 Anna Slanzi
Anna Slanzi Giulia Iannoto
Giulia Iannoto Barbara Rossi
Barbara Rossi Elena Zenaro
Elena Zenaro Gabriela Constantin
Gabriela Constantin- 1Department of Medicine, University of Verona, Verona, Italy
- 2Center for Biomedical Computing (CBMC), University of Verona, Verona, Italy
Neurodegenerative diseases are progressive degenerative conditions characterized by the functional deterioration and ultimate loss of neurons. These incurable and debilitating diseases affect millions of people worldwide, and therefore represent a major global health challenge with severe implications for individuals and society. Recently, several neuroprotective drugs have failed in human clinical trials despite promising pre-clinical data, suggesting that conventional cell cultures and animal models cannot precisely replicate human pathophysiology. To bridge the gap between animal and human studies, three-dimensional cell culture models have been developed from human or animal cells, allowing the effects of new therapies to be predicted more accurately by closely replicating some aspects of the brain environment, mimicking neuronal and glial cell interactions, and incorporating the effects of blood flow. In this review, we discuss the relative merits of different cerebral models, from traditional cell cultures to the latest high-throughput three-dimensional systems. We discuss their advantages and disadvantages as well as their potential to investigate the complex mechanisms of human neurodegenerative diseases. We focus on in vitro models of the most frequent age-related neurodegenerative disorders, such as Parkinson’s disease, Alzheimer’s disease and prion disease, and on multiple sclerosis, a chronic inflammatory neurodegenerative disease affecting young adults.
Introduction
Neurodegenerative diseases are age-related conditions characterized by uncontrolled neuronal death leading to a progressive decline in brain functions. These incurable and debilitating diseases are associated with a wide spectrum of clinical symptoms, including cognitive decline and/or the loss of locomotor functions. The number of affected individuals is growing due to the aging of human populations, and the severe effects of such diseases on the quality of life have increased the burden on healthcare systems worldwide (Heemels, 2016). Dementias in particular are responsible for the greatest burden of age-related neurodegenerative diseases. This is a broad term used to describe a number of conditions characterized by cognitive deficits, including Alzheimer’s disease (AD), vascular dementia, frontotemporal dementia, mixed dementia, and dementia with Lewy bodies. Other neurodegenerative diseases principally affect the locomotor system, including amyotrophic lateral sclerosis, Huntington’s disease, Parkinson’s disease (PD), multiple sclerosis (MS), and spinocerebellar ataxias.
The limited efficacy of drugs for the treatment of neurodegenerative diseases reflects their complex etiology and pathogenesis. In addition to aging, multiple risk factors contribute to susceptibility including environmental triggers and genetic factors. Therefore, more work is required to identify the underlying molecular mechanisms and corresponding pharmacological targets. In addition to the ethical concerns of animal experiments for medical research, the recent failure of several clinical trials targeting neurodegenerative diseases has raised doubts about the translatability of animal disease models to human patients, creating a demand for better research tools in this field (Olanow et al., 2008; Cummings et al., 2014; Schneider et al., 2014; Pfeuffer et al., 2016; Anderson et al., 2017). The development of novel in vitro models with greater physiological relevance may bridge the gap between current pre-clinical animal models and humans, allowing the discovery of promising drug targets that can be tested in future clinical trials. In addition, in vitro testing can reduce the duration and costs of translation by helping to identify the mechanism of action together with any associated risks.
Several in vitro approaches have been developed to understand the etiology and pathogenesis of a broad range of neurodegenerative diseases (Table 1) and we focus on those applied to PD, AD, prion diseases and MS in this review. In 1962 the first CNS organotypic culture was prepared from rat hypophysis tissue (Bousquet and Meunier, 1962). Cells derived from embryonic rat spinal cord and ganglia were subsequently cultured on collagen-coated glass, revealing their potential for organotypic differentiation and bioelectric properties suitable for electrophysiological studies (Crain, 1966). Since then, organotypic cultures have been prepared from brain slices encompassing several cerebral areas, including the hippocampus, substantia nigra, locus coeruleus, striatum, and basal forebrain (Lavail and Wolf, 1973; Whetsell and Schwarcz, 1983; Knopfel et al., 1989; Ostergaard et al., 1995; Robertson et al., 1997). Although tissue explants and organotypic slice cultures faithfully represent the cerebral architecture, they are difficult to prepare and maintain in a viable state, and their inherent variability leads to a lack of reproducibility in experiments (Walsh et al., 2005). The development of immortalized cell lines (Table 1) removed the need to use tissue as a source, but such cell lines often present genetic and metabolic abnormalities compared to normal human cells (Gordon et al., 2014). The advent of human embryonic stem cells (ESCs) and then human induced pluripotent stem cells (iPSCs) (Thomson et al., 1998; Takahashi et al., 2007) provided researchers with the tools to generate multiple differentiated cell types with the same genotype. Methods for the conversion of human somatic cells into iPSCs using retroviral transduction and transcription factors such as OCT4, SOX2, KLF4, and c-MYC have opened new frontiers in the development of in vitro disease models because iPSCs can be derived from patients, providing a source of neurons carrying the same genetic variants associated with pathogenesis in a defined microenvironment (Table 1). In the earliest experiments, iPSCs were cultured in undiversified 2D layers which were of limited value as disease models because they did not recreate authentic interactions between cells. To overcome this drawback, more sophisticated 3D culture models were developed, including spheroids, hydrogels, scaffolds derived from the extracellular matrix (ECM), and organ-like cultures (Fitzgerald et al., 2015). For instance, organoids preserve the cellular interactions that capture key structural and functional aspects of real organs at the micrometer to millimeter scale (Renner et al., 2017). Human brain organoids have recently emerged as invaluable tools to model the pathophysiology of diverse neurodegenerative diseases, facilitating a range of research applications including the analysis of disease mechanisms and progression, drug discovery, drug testing, and cell replacement therapy (Wang, 2018; Costamagna et al., 2019; Logan et al., 2019). The 2D and 3D models developed for the investigation of neurodegenerative diseases are summarized in Table 1.
