 Linda Anna Michelle Kulka
Linda Anna Michelle Kulka Pia-Victoria Fangmann
Pia-Victoria Fangmann Diana Panfilova
Diana Panfilova Heidi Olzscha
Heidi Olzscha- Medical Faculty, Institute of Physiological Chemistry, Martin-Luther-University Halle-Wittenberg, Halle (Saale), Germany
Lysine acetylation is one of the major posttranslational modifications (PTM) in human cells and thus needs to be tightly regulated by the writers of this process, the histone acetyl transferases (HAT), and the erasers, the histone deacetylases (HDAC). Acetylation plays a crucial role in cell signaling, cell cycle control and in epigenetic regulation of gene expression. Bromodomain (BRD)-containing proteins are readers of the acetylation mark, enabling them to transduce the modification signal. HDAC inhibitors (HDACi) have been proven to be efficient in hematologic malignancies with four of them being approved by the FDA. However, the mechanisms by which HDACi exert their cytotoxicity are only partly resolved. It is likely that HDACi alter the acetylation pattern of cytoplasmic proteins, contributing to their anti-cancer potential. Recently, it has been demonstrated that various protein quality control (PQC) systems are involved in recognizing the altered acetylation pattern upon HDACi treatment. In particular, molecular chaperones, the ubiquitin proteasome system (UPS) and autophagy are able to sense the structurally changed proteins, providing additional targets. Recent clinical studies of novel HDACi have proven that proteins of the UPS may serve as biomarkers for stratifying patient groups under HDACi regimes. In addition, members of the PQC systems have been shown to modify the epigenetic readout of HDACi treated cells and alter proteostasis in the nucleus, thus contributing to changing gene expression profiles. Bromodomain (BRD)-containing proteins seem to play a potent role in transducing the signaling process initiating apoptosis, and many clinical trials are under way to test BRD inhibitors. Finally, it has been demonstrated that HDACi treatment leads to protein misfolding and aggregation, which may explain the effect of panobinostat, the latest FDA approved HDACi, in combination with the proteasome inhibitor bortezomib in multiple myeloma. Therefore, proteins of these PQC systems provide valuable targets for precision medicine in cancer. In this review, we give an overview of the impact of HDACi treatment on PQC systems and their implications for malignant disease. We exemplify the development of novel HDACi and how affected proteins belonging to PQC can be used to determine molecular signatures and utilized in precision medicine.
Introduction
Lysine Acetylation and Histones
Lysine acetylation at the ε-amino group is one of the most abundant posttranslational modifications (PTM) in eukaryotic cells. Due to the neutralization of positive charge on lysine residues, acetylated proteins can interact with different molecules and adopt different folds. Thus, reversible lysine acetylation plays a crucial role in many biological processes, including gene expression, chromatin remodeling, cell cycle control, cell signaling and protein quality control (PQC) (Kouzarides, 2000). Protein acetylation is a classic example of a reversible PTM which can accommodate the needs of cells and reflect responses to environmental changes. Traditionally, enzymes which “write” the acetylation mark on lysine residues are termed histone acetyltransferases (HAT), enzymes which remove the acetylation mark from lysine residues in proteins, the erasers, are named histone deacetylases (HDAC) (New et al., 2012). The name “histone” deacetylases reflects the fact that many lysine residues in histones are acetylated and explains to some extent the epigenetic aspect of histone deacetylases and therefore histone deacetylase inhibitors (HDACi). However, it has been demonstrated that thousands of other proteins can be acetylated and deacetylated both in the cytoplasm and in the nucleus (Choudhary et al., 2009). Consequently, most HDACs act on proteins which occur in the nucleus and in the cytoplasm, implying that HDACi can affect not only histones, but all other proteins, depending on their class specificity. Bromodomain (BRD)-containing proteins act as “readers” and recognize acetylation marks. They translate acetylation signals to downstream signaling cascades, leading for example to further histone modifications or chromatin remodeling, finally shaping the cell into diverse phenotypes (Filippakopoulos and Knapp, 2014; Figure 1).
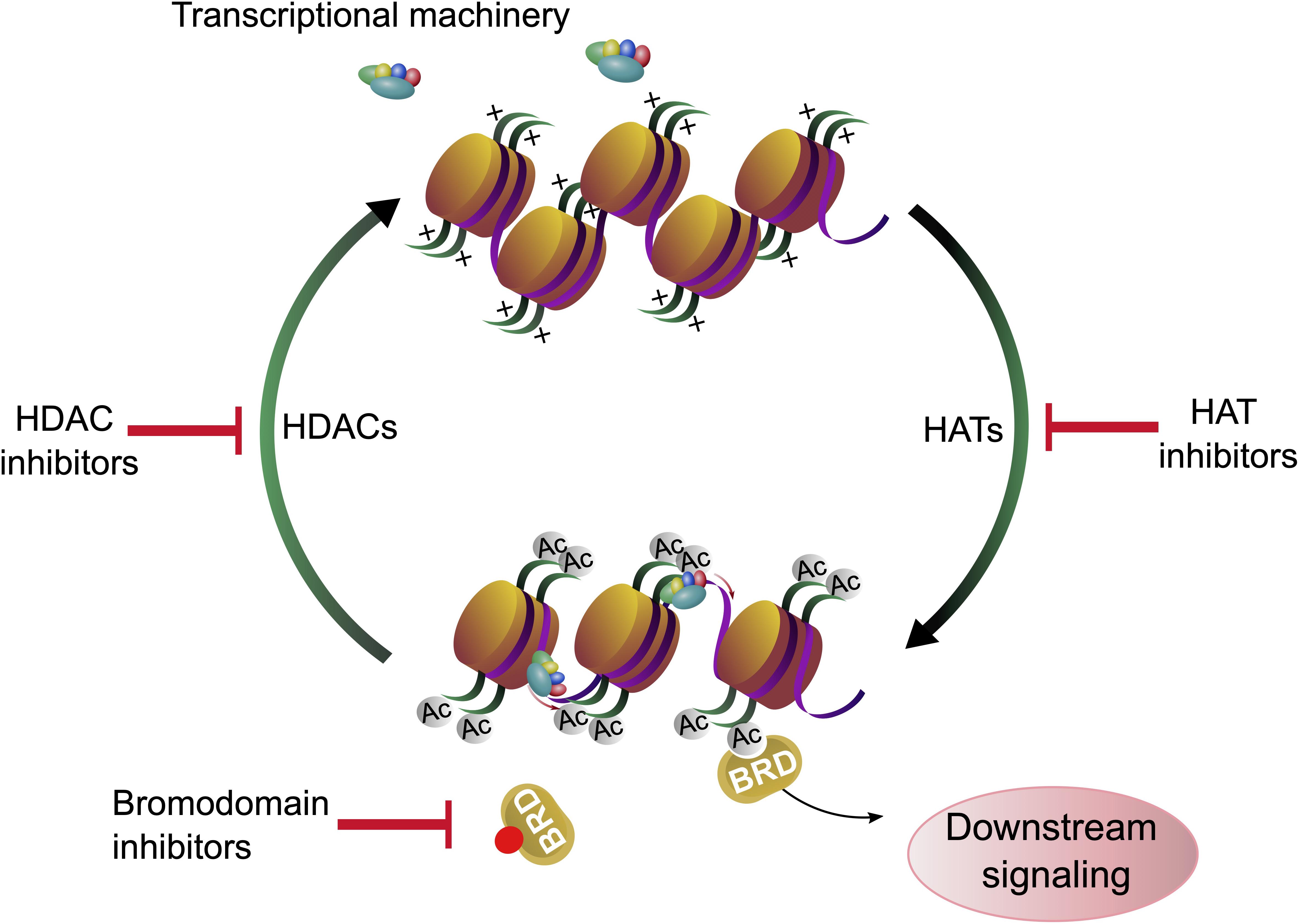
Figure 1. Histone acetylation, deacetylation and chromatin accessibility. Gene expression is regulated by lysine acetylation of histone proteins. Histone acetyl transferases (HATs) catalyze the transfer of acetyl groups onto proteins. Acetylation of histones affects the chromatin structure and can facilitate gene expression. Histone deacetylases (HDACs) remove the acetylation marks from histones. Acetylation and deacetylation can be modulated by histone acetyl transferase inhibitors (HATi) and histone deacetylase inhibitors (HDACi), respectively. The acetylation marks are recognized by bromodomain containing proteins (BRD) whose binding can be blocked by bromodomain inhibitors (BDi).
Histones are proteins, which organize the DNA into a compact form called nucleosome (Olzscha et al., 2015), and the bond strength between histone units and DNA can be determined by the acetylation of lysine residues of histones (Luger and Richmond, 1998). The acetylation of distinct lysine residues of histones (H2A, H2AX, H2B, H3, and H4) has different functions. Generally, histone acetylation is associated with transcriptional activation: If the histones are acetylated at many lysine residues, the nucleosome is present in its open form and genes can be transcribed by RNA-polymerases. It is assumed that the acetylation neutralizes the positive charge of the amino terminus and therefore the binding between histones and DNA is weakened (Annunziato and Hansen, 2018). Consequently, lysine residues regain their positive charge upon deacetylation and the affinity of the negatively charged DNA phosphate backbone to the amino terminus of histones is increased (Shahbazian and Grunstein, 2007). Other functions of lysine acetylation are DNA repair (H2AX on Lys5 and Lys36, H2A on Lys5, H3 on Lys9, 14, 18, 23, 27, 36, 56, and H4 on Lys5, 8, 12, 16, 91), histone deposition (H3 on Lys9,14 and H4 on Lys5, 12), transcriptional elongation (H3 on Lys14 and H4 on Lys8), chromatin assembly (H3 on Lys56), telomeric silencing (H4 on Lys12), chromatin decondensation (H4 on Lys16) and DNA replication (H4 on Lys91) (Koprinarova et al., 2016). Acetylation of H2A and H2B are mostly taking part in transcriptional activation, while acetylation on H2AX, H3 and H4 lysine residues can have different effects. In conclusion, deacetylation of histone proteins with HDACs is required for chromatin remodeling, many downstream processes and regulatory pathways (Wade, 2001; Figure 1).
HDAC Classification and Characterization
Eighteen human HDACs have been described and classified into four groups. We provide only a short introduction about the different HDAC classes, as there are many comprehensive reviews which give overviews about the HDAC classes and also chemical classes of HDACi, for example (New et al., 2012) or (Seto and Yoshida, 2014). The classification in Homo sapiens is based on the HDAC’s homology to yeast proteins (Dokmanovic et al., 2007). HDAC1, 2, 3, and 8 belonging to class I are homolog to the yeast RPD3 protein and are localized in the nucleus; they are involved in cell survival and proliferation. The class II HDACs (HDAC4, 5, 6, 7, 9, and 10) are supposed to play a tissue-specific role (Lagger et al., 2002). They are homolog to the yeast HDAC HDA1 (histone deacetylase 1) and can be found in the nucleus or cytoplasm. HDAC4, 5, 7, and 9 belong to class IIa and contain only one catalytic domain, while class IIb HDACs (6 and 10) have two catalytic domains and can only be detected in the cytoplasm. HDACs of class I and II contain Zn2+ in their catalytic sites, and thus are known as Zn2+-dependent HDACs. The HDACs from class III (SIRT1-7) are homolog to the Sir2 yeast protein. They do not contain Zn2+ in their catalytic sites, but require NAD+ for their enzymatic activity (Bolden et al., 2006). Class IV consists of only one protein, HDAC11. Regions in its catalytic center are similar to both class I and II sequences; hence, it is also classified as Zn2+-dependent HDAC (Gao et al., 2002).
The abundance and enzymatic activity of HDACs in cells is regulated on various levels e.g., by changes in gene expression, protein complex formation, PTMs, subcellular localization and by the availability of metabolic cofactors (Sengupta and Seto, 2004).
HDAC Inhibitors (HDACi)
Histone deacetylase inhibitors suppress HDAC activity. There are six structurally defined classes of HDACi: small molecular weight carboxylates, hydroxamic acids, benzamides, epoxyketones, cyclic peptides and hybrid molecules. They mainly act on HDACs of the classes I, II and IV by binding the Zn2+-containing catalytic domain (Drummond et al., 2005). The first discovered HDACi, the natural antifungal antibiotic trichostatin A (TSA), belongs to hydroxamic acid-type chelators (Yoshida et al., 1990), and the TSA structural analog vorinostat, also known as suberoylanilide hydroxamic acid (SAHA) was the first HDACi being approved by the U.S. Food and Drug Administration (FDA). The other three HDACi approved by the FDA so far are romidepsin, belinostat and panobinostat (Yoon and Eom, 2016). NAD+-dependent class III HDACs are inhibited by NAD+ and its derivates, dehydrocoumarin, splitomycin, 2-OH-naphtaldehyde, sirtinol and M15 (Porcu and Chiarugi, 2005). However, in this review, we focus on the “classic” HDACs belonging to the classes I, II and IV and their respective HDACi.
Vorinostat (Zolinza®) was approved in October 2006 for treatment of advanced primary cutaneous T-cell lymphoma (CTCL) (Mann et al., 2007). Romidepsin (Istodax®) was licensed for CTCL treatment in 2009 (Whittaker et al., 2010), and later, in 2011 for peripheral T-cell lymphoma (PTCL) (Coiffier et al., 2012). Belinostat (Beleodaq®) was approved by the FDA in 2014 for the treatment of PTCL. The fourth approved HDACi panobinostat (Farydak®) was licensed in 2015 for the treatment of multiple myeloma (MM).
As already mentioned, HDACi have a profound effect on the structure of chromatin and therefore on the transcriptional activity of the affected gene chromatin regions. This is why HDACi can be seen as established epigenetic modulators, since they affect the read-out of genes without changing the DNA sequence (Olzscha et al., 2015).
Epigenetics and Cancer
Epigenetics can be defined as inherited changes in phenotypes or entities, which are not encoded in the nucleotide sequence of the organism, but are passed on to daughter cells (Olzscha et al., 2015). Exogenous influences and altered environmental conditions can change epigenetic signatures and may give a hint about the origin of different malignancies, such as cancer or neurological disorders (Tsankova et al., 2007; John and Rougeulle, 2018). One appearance of epigenetics can be biochemical post-replicative modifications of the DNA-sequence, either through alteration of single bases or as described above in proteins (Handy et al., 2011).
Traditionally, cancer has been defined as a group of diseases leading to uncontrolled cell proliferation caused by genetic mutations in tumor-suppressor genes and oncogenes or chromosomal abnormalities (Hanahan and Weinberg, 2000). However, cancer may also be driven by epigenetic changes (Baylin and Ohm, 2006). According to its definition, epigenetic changes can be heritable and also known as epimutations, equivalent to mutations; however, some changes, in particular, histone deacetylation that repress gene expression by wrapping DNA more tightly, are not heritable, but have been also described as “epigenetic” (Berger et al., 2009). Thus, acetylation can influence transcriptional regulation, cell cycle control, apoptosis and autophagy, but also the activity of further proteins that maintain protein homeostasis, which will be described below (Nihira et al., 2017).
Objectives of the Review
Since thousands of proteins can be acetylated by HATs and deacetylated by HDACs, HDACi will not only act on an epigenetic level, but will also influence crucial protein functions, especially in PQC systems. These systems and the underlying effects will be described in this review and how this knowledge is utilized to develop combination therapies of HDACi and modulators of PQC processes. Clinical trials with HDACi alone or in combination are systematically evaluated for their potential to identify novel targets of PQC systems and their effect on epigenetic modulation. We also exemplify the development of novel HDACi which are in clinical trials, provide evidence that PQC systems are involved and how the underlying proteins can be used as biomarkers. Finally, we give an outlook on current and future HDACi development, its impact on proteostasis and how this knowledge can be utilized to improve precision medicine for cancer patients.
HDACs in Epigenetics and Protein Quality Control Systems
The Role of HDACs in Epigenetics and Cancer Cells
Recent studies suggest that cancer cells have increased concentrations of HDACs. For instance, according to clinical and preclinical studies, class I HDACs may stimulate cell proliferation and survival (Yoon and Eom, 2016). It has been shown that HDAC1 is overexpressed in prostate (Halkidou et al., 2004), gastric (Choi et al., 2001), colon (Wilson et al., 2006), and breast (Zhang et al., 2005) carcinomas. HDAC2 is reported to be responsible for the loss of adenomatous polyposis coli (APC) expression in colorectal cancer (CRC) (Zhu et al., 2004) and displays increased expression in cervical (Huang et al., 2005) and gastric (Song et al., 2005) carcinomas. HDAC3 and HDAC6 are also reported to show increased concentration in colon and breast carcinoma cells (Zhang et al., 2004; Wilson et al., 2006).
One of the problems in cancer is the heterogeneity which can occur from different mutations and/or different epigenetic patterns. Genomic variability occurs sometimes even if the cells display similar phenotypes or when there are differences in the phenotype, even though the cells originate from one tumor population (Cantor and Sabatini, 2012). These observations strengthen the hypothesis that epigenetics plays an important role in cancer development. Tumor suppressor genes might be silenced and oncogenes activated upon epigenetic changes without any influence on the genotype (Llinàs-Arias and Esteller, 2017). It is hypothesized that molecular chaperones such as the heat shock protein 90 (HSP90) act as regulators of the genotype-to-phenotype interplay and offer an evolutionary buffer to protect cells from malignant transformation (Whitesell and Lindquist, 2005; Jarosz, 2016).
The first discovered non-histone target of HATs and HDACs was p53 (Sakaguchi et al., 1998; Liu et al., 1999), a tumor suppressor protein that is able to bind DNA. Therefore, it can affect chromatin structure and can epigenetically change gene expression, whereby the binding is regulated by acetylation. Furthermore, acetylated p53 is able to induce apoptosis and autophagy (Fridman and Lowe, 2003; Mrakovcic and Fröhlich, 2018). Apoptosis is a form of highly controlled, energy-dependent programmed cell death, and malignant cells often depend on inherent or acquired mechanisms to resist cell death, which is seen as a hallmark of cancer (Hanahan and Weinberg, 2000). It is still under investigation, whether acetylation alters the interaction with other proteins or whether it results in a conformational change of p53 (Mrakovcic et al., 2018).
Another example for a silenced tumor suppressor in cancer cells is p21, which acts as a cyclin-dependent kinase (CDK) inhibitor. As p21 plays a crucial role in the regulation of CDKs, its expression has a big impact on cancer growth. Silencing of p21 occurs as a result of hypoacetylation of its promotor and consequently, it has been shown that HDAC1 inhibits the promotor by binding at the SP1-site (specificity protein 1) and competes with p53 activating the promoter of p21 (Gui et al., 2004). Using HDACi, there are two different mechanisms known leading to an enhanced expression of p21, with one of them p53-independent and the other p53-dependent. In the p53-independent mechanism, treatment with HDACi results in a release of HDAC1. Therefore, the promotor loses its repression and the gene of p21 is transcribed. The p53-dependent mechanism displays an enhanced expression of p21, as the HDACi induces acetylation of p53 resulting in a higher binding affinity to the p21 promotor (Ocker and Schneider-Stock, 2007).
HDAC1 is also known to bind ETO (eight-twenty-one), which can be fused with AML1 (acute myeloid leukemia 1). This fusion protein AML1-ETO arises as a result of a t(8;21) translocation and it has been shown that HDACi is efficient as a treatment against AML, suggesting a non-epigenetic effect. A study using valproic acid (VPA) as an HDACi reported a dissociation of the AML1-ETO/HDAC1 complex from the AML1-ETO promotor. Other studies reported a proteasomal degradation of AML-ETO after HDACi treatment, again demonstrating the importance of the PQC systems in cancer cells (Hug and Lazar, 2004).
The Role of Protein Quality Control Systems in Cancer Cells
In order to ensure the correct protein folding and protection of the proteome, eukaryotic cells developed a complex PQC system. Molecular chaperones, the ubiquitin proteasome system (UPS) and autophagy form a complex network which maintains the integrity of the proteome (Mogk and Bukau, 2006; Chen et al., 2011; Olzscha, 2019; Figure 2).
