 Lesang Shen1†
Lesang Shen1† Danfeng Lin2†
Danfeng Lin2† Xiaoyi Li3
Xiaoyi Li3 Haijian Wu4
Haijian Wu4 Cameron Lenahan5,6
Cameron Lenahan5,6 Yuanbo Pan5,6
Yuanbo Pan5,6 Weilin Xu5,6
Weilin Xu5,6 Yiding Chen1*
Yiding Chen1* Anwen Shao4*
Anwen Shao4* Jianmin Zhang4
Jianmin Zhang4- 1Department of Breast Surgery, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 2Department of Surgical Oncology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 3Department of Nuclear Medicine and PET-CT Center, The Second Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 4Department of Neurosurgery, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 5Burrell College of Osteopathic Medicine, Las Cruces, NM, United States
- 6Center for Neuroscience Research, School of Medicine, Loma Linda University, Loma Linda, CA, United States
Acute central nervous system (CNS) injuries, such as stroke, traumatic brain injury (TBI), and spinal cord injury (SCI) present a grave health care challenge worldwide due to high morbidity and mortality, as well as limited clinical therapeutic strategies. Established literature has shown that oxidative stress (OS), inflammation, excitotoxicity, and apoptosis play important roles in the pathophysiological processes of acute CNS injuries. Recently, there have been many studies on the topic of ferroptosis, a form of regulated cell death characterized by the accumulation of iron-dependent lipid peroxidation. Some studies have revealed an emerging connection between acute CNS injuries and ferroptosis. Ferroptosis, induced by the abnormal metabolism of lipids, glutathione (GSH), and iron, can accelerate acute CNS injuries. However, pharmaceutical agents, such as iron chelators, ferrostatin-1 (Fer-1), and liproxstatin-1 (Lip-1), can inhibit ferroptosis and may have neuroprotective effects after acute CNS injuries. However, the specific mechanisms underlying this connection has not yet been clearly elucidated. In this paper, we discuss the general mechanisms of ferroptosis and its role in stroke, TBI, and SCI. We also summarize ferroptosis-related drugs and highlight the potential therapeutic strategies in treating various acute CNS injuries. Additionally, this paper suggests a testable hypothesis that ferroptosis may be a novel direction for further research of acute CNS injuries by providing corresponding evidence.
Introduction
Acute CNS injuries, including stroke, TBI, and SCI, are a major burden of morbidity and mortality worldwide (GBD, 2016, 2019). Each year, approximately 80 million individuals in the United States suffer a stroke. Moreover, deaths caused by stroke contribute to nearly 5% of all deaths in the United States. Ischemic stroke accounts for 87% of all strokes, with ICH comprising the remaining 10% (Benjamin et al., 2019). Another neurological disease worth mentioning is TBI, which has a global incidence of more than 50 million cases annually (Maas et al., 2017; Jiang et al., 2019). Regarding the mechanisms associated with acute CNS injuries, previous literature has shown that various mechanisms, including OS, inflammation, excitotoxicity, and apoptosis, play important roles in the pathophysiological processes of acute CNS, and targeting these mechanisms may provide neuroprotection (Roth et al., 2014; Chamorro et al., 2016; Duan et al., 2019; Nazemi et al., 2020). However, there is a lack of effective therapeutic strategies in treating long-term CNS injuries. Patients who survive CNS injuries often have long-term disabilities due to substantial neurological deficits and impaired tissue function, therefore requiring subsequent lifelong care. New therapeutic approaches are urgently required to improve outcomes of patients with acute CNS injuries. In recent years, there has been increasing interest in ferroptosis, suggesting a potential role of ferroptosis in acute CNS injuries and offering opportunities for novel pharmacological interventions, as ferroptosis can be modulated by small molecules (Friedmann Angeli et al., 2014; Lewerenz et al., 2018; Alim et al., 2019).
Ferroptosis, first observed in response to treatment of tumor cells via small-molecule chemical probes, is a newly identified form of regulated cell death characterized by the accumulation of iron-mediated lipid peroxides (Dixon et al., 2012). It differs from other programmed cell deaths (e.g., apoptosis, necrosis, and autophagy) at the morphological, biological, and genetic levels (Dixon et al., 2012). Regarding the function of ferroptosis within the tumor, it is associated with malignant transformation, cancer progression, and drug resistance [for review see Su et al. (2020)]. Moreover, ferroptosis regulation may be useful for anti-cancer therapy (Guo et al., 2019; Su et al., 2020). Although ferroptosis was first defined in cancer cells and has potential in cancer treatment, the latest experimental results have identified its role in the pathophysiology of acute organ injuries, such as acute kidney, lung, and brain injuries (Dixon et al., 2012; Friedmann Angeli et al., 2014; Kenny et al., 2019; Hu et al., 2020; Li Y.C. et al., 2020). More importantly, ferroptosis can cause neuronal cell death and neurological deficits in CNS injuries and human neurodegenerative diseases (Dixon et al., 2012; Morris et al., 2018). Therefore, targeting ferroptosis through effective anti-ferroptotic agents may provide direction for treating acute CNS injuries (Tuo et al., 2017; Zille et al., 2017; Kenny et al., 2019). So, what is the underlying mechanism of ferroptosis, and how does it affect acute CNS injuries?
Discovery and Mechanisms of Ferroptosis
Small-molecule probes are valuable tools for studying different types of regulated cell death (Gangadhar and Stockwell, 2007). During the identification of ferroptosis, there were two important chemical probes. Ferroptosis inducers, erastin, and RSL3, were discovered in a phenotypic small molecule-screening study (Dolma et al., 2003; Yang and Stockwell, 2008). Erastin, a synthetic compound, was capable of inducing non-apoptotic cell death to selectively kill HRAS-mutant engineered cancer cells, and in this process, there was no evidence of caspase activation or apoptotic hallmarks (Dolma et al., 2003; Yagoda et al., 2007). Another compound was RSL3, found in 2008, and capable of triggering a similar form of non-apoptotic and iron-dependent oxidative cell death (Yang and Stockwell, 2008). This erastin- and RSL3-induced cell death did not exhibit the morphological or biochemical features of apoptosis, and inhibition of necroptosis or autophagy had no effect on this mode of cell death (Wolpaw et al., 2011; Dixon et al., 2012; Yang et al., 2014). However, this manner of cell death could be prevented by the iron chelator, DFO, and antioxidants (e.g., Vitamin E) (Dolma et al., 2003; Yagoda et al., 2007; Yang and Stockwell, 2008). Therefore, the term “ferroptosis” was first proposed in 2012 to describe this novel iron-dependent non-apoptotic cell death (Dixon et al., 2012). Hallmark contributions of ferroptosis were well-displayed by Hirschhorn and Stockwell (2019) and Li J. et al. (2020) [for review see Hirschhorn and Stockwell (2019); Li J. et al. (2020)]. Hadian and Stockwell (2020) drew a SnapShot to provide an overview of ferroptosis-related pathways. Although the exact mechanisms of ferroptosis are still being explored, the initiation and execution of ferroptosis involve several biological processes, including lipid, GSH, and iron metabolism, as well as other regulatory processes (Dixon et al., 2012; Figure 1).
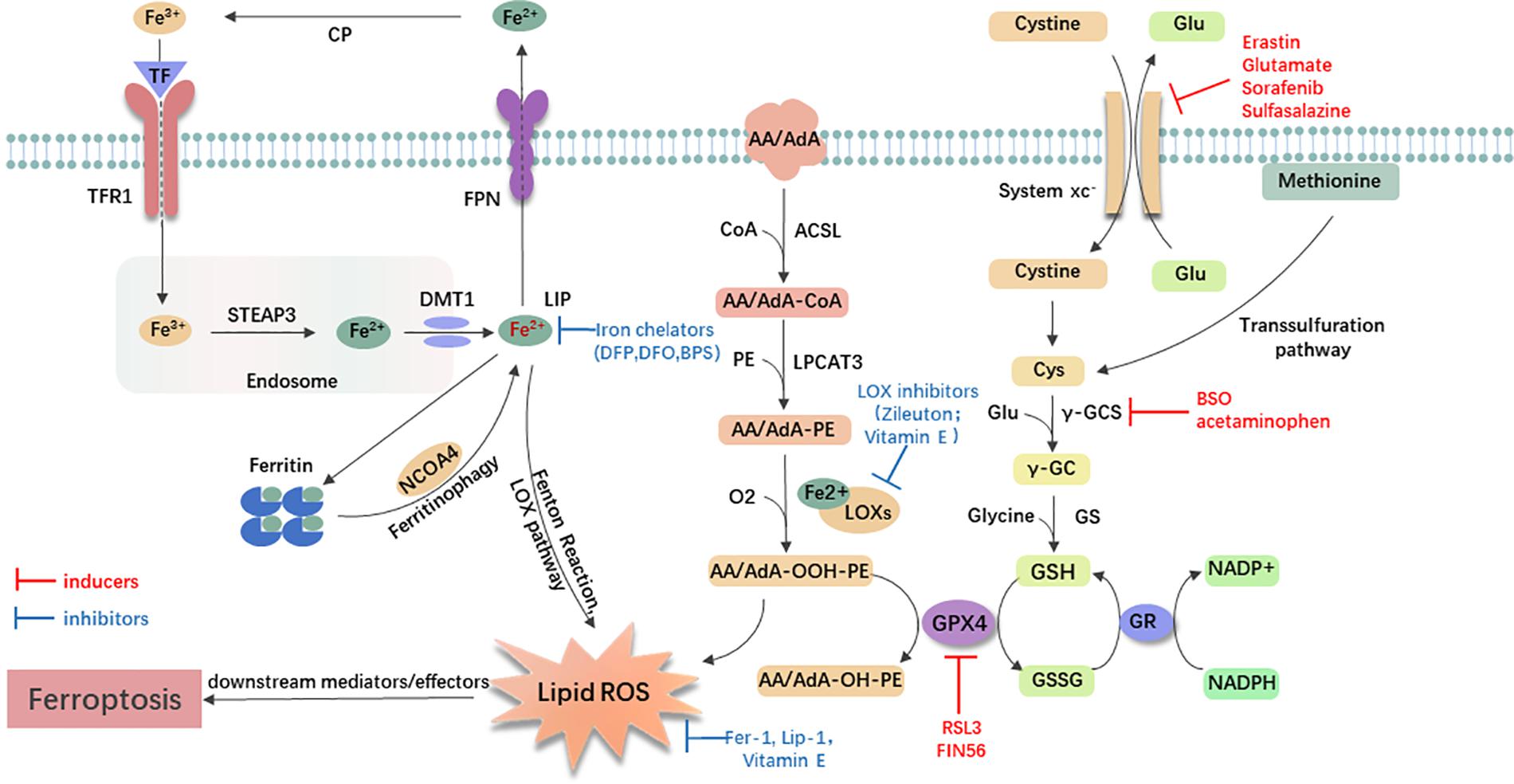
Figure 1. General mechanism of ferroptosis associated with lipid, amino acid, and iron metabolism. The inactive Fe3+ is delivered into the cell by TFR1 and reduced to Fe2+ in the endosome. Then, DMT1 transports Fe2+ to the labile iron pool (LIP). Autophagic degradation of ferritin (ferritinophagy) releases Fe2+ from ferritin, which is mediated by NCOA4. Fe2+ produces lipid ROS via the Fenton reaction and through the LOX pathway. Moreover, ACSL4 is required to activate polyunsaturated fatty acid, especially AA and AdA, to AA/AdA-CoA, then LPCAT3 catalyzes these derivatives and membrane PEs to form AA/AdA-PE, which are further converted into pro-ferroptotic lipid peroxidation under the activity of iron-containing LOXs. In conclusion, the Fenton reaction and oxidation of lipids facilitate the generation of lipid ROS, thus leading to ferroptosis. The system xc- is a cystine/glutamate antiporter. Intracellular cystine is reduced to cysteine for the biosynthesis of GSH. GPX4 converts two GSH molecules to GSSG each catalytic cycle to reduce lipid hydroperoxides, and then GSSG can be recycled back via GSH reductase in an NADPH-dependent manner. Ferroptosis inducers inhibit the GPX4-GSH-cysteine axis, thus inhibiting the reduction of lipid ROS. AA/AdA, arachidonic acid or adrenic acid; AA/AdA-CoA, arachidonic acid or adrenic acid coenzyme A; AA/AdA-PE, arachidonic acid or adrenic acid-phosphatidylethanolamine; AA/AdA-OOH-PE, arachidonic acid or adrenic acid-hydroperoxides-phosphatidylethanolamine; AA/AdA-OH-PE, arachidonic acid or adrenic acid-hydroxides-phosphatidylethanolamine; ACSL4, acyl-CoA synthetase long-chain family member 4; BSO, buthionine sulfoximine; CoA, coenzyme A; Cp, ceruloplasmin; Cys, L-cysteine; DMT1, divalent metal transporter 1; Fer-1, ferrostatin-1; FPN, ferroportin; γ-GC, gamma-glutamylcysteine; γ-GCS, gamma-glutamylcysteine synthetase; Glu, L-glutamate; GPX4, glutathione peroxidase 4; GR, glutathione reductase; GS, glutathione synthetase; GSH, reduced glutathione; GSSG, di-glutathione; LIP, labile iron pool; Lip-1, liproxstatin-1; LOX, lipoxygenase; LPCAT3, lysophosphatidylcholine acyltransferase 3; NCOA4, nuclear receptor coactivator 4; PE, phosphatidylethanolamine; RSL3, RAS-selective lethal 3; STEAP3, 6-transmembrane epithelial antigen of the prostate 3; TF, transferrin; TFR1, transferrin receptor 1.