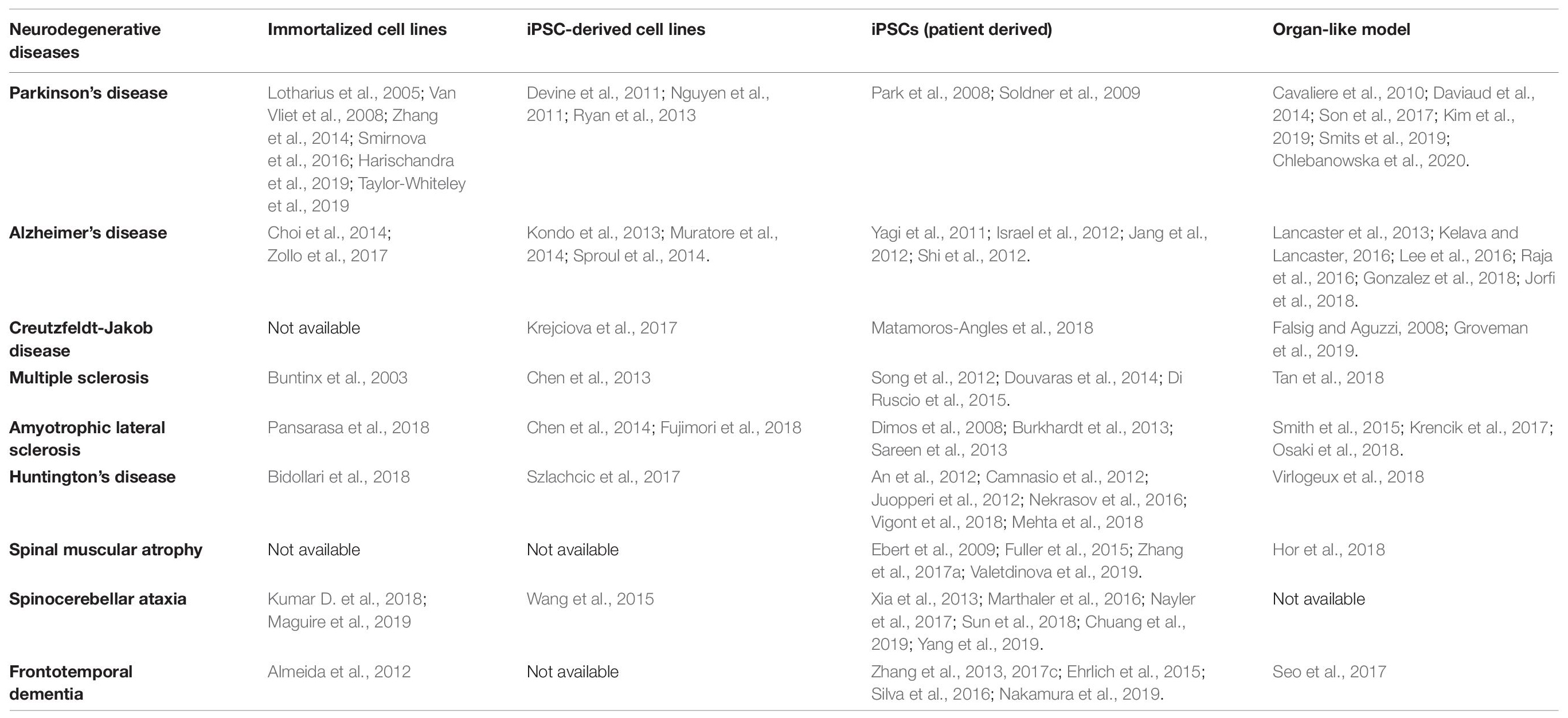
Table 1. Cell culture systems that can be used to study neurodegenerative diseases (iPSC, induced pluripotent stem cell).
Neurons and glial cells (astrocytes, oligodendrocytes, and microglia) cultivated in vitro in static devices such as trans-well systems are useful tools for basic linear kinetic studies during drug discovery. However, the central nervous system (CNS) features structures such as the blood–brain barrier (BBB), which maintains homeostasis between the cerebral vasculature and the brain, and facilitates active interactions between the peripheral circulation and CNS. The BBB comprises specialized microvascular endothelial cells, pericytes, astrocytes and neurons that couple local neuronal functions to local cerebral blood flow and regulate the transport of blood components into and out of the CNS. Impairment of the BBB and the subsequent infiltration of peripheral immune cells through this barrier play important roles in the pathogenesis of several diseases (Liebner et al., 2018). In particular, immune cells exacerbate the pathology in AD patients and in related mouse models (Town et al., 2005; Baik et al., 2014; Zenaro et al., 2015; Pietronigro et al., 2019; Heneka, 2020; McManus and Heneka, 2020), and inflammation can modify or accelerate the progress of PD (Hirsch and Hunot, 2009; Sulzer et al., 2017). Thus, in vitro models should integrate brain organoids with BBB mimics in order to model neurodegenerative diseases more accurately. Furthermore, drug delivery to the CNS under physiological conditions is restricted by the BBB, so the integration of a functional vascular system into the organoid structure would facilitate the discovery of systemic drugs that target neurodegenerative disorders more effectively. In this review, we focus on recent advances in 3D culture systems for the investigation of neurodegenerative diseases, highlighting their strengths, weaknesses and potential future developments. Advances in these recent in vitro techniques will help us to understand the pathophysiology and underlying mechanisms of human neurodegenerative diseases, leading to the development of efficacious new therapies.
