
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 23 June 2020
Sec. Stem Cell Research
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00440
This article is part of the Research TopicInduced Pluripotent Stem Cell-Based Disease Modeling and Drug Discovery: Can We Recapitulate Cardiovascular Disease on a Culture Dish?View all 13 articles
 Min-Su Kim1
Min-Su Kim1 Brandon Fleres2
Brandon Fleres2 Jerrell Lovett2
Jerrell Lovett2 Melissa Anfinson3Sai Suma K. Samudrala3Lauren J. Kelly2
Melissa Anfinson3Sai Suma K. Samudrala3Lauren J. Kelly2 Laura E. Teigen2Matthew Cavanaugh2Maribel Marquez4
Laura E. Teigen2Matthew Cavanaugh2Maribel Marquez4 Aron M. Geurts4
Aron M. Geurts4 John W. Lough3Michael E. Mitchell1Robert H. Fitts2
John W. Lough3Michael E. Mitchell1Robert H. Fitts2 Aoy Tomita-Mitchell1,5*
Aoy Tomita-Mitchell1,5*Hypoplastic left heart syndrome (HLHS) is a clinically and anatomically severe form of congenital heart disease; however, its etiology remains largely unknown. We previously demonstrated that genetic variants in the MYH6 gene are significantly associated with HLHS. Additionally, induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) from an HLHS-affected family trio (affected parent, unaffected parent, affected proband) carrying an MYH6-R443P head domain variant demonstrated dysmorphic sarcomere structure and increased compensatory MYH7 expression. Analysis of iPSC-CMs derived from the HLHS trio revealed that only beta myosin heavy chain expression was observed in CMs carrying the MYH6-R443P variant after differentiation day 15 (D15). Functional assessments performed between D20-D23 revealed that MYH6-R443P variant CMs contracted more slowly (40 ± 2 vs. 47 ± 2 contractions/min, P < 0.05), shortened less (5.6 ± 0.5 vs. 8.1 ± 0.7% of cell length, P < 0.05), and exhibited slower shortening rates (19.9 ± 1.7 vs. 28.1 ± 2.5 μm/s, P < 0.05) and relaxation rates (11.0 ± 0.9 vs. 19.7 ± 2.0 μm/s, P < 0.05). Treatment with isoproterenol had no effect on iPSC-CM mechanics. Using CRISPR/Cas9 gene editing technology, introduction of the R443P variant into the unaffected parent’s iPSCs recapitulated the phenotype of the proband’s iPSC-CMs, and conversely, correction of the R443P variant in the proband’s iPSCs rescued the cardiomyogenic differentiation, sarcomere organization, slower contraction (P < 0.05) and decreased velocity phenotypes (P < 0.0001). This is the first report to identify that cardiac tissues from HLHS patients with MYH6 variants can exhibit sarcomere disorganization in atrial but not ventricular tissues. This new discovery was not unexpected, since MYH6 is expressed predominantly in the postnatal atria in humans. These findings demonstrate the feasibility of employing patient-derived iPSC-CMs, in combination with patient cardiac tissues, to gain mechanistic insight into how genetic variants can lead to HLHS. Results from this study suggest that decreased contractility of CMs due to sarcomere disorganization in the atria may effect hemodynamic changes preventing development of a normal left ventricle.
Hypoplastic left heart syndrome (HLHS) is a severe form of congenital heart disease (CHD) characterized by atresia/stenosis of the aortic and mitral valves, and severe hypoplasia of the left ventricle and aorta (Noonan and Nadas, 1958). HLHS affects more than one in every 4000 live births (Mai et al., 2019). Evidence supporting a genetic basis for HLHS includes observations of familial clustering, high heritability, and its occurrence with specific chromosomal disorders such as Turner and Jacobsen syndromes (Tomita-Mitchell et al., 2016; Lara et al., 2017; Yagi et al., 2018). HLHS exhibits complex genetic inheritance along with an increased frequency of bicuspid aortic valve (BAV) and coarctation of the aorta (CoA) in relatives of HLHS patients (Hinton et al., 2007, 2009). Variants in genes such as GJA1 (Dasgupta et al., 2001), NKX2.5 (Elliott et al., 2003), NOTCH1 (Garg et al., 2005; McBride et al., 2008; Hrstka et al., 2017; Yang et al., 2017), and MYH6 (Theis et al., 2015; Tomita-Mitchell et al., 2016), as well as observations of syndromic or rare copy number variants (CNVs) in cardiomyogenic genes (Grossfeld, 2007; Grossfeld et al., 2009; Tomita-Mitchell et al., 2012; Warburton et al., 2014; Glidewell et al., 2015) have been associated with HLHS. We previously reported that rare variants in MYH6 (alpha myosin heavy chain; α-MHC) were observed in 10% of HLHS patients, and that cardiac transplant-free survival was reduced in HLHS subjects containing MYH6 variants in comparison with HLHS patients without MYH6 variants. Furthermore, cardiac tissue from MYH6 variant carriers exhibited significant upregulation of sarcomere genes, including ACTA1 (actin alpha 1), MYL2 (myosin light chain 2), TNNT2 (cardiac troponin T), and the MYH6 homolog MYH7, which encodes the beta-myosin heavy chain (β-MHC) isoform. Using cardiomyocytes (CMs) derived from induced pluripotent stem cells (iPSCs) obtained from an HLHS patient carrying an MYH6-R443P variant, we discovered that sarcomere structure was dysmorphic (Tomita-Mitchell et al., 2016).
Two myosin heavy chain proteins (α-MHC and β-MHC) are expressed in the human heart. During development, α-MHC is expressed in both the primitive atrium and ventricle until gestational day 35, after which its expression continually decreases in the ventricle (Wessels et al., 1990, 1991). In contrast, β-MHC expression gradually increases in the ventricles throughout gestation and this expression pattern persists throughout adult stages, so that β-MHC is the predominant ventricular isoform, while α-MHC is the predominant atrial isoform in adults (Cummins and Lambert, 1986; Reiser et al., 2001). We previously noted that MYH6 mRNA strongly predominates during the earliest stages of in vitro cardiomyogenesis in H1 human embryonic stem cells wherein MYH6 comprises ∼99% of total MHC transcripts in differentiation day 8 (D8) cultures, and declines to ∼86% at D14 (Kim et al., 2015). This likely reflects a prominent role for α-MHC in nascent myocyte development that may be disrupted by MYH6 variants associated with HLHS.
Fetal heart development relies on proper blood flow, as signaling pathways responsive to shear stress and pressure-related strain both affect cardiac chamber formation. The prevailing hypothesis is that HLHS pathophysiology stems from impaired blood flow through the left ventricle (LV) during cardiogenesis (Fishman et al., 1978; Gruber and Epstein, 2004; deAlmeida et al., 2007). Our findings are consistent with this, as disruptions in an atrial protein such as α-MHC would alter ventricular preload with consequent defective expansion and/or differentiation of cardiomyocytes resulting in a dysmorphic and dysfunctional ventricle (Hove et al., 2003; Burggren et al., 2004; Hierck et al., 2008; McCain and Parker, 2011; Santhanakrishnan and Miller, 2011; Lee et al., 2016; McCormick and Tzima, 2016; Hoog et al., 2018). This is further supported by studies of weak atrium (wea) zebrafish, which harbor myh6 mutations and exhibit defects in both cardiac chambers, including defective atrial contraction along with abnormal sarcomere organization and an underdeveloped ventricle (Berdougo et al., 2003).
Current non-invasive imaging methods allow detection of HLHS as early as 16 weeks gestational age (Friedberg et al., 2009; Galindo et al., 2009), long after the fetal heart is formed and septated at 7–8 weeks. The primitive heart, expressing only MYH6, begins beating even earlier, during the third week of development. Thus, the pathological changes associated with HLHS are likely present long before we are able to detect them. Given these limitations, developmental defects such as HLHS are often investigated using murine models. However, the chamber-specific MHC expression in rodents is the opposite of humans, making them an unsuitable option for our studies. Here we studied human cardiac tissue and patient-specific iPSC-CMs to identify structural characteristics of atrial and ventricular tissue of HLHS patients and contractile properties of human iPSC-CMs with an MYH6 variant. The latter permitted us to monitor early stages of cardiomyogenesis in CMs of HLHS patients in vitro. Our hypothesis is that the MYH6 variant iPSC-CMs exhibit depressed extent and velocity of shortening that can be rescued by correcting the variant in proband-iPSCs using Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 gene editing. Importantly, we show the feasibility of employing iPSC-CMs to ascertain functional consequences of the MYH6-R443P variant, demonstrating that changes in the myosin isoforms confer contractile limitations.
Study subjects have been previously described (Tomita-Mitchell et al., 2016). All subjects provided written informed consent, and procedures were approved by the MCW Institutional Review Board and conformed to the principles in the Declaration of Helsinki.
All CRISPRs were designed as previously described (Mitzelfelt et al., 2017). Briefly, CRISPR target sites within 30 bps of the single nucleotide polymorphism (SNP) MYH6-R443P were identified using ZiFiT Targeter (Version 4.2). Three guide RNAs were cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene, Cambridge, MA, United States) to introduce MYH6-R443P into the heart healthy parent’s iPSCs. CRISPR efficiency was determined using the Cel-1 surveyor assay in HEK293 cells. The variant MYH6-R443P was flanked approximately 60 bp on either side by single strand oligonucleotides (ssODNs) (Table 1). Silent mutations were incorporated to prevent re-cutting by Cas9 by disrupting the target sequence. An additional AAVS1 locus was targeted for incorporation of GFP-puromycin resistance using the AAVS1 Safe Harbor TALE-Nuclease kit (SBI, Palo Alto, CA, United States).

Table 1. Oligonucleotides used for gene editing, sequencing, and genotyping.
To correct the MYH6-R443P variant in proband iPSCs, we designed CRISPR targets (Table 1) to align with the variant allele and purchased synthesized RNA (AltR CRISPR-Cas9 cr RNA, Integrated DNA Technologies, Coralville, IA, United States), RFP-labeled repetitive sequence (AltR CRISPR-Cas9 tracr RNA, Integrated DNA Technologies, Coralville, IA, United States) and purified Cas9 enzyme (40 Um/U1). Additionally, we designed and validated a CRISPR targeting the AAVS1 locus to introduce a puromycin resistance cassette (Integrated DNA Technologies, Coralville, IA, United States).
Prior to transfection, iPSCs were pretreated for 3–4 h with 5 μM Rho-associated kinase (ROCK) inhibitor (Y27632, Stemgent, Lexington, MA, United States), dissociated with accutase (Thermo Fisher Scientific, Waltham, MA, United States) to ensure single cell suspension, and transferred to an electroporation cuvette. For each transfection (D0), 1 μg of the gene-specific CRISPRs, 80 μM of the relevant ssODN, 1 μg of each of the two AAVS1-specific cassettes, and 1 μg AAVS1 donor plasmid were added to 100 μl P4 solution (Lonza, Basel, Switzerland) and electroporated using program CB-150 on a 4D NucleofectorTM (Lonza, Basel, Switzerland) into iPSCs (1 × 106 cells/transfection). iPSCs from each transfection were then seeded into one well of a geltrex-coated 24-well plate (5 × 105 cells/cm2) for recovery in mTeSR1 supplemented with 5 μM ROCK inhibitor. The following day (D1), iPSCs were cultured with mTeSR1. Two days post-transfection (D2), iPSCs from one well of 24-well plate were sub-cultured at a clonal density (∼5 × 104 cells/cm2) into all wells of a 6-well plate pre-seeded with mitomycin C-treated mouse embryonic fibroblast (MEF) feeder cells (Thermo Fisher Scientific, Waltham, MA, United States) in human ESC medium. ESC medium was comprised of Knockout DMEM (Thermo Fisher Scientific, Waltham, MA, United States) supplemented with 20% Knockout Serum Replacement (Thermo Fisher Scientific, Waltham, MA, United States), MEM-NEAA (Thermo Fisher Scientific, Waltham, MA, United States), 2 mM L-glutamine (Thermo Fisher Scientific, Waltham, MA, United States), Penicillin/Streptomycin, 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO, United States), 10 ng/ml human basic fibroblast growth factor (bFGF, Cell Signaling, Danvers, MA, United States), 50 ng/ml L-ascorbic acid (Sigma-Aldrich, St. Louis, MO, United States) and 5 μM ROCK inhibitor. Medium was changed 2 days later (D4) to ESC medium without ROCK inhibitor. Five days post-seeding (D5), medium was changed to puromycin (0.5 g/ml)-supplemented MEF-conditioned medium with bFGF and L-ascorbic acid, which was replaced every 2 days for 1 week. Following maintenance (∼D12), distinct colonies (∼1 mm diameter) were manually transferred to a single well of a 24-well plate pre-coated with geltrex in mTeSR1 medium plus ROCK inhibitor without MEFs. Following genotyping (see below), clones were expanded to a geltrex-coated well of a 12-well plate in mTeSR1 plus ROCK inhibitor and further expanded and frozen in mFreSR freezing medium (STEMCELL Technologies, Vancouver, BC, Canada). Isolated CRISPRed iPSC lines were frequently sub-cloned to ensure homogeneity of the population.