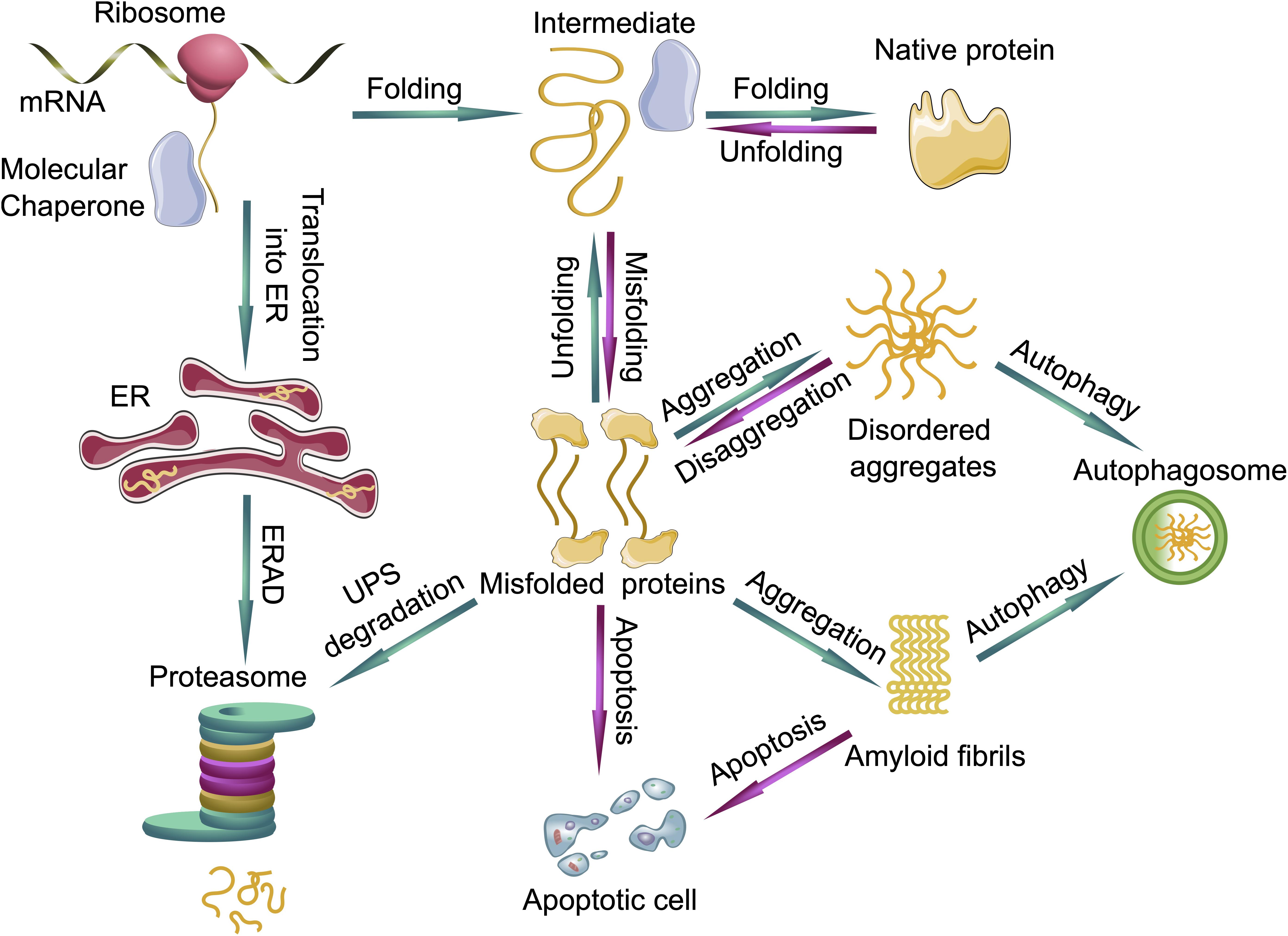
Figure 2. Protein quality control systems and their impact on protein folding, misfolding and aggregation. Molecular chaperones assist in the folding of nascent and unfolded proteins. If the folding fails, the unfolded or misfolded proteins are able to form disordered aggregates or even highly ordered amyloid fibrils. The ubiquitin proteasome system (UPS) can degrade the prefibrillar misfolded proteins from the cytoplasm and misfolded proteins from the endoplasmic reticulum (ER) via endoplasmic reticulum-associated protein degradation (ERAD), whereas larger aggregates can be degraded by autophagy. If protein quality control systems fail, cells can undergo apoptosis. Green arrows represent functioning PQC systems, eliminating cytotoxic species from cells and are pro-survival, whereas magenta arrows represent deleterious events where PQC systems fail, leading to cell death.
Molecular Chaperones
Most molecular chaperones are heat shock proteins and vice versa, they can be ATP-dependent and exert different mechanisms of assistance in protein folding. They are classified due to their sequence homology to specific heat shock proteins and their molecular mass: The HSP100/Clp-family, HSP90-family, HSP70-family, HSP60/GroEL-family and small heat shock proteins (sHSPs) (Jeng et al., 2015). All of them are known to assist proteins to fold correctly, especially complex proteins. They interact specifically with proteins and accelerate the folding, without being part of their final structure (Hartl and Hayer-Hartl, 2009). PTMs can alter protein folding (Santos and Lindner, 2017; Olzscha, 2019), and molecular chaperones are also known to be regulated by PTMs, including acetylation. For instance, it has been demonstrated that romidepsin stabilizes the acetylation of HSP70 leading to an increased binding of oncogenic proteins, which are normally stabilized by HSP90 (Cloutier and Coulombe, 2013). HSP90 is a ubiquitous occurring molecular chaperone, which supports a variety of proteins in their folding process. Accordingly, it affects many cellular processes, such as cell proliferation and signal transduction and plays a crucial role in cancer development (Scroggins et al., 2007).
Transformed cells can adapt metabolites for tumorigenesis, which can lead to further epigenetic modifications and subsequently tumor progression (Pavlova and Thompson, 2016). On the one hand, HSP90 can control this metabolic rewiring (Condelli et al., 2019), on the other hand, it determines the transcription of specific oncogenes. HSP90 can bind the chromatin directly or control the transcription factors of the genes (Khurana and Bhattacharyya, 2015). Thus, HSP90 can be seen as a paradigm for the interplay between molecular chaperones and epigenetics. If HSP90 is hyperacetylated either by knock-down of HDAC6 or by administration of an HDACi, its activity is impaired (Kovacs et al., 2005). This has been demonstrated with the treatment of the pan-HDACi panobinostat (LBH589), an anti-cancer drug approved by the FDA against MM (Yang et al., 2008; FDA, 2015).
Another example is given by the molecular chaperone HSP70, which supports the folding and refolding of proteins and can prevent aggregation or even refold aggregated proteins to a certain extent (Mayer and Bukau, 2005). It has been reported that its promotor is hypermethylated in cancer cells and the expression of HSP70 is enhanced by histone methylation. In a human oral squamous cell carcinoma cell line, the methylation of histone H3 at the lysine residues Lys4 and Lys9 enhanced the expression of HSP70 (Ban et al., 2019).
The Ubiquitin Proteasome System (UPS)
The UPS degrades proteins into oligopeptides (Ravid and Hochstrasser, 2008) and since ubiquitin is attached to lysine residues, it implies that competition with acetylation is generally possible (Caron et al., 2005). The proteasome recognizes the polyubiquitin chain, unfolds the target protein and finally degrades it. Target proteins can be metabolic enzymes, transcription factors and cell cycle regulating proteins, including cyclins and CDK-inhibitors (Schrader et al., 2009). All of these proteins are known to play a crucial role in cancer, for instance, metabolic enzymes are important for maintaining the tumor microenvironment and nutrient availability. In cancer cells, their protein level and occurrence can be altered as a result of mutations and non-genetic changes, including the adaption of metabolic enzymes, which are normally degraded by the UPS (Wegiel et al., 2018). Especially glycolysis and the tricarboxylic acid cycle are well analyzed targets (Yu et al., 2017). As cancer cells have a high proliferation rate, they need high amounts of ATP as energy supply and nutrients, including lipids, nucleotides and amino acids. This higher proliferation rate also led to changes in cell cycle, affecting regulatory proteins such as cyclin and CDK-inhibitors (Deshpande et al., 2005).
Perhaps the best-known protein associated with cancer is the before-mentioned tumor suppression protein p53, which is inactivated in many types of cancer. The concentration of p53 is regulated by polyubiquitination and subsequent proteasomal degradation, in particular by mouse double minute 2 homolog (MDM2) (Patel and Player, 2008), which is a RING (really interesting new gene) E3-ligase. It can form a complex with p300/CBP (CBP, CREB binding protein; CREB, cAMP response-element binding protein) resulting in the polyubiquitination and degradation of p53 (Grossman et al., 1998). Inhibition of the proteasome can prevent this degradation (Harris et al., 2008); however, the p53 gene is often mutated in cancer cells, leading to the conclusion that intervention on the transcriptional level seems to be more promising. However, proteasome inhibitors (PI) were tested together with HDACi and synergistic effects of PIs with HDACi were proven, e.g., the FDA-approved inhibitor bortezomib with the pan-HDACi vorinostat (Johnson, 2015). Thus, the UPS plays a relevant role with regards to the treatment of cancer, and examples of clinical trials in this combination are given in section “HDAC Inhibitors and Proteasome Inhibitors” of this review.
Furthermore, proteasomes degrade proteins that are recognized as misfolded, a process which needs to be distinguished from protein degradation being a regulatory step to control the half-life of a protein (Figure 2). Protein misfolding can occur spontaneously within the cell, or the protein failed to fold correctly after its biogenesis. In case molecular chaperones are unable to assist in protein folding, the misfolded proteins are recognized by specific adapter proteins such as molecular chaperones together with the carboxy terminus of heat shock protein 70-interacting protein (CHIP) and marked for degradation by the UPS (Meccariello et al., 2014). PTMs can be one reason for protein misfolding and in some cases result in aberrant degradation of these proteins via the UPS (Olzscha, 2019).
Glucose-regulated protein 78 (GRP78) is another HSP that plays a crucial role in regulating the unfolded protein response (UPR). This pathway is induced by ER (endoplasmic reticulum) stress. For instance, an increase of accumulated misfolded protein in the ER lumen can lead to ER stress and associated pathways. There are also molecular chaperones present in the ER to prevent misfolding and aggregation of proteins, but they can also fulfill special tasks within the ER. One of the molecular chaperones is binding immunoglobulin protein (BiP), also known as GRP78 (78-kDa glucose-regulated protein). BiP can recognize and bind misfolded proteins in the ER, leading to a dissociation and activation of the protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1) (Warri et al., 2018). ATF6 can induce the Akt/mTOR (mammalian target of rapamycin) pathway and promotes the transcription of different genes, including the autophagy-related genes 12 (ATG12) and 5 (ATG5) regulating autophagy (Yan et al., 2015). Autophagy is another PQC system, which is described below. It is known that different HDACi, for example vorinostat (Kahali et al., 2010), YCW1 and OSU-HDAC2, can induce ER stress causing autophagy (see also section “HDAC Inhibitors Affecting Protein Quality Control Systems”).
Autophagy
Autophagy is another part of the PQC systems, which allows cells to degrade cytoplasmic constituents and to remove unnecessary or dysfunctional proteins (Chun and Kim, 2018). Misfolded and aggregated proteins can be degraded by autophagy, especially, if molecular chaperones or the UPS are not able to cope with the amount of misfolded proteins. These aggregated proteins are capable to form amyloid structures which are the underlying cause for proteinopathies such as Alzheimer’s disease (AD) or amyotrophic lateral sclerosis (ALS). In superoxide dismutase 1 (SOD1) mice, the impairment of the UPR and autophagy was proven to be partly responsible for the pathophysiology of ALS (Ruegsegger and Saxena, 2016).
One can differentiate between macro-, micro- and chaperone-mediated autophagy (CMA) (Glick et al., 2010). Macroautophagy is the main pathway in the cell to degrade damaged cell organelles or aggregated proteins. In the process of engulfment, an autophagosome is built, which is a circular double-membrane structure that encloses the target protein. The autophagosome comes in close proximity to the lysosome and fuses with it, forming the autolysosome, where proteins get hydrolyzed via lysosomal hydrolases in an acidic environment (Feng et al., 2014). During microautophagy, proteins are also degraded via acidic lysosomal hydrolases; however, they are directly engulfed by vesicles originating from lysosomes (Li et al., 2012). During CMA, HSP70 chaperones recognize proteins containing a KFERQ-like motif. This leads to the formation of a CMA-substrate/chaperone complex, which is located to the lysosomal receptor LAMP-2A (lysosome-associated membrane protein). The protein is unfolded and translocated across the lysosomal membrane where it is degraded (Cuervo and Wong, 2014).
Autophagy is regulated by autophagy-related genes (ATG) (Wesselborg and Stork, 2015). In cancer cells, autophagy can be disturbed in a way that either they degrade apoptotic mediators, which would normally kill the cancer cells, or the survival of starving cancer cells is prolonged (Mathew et al., 2007; Fernald and Kurokawa, 2013). However, autophagy acts as a tumor suppressor in non-cancerous cells (Kung et al., 2011). Furthermore, it could be proven that mice are more susceptible to tumorigenesis containing heterozygous beclin 1, a protein, which regulates macroautophagy. If it is overexpressed, tumor development is inhibited, on the other hand, cancer cells utilize autophagy for survival (Boutouja et al., 2017). Autophagy recycles ATP, which is needed by cancer cells in higher amounts, and it could be shown that the inhibition of autophagy genes by treatment with 3-methyladenosine or Atg7-knockdown activates apoptosis in different tumor cell lines, like prostate and colon cancer cells (Bhutia et al., 2013; Li et al., 2018). These results lead to two different strategies in cancer therapy. On the one hand, autophagy is induced, leading to enhanced tumor suppression; on the other hand, autophagy is inhibited and can induce apoptosis.
HDAC Inhibitors Affecting Protein Quality Control Systems in Cancer Treatment
As mentioned in section “The Role of HDACs in Epigenetics and Cancer Cells,” HDACi cause hyperacetylation of histones which is one reason for the induction of apoptosis. As autophagy and apoptosis functionally counteract each other in tumor cells (see section “The Role of Protein Quality Control Systems in Cancer Cells”), one could assume that autophagy is inhibited during treatment with HDACi (Gump and Thorburn, 2011). However, it has been demonstrated that administration of HDACi can also induce autophagy, leading to the paradox situation that autophagy has beneficial effects in the treatment of cancer cells and even facilitates tumor suppression (Zhang et al., 2015). The effect of promoting cell survival or cell death is dependent on the cell type and genetic predisposition of the tumor, as well as the duration and dose of the HDACi. There are many examples described in the literature, one of them is the pan-HDACi panobinostat. On the one hand, panobinostat can inhibit autophagy by increasing the level of acetylation of autophagy-related gene products, for instance ATG7. The acetylation causes a repression of ATG7, which leads to a promotion of apoptosis and decreased autophagy in myeloid leukemia cells (Stankov et al., 2014). On the other hand, it has been reported that autophagy is induced in panobinostat-treated Eμ-myc lymphoma cells, the c-myc gene is here driven by the IgH enhancer. Thereby, an apoptotic protease activating factor 1 (Apaf-1) or caspase-9 deletion has been reported. This deletion causes an apoptosome inactivation and thus a suppression of apoptosis (Mrakovcic et al., 2017, 2018). Furthermore, it has been demonstrated that HDACs themselves can induce autophagy. For example, HDAC6 can induce autophagy as a result of an impaired UPS (Kaliszczak et al., 2018). It binds polyubiquitinated proteins and plays an essential role for the fusion of autophagosomes with lysosomes.
It seems to be a drawback that pan-HDACi not only inhibit histone deacetylation in the nucleus, but also a variety of proteins which can be found in virtually all cellular compartments. However, a greater understanding of the control and homeostasis mechanisms of HDACs is required to enable more effective application of HDACi for the treatment of specific tumor cell types.
HDAC Inhibitors in Preclinical Studies
HDAC Inhibitors Affecting Protein Quality Control Systems
As existing HDACi are mostly pan-HDACi, they do not entail satisfactory specificity. Accordingly, it is of great interest to develop new and more specific inhibitors (see also section “Novel Strategies of HDAC Inhibitors Affecting Protein Quality Control Pathways”). HDAC6 is a potential selective target due to its unique molecular structure with two catalytic domains and its localization in the cytoplasm (Li et al., 2018). Critical substrates with a role in PQC include p300 and HSP90 (Cosenza and Pozzi, 2018). Both are known to play a crucial role in tumorigenesis, showing the importance for the development of specific HDAC6 inhibitors. Tubastatin A is an example for an established HDAC6 inhibitor, often used in pre-clinical studies (Wang et al., 2016). However, a more specific HDAC6 inhibitor (marbostat-100) has been developed and was published in 2018 with a Ki-value of 0.7 nM (Sellmer et al., 2018). In comparison, the value of the most common used selective HDAC6 inhibitor tubastatin A is 10-times higher. A preferred substrate of HDAC6 is α-tubulin; the reverse reaction is catalyzed by the α-tubulin acetyltransferase ATAT1. The deacetylated α-tubulin polymerizes with β-tubulin to form microtubules, components of the cytoskeleton, which play an important role in DNA segregation during mitosis. Inhibition of HDAC6 with marbostat-100 results in hyperacetylated α-tubulin. The specificity of this HDAC6 inhibitor was determined by comparing the enrichment of acetylated histone H3 in marbostat-100 treated cells with entinostat (MS 275) treated cells. It is an HDAC1 and HDAC3 specific inhibitor in phase II clinical studies. This enrichment of acetylated histone H3 could also be detected using the FDA-approved pan-HDACi panobinostat (LBH589) (Grünstein, 2018). It has been demonstrated in different human cell lines and in mice that marbostat-100 is considerably more specific than panobinostat, led more efficiently to hyperacetylation of α-tubulin and displayed only minor proteolytic effects on the target enzyme HDAC6 (Grünstein et al., 2019).
In analogy to marbostat-100, the application of the established HDAC6i tubastatin A led to hyperacetylation of α-tubulin. However, upon oxidative stress, tubastatin A activated the heat shock transcription factor 1 (HSF1) leading to the upregulation of the molecular chaperones HSP70 and HSP25 and increased cell survival (Leyk et al., 2017). Upon proteasomal stress, HDAC6 could initiate autophagy, as it is involved in the transport of ubiquitinated proteins along microtubules (Leyk et al., 2015). This influence of the PQC system has been observed in some preclinical studies of HDACi, which are described below in section “HDAC Inhibitors in Combination With Proteostatic Drugs.”
Two further examples for inhibitors that influence molecular chaperones as well as autophagy are trichostatin A and sodium butyrate. Both can affect the chromatin structure at the site where the gene for HSP70 is located (Chen et al., 2002). HSP70 is a molecular chaperone supporting the folding of many newly synthesized proteins and can recognize the KFERQ motif in proteins resulting in CMA.
In addition to its impact on molecular chaperones and consequently on CMA, another mechanism of action of trichostatin A is known. It has been shown that trichostatin A enhances the ubiquitination of the HAT p300, resulting in its proteasomal degradation. Since it is a co-activator of the expression of the NADPH oxidase 4 (Nox4) (Hakami et al., 2016), an important factor in angiogenesis, cancer cells suffer from oxygen and nutrient deficiency due to its reduced expression in trichostatin A treated cells. Thus, trichostatin A constitutes a paradigm in its ability to impact on molecular chaperones, the UPS and autophagy.
Another example for an inhibitor affecting the ubiquitin-proteasome system is MC1568 (Table 1). It is a class IIa selective HDACi that increases the specific sumoylation of HDAC4. Sumoylation is a PTM using “small ubiquitin-related modifier” (SUMO) to label the target protein and can direct it to different pathways. MC1568 induced HDAC4 down-regulation by increasing its specific sumoylation followed by activation of proteasomal degradation pathways. MC1568 alters not only the pattern of PTMs and activates the degradation of substrates via the UPS, but also changes epigenetic pathways that may be affected by HDAC4 (Scognamiglio et al., 2008).
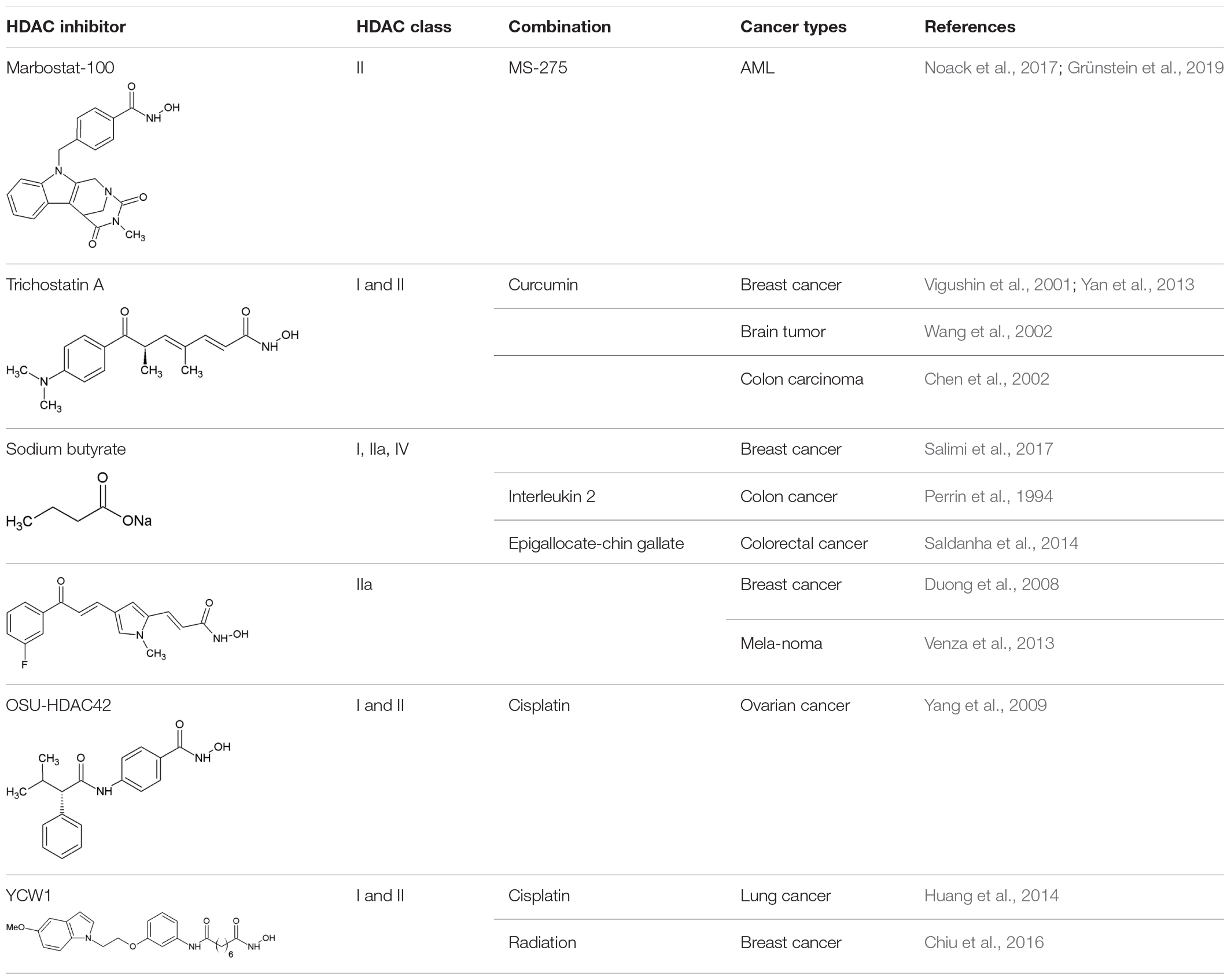
Table 1. Preclinical studies of HDACi and compounds affecting PQC in combinatorial treatment.