Lipid Metabolism Related to Ferroptosis
Lipid metabolism is closely linked to the regulation of ferroptosis. The accumulation of lipid peroxidation seems to be a key process in the execution phase, in which PUFAs play an important role (Stockwell et al., 2017; Wenzel et al., 2017; Yamada et al., 2020). Usually, free PUFAs, especially AA and AdA are esterified to membrane phospholipids [mainly PUFA-containing phosphatidylethanolamines (PEs)]. With the presence of two lipid-metabolic enzymes, ACSL4 and LPCAT3, these membrane phospholipids undergo oxidation to drive ferroptosis (Doll et al., 2017; Kagan et al., 2017). The knockout of ACSL4 or loss of LPCAT3 resulted in significant resistance of certain non-neuronal cells to ferroptosis (Dixon et al., 2015; Yuan et al., 2016; Doll et al., 2017; Kagan et al., 2017). Following the generation of AA/AdA-PE, activated LOXs catalyze AA/AdA-PE into pro-ferroptotic lipid peroxidation AA/AdA-OOH-PE (Yang et al., 2016; Lei et al., 2019). The role of the LOXs in ferroptosis is also supported by a study indicating that genetic depletion or inhibition of LOXs by inhibitors [e.g., zileuton (Liu et al., 2015) and Vitamin E hydroquinone (Hinman et al., 2018)] could protect against ferroptosis in some cell types (Seiler et al., 2008; Yang et al., 2016). Recently, PEBP1 was shown to bind 15-LOX and alter the substrate specificity, changing it from free fatty acid to AA-PE, thereby promoting lipid oxidation (Wenzel et al., 2017). Furthermore, lipid peroxidation is thought to play a role in the final phase of ferroptosis, although the downstream mechanisms remain unclear (Lei et al., 2019). In one hypothesis, lipid peroxides may decompose into reactive toxic aldehydes, such as MDA or 4-HNEs. These decomposed substances react with proteins, nucleic acids, and membrane lipids to initiate ferroptosis (Domingues et al., 2013; Zhong and Yin, 2015). Dixon et al. also favored the hypothesis that showed that increased expression of AKRF1C genes could suppress ferroptosis by encoding aldoketoreductases to detoxify the end-products of lipid peroxidation (Dixon and Stockwell, 2014; Stockwell et al., 2017). As for inhibitors of lipid peroxidation, ferrostatins are the novel synthetic antioxidants that specifically trap lipid radicals and exert anti-ferroptotic function. Fer-1, the first-generation ferrostatin, prevents ferroptosis induced by erastin and RSL3 in HT1080 cells (Dixon et al., 2012). Lip-1 is another recently discovered ferroptosis inhibitor. It can prevent the accumulation of lipid ROS and inhibit erastin- or RSL3- induced ferroptosis in vitro (Friedmann Angeli et al., 2014). In conclusion, AA/AdA-related lipid metabolism can induce ferroptosis, and inhibiting LOXs or lipid peroxidation may have protective effects.
Glutathione Metabolism Related to Ferroptosis
Previous studies have identified that two major mechanisms, the Se-dependent GPX4-GSH-cysteine axis (Friedmann Angeli et al., 2014; Yang et al., 2014; Friedmann Angeli and Conrad, 2018; Ingold et al., 2018) and the FSP1-ubiquinone (CoQ10)-NAD(P)H pathway (Bersuker et al., 2019; Doll et al., 2019), were associated with lipid peroxidation and ferroptotic cell death. Additionally, the FSP1-CoQ10-NAD(P)H pathway is a complementary system to the GPX4-GSH-cysteine axis for controlling ferroptosis. In this axis, key steps include cystine uptake via system xc-, reduction of cystine to cysteine, GSH biosynthesis, and GPX4-mediated reduction of phospholipid hydroperoxides to lipid alcohols. During the process, the cystine/glutamate antiporter (system xc-) which consists of the light-chain subunit xCT (SLC7A11) and the heavy-chain subunit CD98 (SLC3A2) exchanges intracellular glutamate for extracellular cystine at a ratio of 1:1. Cystine is then reduced to cysteine for GSH synthesis [for review see Xie et al. (2016)]. In this regard, several agents [e.g., glutamate and erastin (Dixon et al., 2012), sulfasalazine (Gout et al., 2001), and sorafenib (Dixon et al., 2014)] can inhibit the system xc- to cause the decreased acquisition of precursors and GSH depletion, ultimately leading to ferroptosis. Other agents, including BSO (Sun et al., 2018) and acetaminophen (Lorincz et al., 2015), were observed directly blocking GSH synthesis. Conversely, ferroptosis induced by cystine deprivation can be reversed by reagents that increase the level of intracellular cysteine/cystine. For example, an in vitro study showed that when in the presence of β-mercaptoethanol, the cells were able to constantly utilize cystine through a mixed disulfide of β-mercaptoethanol and cysteine (Ishii et al., 1981). In addition, the loss of cysteinyl-tRNA synthetase, as Hayano et al. (2016) indicated, could trigger the transsulfuration pathway and lead to inhibition of ferroptosis induced by cystine deprivation.
Glutathione peroxidases 4 is a type of selenoprotein that contains one selenocysteine at the active site and seven cysteines. It plays an important role in regulating ferroptosis, and its inhibition promotes ferroptosis (Yang et al., 2016). Regarded as the only GPX that can eliminate biomembrane lipid peroxidation, GPX4 has a unique ability in ferroptosis. It is capable of reducing the toxic, membranous lipid hydroperoxides into non-toxic lipid alcohols (Brigelius-Flohé and Maiorino, 2013; Yang et al., 2014). Increasing GPX4 has been shown to be beneficial in many models of disease by inhibiting ferroptosis (Lan et al., 2020; Shen et al., 2020). However, knockdown or inactivation of GPX4 contributes to the accumulation of lipid peroxidation and initiation of ferroptosis (Park et al., 2019; Ye et al., 2020). For example, RSL3 directly inactivated GPX4 by covalently binding to selenocysteine to trigger ferroptosis (Yang et al., 2014, 2016), and FIN56 promoted degradation of GPX4 (Shimada et al., 2016).
Iron Metabolism Related to Ferroptosis
Besides lipid and GSH metabolism, the essential trace element for life, iron, is indispensable for the execution of ferroptosis (Dixon and Stockwell, 2014). The circulating Fe3+ and TF complex are endocytosed into cells by the membrane protein transferrin receptor 1 (TFR1). In the endosome, Fe3+ is reduced to Fe2+ by STEAP3, and Fe2+ is then released into unstable iron pools mediated by DMT1, or stored in ferritin, which is composed of FTL and FTH1 (Yang and Stockwell, 2008; Dixon et al., 2012). Excessive Fe2+ is exported through the membrane protein FPN and oxidized by ferroxidases, such as ceruloplasmin (Bogdan et al., 2016; Shang et al., 2020). In this process, iron accumulation (Shang et al., 2020) and administration of iron-bound, rather than iron-free TF, promote erastin-induced ferroptosis (Gao et al., 2015). On the contrary, using some iron chelators [e.g., DFP (Wu et al., 2020), DFO (Wu et al., 2018; Chen et al., 2020), and BPS (Codenotti et al., 2018)] may suppress ferroptosis and provide a potential therapeutic approach for diseases. In fact, there are some iron-chelating agents under clinical development for the treatment of cancers [for review see Brown et al. (2020)]. Moreover, inhibition of the IREB2 increases the expression of FTL and FTH1, thus decreasing sensitivity to ferroptosis (Gammella et al., 2015).
Although the importance of intracellular free iron in ferroptosis is confirmed, the regulatory mechanism of iron remains unknown. To date, the evidence has shown that the non-enzymatic free radical chain reaction involving Fenton Chemistry, in which Fe2+ is converted to Fe3+ with increased ROS (Winterbourn, 1995; He et al., 2020), and enzymatic processes (most notably the lipoxygenase pathway, LOXs), contributed to the formation of lipid peroxides in ferroptosis. Moreover, iron may promote ferroptosis through other iron-dependent enzymes, such as HIF-PHDs (Siddiq et al., 2009). Therefore, iron metabolism is one of the mechanisms of ferroptosis, and utilizing iron chelators to decrease iron may be useful for treating diseases.
The Role and Mechanism of Ferroptosis in Acute CNS Injuries
As described above, ferroptosis is an iron-dependent cell death that involves abnormal metabolism of lipids, GSH, and iron. The methods of measurement in evaluating ferroptosis in diverse diseases mainly depend on monitoring the levels of iron and lipid peroxidation, the activity of GPX4, as well as the ability of ferroptosis inhibitors (e.g., iron chelators, LOX inhibitors, and ferrostatins) to reduce cell death [for review see Xie et al. (2016)]. Observations of typical morphological features under a TEM also contribute to the distinguishing characteristics of ferroptosis compared to other cell deaths, both in vitro and vivo (Friedmann Angeli et al., 2014; Alim et al., 2019; Li et al., 2019d). Recently, numerous studies have confirmed the hypothesis of ferroptosis in the pathophysiology of acute CNS injuries, including stroke (Alim et al., 2019; Guan et al., 2019), TBI (Kenny et al., 2019; Xiao et al., 2019; Xie et al., 2019), and SCI (Dinc et al., 2013; Hu et al., 2017). More studies are included in the following text and Figure 2 is a brief summary of ferroptosis in acute CNS injuries.
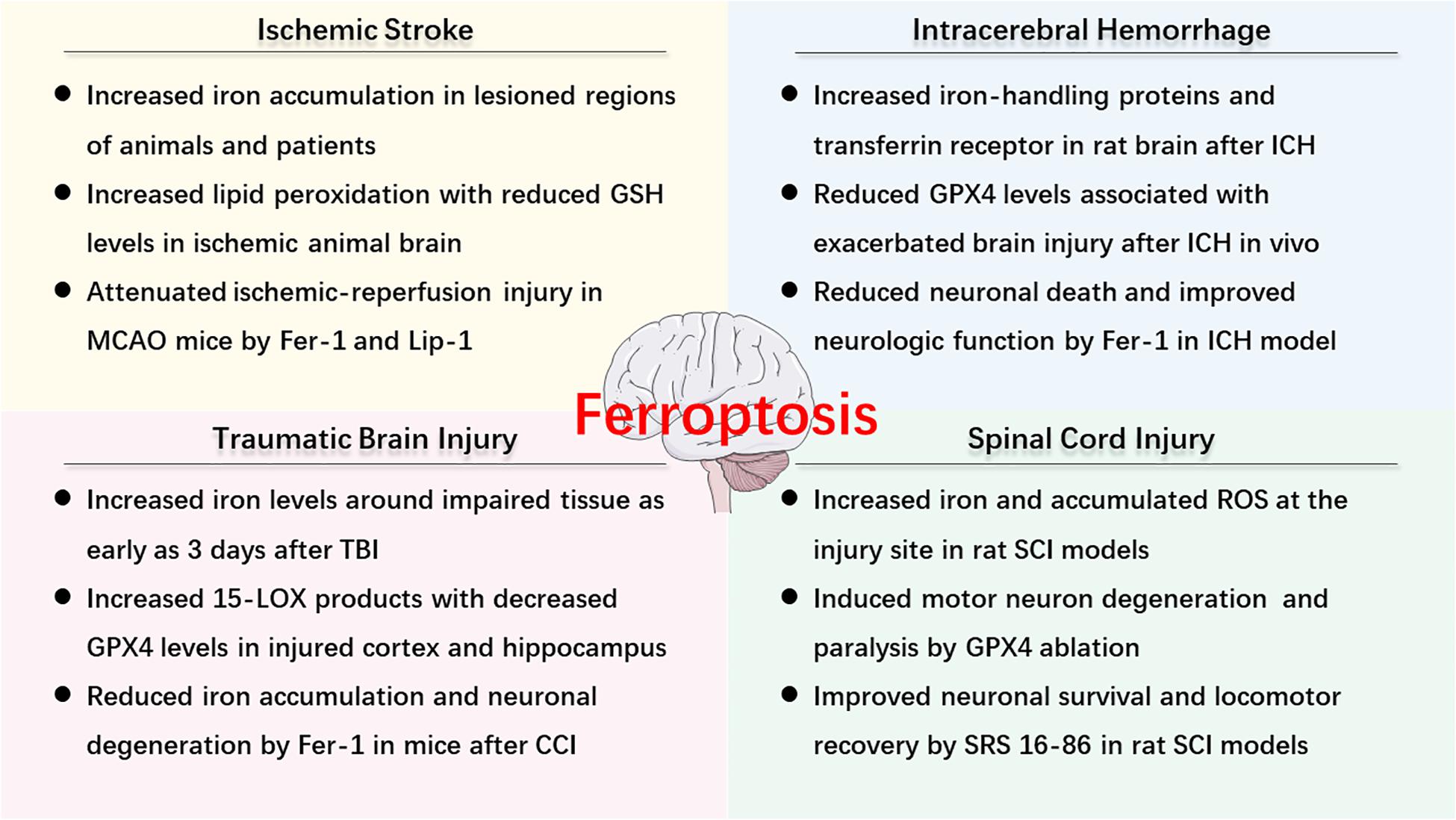
Figure 2. An overview of ferroptosis-associated mechanism and potential therapy in acute CNS injuries.