Parkinson’s Disease
Parkinson’s disease is a movement disorder with a variable etiology. It is defined by deep gray matter volume loss caused by the destruction of neurons in the substantia nigra, leading to dopamine deficiency in the basal ganglia. PD affects 1–2% of individuals above the age of 65, and is the second most common age-related neurodegenerative disorder after AD (Van Den Eeden et al., 2003; Poewe et al., 2017). The hallmark of the disease is a progressive loss of nigrostriatal dopaminergic neurons, which normally present unmyelinated axons and form a large number of synapses (Bolam and Pissadaki, 2012; Kalia and Lang, 2015). This leads to several motor symptoms such as bradykinesia, rigidity, resting tremor, and postural instability. PD is also accompanied by a wide range of non-motor symptoms including sleep disturbance, constipation, dementia, cognitive decline, and olfactory deficits, which severely reduce the quality of life for PD patients (Obeso et al., 2010; Savica et al., 2010; Kalia and Lang, 2015). At the neuropathological level, PD is characterized by the accumulation of protein inclusions within the neuronal cell body and processes, known as Lewy bodies (LBs) and Lewy neurites, respectively (Spillantini et al., 1997). LBs are primarily composed of misfolded and insoluble aggregates of the presynaptic neuronal protein α-synuclein, but this protein also accumulates in other tissues (Kalia and Lang, 2015; Lazaro et al., 2017). Genetic forms of PD can provide information about the neuropathological mechanisms of the disease although they account for only 5–10% of all cases (Kalia and Lang, 2015). However, the molecular basis of neuronal degeneration in PD remains unclear, and current therapeutic strategies are limited to attenuating the motor symptoms (Charvin et al., 2018). Drug candidates that were moderately successful in pre-clinical studies have thus far failed to demonstrate efficacy in phase II or III trials, which is unsurprising given our incomplete knowledge of the pathophysiology and etiology of neurodegenerative diseases. Although patient stratification and the timing and duration of treatment are important factors, another key element required for successful drug development is the availability of robust pre-clinical screening tools for drug validation. Experimental tools such as in vitro 3D cell culture models are therefore needed to facilitate the selection of more promising lead compounds in order to eliminate failures at an earlier stage.
Several culture systems have been developed to study the pathogenesis of PD or to identify promising drug leads, each with advantages and limitations (Table 2). Traditional in vitro cell culture techniques usually involve 2D monolayers in standard tissue-culture plates, Petri dishes or cover slips. These are often based on immortalized cell lines such as human embryonic kidney 293 (HEK293) cells, human neuroglioma (H4) cells, or pheochromocytoma (PC12) cells derived from the rat adrenal medulla. The human neuroblastoma cell line SH-SY5Y is widely used in PD research because it reproduces the dopaminergic phenotype typical of PD pathology (Xicoy et al., 2017). A recent report demonstrated the formation of LB-like inclusions in SH-SY5Y cells cultured on a 3D matrix in serum-free Dulbecco’s modified Eagle’s medium (DMEM) for 7 days to allow differentiation, followed by exposure to recombinant human α-synuclein (Taylor-Whiteley et al., 2019). Although SH-SY5Y cells are widely used in PD research, their limitations include the lack of a standardized protocol to maintain them in culture, which leads to variable cell growth and inconsistent experimental outcomes (Buttiglione et al., 2007; Xicoy et al., 2017). Moreover, the survival of SH-SY5Y cells and their differentiation into neuron-like cells requires ECM proteins, neurotrophic factors and serum, and the use of different serum sources or concentrations can have a particularly significant effect on the experimental results (Encinas et al., 2000; Agholme et al., 2010). Depending on the protocol, SH-SY5Y cells can differentiate into various neuronal cell types in addition to dopaminergic cells, so it is necessary to verify the dopaminergic neuronal phenotype after differentiation by confirming the expression of dopamine and noradrenalin neurotransmitter transporters (Korecka et al., 2013; Xicoy et al., 2017). Given the lack of standardized cultivation methods, it is challenging to use SH-SY5Y-derived dopaminergic neurons for high-throughput cell-based screening assays even though the cells are easy to cultivate.
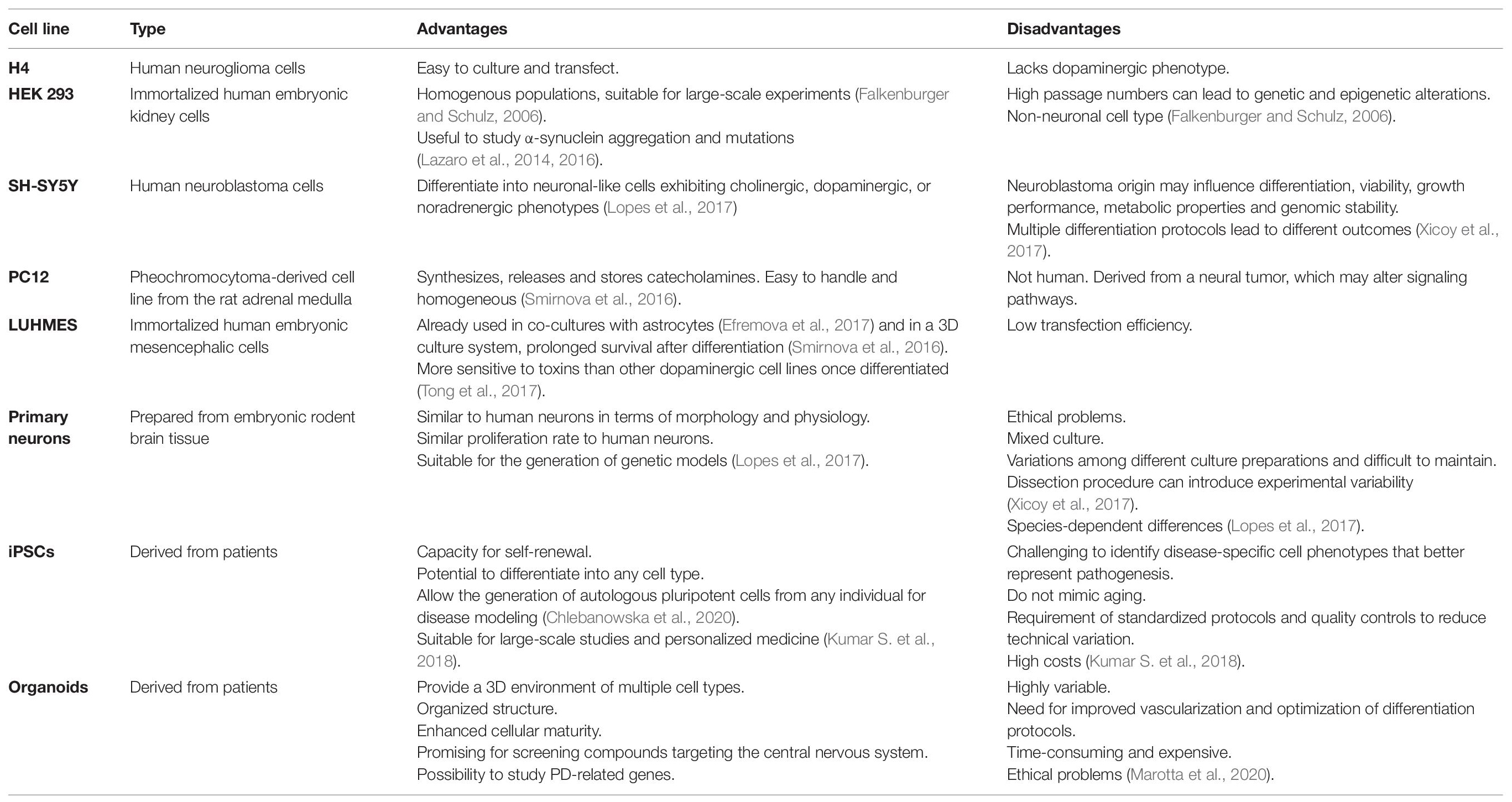
Table 2. Advantages and disadvantages of the different cell lines used to study Parkinson’s disease (iPSC, induced pluripotent stem cell).