Besides trichostatin A, there are other HDACi known to induce autophagy. For instance, OSU-HDAC42 led to downregulation of Akt/mTOR signaling and the induction of the ER stress response to induce autophagy (Liu et al., 2010). On the contrary, triple negative breast cancer cells treated with the HDACi YCW1 showed a downregulation of BNIP3 (Bcl-2/adenovirus E1B 19 kDa protein interacting protein 3) resulting in autophagic cell death. Similarly, treatment of mice with YCW1 led to a decline in lung tumor growth (Huang et al., 2014).
HDAC Inhibitors in Combination With Proteostatic Drugs
The fact that HDACi can influence PQC systems led to a novel strategy where HDACi are used in combination with drugs modulating PQC systems, i.e., PI or modulators of autophagy.
In a study conducted in 2017, trichostatin A was tested in combination with the PI bortezomib for the treatment of ovarian cancer cells and displayed an inhibition of the proliferation of A2780 cells inducing apoptosis. Furthermore, similar results were shown in A2780T cells that are resistant to cytostatic taxanes (Jin et al., 2017). In another attempt, a combination of sodium butyrate was tested with the PIs MG115, MG132, PSI-1, PSI-2, or epoxomicin in human CRC cells (SW48, SW1116, and SW837). In these studies, additive and synergistic anticancer effects, namely growth inhibition and apoptosis, were observed in combination with all tested PIs (Abaza, 2010).
Cell death induced by accelerated autophagy in cancer cells has been shown to be effective in a combined treatment with HDACi and an inducer of autophagy. In Burkitt lymphoma and lymphocyte cell lines, VPA induced autophagosome formation and increased autophagy led to an autophagy-mediated cell death in combination with an mTOR inhibitor (Dong et al., 2013). This experiment has been carried out with the mTOR-specific inhibitor temsirolismus, but as OSU-HDAC42 is known to act as an mTOR inhibitor, it is suspected that OSU-HDAC42 would show similar results (Liu et al., 2010). Furthermore, it has been demonstrated that the protein levels of HDACs, especially HDAC6, are reduced by autophagy after treatment with the HDACi AR42 (OSU-HDAC42). The combination of this HDACi with the kinase inhibitor pazopanib in melanoma cells demonstrated an inhibition of the ATPase activity of the molecular chaperones HSP90 and HSP70. In this setting, HDAC6 could activate HSP90 by deacetylation and the inhibition was enhanced by combined treatment (Booth et al., 2017). Using YCW1 (Table 1) in combination with radiation also demonstrated increased cell death in cancer cells due to ER stress and the induction of autophagy (Chiu et al., 2016). In addition, preclinical studies revealed an enhanced cisplatin effect against YCW1-treated non-small cell lung cancer (NSCLC), whereas cisplatin causes mitochondria-mediated apoptosis (Huang et al., 2014). This highlights again the discrepancy that cancer cells sometimes utilize autophagy to their advantage and sometimes inhibit the autophagic pathway.
Protein ubiquitination can be also mediated by cullin-ring E3 ligases (CRLs), which have to be activated by neddylation with NEDD8 (neural-precursor-cell-expressed developmentally down-regulated 8), another protein similar to ubiquitin acting as PTM (Enchev et al., 2015). Neddylation with NEDD8 is achieved by NEDD8-activating enzymes (NAEs), which are druggable enzymes. It has been shown in several studies that NAE-inhibitors, for instance pevonedistat (MLN4924), can act with other anti-cancer agents including bortezomib in a synergistic manner in MM (Gu et al., 2014). In a different pre-clinical study, it has been demonstrated that the NAE inhibitor pevonedistat acts synergistically with the HDACi belinostat in various AML cell types, especially those with reciprocal effects on homologous recombination (HR) and non-homologous end-joining (NHEJ) mechanisms (Zhou et al., 2016).
HDAC Inhibitors and Bromodomain Inhibitors
There are 61 bromodomains known in the human proteome, integrated in 46 proteins. All of them have a conserved left-handed bundle of four α-helices linked by flexible and variable loops. The best-known proteins containing a bromodomain are part of the bromodomain and extra terminal family (BET). This BRD family is characterized by the presence of two tandem bromodomains (BD1 and BD2) at the N-terminus, an extra terminal domain (ET), and a C-terminal domain (CTD). They play a crucial role in cancer cells, especially in cell proliferation by regulating the expression of oncogenes, for instance c-MYC or nuclear factor κ light chain enhancer of activated B cells (NF-κβ)-dependent genes (Pérez-Salvia and Esteller, 2017).
It has been demonstrated that hyperacetylation of proteins induced by HDACi can result in amyloid-like protein aggregation. This can lead to a reduction of the proteolytic capacity of the UPS, increased autophagy and downregulated translation, summarized as proteostatic failure (Olzscha et al., 2017). Similarly, it has been observed that trichostatin A induced a dramatic increase of the acetylation of tau proteins, which aggregation can be seen under pathological conditions as one of the underlying causes for Alzheimer’s disease (AD) (Cohen et al., 2011). The increase of acetylated tau levels and resulting aggregation is also shown in tubastatin A treated oligodendrocytes, and an alteration of the cell morphology was observed containing a reduced microtubule binding activity of tau (Noack et al., 2014). As described above, tubastatin A is an HDAC6-specific inhibitor; consequently, HDAC6 plays an important role in the cytotoxic accumulation of protein aggregates which may explain the higher level of aggregated tau proteins (Boyault et al., 2007b). Marbostat-100 also inhibits HDAC6, the treatment may have similar effects on the acetylation of the tau protein; however, it remains unclear whether it has an effect on aggregation.
The potential of HDACi-induced aggregation raises the question, how this knowledge can be utilized for benefits in cancer therapy, at the same time preventing adverse side effects and suppressing aggregation. It has been demonstrated that the bromodomain-containing proteins CBP and p300 are involved in the formation of protein aggregates after treatment with HDACi and their depletion results in a reduction of aggregation. This opens a new strategy for cancer treatment without the formation of aggregates. Bromodomain inhibitors are small proteins which can block the binding of bromodomain-containing proteins to acetylated residues and therefore have the potential to reverse the aggregation-induced cytotoxicity and restore proteostasis (Olzscha et al., 2017).
The first published BET inhibitor was (+)-JQ1 tested in NUT (nuclear protein in testis) midline carcinoma cells. NUT can fuse with the bromodomain BRD4 forming the oncoprotein BRD4-NUT, which plays an important role in the differentiation and proliferation of cancer cells. (+)-JQ1 acts as a competitor binding at the acetyl-lysine binding motif and prevents the formation of the fusion protein and the resulting proliferation (Filippakopoulos et al., 2010). It was then tested in many other cancer types, for instance glioblastoma (Cheng et al., 2013), colon cancer (McCleland et al., 2016), lung cancer (Lockwood et al., 2012), Burkitt’s lymphoma (Mertz et al., 2011), and MM (Soodgupta et al., 2015). It led to downregulation of c-MYC, an oncogene responsible for altered transcription and proliferation and showed synergistic effects with the HDACi mocetinostat (Borbely et al., 2015). However, it has never reached a clinical trial, due to its short half-life; therefore, analogs of (+)-JQ1 with a longer half-life were synthesized, one of them is called CPI203 (Alqahtani et al., 2019). It has shown some success in bortezomib-resistant mantle cell lymphoma (Moros et al., 2014) and MM cells (Díaz et al., 2017) in combination with the immunomodulator lenalidomide, whereby reduced c-MYC-levels leading to a downregulation of IRF4 (interferon regulatory factor 4). IRF4 is a transcription factor, which is necessary for the survival of lymphoma and myeloma cells, leading to an induction of apoptosis in these cells. I-BET151 is another BET-inhibitor, which also represses c-MYC in myeloma cells (Chaidos et al., 2014). As a pan-BET inhibitor, it also displayed anti-cancer effects in other types of cancers, for example in medulloblastoma cells by suppressing the Hedgehog-activity or in NUT midline carcinoma (Long et al., 2014).
Other examples where bromodomain inhibitors can influence proteostasis in combination with HDACi are CBP and p300. These transcription modulators are not only bromodomain-containing proteins, they also act as histone acetyltransferases. Thus, they recognize lysine acetylation and may cause further acetylation in histones leading to a relaxation of DNA and an activation of transcription. Two p300/CBP-specific bromodomain inhibitors, I-CBP112 and SGC-CBP30, were investigated in preclinical studies, I-CBP112 for leukemia and prostate cancer (Picaud et al., 2015) and SGC-CBP30 in MM (Hay et al., 2014). I-CBP112 activates the HATs CBP and p300 resulting in a repression of the proliferation in cancer cells (Zucconi et al., 2016). SGC-CBP30 can suppress IRF4 in myeloma cells (Conery et al., 2016). However, currently it is not brought into clinical trial, as it displays a short half-life. Another inhibitor which targets non-BET bromodomains as well as BET-bromodomains is bromosporine (Theodoulou et al., 2016). This pan-BDi reduced the formation of protein aggregates only slightly after HDACi treatment (Olzscha et al., 2017).
There is still an interest in developing more specific and more efficient HDACi for cancer treatment. Most of the current HDACi influence several pathways in the cells, including the PQC system. This can be used by combining HDACi with other cancer treatments, like radiation, PI, bromodomain inhibitors, autophagy- and chaperone-modulating agents. On the other hand, there are also new HDACi broadening the spectrum of molecular actions and therefore their monotherapeutic use has to be reconsidered as a treatment option.
Novel Strategies of HDAC Inhibitors Affecting Protein Quality Control Pathways
Various novel HDACi have undergone pre-clinical and clinical studies over the past 5 years, both HDACi which target specific HDAC classes and HDACs which can be considered as pan-HDACi. CXD101 is a novel class 1-selective HDACi and has shown effects in some hematological malignancies (Eyre et al., 2019). The observed high levels of the proteasomal shuttling factor HR23B indicate a positive outcome, resembling the results of pan-HDACi (Khan et al., 2010; New et al., 2013). In fact, it has been demonstrated that also class I HDACi are able to induce protein aggregation in human cells (Olzscha et al., 2017). As described above, the induced protein aggregation may contribute to the overall cytotoxicity exerted by HDACi and their success in hematological malignancies. Since CBP/p300-specific bromodomain inhibitors are able to partially abrogate this effect (Olzscha et al., 2017), it is likely that the aggregation indirectly affects nuclear proteins and therefore modulates epigenetic regulation in cells. Interestingly, a clinical trial of CXD101 in combination with the tissue-agnostic drug pembrolizumab for relapsed or refractory diffuse large B-cell lymphoma (PLACARD, NCT03873025) is underway, taking the levels of PD-L1 (programmed death-ligand 1) in PD-1 positive cells into account (see also section “Evaluating Alterations of PQC Systems: Precision Medicine Upon HDAC Inhibitor Treatment”).
As mentioned above (section “HDAC Inhibitors in Combination With Proteostatic Drugs”), a common strategy is to apply HDACi in combination therapy with other anti-cancer drugs. However, this strategy faces several problems, including incompatibilities, pharmacokinetic problems when reaching different compartments and unexpected interactions, which may alleviate the activities, but also lead to an increased possibility to generate undesired cytotoxic effects (de Lera and Ganesan, 2016). To overcome some of the problems, several chimeric HDACi have been developed. They consist of hybridized functional groups of an HDACi structure and the respective different group to inhibit or bind to a second target (Nepali et al., 2014). For instance, the inhibitor fimepinostat (CUDC-907), which is a dual HDACi and phosphatidylinositol-3-kinases (PI3K) (Gunst et al., 2019) was tested in a trial in patients with lymphoma (NCT01742988). A striking example of this strategy, which affects epigenetic outcome and PQC pathways, is the generation of chimeras between HDACi and bromodomain inhibitors (BDi). Bromodomains cannot only “read” acetylation marks on proteins, they can also act synergistically with HDACs to guide them to the respective protein and remove the acetylation mark (Olzscha et al., 2015). Many promising examples of BDi have been generated, targeting several bromodomain-containing proteins, including CBP/p300, affecting both chromatin and interacting proteins, including p53 (Picaud et al., 2015). Several chimeric compounds consist of the functional groups of pan-HDACi such as SAHA and different BDi including the BET inhibitors JQ1 and I-BET295 (Atkinson et al., 2014), as well as BRD-4 specific inhibitors (Amemiya et al., 2017). Besides the established functions as epigenetic modulators, some HDACs and bromodomain-containing proteins (BRDs) exert their activities also in the cytoplasm, affecting crucial PQC pathways. Effects of HDACs and BRDs on the protein degradation machinery demonstrate that these mechanisms contribute to the overall cytotoxicity of the single substances or their chimeras (New et al., 2013; Olzscha et al., 2017).
A novel HDACi which affects proteostasis is MPT0G413, a selective HDAC6 inhibitor. This inhibitor did not only inhibit the growth in MM cells, the combination of MPT0G413 and bortezomib enhanced also polyubiquitinated protein accumulation and synergistically reduced MM viability, showing increased caspase-3, caspase-8, and caspase-9 levels (Huang et al., 2019). Since it is an HDAC6 inhibitor, the effects are likely to reflect disturbances in PQC pathways and therefore only indirectly affect epigenetic features in the nucleus.
Clinical Studies: HDAC Inhibitors in Combination With Modifiers of Protein Quality Control
The knowledge about epigenetics has exploded over the past few years, highlighting its importance in crucial functions in the cell such as gene silencing, DNA methylation and histone modification. The observation of aberrant hypermethylation on CpG-rich promoter regions, histone modification, non-coding RNA modification and other epigenetic changes in cancer cells established the research field of the “cancer epigenome” and spurred efforts to investigate appropriate therapies in this newly defined field. During the past few years it has become increasingly apparent that neoplastic cells have a selective advantage not only due to mutations, but also provided by epigenetic changes (Arrowsmith et al., 2012). Histone modification such as HDAC overexpression or altered acetylation levels have been found in prostate, gastric, colorectal, cervical and endometrial cancer (Glozak and Seto, 2007), see also section “HDACS in Epigenetics and Protein Quality Control Systems.” In addition, a negative correlation between HDAC overexpression and overall survival has been described in pancreatic, breast, colorectal, gastric, lung, liver cancer and melanoma (Weichert et al., 2008a; West and Johnstone, 2014; Mottamal et al., 2015; Sarkar et al., 2015). In particular, broad ranges of hematological malignancies appear to be influenced by HDAC alterations. For instance, high expression of HDAC1, 2 and 6 are persistent in patients suffering from CTCL (Marquard et al., 2008). As hematological malignancies were described to be especially sensitive to HDACi therapy, a wide set of clinical trials exploited the research field of monotherapy of HDACi in hematological malignancies with partly successful and promising results (Cashen et al., 2012; Fukutomi et al., 2012; Kirschbaum et al., 2012, 2014; Younes et al., 2012; Platzbecker et al., 2014). Until now, four HDACi are approved by the FDA, as described in the introduction. Although HDACi monotherapy has been described to show promising effects on these types of tumors, further investigations in solid tumors were disappointing, as a large-scale use of HDACi is hampered due to a lack of detailed understanding in molecular mechanisms of HDACi. Several clinical trials revealed that monotherapy with HDACi only showed limited success in solid tumors (Qiu et al., 2013). In addition, some clinical studies had reportedly severe side effects using HDACi in monotherapy, another reason why they were discontinued. Hence, toxicity profiles need to be considered in future clinical trials, especially when combining HDACi with other agents (Subramanian et al., 2010). However, HDACi revealed to function synergistically with a range of other anticancer agents such as immune checkpoint inhibitors, platinum-based chemotherapeutics or tyrosine kinase pathway inhibitors in pre-clinical and clinical studies. Therefore, combination of HDACi with other cancer therapeutics may represent an important direction to enhance their anticancer efficacy and show their full therapeutic potential (Singh et al., 2018; Suraweera et al., 2018).
At present, various clinical trials are in progress, testing different HDACi for both hematological and solid tumors in either mono- or combined therapy regimens (Table 2).
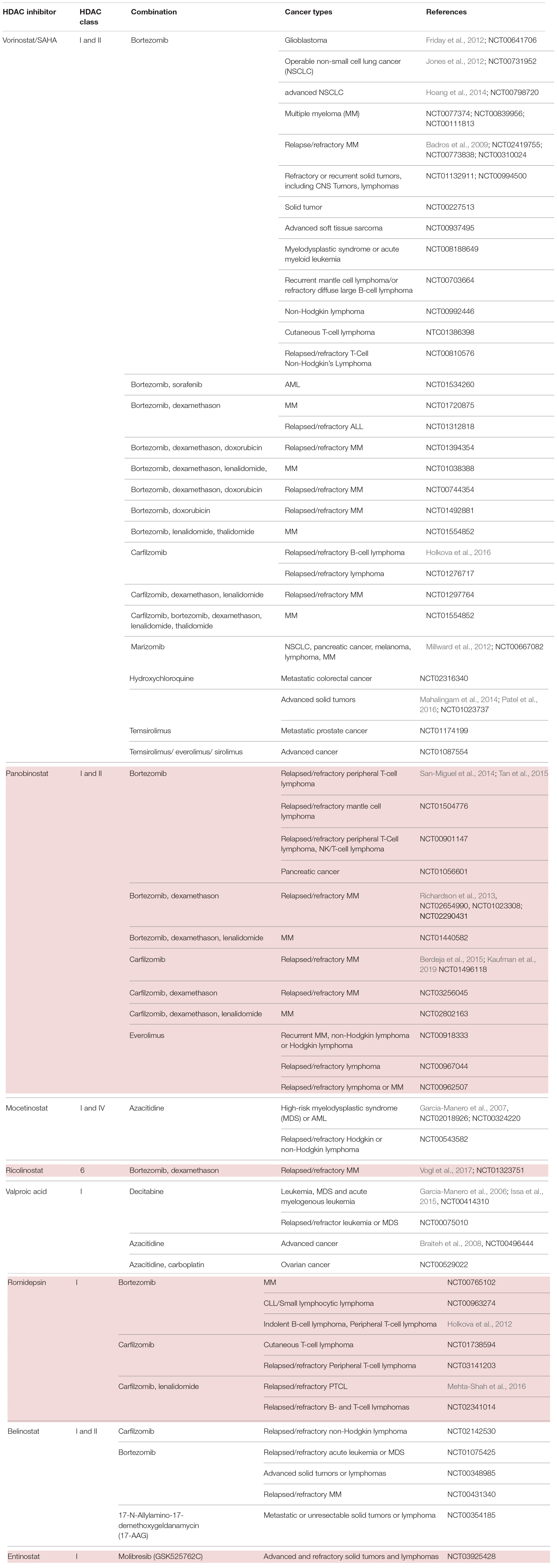
Table 2. HDACi in combination with inhibitors of protein quality control systems in clinical trials.
HDAC Inhibitors and Proteasome Inhibitors
Cancer cells are highly proliferative and show an extensive protein turnover and thus rely heavily on proteasomal degradation of abnormal or mutant proteins (Adams, 2004). Therefore, it can be argued that cancer cells are more dependent on functioning PQC systems such as the UPS, and autophagy than non-transformed cells (Goldberg, 2007). Indeed, several preclinical studies established that proteasome inhibition has a more severe effect on malignant cells than on normal cells (An et al., 1998; Masdehors et al., 2000; Hideshima et al., 2001; LeBlanc et al., 2002). Thus, proteasome inhibition would overload the cancer cell with protein material and accumulation of ubiquitinated proteins, finally causing cell death (Adams, 2004).