To isolate genomic DNA from iPSCs, 30 μl Quick Extract Solution (Epicentre, Madison, WI, United States) was added to each cell pellet and incubated for 15 min at 65°C, followed by 5 min at 95°C. PCR was carried out using gene-specific primers (Table 1), and 2 μl of PCR product were used for further allele-specific SNP genotyping with Quantstudio7 (Thermo Fisher Scientific, Waltham, MA, United States). After confirmation of gene edit, ∼100 ng of PCR product was treated with Exosap-IT (Thermo Fisher Scientific, Waltham, MA, United States) and sent to Retrogen (San Diego, CA, United States) for Sanger sequencing. Clones that were positive for iPSCs containing the edited MYH6-R443P gene were sequenced at the conserved region in MYH7 as well to check the CRISPR’s off-target activity.
Induced pluripotent stem cell (iPSC) lines were generated from dermal fibroblasts donated by HLHS probands and their parents. Fibroblasts were reprogrammed to pluripotent stem cells using Sendai reprogramming as previously described (Tomita-Mitchell et al., 2016). Pluripotency was confirmed with morphological appearance and % of cells exhibiting Oct4-positive immunostaining (99–100%). The cells were karyotypically normal and had the ability to differentiate into multiple lineages (definitive endoderm and cardiomyogenic mesoderm) (Tomita-Mitchell et al., 2016). In short, iPSCs were maintained in a hypoxic incubator at 5% CO2 and 4% O2. All experiments were performed on cells during passages 10–50. One day prior to inducing cardiomyocyte (CM) differentiation, the iPSCs were seeded at a cell density of >1 × 105/cm2 on a 10 μg/cm2 geltrex-coated (Thermo Fisher Scientific, Waltham, MA, United States) tissue culture dish. The cells were cultured in mTeSR1 medium (STEMCELL Technologies, Vancouver, BC, Canada) supplemented with 5 μM ROCK inhibitor. The cells were induced with 10 μM CHIR99021 (Stemgent, Lexington, MA, United States) and 10 ng/ml Activin-A (R&D Systems, Minneapolis, MN, United States) in insulin-free RPMI/B27 (Thermo Fisher Scientific, Waltham, MA, United States). On D3, the medium was exchanged for insulin-free RPMI/B27 with 5 μM IWP (Tocris, Bristol, United Kingdom). Starting at D7, at 2-day intervals the medium was changed to RPMI/B27 with insulin (Thermo Fisher Scientific, Waltham, MA, United States). Cultures were evaluated for % of cardiac troponin T (cTnT) positive cells via flow cytometry and MHC (MF20) expression by immunostaining. The cardiomyocytes were either frozen at D15, or were sub-cultured between D10–D15 after dissociation into single cells with 0.25% trypsin; specifically, cells centrifuged at 1,000 rpm for 5 min were either frozen at a density of >2 × 106 in cryovials with 10% DMSO and 90% FBS, or were re-plated at a density of 2 × 104–5 × 104/cm2 on a geltrex-coated 12 cm2 round cover glass for single cell-based contractility studies, or at >5 × 104/ cm2 for automated contractility video analysis (Pulse Video Analysis, Dana solutions, Palo Alto, CA, United States).
The iPSC-CMs and tissues were co-immunostained with anti-sarcomeric α-actinin (AF594) (Abcam, Cambridge, MA, United States) to image sarcomere organization, and anti-MHC monoclonal MF20 (AF488) (DSHB, University of Iowa, Iowa City, IA, United States) to inform myocyte identity. Sarcomeres in iPSC-CMs were visualized with a Nikon inverted fluorescent microscope at 400x.
All the contractile experiments with iPSC-CMs were conducted in a non-biased blinded fashion. Contractile data were collected from iPSC-CMs at differentiation days D20, D27, D34, and D41. Experiments were conducted over 3 days such that D20 includes D20-D23, D27 (D27–D30), D34 (D34–D37), and D41 (D41–D43). The iPSC-CMs were transferred to a recording chamber containing 1 mM Ca2+ Tyrode’s solution (35°C) mounted on the stage of an inverted microscope (Nikon TE2000). Spontaneously contracting iPSCs were visualized at 40x and contractile events recorded using a MyoCam-S video 250 Hz camera and a CFA300 cell framing adapter (IonOptix, Boston, MA, United States). The left and right edges of iPSC-CMs were defined and contractions per minute (CPM), shortening (μm), % cell shortening, and rates of shortening and relaxation (μm/second) were measured using the IonWizard image detection program (IonOptix, Boston, MA, United States). Ten cells on a 12 cm2 round cover glass were evaluated for 10 contractions each. For a subset of D20 iPSC-CMs, the same parameters were determined for responses to 1 and 2 Hz stimulation or to 1 μM isoproterenol. As an alternative method, an automated contractility software1 (Dana solution, Palo Alto, CA, United States) (Maddah et al., 2014, 2015) was used to study videos of contracting iPSC-CMs.
Action potentials of iPSC-CMs were obtained with microelectrodes (50–70 MΩ) filled with 2.7 M KCl and 10 mM HEPES and recorded at 35°C using Clampex9.2 software (Axon Instruments, Axoclamp-2A, San Jose, CA, United States). For each iPSC-CM measured, the lowest membrane potential (referred to as diastolic potential), threshold potential, peak spike potential, and action potential duration were averaged from 5 action potentials per cell. Action potential duration (APD90) was defined as the time (ms) from the start of the action potential until membrane potential returned to 10% of its peak height.
The iPSC-CMs were plated on an 18 mm2 round cover glass at D15. At D20, the iPSC-CMs were incubated for 45 min at 35°C in 0.5 mM Ca2+ Tyrode’s solution containing 2.5 μM of Fura-2 AM (Invitrogen, Carlsbad, CA, United States). Then, cover glasses were inserted into a quick-release chamber (RC-49MFS, Warner Instruments, Hamden, CT, United States) bathed in 1 mM Ca2+ Tyrode’s solution at 35°C. Whole cell Ca2+ transients were recorded using Fura-2 AM as described previously (Wang and Fitts, 2018). Briefly, Fura-2 AM was excited at 340 and 380 nm switching between wavelengths with a LAMBDA DG4 (Sutter Instrument, Novato, CA, United States) and the fluorescence emission measured at 510 nm. To determine the Ca2+ transient rate of rise and fall and the peak amplitude, the ratio was analyzed with IonWizard and data were expressed as ratio units (peak amplitude) and ratio units/s (rate of transient rise and fall). Ten Ca2+ transients were averaged for every iPSC-CM.
Following contractile studies, iPSC-CMs were scraped from cover glasses into 1 mM Ca2+ Tyrode’s solution and centrifuged for 5 min at 16,100 × g. The pellet of iPSC-CMs was collected in 10 μl SDS sample buffer (10% SDS, 23 mM EDTA, 50% glycerol, 0.4% bromophenol blue, and 5.1% β-mercaptoethanol, pH 6.8) and stored at −80°C until studied.
MHC composition was determined by silver staining 5% SDS-PAGE gels as previously described (Sundberg et al., 2018). Briefly, gels were run for 28–30 h at 4°C to separate α-MHC and β-MHC, and ImageJ (National Institutes of Health) was used to quantify protein expression. Samples were also run for 1 h at 150 V on 12% precast polyacrylamide gels using a Mini-PROTEAN Electrophoresis Cell (Bio-Rad, Hercules, CA, United States) and silver stained to examine lower molecular weight proteins.
Diluted samples were run on a 4–15% gradient gel for 1 h at 150 V and transferred to a 0.2 μm nitrocellulose membrane using a Mini Trans-Blot cell (Bio-Rad, Hercules, CA, United States) at 100 V for 50 min. The gel was run in Tris-glycine running buffer and transferred with running buffer containing 20% methanol. Following transfer, membranes were blocked for 3 h in 5% non-fat dry milk in TBS-0.1% Tween pH 7.6 (TBST) and probed with primary antibodies overnight at 4°C. Primary antibodies for MHC (MF20, 1:800, DSHB), GAPDH (ab181602, 1:1,000, Abcam, Cambridge, United Kingdom), and ventricular myosin light chain-2 (MLC2v) (7C9, 1:2,000, Fisher Scientific, Hampton, NH, United States) were diluted in 5% non-fat dry milk in TBST. Then, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (sc-516132, 1:6,500, Santa Cruz Biotechnology; NBP2-30347H, 1:6,500, Novus Biologicals; G-21234, 1:1,600, Fisher Scientific, Hampton, NH, United States) in 5% non-fat dry milk in TBST for 1 h at room temperature. Protein expression was detected on autoradiography film (Hyblot CL, Fisher Scientific, Hampton, NH, United States) by chemiluminescence. ImageJ (National Institutes of Health) was used for densiometric analysis and protein expression was normalized to GAPDH.
A total of 27 samples from atrial and ventricular tissues were examined, including two 4-day old neonatal atrial tissues, one from an HLHS subject without an MYH6 variant and one from an HLHS subject with the MYH6-R443P variant. The 25 non-neonatal tissues were from subjects between 227 days to 24-years old. These samples were divided into three study groups: control (non-HLHS CHDs and structurally normal cardiac control), HLHS subjects without an MYH6 variant, and HLHS subjects with an MYH6 variant. In each study group, when available, we included atrial and ventricular tissues. In all cases, the samples were pieces of tissue dissected and phenotyped at the time of surgery. They were snap frozen immediately after removal from the patient. Some tissues came at the time of heart implantation, and some came from the time of reparative surgery.
Cryo-sections (10 μm) of the frozen tissues were prepared. The tissues were co-immunostained with anti-sarcomeric α-actinin (AF594) (Abcam, Cambridge, MA, United States) to image sarcomere organization, and anti-MHC monoclonal MF20 (AF488) (DSHB, University of Iowa) to inform myocyte identity. Sarcomeres in tissue sections and iPSC-CMs were visualized with Structured Illumination Microscopy (SIM) at 1000x magnification.
Significant differences between two groups were analyzed through an unpaired two-tailed Student’s t-test using Excel (Microsoft). If the data were not normally distributed, comparisons were made using the non-parametric Wilcoxon signed-rank test. Statistical significance was set at P < 0.05.
When necessary, data were transformed to meet assumptions of normality and homogeneity of variance. For the time course study, a two-way ANOVA was performed to compare iPSC-CM contractility of wild-type (WT) vs. variant (VAR; MYH6-R443P) at timepoints D20, D27, D34, and D41. Post hoc pair-wise comparisons were performed using Tukey’s test. All subsequent studies (action potentials, contractile responses to stimulation, isoproterenol, and Ca2+ transient recordings) were restricted to D20. Data were analyzed with a one-way ANOVA, except for stimulation data, which were analyzed with a two-way ANOVA. Statistics were performed using Minitab v18.1 with significance set at P < 0.05.