In contrast to SH-SY5Y cells, the Lund human mesencephalic (LUHMES) immortalized cell line provides a physiologically relevant system that is compatible with large-scale culture and achieves good batch-to-batch consistency (Zhang et al., 2014; Harischandra et al., 2019). LUHMES cells are derived from healthy 8-week-old human embryonic mesencephalic tissue and are immortalized by inserting the v-myc transgene under the control of a tetracycline-responsive promoter. These cells can be differentiated into mature dopaminergic neurons by adding cAMP, tetracycline and glial cell line-derived neurotrophic factor to the culture medium (Lotharius et al., 2005). LUHMES cells were used to develop a 3D model for neurotoxicity studies by applying the gyratory shaking technique, in which cells are shaken continually to encourage the formation of spherical aggregates containing astrocytes, neurons, oligodendrocytes and microglia (Van Vliet et al., 2008; Smirnova et al., 2016). The advantages of this system include (1) the ability of LUHMES cells to acquire a phenotype that is biochemically and morphologically similar to primary neurons, (2) the formation of a natural ECM that promotes cell–cell interactions, (3) the rapid maturation of the system, with astrocytes, neurons and oligodendrocytes undergoing both myelination and synaptogenesis within 25 days, and (4) the ability of LUHMES cell cultures to be maintained for 2 months while the cell populations continue to mature (Honegger, 2001; Van Vliet et al., 2008).
Primary cultures have the potential to overcome many of the difficulties inherent to cell lines, but isolating and culturing primary dopaminergic neurons from the post-mortem brains of adult/elderly patients is difficult. Therefore, primary dopaminergic neurons are usually obtained from embryonic murine brain tissue, particularly from the central midbrain area, because these cells differentiate rapidly in culture and form neurites and synapses (Gaven et al., 2014; Weinert et al., 2015). The primary neurons in this type of system are often contaminated with glial cells, but this can be regarded as an advantage. For example, this method has been exploited to study the therapeutic effect of microglial modulation in a mixed culture of primary neurons and microglia, thus showing how the microglia and the factors they release into the shared environment may affect neuronal function and survival (Che et al., 2018). Primary cortical neuron cultures have also been used to characterize the transport of synthetic α-synuclein fibrils (Freundt et al., 2012). The authors used a microfluidic device to separate neuronal cell bodies from their axons and from second-order neurons, and demonstrated that α-synuclein is internalized by neurons, can undergo both anterograde and retrograde transport, and can be released from primary neurons to be taken up by second-order neurons.
Although in vitro primary cultures have provided important clues about the mechanisms of PD and potential drug targets, they do not replicate the organization of cells and ECM within the CNS. More advanced systems, which are closer to in vivo animal disease models of PD, include organotypic cultures, 3D cultures and organoids (Chambers et al., 2009; Zhang et al., 2009; Shoji et al., 2016; Chlebanowska et al., 2020). An early example of a murine organotypic culture for PD research modeled nigrostriatal degeneration by cutting brains along the dorsoventral axis and culturing the slices containing cortex, corpus callosum, subventricular zone, striatum and substantia nigra, in order to ensure both the dopaminergic and glutamatergic pathways were represented in each slice (Cavaliere et al., 2010). Nigrostriatal pathway degeneration was induced by mechanical damage, in contrast to other models that used toxins such as 1-methyl-4-phenylpyridinium or 6-hydroxydopamine (Kearns et al., 2006). A more recent ex vivo culture system that models both the early and late stages of PD was prepared by the sagittal dissection of rat brains, allowing the simultaneous observation of cholinergic, GABAergic and dopaminergic neurons and the brain vasculature (Daviaud et al., 2014). Mechanical damage during slice preparation led to progressive degeneration of the nigrostriatal pathway, beginning with dopaminergic degeneration in the striatum and continuing, over the following weeks, with the degeneration of cholinergic and GABAergic neurons (Daviaud et al., 2014). Despite several advantages such as the replication of physiological processes and the well-controlled genetic background, these organotypic models are difficult to reproduce because the dissection procedures require high precision (Stahl et al., 2009). Moreover, the culture conditions required to maintain the brain tissue slices are difficult to standardize, and these models originate from animals with significant differences in neural anatomy, physiology, regulation, gene expression patterns, and drug metabolism compared to humans. The preparation of such primary cultures is expensive and time-consuming, requires significant expertise, and is not suitable for large-scale studies (Lopes et al., 2017).
