
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 12 August 2020
Sec. Stem Cell Research
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00772
This article is part of the Research TopicInduced Pluripotent Stem Cell-Based Disease Modeling and Drug Discovery: Can We Recapitulate Cardiovascular Disease on a Culture Dish?View all 13 articles
 Xiaojing Luo1†
Xiaojing Luo1† Wener Li1†Karolina Künzel1
Wener Li1†Karolina Künzel1 Sarah Henze2
Sarah Henze2 Lukas Cyganek2,3
Lukas Cyganek2,3 Anna Strano1
Anna Strano1 Mareike S. Poetsch1
Mareike S. Poetsch1 Mario Schubert1
Mario Schubert1 Kaomei Guan1,2*
Kaomei Guan1,2*In adult cardiomyocytes (CMs), the type 2 ryanodine receptor (RYR2) is an indispensable Ca2+ release channel that ensures the integrity of excitation-contraction coupling, which is fundamental for every heartbeat. However, the role and importance of RYR2 during human embryonic cardiac development are still poorly understood. Here, we generated two human induced pluripotent stem cell (iPSC)-based RYR2 knockout (RYR2–/–) lines using the CRISPR/Cas9 gene editing technology. We found that RYR2–/–-iPSCs could differentiate into CMs with the efficiency similar to control-iPSCs (Ctrl-iPSCs); however, the survival of iPSC-CMs was markedly affected by the lack of functional RYR2. While Ctrl-iPSC-CMs exhibited regular Ca2+ handling, we observed significantly reduced frequency and intense abnormalities of Ca2+ transients in RYR2–/–-iPSC-CMs. Ctrl-iPSC-CMs displayed sensitivity to extracellular Ca2+ ([Ca2+ ]o) and caffeine in a concentration-dependent manner, while RYR2–/–-iPSC-CMs showed inconsistent reactions to [Ca2+ ]o and were insensitive to caffeine, indicating there is no RYR2-mediated Ca2+ release from the sarcoplasmic reticulum (SR). Instead, compensatory mechanism for calcium handling in RYR2–/–-iPSC-CMs is partially mediated by the inositol 1,4,5-trisphosphate receptor (IP3R). Similar to Ctrl-iPSC-CMs, SR Ca2+ refilling in RYR2–/–-iPSC-CMs is mediated by SERCA. Additionally, RYR2–/–-iPSC-CMs showed a decreased beating rate and a reduced peak amplitude of L-type Ca2+ current. These findings demonstrate that RYR2 is not required for CM lineage commitment but is important for CM survival and contractile function. IP3R-mediated Ca2+ release is one of the major compensatory mechanisms for Ca2+ cycling in human CMs with the RYR2 deficiency.
The type 2 ryanodine receptor (RYR2) represents the major sarcoplasmic reticulum (SR) Ca2+ release channel in adult cardiomyocytes (CMs). It plays an essential role in excitation-contraction coupling, a process by which an electrical signal is converted into a single contraction (Eisner et al., 2017). Upon spontaneous depolarization of membrane potential, voltage-dependent L-type calcium channels (LTCC) are activated, which cause the influx of a small amount of external Ca2+ into the cytosol. The Ca2+ signal is then sensed and amplified by the Ca2+ -sensitive RYR2. The opening of RYR2 causes a substantial release of Ca2+ from the SR into the cytosol and thus elevates intracellular Ca2+ levels. In the end, the binding of Ca2+ to troponin promotes sliding of thick and thin filaments, which results in cardiac contraction (Eisner et al., 2017).
A growing number of studies highlight the importance of RYR2 for the precise functionality of the adult heart. An inducible cardiac-specific Ryr2 knockout (Ryr2–/–) mouse model claimed that RYR2 plays a non-redundant role in the control of heart rate and rhythmicity (Bround et al., 2012). An in-frame deletion of exon-3 in the Ryr2 gene in the mouse (Ex3-del+/−) was associated with bradycardia and death. However, Ex3-del+/− mice did not display some clinical phenotypes of patients with the RYR2 exon-3 deletion, including catecholaminergic polymorphic ventricular tachycardia (Liu et al., 2014).
In the early stage of embryonic heart development in mice, Ca2+ homeostasis is regulated not only by RYR2 but also by LTCC and inositol 1,4,5-trisphosphate receptor (IP3R) (Takeshima et al., 1998; Rosemblit et al., 1999; Kapur and Banach, 2007; Sasse et al., 2007). Ryr2–/– mice possess the ability of repetitive Ca2+ signals and rhythmic contractions at embryonic day E9.5 (Takeshima et al., 1998), when LTCC and IP3R may be responsible for the early Ca2+ cycling (Liu et al., 2002; Kockskamper et al., 2008; Xie et al., 2018). However, as the development of the embryo proceeds, the importance of Ca2+ cycling via SR increases progressively (Liu et al., 2002). Ryr2–/– mice died of cardiac arrest around embryonic day E10 with irregular arranged myocardium and trabeculae (Takeshima et al., 1998).
To date, studies on the expression and function of RYR2 during early embryonic cardiac development are limited to investigations using mouse Ryr2–/– models and mouse embryonic stem cell-derived CMs (mESC-CMs) (Takeshima et al., 1998; Liu et al., 2002; Yang et al., 2002; Fu et al., 2006; Kapur and Banach, 2007; Sasse et al., 2007; Xie et al., 2018). The major challenge to study the origin and development of human embryonic cardiac myocytes is the limitation of a reliable human cell model, which can be used for long-term culture experiments. The appearance of human induced pluripotent stem cell (iPSC) technology has shown its great potential to solve this challenge (Takahashi et al., 2007; Yu et al., 2007). In the iPSC approach, adult somatic cells can be efficiently reprogrammed into pluripotent stem cells by ectopic expression of a set of transcription factors (Takahashi and Yamanaka, 2006). Based on this revolutionary finding, researchers have established the generation of human iPSCs and the efficient differentiation of these cells into iPSC-CMs (Itzhaki et al., 2011; Lian et al., 2013; Cyganek et al., 2018). Previous studies have demonstrated that both RYR2 and IP3R are involved in calcium handling in human ESC- and iPSC-derived CMs (Satin et al., 2008; Itzhaki et al., 2011). However, our knowledge about how these two signaling pathways are cooperated during human embryonic cardiac growth is still not sufficient.
In this study, we used iPSC technology in combination with the CRISPR/Cas9 gene editing technique to investigate the role of RYR2 in differentiation, development, and function of human iPSC-CMs. We hypothesized that the generated RYR2–/–-iPSC-CMs exhibit morphological and physiological abnormalities, providing insights into the function of RYR2 during human embryonic heart development.
Human iPSC lines iWTD2.1 and iBM76.1 were used in this study as controls (Ctrl-iPSCs), which were generated from dermal fibroblasts and mesenchymal stem cells from two healthy donors, respectively, using the STEMCCA lentivirus system, and characterized as previously described (Streckfuss-Bomeke et al., 2013; Cyganek et al., 2018). The study was approved by the Ethics Committee of the University Medical Center Göttingen (approval number: 21/1/11), and carried out in accordance with the approved guidelines. Human iPSCs were cultured in chemically defined E8 medium (Thermo Fischer Scientific) on Geltrex- (Thermo Fischer Scientific) coated cell culture plates at 37°C with 5% CO2. The E8 medium was changed daily and cells at ∼85% confluency were passaged using Versene (Thermo Fischer Scientific).
In order to generate homozygous RYR2 knockout (RYR2–/–) iPSC lines, two guide RNAs (gRNAs) were designed and inserted into the CRISPR/Cas9 plasmid (Sigma-Aldrich), respectively (Supplementary Figure 1A). While gRNA1 (ACGAACTCTTCGTAGTCGAGGG) targets exon 90 (g.741541-741563) of the RYR2 gene, gRNA2 (CCTAGCCTGGTATATGACTATG) targets exon 99 (g.763747-763768). For transfection using the Amaxa Nucleofector II device (Lonza), 2 × 106 iPSCs were collected and re-suspended in a mixture of 82 μl Nucleofector solution and 18 μl supplement 1 (Nucleofector kit 1/2, Lonza) containing 4 μg plasmid. One day after transfection, GFP+ cells were sorted and seeded on Geltrex-coated 96-well plates at a density of 1 × 103 cells/well for expansion. The genomic DNA from the colonies was isolated and purified using the automated Maxwell 16 cell DNA purification kit (Promega) according to the manufacturer’s instruction. For genomic DNA sequencing, the DNA sequence of RYR2 was initially amplified by PCR using the appropriate primer set (Supplementary Table 1A). DNA sequencing of PCR products from CRISPR/Cas9-edited clones was performed by a commercial sequencing facility (Seqlab, Göttingen).
To characterize the pluripotency of the CRISPR/Cas9-edited RYR2–/–-iPSCs, reverse transcription-PCR analysis, immunofluorescence staining, and spontaneous differentiation in vitro were carried out using standard protocols as described earlier (Streckfuss-Bomeke et al., 2013). For detailed description, please see the Supplementary Material.
Directed differentiation of Ctrl- and RYR2–/–-iPSCs into CMs (Ctrl-iPSC-CMs, RYR2–/–-iPSC-CMs, respectively) was induced by modulating WNT signaling as previously described (Lian et al., 2013; Cyganek et al., 2018). Briefly, when monolayer cultures of iPSCs on 12-well plates reached 80–90% confluency, differentiation was initiated by changing the E8 medium to cardio differentiation medium, which was composed of RPMI 1640 with Glutamax and HEPES (Thermo Fischer Scientific), 0.5 mg/ml human recombinant albumin (Sigma-Aldrich) and 0.2 mg/ml L-ascorbic acid 2-phosphate (Sigma-Aldrich). Cells were first treated with 4 μM of the GSK3β inhibitor CHIR99021 (Millipore) for 48 h and then with 5 μM of the WNT signaling inhibitor IWP2 (Millipore) for additional 48 h. Afterward, cells were cultured in cardio differentiation medium for another 4 days. From day 8, cells were cultivated in cardio culture medium containing RPMI 1640 with Glutamax and HEPES, supplemented with 2% B27 (Thermo Fischer Scientific). At day 20, beating iPSC-CMs were detached from the plate by incubating with 2 ml of 1 mg/ml collagenase B (Worthington Biochemical), dissolved in cardio culture medium, for 1 h at 37°C. Floating iPSC-CM sheet was gently transferred into a falcon tube and dissociated with 3 ml of 0.25% Trypsin/EDTA (Thermo Fischer Scientific) for 8 min at 37°C. Digestion was stopped by adding the double volume of the cardio digestion medium (80% cardio culture medium, 20% FCS, and 2 μM Thiazovivin). Cells were centrifuged at 200 g for 5 min, re-suspended in cardio digestion medium, and replated into Geltrex-coated 6-well plates at a density of 800,000 cells/well. Afterward, iPSC-CMs were cultured in cardio culture medium until 90 days.
To investigate the proliferation of iPSC-CMs during long-term culture, both Ctrl- and RYR2–/–-iPSC-CMs were replated at a fixed density of 800,000 cells/well at day 20 post differentiation. Cell number was determined weekly for 10 weeks until CMs reached the age of 90 days. CMs from two randomly selected wells of one differentiation experiment were detached, dissociated, and quantified by counting the cell numbers with a hemocytometer and taking the average.
To assess the metabolic activity of iPSC-CMs, the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrasodium bromide) assay was performed according to the manufacturer’s instructions (MTT Kit CT02, Millipore). Briefly, iPSC-CMs at week 8 after replating were seeded in 48-well plates at a density of 60,000 cells per well, and cultured for another 2 weeks. Afterwards, 50 μg/ml MTT dissolved in 200 μl cardio culture medium was added onto the cells, which were then incubated at 37°C for 2 h. Reaction was stopped by the addition of 200 μl isopropanol supplemented with 0.04 M HCl. Samples were incubated and shaken (300 rpm) for 10 min at room temperature (RT). Absorbance of the formazan at 570 and 630 nm was measured using a plate reader (Biotek Synergy HTX). Cell viability was determined as absorbance (570–630 nm) and normalized to control group.
Ctrl- and RYR2–/–-iPSC-CMs grown on glass coverslips were fixed with 4% paraformaldehyde (PFA; Carl Roth), blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich), and permeabilized with 0.1% Triton X-100 (Carl Roth). Immunofluorescence staining was performed overnight using the following primary antibodies: anti-α-actinin (1:500; mouse monoclonal, IgG1, Sigma-Aldrich), anti-Ki67 (1:400; rabbit polyoclonal, abcam), anti-IP3R (1:100; rabbit polyclonal, Merck Millipore, used for all three subtypes of IP3R), and anti-RYR2 (1:500; rabbit polyclonal, HPA020028; Sigma-Aldrich, used for the full length of RYR2). Afterward, cells were washed three times with PBS and incubated with the corresponding secondary antibodies (1:1000; anti-rabbit Alexa fluor 546, Invitrogen; anti-rabbit Alexa fluor 488, Invitrogen; anti-mouse Alexa fluor 546, Invitrogen; anti-mouse Alexa fluor 488, Invitrogen) for 1 h at RT. Nuclei were co-stained with 4’,6-diamidino-2-phenylindole (DAPI; 0.4 μg/ml; Sigma-Aldrich). Documentation was performed using fluorescence microscopy (Carl Zeiss).
Both Ctrl- and RYR2–/–-iPSC-CMs (90 days old) were scraped off from the culture plates and cell pellets were snap-frozen into liquid nitrogen and stored at –80°C. For cell lysis, frozen cell pellets were resuspended in lysis buffer containing 20 mM Tris/HCl (pH 7.4), 200 mM NaCl, 1 mM Na3VO4, 20 mM NaF, 1% IGEPAL CA-630 (Sigma-Aldrich), 1 mM dithiothreitol (Roth), PhosSTOP phosphatase inhibitor (Roche), and cOmpleteTM protease inhibitor (Roche). After incubation for 30 min on ice, lysates were centrifuged and protein concentration was determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. A total amount of 40 μg protein lysate mixed with SDS loading buffer and DPBS in a volume of 20 μl was denatured for 30 min at 37°C. Afterward, samples were run on a 6–15% polyacrylamide gel with a 5% stacking gel at 250 mA for around 1–2 h and then transferred to PVDF membranes using the Wet/Tank blotting system. Unspecific binding sites on the membrane were blocked with 1% BSA in TBS with 0.1% Tween 20 (TBS-T) or with 5% non-fat dry milk in TBS-T for 1 h at RT. Membranes were incubated with primary antibodies overnight at 4°C. The following primary antibodies were used: anti-RYR2 (1:500; rabbit polyclonal; SAB4502707; Sigma-Aldrich, used for the detection of both the full-length and the truncated RYR2), anti-RYR2 (1:1000; mouse monoclonal, IgG1, MA3-916 C3-33; Thermo Fisher Scientific Pierce antibodies used for the detection of the full length of RYR2), anti-IP3R (1:750; rabbit polyclonal), anti-SERCA2A (sarco/endoplasmic reticulum Ca2+ -ATPase type 2A; 1:1000; mouse monoclonal, IgG2a, Thermo Fisher Scientific), anti-NCX1 (sodium-calcium exchanger type 1; 1:1000; mouse monoclonal, IgG2b, Novus), anti-Cav1.2 (voltage-dependent L-type calcium channel alpha 1C subunit; 1:200; mouse monoclonal, IgG2b, abcam), anti-α-actinin (1:1000; mouse monoclonal, IgG1), anti-eukaryotic elongation factor 2 (EEF2; 1:50,000; rabbit polyclonal, IgG, abcam), anti-cardiac troponin T (cTNT; 1:1000; rabbit polyclonal, IgG, abcam), and anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase; 1:5000; rabbit polyclonal, Thermo Fisher Scientific). The membranes were then incubated with HRP-conjugated goat anti-mouse or anti-rabbit secondary antibodies (1:10,000; Thermo Fisher Scientific) for 1 h at RT. Afterwards, membranes were washed three times with TBS-T. Antigens of interest were detected by chemiluminescence (ECL; GE Healthcare) and visualized using the ChemiDoc MP system. Quantification was performed by calculating the signal intensity with Image Lab software (Bio-Rad).
