 Litian Zhang
Litian Zhang Lukman O. Afolabi
Lukman O. Afolabi Xiaochun Wan
Xiaochun Wan Yang Li
Yang Li Liang Chen
Liang Chen- 1Shenzhen Laboratory of Tumor Cell Biology, Center for Antibody Drug Development, Institute of Biomedicine and Biotechnology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China
- 2Department of Hematology, Zhujiang Hospital, Southern Medical University, Guangzhou, China
- 3University of Chinese Academy of Sciences, Beijing, China
Protein quality control (PQC) is pivotal for eukaryotic cells to eliminate misfolded proteins and maintain cellular homeostasis. A decreased or increased capacity of PQC is associated with various diseases, e.g., neurodegenerative diseases and cancers. Recently, increasing evidences have suggested that tripartite motif-containing family proteins (TRIMs) are the key players in PQC regulation. Most TRIMs are E3 ubiquitin ligases, such as TRIM11/19/25, which, through the ubiquitination modifications, can contribute to effectively remove the cellular misfolded proteins or protein aggregates via the UPS pathway. In this review, we summarized the participation of TRIM members in misfolded protein elimination through distinct pathways, including the ubiquitin–proteasome system (UPS), autophagy system, and ER-associated degradation (ERAD).
Introduction
Proteins are the basic components of cells and are involved in a broad array of cellular processes. As part of the most abundant macromolecules, the cells inevitably need to put a huge strain on the protein production and maintenance of their natural conformations (Dobson, 2003). When cells are in certain physiological states or are exposed to various stress conditions, this leads to a condition in which the correct protein conformations are lost, leading to protein misfolding (Horwich and Weissman, 1997; Balch et al., 2008; Powers and Balch, 2008). Failure to timely remove the misfolded proteins can lead to the generation of proteotoxic stress (Bucciantini et al., 2002; Costanzo and Zurzolo, 2013; Soto and Pritzkow, 2018). Thus, maintaining cellular proteostasis is a requisite for cells to perform their basal function. To achieve this, the cell employs a fairly complex protein quality control (PQC) system that is critical to sequestrate, refold, and degrade any unexpected, accumulated misfolded proteins (Balchin et al., 2016; Sontag et al., 2017). The endoplasmic reticulum (ER) is an important cellular organelle that plays critical roles in the production, processing, and transport of proteins and lipids. It is also the organelle responsible for the maturation of roughly one-half proteins, in which aberrant proteins could be generated particularly under various physiological stress conditions (Wiseman et al., 2007; Wang and Kaufman, 2016). In ER, PQC is also known as ER quality control (ERQC) (Kim et al., 2015), for which nonnative conformational proteins can be refolded and modified following activation of the unfolded protein response (UPR) (Ron and Walter, 2007; Hetz et al., 2015) or eliminated via ER-associated degradation (ERAD) (Hiller et al., 1996). Studies have shown the selective degradation of harmful or exhausted organelles via a specific type of autophagic turnover such as ER-phagy. When the ER becomes overwhelmed and stressed, its fragmented components along with the aberrant protein are delivered to the lysosome where they are degraded via ER-phagy (Grumati et al., 2018).
The cell’s PQC system consists of two separate but collaborated parts: (I) molecular chaperone system, which is constituted by various types of heat shock proteins (HSPs) that function to release and unfold individual misfolded proteins from aggregates (Hartl and Hayer-Hartl, 2002; Sharma et al., 2008; Kim et al., 2013); (II) the degradation system, which relies on the ubiquitin–proteasome system (UPS) and autophagy pathways (Goldberg, 2003; Finley, 2009; Wani et al., 2015). In particular, molecular chaperones – a class of protein family that are evolutionarily conserved and are widely distributed in various organisms – are essential for cell survival, including HSP60, HSP70, HSP100, small HSP, and calnexin (Richter et al., 2010). When a protein is misfolded, molecular chaperones assist in the correct folding of the misfolded protein by reversibly binding to stabilize the unstable intermediates, followed by its release and refolding to its native conformation.
Meanwhile, erroneous protein aggregates that cannot be refolded can be disaggregated by chaperones as well (Saibil, 2013). Molecular chaperone system can also be overstressed, and in such condition, it directs the inundated misfolded proteins or protein aggregates to cellular clearance pathways via the ubiquitin–proteasome pathway or sequestration in autophagosomes (Kaganovich et al., 2008). The UPS and autophagy systems represents two distinct, selective, and well-regulated cellular degradative pathways, with their respective subcellular localization, mechanisms, machinery, and degradative substrates (Mishra et al., 2018). Emerging evidences have shown that these two systems have cross-talk through ubiquitination (Varshavsky, 2017; Goodier et al., 2020), implying that a complementary and synergistic function of the UPS and autophagy systems may exist (Korolchuk et al., 2009; Kwon and Ciechanover, 2017). In addition, these pathways – alone or in cooperation with each other – orchestrate the entire intracellular protein degradation (Wong and Cuervo, 2010; Chhangani et al., 2014).
Ubiquitination is accomplished by three enzymatic steps catalyzed by (1) ubiquitin-activating enzymes (E1s), (2) ubiquitin-conjugating enzymes (E2s), and (3) ubiquitin ligases (E3s). However, the specificity and efficiency of this system (protein ubiquitylation) are largely determined by the E3 ubiquitin ligases that recognize specific substrates (Zheng and Shabek, 2017). Misfolded proteins can be degraded upon the covalent attachment of ubiquitin. Ubiquitination can be either monoubiquitination (addition of a single ubiquitin molecule) or polyubiquitination (addition of a chain of ubiquitin molecules), and the fate of ubiquitinated substrates is determined by the position of the lysine by which polyubiquitination is mediated through K11, K48, K63, etc. (Hjerpe and Rodriguez, 2008; Xu et al., 2009; Sadowski et al., 2012).
For example, the K48-polyubiquitinated substrates are prone to be eliminated by UPS (Grice and Nathan, 2016), while the K63-polyubiquitinated or monoubiquitinated substrates undergo elimination by autophagy (Sun et al., 2018). Hence, the structural complexity of distinct polyubiquitin chains is sufficient to maintain the selectivity and specificity of the UPS and autophagy for each substrate (Alfano et al., 2016). This also suggests that substrates can be recognized through polyubiquitin chains of different topologies, providing degradation signals for distinct protein degradation pathways (Ohtake et al., 2016). Therefore, researches focused on ubiquitin-related enzymes have gained much attention.