To establish reproducible models more closely related to human pathophysiology, iPSC lines have recently emerged as one of the hottest and fastest moving topics in the life sciences. Many researchers have established PD-specific iPSC models by reprogramming somatic cells from PD patients. The first PD-specific iPSC line was derived from a patient affected by a sporadic form of the disease (Park et al., 2008). Since then, iPSC models of PD have been established from patients with susceptibility alleles in genes such as LRRK2, PARKIN, SNCA, GBA, and PINK1.
Mutations in the gene encoding leucine-rich-repeat kinase 2 (LRRK2) correlate with enteric inflammation and reinforce the role of peripheral inflammation in the initiation and/or progression of PD (Devos et al., 2013). Patient-derived iPSCs carrying the autosomal dominant G2019S mutation in the LRRK2 gene have therefore been cultivated as 3D human neuro-ectodermal spheres (hNESs) and human intestinal organoids (hIOs) to study gene expression profiles linked to LRRK2 in the neural and intestinal environments (Son et al., 2017). This revealed a broader alteration in gene expression profiles in the hIOs compared to hNESs, suggesting that LRRK2-G2019S may preferentially trigger the correlated intestinal symptoms of PD. An innovative protocol to generate neural tube lineages (including motor neurons and midbrain dopaminergic neurons) and neural crest lineages has been established using only small molecule neural precursor cells, which are robust, undergo immortal expansion, and do not require cumbersome manual culture and selection steps (Reinhardt et al., 2013). This protocol was recently combined with a microfluidics system to generate a 3D cell culture model based on neurons derived from human neuro-epithelial stem cells (hNESCs) from PD patients carrying the LRRK2-G2019S mutation (Bolognin et al., 2019). These authors generated a mixed population of neural cells that precisely captured the hallmarks of LRRK2 pathogenesis, including degeneration, cell loss, and mitochondrial impairment. The results obtained using this in vitro system showed that the genetic background of PD patients can influence the degeneration of dopaminergic neurons, contributing to phenotypes other than that of the G2019S mutation itself. These data support the use of advanced in vitro models for future patient stratification and personalized drug development (Bolognin et al., 2019). The pioneering model consist in the differentiation of iPSCs into dopaminergic neurons inside a microfluidic bioreactor with a microtiter plate format. Within each bioreactor, cells embedded in Matrigel are loaded in the culture lane and perfused by medium flow. However, this system requires some improvements, including the use of alternatives to Matrigel in order to increase cell viability (Moreno et al., 2015). Recently, a microfluidic bioreactor was used to develop a high-throughput model that integrates laboratory automation technology, resulting in a robotic microfluidic cell culture system named Pelican. Specifically, this new system automates the cell culture protocols for hNESCs, allowing their experimentally reproducible differentiation into dopaminergic neurons. This platform may help to harmonize protocols between laboratories according to the standards of the SiLA consortium1 but it can also be adapted to different experimental conditions (Kane et al., 2019).
A 3D human midbrain organoid (hMO) populated with midbrain dopaminergic neurons (mDANs) derived from iPSCs has recently been described (Smits et al., 2019). The comparison of hMOs from healthy subjects and PD patients with the LRRK2-G2019S mutation revealed a reduction in the number and complexity of mDANs in the disease model, suggesting a neurodevelopmental defect in mDANs expressing LRRK2-G2019S. In addition to organoids derived from the genetic form of PD, the first organoid model of idiopathic PD was recently reported (Chlebanowska et al., 2020). The iPSCs were derived from the peripheral blood mononuclear cells of patients with idiopathic PD and were differentiated into large multicellular organoid-like structures. The authors observed statistical differences in the expression of early and late neuronal markers when comparing organoids prepared from PD patients and healthy volunteers, suggesting that hMOs have a remarkable potential for the investigation of PD pathogenesis in vitro. Overall, neurons derived from iPSCs and organoids from familial and idiopathic PD patients replicate the key pathological phenotypes of PD and are useful tools to study molecular pathways involved in the disease. They are also suitable for target validation, filling the gap between animal models and humans. Such 3D systems are necessary because they form complex structures that cannot be replicated in 2D models (Marotta et al., 2020). However, the application of such 3D models for large-scale studies and the generation of patient-specific organoids remains limited by the laborious procedures and high costs of preparation, and the need for written informed consent and approval from the ethics commission governing the institution conducting the experiments. This will restrict the practical application of patient-derived models for the foreseeable future.