First attempts on using bortezomib, as a single agent PI, showed success in the treatment of relapsed and refractory MM. Various preclinical and clinical trials provided data on significant benefit in patients’ respond and outcome (Orlowski et al., 1998; Hideshima et al., 2001; LeBlanc et al., 2002; Richardson et al., 2005a). The Assessment of Proteasome inhibition for EXtending remissions (APEX) trial confirmed significant benefit in the bortezomib group over the patients treated with dexamethasone in patients with relapsed MM (Richardson et al., 2005b). These findings laid the foundation for the approval of bortezomib by the FDA in 2003. Although thrombocytopenia and peripheral neuropathy were the most frequently associated dose limiting toxicities, the FDA authorized bortezomib for the use in relapsed and refractory myeloma patients who showed no response to two or more prior therapies (Field-Smith et al., 2006). Accordingly, two other substantial drug discoveries were found to have a beneficial effect on MM patients: the immunomodulatory drugs thalidomide and lenalidomide (Dimopoulos et al., 2007; Rajkumar et al., 2008). However, despite all the promising new drug developments and outcomes, a number of patients refractory to prior use of bortezomib, thalidomide, or lenalidomide still only showed poor responses (Kumar et al., 2017). As MM cells have been described to possess an abnormal acetylome, another approach to this group of patients was the implementation of HDACi (Mithraprabhu et al., 2014). In fact, data of preclinical studies demonstrated an anti-proliferative effect of vorinostat, romidepsin, dacinostat and panobinostat resulting in apoptosis of MM cells (Catley et al., 2003; Khan et al., 2004; Campbell et al., 2010; Ocio et al., 2010; Sanchez et al., 2011; Holkova et al., 2012). However, when transferring single agent use of HDACi into clinical trials, only limited effect on MM cells was noted (Richardson et al., 2008; Niesvizky et al., 2011).
Eventually, several preclinical studies postulated synergistic effects of HDACi and proteasomal inhibition, paving the way of combinational therapy of these two agents (Pei et al., 2004; Campbell et al., 2010). The best characterized and coherent explanation of the synergy between PI and HDACi is the dual inhibition of the proteasome and aggresome pathway (Hideshima et al., 2005; Catley et al., 2006). Targeting both of the degradation pathways with bortezomib and HDACi in tumor cells would exponentiate their effect and result in greater accumulation of polyubiquitinated proteins, increased cellular stress and apoptosis. More specifically, despite the fact that proteasome inhibition results in accumulation of ubiquitinated proteins and cell death, malignant cells have shown to evade this life-threatening end by an alternative pathway. Here, malignant cells form aggresomes, engulfing the polyubiquitinated proteins to transport them with the help of HDAC6 via microtubules (Ouyang et al., 2012). Disruption of this alternative pathway was reported using both nonselective and selective HDACi through HDAC6 inhibition thus synergizing with bortezomib and inducing cells to undergo apoptosis in multiple hematologic and epithelial malignancies (Catley et al., 2006; Nawrocki et al., 2006; Heider et al., 2008). In the same vein, the beforementioned observation that pan-HDACi treatment in clinical concentration of human cells led to the formation of amyloid fibrils gave a further proof that HDACi may act synergistically on PQC pathways (Olzscha et al., 2017). On account of the described synergy, dual inhibition could exploit full therapeutic potential of both proteasome and HDAC inhibition.
A hallmark of MM cells is the production of abundant amounts of immunoglobulin which must either be properly folded or degraded. Accordingly, dual disruption in protein degradation seemed to be especially effective in these types of cancer cells (Lee et al., 2003; Obeng et al., 2006). This assumption was substantiated by preclinical data, indicating a combination of PI and HDACi to be an attractive and novel strategy for the treatment of MM (Table 2).
Preliminary data from phase I, II and III studies evaluated success in the treatment regime of panobinostat or vorinostat as HDACi plus bortezomib in patients with relapsed or refractory MM (Badros et al., 2009; San-Miguel et al., 2013). Subsequently, combinational therapy was implemented into clinical settings. Patients with relapsed or refractory MM were examined on the effect of HDACi combined with proteasome inhibition. On the one hand, the phase II VANTAGE trial analyzed vorinostat plus bortezomib (Dimopoulos et al., 2013), whereas on the other hand, the phase II PANORAMA 1 trial tested the combination of panobinostat plus bortezomib and dexamethasone in patients with relapsed and refractory MM (San-Miguel et al., 2011). Despite the fact that VANTAGE displayed prolonged progression-free survival (median PFS of 7.6 vs. 6.8 month) when combining vorinostat and bortezomib, clinical relevance needed to be further examined. The PANORAMA 1 trial was able to show modest overall survival benefit when combinational therapy of panobinostat, bortezomib and dexamethasone was applied (median PFS of 12 vs. 8 month). This led to the approval of panobinostat in 2015 by the FDA. The results of this therapeutic approach were evaluated on bortezomib-refractory patients in the PANORAMA 2 trial (Richardson et al., 2013). According to this trial, it has been proposed that combinational treatment of panobinostat, bortezomib and dexamethasone recaptures response in 34.5% of pre-treated, bortezomib-refractory MM patients. In summary, both results of PANORAMA 1 and 2 are partly coherent with preclinical studies and elucidate considerably the role of panobinostat in combination with bortezomib and dexamethasone, especially in patients with relapsed or bortezomib refractory MM. Furthermore, the results hypothesize HDACi to sensitize patients with bortezomib-resistant MM. Despite the clinical benefit of this combined agent regime, it harbors the danger of poor side effects. Especially the overlapping toxicity profiles make this regime rather toxic. A grade 3 – 4 thrombocytopenia (67%) and gastrointestinal toxicity (diarrhea 25%) indicate a poor safety profile. In order to optimize the safety profile, combinations of panobinostat with second-generation PI were tested at different doses and schedules. Carfilzomib is one example of a second-generation PI and obtained approval by the FDA for the treatment of relapsed and refractory MM, in patients who were given at least two prior therapies (Groen et al., 2019). Combination of panobinostat and carfilzomib was tested in a phase I/II clinical trial in patients with relapsed or refractory MM with promising response rates and an acceptable safety profile (ClinicalTrials.gov identifier: NCT01496118) (Berdeja et al., 2015).
Another idea to address the rather toxic combination of pan-HDACi with PI was to substitute pan-HDACi with class-selective HDACi. In contrast to pan-HDACi, selective class I HDACi rarely induce thrombocytopenia, thus seeming to be more suitable agents. As the key mechanism underlying the synergistic effect of HDACi and PI was mainly explained by HDAC6-dependent aggresome function, it can be argued that class-specific HDACi may still synergize with PI without having a poor safety profile. Therefore, an isoform selective HDAC6 inhibitor, ricolinostat, was introduced in the combinational treatment regime with bortezomib and dexamethasone in relapsed or refractory MM patients (Vogl et al., 2017). This phase I/II study demonstrated that combinational therapy with an isoform selective HDACi shows less severe gastrointestinal, hematologic, and constitutional toxicities in comparison to non-selective HDACi. This raised the idea to test the novel combinational regime in different malignancies. It has been demonstrated that class I HDACi mocetinostat/MGCD0103 has a potent antiproliferative activity in Hodgkin lymphoma (HL) cell lines in an HDAC6-independent manner (Buglio et al., 2010). Regarding these results, mocetinostat was especially interesting to investigate further, as it is a class I HDACi with no effects on HDAC6 (Fournel et al., 2008). The generated data demonstrated that inhibition of class I HDAC by mocetinostat results in an adequate induction of cell death in HL cell lines. On account to that, a broader inhibition of HDACs, including HDAC6, is not needed for a sufficient antiproliferative effect in vitro. Furthermore, they were able to show a synergistic effect of mocetinostat with PI. Mocetinostat induced the expression of various inflammatory cytokines resulting in the activation of NF-κB, which in turn mitigated the killing effect of mocetinostat on tumor cells. As PIs inhibit NF-κB activation, this novel combination would explain how PIs enhance mocetinostat activity, independent of HDAC6. A following phase II trial tested mocetinostat, a class I/IV HDACi for relapsed HL, whereby 85 mg were administered three times per week. 14 of 51 patients (27%) treated with mocetinostat had a complete or partial response whereas only one patient out of 25 (4%) had a partial response on pan-HDACi vorinostat. Single agent use of mocetinostat also induced a reduction in tumor size in more than 4/5 of patients (Younes et al., 2011). Collectively, these data suggest the potential and clinical value of class-specific HDACi in patients with HL. Combination of panobinostat and bortezomib in patients with relapsed or refractory PTCL shows encouraging activity, however displaying a relatively high number of adverse events with 10 out of 25 patients (40%) (Tan et al., 2015).
Noticeably, HDACi and PI have been tested and analyzed in a variety of hematological malignancies. However, their effects are not well investigated in solid tumor malignancies. First attempts in investigating the safety and efficacy of vorinostat and bortezomib were tested 2012 in NSCLC. Here, they examined the two-agent use as induction therapy with an adjacent surgery in patients with NSCLC (Jones et al., 2012). The obtained results showed a decrease in metabolic activity in the tumors. However, due to the short duration of induction treatment, no significant change in tumor size has been observed. A following phase II study testing vorinostat and bortezomib as third-line therapy in patients with advanced NSCLC was terminated at its first temporary analyses due to a lack of anti-tumor activity (Hoang et al., 2014). Nonetheless, they highlighted the relevance of potential biomarkers predicting drug activity and thus driving clinical development.
In another setting, HDACi and PI have been tested in glioblastoma multiforme (GBM) cells, as promising preclinical studies proposed activity against GBM cell lines and glioma models (Eyüpoglu et al., 2005; Ugur et al., 2007; Yin et al., 2007). However, this result was not confirmed in clinical trials. A phase II trial of bortezomib in combination with vorinostat in recurrent glioblastoma had disappointing results and was clinically ineffective (Friday et al., 2012). It should be considered that unlike vorinostat, bortezomib cannot pass an intact blood-brain barrier (BBB) and thus may be the reason for an unsatisfactory result. Since other PIs such as marizomib can cross the BBB, it would eventually be a more beneficial combination with an HDACi in treating tumors beyond the BBB (Gozzetti and Cerase, 2014).
Overall, HDACi in combination with PI showed synergistic effects, which could be validated in several phase I trials in different tumor entities. However, there is still an unmet need of further investigation on molecular mechanisms underlying the combinational treatment regime and especially of the development of predictive biomarkers. These biomarkers (see also section “Evaluating Alterations of PQC Systems: Precision Medicine Upon HDAC Inhibitor Treatment”) would allow clinicians to stratify patients who would benefit from the treatments (Table 2).
HDAC Inhibitors and Modulators of Autophagy
Another approach, going for the same train of thought as seen in the combinational regime of HDACi and PI is the substitution of PI with autophagy inhibition (Table 2). As outlined in section “The Role of Protein Quality Control Systems in Cancer Cells,” autophagy represents a hallmark of the PQC as the proteasome does. Preclinical studies showed a context-dependent effect of autophagy in different states of malignant pathogenesis. As outlined in the section about pre-clinical studies, autophagy has a protective function in premalignant cells. While preventing defective cells from proliferating, it hampers the acquisition of additional mutations that would even promote tumor development. However, looking at advanced cancer cells, autophagy can promote mechanism for oncogenesis. Here, it enhances cell survival under stressful conditions in the tumor microenvironment, such as hypoxia and nutrient deprivation (White, 2012). Besides the already existing endogenous stress, autophagy is even further promoted by anti-cancer treatment, leading to additional protection of tumor development (Janku et al., 2011). This could give a rationale for the poor therapeutic efficacy, as the survival of malignant cells is maintained through autophagy (Carew et al., 2007a, 2012; Amaravadi et al., 2011). Recent investigations confirm the diminished effect of therapies due to autophagy, promoting cancer cell survival (Strait et al., 2002; Duan et al., 2005). Thus, inhibition of autophagy represents a novel strategy to augment cancer treatment efficacy.
HDACi have been described to induce autophagy in many clinical trials, but its full therapeutic potential is hampered due to the protective action of autophagy. Subsequently, disruption of autophagy would boost the pro-apoptotic and cytostatic effects of HDACi. In fact, this mode of action was confirmed by preclinical studies in models of imatinib-resistant chronic myeloid leukemia and colon cancer (Carew et al., 2007b, 2010). In 2014, Mahalingam et al. reported about a phase I clinical trial of the autophagy inhibitor hydroxychloroquine (HCQ) in combination with vorinostat in adult patients with advanced refractory solid malignancies (Mahalingam et al., 2014). In the majority of patients, no significant benefit was observed; only renal cell carcinoma patients had a dramatic and durable response to the novel combination of vorinostat plus hydroxychloroquine (HCQ). Besides the deficient results, the authors accentuated the need of predictive biomarker for assessing clinical sensitivity to autophagy inhibitors in order to optimize drug development. In 2016, Patel et al. revived the scheme and performed a single-arm expansion cohort to assess the efficacy, safety and effects on immunity of vorinostat and HCQ in patients with refractory metastatic CRC (Patel et al., 2016). Results implied no substantial benefit over other oral drug as survival showed to be comparable to other oral drugs for refractory CRC including regorafenib (Grothey et al., 2013). Despite this outcome, vorinostat plus HCQ had a favorable toxicity profile and can be discussed as an alternative treatment for refractory CRC. Subsequently, a randomized phase II trial of vorinostat and HCQ versus regorafenib (a tyrosinkinase inhibitor) is now open to enrollment. Alongside, another phase II trial exhibits the therapeutic benefit on vorinostat plus HCQ over regorafenib in chemo-refractory metastatic colorectal cancer. Results implied that survival of the two treatment regimens showed comparable survival (Arora et al., 2019).
HDAC Inhibitors and HSP90 Inhibitors
Another interesting new idea was to combine HDACi with modulators of molecular chaperones. As outlined in section “The Role of Protein Quality Control Systems in Cancer Cells,” molecular chaperones and particularly HSP90 and HSP70 play not only a role in protein folding, but also in signal transduction and interact in several pathways to maintain cellular protein homeostasis and cell survival (Wiech et al., 1992). HSP90 has been described as a key player to stabilize proteins that are particularly important in cancer cells including BCR-ABL (BCR, breakpoint cluster region; ABL, Abelson), ERB-B2 (erythroblastic oncogene B), proto-oncogene B-Raf (BRAF), AKT Serine/Threonine Kinase (AKT), vascular endothelial growth factor receptor (VEGFR), FMS like tyrosine kinase 3 (FLT3), androgen and estrogen receptors, hypoxia-inducible factor (HIF-1α) and a constantly growing list, affecting a variety of cancer-related functions (Welch and Feramisco, 1982; Powers and Workman, 2006). In some cancerous cell lines, HSP90 has been found in much higher levels compared to normal cells (Kamal et al., 2003) and shows beneficial effects on many oncoproteins (da Rocha Dias et al., 2005; Shimamura et al., 2005). Preclinical studies demonstrated the anti-cancer effect of HSP90 inhibition and suggested its ability could affect several oncogenic signaling pathways simultaneously. Thus, it reduces the likelihood of the tumor acquiring resistance to any single therapeutic pathway and is a major advantage upon other agents (Banerji, 2009). However, HSP90 inhibitors such as 17-AAG have not reached clinical trials beyond phase III, due to minimal effects and toxicity, especially liver toxicity (Hyun et al., 2018). Therefore, the combination with other agents, for instance HDACi, could be a promising alternative, in order to reduce the effective concentration of HSP90 inhibitors. Following this hypothesis, HDAC6 and HSP90 are interactors as HDAC6 can deacetylate HSP90 (Boyault et al., 2007a; Figure 3). Adversely, when inhibiting HDAC6, HSP90 is present in a hyper-acetylated state, losing the bond with the co-chaperone p23 and finally its overall chaperone activity (Kovacs et al., 2005; Aldana-Masangkay and Sakamoto, 2011). This makes the synergistic use a promising anti-cancer treatment strategy (Table 2). Lung cancer has been described to be particularly susceptible to HSP90 inhibition. Overexpressed or mutant ERB-B2 and BRAF are often the driving force in lung cancer development. Interestingly, all of them are degraded with the assistance of HSP90, giving a rationale for the beneficial use of HSP90 inhibitors in lung cancer (Shimamura and Shapiro, 2008; Zismanov et al., 2014). This is now implemented into an ongoing clinical trial with 20 participants aiming to investigate the combined effect of HSP90 inhibitor and PI or HDACi on lung cancer cell fate and ER/Golgi homeostasis (NCT01270399).
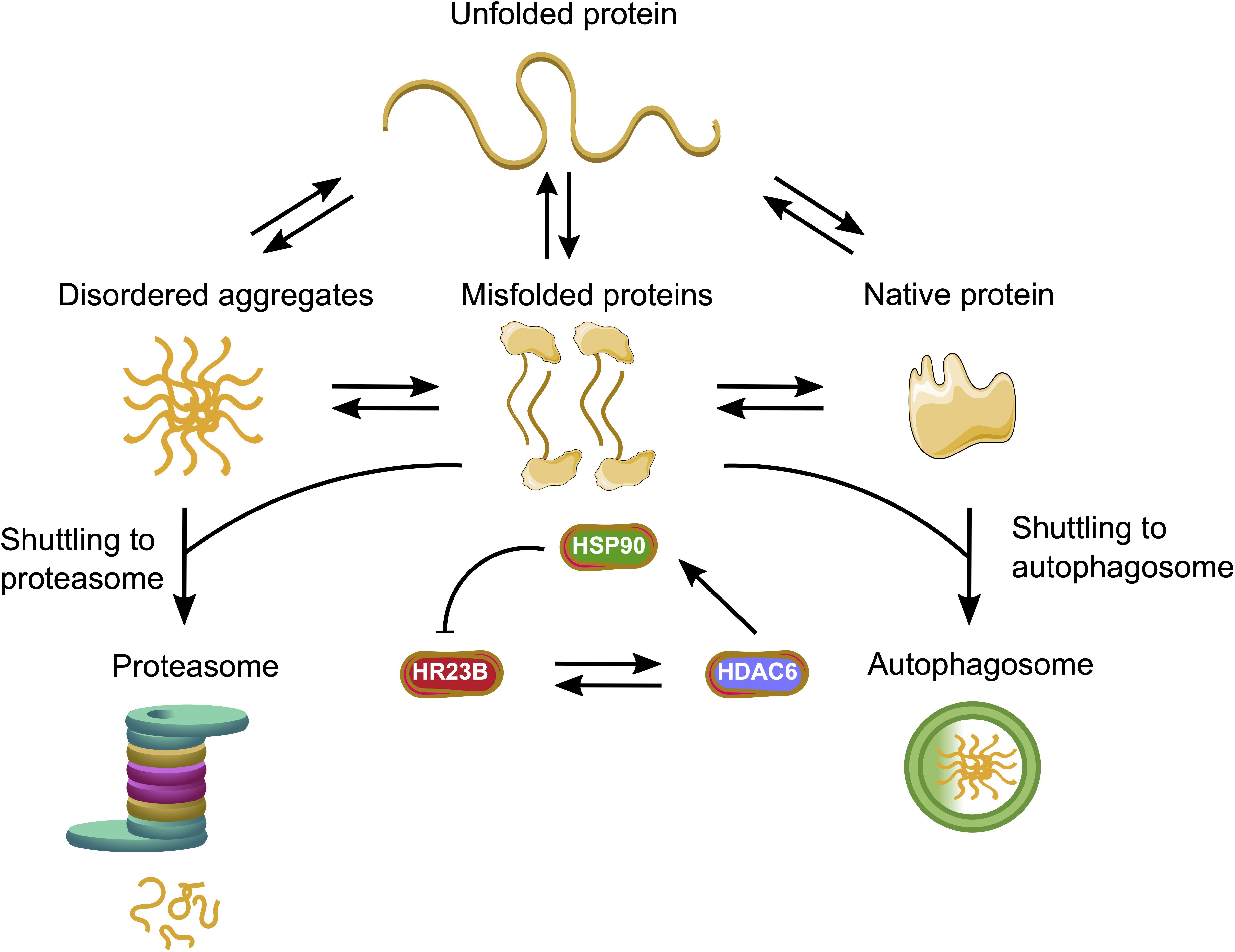
Figure 3. HDACs facilitate the interplay between the major protein quality control systems. HR23B as a proteasome shuttling factor can enhance the proteolysis of ubiquitinated proteins, whereas HDAC6 enables the autophagy process. HDAC6 can either bind HR23B with its BUZ domain or acetylate the molecular chaperone HSP90. The acetylated HSP90 can interact with HR23B and initiate a feedback loop.
HDAC Inhibitors and Bromodomain Inhibitors
This novel combination treatment has not been well investigated in a clinical set-up to this point. Preclinical studies proposed synergistic effects in breast cancer (Rahmani et al., 2003). One phase I trial tested GSK525762C (molibresib besylate) and entinostat in patients with advanced or refractory solid tumors or lymphomas. Results are yet to come.
Overall, epigenetics has become an inevitable part of cancer research. Until now, HDACi have been shown and tested to synergize with a wide range of very different agents. Of particular interest is the synergistic use of HDACi with the inhibition of PQC systems. Herein, progress has been made, implicating that HDACi exhibit their anticancer activity through a multitude of pathways. However, there is still an unmet need for further investigations on detailed mechanistic action of HDACi. The therapeutic effect of HDACi not only depends on the cancer type, but on the stage of cancer, treatment dosage, the individual patient’s biological signature, and other factors. In order to boost the development of HDACi and PQC modulating agents these factors need to be considered. To achieve significant improvement in HDACi therapeutic outcomes, better patient selection and monitoring of biomarkers are strongly required.