The E3 ligases have a large number and has been extensively studied compared with a small number of E1s and E2s (Deshaies and Joazeiro, 2009; Berndsen and Wolberger, 2014; Buetow and Huang, 2016). TRIMs belong to E3s and over 70 members of the TRIMs have been identified in humans and mice (Hatakeyama, 2011). Most of the TRIMs consist of a highly conserved tripartite motif at N-terminus: a RING domain, one or two B-box domain, and a coiled-coil domain (Meroni and Diez-Roux, 2005; James et al., 2007; Li et al., 2014). The RING domain exhibits ubiquitin E3 ligase activity, yet there is still a limited number of TRIMs that are RING-deficient proteins. TRIM proteins can generally form homopolymers and heteropolymers with each other through their coiled-coil domain. The B-box domains are characterized as a universal domain in TRIMs, while their C-terminal domains provide TRIMs diversities. Based on their domain organization, TRIMs are categorized into 11 distinct subgroups (C-I to C-XI) (Figure 1).
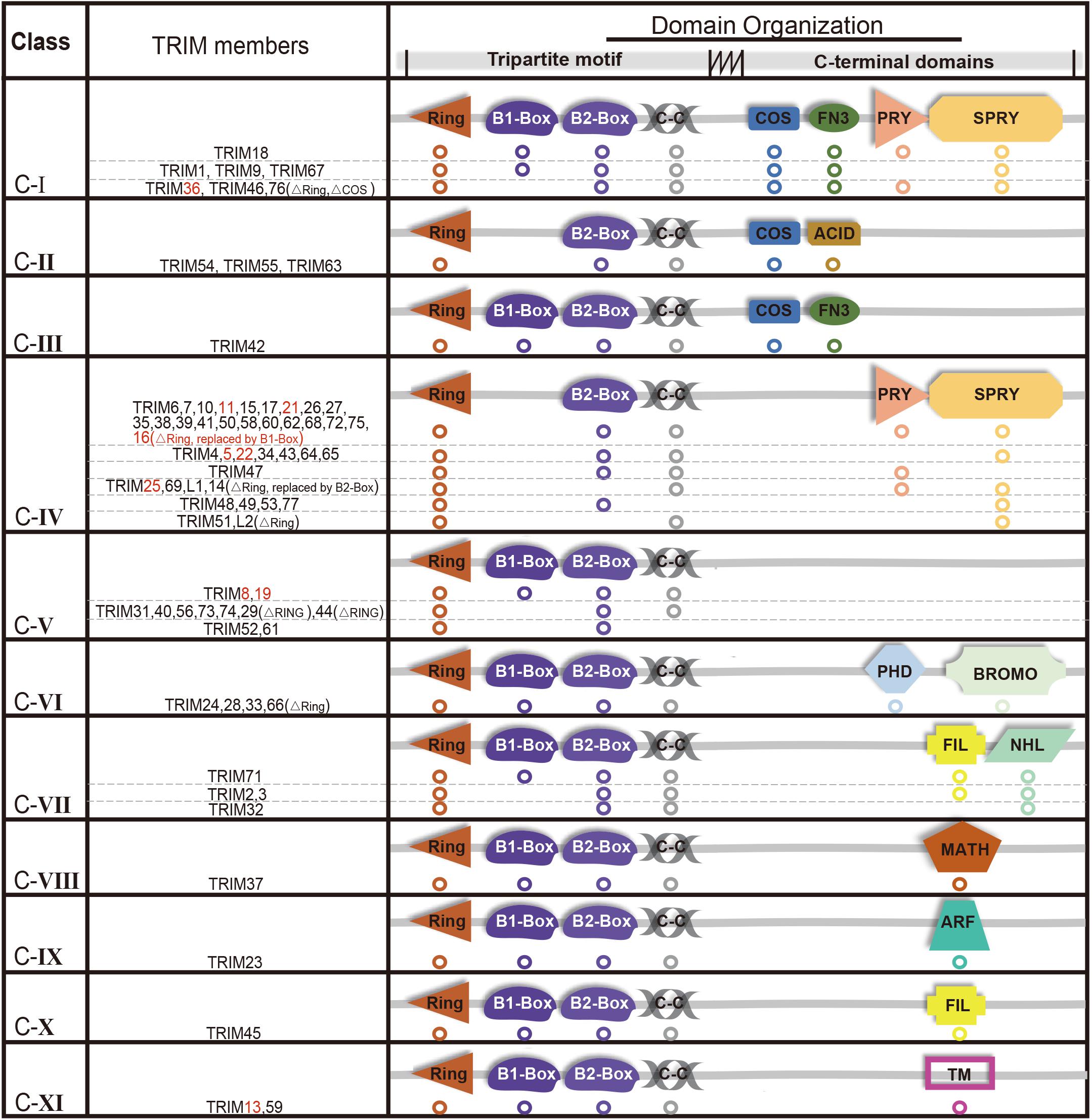
Figure 1. Structure and classification of TRIMs (those mediating degradation of misfolded proteins are marked in red, which are also elaborated in this review). Based on the secondary domain organization, TRIMs are categorized into 11 distinct subgroups (C-I to C-XI). Among 10 TRIMs that are summarized in this review, over one half belongs to C-IV, and sporadic ones to C-I, C-V, or C-XI, respectively. The majority of TRIMS has a highly conserved tripartite motif (RBCC) at N-terminus, while a small number are still missing RING domain. The abbreviations of C-terminal domains are listed as follows: C-terminal subgroup One Signature domain (COS), fibronectin type III repeat domain (FN3), PRY domain, SPRY domain, acid-rich region (ACID), filamin-type IG domain (FIL), NHL domain, PHD domain, bromodomain (BROMO), Meprin and TRAF-homology domain (MATH), ADP-ribosylation factor family domain (ARF), and transmembrane region (TM). Besides, “ΔRING” means the RING domain is absent.
Tripartite motif-containing family proteins play significant roles in various physiological or pathological conditions by acting as ubiquitin E3 ligases, such as cell proliferation and development, DNA damage and repair, neurodegenerative disease, innate immune response, and carcinogenesis (Nisole et al., 2005; Ozato et al., 2008; Hatakeyama, 2017). The mechanisms of TRIMs in misfolded protein clearance remain mysterious, even though it is likely a key function in many of their biological effects. Our group has demonstrated that several TRIM members, including TRIM11/5/25, are crucial for eliminating misfolded proteins (Chen et al., 2017, 2018; Liu et al., 2020). TRIM11 mediates the degradation of misfolded proteins and protein aggregates in the nucleus and cytoplasm through regulating UPS. TRIM5 may mediate protein aggregate degradation through autophagy, and TRIM25 mediates the degradation of misfolded proteins in the ER through ERAD. Other investigators discovered that TRIM13, an ER transmembrane (TM)-anchored E3 ligase, also mediates ERAD particularly for some membrane and secretory proteins from the ER (Tomar et al., 2012). TRIM13 can also orchestrate the initiation of ER-phagy via the N-degron pathway (Ji et al., 2019).