Alzheimer’s Disease
Alzheimer’s disease is a severe neurodegenerative disease characterized by the profound loss of cognitive functions and behavioral abilities, causing progressive deterioration in the patient’s quality of life. The pathogenesis of AD is incompletely understood (Scheltens et al., 2016). Two main forms of AD are recognized: early-onset familial Alzheimer’s disease (EOFAD) and late-onset Alzheimer’s disease (LOAD). EOFAD is relatively uncommon, accounting for less than 5% of all cases. It is diagnosed before the age 65, with an autosomal dominant pattern of inheritance reflecting the presence of one of 200 mutations discovered thus far in three key genes: amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). LOAD is the most common form of AD, also known as sporadic AD, and typically occurs in people of age 65 or older without a family history of dementia (Bekris et al., 2010; Giri et al., 2016). Since 1992, the etiology and pathogenesis of AD has been explained by the amyloid cascade hypothesis, in which the accumulation of pathogenetic amyloid β (Aβ) protein, derived from APP, induces a vicious cycle that triggers the accumulation of neurofibrillary tangles (NFTs), neuronal cell death, and ultimately dementia (Reitz, 2012). The failure of anti-Aβ therapy, which was largely successful in mouse models of AD, helped to revise this hypothesis and define AD as a multifactorial disorder, also highlighting the limitations of animal disease models (Iqbal and Grundke-Iqbal, 2010). Animal models of AD proved to be inaccurate representations of human AD pathology, exclusively mimicking EOFAD (De Strooper, 2014; Henley et al., 2014). Indeed, animal models of AD tend to manifest only some pathological features, lacking important components such as extensive neuronal loss and the development of NFTs. One potential explanation is that rodent tau proteins may not be prone to aggregate formation due to differences in sequence and structure, together with the short life span of mice which does not allow sufficient time for the accumulation of events that take decades in humans (Laferla and Green, 2012). The development of sophisticated 3D in vitro models therefore provides a powerful complementary approach to overcome the limitations of current AD transgenic mice.
Many groups have developed 2D models based on iPSCs (Yagi et al., 2011; Israel et al., 2012; Mohamet et al., 2014; Hu et al., 2015; Moore et al., 2015; Lee et al., 2016; Li et al., 2016). In some cases, iPSCs were prepared from fibroblasts of EOFAD patients with mutations in PSEN1 (A246E) or PSEN2 (N141I), and neurons differentiated from the iPSCs were shown to secrete more Aβ42 than healthy matched controls, elevating the Aβ42 to Aβ40 ratio (Yagi et al., 2011). The reprogramming of fibroblasts from patients with familial and sporadic forms of AD replicated the significantly higher levels of Aβ40, active glycogen synthase kinase-3β (aGSK-3β) and hyperphosphorylated tau protein (Israel et al., 2012). The treatment of neurons derived from these AD patients with β-secretase inhibitors significantly reduced the levels of hyperphosphorylated tau and aGSK-3β, whereas a γ-secretase inhibitor showed no effects compared to controls, suggesting direct crosstalk between APP proteolytic processing during the activation of GSK-3β and tau phosphorylation in human neurons (Israel et al., 2012). CRISPR/Cas9 technology has been combined with iPSCs to generate knock-in human neurons carrying heterozygous or homozygous EOFAD mutations such as APPswe and PSEN1M146V (Paquet et al., 2016). Although these 2D models have revealed some of the key pathophysiological mechanisms of AD, they share with other 2D models the inability to reproduce all disease hallmarks, like any other model generated so far. For example, the levels of Aβ generated by these models were insufficient for the formation of plaques and related pathological features. Furthermore, the use of post-mitotic neurons prevented long-term cultivation and the in vitro models therefore could not replicate the age-dependent pathogenic events in the human AD brain (D’Avanzo et al., 2015). Most importantly, these 2D neuronal cultures lacked the supporting functions of glial cells, which play a key role in the pathogenesis of AD (Nagele et al., 2004; Gonzalez-Reyes et al., 2017).
The limitations of 2D cultures have encouraged the development of 3D models of AD, the first of which was based on an immortalized human neural stem cell line (ReN) containing mutations in the APP (K670N/M671L and V717I) and PSEN1 (ΔE9) genes (Choi et al., 2014). These cells were found to accumulate both senile plaques and NFTs, two hallmarks of the disease not previously observed in 2D cultures or most animal models. Furthermore, ReN cells can differentiate into either neurons or glia, and the presence of Matrigel in the culture medium inhibits Aβ diffusion, promoting aggregation and plaque formation.
Cerebral organoids are the latest avenue for in vitro AD research, allowing the generation of organized structures similar to the human cortex. Human ReN cells were initially used to generate neurospheroids within arrays of microwells, and were found to produce both Aβ plaques and hyperphosphorylated tau protein after 8 weeks in culture (Jorfi et al., 2018). Another recent development is the use of vascularized human cortical organoids (vhCOs), which have the advantage to overcome the limitation of conventional organoids (these often feature necrotic regions due to limited oxygen and nutrient diffusion) by coupling cerebral organoids to a perfusion system that mimics the cortical vasculature (Kelava and Lancaster, 2016). This system is also superior to earlier brain organoids because vhCOs possess classical BBB properties such as the expression of tight junction proteins (ZO-1, occludin, and claudin-5) and nutrient transporters (P-glycoprotein and GLUT1), as well as high trans-endothelial electrical resistance. However, one disadvantage of vhCOs is that they lack blood cells within their vascular structures. In the near future, vhCOs may allow the screening of drugs that regulate the permeability of the BBB, thus providing an authentic model to study the regulation of interactions between the CNS and the periphery (e.g., the trafficking of Aβ and other molecules), as well as the mechanisms involved in the recruitment of leukocytes through the BBB during inflammation (Cakir et al., 2019).
To mimic blood flow, a triculture AD model including neurons, astrocytes and microglia was recently developed using a microfluidics platform (Park et al., 2018). These engineered human neural progenitor cells contained in a two-chamber microfluidic device replicated key features of AD, such as the aggregation of Aβ, the accumulation of hyperphosphorylated tau protein, and neuroinflammation. Interestingly, the authors demonstrated physiologically relevant interactions among neuronal cells, such as microglial recruitment, the release of pro-inflammatory cytokines and chemokines (CCL2, TNFα, and IFNγ) regulated by AD neurons and astrocytes, and microglial neurotoxic activation contributing to neuron/astrocyte damage. In addition to glia, the brain also contains many other non-neuronal cell types that ensure the long-term survival and function of neurons (Zhao et al., 2015). For example, the BBB blocks the passage of many cells and molecules between the CNS and peripheral blood circulation, and oligodendrocytes insulate neuronal axons and promote rapid axonal transmission. However, the contribution of each of these non-neuronal cell types to AD-associated neurodegeneration is not fully understood. A complex 3D model of neural cell culture in a microfluidic system was recently developed by adding brain endothelial cells with a BBB-like phenotype (Shin et al., 2019). This AD platform simulates the cerebral–vascular interface, successfully mimicking several of the vascular phenotypes observed in AD patients, including the greater permeability of the BBB coincident with the downregulation of certain tight junction proteins (claudin-1 and claudin-5), adherens junction proteins and VE-cadherin, as well as the upregulation of matrix-metalloproteinase-2 (MMP-2), the accumulation of reactive oxygen species (ROS), and the aggregation of Aβ on the abluminal side of the BBB endothelium. This experimental system may therefore reveal the physiological and pathological mechanisms of BBB dysfunction in AD and could be used as a standardized drug-screening platform.