To detect protein degradation, Ctrl-, A3 and A5 RYR2–/–-iPSC-CMs (90 days old) were stimulated with 100 nM isoprenaline (Sigma-Aldrich) for 6 h and stepwise treated with the proteasome and calpain inhibitor MG132 (10 μM for 24 h; Sigma-Aldrich) and the autophagy inhibitor bafilomycin A1 (BafA1; 100 nM for 6 h; Sigma-Aldrich) before the cell pellets were collected.
For gene expression analysis, Ctrl-iPSCs, CRISPR-edited A3 and A5 RYR2–/–-iPSCs as well as 90-day-old Ctrl- and RYR2–/–-iPSC-CMs were washed three times with PBS and cell pellets were collected, snap-frozen into liquid nitrogen, and stored at –80°C. Isolation and purification of total RNA were performed using the SV total RNA isolation system (Promega) in accordance to the manufacturer’s instructions. First strand cDNA synthesis was performed using MULV reverse transcriptase (Thermo Fisher Scientific) and Oligo d(T)16 primer (Thermo Fisher Scientific). The expression level of SOX2, OCT4, NANOG, LIN28, and FOXD3 were assessed in Ctrl- and CRISPR-edited iPSC lines by reverse transcription-PCR using the GoTaq DNA polymerase (Promega). In addition, the expression level of RYR2, IP3R1, IP3R2, CACNA1C, TNNT2, and ACTN2 were assessed in Ctrl and RYR2–/–-iPSC-CMs. GAPDH was used as an internal control. Primer sequences, annealing temperature, and cycles used for reverse transcription-PCR analyses are listed in Supplementary Table 1B.
For calcium spark recordings, spontaneously beating Ctrl- and RYR2–/–-iPSC-CMs (around 80-day-old) were dissociated, replated on Geltrex-coated coverslips at a density of 200,000 cells per 6-well and then allowed to recover for at least 10 days in cardio culture medium. Before measurement, cells were loaded with the fluorescent calcium indicator 5 μM fluo-4/AM (Thermo Fisher Scientific) and 0.02% [w/v] pluronic F-127 (Thermo Fisher Scientific) in Tyrode’s solution containing (in mM): NaCl 140, KCl 5.4, CaCl2 1.8, MgCl2 1, HEPES 10, and glucose 10 (pH adjusted to 7.3 with NaOH) for 30 min at RT. Following incubation, the indicator-containing solution was removed and cells were washed twice, and incubated for additional 10 min to allow de-esterification of the indicator. To detect Ca2+ sparks, both Ctrl- and RYR2–/–-iPSC-CMs were pre-treated with 100 nM isoprenaline for 10 min before starting the recordings. Recordings were obtained using a LSM 710 confocal microscopy system in line scan mode (512 pixels, 45 μm, 1057.7 Hz, 20,000 cycles). Cells were incubated in Tyrode’s solution at RT and field stimulated at 0.25 Hz for at least 20 s to bring the cytosolic calcium concentration to a steady state. Fluo-4 was excited at 488 nm and emitted fluorescence was captured at 490–540 nm. Quantification of Ca2+ sparks was performed using the SparkMaster plugin of ImageJ (NIH) and several key parameters were determined: spark frequency (events per 100 μm per second), amplitude (ΔF/F0), full duration at half maximum (FDHM), full width at half maximum (FWHM), and SR Ca2+ leak per cell (spark frequency × amplitude × FDHM × FWHM). As fixed criteria, sparks with minimal amplitude of 0.2 ΔF/F0, minimal width of 0.7 μm, and minimal duration of 7 ms were selected for detailed analysis.
Both Ctrl- and RYR2–/–-iPSC-CMs around day 80 were dissociated and replated on coverslips at a density of 200,000 cells/well. Cells were allowed to recover for at least 10 days post-replating. For measurement, cells were loaded with Fura-2 (Thermo Fisher Scientific) at a final concentration of 5 μM in cardio culture medium for 30 min at 37°C and washed twice with the medium. Prior to measurement, cells were incubated for 10 min to enable complete de-esterification of intracellular Fura-2. Intracellular Ca2+ events were recorded using a 40 × objective on an Olympus IX70 microscope fitted with an IonOptix system (Ionoptix, Milton, MA) at 35°C. Samples were excited at 340 and 380 nm with a switching frequency of 200 Hz and the emitted fluorescence was collected at 510 nm. The cytosolic Ca2+ level was measured as the ratio of fluorescence at 340 and 380 nm (340/380 nm).
Spontaneous whole-cell Ca2+ transients were recorded in normal Tyrode’s solution containing (in mM): NaCl 138, KCl 4, CaCl2 1.8, MgCl2 1, NaH2PO4 0.33, HEPES 10, and glucose 10 (pH adjusted to 7.3 with NaOH). To normalize the Ca2+ transient frequency, Ctrl- and RYR2–/–-iPSC-CMs were field-stimulated using a MyoPacer (Ionoptix, Milton, MA) at a pacing frequency of 0.5 Hz (6 V, 10 ms). Monotonic transient analysis was performed using the LabChart Pro software (ADInstrument) and the following parameters were determined: peak amplitude of Ca2+ transients (the Fura-2 ratio at systole subtracted by the Fura-2 ratio at diastole), decay rate (tau), as well as duration and frequency of Ca2+ transients.
Calcium sensitivity analysis was performed as described previously (Jiang et al., 2007). Briefly, Ctrl- and RYR2–/–-iPSC-CMs loaded with Fura-2 were first exposed to 0 mM Ca2+ Tyrode’s solution until no spontaneous Ca2+ transient was detected and then continuously perfused with Tyrode’s solution containing Ca2+ of increasing concentrations (0, 0.1, 0.2, 0.3, 0.5, 1.0, and 2.0 mM). In the end, caffeine (10 mM; Sigma-Aldrich) was applied to confirm the activity of RYR2.
To examine the caffeine-induced Ca2+ release in iPSC-CMs, cells loaded with Fura-2 were washed with depolarization solution containing (in mM): NaCl 112, KCl 30, CaCl2 1.8, MgCl2 1, NaH2PO4 0.33, HEPES 10, and glucose 10 (pH adjusted to 7.3 with NaOH). Cytosolic Ca2+ level was measured before and after continuous addition of caffeine with increasing concentrations (from 0.025 to 5.0 mM).
To investigate the role of IP3R-mediated Ca2+ release in Ca2+ handling of iPSC-CMs, spontaneous Ca2+ transients were recorded before and after the application of the IP3R antagonists 2-aminoethoxydiphenyl borate (2-APB, 20 μM; Tocris) and Xestospongin C (XeC, 1 μM; abcam). To determine the contribution of SERCA-mediated SR Ca2+ uptake to Ca2+ cycling in iPSC-CMs, spontaneous Ca2+ transients were recorded before and after the application of the SERCA inhibitor thapsigargin (5 μM; Millipore). The changes of Ca2+ transient amplitude and frequency caused by the addition of these inhibitors were determined.
For these experiments, we used an imaging chamber (RC-47FSLP, Warner instruments), which is furnished with a field stimulation and a built-in aspiration port. For fluid control, a 12-valve superfusion system (DAD-VM, ALA Scientific Instruments) combined with a multi-tube pre-heater (MPRE8, Cell MicroControls) was used, which together allows a gentle, rapid, and direct solution change around the measured cells. The overflow solution was removed via the aspiration port by suction with a peristaltic pump (minipuls 3, Gilson).
Ctrl- and RYR2–/–-iPSC-CMs around day 80 were enzymatically singularized into single cells and seeded on coverslips at a density of 30,000 cells per 3.5-cm dish. Whole-cell patch-clamp recording of single cells was conducted using an EPC10 amplifier (HEKA Elektronik), controlled by the PatchMaster software (HEKA Elektronik). Series resistance was compensated by 85%. Pipette potentials (VPip) were corrected for liquid junction potentials. All experiments were performed at RT. Action potential (AP) measurement was performed under current-clamp mode without any current injection in RPMI 1640 basal medium using the pipette solution containing (in mM): KD-gluconate 100, KCl 60, MgATP 4, NaGTP 0.3, Na2-phosphocreatine 5, and HEPES 10 (pH adjusted to 7.2 with KOH). Voltage-clamp measurement of L-type calcium current (ICaL) was conducted in an extracellular solution containing (in mM): NMDG-Cl 140, CaCl2 1.8, MgCl2 1, HEPES 10, and glucose 10 (pH adjusted to 7.3 with HCl) and a pipette solution containing (in mM): CsCl 110, TEA-Cl 20, MgCl2 5, EGTA 10, HEPES 10, and Na2ATP 2 (pH adjusted to 7.2 with CsOH). To elicit ICaL, cells were clamped for 600 ms from the holding potential of –90 mV to test potentials between 70 and –80 mV in steps of –10 mV. The basic cycle length was 3 s.
Results are presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism 5 software with student’s t-test, the one-way ANOVA with the Dunnett’s multiple comparison test or the two-way ANOVA with Sidak’s correction for comparison of more groups and conditions. Results were considered statistically significant when the P-value was <0.05 (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
To edit the RYR2 gene, gRNA1 and gRNA2 were designed to target exon 90 (Figure 1A) and exon 99 (Supplementary Figure 1B) of RYR2, respectively. The success of the CRISPR/cas9 gene editing was screened by DNA sequencing (Figures 1B,C and Supplementary Figures 1C,D). The experiments revealed that the CRISPR/Cas9-targeted clone A3 had a homozygous deletion of one nucleotide (c.12230delC, exon 90), which resulted in a shift of the open reading frame. Consequently, the edited RYR2 gene in A3 clone obtained a premature termination codon (PTC) in the cytosolic domain of RYR2 (L4077Sfs*25) (Figure 1C), leading to a lack of the sequence encoding transmembrane domain, which forms the channel pore. Clone A5 showed homozygous deletion of eight nucleotides (c.14159-14166del, exon 99), leading to a PTC (W4721Dfs*8; Supplementary Figure 1D), and the loss of three transmembrane helical sections and the pore-forming region.
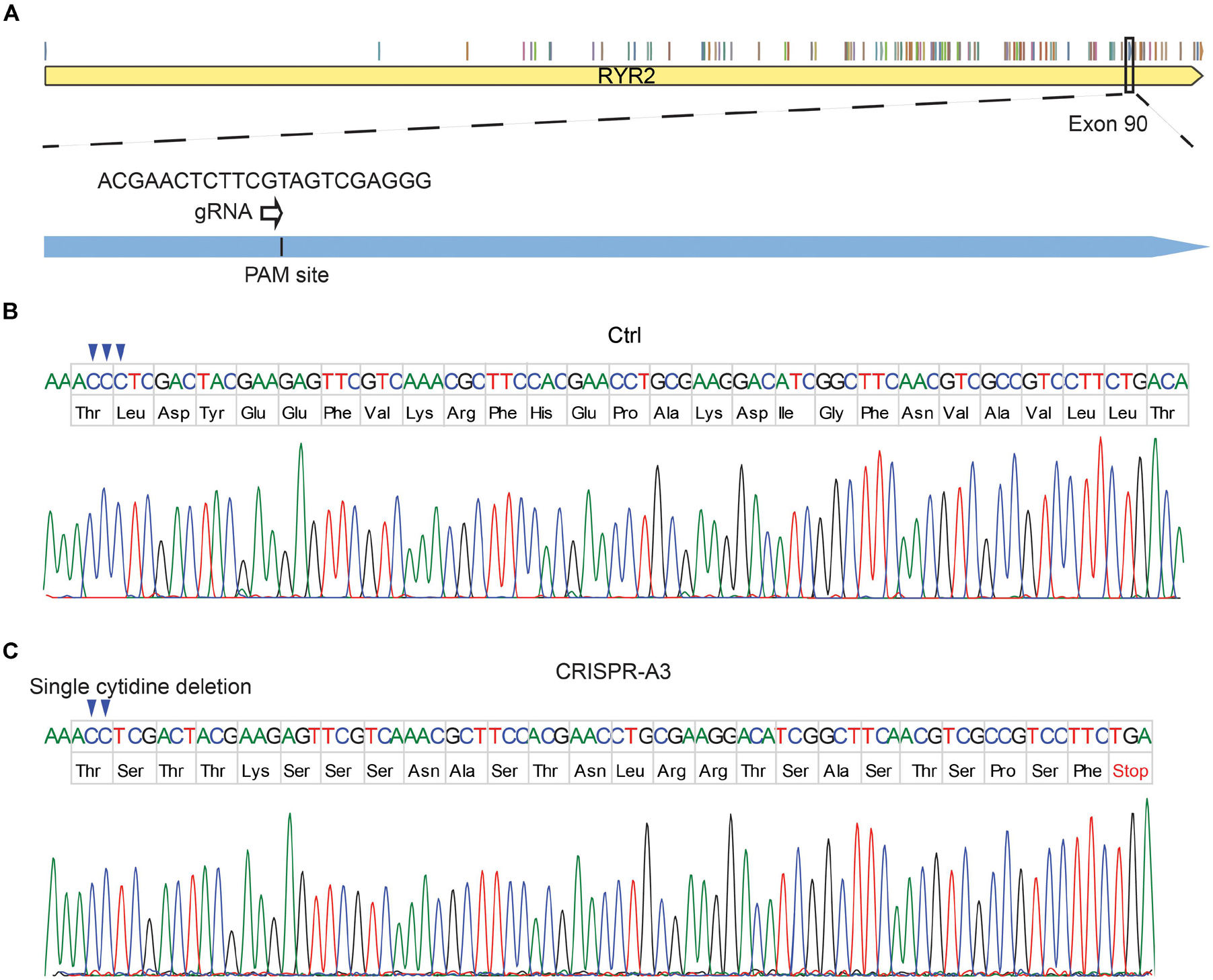
Figure 1. Generation of human homozygous RYR2 knockout iPSCs (RYR2–/–-iPSCs). (A) Illustration of the RYR2 locus and gRNA1 designed for CRISPR/Cas9-mediated gene editing in exon 90 of RYR2. (B,C) DNA sequencing of Ctrl- (B) and CRISPR/Cas9-edited A3 RYR2–/–-iPSC lines (C). The A3 RYR2–/–-iPSC line displayed a single homozygous cytidine deletion in the RYR2 gene, which resulted in a premature termination codon (PTC).
A3 and A5 RYR2–/–-iPSC lines were analyzed for their pluripotency. Both cell lines were positive for the human pluripotency markers OCT4, SOX2, LIN28, NANOG, TRA-1-60, and SSEA4 as demonstrated by immunostaining (Supplementary Figure 1E), similar to their parental Ctrl-iPSCs (Cyganek et al., 2018). In addition, both A3 and A5 RYR2–/–-iPSC lines expressed several pluripotency genes, such as SOX2, OCT4, NANOG, LIN28, and FOXD3 similar to the Ctrl-iPSCs (Supplementary Figure 1F). Differentiation potential of the edited iPSC lines in vitro was studied using the embryoid body-mediated differentiation method. Both A3 and A5 RYR2–/–-iPSCs differentiated into derivatives of three embryonic germ layers, as detected by the expression of genes encoding α-fetoprotein (AFP), cardiac troponin T (TNNT2), and synaptophysin (SYP) (Supplementary Figure 1G). Notably, the embryoid bodies formed from A3 and A5 RYR2–/–-iPSCs grew slower than those from Ctrl-iPSCs and revealed smaller sizes (data not shown).