Here, we summarize the studies of TRIMs in misfolded protein clearance, regarding how TRIMs regulate their downstream pathways and whether they function synergistically or compensate each other. The purpose of this mini-review is to highlight the roles of TRIMs in misfolded protein degradation and describe the internal connections of TRIMs during these processes (Figure 2).
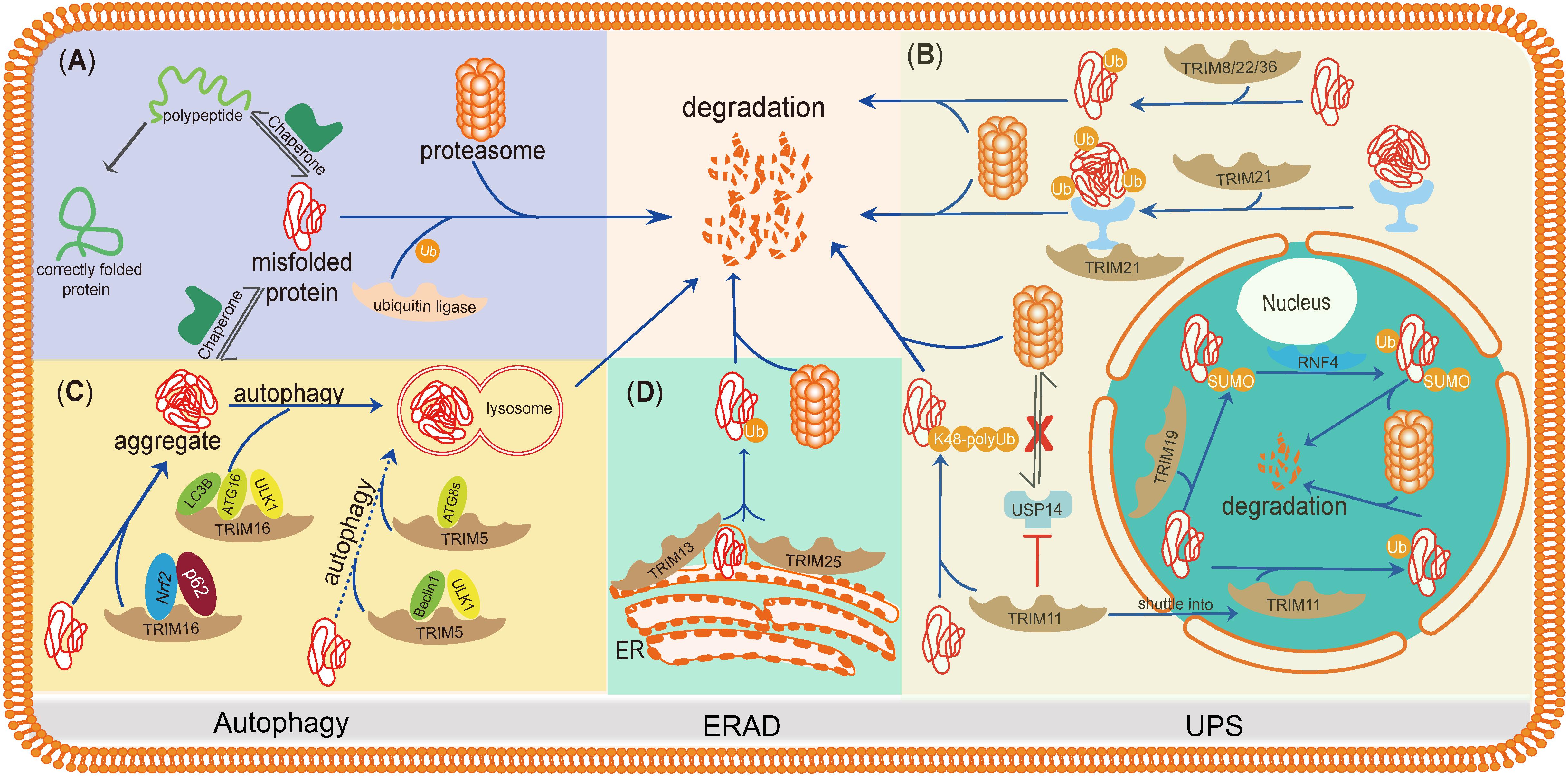
Figure 2. TRIMs mediate degradation of misfolded proteins or protein aggregates through the UPS, autophagy, and ERAD. TRIMs are widely involved in misfolded proteins clearance through distinct mechanisms, and some of them even play dual roles and function in different organelles. (A) The basic mechanism of degradation of misfolded proteins in PQC. (B) TRIM19 (PML), as well as TRIM11, participates in degradation of misfolded proteins in the nucleus via UPS, while the latter also works in cytosol by enhancing the activity of proteasome. TRIM21, as a cytoplasmic Fc receptor, is of high affinity to the complexes composed by aggregates and specific antibody and then activates UPS. Besides, TRIM8/22/36 are also supposed to promote misfolded proteins’ degradation through UPS. (C) TRIM16 promotes misfolded proteins into aggregates by interacting with Nrf2 and p62 and then enhances the biogenesis of autophagosome and autophagy-mediated aggregate removal. TRIM5 acts as an autophagic scaffold protein for Beclin1 and ULK1, which interacts with Atg8s for selective autophagy. (D) Upon ER stress, TRIM25 triggers ERAD to degrade misfolded proteins through ROS-associated pathways potentially. TRIM13, a membrane-bound protein of ER, also contributes to ERAD with its transmembrane domain.
TRIMs in Eliminating Misfolded Proteins via UPS
TRIM19
TRIM19, a typical RBCC ligase without any C-terminal domains, is well known as the promyelocytic leukemia protein (PML), which recruits diverse proteins to assemble PML nuclear bodies (NBs) (Nisole et al., 2013). It has been confirmed that PML NBs are involved in different cellular processes, e.g., antiviral response, DNA damage repair, and PQC (Bernardi and Pandolfi, 2007), implying a potential role of TRIM19 in removing misfolded proteins.