As discussed above for PD models, patient-derived iPSCs have also been used to model AD. The first was a 3D neurospheroid model based on iPSCs derived from the peripheral leukocytes of five patients with sporadic AD (Lee et al., 2016). The authors demonstrated that the administration of BACE1 or γ-secretase inhibited the formation of pathological Aβ peptides. Interestingly, the use of 3D neurospheroids highlighted individual variations in the efficacy of BACE1 that were related to differences in individual genetic backgrounds (APOE genotypes). This 3D model not only provided the basis for more accurate drug-screening methods, but facilitated subsequent studies of individual phenotype variations thus allowing a personalized approach for the treatment of AD (Lee et al., 2016). Recently, a physiological 3D model of AD was described in which neural areas with a cortical-like organization were generated from fibroblast-derived iPSCs donated by adult EOFAD and Down syndrome patients (Gonzalez et al., 2018). These highly reproducible cerebral organoids spontaneously accumulated aggregates of Aβ and hyperphosphorylated tau protein. The analysis of caspase-3 activation indicated a rate of neuronal death proportional to the accumulation of protein aggregates, suggesting that cerebral organoid cultures develop certain neurodegenerative features in common with the AD brain (Gonzalez et al., 2018). Nevertheless, these methods are still limited by their inability to model more complex events including cell-cell interactions and migration in the developing brain, and future research should focus on improvements that accommodate such features.
Oligodendrocyte dysfunction and the loss of white matter during the progression of AD are key features of human patients and rodent models, probably contributing to the neuronal degeneration (Desai et al., 2010; Bartzokis, 2011). The differentiation of iPSCs can be used to produce oligodendrocytes, and recently this has been exploited to generate oligocortical spheroids for the analysis of myelination in the CNS (Hu et al., 2009; Ehrlich et al., 2017; Hubler et al., 2018; Madhavan et al., 2018; Marton et al., 2019). Moreover, neurons do not establish mature synaptic connections in 3D culture models (Huch et al., 2017) and the proportion of cells differs from that in the human brain, mainly comprising neurons with few glial cells and no oligodendrocytes. These sophisticated in vitro culture methods should therefore be improved to represent the cell population found in vivo, which will in turn make it easier to identify the pathological mechanisms underlying AD. The use of AD mini-brains offers the possibility of validating disease mechanisms and should lead to the identification of new pathways contributing to the progression of AD.
Prion Diseases
The hallmark of several age-related neurodegenerative diseases is the formation and aggregation of misfolded proteins in the CNS. PD and AD share key biophysical and biochemical characteristics with prion diseases, a group of neurodegenerative diseases triggered by the misfolding of the cellular prion-related protein (PrPC). The molecular mechanism underlying these diseases is the conformational misfolding of monomeric PrPC to form protein aggregates known as scrapie prion proteins (PrPSc) or proteinaceous infectious particles. The abnormally folded protein is protease resistant and rich in β-strands, enabling the formation of oligomer/fibril structures that are involved in three pathological forms of disease: the sporadic form, also called Creutzfeldt-Jakob disease (CJD), the genetic form, and the acquired form. The genetic forms of prion diseases are caused by mutations in the prion protein gene (PRNP) that favor the abnormal folding events discussed above and are classified as genetic CJD, Gerstmann-Sträussler-Scheinker syndrome, and fatal familial insomnia. The acquired forms of are known as variant CJD (vCJD), iatrogenic CJD (iCJD), and kuru (Collins et al., 2001; Geschwind, 2015; Scheckel and Aguzzi, 2018). However, the most common form affecting humans is sporadic CJD, which accounts for 80–95% of cases. The peculiarity of these diseases is the seeding property of PrPSc, which is able to convert a normal PrPC into another copy of PrPSc with the same seeding capability, leading to the exponential formation of prions (Geschwind, 2015). The pathological hallmarks of prion diseases include neuronal loss, the activation of microglia and astrocytes, spongiform changes, and the formation of PrPSc aggregates and deposits. The pathogenesis of prion diseases is not fully understood and diagnosis is possible only when the disease has already reached an advanced stage (Knight, 2008).
The development of 3D cell culture models based on the differentiation of human iPSCs into neuronal tissue has produced a range of high-throughput platforms to test infection methods and treatments, and to address fundamental questions in prion biology. PrP-expressing human and animal cell lines in 2D culture generally do not establish stable prion infections and do not display cytopathic signs. The first suitable model was based on murine cerebellar organotypic slices, which revealed the amplification of PrPSc following exposure to prions (Falsig and Aguzzi, 2008). Improved models have been developed based on murine neural stem cells, early examples of which included 3D neurospheroids composed of neurons and astrocytes (Collins and Haigh, 2017). The neurospheroids reached maturity after 10 days and could be maintained in culture for up to 1 month. Prions were able to spread and induce toxic changes in the neurospheroids during growth and after differentiation, allowing the investigation of underlying mechanisms (Collins and Haigh, 2017). Despite the advantages of this model, the lack of microglia may influence the experimental outcome. Indeed, prion diseases are normally associated with astrogliosis, which also involves the recruitment of microglia, and their absence or depletion may influence the growth rate of astrocytes (Marella and Chabry, 2004).