Using CRISPR/Cas9 gene editing, the MYH6-R443P variant was introduced into the heart healthy parent’s iPSCs, generating CRISPRed control wild-type (WTcc) lines, as well as lines having one allele (inserted +/VAR) or both alleles inserted (inserted VAR/VAR). The CRISPR target site has one base-pair mismatch, and the sequences of both MYH6 (Figure 1A) and its close homologue MYH7 (Supplementary Figure S1) were confirmed in CRISPRed iPSCs with Sanger sequencing for any off-target effects. The experiments performed in VAR-inserted cells were obtained with only one line per genotype. Following cellular expansion and verification of genotype, iPSCs representing WTcc, inserted +/VAR and inserted VAR/VAR were induced to undergo cardiomyogenic differentiation as we previously described (Kim et al., 2015) (depicted in Figure 1B). The efficiency of cardiomyogenic differentiation during D10–D15 was reduced in both the inserted +/VAR and inserted VAR/VAR iPSC-CMs determined by MF20 immunostaining (Figure 1C) and flow cytometry assessment of % cTnT-positive cells (Figures 1D,E). We also evaluated sarcomere structure in iPSC-CMs at later stages of differentiation in both mass cultured CMs at D50 (Figure 1F) and individual CMs isolated from the culture at D73 (Figure 1G). Over 100 isolated iPSC-CMs of each line were judged for dysmorphic sarcomere organization at D69 and D73 (Figure 1H). “Normal” iPSC-CMs exhibit crisp and elongated ladders with wide Z-bands. Dysmorphic iPSC-CMs are characterized by more than 50% of the myocyte area displaying blurred α-actinin staining and/or sarcomeric ladders with punctate or truncated deposits of α-actinin (Figures 1G,H). iPSC-CMs with the MYH6-R443P variant were substantially disorganized in both the inserted +/VAR and inserted VAR/VAR compared to WTcc-CMs.
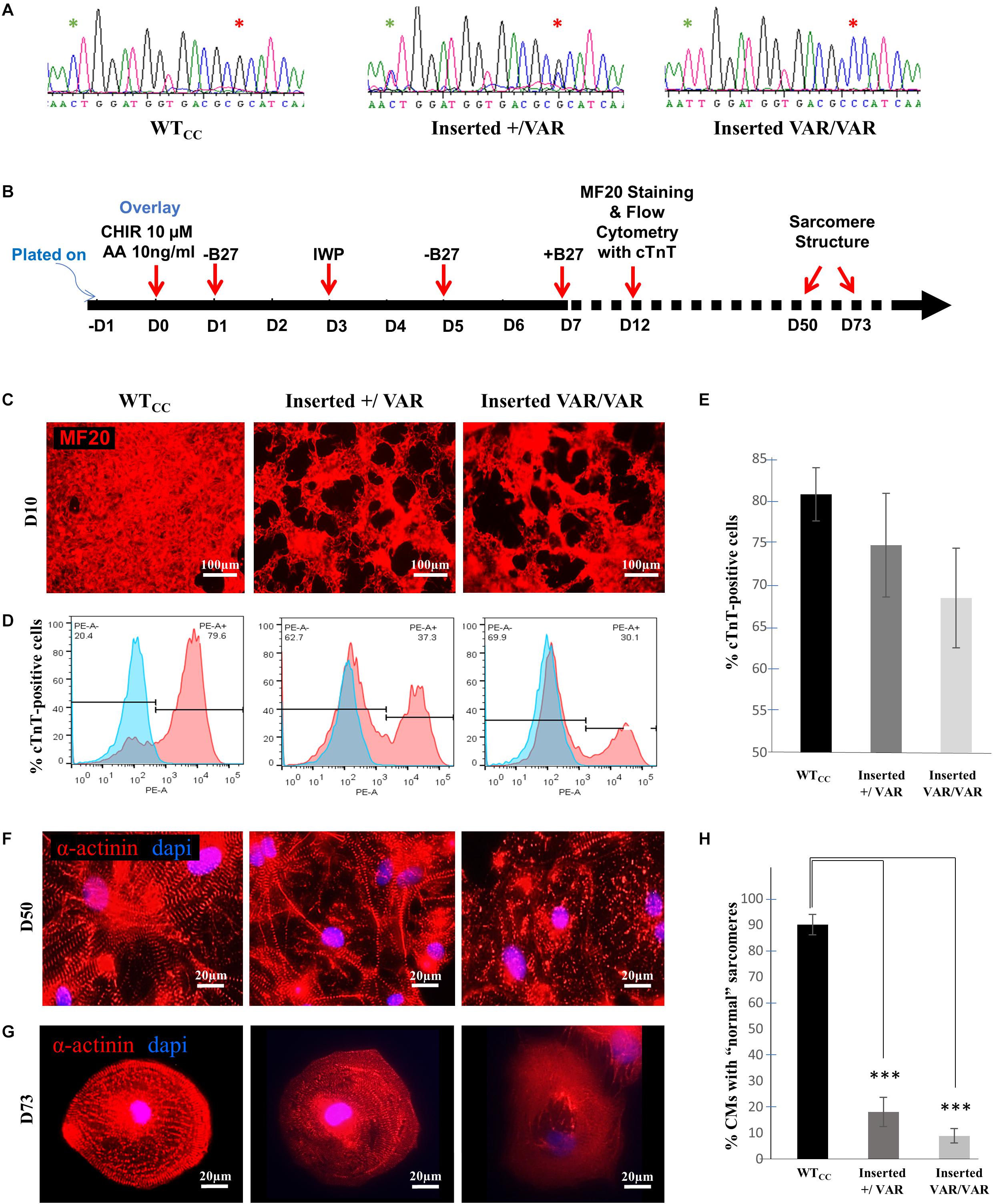
Figure 1. MYH6-R443P VAR-inserted iPSC-CMs recapitulate phenotypes of patient-specific iPSC-CMs. (A) Sequences at the MYH6-R443 locus in VAR-inserted lines. * is R443 locus and * is locus for silencing mutation. (B) Scheme for differentiating CMs using small molecule Gsk3 inhibitor (CHIR99021) with Activin-A and Wnt inhibitor (IWP). (C) Representative immunostaining of VAR-inserted iPSC-CMs with MF20 at D12. (D) Representative flow cytometry of cardiac troponin T (cTnT) positive cells in CRISPRed iPSC-CMs at D12. (E) Flow cytometry of cells cultured in parallel with those in (D), showing % cTnT-positive cells in VAR-inserted iPSC-CMs at D10-15. Data were compiled from 16 replicates in each cell type and 5 separate experiments. (F) Sarcomeres are dysmorphic in VAR-introduced iPSC-CMs as compared to WTCC. α-actinin immunostaining (red) is seen in high density cultures at D50. (G) Comparative sarcomere organization in single cell iPSC-CMs isolated after sub-culturing at a low density at D69-D73. (H) % of cells with normal organized sarcomeres in both VAR-inserted iPSC-CMs, +/VAR and VAR/VAR in parallel to those in (G). WTcc, CRISPRed control wild-type; +/VAR, MYH6-R443P heterozygous inserted; VAR/VAR, MYH6-R443P homozygous inserted. Values are means ± SE. Student’s t-test (two-tailed, equal variance), ***P < 0.0005.
Using CRISPR-Cas9, we corrected the MYH6-R443P variant (corrected WT) in the proband’s iPSCs (Figure 2A). This improved the efficiency of cardiomyogenic differentiation as assessed by MF20 immunostaining (Figure 2B) and by flow cytometric quantitation of the % of cTnT-positive cells (Figures 2C,D). Perhaps most notable was the appearance of the sarcomeres, which were relatively disorganized in CRISPRed control proband (Probandcc) iPSC-CMs expressing the R443P variant (Figure 2E, left) and became organized upon correction in corrected WT iPSC-CMs (Figure 2E, right).
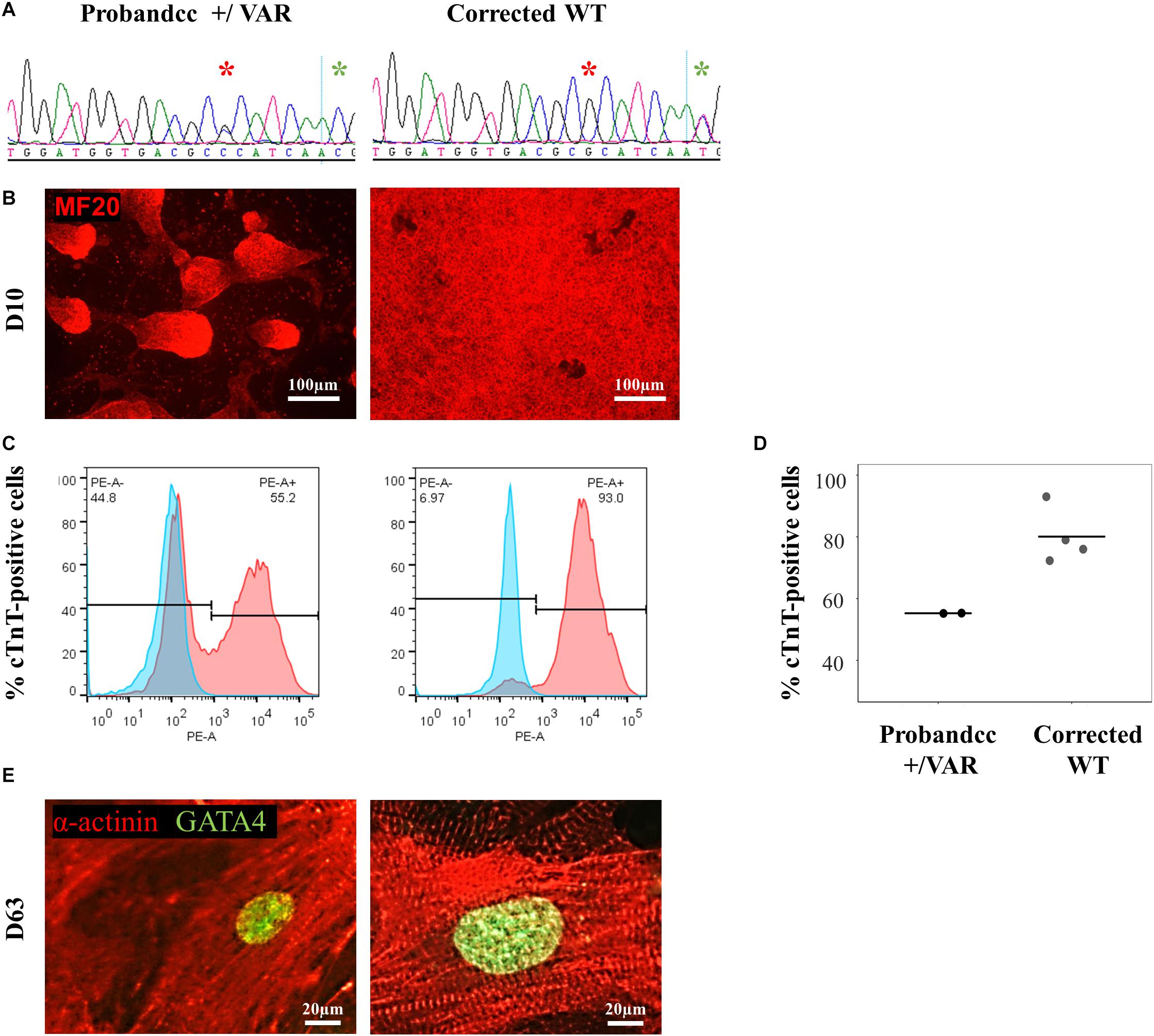
Figure 2. MYH6-R443P VAR-corrected iPSC-CMs rescues the phenotype of proband iPSC-CMs. (A) Sequences at the MYH6-R443P locus in corrected line. * is the R443 locus and * is the locus for a silencing mutation. (B) Representative immunostaining of VAR-corrected iPSC-CMs with MF20 (red) at D10. (C) Representative flow cytometry of cardiac troponin T (cTnT) positive cells in corrected iPSC-CMs at D12. (D) Flow cytometry of cells cultured in parallel with those in (C), showing % cTnT-positive cells at D10. Each dot is a technical replicate (Probandcc n = 2 and corrected WT n = 4). (E) Sarcomeres appear more “normal” in corrected WT iPSC-CMs compared to Probandcc iPSC-CMs. α-actinin immunostaining is seen in red and GATA4 in green in high density cultures at D63. Probandcc, CRISPRed control proband; corrected WT, CRISPRed and corrected MYH6-R443P variant.