The Role and Mechanism of Ferroptosis in Ischemic Stroke
Ischemic stroke occurs when the blood flow to a certain portion of the brain is obstructed secondary to occlusion of cerebral arteries. The following deprivation of oxygen and energy triggers an ischemic cascade, such as OS and inflammation, resulting in neuronal excitotoxicity and cell death (Khoshnam et al., 2017; Li et al., 2019a; Zhang K. et al., 2019). Before ferroptosis was defined, iron accumulation [for review see Selim and Ratan (2004)] had been found in lesioned regions, such as the basal ganglia and the hippocampal area of the brain, and iron overload exaggerated neuronal damage during reperfusion (Dietrich and Bradley, 1988; Kondo et al., 1995; Lipscomb et al., 1998; Park et al., 2011). In MCAO animals, the iron intake was positively associated with the infarct volume (Castellanos et al., 2002; García-Yébenes et al., 2012). Consistently, one in vitro study demonstrated that holo-transferrin increased ROS production, and caused neuronal cell death induced by deprivation of oxygen and glucose (DeGregorio-Rocasolano et al., 2018). The experimental results also illustrated that administration of exogenous apotransferrin reduced brain damage and improved neurological outcomes with decreased lipid peroxidation, supporting the involvement of ferroptosis in ischemia (DeGregorio-Rocasolano et al., 2018). What’s more, the iron levels in the brain increased as humans age (Ward et al., 2014), which may exacerbate ischemic stroke. Recently, the tau-iron interaction has been proposed as an effective modulator of ferroptosis in ischemic stroke. The tau knockout mice were found to have increased protection from ferroptotic cell death following I/R injury, and the benefit of tau knockout was reinstated in older mice using iron-targeting interventions (Tuo et al., 2017). In this regard, iron chelation therapy reduced ischemic damage and improved outcome in mammals after ischemic stroke (Freret et al., 2006; Hanson et al., 2009). Consistently, Speer et al. (2013) found that the iron-dependent HIF-PHDs served as a target of metal chelators in ferroptosis, and the administration of iron chelators could inhibit HIF-PHDs, rather than suppress Fenton’s Reaction or ROS production, providing beneficial effects on subjects.
Additionally, other ferroptosis-associated mechanisms, such as LOXs-mediated pathology (Yang et al., 2016) and the GPX4-GSH-cysteine axis (Cho et al., 2007; Guan et al., 2019), were found to be involved in brain ischemia. MDA, a marker of oxidized lipids, was noticeably elevated, and this change correlated with increased activity of LOXs in an ischemic animal brain (Yigitkanli et al., 2013; Guan et al., 2019). Treatment with 12/15-LOX inhibitor (ML351) was shown to reduce infarct sizes and reperfusion damage in a mouse model (Rai et al., 2014). Moreover, Tuo et al. (2017) observed that brain damage was significantly attenuated by ferroptosis inhibitors, Lip-1, and Fer-1, in an MCAO model. As for the GPX4-GSH-cysteine axis, various groups provide direct evidence supporting that inhibition of system xc- induces ferroptosis and aggravates ischemia. Lan et al. (2020) found that acute cerebral ischemia-induced neuronal ferroptosis and treatment with Naotaifang increased the expression levels of xCT, GPX4 and GSH, and the number of Nissl bodies in MCAO rats. These data suggested that Naotaifang may rescue ischemic stroke by inhibiting ferroptosis through the xCT/GPX4 pathways (Lan et al., 2020). Huang et al. (2019) also observed that inhibition of system xc- with erastin aggravated ferroptosis and augmenter of liver regeneration protected the kidney from ischemia-reperfusion injury in ferroptosis through GSH/GPX system. Guan et al. (2019) and Cho et al. (2007) identified reduced levels of GSH and decreased activities of GPX4 in ischemia. However, many researchers hold opposing views and state that activated system xc- can exacerbate ischemic cerebral injury due to increased glutamate. In their studies, xCT was expressed in significant concentrations in astrocytes in the mouse brain (Jackman et al., 2012; Thorn et al., 2015; Ottestad-Hansen et al., 2018). Increased activity of system xc-(Soria et al., 2014) promoted the release of glutamate which may contribute to excitotoxicity in pathological situations (e.g., oxygen and glucose deprivation), leading to neuronal death (Thorn et al., 2015; Ottestad-Hansen et al., 2018). Hsieh et al. (2017) used both in vitro and in vivo models to reveal that HIF-1α triggered long-lasting glutamate excitotoxicity via activation of system xc- dependent glutamate outflow. HIF-1α conditional knockout mouse had reduced extracellular glutamate in cerebral ischemia-reperfusion, suggesting that system xc- was a promising therapeutic target (Hsieh et al., 2017). Therefore, inhibition of system xc- can induce ferroptosis to promote neuronal death due to GSH depletion and activation of system xc- can also increase neuronal death because of glutamate-associated excitotoxicity. Whether induction of system xc- activity is beneficial or detrimental might depend on the pathway of induction and whether inhibition of system xc-or induction of xCT is the more promising neuroprotective strategy remains to be explored.
Other studies have confirmed the benefits of the necroptosis inhibitor, necrostatin-1, indicating that alternative forms of regulated cell death were involved in ischemic brain injury (Degterev et al., 2005). However, necrostatin-1 was later found to protect against ferroptosis through an unknown target (Friedmann Angeli et al., 2014). These results suggest that there is the direct involvement of ferroptosis in the pathogenesis of ischemic stroke. What about ferroptosis in hemorrhagic stroke?
The Role and Mechanism of Ferroptosis in ICH
After ICH, there is a consequent physical disruption of the neurovascular architecture due to the mass effects and elevated pressure surrounding hemorrhagic sites, inducing primary brain injury. Subsequently, iron accumulates as a result of the degradation of Hb and its metabolite, hemin, which contributes to SBI (Hu et al., 2016). Previous data have identified multiple forms of cell death after ICH, including necrosis, apoptosis, and autophagy (Qureshi et al., 2001, 2003; Wang et al., 2015; Li et al., 2018). To date, multiple laboratories have provided converging lines of evidence that support the role of ferroptosis in ICH with the presence of observed molecular markers and morphological features (Li et al., 2017; Zille et al., 2017; Zhang et al., 2018; Alim et al., 2019), and the underlying mechanisms of ferroptosis in ICH are analogous to those of ischemic stroke. It was identified that iron overload stimulated neuronal ferroptosis, which aggravated brain damage (Wu et al., 2003, 2011). Besides, as one of the major upstream regulators of ferroptosis, GPX4 inactivation also contributes to ICH (Forcina and Dixon, 2019). It was found that levels of GPX4 were reduced and brain injury was exacerbated in a rat model of ICH induced by autologous blood injection, whereas overexpression of GPX4 was able to alleviate SBI and improve neurological outcomes (Zhang et al., 2018). Li et al. (2017) also indicated that administration of Fer-1 reduced Hb-induced cell death and iron deposition, prevented impairment of GPX4 activity in vitro, and improved neurologic function in collagenase-induced ICH models.
Many recent studies have suggested potential approaches to reduce brain damage. Zille et al. (2017) revealed that several ferroptosis inhibitors, including Fer-1, DFO, Trolox (a lipid peroxidation inhibitor), and NAC (a cell-permeable cysteine analog), were able to alleviate Hb- and hemin-induced cell death in vitro. Alim et al. (2019) also supported the role of Fer-1 by showing that inhibition of ferroptosis by Fer-1 exerted a long-term cerebroprotective effect in in vitro and in vivo ICH models. Dharmalingam et al. (2020) synthesized a multifunctional nanoparticle that protected cells from both senescence and ferroptosis, leading to a reduction of hemin/iron-induced toxicity in experimental ICH. In addition, Karuppagounder et al. (2018) found that ALOX5 inhibition could protect against ICH- or hemin-induced ferroptosis in vivo following ICH.
Furthermore, the role of ferroptosis in ICH is supported by altered levels of other ferroptosis-related molecules. For example, phospho-ERK1/2, regarded as a molecular feature of ferroptosis (Yagoda et al., 2007), was significantly increased in mice with ICH, but the MEK inhibitor, U0126, inhibited this type of cell death (Zille et al., 2017). In addition, the expression levels of PTGS2 were also significantly increased in in vitro and in vivo ICH models (Li et al., 2017; Alim et al., 2019). Moreover, PTGS2 has been revealed as part of the downstream signaling pathway of ferroptosis in cancer cells (Yang et al., 2014). Notably, a gene product of PTGS2, known as COX-2, was substantially increased in neurons after ICH, and treatment with Fer-1 could reduce its expression and ICH-induced SBI, implying that COX-2 might be used as a biomarker of ferroptosis (Zhao et al., 2007; Li et al., 2017; Alim et al., 2019). Thus far, there is little evidence that shows the relationship between ferroptosis and subarachnoid hemorrhage. More studies are warranted to investigate this promising topic.
The Role and Mechanism of Ferroptosis in TBI
The previous literature has shown that TBI shares many mechanisms (e.g., OS, inflammation, mitochondrial dysfunction, and neuronal cell death) with stroke (Blennow et al., 2016). In addition to these mechanisms, multiple studies have demonstrated that ferroptosis may contribute to the neuronal cell death and functional outcome in TBI (Ayton et al., 2014; Stockwell et al., 2017). The altered levels of various ferroptosis biomarkers provide evidence of ferroptosis in TBI. Studies have detected elevated iron concentrations around the impaired tissue as early as 3 days after injury, occurring in adult and aged mice models of controlled CCI (Portbury et al., 2016, 2017; Xie et al., 2019). The evidence also suggested that increased iron accumulation was negatively associated with cognitive outcomes in chronic TBI patients (Lu et al., 2015), while iron chelators exhibited neuroprotective effects by diminishing iron-mediated brain damage (Zhang et al., 2013; Khalaf et al., 2018). According to recent studies, ferroptosis was identified in TBI through the detection of ferroptosis-associated molecules, such as 15-LOX and GPX4. For instance, there were enhanced levels of 15-HpETE-PE and 15-LOX2 in the injured cortex and ipsilateral hippocampus, and decreased levels of GPX4 in a pediatric rat CCI model, suggesting that ferroptosis might occur within the first hour after TBI (Wenzel et al., 2017). Researchers also showed a preponderance of 15-LOX products in CCI-injured adult mice, and increased ACSL4 and 15-LOX2 expression in TBI, when compared with naive groups (Kenny et al., 2019). Moreover, numerous experimental and clinical studies observed the increased levels of MDA and 4-HNE in either the injured brain or serum following TBI (Hall et al., 2004; Readnower et al., 2010; Lorente et al., 2015; Xiao et al., 2019; Xie et al., 2019). As for the morphological features, Xie et al. (2019) first verifiably identified the ferroptotic features around brain injury lesions of TBI models at 3 days after CCI. As ferroptosis was shown to participate in TBI by the above work, inhibiting ferroptosis may be useful in treating TBI. For example, treatment with Fer-1 significantly diminished iron accumulation, reduced neuronal cell death, and attenuated neuronal degeneration (Kenny et al., 2019; Xie et al., 2019). More details regarding therapy can be found in the next section.
The Role and Mechanism of Ferroptosis in SCI
In traumatic SCI, the primary injury causes immediate cellular damage and initiates a continuous secondary injury cascade to induce ischemia, inflammation, and cell death [for review see Ahuja et al. (2017)]. Notably, following the rupture of the blood-spinal cord barrier and blood vessels, hemorrhage occurs in the acute phase of SCI and may last for days (Tran et al., 2018). Like other acute CNS diseases, ferroptosis occurs in SCI, and is accompanied by increased iron and accumulated ROS at the site of injury (Liu et al., 2011; Visavadiya et al., 2016; Hao et al., 2017), as well as excessive lipid peroxidation (Dinc et al., 2013; Hu et al., 2017). This phenomenon is more apparent during the first several hours (Liu et al., 2004). In adult mouse models, Chen et al. (2015) observed that conditional ablation of Gpx4 in neurons could induce motor neuron degeneration and cause rapid paralysis, but this result was delayed by supplementation with vitamin E, suggesting that ferroptosis accelerated SCI. Therefore, anti-ferroptotic seems to have potential in SCI, though there are few studies. Feng et al. (2019) established a rat model of DFO and confirmed the positive role of DFO in treating SCI. In the DFO group, there were lower iron concentrations, markedly increased GPX4 expression, and increased neuronal survival (Feng et al., 2019). In the study conducted by Zhang Y. et al. (2019) treatment with the third-generation ferrostatin, SRS 16-86, increased neuronal survival and promoted locomotor recovery in the SCI model, providing potential therapeutic strategies for SCI. Indeed, it is well-known that the excitotoxicity caused by glutamate accumulation preceded neuronal death and reuptake failure of astrocytes, and also induced ferroptotic cell death to stimulate secondary injury after SCI (Dixon et al., 2014; Ahuja et al., 2017). The relationship between ferroptosis or excitotoxicity in SCI requires further studies.