Importantly, using the standard directed differentiation protocol, both A3 and A5 RYR2–/–-iPSCs were able to be successfully differentiated into spontaneously beating CMs (RYR2–/–-iPSC-CMs; Supplementary Videos 1, 2) similar to Ctrl-iPSCs (Supplementary Video 3). The percentage of cTNT-positive CMs at day 20 post differentiation of RYR2–/–-iPSCs (A3: 96.7 ± 0.5%, n = 3; A5: 96.6 ± 0.4%, n = 3) was similar to that of Ctrl-iPSCs (97.7 ± 0.8%, n = 3; Supplementary Figure 2A). These results indicate that loss of RYR2 does not alter the early cardiac commitment of iPSCs and the differentiation efficiency into CMs.
Although RYR2–/–-iPSC-CMs could be cultivated for up to 3 months and remained a high percentage of cTNT-positive cells similar to the Ctrl group (Ctrl: 98.4 ± 0.2%, n = 3; A3: 96.8 ± 2.4%, n = 3; A5: 96.7 ± 0.4%, n = 3; Supplementary Figure 2B), they grew differently with higher number of floating cells and signs of cell death in comparison to Ctrl-iPSC-CMs (Figures 2A,B and Supplementary Figures 2C,D) during long-term culture. To analyze the survival and proliferation rate during long-term culture, both Ctrl- and RYR2–/–-iPSC-CMs were replated at a fixed density of 0.8 million cells/well on day 20 post differentiation. No significant difference in cell numbers was observed between Ctrl- and A3 RYR2–/–-iPSC-CMs during the first five weeks after replating. However, while Ctrl-iPSC-CMs revealed a steady increase in their number until week 8 and remained stable afterward, RYR2–/–-iPSC-CMs showed no increase in their number. At week 10, the number of Ctrl-iPSC-CMs quadrupled to 3.2 ± 0.3 million cells/well, and the number of RYR2–/–-iPSC-CMs only increased to 1.4 ± 0.3 million cells/well, P < 0.0001 (Figure 2C). To analyze whether RYR2–/–-iPSC-CMs have a lower proliferation capacity, Ctrl- and RYR2–/–-iPSC-CMs at week 8 post replating were double immunostained for sarcomeric α-actinin and Ki67, a marker of cell proliferation. The percentage of proliferating CMs was quantified as the number of Ki67-positive nuclei divided by the total number of nuclei. The number of Ki67-positive cells was comparable in Ctrl- and RYR2–/–-iPSC-CMs (Ctrl: 3.9 ± 0.4%; A3-RYR2–/–: 4.6 ± 0.3%; A5-RYR2–/–: 3.6 ± 0.3%; Figures 2D,F,G), suggesting that the lower cell number in RYR2–/–-iPSC-CMs at week 10 is not due to a change in the cell proliferation capacity. Next, we performed the MTT assay to compare the cellular metabolic activity of Ctrl- and RYR2–/–-iPSC-CMs, which reflects the number of viable cells presented. The results showed a significant reduction of cell viability in RYR2–/–-iPSC-CMs compared to Ctrl-CMs (Figure 2E).
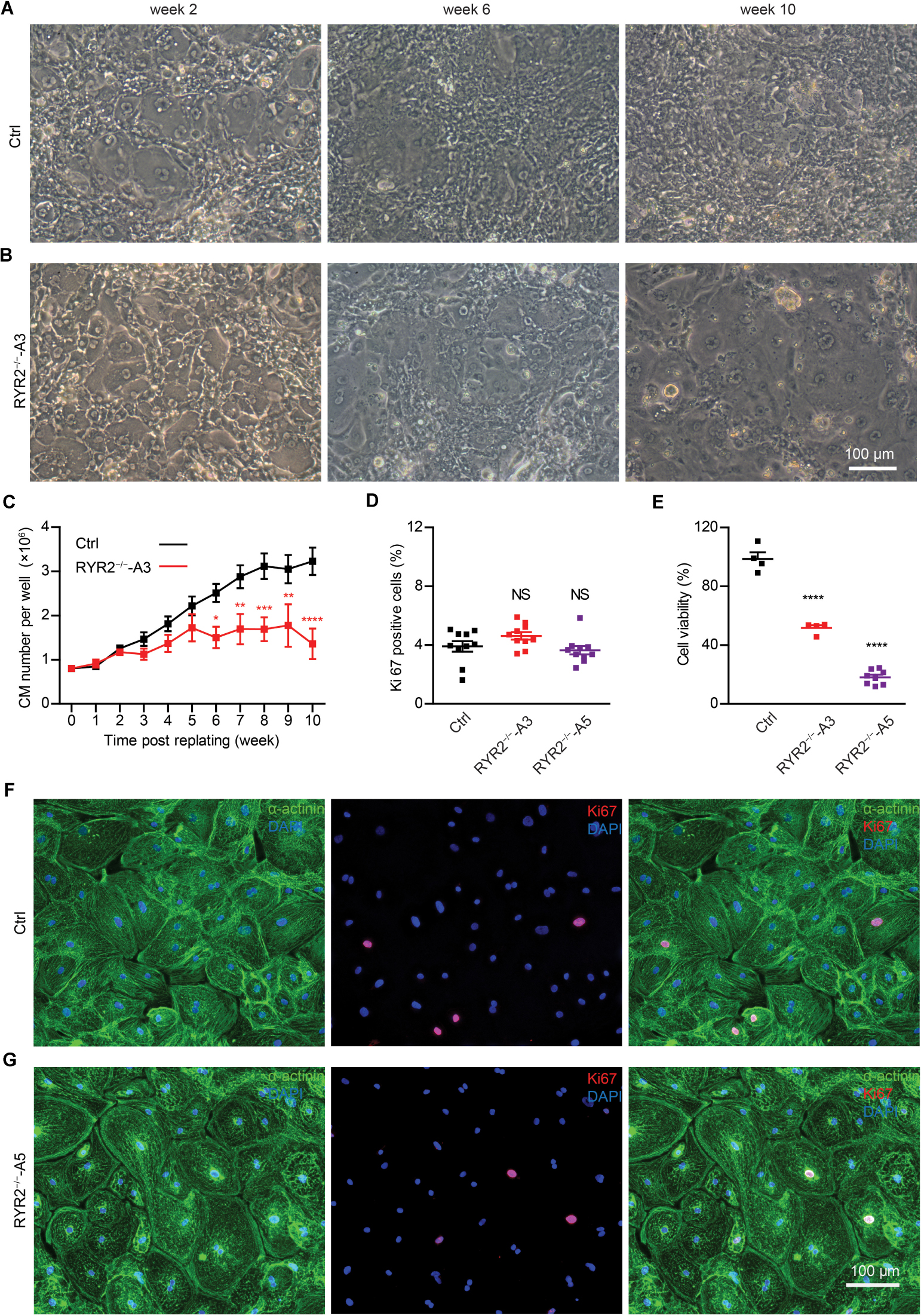
Figure 2. Growth analysis of Ctrl- and RYR2–/–-iPSC-CMs during long-term culture. (A,B) Bright-field images of Ctrl-iPSC-CMs (A) and A3 RYR2–/–-iPSC-CMs (B) after replating at day 20 of differentiation. Ctrl-iPSC-CMs showed an increase in cell density from week 2 to week 10 post replating (A), while RYR2–/–-iPSC-CMs exhibited reduced cell density (B). Scale bar, 100 μm. (C) Quantification of the cardiomyocyte numbers. Ctrl- and A3 RYR2–/–-iPSC-CMs showed distinct growth curves during long-term cell culture. Ctrl-CMs from 2 different iPSC lines and 11 differentiation experiments (iBM76.1: n = 6 and iWTD2.1: n = 5) were analyzed. A3 RYR2–/–-CMs from six differentiation experiments were used. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, RYR2–/– vs. Ctrl by using the two-way ANOVA with the Sidak’s multiple comparison test. (D) Percentage of Ki67-positive CMs (n = 10 samples each for Ctrl, A3 and A5, n = 300–400 cells per sample counted). (E) Cell viability of Ctrl-, A3 and A5 RYR2–/–-iPSC-CMs (Ctrl: n = 4 from two differentiation experiments; A3 RYR2–/–: n = 4 from two differentiation experiments; A5 RYR2–/–: n = 8 from two differentiation experiments). ****P < 0.0001 by using the one-way ANOVA with the Dunnett’s multiple comparison test (D,E). (F,G) Representative immunostaining of Ctrl- and RYR2–/–-iPSC-CMs probing for Ki67 (red) and α-actinin (green). Cells were counterstained with DAPI (blue) to show the nucleus. Scale bar, 100 μm.
To assess whether the RYR2 expression is lost in RYR2–/–-iPSC-CMs, different anti-RYR2 antibodies were applied for western blot analysis. Whereas the mouse monoclonal anti-RYR2 antibody, produced by using canine cardiac Ryr2 as the immunogen, was used to detect the full-length protein of RYR2, the rabbit polyclonal anti-RYR2 antibody (SAB4502707) recognizes both the full-length and the truncated proteins by binding the N-terminal region before the PTC. Neither the full length nor the truncated proteins of RYR2 were detectable in A3 (Figures 3A,B and Supplementary Figure 3A) and A5 RYR2–/–-iPSC-CMs (Supplementary Figure 3B), indicating the loss of functional RYR2 in A3 and A5 RYR2–/–-iPSC-CMs. Immunofluorescence staining using anti-α-actinin and anti-RYR2 antibodies further confirmed the absence of RYR2 in A3 and A5 RYR2–/–-iPSC-CMs (Figure 3D). However, the RYR2 mRNA levels were not altered in A3 and A5 RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs (Figure 4A and Supplementary Figure 3C). To investigate whether the absence of the truncated RYR2 in RYR2–/–-iPSC-CMs might be the result of protein degradation, isoprenaline-stimulated Ctrl- and RYR2–/–-iPSC-CMs were treated with MG132 (10 μM for 24 h) to inhibit the activity of the proteasome and with bafilomycin A1 (BafA1, 100 nM for 6 h) to inhibit the fusion of autophagosomes with lysosomes. Interference of the two major protein degradation pathways did not result in the detection of degraded RYR2 proteins in both A3 (Figure 3C) and A5 RYR2–/–-iPSC-CMs (Supplementary Figure 3D), suggesting that nonsense-mediated mRNA decay leads to no translation of the truncated RYR2 protein in RYR2–/–-iPSC-CMs.
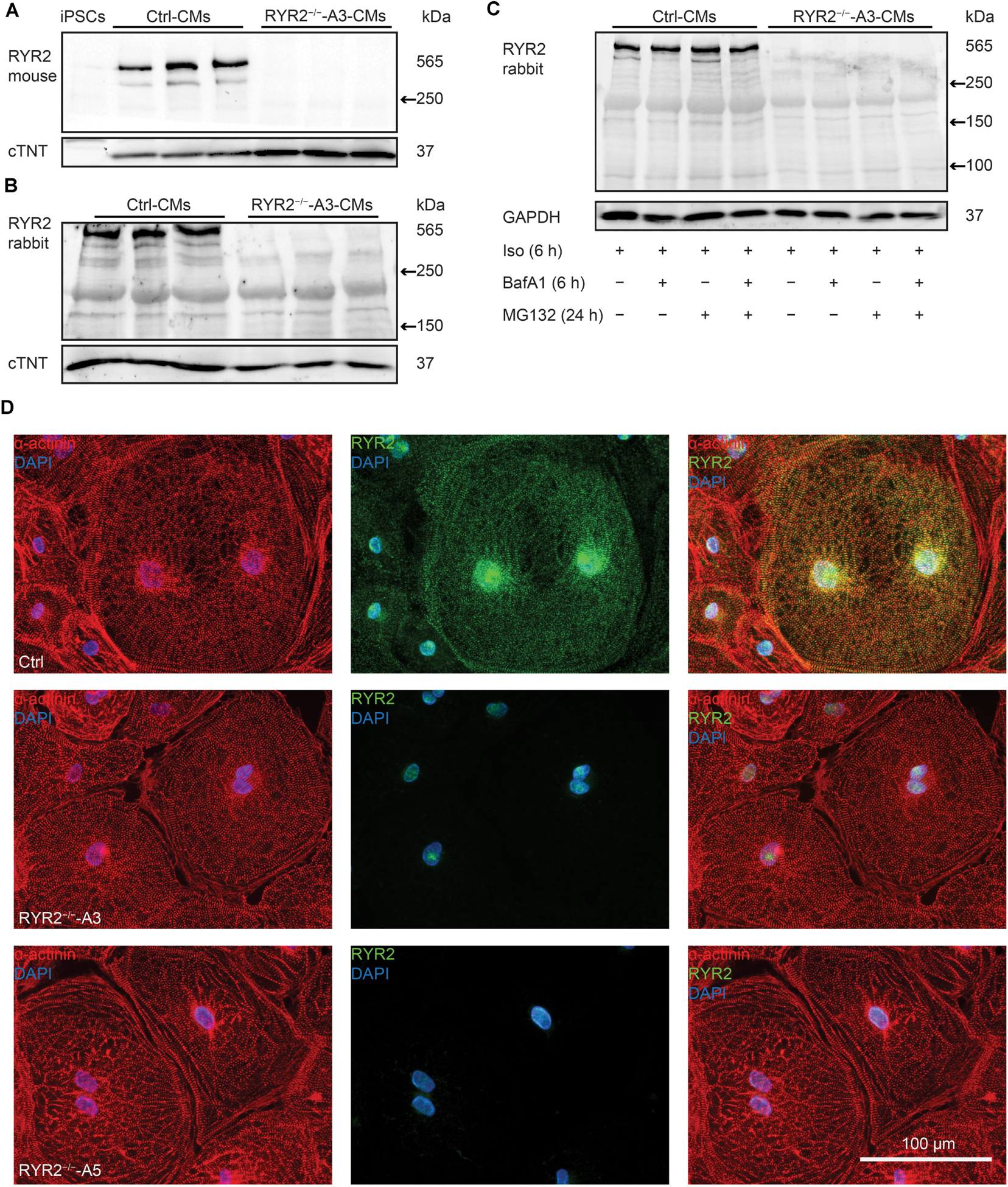
Figure 3. Expression of RYR2 in 3-month-old Ctrl- and RYR2–/–-iPSC-CMs. (A) Western blot analysis of the full-length RYR2 and cardiac troponin T (cTNT) in Ctrl- and A3 RYR2–/–-iPSC-CMs (Ctrl: n = 3 and A3 RYR2–/–: n = 3 independent differentiation experiments). (B) Western blot analysis showing protein expression of N-terminal RYR2 and cTNT in Ctrl- and A3 RYR2–/–-iPSC-CMs (Ctrl: n = 3 and A3 RYR2–/–: n = 3 independent differentiation experiments). (C) Representative western blot analysis of RYR2 in response to treatment with MG132 and/or Bafilomycin A (BafA1) in Ctrl- and RYR2–/–-iPSC-CMs (Ctrl: n = 3, A3 RYR2–/–: n = 3 independent differentiation experiments). No degraded RYR2 proteins were detected in RYR2–/–-iPSC-CMs by inhibiting protein degradation with the proteasome and calpain inhibitor MG132 and the autophagy inhibitor BafA1. (D) Representative immunostaining of Ctrl- and RYR2–/–-iPSC-CMs using antibodies against RYR2 (green) and α-actinin (red). The cells were counterstained with DAPI (blue) to show the nucleus. Scale bar, 100 μm.