Contractile data were collected from WT (heart healthy parent wild-type) and VAR (proband with the MYH6-R443P variant) iPSC-CMs at time points D20, D27, D34, and D41 (Supplementary Table S1). WT iPSC-CMs showed a higher percent shortening and relaxation rate at D20 compared to VAR. However, by D27, there were no significant differences in contractile parameters except values for contraction rate per minute (CPM), which was higher in WT iPSC-CMs (Supplementary Table S1). This indicates that the primary difference in contractile parameters between WT and VAR manifests at relatively early stages. Consequently, the focus of our study shifted to a detailed examination of D20 cells. At D20, the extent of shortening (μm), the % shortening, and the rates of shortening and relaxation were higher in WT iPSC-CMs in comparison with VAR (Table 2). In Figure 3A, the best-fit lines demonstrate the measurement of maximal cell shortening and relaxation rates. For this cell, we measured the following parameters: CPM (WT, 57.2 and VAR, 45.7), shortening (WT, 2.9 μm and VAR, 2.4 μm), percent shortening (WT, 6.7% and VAR, 4.3%), rate of shortening (WT, 28.3 μm/s and VAR 23.2 μm/s) and rate of relaxation (WT, 22.5 μm/s and VAR, 17.4 μm/s). Addition of 1 μM isoproterenol had no effect on iPSC-CMs mechanics of either cell type. However, both cell types responded to 1 Hz stimulation, contracting at 60 ± 4 (WT) and 64 ± 4 (VAR), and while CPM increased further with 2 Hz stimulation, neither cell type was able to respond at 2 Hz (WT, 73 ± 6 and VAR, 76 ± 8). The amplitude of the Ca2+ transient was higher in WT compared to the VAR (Figure 3B) cells, but no differences were observed in the rate of rise or fall of the transient (Supplementary Table S2). Isoproterenol had no effect on the amplitude of the Ca2+ transient in either cell type but did increase the rate of rise and fall of the transient in the VAR (Supplementary Table S2). Figure 3C shows representative action potentials for WT and VAR at D20, followed by a 5 s recording from the same cell. The APD90 for these cells are 298 and 300 ms for WT and VAR, respectively.

Table 2. Summary of contractile differences between WT and VAR iPSC-CMs at D20.
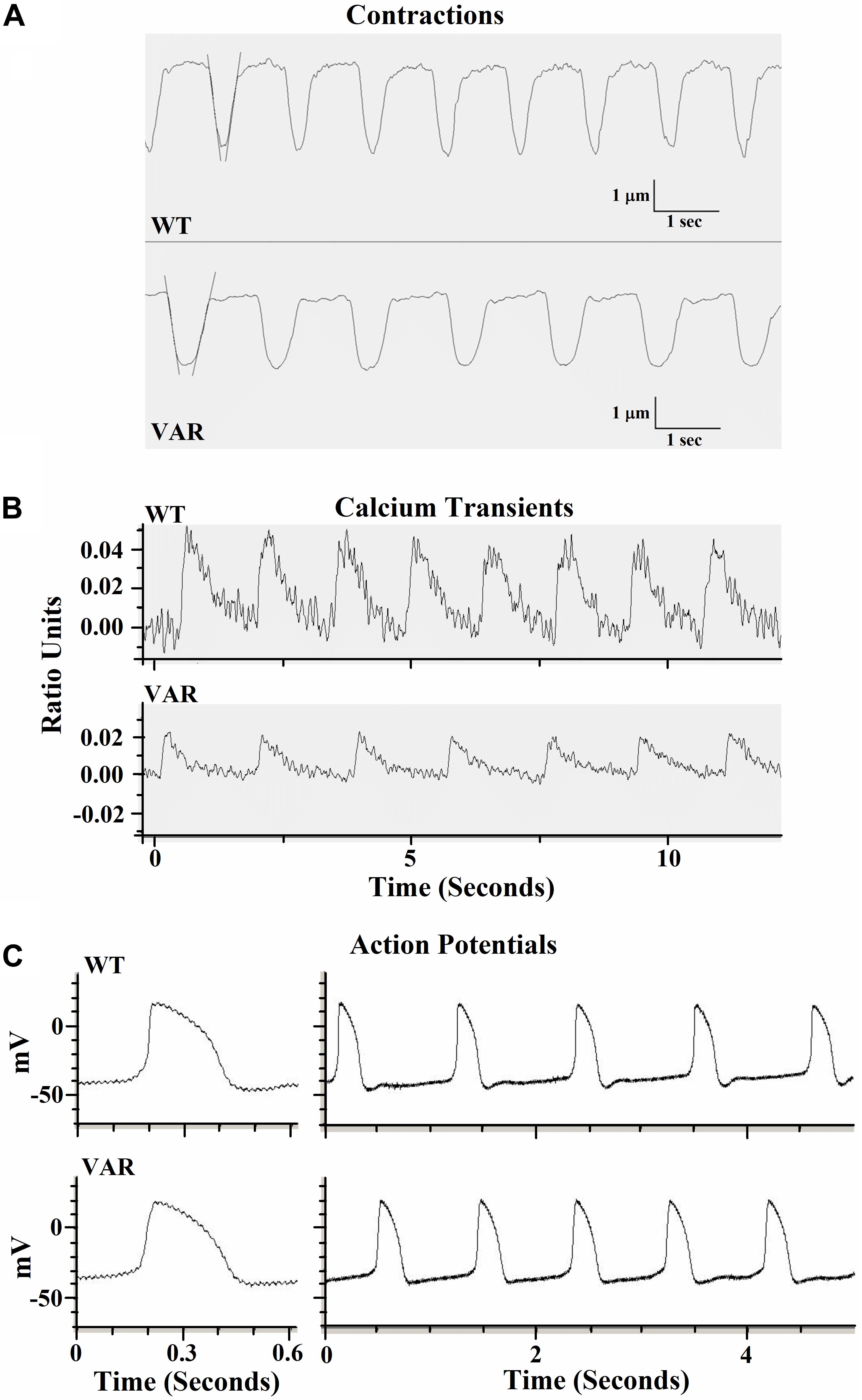
Figure 3. Representative traces of contractile properties of single iPSC-CMs at D20. (A) Spontaneously contracting WT and VAR single iPSC-CMs. Best-fit lines are shown to demonstrate the measurement of maximal cell shortening and relaxation rates (IonOptix). (B) Representative calcium transients of WT and VAR single iPSC-CMs (C) Representative action potentials of WT and VAR single iPSC-CMs, followed by a 5 s recording from the same cell. CPM, contraction per minute; WT, heart-healthy parent wild-type; VAR, proband with MYH6-R443P.
Contractile data revealed that MYH6-R443P variant-inserted iPSC-CMs contracted more slowly, while the % shortening as well as the rates of shortening and relaxation were not different between the WTcc vs. inserted +/VAR and inserted VAR/VAR iPSC-CMs (Table 3 and Supplementary Table S3). All three of the CRISPRed genotypes responded to 1 and 2 Hz stimulation, but the inserted VAR/VAR iPSC-CMs had a higher CPM compared to WTcc at 2 Hz (Supplementary Table S3). The inserted VAR/VAR iPSC-CMs also had a greater shortening rate compared to the inserted +/VAR iPSC-CMs at 1 Hz but not 2 Hz. There were no differences in the Ca2+ transients of the WTcc, inserted +/VAR and inserted VAR/VAR iPSC-CMs (Supplementary Table S4). The mean action potential characteristics for the WT, VAR, WTcc, inserted +/VAR and inserted VAR/VAR were not different (Table 4). This is important as it demonstrates that neither the variant nor the CRISPR technology altered the electrical properties of the sarcolemma membrane.
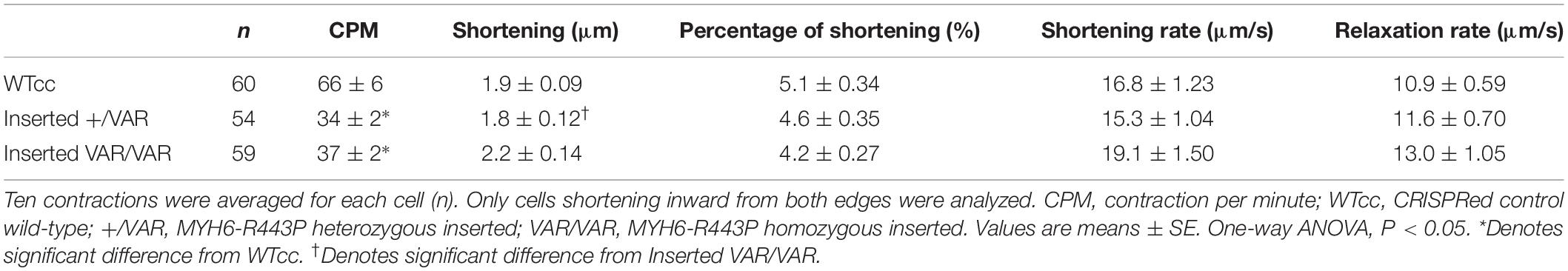
Table 3. Summary of contractile differences between WTcc and VAR-inserted iPSC-CMs at D20.
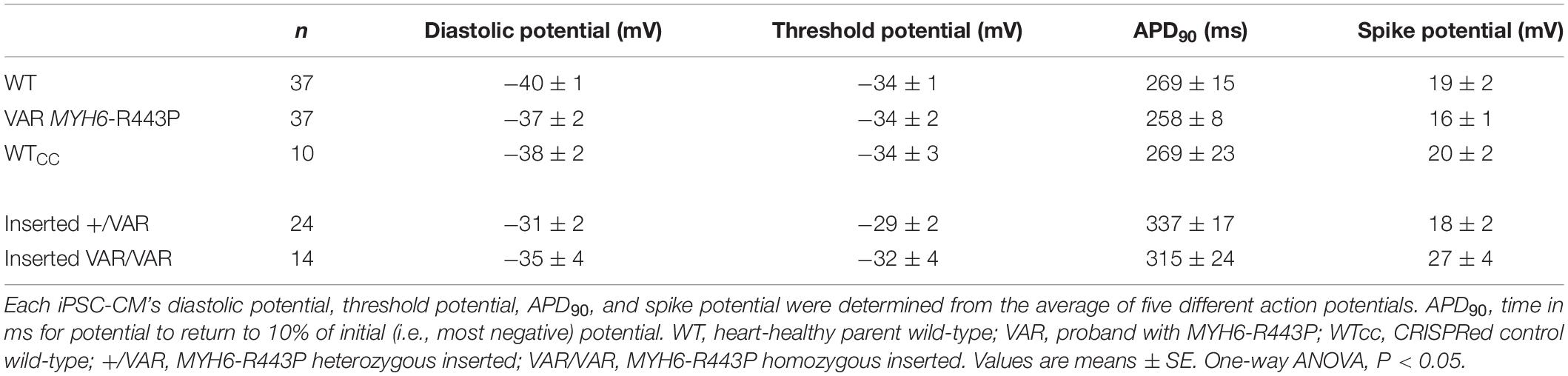
Table 4. Summary of action potentials at D20.
In mass cell cultures, using a separate automated contractility software (see section “Materials and Methods” for details), we also analyzed velocity of corrected WT iPSC-CMs at D20-D22. With this analysis, we obtained similar results as single-cell studies. Corrected WT iPSC-CMs exhibit faster contraction (Figure 4A, P < 0.05) and increased velocity (Figure 4B, P < 0.0001) compared to Probandcc iPSC-CMs.