Potential and Emerging Therapy Targeting Ferroptosis in Acute CNS Injuries
As ferroptosis may be a significant pathogenic pathway in acute CNS injuries, its therapeutic potential should be taken into consideration. Ferroptosis inhibitors (including iron chelators, ferrostatins, liproxstatins, LOX inhibitors, and antioxidants) may prevent iron accumulation or lipid peroxidation, thus offering therapeutic options for treating acute CNS injuries (Table 1).
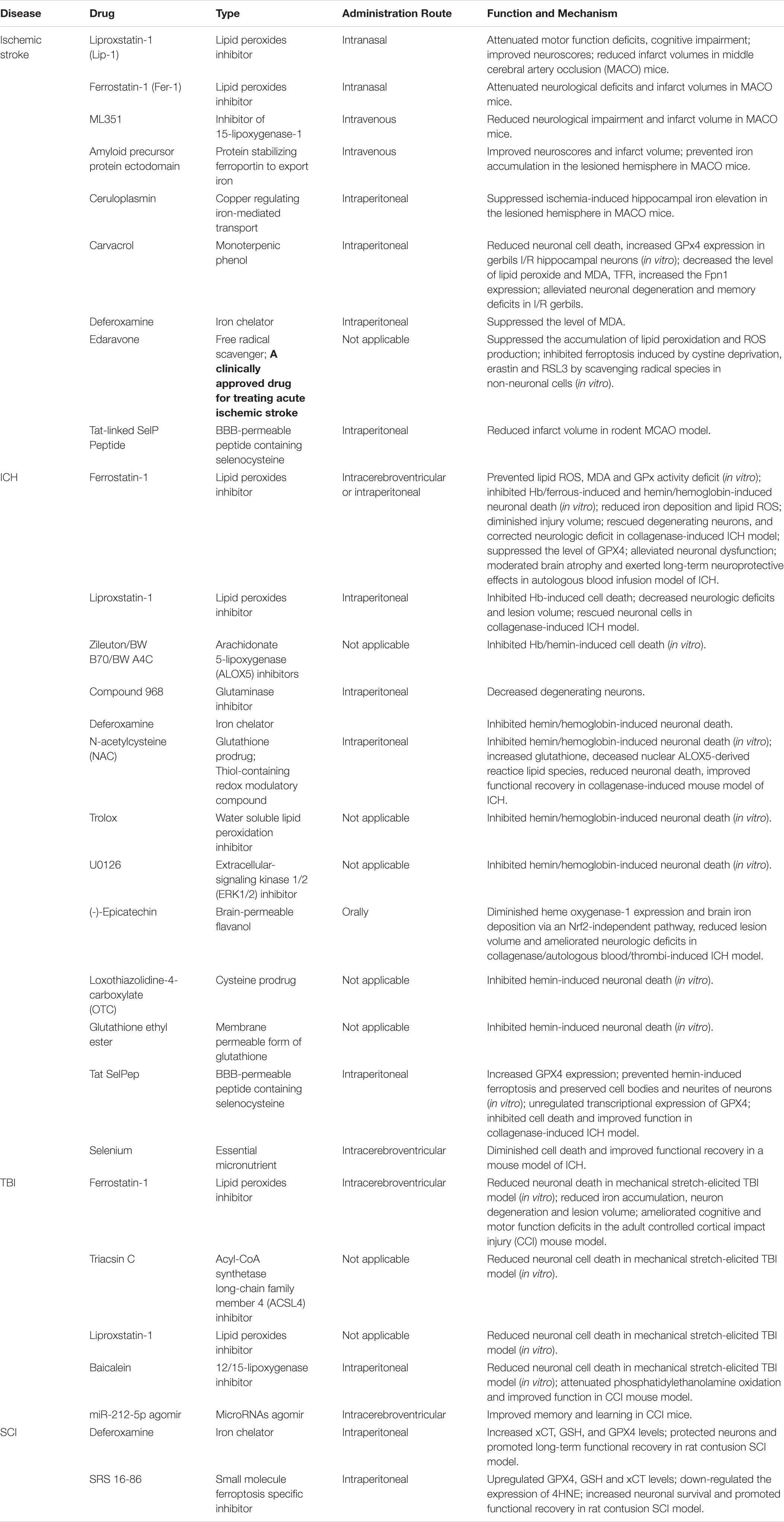
Table 1. Ferroptosis-associated drugs in treating the acute CNS injuries.
Targeting Ferroptosis Therapy in Ischemic Stroke
The mainstay of treatment for acute ischemic stroke is rapid recanalization by mechanical thrombectomy or recombinant tissue plasminogen activator, the only approved thrombolytic agent. It is important to salvage the penumbra, which surrounds the region of the infarct and promotes functional recovery. However, the overall efficacy is limited due to the narrow window of opportunity (Sandercock et al., 2012), but even after timely recanalization, infarct volume often continues to increase in I/R injury (Nour et al., 2013). As previous methods have failed in clinical use, such as blocking excitotoxicity, the role of ferroptosis has been highlighted (Khoshnam et al., 2017; Tuo et al., 2017), and new therapeutic approaches targeting ferroptosis or combined therapies are highly desirable.
As mentioned above, Lip-1 and Fer-1 are both compounds with specific anti-ferroptotic activity. Intranasal administration of Lip-1 and Fer-1, either immediately or 6 h after reperfusion, significantly reduced neuronal damage and functional deficits in MCAO mice, indicating the possible translational value of special exogenous ferroptosis inhibitors (Tuo et al., 2017).
In addition, CoQ10 is an endogenous lipid-soluble antioxidant with established efficacy in suppressing the initiation and amplification of lipid peroxidation (Morris et al., 2013; Viswanathan et al., 2017), presenting a promising candidate for ferroptosis inhibition. Intriguingly, in vivo studies have reported that oral CoQ10 administration markedly improved neurological outcomes in both rat MCAO models and acute ischemic stroke patients (Ramezani et al., 2018; Nasoohi et al., 2019). This neuroprotective benefit of CoQ10 was associated with its anti-apoptotic effect, as the levels of peroxidation products were not altered (Nasoohi et al., 2019). The authors attributed this to a relatively higher dose and multiple potential mechanisms of CoQ10. Of note, it is worth considering whether inhibition of ferroptosis is involved.
Recently, Guan et al. (2019) found that carvacrol, a plant-derived monoterpenic phenol, inhibited hippocampal neuronal damage and reduced functional deficits in gerbils following I/R injury. Furthermore, treatment with carvacrol (100 mg/kg, intraperitoneally) for two consecutive weeks after reperfusion was associated with decreased ROS, reduced iron overload, and increased levels of GPX4, suggesting a possible neuroprotective role of carvacrol via ferroptosis inhibition (Guan et al., 2019). Carvacrol is thought to easily cross the BBB because of the small molecular weight and the lipophilic profile (Savelev et al., 2004). A previous study has proven the benefits of carvacrol when administered intraperitoneally at 2 h after reperfusion. When administered intracerebroventricularly, the treatment window was prolonged to 6 h (Yu et al., 2012). When administered safely, carvacrol may be regarded as a potential therapeutic agent.
Edaravone is an effective radical scavenger that inhibits lipid oxidation by scavenging chain-initiating water-soluble radicals and chain-carrying lipid peroxyl radicals due to its amphiphilicity (Watanabe et al., 2018). There are several papers describing its alleviatory effects on neurological symptoms in ischemia models, and its effective treatment window of at least 3 h after embolism (Nishi et al., 1989; Kawai et al., 1997; Lapchak and Zivin, 2009). In the clinical setting, the appropriate dosage and course of edaravone use for patients suffering from acute ischemic stroke include intravenous administration of 60 mg daily for up to 14 days (Feng et al., 2011). Homma et al. (2019) recently indicated that edaravone participates in rescuing ferroptotic cell death induced by cystine deprivation, erastin, and RSL3 (Homma et al., 2019). In addition, edaravone was confirmed to suppress the accumulation of Fe2+ and lipid peroxidation in vitro, which are known as the metabolic characteristics of ferroptosis.
Targeting Ferroptosis Therapy in ICH
Currently, there are no proven medical or surgical treatments that substantially improve the neurological outcomes in patients with ICH because of multiple underlying mechanisms, including inflammation, excitotoxicity, and OS (Keep et al., 2012). As emerging studies suggest that ferroptosis is involved in SBI after ICH, and contributes to 80% of whole-cell death in vitro (Li et al., 2017; Zille et al., 2017), ferroptosis-based treatments could be highly considered.
Li et al. demonstrated the neuroprotective effects of Fer-1 by striatum injection immediately after and by cerebral ventricular injection 2 h after collagenase-induced ICH [for review see Li et al. (2017)]. Intraperitoneal administration of Fer-1 (a 3-h delay and then once daily) in the autologous blood infusion model of ICH was also shown to improve long-term neurological function (Alim et al., 2019). Moreover, when combined with other inhibitors of either apoptosis or necrosis, Fer-1 was found to be more effective at reducing Hb-induced cell death, which should be further investigated in in vivo models (Li et al., 2017).
Various evidence has shown that iron chelators reduce Hb- and iron-induced neurotoxicity attenuates brain edema, and improve functional neurologic outcomes after ICH (Nakamura et al., 2004; Wu et al., 2012; Hatakeyama et al., 2013). A meta-analysis of 20 studies involving animal models of ICH revealed that DFO was neuroprotective, particularly when administered 2–4 h after ICH induction (Cui et al., 2015), whereas there remains a lack of conclusive clinical evidence regarding iron chelators (Zeng et al., 2018).
N-acetylcysteine (Gilbert et al., 1991; Leslie et al., 1992; Varga et al., 1997) is an FDA-approved cysteine prodrug capable of regulating the activity of system xc- and the biosynthesis of GSH [for review see Berk et al. (2013)]. Interestingly, a recent study reported that systemic administration of NAC post-injury reduced neuronal death and improved behavior following ICH in mice (Karuppagounder et al., 2018). They further pointed out that NAC inhibited hemin- and ICH-induced ferroptosis by neutralizing nuclear ALOX5-derived toxic lipid species (Karuppagounder et al., 2018). This process relied on increased GSH and enhanced activity of GSH-dependent antioxidant enzymes. Considering the poor absorption of direct GSH administration and the insufficient capacity of GSH to cross the BBB (Witschi et al., 1992), NAC may be treated as an adjuvant therapy candidate capable of penetrating the BBB to enter the brain (Farr et al., 2003).
In addition, EC, a brain-permeable flavanol, was shown to reduce early brain injury and improve neurologic deficits in multiple experimental ICH models when administered orally at 3 h post-treatment and subsequent daily administration (Chang et al., 2014). The neuroprotective effects of EC were partially associated with decreased iron deposition and modulation of ferroptosis-related gene expression, indicating the possible ability of EC to inhibit ICH-induced ferroptotic cell death (Chang et al., 2014).
Se is indispensable for the ferroptosis-resistant function of GPX4 (Ingold et al., 2018). It was recently uncovered that Se could amplify an adaptive transcriptional program response to neuronal ferroptosis (Alim et al., 2019), making it a potential therapeutic strategy. Further studies discovered that injection of Se directly into the mouse cerebral ventricle following ICH was associated with elevated GPX4 levels, diminished ferroptotic death, as well as improved functional recovery. Moreover, the researchers developed a peptide (Tat SelPep), which contained a Tat transduction domain combined with selenoprotein P. Intraperitoneal injection of Tat SelPep showed similar effects compared to Se, but with reduced toxicity and a wider treatment window, with benefits shown even at 6 h post-injury (Alim et al., 2019).
Targeting Ferroptosis Therapy in TBI
When contemplating feasible treatments for TBI, researchers focus on secondary events, which cause delayed damage, to provide applicable therapeutic windows for interventions [for review see Lozano et al. (2015)]. Given that there are currently no effective treatments approved by clinical trials for TBI patients (Pearn et al., 2017), there exists a pressing need for developing more innovative methods, such as targeting ferroptotic cell death in a highly regulated manner. The ferroptosis signaling molecules can be prevented as a result of reducing PE oxidation by inhibiting the ability of 15LOX/PEBP1 complexes to produce 15-HpETE-PE, administrating 15LOX inhibitors, or augmenting the GPX4/GSH system to remove oxidized PE products (Wenzel et al., 2017; Kenny et al., 2019).
Baicalein is a polyphenolic antioxidant 12/15-LOX inhibitor and is well-known to exert neuroprotective effects in cerebral ischemia [for review see Liang et al. (2017)]. Recently, Kenny et al. (2019) demonstrated that baicalein decreased the accumulation of pro-ferroptotic PE oxidation, but not pro-apoptotic cardiolipin oxidation after CCI, indicating that the 15-LOX inhibitory effects of baicalein may have an anti-ferroptotic role in TBI. Several studies have also revealed a reduction of functional and histological damage with the immediate administration of baicalein post-injury (Chen et al., 2008; Kenny et al., 2019). With low levels of toxicity and the ability to cross the BBB (Tsai et al., 2002), baicalein offers great promise in clinical settings if the effect of delayed drug delivery is evaluated.
