., Valdivia, C. R., Tester, D. J., Medeiros-Domingo, A., et al. (2010). Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm 7, 771–778. doi: 10.1016/j.hrthm.2010.01.032
Tarradas, A., Selga, E., Beltran-Alvarez, P., Pérez-Serra, A., Riuró, H., Picó, F., et al. (2013). A novel missense mutation, I890T, in the pore region of cardiac sodium channel causes brugada syndrome. PLoS One 8:e0053220. doi: 10.1371/journal.pone.0053220
Taylor, S. C., Berkelman, T., Yadav, G., and Hammond, M. (2013). A defined methodology for reliable quantification of western blot data. Mol. Biotechnol. 55, 217–226. doi: 10.1007/s12033-013-9672-6
Thiffault, I., Wolf, N. I., Forget, D., Guerrero, K., Tran, L. T., Choquet, K., et al. (2015). Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat. Commun. 6, 1–9. doi: 10.1038/ncomms8623
Wallace, R. H., Wang, D. W., Singh, R., Scheffer, I. E., George, A. L., Phillips, H. A., et al. (1998). Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat. Genet. 19, 366–370. doi: 10.1038/1252
Watanabe, H., Koopmann, T. T., Scouarnec, S., Le Yang, T., Ingram, C. R., Schott, J., et al. (2008). Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. Structure 118, 268–275. doi: 10.1172/JCI33891.2260
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4:207. doi: 10.1186/gb-2003-4-3-207
Yuan, L., Koivumäki, J. T., Liang, B., Lorentzen, L. G., Tang, C., Andersen, M. N., et al. (2014). Investigations of the Navβ1b sodium channel subunit in human ventricle; functional characterization of the H162P Brugada syndrome mutant. Am. J. Physiol. Heart Circ. Physiol. 306, 1204–1212. doi: 10.1152/ajpheart.00405.2013
Keywords: NaV1.5, NaV1.1, NaVβ1, NaVβ1b, cardiac arrhythmia, brain hyperexcitability
Citation: Martinez-Moreno R, Selga E, Riuró H, Carreras D, Parnes M, Srinivasan C, Wangler MF, Pérez GJ, Scornik FS and Brugada R (2020) An SCN1B Variant Affects Both Cardiac-Type (NaV1.5) and Brain-Type (NaV1.1) Sodium Currents and Contributes to Complex Concomitant Brain and Cardiac Disorders. Front. Cell Dev. Biol. 8:528742. doi: 10.3389/fcell.2020.528742
Received: 21 January 2020; Accepted: 21 August 2020;
Published: 29 September 2020.
Edited by:
Paola Rizzo, University of Ferrara, ItalyReviewed by:
Thomas Hund, The Ohio State University, United StatesCopyright © 2020 Martinez-Moreno, Selga, Riuró, Carreras, Parnes, Srinivasan, Wangler, Pérez, Scornik and Brugada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillermo J. Pérez, Z3VpbGxlcm1vLnBlcmV6QHVkZy5lZHU=; Fabiana S. Scornik, ZmFiaWFuYXNpbHZpYS5zY29ybmlrQHVkZy5lZHU=; Ramon Brugada, cmJydWdhZGFAaWRpYmdpLm9yZw==
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.