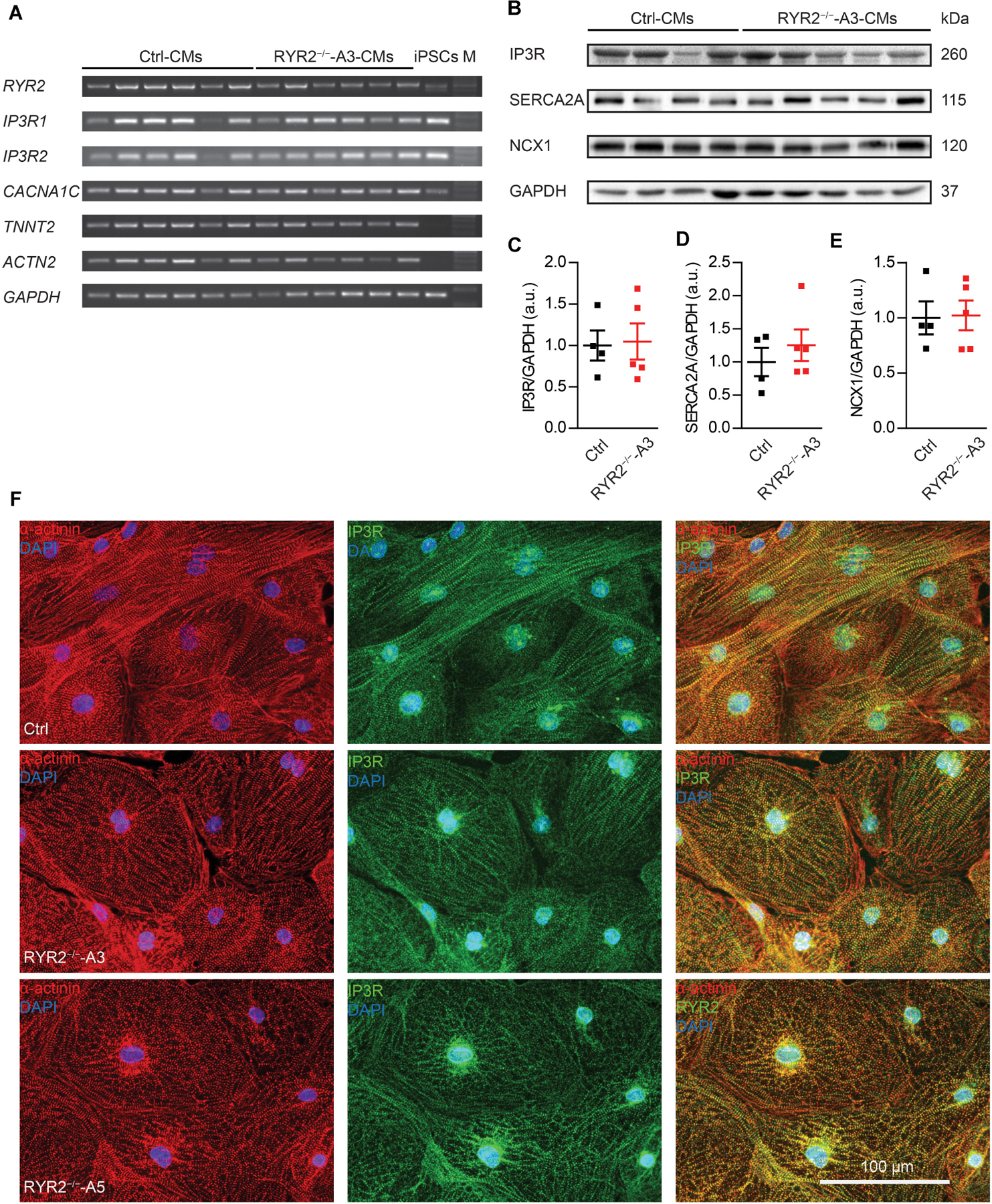
Figure 4. Expression of markers involved in Ca2+ signaling in RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs. (A) Reverse transcription-PCR analyses showing mRNA expression of transcripts for Ca2+ handling-related genes (RYR2, IP3R1, IP3R2, and CACNA1C), genes encoding sarcomeric proteins (TNNT2 and ACTN2), and the housekeeping gene GAPDH in Ctrl- and A3 RYR2–/–-iPSC-CMs (Ctrl: n = 6 and RYR2–/–-A3: n = 6 different differentiation experiments). M: DNA molecular-weight size marker. (B) Western blot showing expression of Ca2+ handling-associated proteins (IP3R, SERCA2A, and NCX1) in both Ctrl- and A3 RYR2–/–-iPSC-CMs. (C–E) Scatter dot plot showing protein levels of IP3R (C), SERCA2A (D), and NCX1 (E) normalized to GAPDH between Ctrl- and A3 RYR2–/–-iPSC-CMs (Ctrl: n = 4 and A3 RYR2–/–: n = 5 different differentiation experiments). (F) Representative immunostaining of Ctrl- and RYR2–/–-iPSC-CMs for IP3R (green) and α-actinin (red). Cells were counterstained with DAPI (blue). Scale bar, 100 μm.
Next, we evaluated the impact of loss of RYR2 on the expression of other cardiac Ca2+ signaling-associated genes in RYR2–/–-iPSC-CMs, such as genes encoding the cardiac IP3R types 1 and 2 (IP3R1, IP3R2) and the voltage-gated calcium channel subunit alpha-1C (CACNA1C). No tendency of increased or decreased expression of tested genes was observed in RYR2–/–-iPSC-CMs in comparison to their corresponding Ctrl-iPSC-CMs (Figure 4A and Supplementary Figure 3C). However, as RYR2–/–-iPSC-CMs remained the ability to spontaneously contract, we hypothesized that RYR2 may not be the only channel promoting intracellular calcium release. To verify potential compensatory mechanisms in response to loss of RYR2, we studied the expression of several Ca2+ -regulation related proteins. Consistent with IP3R mRNA levels, western blot analysis revealed no differences in IP3R protein expression in RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs. In addition, we detected no significant changes in the protein expression of SERCA2A and NCX1 between Ctrl- and RYR2–/–-iPSC-CMs (Figures 4B–E). To study the distribution of IP3R in iPSC-CMs, we conducted immunostaining by using antibodies against IP3R and sarcomeric α-actinin (Figure 4F). IP3R was expressed throughout the cytosol (Figure 4F middle) and partially co-localized with sarcomeric α-actinin (Figure 4F right). The perinuclear region displayed an intensive expression of IP3R. We did not observed differences in the distribution of IP3R between Ctrl- and RYR2–/–-iPSC-CMs.
To determine functional consequences of the loss of RYR2, Ca2+ sparks were measured in Ctrl- and RYR2–/–-iPSC-CMs after pacing at 0.25 Hz, which were pre-treated with 100 nM isoprenaline to activate the β-adrenergic signaling and to promote the occurrence of Ca2+ sparks. In Ctrl-iPSC-CMs, typical Ca2+ sparks occurred randomly throughout the cell (Figure 5A). In contrast, Ca2+ sparks appeared rarely in RYR2–/–-iPSC-CMs (Figure 5B) and showed a significantly lower frequency with smaller amplitude (ΔF/F0) compared to Ctrl-iPSC-CMs (Figure 5C). Additionally, RYR2–/–-iPSC-CMs showed significantly reduced FDHM and FWHM, indicating smaller sizes of the released Ca2+ sparks in RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs. This led to a 95.7 and 94.7% reduction of spontaneous SR Ca2+ leak in A3 and A5 RYR2–/–-iPSC-CMs, respectively, compared to Ctrl-iPSC-CMs (Figure 5C). These data indicate that RYR2 plays a major role in spontaneous Ca2+ leak from the SR at diastole. The remaining but smaller Ca2+ sparks detected in RYR2–/–-iPSC-CMs suggest the existence of some other Ca2+ regulatory channels, which may mediate Ca2+ sparks independent of RYR2.
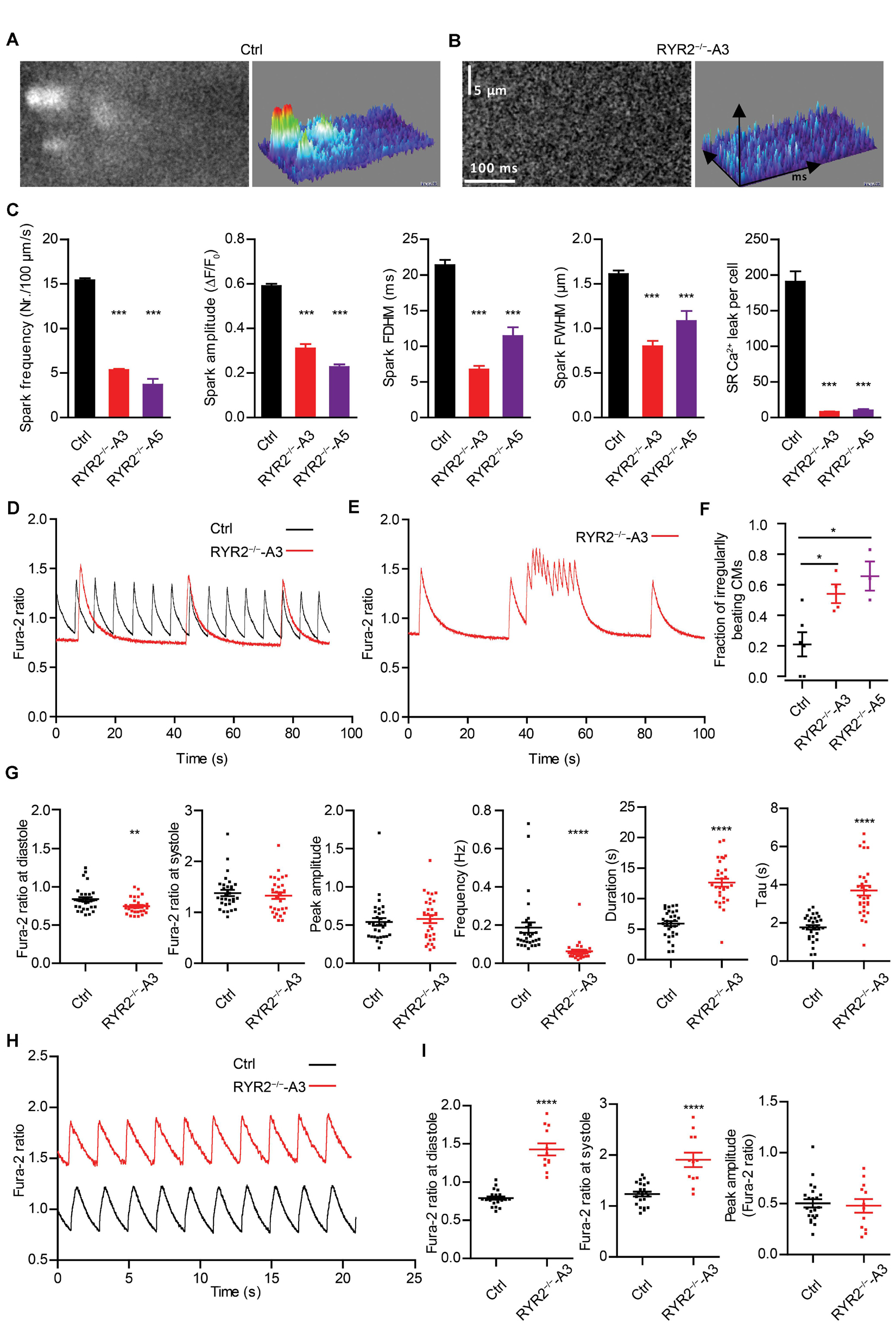
Figure 5. RYR2–/–-iPSC-CMs showed defective Ca2+ cycling. (A,B) Ca2+ sparks in Ctrl- and A3 RYR2–/–-iPSC-CMs during β-adrenergic stimulation. (A) Spontaneous Ca2+ spark images in Ctrl-iPSC-CMs (A; ncell = 358, nspark = 1521) and A3 RYR2–/–-iPSC-CMs (B; ncell = 21, nspark = 38) after treatment with 100 nM isoproterenol. Both Ctrl- and RYR2–/–-iPSC-CMs were paced at 0.25 Hz. (C) Bar graphs summarize the key parameters of Ca2+ sparks in mean ± SEM values for Ctrl- and RYR2–/–-iPSC-CMs: spark frequency (100 μm–1s–1), amplitude (ΔF/F0), FDHM (ms), FWHM (μm) and relative SR Ca2+ leak. For A5 RYR2–/–-iPSC-CMs, ncell = 12, nspark = 20 were analyzed. ***P < 0.001 by using the one-way ANOVA with the Dunnett’s multiple comparison test. (D) Representative Ca2+ transients in spontaneously beating Ctrl- and A3 RYR2–/–-iPSC-CMs. (E) Representative abnormal Ca2+ transients in A3 RYR2–/–-iPSC-CMs. (F) Fractions of irregularly beating Ctrl- and A3- and A5 RYR2–/–-iPSC-CMs (Ctrl: CMs from six differentiation experiments; A3 RYR2–/–: CMs from four differentiation experiments; A5 RYR2–/–: CMs from three differentiation experiments. n > 15 cells per experiment). *P < 0.05 by using the one-way ANOVA with the Dunnett’s multiple comparison test. (G) Scatter dot plot showing the diastolic Ca2+ levels, systolic Ca2+ levels, peak amplitude, frequency and duration of Ca2+ transients, as well as the time constant during the decay of Ca2+ transients (tau) in Ctrl- and A3 RYR2–/–-iPSC-CMs (Ctrl: n = 31 cells from five differentiation experiments; A3 RYR2–/–: n = 32 cells from three differentiation experiments). (H) Representative Ca2+ transients in Ctrl- and A3 RYR2–/–-iPSC-CMs under a normalized field stimulation at a pacing frequency of 0.5 Hz. (I) Scatter dot plot showing the diastolic Ca2+ levels, systolic Ca2+ levels and peak amplitude of paced Ca2+ transients in Ctrl- and A3 RYR2–/–-iPSC-CMs (Ctrl: n = 22 cells from three differentiation experiments; A3 RYR2–/–: n = 12 cells from two differentiation experiments). **P < 0.01; ****P < 0.0001 RYR2–/– vs. Ctrl by using the unpaired Student’s t-test.