As mentioned above, NAC is a precursor for GSH, and it has been shown to confer antioxidant and neuroprotective effects after pre-clinical TBI (Eakin et al., 2014; Senol et al., 2014). As for adult patients, a double-blinded and placebo-controlled study indicated that supplementation of oral NAC had significant short-term benefits on neurological symptoms and sequelae resolution after blast-induced mild TBI (Hoffer et al., 2013). Due to the low bioavailability of NAC, the compound, NACA, was developed with increased membrane permeability, and its neuroprotection was associated with the activation of the Nrf2-antioxidant response elements signaling pathway in a mouse model of TBI (Zhou et al., 2018). It is well-established that Nrf-2 regulates xCT and GPX4, whose inhibition initiates ferroptosis and promotes target genes that mediate the antioxidant and iron metabolic status of cells (Zhou et al., 2018), suggesting another anti-ferroptotic mechanism of NACA.
In a mouse CCI model of TBI, Fer-1 treatment had been injected directly into the cerebral ventricle 0.5 h after injury, causing a reduction in neuronal death and other associated functional defects (Xie et al., 2019). However, more research should be implemented to uncover feasible drug-delivery methods.
Moreover, a recent study demonstrated the role of miR-212-5p in suppressing ferroptosis after TBI, partially by targeting PTGS2 (Xiao et al., 2019). Further results showed that intracerebroventricular injection of miR-212-5p agomir improved spatial memory and learning in CCI mice, suggesting that miR-212-5p may serve as a potential ferroptosis inhibitor to be used in treating TBI. As previously discussed, the oxidation of AA/AdA-PE is a critical step in ferroptosis execution. Therefore, inhibition of ACSL (such as triacsin C and thiazolidinedione) and formation of AA/AdA-esterified PE may also protect against ferroptosis after TBI (Doll et al., 2017; Kagan et al., 2017; Kenny et al., 2019).
Targeting Ferroptosis Therapy in SCI
There are no neuroprotective or neurodegenerative strategies currently approved for acute traumatic SCI, but several are currently undergoing clinical trials (Badhiwala et al., 2018). The concept of “time is spine” is commonly applied in the management of patients with SCI. Since ferroptosis is likely involved in the acutes phases of SCI, therapies targeting ferroptosis are promising (Badhiwala et al., 2018; Zhang Y. et al., 2019).
Targeting iron is one of the treatment strategies. The iron chelator, DFO, reportedly reduced iron accumulation and lipid peroxidation, while modulating the inflammatory response in SCI (Paterniti et al., 2010; Liu et al., 2011; Dinc et al., 2013; Hao et al., 2017). Experimental evidence indicated that DFO improved motor function recovery when injected intraperitoneally post-SCI (Paterniti et al., 2010; Liu et al., 2011; Hao et al., 2017). Moreover, DFO showed neuroprotective effects comparable with methylprednisolone, an effective antioxidant agent that is contentious for the treatment of SCI because of harmful side effects (Dinc et al., 2013; Silva et al., 2014). However, oral treatment of deferasirox, another FDA-approved iron chelator, failed to remove iron from the injured spinal cord, but markedly depleted the systemic iron (Sauerbeck et al., 2013). Considering the detrimental side effects (e.g., anemia) and the absence of potent neuroprotection, systemic administration may not be the ideal approach of iron chelators (Grossman et al., 2012; Sauerbeck et al., 2013).
Besides, previous studies have demonstrated that NAC administration suppressed OS, attenuated neuroinflammation, and improved neuronal survival and neurological recovery following SCI in rodent models (Karalija et al., 2012, 2014; Guo et al., 2015). When administered immediately after SCI, NACA, an amide derivative of NAC, improved mitochondrial function, antioxidant GSH levels, and functional recovery in SCI mice (Patel et al., 2014). These two GSH precursors facilitate the biosynthesis of intracellular GSH. In addition, the GSH antioxidant system plays a pivotal role in the regulation of ferroptosis (Lv et al., 2019).
Recently, Li et al. (2019b) observed that CoQ10, a promising ferroptosis inhibitor previously mentioned, exerted protective effects by decreasing OS partly through activation of the Nrf-2 signaling pathway after SCI. Moreover, Nrf-2 is regarded as a significant mitigator of lipid peroxidation and ferroptosis [for review see Dodson et al. (2019)]. Furthermore, CoQ10 was shown to protect BMSCs from OS, and improved the therapeutic efficacy in combination with BMSC transplantation, suggesting a promising therapy for SCI (Li et al., 2019c).
Besides, post-injury intraperitoneal injection of SRS 16-86 proved to be more potent and stable than Fer-1. It also attenuated the ferroptotic mitochondrial morphology in damaged areas, and improved neurological deficits in SCI model, suggesting the role of ferroptosis-specific inhibitors in the treatment of SCI (Zhang Y. et al., 2019).
Discussion and Perspective: Will Ferroptosis Be the Future Direction?
In this article, we primarily focus on the roles and therapeutic potential of ferroptosis in various acute CNS injury processes, including stroke, TBI, and SCI. Pharmacological effects of multiple inducers and inhibitors of ferroptosis lie at the intersection of lipid, amino acid, and iron metabolism. Although some progress has been made in ferroptosis, there are still controversial questions that have not been fully studied. First, the relationship between ferroptosis and other forms of cell death remains unknown. For example, p53 is an important regulator, both in apoptosis and ferroptosis, while autophagy plays a role in the process of ferroptosis via ferritinophagy. As ferroptosis is involved in acute CNS injuries complicated by necrosis, apoptosis, and autophagy, a head-to-head comparison of individual inhibitors or various combinations of inhibitors is required in further studies. Second, the special molecular markers (e.g., caspase activation for apoptosis or the autophagosome marker, LC3-II, for autophagy) for identifying ferroptosis are still lacking. While the increased mRNA levels of PTGS2 were found in cells undergoing ferroptosis, it did not affect ferroptosis progress (Yang et al., 2014). The specificity of PTGS2 expression or its gene product, COX-2, for ferroptosis needs to be explored in the context of different pathophysiologic processes. Actually, there is copious evidence for the role of COX-2 in several acute neurological disorders (e.g., ischemic and hemorrhagic strokes) (Gong et al., 2001; Tomimoto et al., 2002). The research of additional ferroptosis markers is of great importance for in vivo studies in the future. Moreover, the exact role of iron and the final molecular executor in ferroptosis remains unclear. Considering the complexity of the CNS, the biochemical regulation, as well as the sensitivity of ferroptosis in different cell types (neurons, astrocytes, microglia, or oligodendrocytes), also requires explication.
Potential treatment options targeting ferroptosis (e.g., iron chelators, ferrostatins, NAC, and CoQ10) have shown neuroprotective effects in acute CNS injuries. However, these benefits are largely based on animal models and have not yet translated into clinical application. Furthermore, studies are necessary to clarify the appropriate therapeutic window, clinically feasible routes of administration, and BBB penetration ability of anti-ferroptotic agents. Among the above-mentioned agents, edaravone is the only approved drug with proven clinical efficacy and safety, while others should be explored in further clinical studies. More in-depth and comprehensive research on ferroptosis should be conducted to develop therapeutic methods and eventually alleviate the burden of acute CNS injuries in the future.
Author Contributions
All the authors participated in analyzing and discussing the literature, commenting on, and read and approved the final manuscript. AS and YC supervised the research, led the discussion, and wrote and revised the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (81701144, 81371433, and 81870916), and Major Science and Technology Project in the medical and health of Zhejiang Province (WKJ-ZJ-1615:2016149634).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
4-HNE, 4-hydroxy-2-nonenal; AA, arachidonic acid; ACSL4, acyl-CoA synthetase long-chain family member 4; AdA, adrenic acid; ALOX5, arachidonate 5-lipoxygenase; BBB, blood–brain barrier; BMSC, bone marrow mesenchymal stem cells; BPS, bathophenanthrolinedisulfonic acid; BSO, buthionine sulfoximine; CCI, cortical impact injury; CNS, central nervous system; CoQ10, coenzyme Q10; COX-2, cyclooxygenase-2; DFO, deferoxamine; DFP, deferiprone; DMT1, divalent metal transporter 1; EC, (-)-epicatechin; Fe2+, ferrous iron; Fe3+, ferric iron; Fer-1, ferrostatin-1; FPN, ferroportin; FSP1, ferroptosis suppressor protein 1; FTH1, ferritin heavy chain 1; FTL, ferritin light chain; GPX4, glutathione peroxidases 4; GSH, glutathione; Hb, hemoglobin; HIF-1 α, hypoxia-inducible factor 1 α; HIF-PHD, hypoxia-inducible factor prolyl hydroxylase; I/R, ischemia/reperfusion; ICH, intracerebral hemorrhage; IREB2, iron metabolism essential factor iron response element binding protein 2; Lip-1, liproxstatin-1; LOX, lipoxygenases; LPCAT3, lysophosphatidylcholine acyltransferase 3; MCAO, middle cerebral artery occlusion; MDA, malondialdehyde; NAC, N-acetylcysteine; NACA, N-acetylcysteine amide; Nrf2, nuclear factor erythroid 2-related factor; OS, oxidative stress; PEBP1, phosphatidylethanolamine-binding protein 1; PTGS2, prostaglandin-endoperoxide synthase 2; PUFA, polyunsaturated fatty acid; ROS, reactive oxygen species; RSL3, RAS synthetic lethal 3; SBI, secondary brain injury; SCI, spinal cord injury; Se, selenium; STEAP3, six-transmembrane epithelial antigen of the prostate 3; TBI, traumatic brain injury; TEM, transmission electron microscopy; TF, transferrin; TFR, transferrin receptor.
References
Ahuja, C. S., Wilson, J. R., Nori, S., Kotter, M. R. N., Druschel, C., Curt, A., et al. (2017). Traumatic spinal cord injury. Nat. Rev. Dis. Primers 3:17018.
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 177, 1262–1279.e25. doi: 10.1016/j.cell.2019.03.032
Ayton, S., Zhang, M., Roberts, B. R., Lam, L. Q., Lind, M., McLean, C., et al. (2014). Ceruloplasmin and β-amyloid precursor protein confer neuroprotection in traumatic brain injury and lower neuronal iron. Free Radic. Biol. Med. 69, 331–337. doi: 10.1016/j.freeradbiomed.2014.01.041
Badhiwala, J. H., Ahuja, C. S., and Fehlings, M. G. (2018). Time is spine: a review of translational advances in spinal cord injury. J. Neurosurg. Spine 30, 1–18. doi: 10.3171/2018.9.spine18682
Benjamin, E. J., Muntner, P., Alonso, A., Bittencourt, M. S., Callaway, C. W., Carson, A. P., et al. (2019). Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation 139, e56–e528.
Berk, M., Malhi, G. S., Gray, L. J., and Dean, O. M. (2013). The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol. Sci. 34, 167–177. doi: 10.1016/j.tips.2013.01.001
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi: 10.1038/s41586-019-1705-2
Blennow, K., Brody, D. L., Kochanek, P. M., Levin, H., McKee, A., Ribbers, G. M., et al. (2016). Traumatic brain injuries. Nat. Rev. Dis. Primers 2:16084.
Bogdan, A. R., Miyazawa, M., Hashimoto, K., and Tsuji, Y. (2016). Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem. Sci. 41, 274–286. doi: 10.1016/j.tibs.2015.11.012
Brigelius-Flohé, R., and Maiorino, M. (2013). Glutathione peroxidases. Biochim. Biophys. Acta 1830, 3289–3303.
Brown, R. A. M., Richardson, K. L., Kabir, T. D., Trinder, D., Ganss, R., and Leedman, P. J. (2020). Altered iron metabolism and impact in cancer biology, metastasis, and immunology. Front. Oncol. 10:476. doi: 10.3389/fonc.2020.00476
Castellanos, M., Puig, N., Carbonell, T., Castillo, J., Martinez, J., Rama, R., et al. (2002). Iron intake increases infarct volume after permanent middle cerebral artery occlusion in rats. Brain Res. 952, 1–6. doi: 10.1016/s0006-8993(02)03179-7
Chamorro, A., Dirnagl, U., Urra, X., and Planas, A. M. (2016). Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 15, 869–881. doi: 10.1016/s1474-4422(16)00114-9
Chang, C. F., Cho, S., and Wang, J. (2014). (-)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann. Clin. Transl. Neurol. 1, 258–271. doi: 10.1002/acn3.54
Chen, L., Hambright, W. S., Na, R., and Ran, Q. (2015). Ablation of the Ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 290, 28097–28106. doi: 10.1074/jbc.m115.680090
Chen, S. F., Hsu, C. W., Huang, W. H., and Wang, J. Y. (2008). Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol. 155, 1279–1296. doi: 10.1038/bjp.2008.345
Chen, X., Li, D., Sun, H. Y., Wang, W. W., Wu, H., Kong, W., et al. (2020). Relieving ferroptosis may partially reverse neurodegeneration of the auditory cortex. FEBS J. [Epub ahead of print].