Evaluating Alterations of PQC Systems: Precision Medicine Upon HDAC Inhibitor Treatment
The ability to cost-effectively sequence the human genome and epigenome to apply genetics in drug treatment has changed the approach of cancer treatment in a fundamental way and led to a revolution from “one-size-fits-all” therapy to a more precise therapy approach. It enabled to look at a patient as an individual comprising of a unique set of genes, proteins and environment and mainly formed the term of precision medicine (Figure 4). Here “the specific targeting of molecular abnormalities and the stratification of patients who respond to specific drugs” is in focus (Coyle et al., 2017). This personalized approach to stratify a patient group has attracted attention especially in cancer, where specific information about a patient’s tumor helps diagnose, treatment planning and making of prognoses. Thus, it seemed to be the next logical step in advanced cancer treatment. Alongside with our evolving knowledge about oncogenesis, cancer therapy must also be accompanied by a molecular understanding of both, genetic and epigenetic factors in cancer patients (Hanahan and Weinberg, 2000). Therefore, it is not only important to screen a set of patients’ genes, but it is equally important to predict whether this unique tumor is sensitive to the treatment regime applied. Therefore, two pillars are of importance while developing precision medicine: (1) the individual genome, epigenome, mutations in the cancer and (2) the tumor entity, molecular features and clinical response to cancer therapies (Figure 4). Alongside with DNA sequencing, looking at a patient’s epigenome reinforced the development of precision medicine once again and helped to evaluate new biomarkers. Biomarkers represent a hallmark of precision medicine as they give information on the clinical response to cancer therapies. Recapitulating the development of biomarkers, the term is a portmanteau of “biological marker” that encompasses a wide range of different medical signs. The National Institutes of Health (NIH) biomarkers Definitions Working Group defined a biomarker as “a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions. Molecular, histologic, radiographic, or physiologic characteristics are types of biomarkers, but a biomarker is not an assessment of how an individual feels, functions, or survives.” (Biomarkers Definitions Working Group, 2001).
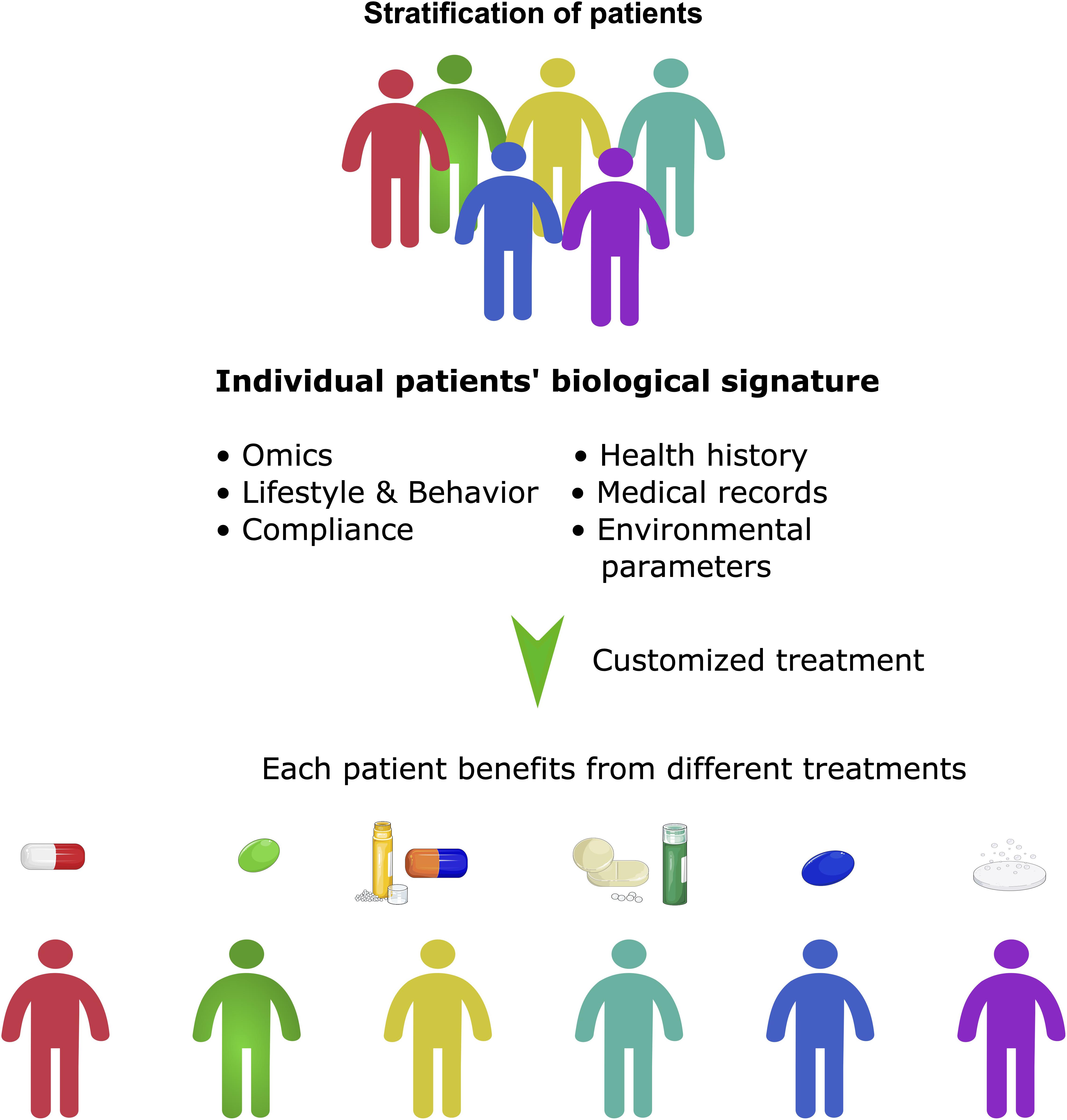
Figure 4. Precision medicine beyond stratification of patients. Precision medicine can be seen as a more holistic approach than personalized medicine. It takes many factors into account, not only the stratification of patients, but also their molecular signature, social and environmental factors and lifestyle. It often involves the application of pan-omic analyses and systems biology to determine the cause of an individual patient’s disease at the molecular level and then to utilize different targeted treatments.
Nowadays a wide range of biomarkers and their informative value have been defined and a glossary of terms and definitions has been developed by the FDA-NIH Biomarker Working Group called BEST (Biomarkers, Endpoints, and other Tools). A predictive biomarker has been defined as “a biomarker used to identify individuals who are more likely than similar individuals without the biomarker to experience a favorable or unfavorable effect from exposure to a medical product or an environmental agent.” On the other hand, a prognostic biomarker has been described as “a biomarker used to identify likelihood of a clinical event, disease recurrence or progression in patients who have the disease or medical condition of interest” (Cagney et al., 2018). In order to clarify the distinction between those two forms of biomarkers, a prognostic biomarker states distinct disease outcome, whereas predictive biomarkers differentiate between patients who will react or not react to the therapy (Califf, 2018). As described in section “HDACS in Epigenetics and Protein Quality Control Systems” and “Clinical Studies: HDAC Inhibitors in Combination With Modifiers of Protein Quality Control,” epigenetic approaches in cancer treatment and especially HDACi have been implemented and tested in several preclinical and clinical trials. However, open questions on how HDACi function and operate antitumor activity, especially the pathways that are directly linked to HDAC inhibition and tumor cell proliferation, remain to be answered. Due to the paucity of information on HDACi function, it is of importance to detect biomarkers determining the accessibility of tumors during the HDACi treatment regime.
A viable biomarker could detect tumor types that are likely to undergo a favorable clinical response under HDACi therapy. Indeed, the proteasome shuttling factor HR23B seems to play a key role in HDACi-induced apoptosis. In order to identify genes that have an impact on the sensitivity of tumor cells to HDACi, a genome-wide loss-of-function screen was performed. Results revealed not only the role of the UPS in HDACi-induced apoptosis but also the potential of HR23B as a possible biomarker. It has been demonstrated in cells treated with pan-HDACi that HR23B is a sensitivity determinant for HDACi. Therefore, the hypothesis has been proposed that HR23B could function as a biomarker in order to identify tumors that would react favorably to HDACi (Fotheringham et al., 2009). CTCL patients who were treated with vorinostat, showed a positive correlation between HR23B expression levels and therapeutic response. It is therefore likely that HR23B can serve as a predictive biomarker for identifying CTCL patients that respond favorably to HDACi (Khan et al., 2010). The role of HR23B in regulating the biological outcome of treatment with HDACi was then further investigated. Two correlated effects of HR23B in HDACi treated cells were shown: autophagy and apoptosis. While high levels of HR23B cause cells treated with HDACi to undergo apoptosis, low levels of HR23B expression were associated with autophagy (Figure 3). Thus, it was proven that HR23B impacts on the therapy efficacy, as it regulates the switch between apoptosis and autophagy (New et al., 2013). In summary, HR23B represents a promising predictive biomarker and patients with high levels of HR23B would be stratified into a sub-group that would benefit from HDACi therapy, for instance in the PLACARD-trial (NCT03873025) for relapsed or refractory diffuse large B-cell lymphoma (see also section “Novel Strategies of HDAC Inhibitors Affecting Protein Quality Control Pathways”).
Since HDACs have been described to play a role in tumorigenesis, it seems reasonable to measure levels of HDAC enzymes and predict responsive tumor types. It has been demonstrated that specific HDAC isoform expression could be a predictive biomarker. In this study, the influence on knockdown of HDAC1, 2 and 3 isoforms in human cancer cell lines, treated with two unrelated HDACi (belinostat and VPA) have been analyzed. While knockdown of HDAC1 resulted in an increased resistance to belinostat in HeLa cells, no influence was seen in response to either HDAC2 or 3 knockdowns or under VPA treatment. These data suggest that HDAC1 knockdown reduces sensitivity to the HDACi belinostat and in turn high levels of HDAC1 correlate with sensitivity to belinostat treatment (Dejligbjerg et al., 2008). According to these observations, stratification of patients due to their HDAC expression pattern was suggested in colon cancer cell culture models. The specific characterization of class I HDAC isoforms might allow the prediction of individual patient’s prognosis. They observed a negative correlation between HDAC2 expression level and reduced patient survival in patients with CRC (Weichert et al., 2008c). Therefore, they point out how evaluation of HDAC expression profiles would benefit selecting patient populations before HDACi treatment. Similar conclusions were made looking at the class I HDAC expression levels in prostate carcinomas. 192 prostate carcinomas were analyzed using immunohistochemistry and put into subjection to pathological parameters. Again, high expression of HDAC1 and HDAC2 correlated with tumor de-differentiation (Weichert et al., 2008b).
Until now, CTCL represents the malignancy most sensitive to HDACi treatment. Therefore, it is of interest to analyze HDAC profiles in patients suffering from CTCL. In fact, taken 73 CTCL biopsies and analyzing HDAC1, HDAC2, HDAC6, and histone H4 acetylation demonstrated that especially HDAC6 expression correlates with a favorable outcome in CTCL (Marquard et al., 2008). Further studies on HDAC expression levels were performed and implied that depending on the specific tumor entity, different HDAC expression profiles can be observed (Sasaki et al., 2004; Zhang et al., 2004). As the proteome of different tumors can be modified by different HDACs, a comprehensive analysis that could be implemented into clinical routines is desirable. Biomarkers that can be easily detected in peripheral blood mononuclear cells are H3 and H4. They reflect histone acetylation as they are directly modified and regulated by HDACs. Preclinical studies showed a time- and concentration-dependent correlation when histone acetylation is inhibited by HDACi on H3 and H4 (Plumb et al., 2003). However, histone acetylation should only be seen as a surrogate for HDAC inhibition as it does not have the diagnostic value to reflect tumor response. A phase I trial demonstrated the limited validity of H4 measurement. Here, belinostat/PXD101 was used and histone acetylation showed to return to the initial levels within a period of 2 h after drug infusion and displayed to plateau at the maximum tolerated dose (Steele et al., 2008).
As HDACi also intervene in transcriptional regulation, a gene set analysis could give information on HDACi response. Indeed, molecular profiling has been shown to be value in predicting sensitivity during HDACi therapy. In one study, nine genes were tested and identified in NSCLC cell lines under vorinostat and trichostatin A treatment. Three genes were highly associated with drug activity: NAD(P)H quinone dehydrogenase 1 (NQO1), sec homolog A (SEC23A), and proteasome activator subunit 2 (PSME2) (Miyanaga et al., 2008). Further investigations need to be done in order to confirm this nine-gene signature in predicting drug sensitivity. A similar study investigated genes regulated by panobinostat in CTCL patients. In time intervals of 0, 4, 8, and 24 h after panobinostat administration, microarray gene expression profiling was realized. Over time, separate unique gene profiles were reported and 23 genes showed statistical significance. Out of these 23 genes, 4 genes were particularly interesting: guanylate cyclase 1A3 (GUCY1A3), endothelial Tie2/tek ligands angiopoietin-1 (ANGPT1), both associated with angiogenesis and two cell cycle progression genes, transcription factor COUP-TFII (NR2F2) and CCND1 (Ellis et al., 2008). However, a larger study should be executed in order to make a valid statement, thereby also concentrating on cyclin D1 (CCND1), as it is known to be commonly down-regulated by various HDACi (Johnstone, 2002). One challenge to overcome is that due to the wide activity profile of HDACs, gene signatures are likely to vary tremendously depending on tumor type, inhibitor type and concentration. In addition, genes having a prognostic value would be more reasonable to identify than ones that have a response signature.
The future of precision medicine in cancer treatment is highly exciting and promising, although many challenges remain to be solved. Especially biomarkers gain increased attention in order to stratify patients and tumors into sub-groups that are sensitive to HDACi treatment and monitor targeted modulations.
Conclusion
Over the last decade, we have seen that our scientific knowledge of epigenetics and in particular of HDAC inhibition has been translated into clinical benefit for cancer patients. Epigenetic therapy, such as HDACi, has provided a proof-of-concept of their clinical efficacy. However, one obvious observation made with HDACi and with other epigenetic modifiers such as hypomethylating agents including the DNA methyltransferase inhibitors azacytidine (AZA) or decitabine is that they prove clinical efficacy as single agents in hematological malignancies rather than in solid tumors (Sigalotti et al., 2007). The reasons for this observed discrepancy between hematological malignancies and solid malignancies is still unclear, since many preclinical studies of HDACi alone or in combination with different anti-tumor agents demonstrated their potential to inhibit cell proliferation or even induce apoptosis. One may speculate that the general high cell proliferation in hematological cells foster the effect of epigenetic drugs.
With these observations in mind, combination therapies of HDACi with drugs acting on PQC mechanisms seem to be rational, if cells with a high protein turnover are targeted. A paradigm for this concept is the combination therapy with HDACi and proteasome inhibitors. As outlined in the section “HDAC Inhibitors and Proteasome Inhibitors,” this strategy is proven for MM and is currently tested in various studies for different tumor types and drugs. Our prediction would be that this concept will be successful for those tumor types where a high protein turnover can be observed. This is not only the case in MM with the biogenesis of antibodies, but different cell types excreting proteins such as hepatocytes and cells of the intestine may be more susceptible to drugs acting on PQC systems. Indeed, it has been demonstrated that the half-life of indispensable scaffold proteins in hepatocytes is not necessarily shorter than in highly dividing monocytes (Mathieson et al., 2018). Interestingly, proteins of the UPS are highly abundant in the nucleus. This may reflect the fact that the nucleus is especially vulnerable to imbalances in proteostasis, on the other hand, they may be involved in epigenetic phenomena as well as in DNA damage repair. For instance, the before-mentioned proteasomal shuttling factor HR23B, which plays a role as a potential biomarker in HDACi therapy, is known to mediate DNA damage response (Sugasawa et al., 1998). Usually, transcription factors are rapidly turned over and disturbances in the nuclear UPS will result in differential gene expression, similar to an epigenetic outcome.
As described in sections “HDAC Inhibitors and Bromodomain Inhibitors” and “Novel Strategies of HDAC Inhibitors Affecting Protein Quality Control Pathways,” it has been demonstrated that treatment of cells with clinical concentration of HDACi leads to protein misfolding and aggregation (Olzscha et al., 2017). One could assume that inhibition of molecular chaperones may therefore increase the cytotoxic effects of HDACi. Indeed, some clinical studies provided evidence that this combination therapy may be beneficial for patients with solid tumors, for instance the combination therapy of the HDACi belinostat with the HSP90 inhibitor 17-N-Allylamino-17-demethoxygeldanamycin (17-AAG) (NCT00354185). In order to understand the mechanisms of action, more research is necessary to elucidate the influence of aberrant PTMs on protein folding as well as deciphering which and how molecular chaperones could recognize them.
One of the future challenges will be to identify different tumor types and choose the right therapy for the right patient at the right time. The concept of precision medicine aims not only for stratification of patients; it ensures that each patient benefits from individualized treatment. In order to achieve that, companion diagnostic biomarkers, molecular signatures obtained by genomic and proteomic profiling as well as the health history of the patient should be taken into account (Figure 4). However, a prerequisite for this tailored therapy is a deeper knowledge about the involved pathways of the known HDACs in H. sapiens and the involved mechanisms of proteostasis. More basic research is needed to understand how the changing acetylome landscape affects PQC upon HDACi treatment.
Author Contributions
LK, P-VF, DP, and HO wrote the manuscript. DP created the figures. HO designed the concept and supervised the work.
Funding
LK and HO were supported by the DFG, RTK 2155 (ProMoAge) and P-VF by the Wilhelm Roux Program HaPKoM (FKZ PK28).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge Rüdiger Horstkorte and Thorsten Pfirrmann for critically reading the manuscript.
Abbreviations
ABL, Abelson leukemia; AD, Alzheimer’s disease; AKT, AKT Serine/Threonine Kinase; ALS, amyotrophic lateral sclerosis; AML, acute myeloid leukemia; ANGPT1, endothelial Tie2/tek ligands angiopoietin-1; Apaf-1, apoptotic protease activating factor 1; ATAT1, alpha-tubulin N-acetyltransferase; ATF6, activating transcription factor 6; ATG, autophagy-related genes; ATP, adenosine triphosphate; BBB, blood-brain barrier; Bcl-2, B-cell lymphoma 2; BCR-ABL, breakpoint cluster region protein Abelson murine leukemia viral oncogene homolog; BET, bromodomain and extra-terminal domain; BiP, binding immunoglobulin protein; BNIP3, Bcl-2/adenovirus E1B 19 kDa protein-interacting protein 3; BCR, breakpoint cluster region; BRAF, proto-oncogene B-Raf; BRD, bromodomain; cAMP, cyclic adenosine monophosphate; CBP, CREB binding protein; CCND1, cyclin D1; CDK, cyclin-dependent kinase; CHIP, carboxy terminus of heat shock protein 70-interacting protein; CMA, chaperone-mediated autophagy; c-Met, tyrosine-protein kinase Met; CRC, colorectal cancer; CREB, cAMP response element-binding protein; CRL, cullin-ring E3 ligases; CTCL, cutaneous T cell lymphoma; DNA, deoxyribonucleic acid; ER, endoplasmic reticulum; ERB-B2, Erb-B2 receptor tyrosine kinase 2; ETO, eight-twenty-one; FDA, U.S Food and Drug Administration; FLT3, FMS like tyrosine kinase 3; GBM, glioblastoma; GRP78, Glucose-regulated protein 78; GUCY1A3, guanylate cyclase 1A3; H3, histone H3; H4, histone H4; HCQ, hydroxychloroquine; HDA1, histone deacetylase 1; HDAC, histone deacetylase; HDACi, histone deacetylase inhibitor; HL, Hodgkin lymphoma; HR, homologous recombination; HR23B, nucleotide excision repair protein homolog B; HSP, heat shock protein; HSP70, heat shock protein 70; HSP90, heat shock protein 90 hypoxia-inducible factor (HIF)-1 α; IRE1, inositol-requiring enzyme 1; IRF4, interferon regulatory factor 4; Ki, inhibitory constant; MDM2, mouse double minute 2; MM, multiple myeloma; mTOR, mammalian target of rapamycin; MYC, myelocytomatosis; NADPH, nicotinamide adenine dinucleotide phosphate; NAE, NEDD8-activating enzymes; NEDD8, neural-precursor-cell-expressed developmentally down-regulated 8; NF- κ B, nuclear factor “kappa-light-chain-enhancer” of activated B-cells; NHEJ, non-homologous end-joining recombination; NIH, National Institutes of Health; Nox4, NADPH oxidase 4; NQO1, NAD(P)H quinone dehydrogenase 1; NR2F2, nuclear receptor subfamily 2 group F member 2; NSCLC, non-small-cell lung cancer; NUT, nuclear protein in testis; PERK, the protein kinase RNA-like endoplasmic reticulum kinase; PFS, progression-free survival; PI, proteasome inhibitor; PQC, protein quality control; PSME2, proteasome activator subunit 2; PTCL, peripheral T-cell lymphoma; PTM, posttranslational modification; RING, really interesting new gene; SAHA, suberoylanilide hydroxamic acid; SEC23A, sec homolog A; COPII, coat complex component; sHSP, small heat shock protein; SOD1, superoxide dismutase 1; SP1, specific protein 2; SUMO, small ubiquitin like modifier; UPR, unfolded protein response; UPS, ubiquitin proteasome system; TSA, trichostatin A; VEGFR, vascular endothelial growth factor receptor; VPA, valproic acid.