To further investigate whether normal Ca2+ homeostasis was affected in RYR2–/–-iPSC-CMs, spontaneous Ca2+ transients were assessed. Although we detected spontaneous Ca2+ transients in both Ctrl- and RYR2–/–-iPSC-CMs (Figure 5D), 54.1 ± 6.1% of A3 and 65.7 ± 9.6% of A5 RYR2–/–-iPSC-CMs showed abnormalities in Ca2+ handling (Figures 5E,F). These irregularities were mainly detectable as highly frequent Ca2+ release events at elevated diastolic Ca2+ levels (Figure 5E), which were not observed in Ctrl-iPSC-CMs. Furthermore, RYR2–/–-iPSC-CMs showed significantly lower diastolic Ca2+ levels, but no significant differences in systolic Ca2+ levels and peak amplitude of Ca2+ transients in comparison to Ctrl-iPSC-CMs (Figure 5G and Supplementary Figure 3E). We also observed a significant decrease in the frequency of Ca2+ transients and an increase in the duration of Ca2+ transients in RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs (Figure 5G and Supplementary Figure 3E), indicating that loss of RYR2 results in reduced but prolonged contraction-relaxation cycles. Moreover, the time constant (tau) during the decay of Ca2+ transients was significantly increased in RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs (Figure 5G and Supplementary Figure 3E). By applying a field stimulation, we normalized the Ca2+ cycling of Ctrl- and RYR2–/–-iPSC-CMs to 0.5 Hz. Paced Ca2+ transients in A3 RYR2–/–-iPSC-CMs revealed a comparable peak amplitude (Figures 5H,I). However, the diastolic level of Ca2+ transients after pacing was much higher in RYR2–/–-iPSC-CMs than in non-paced RYR2–/–-iPSC-CMs as well as in Ctrl-iPSC-CMs (Figures 5H,I). Together with the increased time constant of spontaneous Ca2+ transients in RYR2–/–-iPSC-CMs (Figure 5G and Supplementary Figure 3E), these data suggest that the efficiency of Ca2+ removal from the cytosol either by reuptake into the SR or by pumping Ca2+ out of the cell is lower in RYR2–/–-iPSC-CMs than in Ctrl-iPSC-CMs.
To evaluate the relationship between extracellular Ca2+ ([Ca2+ ]o) levels and the occurrence of Ca2+ transients in iPSC-CMs, we measured Ca2+ events under different [Ca2+ ]o concentrations using the fluorescent Ca2+ dye Fura-2 in combination with Ca2+ imaging. To this end, spontaneously beating iPSC-CMs were first washed with Tyrode’s solution containing no Ca2+ until no spontaneous Ca2+ transient was detectable. Afterward, cytosolic Ca2+ changes were monitored in response to continuous perfusion with increasing Ca2+ concentrations (from 0 to 2.0 mM) and addition of 10 mM caffeine at the end. To quantify the fraction of Ca2+ -oscillating cells for each condition, the number of iPSC-CMs, which displayed spontaneous Ca2+ transients at each [Ca2+ ]o concentration was determined. As shown in Figure 6A, no Ca2+ transients in Ctrl-iPSC-CMs were detected with [Ca2+ ]o lower than 0.5 mM. Raising [Ca2+ ]o concentrations stepwise from 0.3 to 2.0 mM concomitantly increased the amplitude and frequency of Ca2+ transients in Ctrl-iPSC-CMs (Figures 6A,C), as well as the fraction of Ca2+ -oscillating cells (Figure 6D). In contrast, RYR2–/–-iPSC-CMs responded differently to increased [Ca2+ ]o concentrations (Figures 6B–D). While 6 out of 16 A3 RYR2–/–-iPSC-CMs did not display any Ca2+ transients from 0.1 to 2 mM [Ca2+ ]o, the remaining 10 cells revealed Ca2+ transients already at 0.1 or 0.2 mM [Ca2+ ]o (Figure 6B). Similar results were observed in A5 RYR2–/–-iPSC-CMs; 5 out of 12 cells showed no Ca2+ transients under all conditions while the remaining 7 cells exhibited Ca2+ transients already at 0.1 mM [Ca2+ ]o (Figure 6D). Furthermore, both A3 and A5 RYR2–/–-iPSC-CMs showed no caffeine-induced Ca2+ release as observed in Ctrl-iPSC-CMs (Figures 6A,B), which confirms the absence of functional RYR2.
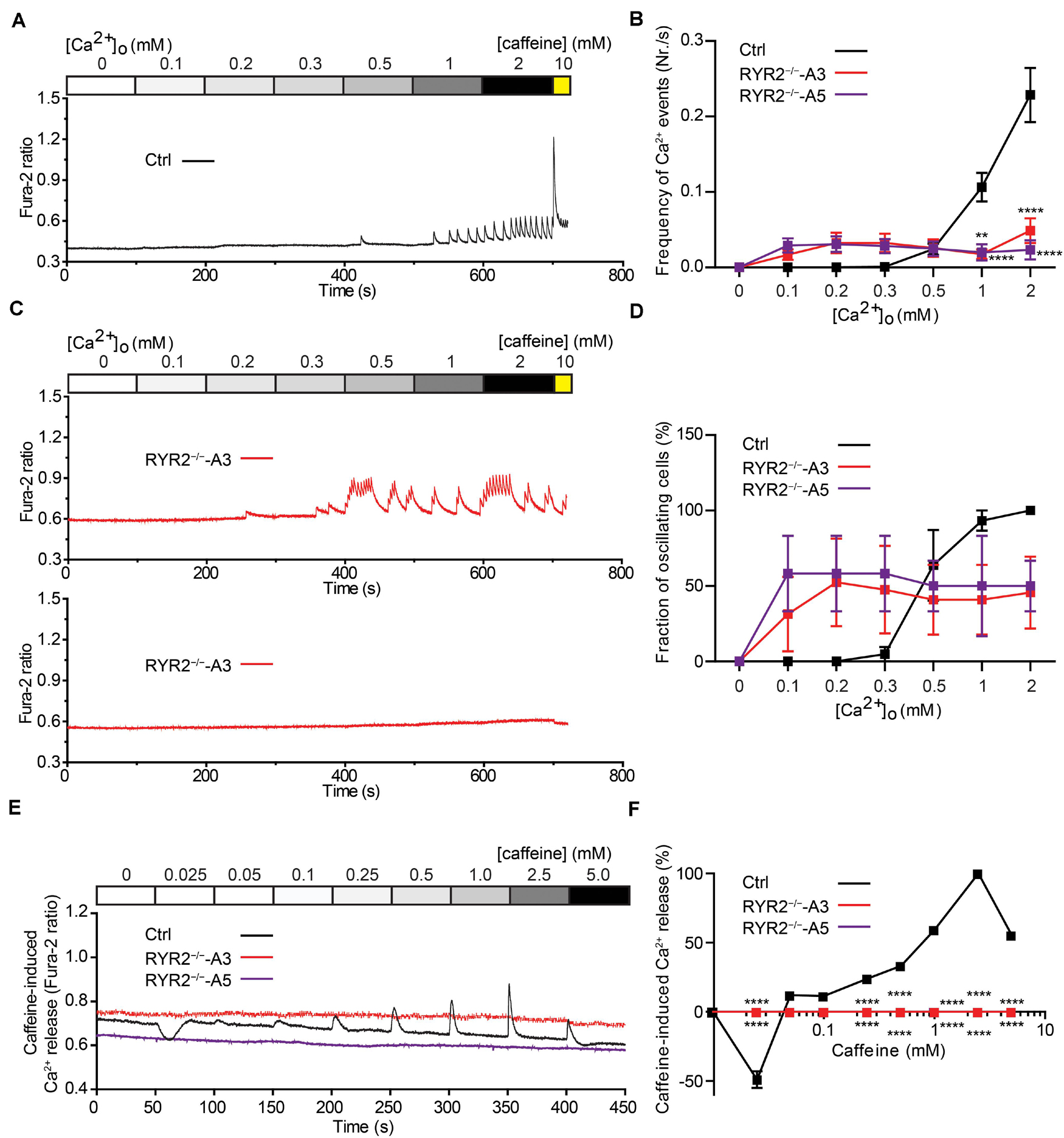
Figure 6. Sensitivity of Ctrl- and RYR2–/–-iPSC-CMs to extracellular calcium and caffeine. (A,B) Cytosolic Ca2+ dynamics in Ctrl-iPSC-CMs (A) and A3 RYR2–/–-iPSC-CMs (B) perfused with increasing concentrations of extracellular Ca2+ ([Ca2+ ]o). Cytosolic Ca2+ dynamics in A3 RYR2–/–-iPSC-CMs (B) were inconsistent from cell to cell. (C) Frequency of Ca2+ events detected in Ctrl- and RYR2–/–-iPSC-CMs under different [Ca2+ ]o. (D) Percentage of CMs displaying Ca2+ events under different [Ca2+ ]o concentrations among the three groups (Ctrl: n = 20 cells from four differentiation experiments; A3 RYR2–/–: n = 15 cells from three differentiation experiments; A5 RYR2–/–: n = 12 cells from two differentiation experiments). (E) Representative traces of caffeine-induced Ca2+ release in Ctrl- and RYR2–/–-iPSC-CMs. (F) Relationship between Ca2+ release and caffeine concentration in CMs from the two groups. The amplitude of each caffeine-induced Ca2+ release was normalized to the maximum peak for each experiment (Ctrl: n = 20 cells from four differentiation experiments; A3 RYR2–/–: n = 6 cells from two differentiation experiments; A5 RYR2–/–: n = 6 cells from two differentiation experiments). **P < 0.01, ****P < 0.0001 RYR2–/– vs. Ctrl by the two-way ANOVA with the Sidak’s multiple comparison test.
Caffeine, a pharmacological agonist of RYR2 (Porta et al., 2011), is commonly used to monitor RYR2-mediated Ca2+ release. To determine the impact of loss of RYR2 in iPSC-CMs, we measured cytosolic Ca2+ levels in Ctrl- and RYR2–/–-iPSC-CMs before and after continuously supplementing caffeine with increasing concentrations (from 0.025 to 5.0 mM). As shown in Figure 6E, low concentration of caffeine (0.025 mM) resulted in a reduction of cytosolic Ca2+ levels in Ctrl-iPSC-CMs, while the amplitudes of caffeine-induced Ca2+ release progressively increased under the caffeine treatment at concentrations from 0.05 to 2.5 mM and declined at the concentration of 5 mM (Figure 6F). We believe that the decrease in caffeine-induced Ca2+ release at 5 mM caffeine might be the result of depletion of internal Ca2+ stores in the SR due to previous caffeine supplementation. In contrast to Ctrl-iPSC-CMs, caffeine treatment did not result in any change of cytosolic Ca2+ levels in both A3 and A5 RYR2–/–-iPSC-CMs (Figures 6E,F), indicating that no functional RYR2 is present in RYR2–/–-iPSC-CMs and that Ca2+ transients observed in RYR2–/–-iPSC-CMs (Figures 5D,E, 6B) are not caused by Ca2+ release via RYR2, rather via other mechanisms.
Given that RYR2–/–-iPSC-CMs can spontaneously beat and release Ca2+ sparks and Ca2+ transients without RYR2 protein expression, we believe that these cells possess an alternative Ca2+ regulatory mechanism to compensate for the missing RYR2. IP3R-involved Ca2+ signaling has been discovered to play an important role during the process of cardiac development (Rosemblit et al., 1999; Poindexter et al., 2001). Therefore, we studied the effect of IP3R blockade on Ca2+ handling in iPSC-CMs. To this end, spontaneous Ca2+ transients in Ctrl- and A3 RYR2–/–-iPSC-CMs were measured before and after the application of 20 μM 2-APB. As shown in Figures 7A–C, 2-APB treatment for 500 s completely and reversibly blocked all Ca2+ transients in 10 out of 14 tested A3 RYR2–/–-iPSC-CMs (Figures 7B,C), while Ctrl-iPSC-CMs (10 out of 13) displayed intensive but slightly smaller Ca2+ transients (Figures 7A,C). Although 2-APB application decreased Ca2+ transient amplitude in Ctrl-iPSC-CMs (Figure 7D), it did not reduce the beating frequency of these cells (Figure 7E). On the contrary, in A3 RYR2–/–-iPSC-CMs, the amplitude and frequency of Ca2+ transients during 2-APB treatment for 100∼300 s declined to 54.7 and 49.1%, respectively, whereas the inhibitory effects increased as treatment lasted longer (300–500 s): the amplitude and frequency decreased to 20.1 and 12.9%, respectively (Figures 7D,E). Previous studies reported that 2-APB is also a blocker of store-operated Ca2+ entry independently of the function of IP3R (Iwasaki et al., 2001; Bootman et al., 2002). Therefore, we used the IP3R inhibitor Xestospongin C (XeC) to confirm our hypothesis. As shown in Figures 7F–J, the Ca2+ transients in 12 out of 13 A3 and 8 out of 10 A5 RYR2–/–-iPSC-CMs were completely and reversibly blocked after the addition of 1 μM XeC (Figures 7G,H), while the majority of Ctrl-iPSC-CMs (12 out of 14) still showed Ca2+ transients (Figures 7F,H). Additionally, RYR2–/–-iPSC-CMs showed a faster reaction to XeC (1 μM) than 2-APB (20 μM), as 200 s of XeC (1 μM) treatment was sufficient to block the Ca2+ transients in these cells. These observations imply that an IP3-gated Ca2+ pool is functional in iPSC-CMs, which is dominant in the modulation of Ca2+ handling in RYR2–/–-iPSC-CMs.
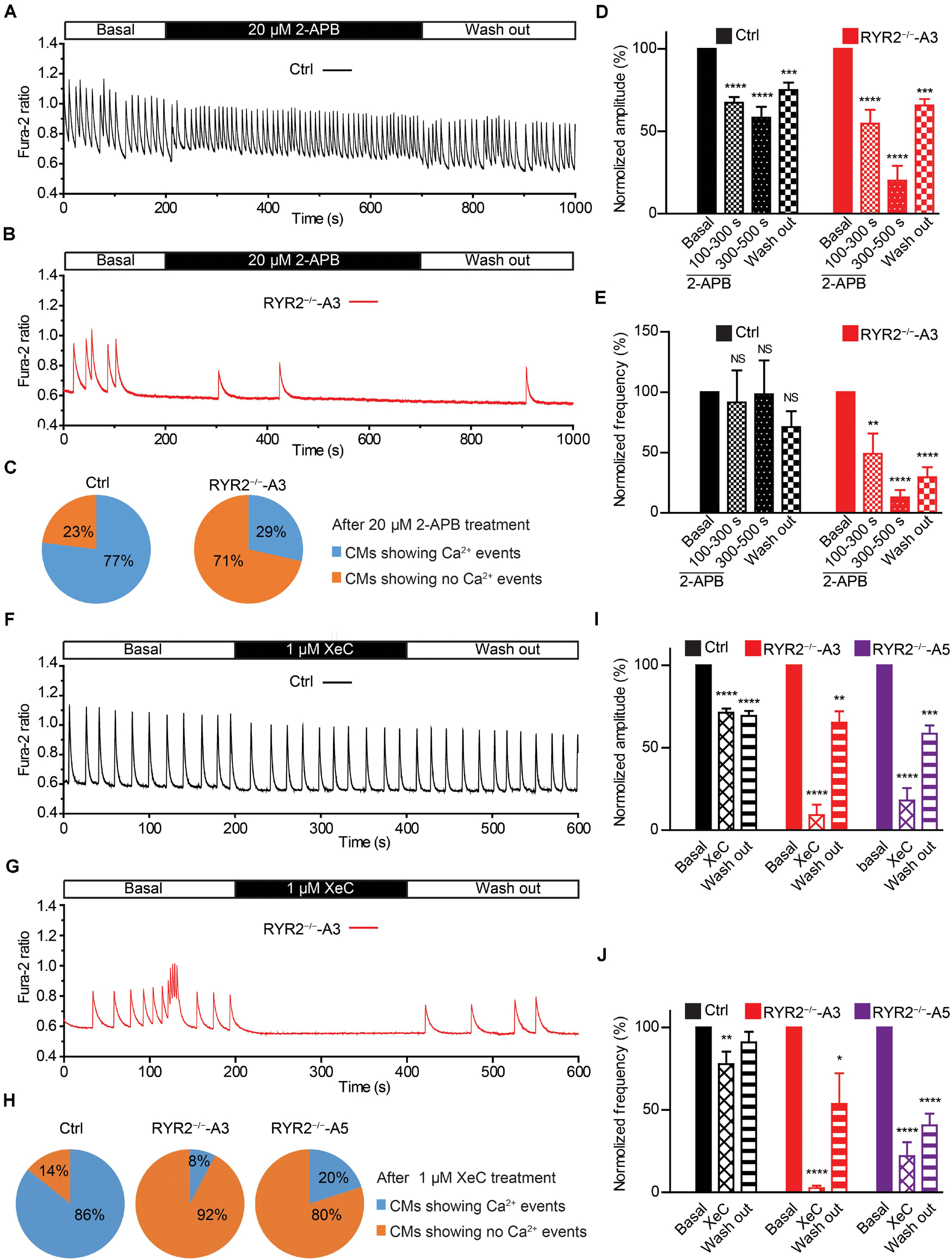
Figure 7. Contributions of IP3R-mediated Ca2+ release to spontaneous Ca2+ transients in Ctrl- and RYR2–/–-iPSC-CMs. (A,B) Representative cytosolic Ca2+ dynamics in Ctrl- (A) and RYR2–/–-iPSC-CMs (B) before and after the addition of IP3R antagonist 2-APB (20 μM), and after the washout of the drug. (C) Pie charts depict the percentage of cells maintaining Ca2+ transients after the treatment with 2-APB for 500 s. (D,E) Changes of Ca2+ transient amplitude (D) and frequency (E) after the treatment and the following removal of 2-APB, which were normalized to those from the same cell under the basal condition (Ctrl: n = 13 cells from three differentiation experiments; A3 RYR2–/–: n = 14 cells from three differentiation experiments). (F,G) Representative cytosolic Ca2+ dynamics in Ctrl- (F) and RYR2–/–-iPSC-CMs (G) before and after the addition of IP3R inhibitor Xestospongin C (XeC;1 μM), and after the washout of the drug. (H) Pie charts depict the percentage of cells maintaining Ca2+ transients after the treatment with XeC for 200 s. (I,J) Changes of Ca2+ transient amplitude (I) and frequency (J) after the treatment and following the washout of XeC, which were normalized to those from the same cell under the basal condition (Ctrl: n = 14 cells from three differentiation experiments; A3 RYR2–/–: n = 13 cells from three differentiation experiments; A5 RYR2–/–: n = 10 cells from two differentiation experiments). *P < 0.05; **P < 0.01; ***P < 0.001;****P < 0.0001 by using the one-way ANOVA with the Dunnett’s multiple comparison test.