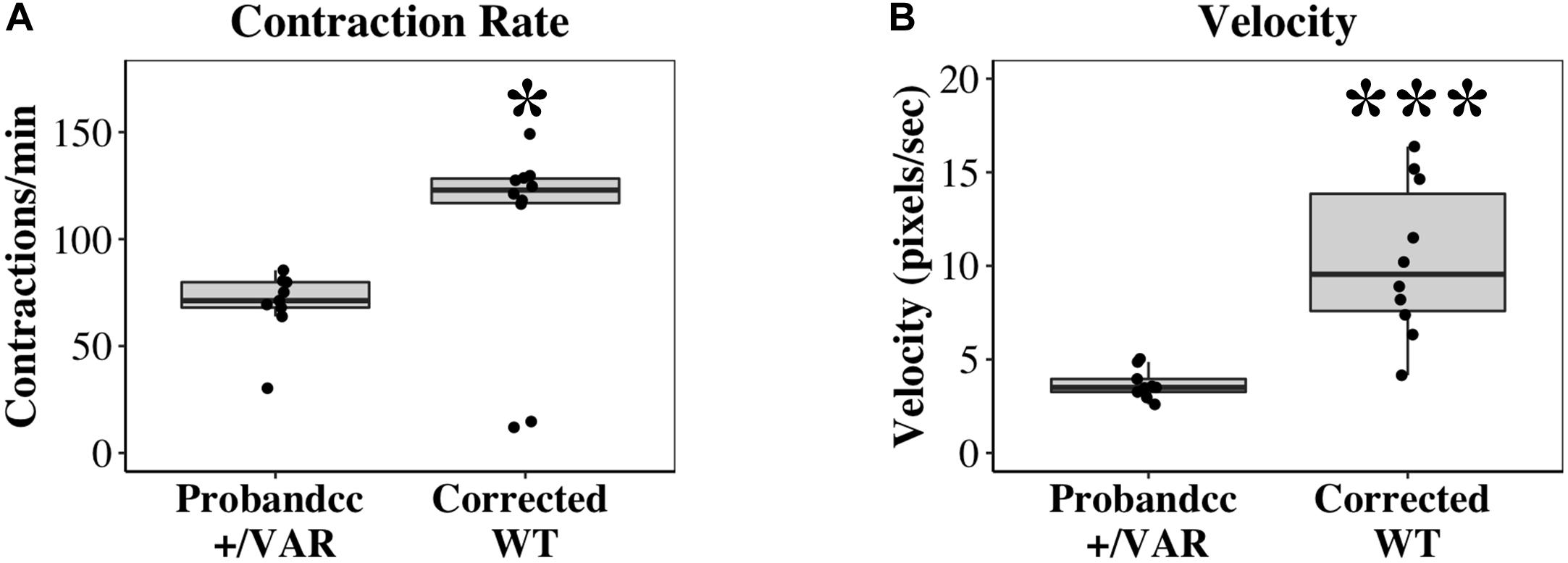
Figure 4. Contractile differences between Probandcc and corrected WT iPSC-CMs at D20-D22. (A) Contraction rate and (B) Velocity of iPSC-CMs were recorded at D20-D22 using an automated contractility software with four technical replicates for each genotype (Probandcc n = 9 and corrected WT n = 10). Probandcc, CRISPRed control proband; corrected WT, CRISPRed and corrected MYH6-R443P variant. The lines in the box plots indicate median, with maximum and minimum calculated values shown as whiskers. Each dot is a technical replicate. Wilcoxon signed-rank test, *P < 0.05 and ***P < 0.001.
Both α-MHC and β-MHC were present in WT iPSC-CMs throughout the time course of D15-D34, while VAR iPSC-CMs showed the presence of only β-MHC after D15 (Figure 5A). The 12% SDS gel (Supplementary Figure S2) shows that both cell types contain multiple proteins known to exist in cardiac muscle, including actin and MLC2v. Western blot analyses confirmed the presence of MHC and MLC2v in all iPSC-CM samples and densitometric analyses found no difference between cell types in the expression of either total MHC (both α- and β- isoforms of MHC) or MLC2v (Figure 5B).
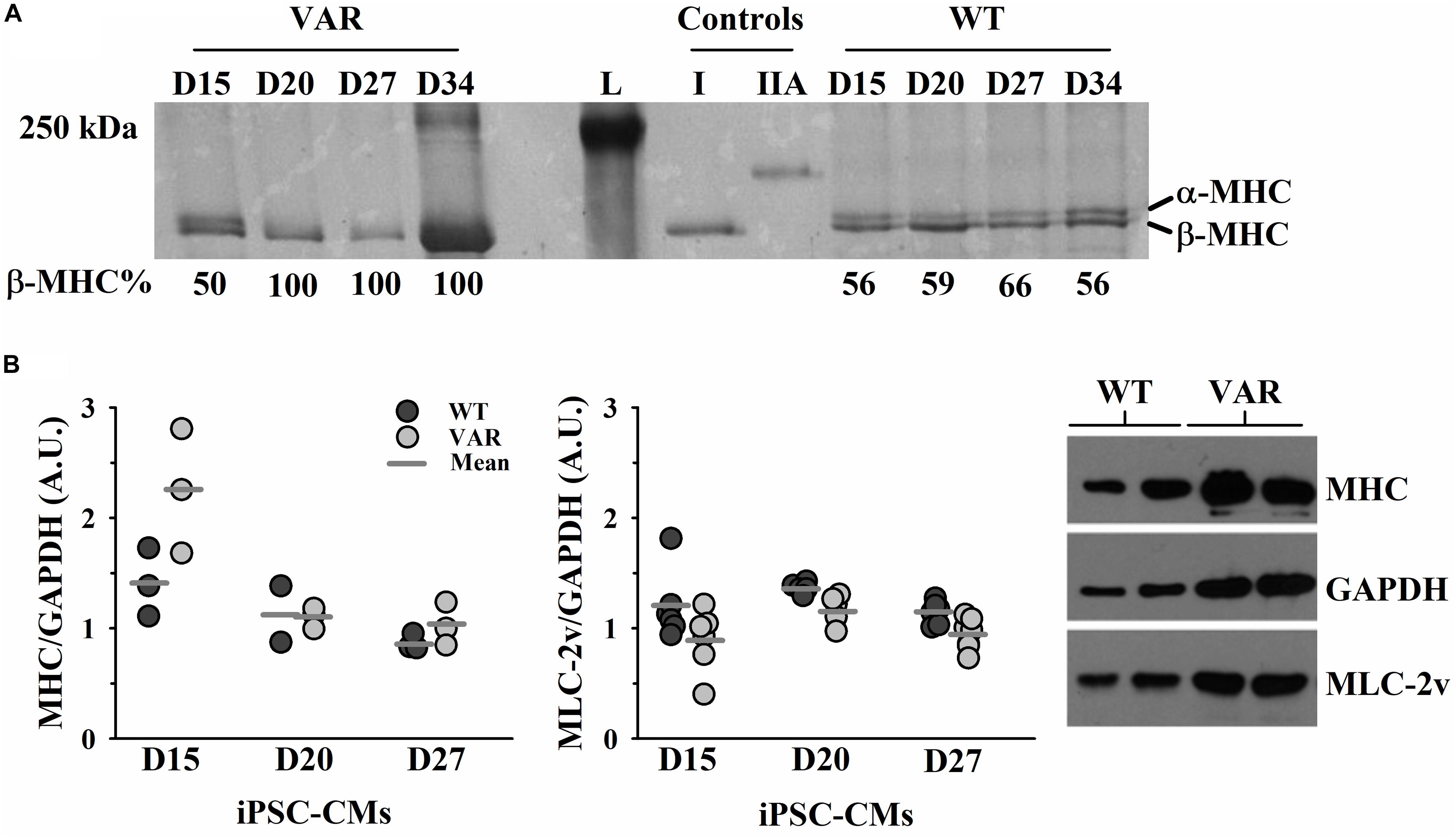
Figure 5. Upregulation of β-MHC in VAR at D15-D34. (A) Silver stained 5% SDS PAGE gel with each lane corresponding to the differentiation days and % of β-MHC expression listed beneath, as determined by densitometric analysis. (B) Protein expression in WT and VAR at D15, D20 and D27, as determined by densitometric analysis. Left: WT and VAR expression of MHC (left) and MLC2v (right), normalized to GAPDH. Values are means ± SEM of three technical replicates for each day and cell type. Right: Representative Western blot showing MHC, MLC2v, and GAPDH expression in WT and VAR at D20. L, ladder; I, type I skeletal muscle fiber; IIA, type IIA skeletal muscle fiber; WT, heart-healthy parent wild-type; VAR, proband with MYH6-R443P.
We observed a dysmorphic sarcomere phenotype in cardiac tissues from HLHS subjects with MYH6 variants. Detailed sample information is listed in Table 5 and Supplementary Table S5. We also identified combined annotation dependent depletion (CADD) scores for variant pathogenicity for all the MYH6 variants (GRCh38-v1.6 for SNP and GRCh38-v1.3 for deletion): 26.5 for MYH6-R443P (Figure 6I), 25.9 for MYH6-K849- (Figure 6J), 33 for MYH6-E1503V (Figure 6K), and 22.9 for MYH6-S385L/25.2 for M436V compound heterozygous variant (Figure 6L; Rentzsch et al., 2019).
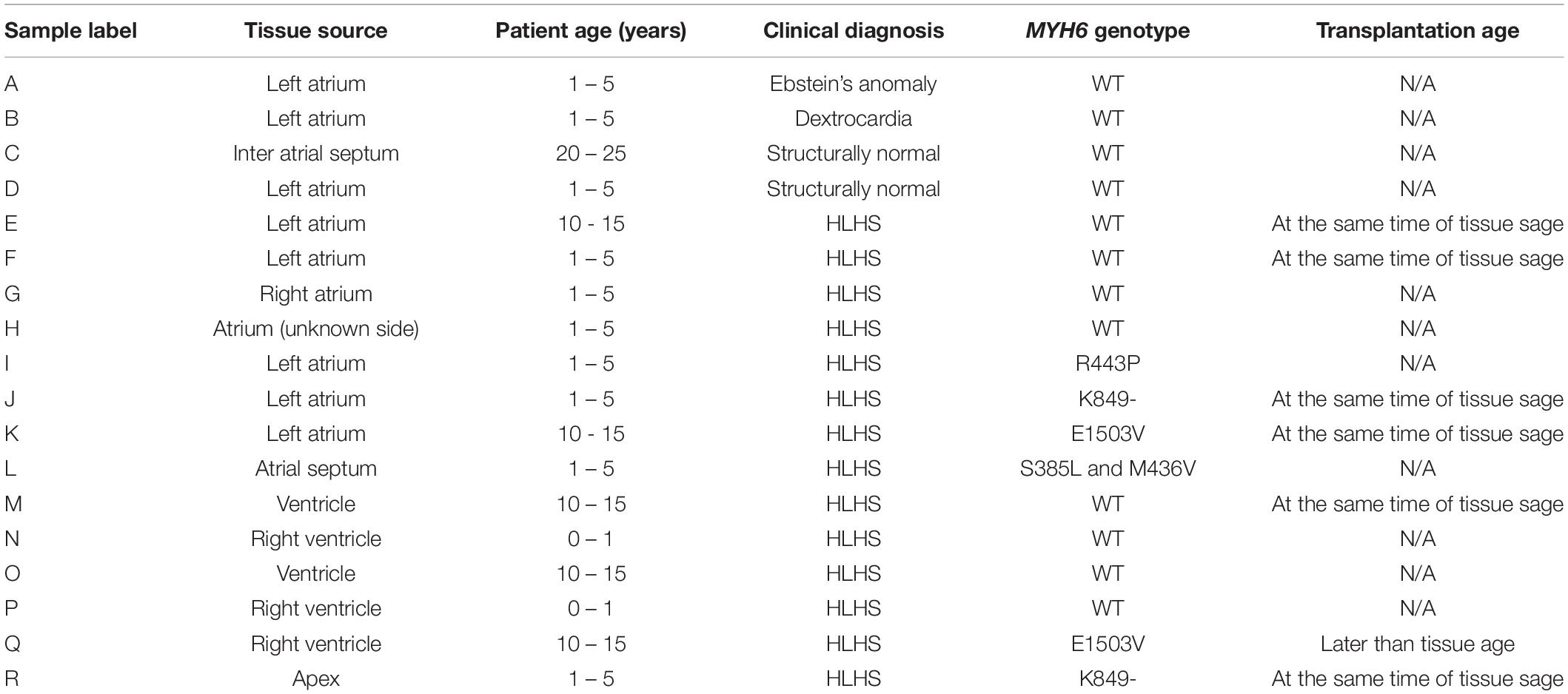
Table 5. Tissue characteristics for Figure 6.