References
Abaza, M. S. I. (2010). Augmentation of the anticancer effects of proteasome inhibitors by combination with sodium butyrate in human colorectal cancer cells. Exp. Therap. Med. 1, 675–693. doi: 10.3892/etm_00000106
Adams, J. (2004). The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer 4, 349–360. doi: 10.1038/nrc1361
Aldana-Masangkay, G. I., and Sakamoto, K. M. (2011). The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011:875824. doi: 10.1155/2011/875824
Alqahtani, A., Choucair, K., Ashraf, M., Hammouda, D. M., Alloghbi, A., Khan, T., et al. (2019). Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future Sci. OA 5:FSO372. doi: 10.4155/fsoa-2018-0115
Amaravadi, R. K., Lippincott-Schwartz, J., Yin, X.-M., Weiss, W. A., Takebe, N., Timmer, W., et al. (2011). Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 17, 654–666. doi: 10.1158/1078-0432.CCR-10-2634
Amemiya, S., Yamaguchi, T., Hashimoto, Y., and Noguchi-Yachide, T. (2017). Synthesis and evaluation of novel dual BRD4/HDAC inhibitors. Bioorg. Med. Chem. 25, 3677–3684. doi: 10.1016/j.bmc.2017.04.043
An, B., Goldfarb, R. H., Siman, R., and Dou, Q. P. (1998). Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 5, 1062–1075. doi: 10.1038/sj.cdd.4400436
Annunziato, A. T., and Hansen, J. C. (2018). Role of histone acetylation in the assembly and modulation of chromatin structures. Gene Expres. 9, 37–61. doi: 10.3727/000000001783992687
Arora, S. P., Tenner, L. L., Sarantopoulos, J., Morris, J. L., Longoria, L., Liu, Q., et al. (2019). Modulation of autophagy: a phase II study of vorinostat (VOR) plus hydroxychloroquine (HCQ) vs regorafenib (RGF) in chemo-refractory metastatic colorectal cancer (mCRC). J. Clin. Oncol. 37, 3551–3551. doi: 10.1200/JCO.2019.37.15_suppl.3551
Arrowsmith, C. H., Bountra, C., Fish, P. V., Lee, K., and Schapira, M. (2012). Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 11, 384–400. doi: 10.1038/nrd3674
Atkinson, S. J., Soden, P. E., Angell, D. C., Bantscheff, M., Chung, C. W., Giblin, K. A., et al. (2014). The structure based design of dual HDAC/BET inhibitors as novel epigenetic probes. Medchemcomm 5, 342–351. doi: 10.1039/c3md00285c
Badros, A., Burger, A. M., Philip, S., Niesvizky, R., Kolla, S. S., Goloubeva, O., et al. (2009). Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer Res. 15, 5250–5257. doi: 10.1158/1078-0432.Ccr-08-2850
Ban, H. S., Han, T.-S., Hur, K., and Cho, H.-S. (2019). Epigenetic alterations of heat shock proteins (HSPs) in Cancer. Intern. J. Mol. Sci. 20:758. doi: 10.3390/ijms20194758
Banerji, U. (2009). Heat shock protein 90 as a drug target: some like it hot. Clin. Cancer Res. 15:9. doi: 10.1158/1078-0432.CCR-08-0132
Baylin, S. B., and Ohm, J. E. (2006). Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 6, 107–116. doi: 10.1038/nrc1799
Berdeja, J. G., Hart, L. L., Mace, J. R., Arrowsmith, E. R., Essell, J. H., Owera, R. S., et al. (2015). Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica 100, 670–676. doi: 10.3324/haematol.2014.119735
Berger, S. L., Kouzarides, T., Shiekhattar, R., and Shilatifard, A. (2009). An operational definition of epigenetics. Genes Dev. 23, 781–783. doi: 10.1101/gad.1787609
Bhutia, S. K., Mukhopadhyay, S., Sinha, N., Das, D. N., Panda, P. K., Patra, S. K., et al. (2013). Autophagy: cancer’s friend or foe? Adv. Cancer Res. 118, 61–95. doi: 10.1016/B978-0-12-407173-5.00003-0
Biomarkers Definitions Working Group (2001). Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin. Pharmacol. Therap. 69, 89–95. doi: 10.1067/mcp.2001.113989
Bolden, J. E., Peart, M. J., and Johnstone, R. W. (2006). Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 5, 769–784. doi: 10.1038/nrd2133
Booth, L., Roberts, J. L., Sander, C., Lee, J., Kirkwood, J. M., Poklepovic, A., et al. (2017). The HDAC inhibitor AR42 interacts with pazopanib to kill trametinib/dabrafenib-resistant melanoma cells in vitro and in vivo. Oncotarget 8, 16367–16386. doi: 10.18632/oncotarget.14829
Borbely, G., Haldosen, L.-A., Dahlman-Wright, K., and Zhao, C. (2015). Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 6, 33623–33635. doi: 10.18632/oncotarget.5601
Boutouja, F., Brinkmeier, R., Mastalski, T., El Magraoui, F., and Platta, H. W. (2017). Regulation of the tumor-suppressor BECLIN 1 by distinct ubiquitination cascades. Intern. J. Mol. Sci. 18:541. doi: 10.3390/ijms18122541
Boyault, C., Sadoul, K., Pabion, M., and Khochbin, S. (2007a). HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 26, 5468–5476. doi: 10.1038/sj.onc.1210614
Boyault, C., Zhang, Y., Fritah, S., Caron, C., Gilquin, B., Kwon, S. H., et al. (2007b). HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 21, 2172–2181. doi: 10.1101/gad.436407
Braiteh, F., Soriano, A. O., Garcia-Manero, G., Hong, D., Johnson, M. M., Silva, L. D. P., et al. (2008). Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin. Cancer Res. 14, 6296–6301. doi: 10.1158/1078-0432.CCR-08-1247
Buglio, D., Mamidipudi, V., Khaskhely, N. M., Brady, H., Heise, C., Besterman, J., et al. (2010). The class-I HDAC inhibitor MGCD0103 induces apoptosis in Hodgkin lymphoma cell lines and synergizes with proteasome inhibitors by an HDAC6-independent mechanism. Br. J. Haematol. 151, 387–396. doi: 10.1111/j.1365-2141.2010.08342.x
Cagney, D. N., Sul, J., Huang, R. Y., Ligon, K. L., Wen, P. Y., and Alexander, B. M. (2018). The FDA NIH biomarkers, endpointS, and other tools (BEST) resource in neuro-oncology. Neuro. Oncol. 20, 1162–1172. doi: 10.1093/neuonc/nox242
Califf, R. M. (2018). Biomarker definitions and their applications. Exp. Biol. Med. 243, 213–221. doi: 10.1177/1535370217750088
Campbell, R. A., Sanchez, E., Steinberg, J., Shalitin, D., Li, Z.-W., Chen, H., et al. (2010). Vorinostat enhances the antimyeloma effects of melphalan and bortezomib. Eur. J. Haematol. 84, 201–211. doi: 10.1111/j.1600-0609.2009.01384.x
Cantor, J. R., and Sabatini, D. M. (2012). Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898. doi: 10.1158/2159-8290.CD-12-0345
Carew, J. S., Kelly, K. R., and Nawrocki, S. T. (2012). Autophagy as a target for cancer therapy: new developments. Cancer Manag. Res. 4, 357–365. doi: 10.2147/CMAR.S26133
Carew, J. S., Medina, E. C., Esquivel, J. A. II, Mahalingam, D., Swords, R., Kelly, K., et al. (2010). Autophagy inhibition enhances vorinostat-induced apoptosis via ubiquitinated protein accumulation. J. Cell. Mol. Med. 14, 2448–2459. doi: 10.1111/j.1582-4934.2009.00832.x
Carew, J. S., Nawrocki, S. T., and Cleveland, J. L. (2007a). Modulating autophagy for therapeutic benefit. Autophagy 3, 464–467. doi: 10.4161/auto.4311
Carew, J. S., Nawrocki, S. T., Kahue, C. N., Zhang, H., Yang, C., Chung, L., et al. (2007b). Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 110, 313–322. doi: 10.1182/blood-2006-10-050260
Caron, C., Boyault, C., and Khochbin, S. (2005). Regulatory cross-talk between lysine acetylation and ubiquitination: role in the control of protein stability. Bioessays 27, 408–415. doi: 10.1002/bies.20210
Cashen, A., Juckett, M., Jumonville, A., Litzow, M., Flynn, P. J., Eckardt, J., et al. (2012). Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann. Hematol. 91, 33–38. doi: 10.1007/s00277-011-1240-1
Catley, L., Weisberg, E., Kiziltepe, T., Tai, Y. T., Hideshima, T., Neri, P., et al. (2006). Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood 108, 3441–3449. doi: 10.1182/blood-2006-04-016055
Catley, L., Weisberg, E., Tai, Y. T., Atadja, P., Remiszewski, S., Hideshima, T., et al. (2003). NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood 102, 2615–2622. doi: 10.1182/blood-2003-01-0233
Chaidos, A., Caputo, V., Gouvedenou, K., Liu, B., Marigo, I., Chaudhry, M. S., et al. (2014). Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood 123, 697–705. doi: 10.1182/blood-2013-01-478420
Chen, B., Retzlaff, M., Roos, T., and Frydman, J. (2011). Cellular strategies of protein quality control. Cold Spring Harb. Perspect. Biol. 3:a004374. doi: 10.1101/cshperspect.a004374
Chen, T., Sun, H., Lu, J., Zhao, Y., Tao, D., Li, X., et al. (2002). Histone acetylation is involved in hsp70 gene transcription regulation in Drosophila melanogaster. Archiv. Biochem. Biophys. 408, 171–176. doi: 10.1016/s0003-9861(02)00564-7
Cheng, Z., Gong, Y., Ma, Y., Lu, K., Lu, X., Pierce, L. A., et al. (2013). Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 19, 1748–1759. doi: 10.1158/1078-0432.CCR-12-3066
Chiu, H.-W., Yeh, Y.-L., Wang, Y.-C., Huang, W.-J., Ho, S.-Y., Lin, P., et al. (2016). Combination of the novel histone deacetylase inhibitor YCW1 and radiation induces autophagic cell death through the downregulation of BNIP3 in triple-negative breast cancer cells in vitro and in an orthotopic mouse model. Mol. Cancer 15:46. doi: 10.1186/s12943-016-0531-5
Choi, J. H., Kwon, H. J., Yoon, B. I., Kim, J. H., Han, S. U., Joo, H. J., et al. (2001). Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn. J. Cancer Res. 92, 1300–1304. doi: 10.1111/j.1349-7006.2001.tb02153.x
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
Chun, Y., and Kim, J. (2018). Autophagy: an essential degradation program for cellular homeostasis and life. Cells 7:278. doi: 10.3390/cells7120278
Cloutier, P., and Coulombe, B. (2013). Regulation of molecular chaperones through post-translational modifications: decrypting the chaperone code. Biochim. Biophys. Acta 1829, 443–454. doi: 10.1016/j.bbagrm.2013.02.010
Cohen, T. J., Guo, J. L., Hurtado, D. E., Kwong, L. K., Mills, I. P., Trojanowski, J. Q., et al. (2011). The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2:252. doi: 10.1038/ncomms1255
Coiffier, B., Pro, B., Prince, H. M., Foss, F., Sokol, L., Greenwood, M., et al. (2012). Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 30, 631–636. doi: 10.1200/JCO.2011.37.4223
Condelli, V., Crispo, F., Pietrafesa, M., Lettini, G., Matassa, D. S., Esposito, F., et al. (2019). HSP90 molecular chaperones, metabolic rewiring, and epigenetics: impact on tumor progression and perspective for anticancer therapy. Cells 8:532. doi: 10.3390/cells8060532
Conery, A. R., Centore, R. C., Neiss, A., Keller, P. J., Joshi, S., Spillane, K. L., et al. (2016). Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 5:483. doi: 10.7554/eLife.10483
Cosenza, M., and Pozzi, S. (2018). The therapeutic strategy of HDAC6 inhibitors in lymphoproliferative disease. Int. J. Mol. Sci. 19:337. doi: 10.3390/ijms19082337
Coyle, K. M., Boudreau, J. E., and Marcato, P. (2017). Genetic mutations and epigenetic modifications: driving cancer and informing precision medicine. Biomed. Res. Intern. 2017:9620870. doi: 10.1155/2017/9620870
Cuervo, A. M., and Wong, E. (2014). Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 24, 92–104. doi: 10.1038/cr.2013.153
da Rocha Dias, S., Friedlos, F., Light, Y., Springer, C., Workman, P., and Marais, R. (2005). Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 65, 10686–10691. doi: 10.1158/0008-5472.Can-05-2632
de Lera, A. R., and Ganesan, A. (2016). Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin. Epigen. 8:105. doi: 10.1186/s13148-016-0271-9
Dejligbjerg, M., Grauslund, M., Litman, T., Collins, L., Qian, X., Jeffers, M., et al. (2008). Differential effects of class I isoform histone deacetylase depletion and enzymatic inhibition by belinostat or valproic acid in HeLa cells. Mol. Cancer 7:70. doi: 10.1186/1476-4598-7-70
Deshpande, A., Sicinski, P., and Hinds, P. W. (2005). Cyclins and cdks in development and cancer: a perspective. Oncogene 24, 2909–2915. doi: 10.1038/sj.onc.1208618
Díaz, T., Rodríguez, V., Lozano, E., Mena, M.-P., Calderón, M., Rosiñol, L., et al. (2017). The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity in in vitro and in vivo models of multiple myeloma by blockade of Ikaros and MYC signaling. Haematologica 102, 1776–1784. doi: 10.3324/haematol.2017.164632
Dimopoulos, M., Siegel, D. S., Lonial, S., Qi, J., Hajek, R., Facon, T., et al. (2013). Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 14, 1129–1140. doi: 10.1016/S1470-2045(13)70398-X
Dimopoulos, M., Spencer, A., Attal, M., Prince, H. M., Harousseau, J. L., Dmoszynska, A., et al. (2007). Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N. Engl. J. Med. 357, 2123–2132. doi: 10.1056/NEJMoa070594
Dokmanovic, M., Clarke, C., and Marks, P. A. (2007). Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res. MCR 5, 981–989. doi: 10.1158/1541-7786.MCR-07-0324
Dong, L. H., Cheng, S., Zheng, Z., Wang, L., Shen, Y., Shen, Z. X., et al. (2013). Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. J. Hematol. Oncol. 6:53. doi: 10.1186/1756-8722-6-53
Drummond, D. C., Noble, C. O., Kirpotin, D. B., Guo, Z., Scott, G. K., and Benz, C. C. (2005). Clinical development of histone deacetylase inhibitors as anticancer agents. Annu. Rev. Pharmacol. Toxicol. 45, 495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825
Duan, H., Heckman, C. A., and Boxer, L. M. (2005). Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol. Cell. Biol. 25, 1608–1619. doi: 10.1128/MCB.25.5.1608-1619.2005
Duong, V., Bret, C., Altucci, L., Mai, A., Duraffourd, C., Loubersac, J., et al. (2008). Specific activity of class II histone deacetylases in human breast cancer cells. Mol. Cancer Res. MCR 6, 1908–1919. doi: 10.1158/1541-7786.MCR-08-0299
Ellis, L., Pan, Y., Smyth, G. K., George, D. J., McCormack, C., Williams-Truax, R., et al. (2008). Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clin. Cancer Res. 14, 4500–4510. doi: 10.1158/1078-0432.Ccr-07-4262
Enchev, R. I., Schulman, B. A., and Peter, M. (2015). Protein neddylation: beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 16, 30–44. doi: 10.1038/nrm3919
Eyre, T. A., Collins, G. P., Gupta, A., Coupe, N., Sheikh, S., Whittaker, J., et al. (2019). A phase 1 study to assess the safety, tolerability, and pharmacokinetics of CXD101 in patients with advanced cancer. Cancer 125, 99–108. doi: 10.1002/cncr.31791
Eyüpoglu, I. Y., Hahnen, E., Buslei, R., Siebzehnrübl, F. A., Savaskan, N. E., Lüders, M., et al. (2005). Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. J. Neurochem. 93, 992–999. doi: 10.1111/j.1471-4159.2005.03098.x
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Fernald, K., and Kurokawa, M. (2013). Evading apoptosis in cancer. Trends Cell Biol. 23, 620–633. doi: 10.1016/j.tcb.2013.07.006
Field-Smith, A., Morgan, G. J., and Davies, F. E. (2006). Bortezomib (Velcadetrade mark) in the treatment of multiple myeloma. Therap. Clin. Risk Manag. 2, 271–279. doi: 10.2147/tcrm.2006.2.3.271
Filippakopoulos, P., and Knapp, S. (2014). Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356. doi: 10.1038/nrd4286
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O., et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. doi: 10.1038/nature09504
Fotheringham, S., Epping, M. T., Stimson, L., Khan, O., Wood, V., Pezzella, F., et al. (2009). Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell 15, 57–66. doi: 10.1016/j.ccr.2008.12.001
Fournel, M., Bonfils, C., Hou, Y., Yan, P. T., Trachy-Bourget, M. C., Kalita, A., et al. (2008). MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 7, 759–768. doi: 10.1158/1535-7163.MCT-07-2026
Friday, B. B., Anderson, S. K., Buckner, J., Yu, C., Giannini, C., Geoffroy, F., et al. (2012). Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro. Oncol. 14, 215–221. doi: 10.1093/neuonc/nor198
Fridman, J. S., and Lowe, S. W. (2003). Control of apoptosis by p53. Oncogene 22, 9030–9040. doi: 10.1038/sj.onc.1207116
Fukutomi, A., Hatake, K., Matsui, K., Sakajiri, S., Hirashima, T., Tanii, H., et al. (2012). A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Invest. New Drugs 30, 1096–1106. doi: 10.1007/s10637-011-9666-9
Gao, L., Cueto, M. A., Asselbergs, F., and Atadja, P. (2002). Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 277, 25748–25755. doi: 10.1074/jbc.M111871200
Garcia-Manero, G., Kantarjian, H. M., Sanchez-Gonzalez, B., Yang, H., Rosner, G., Verstovsek, S., et al. (2006). Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in patients with leukemia. Blood 108, 3271–3279. doi: 10.1182/blood-2006-03-009142
Garcia-Manero, G., Yang, A. S., Klimek, V., Luger, S., Newsome, W. M., Berman, N., et al. (2007). Phase I/II study of a novel oral isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 in combination with azacitidine in patients (pts) with high-risk myelodysplastic syndrome (MDS) or acute myelogenous leukemia (AML). J. Clin. Oncol. 25, 7062–7062. doi: 10.1200/jco.2007.25.18_suppl.7062
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Glozak, M. A., and Seto, E. (2007). Histone deacetylases and cancer. Oncogene 26, 5420–5432. doi: 10.1038/sj.onc.1210610
Goldberg, A. L. (2007). Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem. Soc. Trans. 35(Pt 1), 12–17. doi: 10.1042/bst0350012
Gozzetti, A., and Cerase, A. (2014). Novel agents in CNS myeloma treatment. Cent. Nerv. Syst. Agents Med. Chem. 14, 23–27. doi: 10.2174/1871524914999140818111514
Groen, K., van de Donk, N., Stege, C., Zweegman, S., and Nijhof, I. S. (2019). Carfilzomib for relapsed and refractory multiple myeloma. Cancer Manag. Res. 11, 2663–2675. doi: 10.2147/CMAR.S150653
Grossman, S. R., Perez, M., Kung, A. L., Joseph, M., Mansur, C., Xiao, Z.-X., et al. (1998). p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol. Cell 2, 405–415. doi: 10.1016/S1097-2765(00)80140-9
Grothey, A., Van Cutsem, E., Sobrero, A., Siena, S., Falcone, A., Ychou, M., et al. (2013). Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 381, 303–312. doi: 10.1016/S0140-6736(12)61900-X
Grünstein, E. (2018). Selektive HDAC6-Inhibitoren: Synthese und Biologische Testung. Dissertation, Universität Regensburg, Germany.
Grünstein, E., Sellmer, A., and Mahboobi, S. (2019). Enantioselective synthesis and biological investigation of tetrahydro-β-carboline-based HDAC6 inhibitors with improved solubility. Archiv. Pharm. 352:e1900026. doi: 10.1002/ardp.201900026
Gu, Y., Kaufman, J. L., Bernal, L., Torre, C., Matulis, S. M., Harvey, R. D., et al. (2014). MLN4924, an NAE inhibitor, suppresses AKT and mTOR signaling via upregulation of REDD1 in human myeloma cells. Blood 123, 3269–3276. doi: 10.1182/blood-2013-08-521914
Gui, C.-Y., Ngo, L., Xu, W. S., Richon, V. M., and Marks, P. A. (2004). Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. U.S.A. 101, 1241–1246. doi: 10.1073/pnas.0307708100
Gump, J. M., and Thorburn, A. (2011). Autophagy and apoptosis: what is the connection? Trends Cell Biol. 21, 387–392. doi: 10.1016/j.tcb.2011.03.007
Gunst, J. D., Kjaer, K., Olesen, R., Rasmussen, T. A., Ostergaard, L., Denton, P. W., et al. (2019). Fimepinostat, a novel dual inhibitor of HDAC and PI3K, effectively reverses HIV-1 latency ex vivo without T cell activation. J. Virus Erad. 5, 133–137.