We next studied the contribution of another important Ca2+ handling protein located on the SR membrane, namely SERCA. To this end, we recorded spontaneous Ca2+ transients in Ctrl- and RYR2–/–-iPSC-CMs before and after the application of 5 μM thapsigargin, a specific SERCA inhibitor. It turned out that thapsigargin treatment for 500 s was sufficient to block all Ca2+ transients in most of Ctrl- (12 out of 14), A3 (8 out of 10) and A5 RYR2–/–-iPSC-CMs (7 out of 8) in a time-dependent manner (Figures 8A–E). The amplitude and frequency of Ca2+ transients in Ctrl-iPSC-CMs declined to 21.8 and 13.2%, respectively, during thapsigargin treatment for 300∼500 s, which was comparable with the changes found in RYR2–/–-iPSC-CMs (Figures 8D,E). These findings indicate that the refilling of SR Ca2+ content by SERCA pumps is the main mechanism in both Ctrl- and RYR2–/–-iPSC-CMs with no significant differences.
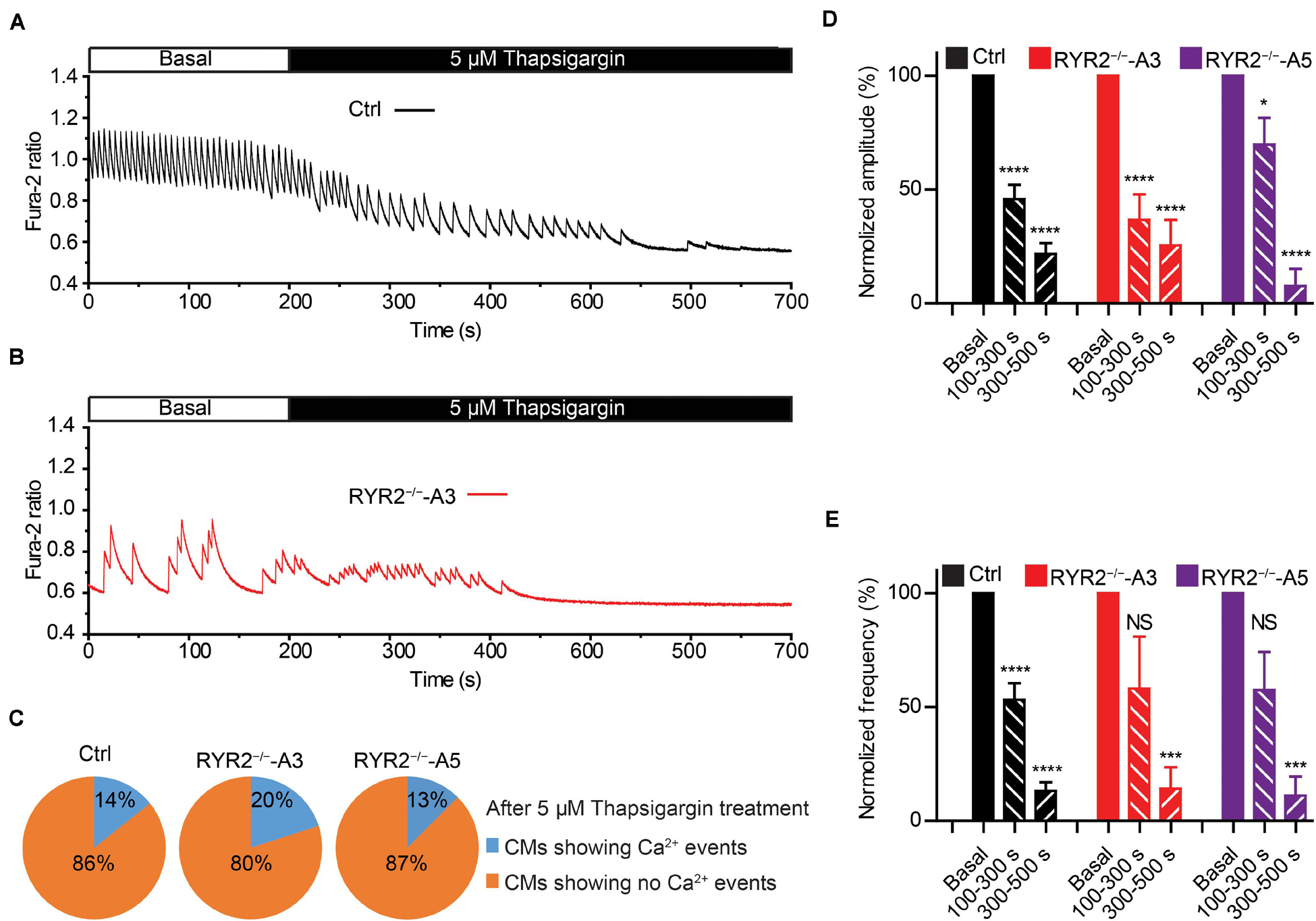
Figure 8. Contributions of SERCA-mediated SR Ca2+ uptake to spontaneous Ca2+ transients in Ctrl- and RYR2–/–-iPSC-CMs. (A,B) Representative cytosolic Ca2+ dynamics in Ctrl- (A) and RYR2–/–-iPSC-CMs (B) before and after the addition of SERCA inhibitor thapsigargin (5 μM). (C) Pie charts depict the percentage of cells maintaining Ca2+ transients after the treatment with thapsigargin for 500 s. (D,E) Changes of Ca2+ transient amplitude (D) and frequency (E) after the treatment with thapsigargin, which were normalized to those from the same cell under the basal condition. (Ctrl: n = 14 cells from three differentiation experiments; A3 RYR2–/–: n = 10 cells from three differentiation experiments; A5 RYR2–/–: n = 8 cells from two differentiation experiments). *P < 0.05; ***P < 0.001; ****P < 0.0001 by using the one-way ANOVA with the Dunnett’s multiple comparison test.
It is well-known that LTCC are the main transporter for trans-sarcolemmal Ca2+ influx responsible for the activation of RYR2. To understand whether loss of RYR2 function affects LTCC, we analyzed ICaL in Ctrl- and RYR2–/–-iPSC-CMs. We found that ICaL density was significantly lower in RYR2–/–-iPSC-CMs (Figures 9A–D). Compared to 3-month-old Ctrl-iPSC-CMs, both A3 and A5 RYR2–/–-iPSC-CMs at the same age presented a reduction of peak current density at 0 mV by half (Figure 9D). No significant difference in I-V curve of ICaL between A3 and A5 RYR2–/–-iPSC-CMs was detectable. In addition, a similar tendency was also found in 1-month-old RYR2–/–-iPSC-CMs (Figure 9C). To investigate whether the loss of RYR2 has an effect on the protein expression of L-type Ca2+ channel Cav1.2 in iPSC-CMs, we performed western blot analysis and found no differences between Ctrl- and RYR2–/–-iPSC-CMs (Figures 9E–G). These data suggest that the modulation of the LTCC (for example, calmodulin binding, or phosphorylation) might be altered in RYR2–/–-iPSC-CMs, leading to the inactivation of the channel or to a reduced open probability of the channel.
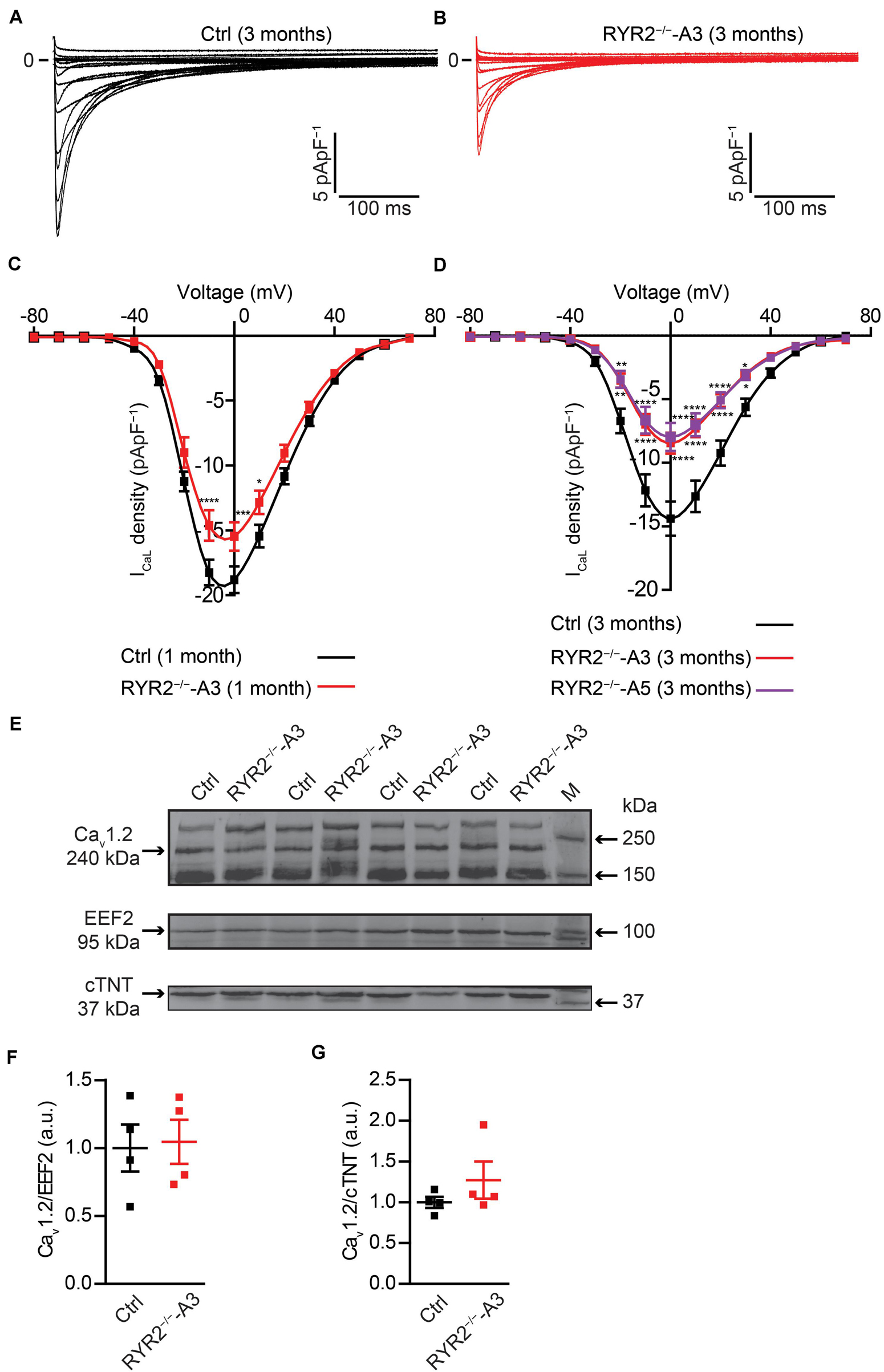
Figure 9. Electrophysiological analysis of ICaL in Ctrl- and RYR2–/–-iPSC-CMs. (A,B) Representative ICaL traces of 3-month-old Ctrl- (A) and A3 RYR2–/–-iPSC-CMs (B). (C) Average current-voltage (I–V) curves for peak ICaL in 1-month-old Ctrl- and RYR2–/–-iPSC-CMs (Ctrl: n = 12 cells from three differentiation experiments; RYR2–/–: n = 15 cells from two differentiation experiments). (D) Average current-voltage (I–V) curves for peak ICaL in 3-month-old Ctrl- and RYR2–/–-iPSC-CMs (Ctrl: n = 32 cells from four differentiation experiments; A3 RYR2–/–: n = 24 cells from three differentiation experiments; A5 RYR2–/–: n = 18 cells from two differentiation experiments). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 RYR2–/– vs. Ctrl by the two-way ANOVA with Sidak’s multiple comparison test. (E–G) Western blot analysis of the voltage-dependent calcium channel alpha 1C subunit (Cav1.2) in Ctrl- and A3 RYR2–/–-iPSC-CMs. Protein levels of Cav1.2 normalized to EEF2 (F) and cTNT (G) in Ctrl- and A3 RYR2–/–-iPSC-CMs. Ctrl: n = 4 and A3 RYR2–/–: n = 4 independent differentiation experiments.
Calcium-induced calcium release is fundamental for excitation-contraction coupling, in which RYR2 plays an important role. To investigate if RYR2–/–-iPSC-CMs are still able to generate cardiac action potentials (APs), we recorded APs of Ctrl- and RYR2–/–-iPSC-CMs at single-cell level (Figures 10A,B) and analyzed main AP parameters, including resting membrane potential (RMP), AP amplitude (APA), upstroke velocity (Vmax), and AP frequency (Figures 10C–F). While Ctrl-iPSC-CMs showed regular spontaneous APs with typical ventricular-like AP morphology, APs in both A3 and A5 RYR2–/–-iPSC-CMs remained mainly abnormal, as indicated by more positive RMP (Figure 10C), smaller APA (Figure 10D), reduced Vmax (Figure 10E), as well as lower and irregular beating frequency (Figures 10B,F).