Cho, S., Szeto, H. H., Kim, E., Kim, H., Tolhurst, A. T., and Pinto, J. T. (2007). A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J. Biol. Chem. 282, 4634–4642. doi: 10.1074/jbc.m609388200
Codenotti, S., Poli, M., Asperti, M., Zizioli, D., Marampon, F., and Fanzani, A. (2018). Cell growth potential drives ferroptosis susceptibility in rhabdomyosarcoma and myoblast cell lines. J. Cancer Res. Clin. Oncol. 144, 1717–1730. doi: 10.1007/s00432-018-2699-0
Cui, H.-J., He, H.-Y., Yang, A. L., Zhou, H.-J., Wang, C., Luo, J.-K., et al. (2015). Efficacy of deferoxamine in animal models of intracerebral hemorrhage: a systematic review and stratified meta-analysis. PLoS One 10:e0127256. doi: 10.1371/journal.pone.0127256
DeGregorio-Rocasolano, N., Marti-Sistac, O., Ponce, J., Castello-Ruiz, M., Millan, M., Guirao, V., et al. (2018). Iron-loaded transferrin (Tf) is detrimental whereas iron-free Tf confers protection against brain ischemia by modifying blood Tf saturation and subsequent neuronal damage. Redox Biol. 15, 143–158. doi: 10.1016/j.redox.2017.11.026
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119. doi: 10.1038/nchembio711
Dharmalingam, P., Talakatta, G., Mitra, J., Wang, H., Derry, P. J., Nilewski, L. G., et al. (2020). Pervasive genomic damage in experimental intracerebral hemorrhage: therapeutic potential of a mechanistic-based carbon nanoparticle. ACS Nano 14, 2827–2846. doi: 10.1021/acsnano.9b05821
Dietrich, R. B., and Bradley, W. G. (1988). Iron accumulation in the basal ganglia following severe ischemic-anoxic insults in children. Radiology 168, 203–206. doi: 10.1148/radiology.168.1.3380958
Dinc, C., Iplikcioglu, A. C., Atabey, C., Eroglu, A., Topuz, K., Ipcioglu, O., et al. (2013). Comparison of deferoxamine and methylprednisolone: protective effect of pharmacological agents on lipid peroxidation in spinal cord injury in rats. Spine 38, E1649–E1655.
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Dixon, S. J., Patel, D., Welsch, M., Skouta, R., Lee, E., Hayano, M., et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3:e02523.
Dixon, S. J., and Stockwell, B. R. (2014). The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 10, 9–17. doi: 10.1038/nchembio.1416
Dixon, S. J., Winter, G. E., Musavi, L. S., Lee, E. D., Snijder, B., Rebsamen, M., et al. (2015). Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem. Biol. 10, 1604–1609. doi: 10.1021/acschembio.5b00245
Dodson, M., Castro-Portuguez, R., and Zhang, D. D. (2019). NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23:101107. doi: 10.1016/j.redox.2019.101107
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi: 10.1038/s41586-019-1707-0
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi: 10.1038/nchembio.2239
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3, 285–296. doi: 10.1016/s1535-6108(03)00050-3
Domingues, R. M., Domingues, P., Melo, T., érez-Sala, D. P., Reis, A., and Spickett, C. M. (2013). Spickett, lipoxidation adducts with peptides and proteins: deleterious modifications or signaling mechanisms? J. Proteom. 92, 110–131. doi: 10.1016/j.jprot.2013.06.004
Duan, J. L., Cui, J., Yang, Z. F., Guo, C., Cao, J. Y., Xi, M. M., et al. (2019). Neuroprotective effect of Apelin 13 on ischemic stroke by activating AMPK/GSK-3 beta/Nrf2 signaling. J. Neuroinflamm. 16:24.
Eakin, K., Baratz-Goldstein, R., Pick, C. G., Zindel, O., Balaban, C. D., Hoffer, M. E., et al. (2014). Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One 9:e90617. doi: 10.1371/journal.pone.0090617
Farr, S. A., Poon, H. F., Dogrukol-Ak, D., Drake, J., Banks, W. A., Eyerman, E., et al. (2003). The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J. Neurochem. 84, 1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x
Feng, S., Yang, Q., Liu, M., Li, W., Yuan, W., Zhang, S., et al. (2011). Edaravone for acute ischaemic stroke. Cochrane Database Syst Rev 12:CD007230.
Feng, S.-Q., Yao, X., Zhang, Y., Hao, J., Duan, H.-Q., Zhao, C.-X., et al. (2019). Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural. Regen. Res. 14, 532–541.
Forcina, G. C., and Dixon, S. J. (2019). GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics 19:e1800311.
Freret, T., Valable, S., Chazalviel, L., Saulnier, R., Mackenzie, E. T., Petit, E., et al. (2006). Delayed administration of deferoxamine reduces brain damage and promotes functional recovery after transient focal cerebral ischemia in the rat. Eur. J. Neurosci. 23, 1757–1765. doi: 10.1111/j.1460-9568.2006.04699.x
Friedmann Angeli, J. P., and Conrad, M. (2018). Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 127, 153–159. doi: 10.1016/j.freeradbiomed.2018.03.001
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell. Biol. 16, 1180–1191. doi: 10.1038/ncb3064
Gammella, E., Recalcati, S., Rybinska, I., Buratti, P., and Cairo, G. (2015). Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms. Oxid. Med. Cell. Longev. 2015:230182.
Gangadhar, N. M., and Stockwell, B. R. (2007). Chemical genetic approaches to probing cell death. Curr. Opin. Chem. Biol. 11, 83–87. doi: 10.1016/j.cbpa.2006.11.033
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 59, 298–308. doi: 10.1016/j.molcel.2015.06.011
García-Yébenes, I., Sobrado, M., Moraga, A., Zarruk, J. G., Romera, V. G., Pradillo, J. M., et al. (2012). Iron overload, measured as serum ferritin, increases brain damage induced by focal ischemia and early reperfusion. Neurochem. Int. 61, 1364–1369. doi: 10.1016/j.neuint.2012.09.014
GBD (2016). Global, regional, and national burden of stroke, 1990-2016: a systematic analysis for the Global Burden of disease study 2016. Lancet Neurol. 18, 439–458.
GBD (2019). Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990-2016: a systematic analysis for the Global Burden of disease study 2016. Lancet Neurol. 18, 56–87.
Gilbert, K. R., Aizenman, E., and Reynolds, I. J. (1991). Oxidized glutathione modulates N-Methyl-D-aspartate-induced and depolarization-induced increases in intracellular Ca2+ in cultured rat forebrain neurons. Neurosci. Lett. 133, 11–14. doi: 10.1016/0304-3940(91)90045-u
Gong, C., Ennis, S. R., Hoff, J. T., and Keep, R. F. (2001). Inducible cyclooxygenase-2 expression after experimental intracerebral hemorrhage. Brain Res. 901, 38–46. doi: 10.1016/s0006-8993(01)02186-2
Gout, P. W., Buckley, A. R., Simms, C. R., and Bruchovsky, N. (2001). Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)(-) cystine transporter: a new action for an old drug. Leukemia 15, 1633–1640. doi: 10.1038/sj.leu.2402238
Grossman, R. G., Frankowski, R. F., Burau, K. D., Toups, E. G., Crommett, J. W., Johnson, M. M., et al. (2012). Incidence and severity of acute complications after spinal cord injury. J. Neurosurg. Spine 17, 119–128.
Guan, X., Li, X., Yang, X., Yan, J., Shi, P., Ba, L., et al. (2019). The neuroprotective effects of carvacrol on ischemia/reperfusion-induced hippocampal neuronal impairment by ferroptosis mitigation. Life Sci. 235:116795. doi: 10.1016/j.lfs.2019.116795
Guo, J., Li, Y., Chen, Z., He, Z., Zhang, B., Li, Y., et al. (2015). N-acetylcysteine treatment following spinal cord trauma reduces neural tissue damage and improves locomotor function in mice. Mol. Med. Rep. 12, 37–44. doi: 10.3892/mmr.2015.3390
Guo, Y., Liu, X., Liu, D., Li, K., Wang, C., Liu, Y., et al. (2019). Inhibition of BECN1 suppresses lipid peroxidation by increasing system Xc(-) activity in early brain injury after subarachnoid hemorrhage. J. Mol. Neurosci. 67, 622–631. doi: 10.1007/s12031-019-01272-5
Hadian, K., and Stockwell, B. R. (2020). SnapShot: ferroptosis. Cell 181, 1188–1188.e1. doi: 10.1016/j.cell.2020.04.039
Hall, E. D., Detloff, M. R., Johnson, K., and Kupina, N. C. (2004). Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J. Neurotrauma 21, 9–20. doi: 10.1089/089771504772695904
Hanson, L. R., Roeytenberg, A., Martinez, P. M., Coppes, V. G., Sweet, D. C., Rao, R. J., et al. (2009). Intranasal deferoxamine provides increased brain exposure and significant protection in rat ischemic stroke. J. Pharmacol. Exp. Ther. 330, 679–686. doi: 10.1124/jpet.108.149807
Hao, J., Li, B., Duan, H.-Q., Zhao, C.-X., Zhang, Y., Sun, C., et al. (2017). Mechanisms underlying the promotion of functional recovery by deferoxamine after spinal cord injury in rats. Neural. Regen. Res. 12, 959–968.
Hatakeyama, T., Okauchi, M., Hua, Y., Keep, R. F., and Xi, G. (2013). Deferoxamine reduces neuronal death and hematoma lysis after intracerebral hemorrhage in aged rats. Transl. Stroke Res. 4, 546–553. doi: 10.1007/s12975-013-0270-5
Hayano, M., Yang, W. S., Corn, C. K., Pagano, N. C., and Stockwell, B. R. (2016). Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278. doi: 10.1038/cdd.2015.93
He, Y. J., Liu, X. Y., Xing, L., Wan, X., Chang, X., and Jiang, H. L. (2020). Fenton reaction-independent ferroptosis therapy via glutathione and iron redox couple sequentially triggered lipid peroxide generator. Biomaterials 241:119911. doi: 10.1016/j.biomaterials.2020.119911
Hinman, A., Holst, C. R., Latham, J. C., Bruegger, J. J., Ulas, G., McCusker, K. P., et al. (2018). Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS One 13:e0201369. doi: 10.1371/journal.pone.0201369
Hirschhorn, T., and Stockwell, B. R. (2019). The development of the concept of ferroptosis. Free Radic. Biol. Med. 133, 130–143. doi: 10.1016/j.freeradbiomed.2018.09.043
Hoffer, M. E., Balaban, C., Slade, M. D., Tsao, J. W., and Hoffer, B. (2013). Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PLoS One 8:e54163. doi: 10.1371/journal.pone.0054163
Homma, T., Kobayashi, S., Sato, H., and Fujii, J. (2019). Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp. Cell Res. 384:111592. doi: 10.1016/j.yexcr.2019.111592
Hsieh, C. H., Lin, Y. J., Chen, W. L., Huang, Y. C., Chang, C. W., Cheng, F. C., et al. (2017). HIF-1alpha triggers long-lasting glutamate excitotoxicity via system xc(-) in cerebral ischaemia-reperfusion. J. Pathol. 241, 337–349. doi: 10.1002/path.4838
Hu, W., Wang, H., Liu, Z., Liu, Y., Wang, R., Luo, X., et al. (2017). Neuroprotective effects of lycopene in spinal cord injury in rats via antioxidative and anti-apoptotic pathway. Neurosci. Lett. 642, 107–112. doi: 10.1016/j.neulet.2017.02.004
Hu, X., Tao, C., Gan, Q., Zheng, J., Li, H., and You, C. (2016). Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid. Med. Cell. Longev. 2016:215391.
Hu, Z. X., Zhang, H., Yi, B., Yang, S. K., Liu, J., Hu, J., et al. (2020). VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 11:73.
Huang, L. L., Liao, X. H., Sun, H., Jiang, X., Liu, Q., and Zhang, L. (2019). Augmenter of liver regeneration protects the kidney from ischaemia-reperfusion injury in ferroptosis. J. Cell. Mol. Med. 23, 4153–4164. doi: 10.1111/jcmm.14302
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2018). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422.e21. doi: 10.1016/j.cell.2017.11.048
Ishii, T., Bannai, S., and Sugita, Y. (1981). Mechanism of growth stimulation of L1210 cells by 2-mercaptoethanol in vitro. Role of the mixed disulfide of 2-mercaptoethanol and cysteine. J. Biol. Chem. 256, 12387–12392.
Jackman, N. A., Melchior, S. E., Hewett, J. A., and Hewett, S. J. (2012). Non-cell autonomous influence of the astrocyte system xc- on hypoglycaemic neuronal cell death. ASN Neuro 4:e00074.
Jiang, J. Y., Gao, G. Y., Feng, J. F., Mao, Q., Chen, L. G., Yang, X. F., et al. (2019). Traumatic brain injury in China. Lancet Neurol. 18, 286–295.
Kagan, V. E., Mao, G., Qu, F., Angeli, J. P., Doll, S., Croix, C. S., et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90. doi: 10.1038/nchembio.2238
Karalija, A., Novikova, L. N., Kingham, P. J., Wiberg, M., and Novikov, L. N. (2012). Neuroprotective effects of N-acetyl-cysteine and acetyl-L-carnitine after spinal cord injury in adult rats. PLoS One 7:e41086. doi: 10.1371/journal.pone.0041086
Karalija, A., Novikova, L. N., Kingham, P. J., Wiberg, M., and Novikov, L. N. (2014). The effects of N-acetyl-cysteine and acetyl-L-carnitine on neural survival, neuroinflammation and regeneration following spinal cord injury. Neuroscience 269, 143–151. doi: 10.1016/j.neuroscience.2014.03.042
Karuppagounder, S. S., Alin, L., Chen, Y., Brand, D., Bourassa, M. W., Dietrich, K., et al. (2018). N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E2 to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 84, 854–872. doi: 10.1002/ana.25356
Kawai, H., Nakai, H., Suga, M., Yuki, S., Watanabe, T., and Saito, K. I. (1997). Effects of a novel free radical scavenger, MCl-186, on ischemic brain damage in the rat distal middle cerebral artery occlusion model. J. Pharmacol. Exp. Ther. 281, 921–927.