A polyQ stretch expansion in the nucleus of spinal or cerebellar neurons is pathologically associated with one of the neurodegenerative diseases spinocerebellar ataxias (SCAs) (Janer et al., 2006). SCA1 gene Ataxin-1 encodes pathogenic protein Atxn1-82Q, but not the normal protein Atxn1-30Q, forming protein aggregates in the nucleus, which could be co-localized and decreased by TRIM19 (Guo et al., 2014). Notably, more distinct sites on TRIM19 were identified to directly recognize multiple misfolded proteins [i.e., the CC region of the TRIM/RBCC motif (SRS1) and the 63 amino acid residues at its C terminus (SRS2)] in the nucleus. Mechanistically, TRIM19 first acts as a small ubiquitin-like modifier (SUMO) E3 ligase by conjugating SUMO to misfolded proteins, which are recognized and ubiquitinated by a SUMO-targeted ubiquitin ligase called RING finger protein 4 (RNF4). Through the sequential SUMOylation and ubiquitination, misfolded proteins in cancer cells are efficiently degraded via the proteasome (Guo et al., 2014); furthermore, the capacity of TRIM19 to mediate the degradation of Atxn1-82Q protein aggregates in differentiated acute promyelocytic leukemia (APL) cells was found to be markedly reduced (Chen et al., 2017). Thus, the function of TRIM19 provides a new insight, suggesting that maintenance of malignant phenotypes may rely on effective clearance of misfolded proteins.
TRIM11
TRIM11 is structurally characterized by a typical TRIM/RBCC (RING, B-box, and coiled-coil) motif at its N-terminus and a PRY/SPRY domain at its C-terminus. Extensive studies have demonstrated the critical roles of TRIM11 in the processes of neurogenesis as well as oncogenesis (Niikura et al., 2003; Tuoc and Stoykova, 2008; Wang et al., 2016).
The expression of all TRIM genes in the human mammary epithelial cell (HMEC) transformation model was analyzed, and the results revealed that TRIM11 is amplified in a series of cancer cell lines and tissues, e.g., breast carcinoma, gliomas, and lung cancer. TRIM11 promotes the degradation of misfolded and aggregated proteins in tumor cells’ nucleus or cytosol; for instance, forced expression of TRIM11 was shown to reduce the inclusions formed by Atxn1-82Q and Httax1p-97QP (Chen et al., 2017).
Notably, the RING domain of TRIM11 is required to perform its functions. On one hand, this domain catalyzes the conjugation of K48-polyubiquitin to various types of substrates, thus, ensuring adequate concentration of substrate for UPS (Chen et al., 2017, 2018). On the other hand, it directly enhances the proteasome activity by interacting with the ubiquitin-like (UBL) domain of ubiquitin-specific protease 14 (also called USP14, which removes ubiquitin chains from substrates) which deterred the interaction between USP14 and the 19S subunit of the proteasome (Chen et al., 2018). Collectively, these studies revealed that TRIM11 effectively clears misfolded proteins through promoting the activity of proteasome.
TRIM21
TRIM21/Ro52 is another member of the TRIM protein family that possesses a PRY/SPRY domain at its C-terminus, along with a typical RBCC motif at the N-terminus modulating its ubiquitin E3 ligase activity (Takahata et al., 2008). Reports have shown that TRIM21 has a high affinity to immunoglobulins (Keeble et al., 2008); thus, it acts mainly as an Fc receptor in the cytoplasm, to recognize the antigen–antibody complex (Kimura et al., 2015; Bottermann et al., 2018).
During immune defensive response against viral or bacterial infection, TRIM21 triggers pathogen neutralization by mediating UPS and valosin-containing protein (VCP, a molecular unfoldase) to eliminate such pathogens (McEwan et al., 2012). Similar to pathogen infection, misfolded tau proteins can be assembled, transferred, and spread in neurons leading to neuron cell apoptosis and induction of neurodegenerative disease. The accumulation of tau proteins has been associated with both acute neurological dysfunctions such as traumatic brain injury (TBI) and chronic neurodegenerative disorders like Alzheimer’s disease (Bloom, 2014; Edwards et al., 2020).
In addition, robust cis p-tau proteins that are found in TBI models were targeted and ablated by monoclonal antibodies (mAbs) (Kondo et al., 2015). These mAbs gained entry into the neurons via TRIM21 and then neutralized the tau aggregates, which was accompanied by TRIM21 recruitment (McEwan et al., 2017). Therefore, it inspires us to consider whether there are more similarities between invasion of pathogen and aggregation of misfolded proteins, and it raises our interest in investigating the critical roles of TRIM21 in several neuron system diseases via UPS.
TRIM8, TRIM22, and TRIM36
TRIM8, TRIM22, and TRIM36 belong to a different subclass of TRIM family. They can be significantly upregulated during cell transformation and effectively promote the elimination of misfolded proteins in transformed cells (Chen et al., 2017). This class of TRIMs was observed to be upregulated along with the proteasome in tumor cells during cell transformation (Chen et al., 2017). Hence, we propose that TRIM8/22/36 enhance misfolded protein degradation probably through the UPS pathway; however, the mechanism through which they perform this function needs further investigation.
TRIMs in Eliminating Misfolded Proteins/Aggregates via Autophagy
TRIM16
TRIM16 (also termed as estrogen-responsive B box protein-EBBP) has a missing RING domain at the N-terminus but harbors two B-box domains that possess RING-like folds, which still has the E3 ligase activity, a coiled-coil region, and a C-terminal PRY-SPRY domain.
TRIM16 was reported to bolster the turnover of stress-induced misfolded proteins by positively regulating the nuclear factor erythroid 2-related factor 2 (Nrf2) and autophagy signaling pathways (Jena et al., 2018b, 2019). TRIM16 binds to Nrf2, inducing K63-ubiquitination of Nrf2 and enhancing p62-Kelch-like ECH-associated protein 1 (KEAP1) interaction, which displaces Nrf2 from KEAP1 and prevents Nrf2 from proteasomal degradation (Jena et al., 2018a). As a positive feedback loop, the activated Nrf2 then converts misfolded proteins into aggregates by induction of p62, TRIM16, and other ubiquitin system genes. Recent studies demonstrated that TRIM16 is capable of assembling the autophagy machinery that contains TRIM16 (which acts as a scaffold protein), unc-51 Like Autophagy Activating Kinase 1 (ULK1), ATG16, and LC3B, which governed autophagosome biogenesis, and thus accelerate protein aggregates sequestration and clearance (Jena et al., 2018a; New and Thomas, 2019). Taken together, TRIM16 acts as a central player in eliminating misfolded proteins/protein aggregates during exposure to oxidative/proteotoxic stress by modulating the autophagy pathway.
TRIM5
TRIM5 has a similar structure to TRIM11 except for the absence of a PRY domain at its C-terminus. As an innate immune activator and a ubiquitin E3 ligase, TRIM5 is highly relevant in retroviral restriction such as anti-HIV infection (Pertel et al., 2011; Fletcher et al., 2018).