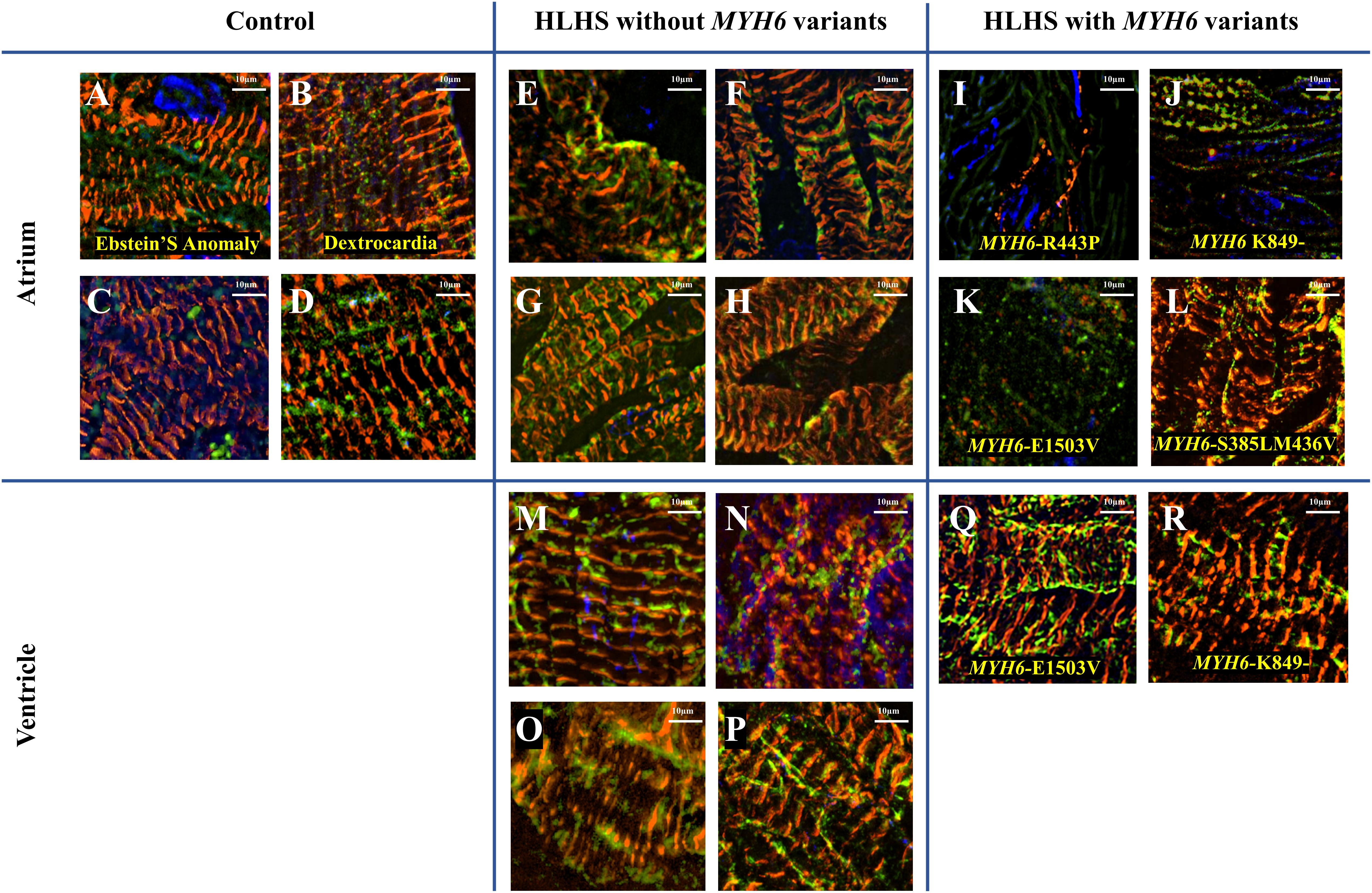
Figure 6. Dysmorphic sarcomeres were observed in atrial tissues from HLHS patients with MYH6 variants, but not in ventricles. Sarcomeric α-actinin is in red, MF20 in green and dapi in blue. (A–D) Atrial tissues from HLHS free patients without MYH6 variants. (E–H) Atrial tissues from HLHS patients without MYH6 variants. (I–L) Atrial tissues from HLHS patients with MYH6 variants. (M–P) Ventricular tissues from HLHS patients without MYH6 variants. (Q,R) Ventricular tissues from HLHS patients with MYH6 variants.
Sarcomeres in the atria and ventricles of healthy (Figures 6A–D) and HLHS tissues without MYH6 variants (Figures 6E–H and Supplementary Figures S4A–D,G) were organized except two samples, which are further described below. This was also the case in atrial tissues of other CHD subjects, including Ebstein’s Anomaly (Figure 6A) and Dextrocardia (Figure 6B). The organized sarcomere structure in HLHS patients without the MYH6 variant is clearly seen by comparing the atrial tissue in Figure 6E with the ventricular tissue in Figure 6M, both obtained at the time of surgery from the same HLHS subject. Notably, 3 of the 4 atrial tissues from HLHS subjects possessing the MYH6 variant (Figures 6I–L) exhibited poor sarcomere organization, while ventricular tissues showed normal structure (Figures 6Q,R). Sarcomeres in atrial tissues from HLHS subjects with MYH6 variants are absent, or if detectable are disorganized [Figure 6I (MYH6-R443P), Figure 6J (MYH6-K849-), Figure 6K (MYH6-E1503V)]. The MYH6-R443P variant is in the head domain whereas MYH6-K849- and MYH6-E1503V are in the tail domain. The atrial and the ventricular tissues in Figures 6J,R are from a HLHS patient with MYH6- K849-, while the atrial and the ventricular tissues in Figures 6K,Q are from a HLHS patient with MYH6-E1503V. At the neonatal stage, sarcomeres are organized in both atrial tissues from HLHS patients with and without MYH6 variants (Supplementary Figure S3). The atrial restriction of this sarcomere phenotype corresponds to the predominance of MYH6 expression in the atria after the neonatal stage. The samples in Supplementary Figure S3B were from the same HLHS patient with MYH6-R443P (Figure 6I). Additional samples exhibited disrupted sarcomeres in HLHS subjects without an MYH6 variant, one carrying a Dystrophin hemizygous variant (DMD-I228N, scaled CADD-score 26.6) (Supplementary Figure S4E) while the other HLHS subject did not reveal any sarcomere gene variants from whole exome sequencing (Supplementary Figure S4F).
HLHS etiology remains incomplete, in part due to its diverse phenotype and complex genetic inheritance, as well as environmental factors. To understand the molecular and cellular mechanisms of HLHS, we investigated patient-specific HLHS etiology using an iPSC-CM model and examined cardiac tissue from HLHS patients with sarcomere immunostaining. To further demonstrate the impact of the MYH6-R443P variant on HLHS, we used CRISPR/Cas9 gene editing to generate isogenic controls. Insertion of the MYH6-R443P variant into the heart healthy parent’s iPSCs recapitulated the dysmorphic sarcomere phenotype (Figure 1) and contractile dysfunction (compare Tables 2, 3). Correcting the MYH6-R443P variant in the proband’s iPSCs rescued the phenotypes of decreased CM differentiation efficiency, dysmorphic sarcomeres, and abnormal CPM and velocity (Figure 4).
Based on functional analysis of iPSC-CMs with the MYH6-R443P variant, the primary contractile defects were reduced CPM, impaired cell shortening (expressed as a % of cell length), and decreased rates of shortening and relaxation. These defects occurred early (by D20) and likely resulted from reduced development of the SR including the SERCA pump and the increased expression of the slow β-MHC isoform in the variant iPSC-CMs (Figure 5A). The reduced SR SERCA pump activity would explain the slower relaxation rates in the VAR. Regarding contraction rate, the β-MHC isoform is known to have a lower ATPase activity which limits cross-bridge cycle speed, and skeletal fibers containing this isoform are 3–4 times slower than α-MHC fast fibers (Schluter and Fitts, 1994; Fitts, 2008). The reduced contractile function in the MYH6-R443P variant compared to WT iPSC-CMs at D20 may be due to delayed development, as by D27 there were no differences in the contractility between the WT and VAR. Beyond D27, the loss of difference between cell types in contractile function was likely at least in part due to delayed development of the VAR particularly the SR. This is best demonstrated by the relaxation rate where at D20 the WT was twofold higher but by D27 the VAR relaxation rate had increased to match the WT (Supplementary Table S1). Despite no differences in shortening rate between WT and VAR past D20, the β-MHC content remained high in the VAR from D21 through D34 (Figure 5). We know iPSC-CMs are immature at D20, which may explain why cell contractility was not increased by adrenergic stimulation with isoproterenol (Supplementary Table S2). This lack of response most likely indicates immaturity of the sarcoplasmic reticulum, but also could indicate lack of the ß1 receptor on the surface membrane (Pillekamp et al., 2012).
The MYH6-R443P variant showed a reduced amplitude of the Ca2+ transients upon activation compared to WT, but both showed diminished Ca2+ release compared to adult rat ventricular cardiac cells (Wang and Fitts, 2017). It is likely that the reduced Ca2+ release relates to lower sarcoplasmic reticulum content and not to differences in surface membrane activation, as the recorded action potentials were identical between the WT and VAR. Differences in membrane excitability at D20 were noted, indicated by lower CPM in the VAR compared to the WT. Since the lowest membrane potential (referred to as diastolic potential) and the threshold potential were not different between cell types, this suggests that the rate of spontaneous depolarization to threshold may have been faster in the WT. However, our recordings did not show an obvious difference in the rate of depolarization to threshold between cell types (Figure 3 and Table 4). This may be because the difference is too small for detection during the single-cell level experiments performed at D20. Sorting out differences between the WT and VAR iPSC-CMs will require additional studies of organelle function (particularly the sarcoplasmic reticulum and myofilaments).
The basic contractile unit of striated (cardiac and skeletal) muscle is the sarcomere, a highly ordered structure containing contractile and regulatory proteins. Disruptions in sarcomere proteins have been implicated in many cardiac diseases (Redwood et al., 1999; Seidman and Seidman, 2001; Richard et al., 2003; Laing and Nowak, 2005). Here, we observed dysmorphic sarcomeres in tissues from HLHS patients with certain MYH6 variants and these changes were phenocopied in iPSC-CMs generated from a HLHS patient with the MYH6-R443P variant. Sarcomere disorganization in cardiac tissues from three out of four HLHS patients with MYH6 variants was observed in atrial but not in ventricular tissues (Figure 6). This result was expected as α-MHC protein expression is predominantly in the postnatal atria in humans. Likely, the in vivo contractile dysfunction is mainly in the atria, with the hypoplastic ventricular phenotype representing a developmental response resultant from diminished inflow pulsatility. Adding to the idea that poorly organized sarcomeres might contribute to the development of HLHS, atrial tissues from a HLHS patient with a costameric variant in Dystrophin (DMD) also showed sarcomere disruption (Supplementary Figure S4E). Furthermore, we observed that variants with the highest scaled CADD-scores: MYH6-R443P (26.5), MYH6-K849- (25.9), MYH6-E1503V (33) and DMD-I288N (26.6) had the most apparent disruptions in sarcomere structure in atrial tissues ex vivo. One limitation was lack of paired controls, however tissue samples showed convincing evidence of abnormalities (Figure 6).