Keep, R. F., Hua, Y., and Xi, G. (2012). Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 11, 720–731. doi: 10.1016/s1474-4422(12)70104-7
Kenny, E. M., Fidan, E., Yang, Q., Anthonymuthu, T. S., New, L. A., Meyer, E. A., et al. (2019). Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit. Care Med. 47, 410–418. doi: 10.1097/ccm.0000000000003555
Khalaf, S., Ahmad, A. S., Chamara, K. V. D. R., and Doré, S. (2018). Unique properties associated with the brain penetrant iron chelator HBED reveal remarkable beneficial effects after brain trauma. J. Neurotrauma 36, 43–53. doi: 10.1089/neu.2017.5617
Khoshnam, S. E., Winlow, W., Farzaneh, M., Farbood, Y., and Moghaddam, H. F. (2017). Pathogenic mechanisms following ischemic stroke. Neurol. Sci. 38, 1167–1186. doi: 10.1007/s10072-017-2938-1
Kondo, Y., Ogawa, N., Asanuma, M., Ota, Z., and Mori, A. (1995). Regional differences in late-onset iron deposition, ferritin, transferrin, astrocyte proliferation, and microglial activation after transient forebrain ischemia in rat brain. J. Cereb. Blood Flow Metab. 15, 216–226. doi: 10.1038/jcbfm.1995.27
Lan, B., Ge, J. W., Cheng, S. W., Zheng, X. L., Liao, J., He, C., et al. (2020). Extract of Naotaifang, a compound Chinese herbal medicine, protects neuron ferroptosis induced by acute cerebral ischemia in rats. J. Integr. Med. [Epub ahead of print].
Lapchak, P. A., and Zivin, J. A. (2009). The lipophilic multifunctional antioxidant edaravone (radicut) improves behavior following embolic strokes in rabbits: a combination therapy study with tissue plasminogen activator. Exp. Neurol. 215, 95–100. doi: 10.1016/j.expneurol.2008.09.004
Lei, P., Bai, T., and Sun, Y. (2019). Mechanisms of Ferroptosis and relations with regulated cell death: a review. Front. Physiol. 10:139. doi: 10.3389/fphys.2019.00139
Leslie, S. W., Brown, L. M., Trent, R. D., Lee, Y. H., Morris, J. L., Jones, T. W., et al. (1992). Stimulation of N-Methyl-D-aspartate receptor-mediated calcium entry into dissociated neurons by reduced and oxidized glutathione. Mol. Pharmacol. 41, 308–314.
Lewerenz, J., Ates, G., Methner, A., Conrad, M., and Maher, P. (2018). Oxytosis/Ferroptosis-(Re-) Emerging roles for oxidative stress-dependent non-apoptotic cell death in diseases of the central nervous system. Front. Neurosci. 12:214. doi: 10.3389/fnins.2018.00214
Li, J., Cao, F., Yin, H. L., Huang, Z. J., Lin, Z. T., Mao, N., et al. (2020). Ferroptosis: past, present and future. Cell Death Dis. 11:88.
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2:e90777.
Li, Q., Weiland, A., Chen, X., Lan, X., Han, X., Durham, F., et al. (2018). Ultrastructural characteristics of neuronal death and white matter injury in mouse brain tissues after intracerebral hemorrhage: coexistence of ferroptosis, autophagy, and necrosis. Front. Neurol. 9:581. doi: 10.3389/fneur.2018.00581
Li, S., Jiang, D., Rosenkrans, Z. T., Barnhart, T. E., Ehlerding, E. B., Ni, D., et al. (2019a). Aptamer-conjugated framework nucleic acids for the repair of cerebral ischemia-reperfusion injury. Nano Lett. 19, 7334–7341. doi: 10.1021/acs.nanolett.9b02958
Li, X., Zhan, J., Hou, Y., Chen, S., Hou, Y., Xiao, Z., et al. (2019b). Coenzyme Q10 suppresses oxidative stress and apoptosis via activating the Nrf-2/NQO-1 and NF-κB signaling pathway after spinal cord injury in rats. Am. J. Transl. Res. 11, 6544–6552.
Li, X., Zhan, J., Hou, Y., Hou, Y., Chen, S., Luo, D., et al. (2019c). Coenzyme Q10 regulation of apoptosis and oxidative stress in H2O2 induced BMSC death by modulating the Nrf-2/NQO-1 signaling pathway and its application in a model of spinal cord injury. Oxid. Med. Cell. Longev. 2019:6493081.
Li, X., Zhuang, X., and Qiao, T. (2019d). Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem. Biophys. Res. Commun. 519, 240–245. doi: 10.1016/j.bbrc.2019.08.165
Li, Y. C., Cao, Y. M., Xiao, J., Shang, J. W., Tan, Q., Ping, F., et al. (2020). Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ.
Liang, W., Huang, X., and Chen, W. (2017). The effects of baicalin and baicalein on cerebral ischemia: a review. Aging Dis. 8, 850–867.
Lipscomb, D. C., Gorman, L. G., Traystman, R. J., and Hurn, P. D. (1998). Low molecular weight iron in cerebral ischemic acidosis in vivo. Stroke 29, 487–492.
Liu, J., Tang, T., and Yang, H. (2011). Protective effect of deferoxamine on experimental spinal cord injury in rat. Injury 42, 742–745. doi: 10.1016/j.injury.2010.08.028
Liu, J.-B., Tang, T.-S., and Xiao, D.-S. (2004). Changes of free iron contents and its correlation with lipid peroxidation after experimental spinal cord injury. Chin J. Traumatol. 7, 229–232.
Liu, Y., Wang, W., Li, Y., Xiao, Y., Cheng, J., and Jia, J. (2015). The 5-Lipoxygenase inhibitor zileuton confers neuroprotection against glutamate oxidative damage by inhibiting ferroptosis. Biol. Pharm. Bull. 38, 1234–1239. doi: 10.1248/bpb.b15-00048
Lorente, L., Martín, M. M., Abreu-González, P., Ramos, L., Argueso, M., Cáceres, J. J., et al. (2015). Association between serum malondialdehyde levels and mortality in patients with severe brain trauma injury. J. Neurotrauma 32, 1–6. doi: 10.1089/neu.2014.3456
Lorincz, T., Jemnitz, K., Kardon, T., Mandl, J., and Szarka, A. (2015). Ferroptosis is involved in acetaminophen induced cell death. Pathol. Oncol. Res. 21, 1115–1121. doi: 10.1007/s12253-015-9946-3
Lozano, D., Gonzales-Portillo, G. S., Acosta, S., de la Pena, I., Tajiri, N., Kaneko, Y., et al. (2015). Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 11, 97–106.
Lu, L., Cao, H., Wei, X., Li, Y., and Li, W. (2015). Iron deposition is positively related to cognitive impairment in patients with chronic mild traumatic brain injury: assessment with susceptibility weighted imaging. BioMed Res. Int. 2015:470676.
Lv, H., Zhen, C., Liu, J., Yang, P., Hu, L., and Shang, P. (2019). Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid. Med. Cell. Longev. 2019:3150145.
Maas, A. I. R., Menon, D. K., Adelson, P. D., Andelic, N., Bell, M. J., Belli, A., et al. (2017). In, and investigators, traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 16, 987–1048.
Morris, G., Anderson, G., Berk, M., and Maes, M. (2013). Coenzyme Q10 depletion in medical and neuropsychiatric disorders: potential repercussions and therapeutic implications. Mol. Neurobiol. 48, 883–903. doi: 10.1007/s12035-013-8477-8
Morris, G., Berk, M., Carvalho, A. F., Maes, M., Walker, A. J., and Puri, B. K. (2018). Why should neuroscientists worry about iron? The emerging role of ferroptosis in the pathophysiology of neuroprogressive diseases. Behav. Brain Res. 341, 154–175. doi: 10.1016/j.bbr.2017.12.036
Nakamura, T., Keep, R. F., Hua, Y., Schallert, T., Hoff, J. T., and Xi, G. (2004). Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J. Neurosurg. 100, 672–678. doi: 10.3171/jns.2004.100.4.0672
Nasoohi, S., Simani, L., Khodagholi, F., Nikseresht, S., Faizi, M., and Naderi, N. (2019). Coenzyme Q10 supplementation improves acute outcomes of stroke in rats pretreated with atorvastatin. Nutr. Neurosci. 22, 264–272. doi: 10.1080/1028415x.2017.1376928
Nazemi, Z., Nourbakhsh, M. S., Kiani, S., Heydari, Y., Ashtiani, M. K., Daemi, H., et al. (2020). Co-delivery of minocycline and paclitaxel from injectable hydrogel for treatment of spinal cord injury. J. Control Release 321, 145–158. doi: 10.1016/j.jconrel.2020.02.009
Nishi, H., Watanabe, T., Sakurai, H., Yuki, S., and Ishibashi, A. (1989). Effect of MCI-186 on brain edema in rats. Stroke 20, 1236–1240. doi: 10.1161/01.str.20.9.1236
Nour, M., Scalzo, F., and Liebeskind, D. S. (2013). Ischemia-reperfusion injury in stroke. Interv. Neurol. 1, 185–199.
Ottestad-Hansen, S., Hu, Q. X., Follin-Arbelet, V. V., Bentea, E., Sato, H., Massie, A., et al. (2018). The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia 66, 951–970. doi: 10.1002/glia.23294
Park, T. J., Park, J. H., Lee, G. S., Lee, J. Y., Shin, J. H., Kim, M. W., et al. (2019). Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 10:835.
Park, U. J., Lee, Y. A., Won, S. M., Lee, J. H., Kang, S.-H., Springer, J. E., et al. (2011). Blood-derived iron mediates free radical production and neuronal death in the hippocampal CA1 area following transient forebrain ischemia in rat. Acta Neuropathol. 121, 459–473. doi: 10.1007/s00401-010-0785-8
Patel, S. P., Sullivan, P. G., Pandya, J. D., Goldstein, G. A., VanRooyen, J. L., Yonutas, H. M., et al. (2014). N-acetylcysteine amide preserves mitochondrial bioenergetics and improves functional recovery following spinal trauma. Exp. Neurol. 257, 95–105. doi: 10.1016/j.expneurol.2014.04.026
Paterniti, I., Mazzon, E., Emanuela, E., Paola, R. D., Galuppo, M., Bramanti, P., et al. (2010). Modulation of inflammatory response after spinal cord trauma with deferoxamine, an iron chelator. Free Radic. Res. 44, 694–709. doi: 10.3109/10715761003742993
Pearn, M. L. I, Niesman, R., Egawa, J., Sawada, A., Almenar-Queralt, A., Shah, S. B., et al. (2017). Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell. Mol. Neurobiol. 37, 571–585. doi: 10.1007/s10571-016-0400-1
Portbury, S. D., Hare, D. J., Sgambelloni, C., Finkelstein, D. I., and Adlard, P. A. (2016). A time-course analysis of changes in cerebral metal levels following a controlled cortical impact. Metallomics 8, 193–200. doi: 10.1039/c5mt00234f
Portbury, S. D., Hare, D. J., Sgambelloni, C. J., Bishop, D. P., Finkelstein, D. I., Doble, P. A., et al. (2017). Age modulates the injury-induced metallomic profile in the brain. Metallomics 9, 402–410. doi: 10.1039/c6mt00260a
Qureshi, A. I., Ling, G. S., Khan, J., Suri, M. F., Miskolczi, L., Guterman, L. R., et al. (2001). Quantitative analysis of injured, necrotic, and apoptotic cells in a new experimental model of intracerebral hemorrhage. Crit. Care Med. 29, 152–157. doi: 10.1097/00003246-200101000-00030
Qureshi, A. I., Suri, M. F. K., Ostrow, P. T., Kim, S. H., Ali, Z., Shatla, A. A., et al. (2003). Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery 52, 1041–1047.
Rai, G., Joshi, N., Jung, J. E., Liu, Y., Schultz, L., Yasgar, A., et al. (2014). Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J. Med. Chem. 57, 4035–4048. doi: 10.1021/jm401915r
Ramezani, M., Sahraei, Z., Simani, L., Heydari, K., and Shahidi, F. (2018). Coenzyme Q10 supplementation in acute ischemic stroke: is it beneficial in short-term administration? Nutr. Neurosci. 1–6. doi: 10.1080/1028415x.2018.1541269 [Epub ahead of print].