TRIM5 can be significantly upregulated during cell transformation to eliminate misfolded proteins/aggregates effectively (Chen et al., 2017). Mandell et al. (2014) screened all TRIMs involved in autophagy and identified that TRIM5 acts as a scaffold protein that interacts with the key members of autophagy like protein ULK1 and Beclin1. TRIM5 also served as a selective autophagic receptor mediating substrates’ autophagic degradation (Mandell et al., 2014). Thus, TRIM5 promotes the removal of misfolded proteins/aggregates most likely via the autophagy pathway.
TRIMs in Eliminating Misfolded Proteins via ERAD
TRIM25
TRIM25, known as the estrogen-responsive finger protein (EFP), possesses a typical N-terminal TRIM/RBCC motif and a C-terminal PRY/SPRY domain that has been extensively studied in innate immunity (Zou and Zhang, 2006; Versteeg et al., 2013). Previous studies showed that TRIM25 mainly acted as an E3 ligase and mediated ubiquitination of various key molecular biomarkers such as retinoic acid-inducible gene 1 (RIG-I) for antiviral response and peroxisome proliferator-activated receptor-gamma (PPARγ) for metabolism (Gack et al., 2007; Lee et al., 2018).
Since the ER is fundamental for protein and lipid biosynthesis as well as protein posttranslational processing and transportation, various physiological or pathological processes can lead to an accumulation of misfolded proteins in the ER resulting in a condition called ER stress (Wu and Kaufman, 2006). To cope with ER stress, cells rely on ERQC systems such as the UPR and ER-associated protein degradation (ERAD) (Guerriero and Brodsky, 2012; Hetz and Saxena, 2017). Recently, our group identified that TRIM25 is significantly induced upon ER stress, which promotes ERAD and finally restores ER homeostasis (Liu et al., 2020). Moreover, we found that TRIM25 expression is associated with hepatocellular carcinoma (HCC) progression and high TRIM25 expression correlates with poor patient survival in HCC (Liu et al., 2020). This reveals that TRIM25 modulates the ER homeostasis and could serve as a potential target for HCC therapy. In summary, TRIM25 is a novel ERQC player, mediating misfolded protein clearance via the ERAD.
TRIM13
TRIM13 is another member of the TRIM protein family that is encoded by the Ret Finger Protein 2 (RFP2) gene that consists of RING, B-box, and coiled-coil domains. Besides, TRIM13 also contains a TM domain, which is required for its ER localization (Lerner et al., 2007). Interestingly, this TM domain is only found in TRIM13 and TRIM59 among all the TRIM members.
Reports have shown that TRIM13 can recognize ERAD substrate CD3δ and degraded it via UPS, which relies on its RING domain (Lerner et al., 2007). In a condition of ER stress, TRIM13 also contributes to autophagosome biogenesis through its strong interaction with p62 and double FYVE-containing protein 1 (DFCP1). The deletion construct of the coiled-coil domain of TRIM13 represses autophagy regulation (Tomar et al., 2012). Additionally, TRIM13 has been found to regulate the activation of caspase-8 to induce autophagy, leading to tumor cell death in tumors (Tomar et al., 2013). These studies enrich our understanding of the critical roles played by TRIM13 in ERAD and suggest that TRIM13 may be a tumor suppressor.
Discussion and Future Prospects
In this review, we summarized the defined roles of TRIM proteins in misfolded protein clearance. These TRIM members can be localized at the nucleus, cytoplasm, or ER and facilitate the degradation of misfolded and aggregated proteins through distinct pathways, including the UPS, autophagy, and ERAD. Recent studies revealed that TRIM5 and TRIM16, serve as autophagy receptors for cargo recruitment providing the possibility that TRIM members can collaborate to regulate autophagy-mediated misfolded proteins’ clearance (Kimura et al., 2017; New and Thomas, 2019). In previous studies, TRIM5 has been shown to shuttle into the nucleus, where it forms heterodimer with TRIM19 (PML) or TRIM16 resulting in PML NBs (Stremlau et al., 2004; Diaz-Griffero et al., 2011; Bell et al., 2012); thus, indicating a synergistic effect of autophagy and UPS on misfolded protein removal. In addition, the complex of MAGE-A3/6-TRIM28 could target 5′ AMP-activated protein kinase (AMPK) for ubiquitination and proteasome-mediated degradation resulting in significant autophagy inhibition (Pineda et al., 2015). Together, these reports suggest a positive feedback loop and the existence of a cross-talk between the UPS and autophagy pathways. Thus, it is imperative to investigate the cooperation among the TRIM protein members in the degradation of misfolded protein via the UPS, autophagy, or their combination.
Of note, TRIM proteins not only act as E3 ligases but also may have extensive, yet unidentified roles. For example, TRIM19 (PML) possesses SUMOylation ligase activity in the nucleus (Guo et al., 2014), and other TRIMs with a SUMOylation ligase activity include TRIM5, TRIM27, and TRIM36 (Chu and Yang, 2011). Misfolded proteins may undergo sequential SUMOylation and ubiquitination mediated by one or several TRIM members. Moreover, the association between SUMOylation and ubiquitination and their roles in misfolded protein degradation provides a better understanding of those TRIMs containing TRIM19-like RBCC structure and is worth further exploration (Wang et al., 2018).
We have proven that the effective removal of misfolded proteins is required for oncogenesis and that TRIM members (like TRIM11) are upregulated and play critical roles during oncogenic transformation (Chen et al., 2017); this suggests that TRIM proteins can serve as novel therapeutic targets for clinical treatment of cancer. An approach has recently been developed to acutely and rapidly degrade endogenous proteins, which is based on TRIM21 and UPS pathway (Clift et al., 2017, 2018). This method, which can also precisely target the aberrant proteins in the cell, may contribute to developing other novel therapeutics for diseases associated with misfolded proteins such as cancers and neurodegenerative diseases.
To conclude, TRIM proteins are involved in misfolded protein clearance through distinct mechanisms, and the identification of other TRIM members capable of efficient misfolded proteins removal is imperative. More importantly, there exists a strong connection between TRIM proteins and proteostasis – a condition that can affect tumorigenesis and neurodegeneration. It is of key importance to further elucidate the relationships and dynamics between TRIMs and proteostasis as this is crucial for the development of novel therapeutic intervention by targeting relevant TRIMs in cancer, neurodegenerative diseases, and other associated pathologies.