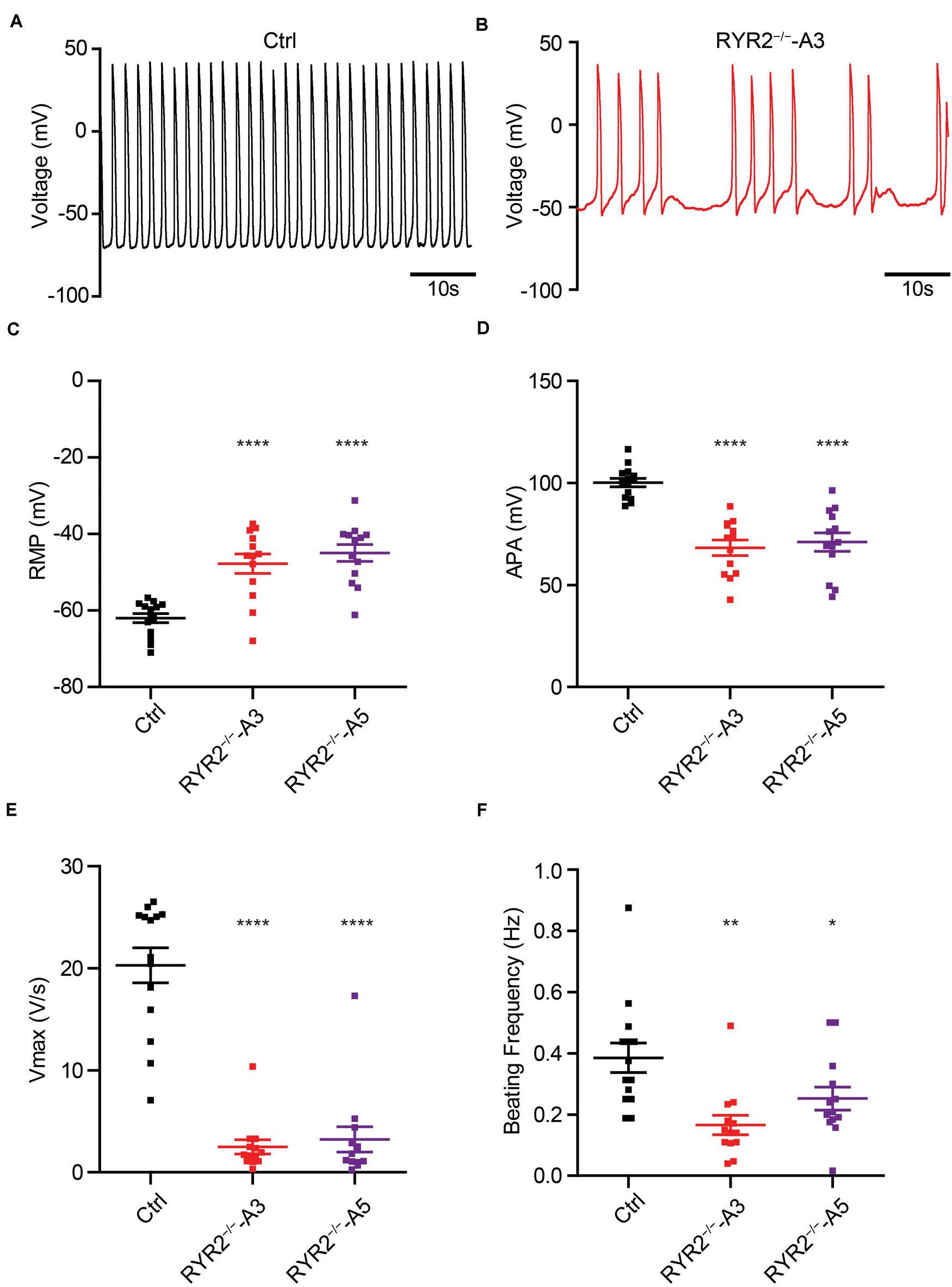
Figure 10. Analysis of action potentials in Ctrl- and RYR2–/–-iPSC-CMs. (A,B) Representative traces of APs revealed normal beating rhythm in Ctrl-iPSC-CMs (A) and arrhythmic beating in A3 RYR2–/–-iPSC-CMs (B). (C–F) Scatter dot plot comparing RMP (C), APA (D), maximal upstroke velocity Vmax (E), and beating frequency (F) between Ctrl- and RYR2–/–-iPSC-CMs (Ctrl: n = 12 cells from three differentiation experiments; A3 RYR2–/–: n = 13 cells from two differentiation experiments; A5 RYR2–/–: n = 13 cells from two differentiation experiments). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 by the one-way ANOVA with the Dunnett’s multiple comparison test.
In this study, we have generated two iPSC-based RYR2 knockout lines using the CRISPR/Cas9 gene-editing technology to study the physiological role of RYR2 in human iPSC-CMs. Our data reveal that (i) RYR2 is not essential for the early commitment of iPSCs into the cardiac lineage, while RYR2 is important for the viability and function of iPSC-CMs; (ii) RYR2-mediated Ca2+ release is essential for the maintenance of normal Ca2+ transients, which directly modulates the beating rate of iPSC-CMs; (iii) Ca2+ handling in iPSC-CMs is sensitive to [Ca2+ ]o, which mainly depends on RYR2; (iv) in the absence of functional RYR2, RYR2–/–-iPSC-CMs generate Ca2+ sparks and transients via compensative IP3R-mediated pathway; and v) refilling of SR Ca2+ store by SERCA pumps is required for the Ca2+ cycling in RYR2–/–-iPSC-CMs similar to Ctrl-iPSC-CMs. We conclude that SR Ca2+ release via RYR2 is critical for the survival and whole-cell Ca2+ handling of iPSC-CMs. When the function of RYR2 is lost, IP3R-mediated Ca2+ release from the SR compensates the RYR2 function partially.
So far, the importance of RYR2 in developing fetal heart has mainly been investigated in animal models. As documented previously, the homozygous deletion of the mouse Ryr2 gene is associated with embryonic cardiac arrest at around E10.5, even the looped heart tubes were formed at E8.5 and spontaneous heartbeat was observed at E9.5 (Takeshima et al., 1998). Cardiomyocyte-specific deletion of Ryr2 in mice caused calpain-10 dependent programmed cell death in vivo (Bround et al., 2013). Moreover, homozygous deletion of exon-3 in mouse Ryr2 gene led to embryonic lethality (Liu et al., 2014). In this study, we performed the first in vitro study to investigate the effect of homozygous knockout of RYR2 on the survival and function of human iPSC-CMs that are structurally and functionally similar to human embryonic CMs (Kolanowski et al., 2017). Generated human RYR2–/–-iPSCs retained stem cell morphology and pluripotency and were able to differentiate into spontaneously beating CMs, which is in line with the previous study using Ryr2–/–mESCs (Yang et al., 2002). We detected no differences in the proliferation capacity of RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs. However, RYR2–/–-iPSC-CMs displayed a tremendously low cell viability during long-term culture, which resulted from increased cell death, indicating that RYR2 is critical for the survival of human embryonic CMs.
The most striking finding of the current study is that RYR2–/–-iPSC-CMs developed an alternative mechanism for Ca2+ handling to compensate for the missing of RYR2. In functional CMs, Ca2+ enters the cell through LTCC and activates RYR2, which results in Ca2+ release from the SR into the cytosol (Sham et al., 1995; Adachi-Akahane et al., 1996). It was reported that knockout of Ryr2 in mESC-derived CMs resulted in the absence of Ca2+ release from the SR and an increase of ICaL (Fu et al., 2006). Inhibition of SERCA by thapsigargin hardly affected the dynamics of Ca2+ transients in both Ryr2–/– mESC-CMs and embryonic Ryr2–/– CMs (Liu et al., 2002; Guo and Yang, 2009). Ca2+ transients and CM contraction in the early embryonic stage of the mouse were attained by a compensatory Ca2+ influx into the cytosol via LTCC, accompanied by reduced Ca2+ release-induced Ca2+ channel inactivation (Liu et al., 2002; Guo and Yang, 2009). In contrast to these data, both 1- and 3-month-old RYR2–/–-iPSC-CMs in the current study displayed reduced ICaL, indicating that trans-sarcolemmal Ca2+ influx via LTCC during each AP was lower in RYR2–/–-iPSC-CMs compared to Ctrl-iPSC-CMs. Moreover, we noticed that treatment of iPSC-CMs with the SERCA inhibitor thapsigargin decreased the amplitude of Ca2+ transients in both Ctrl- and RYR2–/–-iPSC-CMs in a similar time-dependent manner, resulting in a complete inhibition of Ca2+ transients over long time treatment. Therefore, we assume that the differences observed between human iPSC-CMs and mESC-CMs might be a result of different species. Interestingly, a recent study demonstrated that mitochondrial Ca2+ flux modulates the spontaneous electrical activity in Ryr2–/– mESC-CMs, which depends on IP3R-mediated SR Ca2+ release (Xie et al., 2018).
IP3R shares structural and functional similarities to RYRs. IP3-operated releasable Ca2+ store has been demonstrated to be expressed and functional in human ESC- and iPSC-CMs (Satin et al., 2008; Sedan et al., 2008; Itzhaki et al., 2011). In our study, although IP3R gene and protein expression was not altered in RYR2–/–-iPSC-CMs, we discovered an increased sensitivity to the IP3R inhibitors 2-APB and XeC in iPSC-CMs. Treatment of Ctrl-iPSC-CMs with 20 μM 2-APB or 1 μM XeC, respectively, showed minor or nearly no effect on the Ca2+ transient frequency, while both inhibitors completely and reversibly blocked all Ca2+ events in more than 70% RYR2–/–-iPSC-CMs. These data demonstrate that the dependency of iPSC-CMs on IP3R-mediated Ca2+ release is significantly strengthened in the absence of functional RYR2. The finding of declined Ca2+ transient amplitude in Ctrl-iPSC-CMs after 2-APB application is consistent with a previous report showing that IP3-releasable Ca2+ pool contributes to whole-cell intracellular Ca2+ transients in human iPSC-CMs (Itzhaki et al., 2011). To maintain sustainable Ca2+ cycling, released Ca2+ must be removed from the cytosol. In adult human CMs, 70% of cytosolic Ca2+ during CM contraction is pumped back to the SR via SERCA to decrease intracellular Ca2+ level and to regulate SR Ca2+ load (Bers, 2002). SERCA-mediated Ca2+ uptake is required for Ca2+ handling in human iPSC-CMs (Itzhaki et al., 2011). These findings indicate that IP3R-mediated Ca2+ release and SERCA-mediated SR Ca2+ uptake are one of the major compensatory mechanisms for Ca2+ handling in human CMs when the RYR2 function is lost. Future studies should investigate whether IP3R-dependent mitochondrial Ca2+ flux modulates spontaneous electrical activity in human RYR2–/–-iPSC-CMs and whether IP3R-mediated nuclear Ca2+ signaling is involved in the activation of nuclear signal transduction for CM survival (Garcia and Boehning, 2017). Previous studies demonstrated that the store-operated calcium entry (SOCE) is present in embryonic cardiomyocytes and induces significant rise in Ca2+ entry when the depletion of SR Ca2+ stores occurs (Huang et al., 2006; Avila-Medina et al., 2018). In the future, it is also necessary to figure out whether SOCE plays an important role in Ca2+ handling in human CMs lacking functional RYR2, either by abolishing the SOCE response with known SOCE inhibitors (BTP-2 and SKF-96365) or by targeted knockdown of Orai1 with RNAi in RYR2–/–-iPSCs.
In the present study, we identified abnormalities of Ca2+ transients in more than 50% of RYR2–/–-iPSC-CMs. The abnormal Ca2+ transients were found only in a small number of Ctrl-iPSC-CMs, which is consistent with the results of other studies (Maizels et al., 2017) and may be explained by an inhomogeneous maturity of iPSC-CMs (Ronaldson-Bouchard et al., 2018). Calcium spark, an elementary event of Ca2+ release from SR, is believed to be mainly mediated by the spontaneous opening of one or a few RYR2 channels and contributes to the diastolic Ca2+ levels. In RYR2–/–-iPSC-CMs, β-adrenergic stimulation induced less generation of Ca2+ sparks with smaller size and amplitude compared to Ctrl-iPSC-CMs. This might explain the lower diastolic Ca2+ levels observed in spontaneously beating RYR2–/–-iPSC-CMs. Furthermore, Ca2+ transients in RYR2–/–-iPSC-CMs showed a comparable peak amplitude, but longer duration and slower tau compared to Ctrl-iPSC-CMs, suggesting that a higher amount of Ca2+ in one Ca2+ transient are released from the SR in RYR2–/–-iPSC-CMs. When RYR2–/–-iPSC-CMs are paced at 0.5 Hz, Ca2+ transients displayed comparable morphology and similar peak amplitude, but significantly elevated diastolic and systolic Ca2+ levels compared to Ctrl-iPSC-CMs. These findings suggest that Ca2+ cannot be removed rapidly and efficiently from the cytosol. Given that the SERCA-mediated refilling of SR Ca2+ is not significantly affected in RYR2–/–-iPSC-CMs, future studies should focus on the investigation of other mechanisms for Ca2+ removal from the cytosol, especially the function of NCX. Previous studies have shown that the majority of Ca2+ removal (about 75%) is accomplished by SERCA2A and by the sarcolemmal NCX (about 25%) in human cardiomyocytes. Further studies should focus on the investigation of the localization and activity of NCX in RYR2–/–-iPSC-CMs.
Since Ca2+ plays a critical role in the formation of Ca2+ transients, which are instrumental for subsequent cardiac contraction, we investigated the relationship between [Ca2+ ]o levels and Ca2+ transients in iPSC-CMs with RYR2 deficiency. Our results revealed that 3-month-old iPSC-CMs are sensitive toward [Ca2+ ]o at 0.5 mM or higher, with increased frequency and amplitude of Ca2+ transients, as well as increased fraction of Ca2+ -oscillating cells in response to elevated [Ca2+ ]o. These findings indicate a Ca2+ concentration dependent opening of RYR2 in iPSC-CMs, which is in line with the previous studies showing that human embryonic kidney (HEK) 293 cells expressing wild-type RYR2 displayed Ca2+ oscillations under [Ca2+ ]o at 0.3 mM or higher and in a concentration dependent manner (Jiang et al., 2007). In contrast, RYR2–/–-iPSC-CMs reacted differently to the gradually increased [Ca2+ ]o concentrations, showing Ca2+ transients already at very low [Ca2+ ]o concentrations (0.1–0.2 mM), suggesting the opening of IP3R occurred at lower intracellular Ca2+ concentration than RYR2. In RYR2–/–-iPSC-CMs, spontaneous Ca2+ transients with lower frequency in comparison to Ctrl-iPSC-CMs were detected, which is in line with the observation in Ryr2–/– mESC-CMs (Fu et al., 2006). Our data support the hypothesis by Korhonen et al. (2010) demonstrating that the developing CMs might lose the capability to generate homogeneous Ca2+ signals at a high frequency in response to the deficiency of the functional Ca2+ release channel RYR2. AP measurements revealed that loss of RYR2 resulted in a lower beating frequency in comparison to Ctrl-iPSC-CMs. These data are consistent with studies performed in mESC-derived CMs, showing similar decline in the beating frequency of Ryr2–/– mESC-derived CMs (Yang et al., 2002). In addition, cardiomyocyte-specific loss of Ryr2 in mice resulted in arrhythmogenic events, such as bradycardia and tachycardia (Bround et al., 2012). As RYR2–/–-iPSC-CM also displayed disturbances in the rhythmicity, manifesting bradycardia, we believe that RYR2 is a critical player in the regulation of heart rate.