Investigating HLHS etiology requires a physiological system of electrically and mechanically connected iPSC-CMs. One concern with employing iPSCs is the clonal variability; to reduce variability, we conducted all experiments multiple times (as indicated in each figure legend) in a non-biased, blinded fashion. A WT line was always differentiated along with experimental lines to compare differences among cell types at each differentiation stage. Additionally, isogenic MYH6-R443P cell lines were created using CRISPR/Cas9 gene editing and these lines recapitulated the phenotypes of patient-specific MYH6-R443P iPSC-CMs. An additional limitation is the heterogenous population of iPSC-CMs, including both atrial and ventricular cells. There are MLC-2a, MLC-2v, as well as double-positive iPSC-CMs in our cultures upon immunostaining at D31-D35 (data not shown). As our protein expression (Figure 5) and automated contractility assays (Figure 4) were analyzed in masse, they may underestimate the true heterogeneity of our cells. While we have a relatively pure population of cTnT positive CMs that express our cardiac gene of interest, MYH6, single-cell analyses similar to those completed by other groups may help further clarify their composition (Weber et al., 2016).
Despite being relatively immature, iPSC-CMs expressing the MYH6-R443P allele showed morphological and physiological differences from WT. iPSC-CMs could be used for clinical applications such as pharmacological drug testing, but these applications require mature CMs. A number of strategies have been employed to promote the maturity of iPSC-CMs, including: the administration of biochemical agents [adrenergic receptor agonists, Triiodothyronine (Yang et al., 2014b; Jackman et al., 2018), insulin-like growth factor 1 or microRNA (mir1 or mir208)], altering substrate stiffness or utilizing molds for cell-patterning (Ribeiro et al., 2015, 2016, 2017; Carson et al., 2016; Jung et al., 2016), application of mechanical stimuli (Hirt et al., 2014), and co-culture with different cell types to mimic human tissue, including bioengineering of 3D tissue (Yang et al., 2014a; Ronaldson-Bouchard et al., 2018). iPSC-CM maturation can then be confirmed using a multiparametric quality assessment, including gene expression profiling and structural, electrophysiological, and contractile measurements (Sheehy et al., 2014).
We showed contractile and sarcomere organization differences between WT and MYH6-R443P iPSC-CMs, however, how the early stage CM-phenotype affects cardiac morphogenesis in vivo and how this phenotype progresses to adulthood is complex and not understood. To investigate this progression in vivo it may be beneficial to use an animal model in parallel with iPSC-CMs. While murine models have opposite chamber-specific expression of Myh6 and Myh7, zebrafish may be a suitable alternative for studying MYH6-associated CHD despite having a two-chambered heart. Importantly, zebrafish chamber-specific MHC expression is the same as humans. The phenotype of weak atrium (wea) myh6-mutant zebrafish exhibits a hypoplastic ventricle (Berdougo et al., 2003).
Our study proposes a new potential mechanism for the development of HLHS related to MYH6-R443P: sarcomere disorganization likely causes decreased atrial contractility and results in hypoplastic left ventricular development. The failing structural and contractile changes of the atrial sarcomere may effect hemodynamic changes that prevent the development of a normal left ventricle and downstream structures including the mitral valve, the aortic valve and the aorta. In addition, postnatally, atrial dysfunction due to sarcomere disorganization in single ventricle patients may result in decreased atrial kick, which would be expected to lead to heart failure over time. Further studies are needed to test potential ways to correct these contractile changes. It is possible that drugs targeting contractility may be able to alleviate some of the contractile changes due to the MYH6-R443P variant. Ultimately, we may be able to alter the developmental pathways and minimize or prevent the development of MYH6 variant associated HLHS.
All datasets generated for this study are included in the article/Supplementary Material. Additional raw data will be made available by the authors, without undue reservation, to any qualified researcher.
This study is in accordance with the principles outlined in the Declaration of Helsinki and institutionally approved research (IRB) protocols by the Children’s Hospital of Wisconsin (CHW, Milwaukee, WI, United States). Subjects were consented through the CHD Tissue Bank (IRB #CHW 06/229, GC 300) and the Wisconsin Pediatric Cardiac Registry (IRB #CHW 09/91, GC 889), IRB-approved research databases housed at CHW prior to inclusion in the study. Both biorepositories provided all cardiac tissue as well as iPSCs, from patients and family members, with associated clinical outcome variables.
AT-M and RF designed and directed the project. AT-M, RF, MEM, JWL, and AG were involved in planning and supervising the work. M-SK, RF, BF, JL, MC, MA, SS, LK, LT, and MM processed the experimental data, performed the analysis, and interpreted the data. M-SK took the lead in writing manuscript with support from AT-M, RF, BF, JL, LK, LT, MM, MA, and SS. All authors discussed the results and commented on the manuscript.
This work was supported by Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin Grant, Clinical and Translational Science Institute from the National Institutes of Health (NIH) CTSA award: UL1TR001436, Greater Milwaukee Foundation and the Little Hearts for Life Foundation, the Medical College of Wisconsin’s Department of Surgery, and the Herma Heart Institute/Children’s Research Institute.
AT-M and MEM are cofounders of TAI Diagnostics (Milwaukee, WI, United States), a biotechnology company involved in transplant diagnostics, and members of its scientific advisory board.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the families, physicians, and clinical care team of the Herma Heart Institute from CHW, Mary Krolikowski for tremendous IRB support, the outstanding cardiothoracic surgical team including M. E. Barnes, J. L. Dunham-Ingle, T. A. Fehrenbacher, M. R. Madrzak, L. E. May, D. Semerda, R. G. Smith, and the operating room staff for making this study possibly. We gratefully acknowledge M. Goetsch, H.-L. Liang, and L. Armitage for helpful support and the excellent technical assistance from the Children’s Research Institute (CRI) Pathology Core. We also thank M. Gryzbowski for expertise that enabled use of CRISPR/Cas9 technique.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2020.00440/full#supplementary-material
Berdougo, E., Coleman, H., Lee, D. H., Stainier, D. Y., and Yelon, D. (2003). Mutation of weak atrium/atrial myosin heavy chain disrupts atrial function and influences ventricular morphogenesis in zebrafish. Development 130, 6121–6129. doi: 10.1242/dev.00838
Burggren, W., Khorrami, S., Pinder, A., and Sun, T. (2004). Body, eye, and chorioallantoic vessel growth are not dependent on cardiac output level in day 3-4 chicken embryos. Am. J. Physiol. Regul. Integr. Comp. Physiol. 287, R1399–R1406. doi: 10.1152/ajpregu.00086.2004
Carson, D., Hnilova, M., Yang, X., Nemeth, C. L., Tsui, J. H., Smith, A. S., et al. (2016). Nanotopography-induced structural anisotropy and sarcomere development in human cardiomyocytes derived from induced pluripotent stem cells. ACS Appl. Mater. Interfaces 8, 21923–21932. doi: 10.1021/acsami.5b11671
Cummins, P., and Lambert, S. J. (1986). Myosin transitions in the bovine and human heart. A developmental and anatomical study of heavy and light chain subunits in the atrium and ventricle. Circ. Res. 58, 846–858. doi: 10.1161/01.res.58.6.846
Dasgupta, C., Martinez, A. M., Zuppan, C. W., Shah, M. M., Bailey, L. L., and Fletcher, W. H. (2001). Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE). Mutat. Res. 479, 173–186. doi: 10.1016/s0027-5107(01)00160-9
deAlmeida, A., McQuinn, T., and Sedmera, D. (2007). Increased ventricular preload is compensated by myocyte proliferation in normal and hypoplastic fetal chick left ventricle. Circ. Res. 100, 1363–1370. doi: 10.1161/01.RES.0000266606.88463.cb
Elliott, D. A., Kirk, E. P., Yeoh, T., Chandar, S., McKenzie, F., Taylor, P., et al. (2003). Cardiac homeobox gene NKX2-5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J. Am. Coll. Cardiol. 41, 2072–2076.
Fishman, N. H., Hof, R. B., Rudolph, A. M., and Heymann, M. A. (1978). Models of congenital heart disease in fetal lambs. Circulation 58, 354–364. doi: 10.1161/01.cir.58.2.354
Fitts, R. H. (2008). The cross-bridge cycle and skeletal muscle fatigue. J. Appl. Physiol. 104, 551–558. doi: 10.1152/japplphysiol.01200.2007
Friedberg, M. K., Silverman, N. H., Moon-Grady, A. J., Tong, E., Nourse, J., Sorenson, B., et al. (2009). Prenatal detection of congenital heart disease. J. Pediatr. 155, 26–31, 31.e1. doi: 10.1016/j.jpeds.2009.01.050
Galindo, A., Nieto, O., Villagra, S., Graneras, A., Herraiz, I., and Mendoza, A. (2009). Hypoplastic left heart syndrome diagnosed in fetal life: associated findings, pregnancy outcome and results of palliative surgery. Ultrasound Obstet. Gynecol. 33, 560–566. doi: 10.1002/uog.6355
Garg, V., Muth, A. N., Ransom, J. F., Schluterman, M. K., Barnes, R., King, I. N., et al. (2005). Mutations in NOTCH1 cause aortic valve disease. Nature 437, 270–274. doi: 10.1038/nature03940
Glidewell, S. C., Miyamoto, S. D., Grossfeld, P. D., Clouthier, D. E., Coldren, C. D., Stearman, R. S., et al. (2015). Transcriptional impact of rare and private copy number variants in hypoplastic left heart syndrome. Clin. Transl. Sci. 8, 682–689. doi: 10.1111/cts.12340
Grossfeld, P. (2007). Hypoplastic left heart syndrome: new insights. Circ. Res. 100, 1246–1248. doi: 10.1161/01.RES.0000268192.20525.c2
Grossfeld, P., Ye, M., and Harvey, R. (2009). Hypoplastic left heart syndrome: new genetic insights. J. Am. Coll. Cardiol. 53, 1072–1074. doi: 10.1016/j.jacc.2008.12.024
Gruber, P. J., and Epstein, J. A. (2004). Development gone awry: congenital heart disease. Circ. Res. 94, 273–283. doi: 10.1161/01.RES.0000116144.43797.3B
Hierck, B. P., Van der Heiden, K., Poelma, C., Westerweel, J., and Poelmann, R. E. (2008). Fluid shear stress and inner curvature remodeling of the embryonic heart. Choosing the right lane! ScientificWorldJournal 8, 212–222. doi: 10.1100/tsw.2008.42
Hinton, R. B. Jr., Martin, L. J., Tabangin, M. E., Mazwi, M. L., Cripe, L. H., and Benson, D. W. (2007). Hypoplastic left heart syndrome is heritable. J. Am. Coll. Cardiol. 50, 1590–1595. doi: 10.1016/j.jacc.2007.07.021
Hinton, R. B., Martin, L. J., Rame-Gowda, S., Tabangin, M. E., Cripe, L. H., and Benson, D. W. (2009). Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J. Am. Coll. Cardiol. 53, 1065–1071. doi: 10.1016/j.jacc.2008.12.023
Hirt, M. N., Boeddinghaus, J., Mitchell, A., Schaaf, S., Bornchen, C., Muller, C., et al. (2014). Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J. Mol. Cell. Cardiol. 74, 151–161. doi: 10.1016/j.yjmcc.2014.05.009