Author Contributions
LZ, LA, XW, YL, and LC contributed to the acquisition of data, interpretation of data, and drafting the article. LZ and LC contributed to analyze the data and wrote the manuscript. All authors contributed to the manuscript revision and read and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (31801186) and SIAT Innovation Program for Excellent Young Researchers (201801).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alfano, C., Faggiano, S., and Pastore, A. (2016). The Ball and Chain of Polyubiquitin Structures. Trends Biochem. Sci. 41, 371–385. doi: 10.1016/j.tibs.2016.01.006
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354
Bell, J. L., Malyukova, A., Holien, J. K., Koach, J., Parker, M. W., Kavallaris, M., et al. (2012). TRIM16 acts as an E3 ubiquitin ligase and can heterodimerize with other TRIM family members. PLoS One 7:e37470. doi: 10.1371/journal.pone.0037470
Bernardi, R., and Pandolfi, P. P. (2007). Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8, 1006–1016. doi: 10.1038/nrm2277
Berndsen, C. E., and Wolberger, C. (2014). New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 21, 301–307. doi: 10.1038/nsmb.2780
Bloom, G. S. (2014). Amyloid-beta and tau: the trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Bottermann, M., Foss, S., van Tienen, L. M., Vaysburd, M., Cruickshank, J., O’Connell, K., et al. (2018). TRIM21 mediates antibody inhibition of adenovirus-based gene delivery and vaccination. Proc. Natl. Acad. Sci. U.S.A. 115, 10440–10445. doi: 10.1073/pnas.1806314115
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., et al. (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. doi: 10.1038/416507a
Buetow, L., and Huang, D. T. (2016). Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 17, 626–642. doi: 10.1038/nrm.2016.91
Chen, L., Brewer, M. D., Guo, L., Wang, R., Jiang, P., and Yang, X. (2017). Enhanced Degradation of misfolded proteins promotes tumorigenesis. Cell Rep. 18, 3143–3154. doi: 10.1016/j.celrep.2017.03.010
Chen, L., Zhu, G., Johns, E. M., and Yang, X. (2018). TRIM11 activates the proteasome and promotes overall protein degradation by regulating USP14. Nat. Commun. 9:1223. doi: 10.1038/s41467-018-03499-z
Chhangani, D., Chinchwadkar, S., and Mishra, A. (2014). Autophagy coupling interplay: can improve cellular repair and aging? Mol. Neurobiol. 49, 1270–1281. doi: 10.1007/s12035-013-8599-z
Chu, Y., and Yang, X. (2011). SUMO E3 ligase activity of TRIM proteins. Oncogene 30, 1108–1116. doi: 10.1038/onc.2010.462
Clift, D., McEwan, W. A., Labzin, L. I., Konieczny, V., Mogessie, B., James, L. C., et al. (2017). A method for the acute and rapid degradation of endogenous proteins. Cell 171, 1692–1706.e1618. doi: 10.1016/j.cell.2017.10.033
Clift, D., So, C., McEwan, W. A., James, L. C., and Schuh, M. (2018). Acute and rapid degradation of endogenous proteins by Trim-Away. Nat. Protoc. 13, 2149–2175. doi: 10.1038/s41596-018-0028-23
Costanzo, M., and Zurzolo, C. (2013). The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem. J. 452, 1–17. doi: 10.1042/BJ20121898
Deshaies, R. J., and Joazeiro, C. A. (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434. doi: 10.1146/annurev.biochem.78.101807.093809
Diaz-Griffero, F., Gallo, D. E., Hope, T. J., and Sodroski, J. (2011). Trafficking of some old world primate TRIM5alpha proteins through the nucleus. Retrovirology 8| :38. doi: 10.1186/1742-4690-8-38
Edwards, G. III, Zhao, J., Dash, P. K., Soto, C., and Moreno-Gonzalez, I. (2020). Traumatic brain injury induces tau aggregation and spreading. J. Neurotrauma 37, 80–92. doi: 10.1089/neu.2018.6348
Finley, D. (2009). Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513. doi: 10.1146/annurev.biochem.78.081507.101607
Fletcher, A. J., Vaysburd, M., Maslen, S., Zeng, J., Skehel, J. M., Towers, G. J., et al. (2018). Trivalent RING Assembly on Retroviral Capsids Activates TRIM5 Ubiquitination and Innate Immune Signaling. Cell Host Microbe 24, 761–775.e766. doi: 10.1016/j.chom.2018.10.007
Gack, M. U., Shin, Y. C., Joo, C. H., Urano, T., Liang, C., Sun, L., et al. (2007). TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446, 916–920. doi: 10.1038/nature05732
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. doi: 10.1038/nature02263
Goodier, J. L., Soares, A. O., Pereira, G. C., DeVine, L. R., Sanchez, L., Cole, R. N., et al. (2020). C9orf72-associated SMCR8 protein binds in the ubiquitin pathway and with proteins linked with neurological disease. Acta Neuropathol. Commun. 8:110. doi: 10.1186/s40478-020-00982-x
Grice, G. L., and Nathan, J. A. (2016). The recognition of ubiquitinated proteins by the proteasome. Cell Mol. Life Sci. 73, 3497–3506. doi: 10.1007/s00018-016-2255-5
Grumati, P., Dikic, I., and Stolz, A. (2018). ER-phagy at a glance. J. Cell Sci. 131:jcs217364. doi: 10.1242/jcs.217364
Guerriero, C. J., and Brodsky, J. L. (2012). The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576. doi: 10.1152/physrev.00027.2011
Guo, L., Giasson, B. I., Glavis-Bloom, A., Brewer, M. D., Shorter, J., Gitler, A. D., et al. (2014). A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 55, 15–30. doi: 10.1016/j.molcel.2014.04.030
Hartl, F. U., and Hayer-Hartl, M. (2002). Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295, 1852–1858. doi: 10.1126/science.1068408
Hatakeyama, S. (2017). TRIM family proteins: roles in Autophagy. Immunity, and Carcinogenesis. Trends Biochem. Sci. 42, 297–311. doi: 10.1016/j.tibs.2017.01.002
Hetz, C., Chevet, E., and Oakes, S. A. (2015). Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838. doi: 10.1038/ncb3184
Hetz, C., and Saxena, S. (2017). ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491. doi: 10.1038/nrneurol.2017.99
Hiller, M. M., Finger, A., Schweiger, M., and Wolf, D. H. (1996). ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science 273, 1725–1728.