Furthermore, our study provides the first comprehensive assessment regarding the sensitivity of iPSC-CMs to caffeine, a pharmacological agonist of cardiac RYR2 Ca2+ release channels. Continuously exposing Ctrl-iPSC-CMs to caffeine with increasing concentrations resulted in a series of cytosolic Ca2+ changes, while low concentration of caffeine (0.025 mM) decreased cytosolic Ca2+ levels. Under the stimulation of 0.025 mM caffeine, HEK 293 cells expressing wild-type RYR2 presented a caffeine-induced Ca2+ release (Liu et al., 2010; Roston et al., 2017). This difference may be caused by the different expression and assembling of Ca2+ handling elements in HEK cells compared to iPSC-CMs. Interestingly, HEK 293 cells transfected with wild-type RYR2 alone or with wild-type and I4855M mutant RYR2 together showed a slight decrease of cytosolic Ca2+ levels at 0.025 mM caffeine (Liu et al., 2010; Roston et al., 2017), which were normally considered an artifact. However, this phenomenon was not found in RYR2–/–-iPSC-CMs, suggesting that the decrease of cytosolic Ca2+ levels might be related to RYR2 opening. We assume that such a low concentration of caffeine might result in a small amount of Ca2+ release from the SR in Ctrl-iPSC-CMs, which might reach a threshold of NCX activation, leading to slow extrusion of calcium from the cytosol via NCX. Further study should be performed to investigate whether the NCX inhibitor can block the reduction of cytosolic Ca2+ levels at a low concentration of caffeine in Ctrl-iPSC-CMs. Importantly, no changes in cytosolic Ca2+ levels were detected in RYR2–/–-iPSC-CMs treated with caffeine at low or high concentrations, which indicate that IP3R-mediated SR Ca2+ release in RYR2–/–-iPSC-CMs is not sensitive to caffeine.
In conclusion, we generated two homozygous RYR2–/–-iPSC lines with normal capacity to differentiate into spontaneously beating CMs. However, RYR2–/–-iPSC-CMs in long-term culture showed enhanced cell death, indicating that RYR2 is critical for the survival but less important for the initiation of contraction of iPSC-CMs. Moreover, our study demonstrates that RYR2 plays a vital role in the maintenance of rhythmic heart beating and normal Ca2+ handling. Loss of functional RYR2 strengthens the dependency of iPSC-CMs on IP3R-related pathway, which combines with SERCA pumps to make critical contributions to the Ca2+ cycling in RYR2–/–-iPSC-CMs. As RYR2 is crucial for the development and function of CMs, the availability of human RYR2–/–-iPSC lines represent a useful tool for the in vitro study of early human heart development and molecular mechanism of Ca2+ cycling in human CMs.
All datasets generated for this study are included in the article/Supplementary Material.
The studies involving the use of human induced pluripotent stem cells were reviewed and approved by the Ethics Committee of the University Medical Center Göttingen (approval number: 21/1/11), and carried out in accordance with the approved guidelines. The patients/participants provided their written informed consent to participate in this study.
XL, WL, and KG conceived the study and designed experiments, contributed to interpretation of the data, and wrote the manuscript. XL, WL, KK, SH, LC, AS, MP, and MS performed experiments and acquisition of the data. XL, WL, KK, SH, AS, MS, and KG analyzed the data. All authors contributed to the article and approved the submitted version.
The authors would like to express great appreciation to the E-Rare-3 grant (JTC 2015) and the DFG (GU 595/3-1 and SFB1002 A04 to KG) for the financial support of this work. This work was also supported by the Free State of Saxony and the European Union EFRE (SAB projects “PhänoKard,” “HERMES,” and “PhenoCor” to KG) and the DFG Project Number 288034826–IRTG 2251: Immunological and Cellular Strategies in Metabolic Disease.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Jessie Pöche, Konstanze Fischer, Ying Ulbricht, Judith Müller, and Romy Kempe for their excellent technical assistances and Silke Tulok (CFCI TU Dresden) for assistance in LSM microscopy. We acknowledge the support of the Open Access Funding by the Publication Fund of the TU Dresden.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.00772/full#supplementary-material
Adachi-Akahane, S., Cleemann, L., and Morad, M. (1996). Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. J. Gen. Physiol. 108, 435–454. doi: 10.1085/jgp.108.5.435
Avila-Medina, J., Mayoral-Gonzalez, I., Dominguez-Rodriguez, A., Gallardo-Castillo, I., Ribas, J., Ordonez, A., et al. (2018). The complex role of store operated calcium entry pathways and related proteins in the function of cardiac, skeletal and vascular smooth muscle cells. Front. Physiol. 9:257. doi: 10.3389/fphys.2018.00257
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bootman, M. D., Collins, T. J., Mackenzie, L., Roderick, H. L., Berridge, M. J., and Peppiatt, C. M. (2002). 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16, 1145–1150. doi: 10.1096/fj.02-0037rev
Bround, M. J., Asghari, P., Wambolt, R. B., Bohunek, L., Smits, C., Philit, M., et al. (2012). Cardiac ryanodine receptors control heart rate and rhythmicity in adult mice. Cardiovasc. Res. 96, 372–380. doi: 10.1093/cvr/cvs260
Bround, M. J., Wambolt, R., Luciani, D. S., Kulpa, J. E., Rodrigues, B., Brownsey, R. W., et al. (2013). Cardiomyocyte ATP production, metabolic flexibility, and survival require calcium flux through cardiac ryanodine receptors in vivo. J. Biol. Chem. 288, 18975–18986. doi: 10.1074/jbc.M112.427062
Cyganek, L., Tiburcy, M., Sekeres, K., Gerstenberg, K., Bohnenberger, H., Lenz, C., et al. (2018). Deep phenotyping of human induced pluripotent stem cell-derived atrial and ventricular cardiomyocytes. JCI Insight 3:e99941. doi: 10.1172/jci.insight.99941
Eisner, D. A., Caldwell, J. L., Kistamas, K., and Trafford, A. W. (2017). Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195. doi: 10.1161/CIRCRESAHA.117.310230
Fu, J. D., Li, J., Tweedie, D., Yu, H. M., Chen, L., Wang, R., et al. (2006). Crucial role of the sarcoplasmic reticulum in the developmental regulation of Ca2+ transients and contraction in cardiomyocytes derived from embryonic stem cells. FASEB J. 20, 181–183. doi: 10.1096/fj.05-4501fje
Garcia, M. I., and Boehning, D. (2017). Cardiac inositol 1,4,5-trisphosphate receptors. Biochim. Biophys. Acta Mol. Cell Res. 1864, 907–914. doi: 10.1016/j.bbamcr.2016.11.017
Guo, A., and Yang, H. T. (2009). Ca2+ removal mechanisms in mouse embryonic stem cell-derived cardiomyocytes. Am. J. Physiol. Cell Physiol. 297, C732–C741. doi: 10.1152/ajpcell.00025.2009
Huang, J., van Breemen, C., Kuo, K. H., Hove-Madsen, L., and Tibbits, G. F. (2006). Store-operated Ca2+ entry modulates sarcoplasmic reticulum Ca2+ loading in neonatal rabbit cardiac ventricular myocytes. Am. J. Physiol. Cell Physiol. 290, C1572–C1582. doi: 10.1152/ajpcell.00226.2005
Itzhaki, I., Rapoport, S., Huber, I., Mizrahi, I., Zwi-Dantsis, L., Arbel, G., et al. (2011). Calcium handling in human induced pluripotent stem cell derived cardiomyocytes. PLoS One 6:e18037. doi: 10.1371/journal.pone.0018037
Iwasaki, H., Mori, Y., Hara, Y., Uchida, K., Zhou, H., and Mikoshiba, K. (2001). 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Recept. Channels 7, 429–439.
Jiang, D., Chen, W., Wang, R., Zhang, L., and Chen, S. R. (2007). Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc. Natl. Acad. Sci. U.S.A. 104, 18309–18314. doi: 10.1073/pnas.0706573104
Kapur, N., and Banach, K. (2007). Inositol-1,4,5-trisphosphate-mediated spontaneous activity in mouse embryonic stem cell-derived cardiomyocytes. J. Physiol. 581(Pt 3), 1113–1127. doi: 10.1113/jphysiol.2006.125955
Kockskamper, J., Zima, A. V., Roderick, H. L., Pieske, B., Blatter, L. A., and Bootman, M. D. (2008). Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J. Mol. Cell Cardiol. 45, 128–147. doi: 10.1016/j.yjmcc.2008.05.014
Kolanowski, T. J., Antos, C. L., and Guan, K. (2017). Making human cardiomyocytes up to date: derivation, maturation state and perspectives. Int. J. Cardiol. 241, 379–386. doi: 10.1016/j.ijcard.2017.03.099
Korhonen, T., Rapila, R., Ronkainen, V. P., Koivumaki, J. T., and Tavi, P. (2010). Local Ca2+ releases enable rapid heart rates in developing cardiomyocytes. J. Physiol. 588(Pt 9), 1407–1417. doi: 10.1113/jphysiol.2009.185173
Lian, X., Zhang, J., Azarin, S. M., Zhu, K., Hazeltine, L. B., Bao, X., et al. (2013). Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat. Protoc. 8, 162–175. doi: 10.1038/nprot.2012.150
Liu, W., Yasui, K., Opthof, T., Ishiki, R., Lee, J. K., Kamiya, K., et al. (2002). Developmental changes of Ca(2+) handling in mouse ventricular cells from early embryo to adulthood. Life Sci. 71, 1279–1292. doi: 10.1016/s0024-3205(02)01826-x
Liu, Y., Wang, R., Sun, B., Mi, T., Zhang, J., Mu, Y., et al. (2014). Generation and characterization of a mouse model harboring the exon-3 deletion in the cardiac ryanodine receptor. PLoS One 9:e95615. doi: 10.1371/journal.pone.0095615
Liu, Z., Wang, R., Tian, X., Zhong, X., Gangopadhyay, J., Cole, R., et al. (2010). Dynamic, inter-subunit interactions between the N-terminal and central mutation regions of cardiac ryanodine receptor. J. Cell Sci. 123(Pt 10), 1775–1784. doi: 10.1242/jcs.064071
Maizels, L., Huber, I., Arbel, G., Tijsen, A. J., Gepstein, A., Khoury, A., et al. (2017). Patient-specific drug screening using a human induced pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia type 2. Circ. Arrhythm. Electrophysiol. 10:e004725. doi: 10.1161/CIRCEP.116.004725
Poindexter, B. J., Smith, J. R., Buja, L. M., and Bick, R. J. (2001). Calcium signaling mechanisms in dedifferentiated cardiac myocytes: comparison with neonatal and adult cardiomyocytes. Cell Calcium. 30, 373–382. doi: 10.1054/ceca.2001.0249
Porta, M., Zima, A. V., Nani, A., Diaz-Sylvester, P. L., Copello, J. A., Ramos-Franco, J., et al. (2011). Single ryanodine receptor channel basis of caffeine’s action on Ca2+ sparks. Biophys. J. 100, 931–938. doi: 10.1016/j.bpj.2011.01.017
Ronaldson-Bouchard, K., Ma, S. P., Yeager, K., Chen, T., Song, L., Sirabella, D., et al. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. doi: 10.1038/s41586-018-0016-3
Rosemblit, N., Moschella, M. C., Ondriasova, E., Gutstein, D. E., Ondrias, K., and Marks, A. R. (1999). Intracellular calcium release channel expression during embryogenesis. Dev. Biol. 206, 163–177. doi: 10.1006/dbio.1998.9120
Roston, T. M., Guo, W., Krahn, A. D., Wang, R., Van Petegem, F., Sanatani, S., et al. (2017). A novel RYR2 loss-of-function mutation (I4855M) is associated with left ventricular non-compaction and atypical catecholaminergic polymorphic ventricular tachycardia. J. Electrocardiol. 50, 227–233. doi: 10.1016/j.jelectrocard.2016.09.006
Sasse, P., Zhang, J., Cleemann, L., Morad, M., Hescheler, J., and Fleischmann, B. K. (2007). Intracellular Ca2+ oscillations, a potential pacemaking mechanism in early embryonic heart cells. J. Gen. Physiol. 130, 133–144. doi: 10.1085/jgp.200609575
Satin, J., Itzhaki, I., Rapoport, S., Schroder, E. A., Izu, L., Arbel, G., et al. (2008). Calcium handling in human embryonic stem cell-derived cardiomyocytes. Stem Cells 26, 1961–1972. doi: 10.1634/stemcells.2007-2591
Sedan, O., Dolnikov, K., Zeevi-Levin, N., Leibovich, N., Amit, M., Itskovitz-Eldor, J., et al. (2008). 1,4,5-Inositol trisphosphate-operated intracellular Ca(2+) stores and angiotensin-II/endothelin-1 signaling pathway are functional in human embryonic stem cell-derived cardiomyocytes. Stem Cells 26, 3130–3138. doi: 10.1634/stemcells.2008-0777
Sham, J. S., Cleemann, L., and Morad, M. (1995). Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 92, 121–125. doi: 10.1073/pnas.92.1.121
Streckfuss-Bomeke, K., Wolf, F., Azizian, A., Stauske, M., Tiburcy, M., Wagner, S., et al. (2013). Comparative study of human-induced pluripotent stem cells derived from bone marrow cells, hair keratinocytes, and skin fibroblasts. Eur. Heart J. 34, 2618–2629. doi: 10.1093/eurheartj/ehs203
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Takeshima, H., Komazaki, S., Hirose, K., Nishi, M., Noda, T., and Iino, M. (1998). Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. EMBO J. 17, 3309–3316. doi: 10.1093/emboj/17.12.3309
Xie, A., Zhou, A., Liu, H., Shi, G., Liu, M., Boheler, K. R., et al. (2018). Mitochondrial Ca2+ flux modulates spontaneous electrical activity in ventricular cardiomyocytes. PLoS One 13:e0200448. doi: 10.1371/journal.pone.0200448
Yang, H. T., Tweedie, D., Wang, S., Guia, A., Vinogradova, T., Bogdanov, K., et al. (2002). The ryanodine receptor modulates the spontaneous beating rate of cardiomyocytes during development. Proc. Natl. Acad. Sci. U.S.A. 99, 9225–9230. doi: 10.1073/pnas.142651999
Keywords: ryanodine receptor 2, induced pluripotent stem cell-derived cardiomyocytes, CRISPR/Cas9, calcium handling, inositol 1,4,5-trisphosphate receptor
Citation: Luo X, Li W, Künzel K, Henze S, Cyganek L, Strano A, Poetsch MS, Schubert M and Guan K (2020) IP3R-Mediated Compensatory Mechanism for Calcium Handling in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes With Cardiac Ryanodine Receptor Deficiency. Front. Cell Dev. Biol. 8:772. doi: 10.3389/fcell.2020.00772
Received: 13 January 2020; Accepted: 22 July 2020;
Published: 12 August 2020.
Edited by:
Shinsuke Yuasa, Keio University, JapanReviewed by:
Keiko Uchida, Keio University, JapanCopyright © 2020 Luo, Li, Künzel, Henze, Cyganek, Strano, Poetsch, Schubert and Guan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaomei Guan, a2FvbWVpLmd1YW5AdHUtZHJlc2Rlbi5kZQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.