Hoog, T. G., Fredrickson, S. J., Hsu, C. W., Senger, S. M., Dickinson, M. E., and Udan, R. S. (2018). The effects of reduced hemodynamic loading on morphogenesis of the mouse embryonic heart. Dev. Biol. 442, 127–137. doi: 10.1016/j.ydbio.2018.07.007
Hove, J. R., Koster, R. W., Forouhar, A. S., Acevedo-Bolton, G., Fraser, S. E., and Gharib, M. (2003). Intracardiac fluid forces are an essential epigenetic factor for embryonic cardiogenesis. Nature 421, 172–177. doi: 10.1038/nature01282
Hrstka, S. C., Li, X., and Nelson, T. J., Wanek Program Genetics Pipeline Group (2017). NOTCH1-dependent nitric oxide signaling deficiency in hypoplastic left heart syndrome revealed through patient-specific phenotypes detected in bioengineered cardiogenesis. Stem Cells 35, 1106–1119. doi: 10.1002/stem.2582
Jackman, C., Li, H., and Bursac, N. (2018). Long-term contractile activity and thyroid hormone supplementation produce engineered rat myocardium with adult-like structure and function. Acta Biomater. 78, 98–110. doi: 10.1016/j.actbio.2018.08.003
Jung, G., Fajardo, G., Ribeiro, A. J., Kooiker, K. B., Coronado, M., Zhao, M., et al. (2016). Time-dependent evolution of functional vs. remodeling signaling in induced pluripotent stem cell-derived cardiomyocytes and induced maturation with biomechanical stimulation. FASEB J. 30, 1464–1479. doi: 10.1096/fj.15-280982
Kim, M. S., Horst, A., Blinka, S., Stamm, K., Mahnke, D., Schuman, J., et al. (2015). Activin-A and Bmp4 levels modulate cell type specification during CHIR-induced cardiomyogenesis. PLoS One 10:e0118670. doi: 10.1371/journal.pone.0118670
Laing, N. G., and Nowak, K. J. (2005). When contractile proteins go bad: the sarcomere and skeletal muscle disease. Bioessays 27, 809–822. doi: 10.1002/bies.20269
Lara, D. A., Ethen, M. K., Canfield, M. A., Nembhard, W. N., and Morris, S. A. (2017). A population-based analysis of mortality in patients with Turner syndrome and hypoplastic left heart syndrome using the Texas Birth Defects Registry. Congenit. Heart Dis. 12, 105–112. doi: 10.1111/chd.12413
Lee, J., Fei, P., Packard, R. R., Kang, H., Xu, H., Baek, K. I., et al. (2016). 4-Dimensional light-sheet microscopy to elucidate shear stress modulation of cardiac trabeculation. J. Clin. Invest. 126, 1679–1690. doi: 10.1172/JCI83496
Maddah, M., Heidmann, J. D., Mandegar, M. A., Walker, C. D., Bolouki, S., Conklin, B. R., et al. (2015). A non-invasive platform for functional characterization of stem-cell-derived cardiomyocytes with applications in cardiotoxicity testing. Stem Cell Rep. 4, 621–631. doi: 10.1016/j.stemcr.2015.02.007
Maddah, M., Shoukat-Mumtaz, U., Nassirpour, S., and Loewke, K. (2014). A system for automated, noninvasive, morphology-based evaluation of induced pluripotent stem cell cultures. J. Lab. Autom. 19, 454–460. doi: 10.1177/2211068214537258
Mai, C. T., Isenburg, J. L., Canfield, M. A., Meyer, R. E., Correa, A., Alverson, C. J., et al. (2019). National population-based estimates for major birth defects, 2010-2014. Birth Defects Res. 111, 1420–1435. doi: 10.1002/bdr2.1589
McBride, K. L., Riley, M. F., Zender, G. A., Fitzgerald-Butt, S. M., Towbin, J. A., Belmont, J. W., et al. (2008). NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand-induced signaling. Hum. Mol. Genet. 17, 2886–2893. doi: 10.1093/hmg/ddn187
McCain, M. L., and Parker, K. K. (2011). Mechanotransduction: the role of mechanical stress, myocyte shape, and cytoskeletal architecture on cardiac function. Pflugers Arch. 462, 89–104. doi: 10.1007/s00424-011-0951-4
McCormick, M. E., and Tzima, E. (2016). Pulling on my heartstrings: mechanotransduction in cardiac development and function. Curr. Opin. Hematol. 23, 235–242. doi: 10.1097/MOH.0000000000000240
Mitzelfelt, K. A., McDermott-Roe, C., Grzybowski, M. N., Marquez, M., Kuo, C. T., Riedel, M., et al. (2017). Efficient precision genome editing in iPSCs via genetic co-targeting with selection. Stem Cell Rep. 8, 491–499. doi: 10.1016/j.stemcr.2017.01.021
Noonan, J. A., and Nadas, A. S. (1958). The hypoplastic left heart syndrome; an analysis of 101 cases. Pediatr. Clin. North Am. 5, 1029–1056.
Pillekamp, F., Haustein, M., Khalil, M., Emmelheinz, M., Nazzal, R., Adelmann, R., et al. (2012). Contractile properties of early human embryonic stem cell-derived cardiomyocytes: beta-adrenergic stimulation induces positive chronotropy and lusitropy but not inotropy. Stem Cells Dev. 21, 2111–2121. doi: 10.1089/scd.2011.0312
Redwood, C. S., Moolman-Smook, J. C., and Watkins, H. (1999). Properties of mutant contractile proteins that cause hypertrophic cardiomyopathy. Cardiovasc. Res. 44, 20–36. doi: 10.1016/s0008-6363(99)00213-8
Reiser, P. J., Portman, M. A., Ning, X. H., and Schomisch Moravec, C. (2001). Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am. J. Physiol. Heart Circ. Physiol. 280, H1814–H1820. doi: 10.1152/ajpheart.2001.280.4.H1814
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., and Kircher, M. (2019). CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894. doi: 10.1093/nar/gky1016
Ribeiro, A. J. S., Schwab, O., Mandegar, M. A., Ang, Y. S., Conklin, B. R., Srivastava, D., et al. (2017). Multi-imaging method to assay the contractile mechanical output of micropatterned human iPSC-Derived cardiac myocytes. Circ. Res. 120, 1572–1583. doi: 10.1161/CIRCRESAHA.116.310363
Ribeiro, A. J., Ang, Y. S., Fu, J. D., Rivas, R. N., Mohamed, T. M., Higgs, G. C., et al. (2015). Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. U.S.A. 112, 12705–12710. doi: 10.1073/pnas.1508073112
Ribeiro, A. J., Denisin, A. K., Wilson, R. E., and Pruitt, B. L. (2016). For whom the cells pull: Hydrogel and micropost devices for measuring traction forces. Methods 94, 51–64. doi: 10.1016/j.ymeth.2015.08.005
Richard, P., Charron, P., Carrier, L., Ledeuil, C., Cheav, T., Pichereau, C., et al. (2003). Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107, 2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54
Ronaldson-Bouchard, K., Ma, S. P., Yeager, K., Chen, T., Song, L., Sirabella, D., et al. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556, 239–243. doi: 10.1038/s41586-018-0016-3
Santhanakrishnan, A., and Miller, L. A. (2011). Fluid dynamics of heart development. Cell Biochem. Biophys. 61, 1–22. doi: 10.1007/s12013-011-9158-8
Schluter, J. M., and Fitts, R. H. (1994). Shortening velocity and ATPase activity of rat skeletal muscle fibers: effects of endurance exercise training. Am. J. Physiol. 266(6 Pt 1), C1699–C1713. doi: 10.1152/ajpcell.1994.266.6.C1699
Seidman, J. G., and Seidman, C. (2001). The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104, 557–567. doi: 10.1016/s0092-8674(01)00242-2
Sheehy, S. P., Pasqualini, F., Grosberg, A., Park, S. J., Aratyn-Schaus, Y., and Parker, K. K. (2014). Quality metrics for stem cell-derived cardiac myocytes. Stem Cell Rep. 2, 282–294. doi: 10.1016/j.stemcr.2014.01.015
Sundberg, C. W., Hunter, S. K., Trappe, S. W., Smith, C. S., and Fitts, R. H. (2018). Effects of elevated H(+) and Pi on the contractile mechanics of skeletal muscle fibres from young and old men: implications for muscle fatigue in humans. J. Physiol. 596, 3993–4015. doi: 10.1113/jp276018
Theis, J. L., Zimmermann, M. T., Evans, J. M., Eckloff, B. W., Wieben, E. D., Qureshi, M. Y., et al. (2015). Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circ. Cardiovasc. Genet. 8, 564–571. doi: 10.1161/CIRCGENETICS.115.001070
Tomita-Mitchell, A., Mahnke, D. K., Struble, C. A., Tuffnell, M. E., Stamm, K. D., Hidestrand, M., et al. (2012). Human gene copy number spectra analysis in congenital heart malformations. Physiol. Genomics 44, 518–541. doi: 10.1152/physiolgenomics.00013.2012
Tomita-Mitchell, A., Stamm, K. D., Mahnke, D. K., Kim, M. S., Hidestrand, P. M., Liang, H. L., et al. (2016). Impact of MYH6 variants in hypoplastic left heart syndrome. Physiol. Genomics 48, 912–921. doi: 10.1152/physiolgenomics.00091.2016
Wang, X., and Fitts, R. H. (2017). Ventricular action potential adaptation to regular exercise: role of beta-adrenergic and KATP channel function. J. Appl. Physiol. 123, 285–296. doi: 10.1152/japplphysiol.00197.2017
Wang, X., and Fitts, R. H. (2018). Effects of regular exercise on ventricular myocyte biomechanics and KATP channel function. Am. J. Physiol. Heart Circ. Physiol. 315, H885–H896. doi: 10.1152/ajpheart.00130.2018
Warburton, D., Ronemus, M., Kline, J., Jobanputra, V., Williams, I., Anyane-Yeboa, K., et al. (2014). The contribution of de novo and rare inherited copy number changes to congenital heart disease in an unselected sample of children with conotruncal defects or hypoplastic left heart disease. Hum. Genet. 133, 11–27. doi: 10.1007/s00439-013-1353-9
Weber, N., Schwanke, K., Greten, S., Wendland, M., Iorga, B., Fischer, M., et al. (2016). Stiff matrix induces switch to pure beta-cardiac myosin heavy chain expression in human ESC-derived cardiomyocytes. Basic Res. Cardiol. 111:68.
Wessels, A., Vermeulen, J. L., Viragh, S., and Moorman, A. F. (1990). The ontogenesis of myosin heavy chain isoforms in the developing human hearta. Ann. N. Y. Acad. Sci. 588, 461–464. doi: 10.1111/j.1749-6632.1990.tb13264.x
Wessels, A., Vermeulen, J. L., Viragh, S., Kalman, F., Lamers, W. H., and Moorman, A. F. (1991). Spatial distribution of “tissue-specific” antigens in the developing human heart and skeletal muscle. II. An immunohistochemical analysis of myosin heavy chain isoform expression patterns in the embryonic heart. Anat. Rec. 229, 355–368. doi: 10.1002/ar.1092290309
Yagi, H., Liu, X., Gabriel, G. C., Wu, Y., Peterson, K., Murray, S. A., et al. (2018). The genetic landscape of hypoplastic left heart syndrome. Pediatr. Cardiol. 39, 1069–1081. doi: 10.1007/s00246-018-1861-4
Yang, C., Xu, Y., Yu, M., Lee, D., Alharti, S., Hellen, N., et al. (2017). Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum. Mol. Genet. 26, 3031–3045. doi: 10.1093/hmg/ddx140
Yang, X., Pabon, L., and Murry, C. E. (2014a). Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ. Res. 114, 511–523. doi: 10.1161/CIRCRESAHA.114.300558
Keywords: HLHS, hypoplastic left heart syndrome, iPSC-cardiomyocytes, MYH6, contractility, CRISPR/Cas9, sarcomere
Citation: Kim M-S, Fleres B, Lovett J, Anfinson M, Samudrala SSK, Kelly LJ, Teigen LE, Cavanaugh M, Marquez M, Geurts AM, Lough JW, Mitchell ME, Fitts RH and Tomita-Mitchell A (2020) Contractility of Induced Pluripotent Stem Cell-Cardiomyocytes With an MYH6 Head Domain Variant Associated With Hypoplastic Left Heart Syndrome. Front. Cell Dev. Biol. 8:440. doi: 10.3389/fcell.2020.00440
Received: 14 January 2020; Accepted: 11 May 2020;
Published: 23 June 2020.
Edited by:
Shinsuke Yuasa, Keio University, JapanReviewed by:
Shijun Hu, Soochow University, ChinaCopyright © 2020 Kim, Fleres, Lovett, Anfinson, Samudrala, Kelly, Teigen, Cavanaugh, Marquez, Geurts, Lough, Mitchell, Fitts and Tomita-Mitchell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aoy Tomita-Mitchell, YW1pdGNoZWxsQG1jdy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.