Hakami, N. Y., Dusting, G. J., and Peshavariya, H. M. (2016). Trichostatin A, a histone deacetylase inhibitor suppresses NADPH Oxidase 4-Derived Redox Signalling and Angiogenesis. J. Cell. Mol. Med. 20, 1932–1944. doi: 10.1111/jcmm.12885
Halkidou, K., Gaughan, L., Cook, S., Leung, H. Y., Neal, D. E., and Robson, C. N. (2004). Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59, 177–189. doi: 10.1002/pros.20022
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57–70. doi: 10.1016/s0092-8674(00)81683-9
Handy, D. E., Castro, R., and Loscalzo, J. (2011). Epigenetic modifications: basic mechanisms and role in cardiovascular Disease. Circulation 123, 2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839
Harris, G. F., Anderson, M. E., and Lee, J. H. (2008). The effect of proteasome inhibition on p53 degradation and proliferation in tonsil epithelial cells. Archiv. Otolaryngol. Head Neck Surg. 134, 157–163. doi: 10.1001/archoto.2007.37
Hartl, F. U., and Hayer-Hartl, M. (2009). Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 16, 574–581. doi: 10.1038/nsmb.1591
Hay, D. A., Fedorov, O., Martin, S., Singleton, D. C., Tallant, C., Wells, C., et al. (2014). Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 136, 9308–9319. doi: 10.1021/ja412434f
Heider, U., von Metzler, I., Kaiser, M., Rosche, M., Sterz, J., Rötzer, S., et al. (2008). Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in mantle cell lymphoma. Eur. J. Haematol. 80, 133–142. doi: 10.1111/j.1600-0609.2007.00995.x
Hideshima, T., Bradner, J. E., Wong, J., Chauhan, D., Richardson, P., Schreiber, S. L., et al. (2005). Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 102, 8567–8572. doi: 10.1073/pnas.0503221102
Hideshima, T., Richardson, P., Chauhan, D., Palombella, V. J., Elliott, P. J., Adams, J., et al. (2001). The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 61, 3071–3076.
Hoang, T., Campbell, T. C., Zhang, C., Kim, K., Kolesar, J. M., Oettel, K. R., et al. (2014). Vorinostat and bortezomib as third-line therapy in patients with advanced non-small cell lung cancer: a Wisconsin oncology network phase II study. Invest. New Drugs 32, 195–199. doi: 10.1007/s10637-013-9980-5
Holkova, B., Kmieciak, M., Bose, P., Yazbeck, V. Y., Barr, P. M., Tombes, M. B., et al. (2016). Phase 1 trial of carfilzomib (PR-171) in combination with vorinostat (SAHA) in patients with relapsed or refractory B-cell lymphomas. Leukemia Lymph. 57, 635–643. doi: 10.3109/10428194.2015.1075019
Holkova, B., Shea, T. C., Bose, P., Tombes, M. B., Shrader, E., Wan, W., et al. (2012). Phase I study of bortezomib and romidepsin in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma, indolent b-cell lymphoma, or peripheral T-Cell lymphoma. Blood 120, 1794–1794. doi: 10.1182/blood.V120.21.1794.1794
Huang, B. H., Laban, M., Leung, C. H.-W., Lee, L., Lee, C. K., Salto-Tellez, M., et al. (2005). Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 12, 395–404. doi: 10.1038/sj.cdd.4401567
Huang, F. I., Wu, Y. W., Sung, T. Y., Liou, J. P., Lin, M. H., Pan, S. L., et al. (2019). MPT0G413, A novel HDAC6-selective inhibitor, and bortezomib synergistically exert anti-tumor activity in multiple myeloma cells. Front. Oncol. 9:249. doi: 10.3389/fonc.2019.00249
Huang, W.-J., Tang, Y.-A., Chen, M.-Y., Wang, Y.-J., Hu, F.-H., Wang, T.-W., et al. (2014). A histone deacetylase inhibitor YCW1 with antitumor and antimetastasis properties enhances cisplatin activity against non-small cell lung cancer in preclinical studies. Cancer Lett. 346, 84–93. doi: 10.1016/j.canlet.2013.12.016
Hug, B. A., and Lazar, M. A. (2004). ETO interacting proteins. Oncogene 23, 4270–4274. doi: 10.1038/sj.onc.1207674
Hyun, S. Y., Le, H. T., Nguyen, C. T., Yong, Y. S., Boo, H. J., Lee, H. J., et al. (2018). Development of a novel Hsp90 inhibitor NCT-50 as a potential anticancer agent for the treatment of non-small cell lung cancer. Sci. Rep. 8:13924. doi: 10.1038/s41598-018-32196-6
Issa, J. P., Garcia-Manero, G., Huang, X., Cortes, J., Ravandi, F., Jabbour, E., et al. (2015). Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer 121, 556–561. doi: 10.1002/cncr.29085
Janku, F., McConkey, D. J., Hong, D. S., and Kurzrock, R. (2011). Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 8, 528–539. doi: 10.1038/nrclinonc.2011.71
Jarosz, D. (2016). Hsp90: a global regulator of the genotype-to-phenotype map in cancers. Adv. Cancer Res. 129, 225–247. doi: 10.1016/bs.acr.2015.11.001
Jeng, W., Lee, S., Sung, N., Lee, J., and Tsai, F. T. F. (2015). Molecular chaperones: guardians of the proteome in normal and disease states. F1000Research 4:214. doi: 10.12688/f1000research.7214.1
Jin, X., Fang, Y., Hu, Y., Chen, J., Liu, W., Chen, G., et al. (2017). Synergistic activity of the histone deacetylase inhibitor trichostatin A and the proteasome inhibitor PS-341 against taxane-resistant ovarian cancer cell lines. Oncol. Lett. 13, 4619–4626. doi: 10.3892/ol.2017.6032
John, R. M., and Rougeulle, C. (2018). Developmental epigenetics: phenotype and the flexible epigenome. Front. Cell Dev. Biol. 6:130. doi: 10.3389/fcell.2018.00130
Johnson, D. E. (2015). The ubiquitin-proteasome system: opportunities for therapeutic intervention in solid tumors. Endocrine Relat. Cancer 22, T1–T17. doi: 10.1530/ERC-14-0005
Johnstone, R. W. (2002). Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 1, 287–299. doi: 10.1038/nrd772
Jones, D. R., Moskaluk, C. A., Gillenwater, H. H., Petroni, G. R., Burks, S. G., Philips, J., et al. (2012). Phase I trial of induction histone deacetylase and proteasome inhibition followed by surgery in non-small-cell lung cancer. J. Thorac. Oncol. 7, 1683–1690. doi: 10.1097/JTO.0b013e318267928d
Kahali, S., Sarcar, B., Fang, B., Williams, E. S., Koomen, J. M., Tofilon, P. J., et al. (2010). Activation of the unfolded protein response contributes toward the antitumor activity of vorinostat. Neoplasia 12, 80–86. doi: 10.1593/neo.91422
Kaliszczak, M., van Hechanova, E., Li, Y., Alsadah, H., Parzych, K., Auner, H. W., et al. (2018). The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death. Br. J. Cancer 119, 1278–1287. doi: 10.1038/s41416-018-0232-5
Kamal, A., Thao, L., Sensintaffar, J., Zhang, L., Boehm, M. F., Fritz, L. C., et al. (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410. doi: 10.1038/nature01913
Kaufman, J. L., Mina, R., Jakubowiak, A. J., Zimmerman, T. L., Wolf, J. J., Lewis, C., et al. (2019). Combining carfilzomib and panobinostat to treat relapsed/refractory multiple myeloma: results of a multiple myeloma research consortium phase I study. Blood Cancer J. 9:3. doi: 10.1038/s41408-018-0154-8
Khan, O., Fotheringham, S., Wood, V., Stimson, L., Zhang, C., Pezzella, F., et al. (2010). HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc. Natl. Acad. Sci. U.S.A. 107, 6532–6537. doi: 10.1073/pnas.0913912107
Khan, S. B., Maududi, T., Barton, K., Ayers, J., and Alkan, S. (2004). Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br. J. Haematol. 125, 156–161. doi: 10.1111/j.1365-2141.2004.04882.x
Khurana, N., and Bhattacharyya, S. (2015). Hsp90, the concertmaster: tuning transcription. Front. Oncol. 5:100. doi: 10.3389/fonc.2015.00100
Kirschbaum, M. H., Foon, K. A., Frankel, P., Ruel, C., Pulone, B., Tuscano, J. M., et al. (2014). A phase 2 study of belinostat (PXD101) in patients with relapsed or refractory acute myeloid leukemia or patients over the age of 60 with newly diagnosed acute myeloid leukemia: a california cancer consortium study. Leukemia Lymph. 55, 2301–2304. doi: 10.3109/10428194.2013.877134
Kirschbaum, M. H., Goldman, B. H., Zain, J. M., Cook, J. R., Rimsza, L. M., Forman, S. J., et al. (2012). A phase 2 study of vorinostat for treatment of relapsed or refractory hodgkin lymphoma: southwest oncology group study S0517. Leuk. Lymph. 53, 259–262. doi: 10.3109/10428194.2011.608448
Koprinarova, M., Schnekenburger, M., and Diederich, M. (2016). Role of histone acetylation in cell cycle regulation. Curr. Top. Med. Chem. 16, 732–744. doi: 10.2174/1568026615666150825140822
Kouzarides, T. (2000). Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179. doi: 10.1093/emboj/19.6.1176
Kovacs, J. J., Murphy, P. J. M., Gaillard, S., Zhao, X., Wu, J.-T., Nicchitta, C. V., et al. (2005). HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607. doi: 10.1016/j.molcel.2005.04.021
Kumar, S. K., Dimopoulos, M. A., Kastritis, E., Terpos, E., Nahi, H., Goldschmidt, H., et al. (2017). Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia 31, 2443–2448. doi: 10.1038/leu.2017.138
Kung, C.-P., Budina, A., Balaburski, G., Bergenstock, M. K., and Murphy, M. (2011). Autophagy in tumor suppression and cancer therapy. Crit. Rev. Eukary. Gene Exp. 21, 71–100. doi: 10.1615/critreveukargeneexpr.v21.i1.50
Lagger, G., O’Carroll, D., Rembold, M., Khier, H., Tischler, J., Weitzer, G., et al. (2002). Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 21, 2672–2681. doi: 10.1093/emboj/21.11.2672
LeBlanc, R., Catley, L. P., Hideshima, T., Lentzsch, S., Mitsiades, C. S., Mitsiades, N., et al. (2002). Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 62, 4996–5000.
Lee, A.-H., Iwakoshi, N. N., Anderson, K. C., and Glimcher, L. H. (2003). Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. U.S.A. 100, 9946–9951. doi: 10.1073/pnas.1334037100
Leyk, J., Daly, C., Janssen-Bienhold, U., Kennedy, B. N., and Richter-Landsberg, C. (2017). HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 8:e3028. doi: 10.1038/cddis.2017.415
Leyk, J., Goldbaum, O., Noack, M., and Richter-Landsberg, C. (2015). Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J. Mol. Neurosci. MN 55, 1031–1046. doi: 10.1007/s12031-014-0460-y
Li, T., Zhang, C., Hassan, S., Liu, X., Song, F., Chen, K., et al. (2018). Histone deacetylase 6 in cancer. J. Hematol. Oncol. 11:111. doi: 10.1186/s13045-018-0654-9
Li, W.-W., Li, J., and Bao, J.-K. (2012). Microautophagy: lesser-known self-eating. Cell. Mol. Life Sci. 69, 1125–1136. doi: 10.1007/s00018-011-0865-5
Liu, L., Scolnick, D. M., Trievel, R. C., Zhang, H. B., Marmorstein, R., Halazonetis, T. D., et al. (1999). p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19, 1202–1209. doi: 10.1128/mcb.19.2.1202
Liu, Y.-L., Yang, P.-M., Shun, C.-T., Wu, M.-S., Weng, J.-R., and Chen, C.-C. (2010). Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 6, 1057–1065. doi: 10.4161/auto.6.8.13365
Llinàs-Arias, P., and Esteller, M. (2017). Epigenetic inactivation of tumour suppressor coding and non-coding genes in human cancer: an update. Open Biol. 7:152. doi: 10.1098/rsob.170152
Lockwood, W. W., Zejnullahu, K., Bradner, J. E., and Varmus, H. (2012). Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. U.S.A. 109, 19408–19413. doi: 10.1073/pnas.1216363109
Long, J., Li, B., Rodriguez-Blanco, J., Pastori, C., Volmar, C.-H., Wahlestedt, C., et al. (2014). The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J. Biol. Chem. 289, 35494–35502. doi: 10.1074/jbc.M114.595348
Luger, K., and Richmond, T. J. (1998). The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 8, 140–146. doi: 10.1016/S0959-437X(98)80134-2
Mahalingam, D., Mita, M., Sarantopoulos, J., Wood, L., Amaravadi, R. K., Davis, L. E., et al. (2014). Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 10, 1403–1414. doi: 10.4161/auto.29231
Mann, B. S., Johnson, J. R., Cohen, M. H., Justice, R., and Pazdur, R. (2007). FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncol. 12, 1247–1252. doi: 10.1634/theoncologist.12-10-1247
Marquard, L., Gjerdrum, L. M., Christensen, I. J., Jensen, P. B., Sehested, M., and Ralfkiaer, E. (2008). Prognostic significance of the therapeutic targets histone deacetylase 1, 2, 6 and acetylated histone H4 in cutaneous T-cell lymphoma. Histopathology 53, 267–277. doi: 10.1111/j.0309-0167.2008.03109.x
Masdehors, P., Merle-Beral, H., Maloum, K., Omura, S., Magdelenat, H., and Delic, J. (2000). Deregulation of the ubiquitin system and p53 proteolysis modify the apoptotic response in B-CLL lymphocytes. Blood 96, 269–274.
Mathew, R., Karantza-Wadsworth, V., and White, E. (2007). Role of autophagy in cancer. Nat. Rev. Cancer 7, 961–967. doi: 10.1038/nrc2254
Mathieson, T., Franken, H., Kosinski, J., Kurzawa, N., Zinn, N., Sweetman, G., et al. (2018). Systematic analysis of protein turnover in primary cells. Nat. Commun. 9:689. doi: 10.1038/s41467-018-03106-1
Mayer, M. P., and Bukau, B. (2005). Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684. doi: 10.1007/s00018-004-4464-6
McCleland, M. L., Mesh, K., Lorenzana, E., Chopra, V. S., Segal, E., Watanabe, C., et al. (2016). CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Invest. 126, 639–652. doi: 10.1172/JCI83265
Meccariello, R., Chianese, R., Ciaramella, V., Fasano, S., and Pierantoni, R. (2014). Molecular chaperones, cochaperones, and ubiquitination/deubiquitination system: involvement in the production of high quality spermatozoa. Biomed. Res. Intern. 2014:10. doi: 10.1155/2014/561426
Mehta-Shah, N., Moskowitz, A. J., Lunning, M., Lynch, P., Scheuerman, M., Kumar, A., et al. (2016). A Phase Ib/IIa trial of the combination of romidepsin, lenalidomide and carfilzomib in patients with relapsed/refractory lymphoma shows complete responses in relapsed and refractory T-Cell lymphomas. Blood 128, 2991–2991. doi: 10.1182/blood.V128.22.2991.2991
Mertz, J. A., Conery, A. R., Bryant, B. M., Sandy, P., Balasubramanian, S., Mele, D. A., et al. (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U.S.A. 108, 16669–16674. doi: 10.1073/pnas.1108190108
Millward, M., Price, T., Townsend, A., Sweeney, C., Spencer, A., Sukumaran, S., et al. (2012). Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination. Invest. New Drugs 30, 2303–2317. doi: 10.1007/s10637-011-9766-6
Mithraprabhu, S., Kalff, A., Chow, A., Khong, T., and Spencer, A. (2014). Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 9, 1511–1520. doi: 10.4161/15592294.2014.983367
Miyanaga, A., Gemma, A., Noro, R., Kataoka, K., Matsuda, K., Nara, M., et al. (2008). Antitumor activity of histone deacetylase inhibitors in non-small cell lung cancer cells: development of a molecular predictive model. Mol. Cancer Ther. 7, 1923–1930. doi: 10.1158/1535-7163.Mct-07-2140
Mogk, A., and Bukau, B. (2006). Protein quality control systems: mechanisms and applications. Microb. Cell Factor. 5, (Suppl. 1):S8. doi: 10.1186/1475-2859-5-S1-S8
Moros, A., Rodríguez, V., Saborit-Villarroya, I., Montraveta, A., Balsas, P., Sandy, P., et al. (2014). Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 28, 2049–2059. doi: 10.1038/leu.2014.106
Mottamal, M., Zheng, S., Huang, T. L., and Wang, G. (2015). Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 20, 3898–3941. doi: 10.3390/molecules20033898
Mrakovcic, M., Bohner, L., Hanisch, M., and Fröhlich, L. F. (2018). Epigenetic targeting of autophagy via HDAC inhibition in tumor cells: role of p53. Intern. J. Mol. Sci. 19:952. doi: 10.3390/ijms19123952
Mrakovcic, M., and Fröhlich, L. F. (2018). p53-mediated molecular control of autophagy in tumor cells. Biomolecules 8:14. doi: 10.3390/biom8020014
Mrakovcic, M., Kleinheinz, J., and Fröhlich, L. F. (2017). Histone deacetylase inhibitor-induced autophagy in tumor cells: implications for p53. Intern. J. Mol. Sci. 18:883. doi: 10.3390/ijms18091883
Nawrocki, S. T., Carew, J. S., Pino, M. S., Highshaw, R. A., Andtbacka, R. H. I., Dunner, K., et al. (2006). Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 66, 3773–3781. doi: 10.1158/0008-5472.CAN-05-2961
Nepali, K., Sharma, S., Sharma, M., Bedi, P. M., and Dhar, K. L. (2014). Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 77, 422–487. doi: 10.1016/j.ejmech.2014.03.018
New, M., Olzscha, H., and La Thangue, N. B. (2012). HDAC inhibitor-based therapies: can we interpret the code? Mol. Oncol. 6, 637–656. doi: 10.1016/j.molonc.2012.09.003
New, M., Olzscha, H., Liu, G., Khan, O., Stimson, L., McGouran, J., et al. (2013). A regulatory circuit that involves HR23B and HDAC6 governs the biological response to HDAC inhibitors. Cell Death Differ. 20, 1306–1316. doi: 10.1038/cdd.2013.47
Niesvizky, R., Ely, S., Mark, T., Aggarwal, S., Gabrilove, J. L., Wright, J. J., et al. (2011). Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer 117, 336–342. doi: 10.1002/cncr.25584
Nihira, N. T., Ogura, K., Shimizu, K., North, B. J., Zhang, J., Gao, D., et al. (2017). Acetylation-dependent regulation of MDM2 E3 ligase activity dictates its oncogenic function. Sci. Signal. 10:aai8026. doi: 10.1126/scisignal.aai8026
Noack, K., Mahendrarajah, N., Hennig, D., Schmidt, L., Grebien, F., Hildebrand, D., et al. (2017). Analysis of the interplay between all-trans retinoic acid and histone deacetylase inhibitors in leukemic cells. Archiv. Toxicol. 91, 2191–2208. doi: 10.1007/s00204-016-1878-5
Noack, M., Leyk, J., and Richter-Landsberg, C. (2014). HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia 62, 535–547. doi: 10.1002/glia.22624
Obeng, E. A., Carlson, L. M., Gutman, D. M., Harrington, W. J. Jr., and Lee, K. P. (2006). Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107, 4907–4916. doi: 10.1182/blood-2005-08-3531
Ocio, E. M., Vilanova, D., Atadja, P., Maiso, P., Crusoe, E., Fernandez-Lazaro, D., et al. (2010). In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica 95, 794–803. doi: 10.3324/haematol.2009.015495
Ocker, M., and Schneider-Stock, R. (2007). Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Intern. J. Biochem. Cell Biol. 39, 1367–1374. doi: 10.1016/j.biocel.2007.03.001
Olzscha, H. (2019). Posttranslational modifications and proteinopathies: how guardians of the proteome are defeated. Biol. Chem. 400, 895–915. doi: 10.1515/hsz-2018-0458
Olzscha, H., Fedorov, O., Kessler, B. M., Knapp, S., and La Thangue, N. B. (2017). CBP/p300 bromodomains regulate amyloid-like protein aggregation upon aberrant lysine acetylation. Cell Chem. Biol. 24, 9–23. doi: 10.1016/j.chembiol.2016.11.009
Olzscha, H., Sheikh, S., and La Thangue, N. B. (2015). Deacetylation of chromatin and gene expression regulation: a new target for epigenetic therapy. Crit. Rev. Oncog. 20, 1–17. doi: 10.1615/critrevoncog.2014012463
Orlowski, R. Z., Eswara, J. R., Lafond-Walker, A., Grever, M. R., Orlowski, M., and Dang, C. V. (1998). Tumor growth inhibition induced in a murine model of human Burkitt’s lymphoma by a proteasome inhibitor. Cancer Res. 58, 4342–4348.