Hjerpe, R., and Rodriguez, M. S. (2008). Alternative UPS drug targets upstream the 26S proteasome. Int. J. Biochem. Cell Biol. 40, 1126–1140. doi: 10.1016/j.biocel.2007.11.021
Horwich, A. L., and Weissman, J. S. (1997). Deadly conformations–protein misfolding in prion disease. Cell 89, 499–510. doi: 10.1016/s0092-8674(00)80232-9
James, L. C., Keeble, A. H., Khan, Z., Rhodes, D. A., and Trowsdale, J. (2007). Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc. Natl. Acad. Sci. U.S.A. 104, 6200–6205. doi: 10.1073/pnas.0609174104
Janer, A., Martin, E., Muriel, M. P., Latouche, M., Fujigasaki, H., Ruberg, M., et al. (2006). PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. J. Cell Biol. 174, 65–76. doi: 10.1083/jcb.200511045
Jena, K. K., Kolapalli, S. P., Mehto, S., Nath, P., Das, B., Sahoo, P. K., et al. (2018a). TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 37:e98358. doi: 10.15252/embj.201798358
Jena, K. K., Mehto, S., Kolapalli, S. P., Nath, P., Chauhan, S., and Chauhan, S. (2018b). TRIM16 employs NRF2, ubiquitin system and aggrephagy for safe disposal of stress-induced misfolded proteins. Cell Stress 2, 365–367. doi: 10.15698/cst2018.12.169
Jena, K. K., Mehto, S., Kolapalli, S. P., Nath, P., Sahu, R., Chauhan, N. R., et al. (2019). TRIM16 governs the biogenesis and disposal of stress-induced protein aggregates to evade cytotoxicity: implication for neurodegeneration and cancer. Autophagy 15, 924–926. doi: 10.1080/15548627.2019.1586251
Ji, C. H., Kim, H. Y., Heo, A. J., Lee, S. H., Lee, M. J., Kim, S. B., et al. (2019). The N-Degron Pathway Mediates ER-phagy. Mol. Cell 75, 1058–1072.e1059. doi: 10.1016/j.molcel.2019.06.028
Kaganovich, D., Kopito, R., and Frydman, J. (2008). Misfolded proteins partition between two distinct quality control compartments. Nature 454, 1088–1095. doi: 10.1038/nature07195
Keeble, A. H., Khan, Z., Forster, A., and James, L. C. (2008). TRIM21 is an IgG receptor that is structurally, thermodynamically, and kinetically conserved. Proc. Natl. Acad. Sci. U.S.A. 105, 6045–6050. doi: 10.1073/pnas.0800159105
Kim, H., Bhattacharya, A., and Qi, L. (2015). Endoplasmic reticulum quality control in cancer: friend or foe. Semin. Cancer Biol. 33, 25–33. doi: 10.1016/j.semcancer.2015.02.003
Kim, Y. E., Hipp, M. S., Bracher, A., Hayer-Hartl, M., and Hartl, F. U. (2013). Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355. doi: 10.1146/annurev-biochem-060208-092442
Kimura, T., Jain, A., Choi, S. W., Mandell, M. A., Schroder, K., Johansen, T., et al. (2015). TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 210, 973–989. doi: 10.1083/jcb.201503023
Kimura, T., Jia, J., Kumar, S., Choi, S. W., Gu, Y., Mudd, M., et al. (2017). Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 36, 42–60. doi: 10.15252/embj.201695081
Kondo, A., Shahpasand, K., Mannix, R., Qiu, J., Moncaster, J., Chen, C. H., et al. (2015). Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523, 431–436. doi: 10.1038/nature14658
Korolchuk, V. I., Mansilla, A., Menzies, F. M., and Rubinsztein, D. C. (2009). Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527. doi: 10.1016/j.molcel.2009.01.021
Kwon, Y. T., and Ciechanover, A. (2017). The Ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 42, 873–886. doi: 10.1016/j.tibs.2017.09.002
Lee, J. M., Choi, S. S., Lee, Y. H., Khim, K. W., Yoon, S., Kim, B.-G., et al. (2018). The E3 ubiquitin ligase TRIM25 regulates adipocyte differentiation via proteasome-mediated degradation of PPARγ. Exp. Mol. Med. 50:135. doi: 10.1038/s12276-018-0162-6
Lerner, M., Corcoran, M., Cepeda, D., Nielsen, M. L., Zubarev, R., Ponten, F., et al. (2007). The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Mol. Biol. Cell 18, 1670–1682. doi: 10.1091/mbc.e06-03-0248
Li, Y., Wu, H., Wu, W., Zhuo, W., Liu, W., Zhang, Y., et al. (2014). Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res. 24, 762–765. doi: 10.1038/cr.2014.46
Liu, Y., Tao, S., Liao, L., Li, Y., Li, H., Li, Z., et al. (2020). TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 11:348. doi: 10.1038/s41467-019-14190-2
Mandell, M. A., Jain, A., Arko-Mensah, J., Chauhan, S., Kimura, T., Dinkins, C., et al. (2014). TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 30, 394–409. doi: 10.1016/j.devcel.2014.06.013
McEwan, W. A., Falcon, B., Vaysburd, M., Clift, D., Oblak, A. L., Ghetti, B., et al. (2017). Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci.U.S.A. 114, 574–579. doi: 10.1073/pnas.1607215114
McEwan, W. A., Hauler, F., Williams, C. R., Bidgood, S. R., Mallery, D. L., Crowther, R. A., et al. (2012). Regulation of virus neutralization and the persistent fraction by TRIM21. J. Virol. 86, 8482–8491. doi: 10.1128/jvi.00728-12
Meroni, G., and Diez-Roux, G. (2005). TRIM/RBCC, a novel class of ‘single protein RING finger’. E3 ubiquitin ligases. Bioessays 27, 1147–1157. doi: 10.1002/bies.20304
Mishra, R., Upadhyay, A., Prajapati, V. K., and Mishra, A. (2018). Proteasome-mediated proteostasis: novel medicinal and pharmacological strategies for diseases. Med. Res. Rev. 38, 1916–1973. doi: 10.1002/med.21502
New, J., and Thomas, S. M. (2019). Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy 15, 1682–1693. doi: 10.1080/15548627.2019.1596479
Niikura, T., Hashimoto, Y., Tajima, H., Ishizaka, M., Yamagishi, Y., Kawasumi, M., et al. (2003). A tripartite motif protein TRIM11 binds and destabilizes Humanin, a neuroprotective peptide against Alzheimer’s disease-relevant insults. Eur. J. Neurosci. 17, 1150–1158. doi: 10.1046/j.1460-9568.2003.02553.x
Nisole, S., Maroui, M. A., Mascle, X. H., Aubry, M., and Chelbi-Alix, M. K. (2013). Differential roles of PML isoforms. Front. Oncol. 3:125. doi: 10.3389/fonc.2013.00125
Nisole, S., Stoye, J. P., and Saib, A. (2005). TRIM family proteins: retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 3, 799–808. doi: 10.1038/nrmicro1248