Readnower, R. D., Chavko, M., Adeeb, S., Conroy, M. D., Pauly, J. R., McCarron, R. M., et al. (2010). Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 88, 3530–3539. doi: 10.1002/jnr.22510
Roth, T. L., Nayak, D., Atanasijevic, T., Koretsky, A. P., Latour, L. L., and McGavern, D. B. (2014). Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228. doi: 10.1038/nature12808
Sandercock, P., Wardlaw, J. M., Lindley, R. I., Dennis, M., Cohen, G., Murray, G., et al. (2012). The benefits and harms of intravenous thrombolysis with recombinant tissue plasminogen activator within 6 h of acute ischaemic stroke (the third international stroke trial [IST-3]): a randomised controlled trial. Lancet 379, 2352–2363. doi: 10.1016/s0140-6736(12)60768-5
Sauerbeck, A., Schonberg, D. L., Laws, J. L., and McTigue, D. M. (2013). Systemic iron chelation results in limited functional and histological recovery after traumatic spinal cord injury in rats. Exp. Neurol. 248, 53–61. doi: 10.1016/j.expneurol.2013.05.011
Savelev, S. U., Okello, E. J., and Perry, E. K. (2004). Butyryl- and acetyl-cholinesterase inhibitory activities in essential oils of Salvia species and their constituents. Phytother. Res. 18, 315–324. doi: 10.1002/ptr.1451
Seiler, A., Schneider, M., Förster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248. doi: 10.1016/j.cmet.2008.07.005
Selim, M. H., and Ratan, R. R. (2004). The role of iron neurotoxicity in ischemic stroke. Ageing Res. Rev. 3, 345–353. doi: 10.1016/j.arr.2004.04.001
Senol, N., Nazıroglu, M., and Yürüker, V. (2014). N-acetylcysteine and selenium modulate oxidative stress, antioxidant vitamin and cytokine values in traumatic brain injury-induced rats. Neurochem. Res. 39, 685–692. doi: 10.1007/s11064-014-1255-9
Shang, Y., Luo, M., Yao, F., Wang, S., Yuan, Z., and Yang, Y. (2020). Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal. 72:109633. doi: 10.1016/j.cellsig.2020.109633
Shen, L. D., Qi, W. H., Bai, J. J., Zuo, C. Y., Bai, D. L., Gao, W. D., et al. (2020). Resibufogenin inhibited colorectal cancer cell growth and tumorigenesis through triggering ferroptosis and ROS production mediated by GPX4 inactivation. Anat. Rec. [Epub ahead of print].
Shimada, K., Skouta, R., Kaplan, A., Yang, W. S., Hayano, M., Dixon, S. J., et al. (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503. doi: 10.1038/nchembio.2079
Siddiq, A., Aminova, L. R., Troy, C. M., Suh, K., Messer, Z., Semenza, G. L., et al. (2009). Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J. Neurosci. 29, 8828–8838. doi: 10.1523/jneurosci.1779-09.2009
Silva, N. A., Sousa, N., Reis, R. L., and Salgado, A. J. (2014). From basics to clinical: a comprehensive review on spinal cord injury. Prog. Neurobiol. 114, 25–57. doi: 10.1016/j.pneurobio.2013.11.002
Soria, F. N., Perez-Samartin, A., Martin, A., Gona, K. B., Llop, J., Szczupak, B., et al. (2014). Extrasynaptic glutamate release through cystine/glutamate antiporter contributes to ischemic damage. J. Clin. Invest. 124, 3645–3655. doi: 10.1172/jci71886
Speer, R. E., Karuppagounder, S. S., Basso, M., Sleiman, S. F., Kumar, A., Brand, D., et al. (2013). Hypoxia-inducible factor prolyl hydroxylases as targets for neuroprotection by “antioxidant” metal chelators: from ferroptosis to stroke. Free Radic. Biol. Med. 62, 26–36. doi: 10.1016/j.freeradbiomed.2013.01.026
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Su, Y., Zhao, B., Zhou, L., Zhang, Z., Shen, Y., Lv, H., et al. (2020). Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 483, 127–136. doi: 10.1016/j.canlet.2020.02.015
Sun, Y., Zheng, Y., Wang, C., and Liu, Y. (2018). Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 9:753.
Thorn, T. L., He, Y., Jackman, N. A., Lobner, D., Hewett, J. A., and Hewett, S. J. (2015). A cytotoxic co-operative interaction between energy deprivation and glutamate release from system x(c)(-) mediates aglycemic neuronal cell death. ASN Neuro 7:759091415614301.
Tomimoto, H., Shibata, M., Ihara, M., Akiguchi, I., Ohtani, R., and Budka, H. (2002). A comparative study on the expression of cyclooxygenase and 5-lipoxygenase during cerebral ischemia in humans. Acta Neuropathol. 104, 601–607. doi: 10.1007/s00401-002-0590-0
Tran, A. P., Warren, P. M., and Silver, J. (2018). The biology of regeneration failure and success after spinal cord injury. Physiol. Rev. 98, 881–917. doi: 10.1152/physrev.00017.2017
Tsai, T. H., Liu, S. C., Tsai, P. L., Ho, L. K., Shum, A. Y. C., and Chen, C. F. (2002). The effects of the cyclosporin A, a P-glycoprotein inhibitor, on the pharmacokinetics of baicalein in the rat: a microdialysis study. Br. J. Pharmacol. 137, 1314–1320. doi: 10.1038/sj.bjp.0704959
Tuo, Q. Z., Lei, P., Jackman, K. A., Li, X. L., Xiong, H., Li, X. L., et al. (2017). Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 22, 1520–1530. doi: 10.1038/mp.2017.171
Varga, V., Jenei, Z., Janaky, R., Saransaari, P., and Oja, S. S. (1997). Glutathione is an endogenous ligand of rat brain N-methyl-D-aspartate (n.d.) and 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem. Res. 22, 1165–1171.
Visavadiya, N. P., Patel, S. P., VanRooyen, J. L., Sullivan, P. G., and Rabchevsky, A. G. (2016). Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol. 8, 59–67. doi: 10.1016/j.redox.2015.12.011
Viswanathan, V. S., Ryan, M. J., Dhruv, H. D., Gill, S., Eichhoff, O. M., Seashore-Ludlow, B., et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457.
Wang, K.-Y., Wu, C.-H., Zhou, L.-Y., Yan, X.-H., Yang, R.-L., Liao, L.-M., et al. (2015). Ultrastructural changes of brain tissues surrounding hematomas after intracerebral hemorrhage. Eur. Neurol. 74, 28–35. doi: 10.1159/000434631
Ward, R. J., Zucca, F. A., Duyn, J. H., Crichton, R. R., and Zecca, L. (2014). The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 13, 1045–1060. doi: 10.1016/s1474-4422(14)70117-6
Watanabe, K., Tanaka, M., Yuki, S., Hirai, M., and Yamamoto, Y. (2018). How is edaravone effective against acute ischemic stroke and amyotrophic lateral sclerosis? J. Clin. Biochem. Nutr. 62, 20–38. doi: 10.3164/jcbn.17-62
Wenzel, S. E., Tyurina, Y. Y., Zhao, J., St Croix, C. M., Dar, H. H., Mao, G., et al. (2017). PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell 171, 628–641.e26. doi: 10.1016/j.cell.2017.09.044
Winterbourn, C. C. (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 8, 969–974. doi: 10.1016/0378-4274(95)03532-x
Witschi, A., Reddy, S., Stofer, B., and Lauterburg, B. H. (1992). The systemic availability of oral glutathione. Eur. J. Clin. Pharmacol. 43, 667–669. doi: 10.1007/bf02284971
Wolpaw, A. J., Shimada, K., Skouta, R., Welsch, M. E., Akavia, U. D., Pe’er, D., et al. (2011). Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 108, E771–E780.
Wu, C., Zhao, W., Yu, J., Li, S., Lin, L., and Chen, X. (2018). Induction of ferroptosis and mitochondrial dysfunction by oxidative stress in PC12 cells. Sci. Rep. 8:574.
Wu, H., Wu, T., Li, M., and Wang, J. (2012). Efficacy of the lipid-soluble iron chelator 2,2’-dipyridyl against hemorrhagic brain injury. Neurobiol. Dis. 45, 388–394. doi: 10.1016/j.nbd.2011.08.028
Wu, H., Wu, T., Xu, X., Wang, J., and Wang, J. (2011). Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 31, 1243–1250. doi: 10.1038/jcbfm.2010.209
Wu, J., Hua, Y., Keep, R. F., Nakamura, T., Hoff, J. T., and Xi, G. (2003). Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke 34, 2964–2969. doi: 10.1161/01.str.0000103140.52838.45
Wu, J., Yang, J. J., Cao, Y., Li, H., Zhao, H., Yang, S., et al. (2020). Iron overload contributes to general anaesthesia-induced neurotoxicity and cognitive deficits. J. Neuroinflamm. 17:110.
Xiao, X., Jiang, Y., Liang, W., Wang, Y., Cao, S., Yan, H., et al. (2019). miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol. Brain 12:78.
Xie, B. S., Wang, Y. Q., Lin, Y., Mao, Q., Feng, J. F., Gao, G. Y., et al. (2019). Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci. Ther. 25, 465–475. doi: 10.1111/cns.13069
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: process and function. Cell Death Differ. 23, 369–379. doi: 10.1038/cdd.2015.158
Yagoda, N., von Rechenberg, M., Zaganjor, E., Bauer, A. J., Yang, W. S., Fridman, D. J., et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868.
Yamada, N., Karasawa, T., Kimura, H., Watanabe, S., Komada, T., Kamata, R., et al. (2020). Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 11:144.
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U.S.A. 113, E4966–E4975.
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 15, 234–245. doi: 10.1016/j.chembiol.2008.02.010
Ye, L. F., Chaudhary, K. R., Zandkarimi, F., Harken, A. D., Kinslow, C. J., Upadhyayula, P. S., et al. (2020). Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem. Biol. 15, 469–484. doi: 10.1021/acschembio.9b00939
Yigitkanli, K., Pekcec, A., Karatas, H., Pallast, S., Mandeville, E., Joshi, N., et al. (2013). Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann. Neurol. 73, 129–135. doi: 10.1002/ana.23734
Yu, H., Zhang, Z. L., Chen, J., Pei, A., Hua, F., Qian, X., et al. (2012). Carvacrol, a food-additive, provides neuroprotection on focal cerebral ischemia/reperfusion injury in mice. PLoS One 7:e33584. doi: 10.1371/journal.pone.0033584
Yuan, H., Li, X., Zhang, X., Kang, R., and Tang, D. (2016). Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 478, 1338–1343. doi: 10.1016/j.bbrc.2016.08.124
Zeng, L., Tan, L., Li, H., Zhang, Q., Li, Y., and Guo, J. (2018). Deferoxamine therapy for intracerebral hemorrhage: a systematic review. PLoS One 13:e0193615. doi: 10.1371/journal.pone.0193615
Zhang, K., Tu, M., Gao, W., Cai, X., Song, F., Chen, Z., et al. (2019). Hollow prussian blue nanozymes drive neuroprotection against ischemic stroke via attenuating oxidative stress, counteracting inflammation, and suppressing cell apoptosis. Nano Lett. 19, 2812–2823. doi: 10.1021/acs.nanolett.8b04729
Zhang, L., Hu, R., Li, M., Li, F., Meng, H., Zhu, G., et al. (2013). Deferoxamine attenuates iron-induced long-term neurotoxicity in rats with traumatic brain injury. Neurol. Sci. 34, 639–645. doi: 10.1007/s10072-012-1090-1
Zhang, Y., Sun, C., Zhao, C., Hao, J., Zhang, Y., Fan, B., et al. (2019). Ferroptosis inhibitor SRS 16-86 attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury. Brain Res. 1706, 48–57. doi: 10.1016/j.brainres.2018.10.023
Zhang, Z., Wu, Y., Yuan, S., Zhang, P., Zhang, J., Li, H., et al. (2018). Glutathione peroxidase 4 participates in secondary brain injury through mediating ferroptosis in a rat model of intracerebral hemorrhage. Brain Res. 1701, 112–125. doi: 10.1016/j.brainres.2018.09.012
Zhao, X., Zhang, Y., Strong, R., Zhang, J., Grotta, J. C., and Aronowski, J. (2007). Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. J. Neurochem. 101, 652–663. doi: 10.1111/j.1471-4159.2006.04414.x
Zhong, H., and Yin, H. (2015). Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol. 4, 193–199. doi: 10.1016/j.redox.2014.12.011
Zhou, Y., Wang, H.-D., Zhou, X.-M., Fang, J., Zhu, L., and Ding, K. (2018). N-acetylcysteine amide provides neuroprotection via Nrf2-ARE pathway in a mouse model of traumatic brain injury. Drug Des. Dev. Ther. 12, 4117–4127. doi: 10.2147/dddt.s179227
Keywords: ferroptosis, iron metabolism, lipid metabolism, stroke, traumatic brain injury, spinal cord injury, therapy
Citation: Shen L, Lin D, Li X, Wu H, Lenahan C, Pan Y, Xu W, Chen Y, Shao A and Zhang J (2020) Ferroptosis in Acute Central Nervous System Injuries: The Future Direction? Front. Cell Dev. Biol. 8:594. doi: 10.3389/fcell.2020.00594
Received: 11 March 2020; Accepted: 18 June 2020;
Published: 15 July 2020.
Edited by:
Alexander A. Mongin, Albany Medical College, United StatesReviewed by:
Sandra Hewett, Syracuse University, United StatesChunying Li, Georgia State University, United States
Copyright © 2020 Shen, Lin, Li, Wu, Lenahan, Pan, Xu, Chen, Shao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anwen Shao, MjExMTgxMTZAemp1LmVkdS5jbg==; YW53ZW5zaGFvQHNpbmEuY29t; Yiding Chen, eWRjaGVuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work