Ouyang, H., Ali, Y. O., Ravichandran, M., Dong, A., Qiu, W., MacKenzie, F., et al. (2012). Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 287, 2317–2327. doi: 10.1074/jbc.M111.273730
Patel, S., Hurez, V., Nawrocki, S. T., Goros, M., Michalek, J., Sarantopoulos, J., et al. (2016). Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget 7, 59087–59097. doi: 10.18632/oncotarget.10824
Patel, S., and Player, M. R. (2008). Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin. Invest. Drugs 17, 1865–1882. doi: 10.1517/13543780802493366
Pavlova, N. N., and Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47. doi: 10.1016/j.cmet.2015.12.006
Pei, X.-Y., Dai, Y., and Grant, S. (2004). Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 10, 3839–3852. doi: 10.1158/1078-0432.CCR-03-0561
Pérez-Salvia, M., and Esteller, M. (2017). Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics 12, 323–339. doi: 10.1080/15592294.2016.1265710
Perrin, P., Cassagnau, E., Burg, C., Patry, Y., Vavasseur, F., Harb, J., et al. (1994). An interleukin 2/sodium butyrate combination as immunotherapy for rat colon cancer peritoneal carcinomatosis. Gastroenterology 107, 1697–1708. doi: 10.1016/0016-5085(94)90810-9
Picaud, S., Fedorov, O., Thanasopoulou, A., Leonards, K., Jones, K., Meier, J., et al. (2015). Generation of a selective small molecule inhibitor of the cbp/p300 bromodomain for leukemia therapy. Cancer Res. 75, 5106–5119. doi: 10.1158/0008-5472.CAN-15-0236
Platzbecker, U., Al-Ali, H. K., Gattermann, N., Haase, D., Janzen, V., Krauter, J., et al. (2014). Phase 2 study of oral panobinostat (LBH589) with or without erythropoietin in heavily transfusion-dependent IPSS low or int-1 MDS patients. Leukemia 28, 696–698. doi: 10.1038/leu.2013.325
Plumb, J. A., Finn, P. W., Williams, R. J., Bandara, M. J., Romero, M. R., Watkins, C. J., et al. (2003). Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Therap. 2:721.
Porcu, M., and Chiarugi, A. (2005). The emerging therapeutic potential of sirtuin-interacting drugs: from cell death to lifespan extension. Trends Pharmacol. Sci. 26, 94–103. doi: 10.1016/j.tips.2004.12.009
Powers, M. V., and Workman, P. (2006). Targeting of multiple signalling pathways by heat shock protein 90 molecular chaperone inhibitors. Endocr. Relat. Cancer 13, (Suppl. 1), S125–S135. doi: 10.1677/erc.1.01324
Qiu, T., Zhou, L., Zhu, W., Wang, T., Wang, J., Shu, Y., et al. (2013). Effects of treatment with histone deacetylase inhibitors in solid tumors: a review based on 30 clinical trials. Future Oncol. 9, 255–269. doi: 10.2217/fon.12.173
Rahmani, M., Yu, C., Dai, Y., Reese, E., Ahmed, W., Dent, P., et al. (2003). Coadministration of the heat shock protein 90 antagonist 17-allylamino- 17-demethoxygeldanamycin with suberoylanilide hydroxamic acid or sodium butyrate synergistically induces apoptosis in human leukemia cells. Cancer Res. 63, 8420–8427.
Rajkumar, S. V., Rosinol, L., Hussein, M., Catalano, J., Jedrzejczak, W., Lucy, L., et al. (2008). Multicenter, randomized, double-blind, placebo-controlled study of thalidomide plus dexamethasone compared with dexamethasone as initial therapy for newly diagnosed multiple myeloma. J. Clin. Oncol. 26, 2171–2177. doi: 10.1200/jco.2007.14.1853
Ravid, T., and Hochstrasser, M. (2008). Degradation signal diversity in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 9, 679–690. doi: 10.1038/nrm2468
Richardson, P., Mitsiades, C., Colson, K., Reilly, E., McBride, L., Chiao, J., et al. (2008). Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk. Lymph. 49, 502–507. doi: 10.1080/10428190701817258
Richardson, P., Sonneveld, P., Schuster, M., Irwin, D., Stadtmauer, E., Facon, T., et al. (2005a). Bortezomib continues demonstrates superior efficacy compared with high-dose dexamethasone in relapsed multiple myeloma: updated results of the APEX trail. Blood 106, 2547–2547. doi: 10.1182/blood.V106.11.2547.2547
Richardson, P. G., Sonneveld, P., Schuster, M. W., Irwin, D., Stadtmauer, E. A., Facon, T., et al. (2005b). Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 352, 2487–2498. doi: 10.1056/NEJMoa043445
Richardson, P. G., Schlossman, R. L., Alsina, M., Weber, D. M., Coutre, S. E., Gasparetto, C., et al. (2013). PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood 122, 2331–2337. doi: 10.1182/blood-2013-01-481325
Ruegsegger, C., and Saxena, S. (2016). Proteostasis impairment in ALS. Brain Res. 1648(Pt B), 571–579. doi: 10.1016/j.brainres.2016.03.032
Sakaguchi, K., Herrera, J. E., Saito, S., Miki, T., Bustin, M., Vassilev, A., et al. (1998). DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12, 2831–2841. doi: 10.1101/gad.12.18.2831
Saldanha, S. N., Kala, R., and Tollefsbol, T. O. (2014). Molecular mechanisms for inhibition of colon cancer cells by combined epigenetic-modulating epigallocatechin gallate and sodium butyrate. Exper. Cell Res. 324, 40–53. doi: 10.1016/j.yexcr.2014.01.024
Salimi, V., Shahsavari, Z., Safizadeh, B., Hosseini, A., Khademian, N., and Tavakoli-Yaraki, M. (2017). Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids Health Dis. 16:208. doi: 10.1186/s12944-017-0593-4
Sanchez, E., Shen, J., Steinberg, J., Li, M., Wang, C., Bonavida, B., et al. (2011). The histone deacetylase inhibitor LBH589 enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Leuk. Res. 35, 373–379. doi: 10.1016/j.leukres.2010.06.026
San-Miguel, J. F., Hungria, V. T., Yoon, S. S., Beksac, M., Dimopoulos, M. A., Elghandour, A., et al. (2014). Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 15, 1195–1206. doi: 10.1016/s1470-2045(14)70440-1
San-Miguel, J. F., Lonial, S., Hungria, V., Moreau, P., Einsele, H., Lee, J. H., et al. (2011). PANORAMA1: a randomized, double-blind, placebo controlled phase III study of panobinostat in combination with bortezomib and dexamethasone in patients with relapsed multiple myeloma. J. Clin. Oncol. 29:TS227. doi: 10.1200/jco.2011.29.15_suppl.tps227
San-Miguel, J. F., Richardson, P. G., Gunther, A., Sezer, O., Siegel, D., Blade, J., et al. (2013). Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. J. Clin. Oncol. 31, 3696–3703. doi: 10.1200/jco.2012.46.7068
Santos, A. L., and Lindner, A. B. (2017). Protein posttranslational modifications: roles in aging and age-related disease. Oxidat. Med. Cell. Longev. 2017:5716409. doi: 10.1155/2017/5716409
Sarkar, D., Leung, E. Y., Baguley, B. C., Finlay, G. J., and Askarian-Amiri, M. E. (2015). Epigenetic regulation in human melanoma: past and future. Epigenetics 10, 103–121. doi: 10.1080/15592294.2014.1003746
Sasaki, H., Moriyama, S., Nakashima, Y., Kobayashi, Y., Kiriyama, M., Fukai, I., et al. (2004). Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer 46, 171–178. doi: 10.1016/j.lungcan.2004.03.021
Schrader, E. K., Harstad, K. G., and Matouschek, A. (2009). Targeting proteins for degradation. Nat. Chem. Biol. 5, 815–822. doi: 10.1038/nchembio.250
Scognamiglio, A., Nebbioso, A., Manzo, F., Valente, S., Mai, A., and Altucci, L. (2008). HDAC-class II specific inhibition involves HDAC proteasome-dependent degradation mediated by RANBP2. Biochim. Biophys. Acta 1783, 2030–2038. doi: 10.1016/j.bbamcr.2008.07.007
Scroggins, B. T., Robzyk, K., Wang, D., Marcu, M. G., Tsutsumi, S., Beebe, K., et al. (2007). An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25, 151–159. doi: 10.1016/j.molcel.2006.12.008
Sellmer, A., Stangl, H., Beyer, M., Grünstein, E., Leonhardt, M., Pongratz, H., et al. (2018). Marbostat-100 defines a new class of potent and selective antiinflammatory and antirheumatic histone deacetylase 6 inhibitors. J. Med. Chem. 61, 3454–3477. doi: 10.1021/acs.jmedchem.7b01593
Sengupta, N., and Seto, E. (2004). Regulation of histone deacetylase activities. J. Cell. Biochem. 93, 57–67. doi: 10.1002/jcb.20179
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6:a018713. doi: 10.1101/cshperspect.a018713
Shahbazian, M. D., and Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. doi: 10.1146/annurev.biochem.76.052705.162114
Shimamura, T., Lowell, A. M., Engelman, J. A., and Shapiro, G. I. (2005). Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 65, 6401–6408. doi: 10.1158/0008-5472.Can-05-0933
Shimamura, T., and Shapiro, G. I. (2008). Heat shock protein 90 inhibition in lung cancer. J. Thorac. Oncol. 3(6 Suppl. 2), S152–S159. doi: 10.1097/JTO.0b013e318174ea3a
Sigalotti, L., Fratta, E., Coral, S., Cortini, E., Covre, A., Nicolay, H. J., et al. (2007). Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J. Cell Physiol. 212, 330–344. doi: 10.1002/jcp.21066
Singh, A. K., Bishayee, A., and Pandey, A. K. (2018). Targeting histone deacetylases with natural and synthetic agents: an emerging anticancer strategy. Nutrients 10:731. doi: 10.3390/nu10060731
Song, J., Noh, J. H., Lee, J. H., Eun, J. W., Ahn, Y. M., Kim, S. Y., et al. (2005). Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 113, 264–268. doi: 10.1111/j.1600-0463.2005.apm_04.x
Soodgupta, D., Pan, D., Cui, G., Senpan, A., Yang, X., Lu, L., et al. (2015). Small molecule MYC inhibitor conjugated to integrin-targeted nanoparticles extends survival in a mouse model of disseminated multiple myeloma. Mol. Cancer Therap. 14, 1286–1294. doi: 10.1158/1535-7163.MCT-14-0774-T
Stankov, M. V., El Khatib, M., Kumar Thakur, B., Heitmann, K., Panayotova-Dimitrova, D., Schoening, J., et al. (2014). Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 28, 577–588. doi: 10.1038/leu.2013.264
Steele, N. L., Plumb, J. A., Vidal, L., Tjornelund, J., Knoblauch, P., Rasmussen, A., et al. (2008). A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res. 14, 804–810. doi: 10.1158/1078-0432.Ccr-07-1786
Strait, K. A., Dabbas, B., Hammond, E. H., Warnick, C. T., Iistrup, S. J., and Ford, C. D. (2002). Cell cycle blockade and differentiation of ovarian cancer cells by the histone deacetylase inhibitor trichostatin A are associated with changes in p21, Rb, and Id proteins. Mol. Cancer Therap. 1, 1181–1190.
Subramanian, S., Bates, S. E., Wright, J. J., Espinoza-Delgado, I., and Piekarz, R. L. (2010). Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals 3, 2751–2767. doi: 10.3390/ph3092751
Sugasawa, K., Ng, J. M., Masutani, C., Iwai, S., van der Spek, P. J., Eker, A. P., et al. (1998). Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 2, 223–232. doi: 10.1016/s1097-2765(00)80132-x
Suraweera, A., O’Byrne, K. J., and Richard, D. J. (2018). Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front. Oncol. 8:92. doi: 10.3389/fonc.2018.00092
Tan, D., Phipps, C., Hwang, W. Y., Tan, S. Y., Yeap, C. H., Chan, Y. H., et al. (2015). Panobinostat in combination with bortezomib in patients with relapsed or refractory peripheral T-cell lymphoma: an open-label, multicentre phase 2 trial. Lancet Haematol. 2, e326–e333. doi: 10.1016/s2352-3026(15)00097-6
Theodoulou, N. H., Tomkinson, N. C. O., Prinjha, R. K., and Humphreys, P. G. (2016). Progress in the development of non-BET bromodomain chemical probes. Chemmedchem 11, 477–487. doi: 10.1002/cmdc.201500540
Tsankova, N., Renthal, W., Kumar, A., and Nestler, E. J. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8, 355–367. doi: 10.1038/nrn2132
Ugur, H. C., Ramakrishna, N., Bello, L., Menon, L. G., Kim, S.-K., Black, P. M., et al. (2007). Continuous intracranial administration of suberoylanilide hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic glioma model. J. Neuro Oncol. 83, 267–275. doi: 10.1007/s11060-007-9337-z
Venza, I., Visalli, M., Oteri, R., Cucinotta, M., Teti, D., and Venza, M. (2013). Class II-specific histone deacetylase inhibitors MC1568 and MC1575 suppress IL-8 expression in human melanoma cells. Pigment Cell Melan. Res. 26, 193–204. doi: 10.1111/pcmr.12049
Vigushin, D. M., Ali, S., Pace, P. E., Mirsaidi, N., Ito, K., Adcock, I., et al. (2001). Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 7, 971–976.
Vogl, D. T., Raje, N., Jagannath, S., Richardson, P., Hari, P., Orlowski, R., et al. (2017). Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin. Cancer Res. 23, 3307–3315. doi: 10.1158/1078-0432.Ccr-16-2526
Wade, P. A. (2001). Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum. Mol. Genet. 10, 693–698. doi: 10.1093/hmg/10.7.693
Wang, Z., Leng, Y., Wang, J., Liao, H. M., Bergman, J., Leeds, P., et al. (2016). Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of alpha-tubulin acetylation and FGF-21 up-regulation. Sci. Rep. 6:19626. doi: 10.1038/srep19626
Wang, Z.-M., Hu, J., Zhou, D., Xu, Z.-Y., Panasci, L. C., and Chen, Z.-P. (2002). Trichostatin A inhibits proliferation and induces expression of p21WAF and p27 in human brain tumor cell lines. Chin. J. Cancer 21, 1100–1105.
Warri, A., Cook, K. L., Hu, R., Jin, L., Zwart, A., Soto-Pantoja, D. R., et al. (2018). Autophagy and unfolded protein response (UPR) regulate mammary gland involution by restraining apoptosis-driven irreversible changes. Cell Death Discov. 4:40. doi: 10.1038/s41420-018-0105-y
Wegiel, B., Vuerich, M., Daneshmandi, S., and Seth, P. (2018). Metabolic switch in the tumor microenvironment determines immune responses to anti-cancer therapy. Front. Oncol. 8:284. doi: 10.3389/fonc.2018.00284
Weichert, W., Röske, A., Gekeler, V., Beckers, T., Ebert, M. P. A., Pross, M., et al. (2008a). Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 9, 139–148. doi: 10.1016/S1470-2045(08)70004-4
Weichert, W., Roske, A., Gekeler, V., Beckers, T., Stephan, C., Jung, K., et al. (2008b). Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 98, 604–610. doi: 10.1038/sj.bjc.6604199
Weichert, W., Roske, A., Niesporek, S., Noske, A., Buckendahl, A. C., Dietel, M., et al. (2008c). Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 14, 1669–1677. doi: 10.1158/1078-0432.Ccr-07-0990
Welch, W. J., and Feramisco, J. R. (1982). Purification of the major mammalian heat shock proteins. J. Biol. Chem. 257, 14949–14959.
Wesselborg, S., and Stork, B. (2015). Autophagy signal transduction by ATG proteins: from hierarchies to networks. Cell. Mol. Life Sci. 72, 4721–4757. doi: 10.1007/s00018-015-2034-8
West, A. C., and Johnstone, R. W. (2014). New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 124, 30–39. doi: 10.1172/JCI69738
White, E. (2012). Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 12, 401–410. doi: 10.1038/nrc3262
Whitesell, L., and Lindquist, S. L. (2005). HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772. doi: 10.1038/nrc1716
Whittaker, S. J., Demierre, M.-F., Kim, E. J., Rook, A. H., Lerner, A., Duvic, M., et al. (2010). Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 28, 4485–4491. doi: 10.1200/JCO.2010.28.9066
Wiech, H., Buchner, J., Zimmermann, R., and Jakob, U. (1992). Hsp90 chaperones protein folding in vitro. Nature 358, 169–170. doi: 10.1038/358169a0
Wilson, A. J., Byun, D.-S., Popova, N., Murray, L. B., L’Italien, K., Sowa, Y., et al. (2006). Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 281, 13548–13558. doi: 10.1074/jbc.M510023200
Yan, G., Graham, K., and Lanza-Jacoby, S. (2013). Curcumin enhances the anticancer effects of trichostatin a in breast cancer cells. Mol. Carcinogen. 52, 404–411. doi: 10.1002/mc.21875
Yan, M. M., Ni, J. D., Song, D., Ding, M., and Huang, J. (2015). Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol. Lett. 10, 1959–1969. doi: 10.3892/ol.2015.3508
Yang, Y., Rao, R., Shen, J., Tang, Y., Fiskus, W., Nechtman, J., et al. (2008). Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 68, 4833–4842. doi: 10.1158/0008-5472.CAN-08-0644
Yang, Y.-T., Balch, C., Kulp, S. K., Mand, M. R., Nephew, K. P., and Chen, C.-S. (2009). A rationally designed histone deacetylase inhibitor with distinct antitumor activity against ovarian cancer. Neoplasia 11, 552–563. doi: 10.1593/neo.09204
Yin, D., Ong, J. M., Hu, J., Desmond, J. C., Kawamata, N., Konda, B. M., et al. (2007). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clin. Cancer Res. 13, 1045–1052. doi: 10.1158/1078-0432.CCR-06-1261
Yoon, S., and Eom, G. H. (2016). HDAC and HDAC inhibitor: from cancer to cardiovascular diseases. Chonnam Med. J. 52, 1–11. doi: 10.4068/cmj.2016.52.1.1
Yoshida, M., Kijima, M., Akita, M., and Beppu, T. (1990). Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J. Biol. Chem. 265, 17174–17179.
Younes, A., Oki, Y., Bociek, R. G., Kuruvilla, J., Fanale, M., Neelapu, S., et al. (2011). Mocetinostat for relapsed classical Hodgkin’s lymphoma: an open-label, single-arm, phase 2 trial. Lancet Oncol. 12, 1222–1228. doi: 10.1016/S1470-2045(11)70265-0
Younes, A., Sureda, A., Ben-Yehuda, D., Zinzani, P. L., Ong, T. C., Prince, H. M., et al. (2012). Panobinostat in patients with relapsed/refractory Hodgkin’s lymphoma after autologous stem-cell transplantation: results of a phase II study. J. Clin. Oncol. 30, 2197–2203. doi: 10.1200/jco.2011.38.1350
Yu, L., Chen, X., Sun, X., Wang, L., and Chen, S. (2017). The glycolytic switch in tumors: how many players are involved? J. Cancer 8, 3430–3440. doi: 10.7150/jca.21125
Zhang, J., Ng, S., Wang, J., Zhou, J., Tan, S.-H., Yang, N., et al. (2015). Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 11, 629–642. doi: 10.1080/15548627.2015.1023981
Zhang, Z., Yamashita, H., Toyama, T., Sugiura, H., Ando, Y., Mita, K., et al. (2005). Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res. Treat. 94, 11–16. doi: 10.1007/s10549-005-6001-1
Zhang, Z., Yamashita, H., Toyama, T., Sugiura, H., Omoto, Y., Ando, Y., et al. (2004). HDAC6 expression is correlated with better survival in breast cancer. Clin. Cancer Res. 10, 6962–6968. doi: 10.1158/1078-0432.CCR-04-0455
Zhou, L., Chen, S., Zhang, Y., Kmieciak, M., Leng, Y., Li, L., et al. (2016). The NAE inhibitor pevonedistat interacts with the HDAC inhibitor belinostat to target AML cells by disrupting the DDR. Blood 127, 2219–2230. doi: 10.1182/blood-2015-06-653717
Zhu, P., Martin, E., Mengwasser, J., Schlag, P., Janssen, K.-P., and Göttlicher, M. (2004). Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 5, 455–463. doi: 10.1016/s1535-6108(04)00114-x
Zismanov, V., Drucker, L., and Gottfried, M. (2014). Combined inhibition of Hsp90 and the proteasome affects NSCLC proteostasis and attenuates cell migration. Anti Cancer Drugs 25, 998–1006. doi: 10.1097/cad.0000000000000140
Keywords: autophagy, bromodomain-containing protein, epigenetic drug, histone deacetylase inhibitor, molecular chaperone, precision medicine, protein quality control, ubiquitin proteasome system
Citation: Kulka LAM, Fangmann P-V, Panfilova D and Olzscha H (2020) Impact of HDAC Inhibitors on Protein Quality Control Systems: Consequences for Precision Medicine in Malignant Disease. Front. Cell Dev. Biol. 8:425. doi: 10.3389/fcell.2020.00425
Received: 21 March 2020; Accepted: 07 May 2020;
Published: 03 June 2020.
Edited by:
Christiane Pienna Soares, São Paulo State University, BrazilReviewed by:
Gaël Roué, Vall d’Hebron Institute of Oncology, SpainXiangbo Wan, The Sixth Affiliated Hospital of Sun Yat-sen University, China
Brian William Dymock, UniQuest Pty Ltd., Queensland, Australia
Copyright © 2020 Kulka, Fangmann, Panfilova and Olzscha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heidi Olzscha, SGVpZGkuT2x6c2NoYUBtZWRpemluLnVuaS1oYWxsZS5kZQ==