Ohtake, F., Saeki, Y., Ishido, S., Kanno, J., and Tanaka, K. (2016). The K48-K63 branched ubiquitin chain regulates NF-kappaB signaling. Mol. Cell 64, 251–266. doi: 10.1016/j.molcel.2016.09.014
Ozato, K., Shin, D. M., Chang, T. H., and Morse, H. C. (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860. doi: 10.1038/nri2413
Pertel, T., Hausmann, S., Morger, D., Zuger, S., Guerra, J., Lascano, J., et al. (2011). TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365. doi: 10.1038/nature09976
Pineda, C. T., Ramanathan, S., Fon Tacer, K., Weon, J. L., Potts, M. B., Ou, Y. H., et al. (2015). Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728. doi: 10.1016/j.cell.2015.01.034
Powers, E. T., and Balch, W. E. (2008). Costly mistakes: translational infidelity and protein homeostasis. Cell 134, 204–206. doi: 10.1016/j.cell.2008.07.005
Richter, K., Haslbeck, M., and Buchner, J. (2010). The heat shock response: life on the verge of death. Mol. Cell 40, 253–266. doi: 10.1016/j.molcel.2010.10.006
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529. doi: 10.1038/nrm2199
Sadowski, M., Suryadinata, R., Tan, A. R., Roesley, S. N., and Sarcevic, B. (2012). Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life 64, 136–142. doi: 10.1002/iub.589
Saibil, H. (2013). Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14, 630–642. doi: 10.1038/nrm3658
Sharma, S., Chakraborty, K., Müller, B. K., Astola, N., Tang, Y.-C., Lamb, D. C., et al. (2008). Monitoring protein conformation along the pathway of chaperonin-assisted folding. Cell 133, 142–153. doi: 10.1016/j.cell.2008.01.048
Sontag, E. M., Samant, R. S., and Frydman, J. (2017). Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 86, 97–122. doi: 10.1146/annurev-biochem-060815-014616
Soto, C., and Pritzkow, S. (2018). Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 21, 1332–1340. doi: 10.1038/s41593-018-0235-9
Stremlau, M., Owens, C. M., Perron, M. J., Kiessling, M., Autissier, P., and Sodroski, J. (2004). The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853. doi: 10.1038/nature02343
Sun, D., Wu, R., Zheng, J., Li, P., and Yu, L. (2018). Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 28, 405–415. doi: 10.1038/s41422-018-0017-7
Takahata, M., Bohgaki, M., Tsukiyama, T., Kondo, T., Asaka, M., and Hatakeyama, S. (2008). Ro52 functionally interacts with IgG1 and regulates its quality control via the ERAD system. Mol. Immunol. 45, 2045–2054. doi: 10.1016/j.molimm.2007.10.023
Tomar, D., Prajapati, P., Sripada, L., Singh, K., Singh, R., Singh, A. K., et al. (2013). TRIM13 regulates caspase-8 ubiquitination, translocation to autophagosomes and activation during ER stress induced cell death. Biochim. Biophys. Acta 1833, 3134–3144. doi: 10.1016/j.bbamcr.2013.08.021
Tomar, D., Singh, R., Singh, A. K., Pandya, C. D., and Singh, R. (2012). TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim. Biophys. Acta 1823, 316–326. doi: 10.1016/j.bbamcr.2011.11.015
Tuoc, T. C., and Stoykova, A. (2008). Trim11 modulates the function of neurogenic transcription factor Pax6 through ubiquitin-proteosome system. Genes Dev. 22, 1972–1986. doi: 10.1101/gad.471708
Varshavsky, A. (2017). The Ubiquitin system, autophagy, and regulated protein degradation. Annu. Rev. Biochem. 86, 123–128. doi: 10.1146/annurev-biochem-061516-044859
Versteeg, G. A., Rajsbaum, R., Sanchez-Aparicio, M. T., Maestre, A. M., Valdiviezo, J., Shi, M., et al. (2013). The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38, 384–398. doi: 10.1016/j.immuni.2012.11.013
Wang, M., and Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. doi: 10.1038/nature17041
Wang, P., Benhenda, S., Wu, H., Lallemand-Breitenbach, V., Zhen, T., Jollivet, F., et al. (2018). RING tetramerization is required for nuclear body biogenesis and PML sumoylation. Nat. Commun. 9:1277. doi: 10.1038/s41467-018-03498-0
Wang, X., Shi, W., Shi, H., Lu, S., Wang, K., Sun, C., et al. (2016). TRIM11 overexpression promotes proliferation, migration and invasion of lung cancer cells. J. Exp. Clin. Cancer Res. 35:100. doi: 10.1186/s13046-016-0379-y
Wani, W. Y., Boyer-Guittaut, M., Dodson, M., Chatham, J., Darley-Usmar, V., and Zhang, J. (2015). Regulation of autophagy by protein post-translational modification. Lab Invest. 95, 14–25. doi: 10.1038/labinvest.2014.131
Wiseman, R. L., Powers, E. T., Buxbaum, J. N., Kelly, J. W., and Balch, W. E. (2007). An adaptable standard for protein export from the endoplasmic reticulum. Cell 131, 809–821. doi: 10.1016/j.cell.2007.10.025
Wong, E., and Cuervo, A. M. (2010). Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb. Perspect. Biol. 2:a006734. doi: 10.1101/cshperspect.a006734
Wu, J., and Kaufman, R. J. (2006). From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 13, 374–384. doi: 10.1038/sj.cdd.4401840
Xu, P., Duong, D. M., Seyfried, N. T., Cheng, D., Xie, Y., Robert, J., et al. (2009). Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145. doi: 10.1016/j.cell.2009.01.041
Zheng, N., and Shabek, N. (2017). Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 86, 129–157. doi: 10.1146/annurev-biochem-060815-014922
Keywords: TRIMs, misfolded proteins, aggregates, degradation, UPS, autophagy, ERAD
Citation: Zhang L, Afolabi LO, Wan X, Li Y and Chen L (2020) Emerging Roles of Tripartite Motif-Containing Family Proteins (TRIMs) in Eliminating Misfolded Proteins. Front. Cell Dev. Biol. 8:802. doi: 10.3389/fcell.2020.00802
Received: 16 June 2020; Accepted: 28 July 2020;
Published: 25 August 2020.
Edited by:
Lawrence H. Boise, Emory University, United StatesReviewed by:
Arun Upadhyay, Northwestern University, United StatesElodie Lafont, INSERM U1242 Laboratoire COSS, France
Copyright © 2020 Zhang, Afolabi, Wan, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Li, eWFuZy5saUBzaWF0LmFjLmNu; Liang Chen, bGlhbmcuY2hlbkBzaWF0LmFjLmNu