Recently, cerebral organoids derived from human iPSCs were established for the first time as a 3D model to study genetic prion diseases in individuals with a predisposition caused by the mutation PRNP-Y218N (Matamoros-Angles et al., 2018). Although the cultures were characterized by astrogliosis and tau hyperphosphorylation, this model failed to replicate PrPSc accumulation, and neurons generated from human iPSCs with or without the Y218N mutation were unable to propagate infection following inoculation with human PrPSc from either spontaneous CJD or PRNP-Y218N individuals (Matamoros-Angles et al., 2018). To replicate human prion infection and pathogenesis, cerebral organoids were inoculated with brain homogenates from post-mortem samples of patients affected by different forms of spontaneous CJD, revealing that seeding activity was influenced by the spontaneous CJD subtype (Groveman et al., 2019). This new in vitro 3D model for the investigation of human prion disease provides insight into the pathological events caused by different human prion subtypes, and offers a promising platform for drug discovery using a relevant human tissue background. Although cerebral organoids can be maintained in culture for a long time, their limitations include structural heterogeneity, the lack of vascularization, and the absence of non-neuronal cells such as endothelial cells and microglia.
Multiple Sclerosis
Multiple sclerosis is the most common chronic inflammatory, demyelinating and neurodegenerative disease of the CNS in young adults, with an onset at 20–40 years of age and a higher prevalence in women (Compston and Coles, 2008). MS neuropathology involves the appearance of focal plaques containing demyelinated axons, proliferating astrocytes, activated microglia, infiltrating lymphocytes and macrophages, and a reduced population of oligodendrocytes, typically located around post-capillary venules characterized by a breakdown of the BBB (Mahad et al., 2015; Huang et al., 2017; Stys and Tsutsui, 2019). Clinical symptoms of MS include motor dysfunction, fatigue, tremor, nystagmus, acute paralysis, loss of coordination or balance, numbness, disturbed speech and vision, and cognitive impairment (Ghasemi et al., 2017). The disease usually begins with a primary relapsing-remitting phase (RRMS) during which the symptoms are intermittent. Over the next 10–15 years, this transitions to a secondary progressive phase (SPMS) with continuous deterioration. But ∼15% of cases are classed as primary progressive (PPMS), in which the disease progression is relentless from the onset (Mahad et al., 2015; Huang et al., 2017). MS is a heterogeneous, multifactorial, immune-mediated disease that is influenced by both genetic and environmental factors. Two main hypotheses have been proposed for the pathogenetic mechanism: (1) the “outside-in” hypothesis, mostly based on findings from experimental animal models of MS, states that the CNS is invaded by auto-reactive T cells activated in the periphery, contributing to inflammation, BBB leakage and tissue damage (Lucchinetti et al., 2000; Frohman et al., 2006); and (2) the “inside-out” hypothesis states that MS is a primary degenerative disease but its severity increases following an amplification of the immune response (Henderson et al., 2009). In the latter case, the trigger may be a primary defect in the oligodendrocytes, such as a mutation that causes them to die off, resulting in the activation of microglial cells (Hemmer et al., 2015). Drugs that promote the remyelination of damaged axons are required to overcome the neurodegenerative phase of MS. However, the molecular and cellular basis of the myelin repair deficiency during the progression of MS are still unclear.
Given the complexity of the neurodegenerative mechanism during the progression of MS, rodent 3D organotypic brain slices have been widely used to study pathogenesis because they contain different regions of the CNS, allowing experimental manipulations that cannot be easily made in vivo. Organotypic brain slices can be maintained ex vivo for several months and allow researchers to study the physiological process of myelination in a system where complex cell–cell relationships are preserved (Schnadelbach et al., 2001). The tissue slices are often placed on porous membranes in cell culture dishes and cultured at an air–liquid interface, with the medium in contact with the permeable membrane (Gogolla et al., 2006). The slices can be demyelinated in vitro by applying toxins or an immune challenge, but undergo spontaneous remyelination (Birgbauer et al., 2004; Zhang et al., 2017b). This in vitro system can also be used to screen promising drugs that promote remyelination (Zhang et al., 2011; Doussau et al., 2017; Tan et al., 2018).
Although rodent brain slices have been widely used in MS research, several compounds that promote remyelination and neuronal survival in rodent models of MS have failed when tested in human clinical trials, highlighting the inability of animal models to completely replicate the complexities of the human disease phenotype (t Hart et al., 2011). As described above for PD, AD and prion diseases, new in vitro tools based on 3D cultures are therefore required to replicate the pathophysiology of human MS and identify the most promising drug targets. Culture systems based on human oligodendrocytes or oligodendrocyte precursor cells (OPCs) have been developed to address the deficiencies of animal MS models. These are preferred because they have the ability to differentiate and generate myelin in vitro in the absence of signals from axons (Bechler et al., 2015). Protocols based on OPCs have been particularly successful because they can generate mature myelinating oligodendrocytes (Dugas and Emery, 2013; Barateiro and Fernandes, 2014; Chew et al., 2014). Mouse, rat and human OPCs differ in terms of their longevity in culture, with rat OPCs proving the easiest to isolate and maintain whereas human OPCs take the longest to develop the classical mature oligodendrocyte phenotype. Moreover, human OPCs must be isolated from brain biopsies, with the attendant issues surrounding ethical clearance. For these reasons, human oligodendrocyte cultures are usually initiated using OPCs generated de novo from the differentiation of human embryonic stem cells, iPSCs (Hu et al., 2009), fetal cortical neurospheroid-derived cells, or umbilical cord-derived stem cells (Chen et al., 2013; Leite et al., 2014).
The use of stem cells raises the possibility that 3D cultures could be generated from the cells of patients carrying specific MS susceptibility alleles, allowing the analysis of genetic mechanisms and the development of personalized therapies. Dermal fibroblasts from a 35-year-old patient affected by RRMS were reprogrammed; these were named MS iPSCs and were successfully differentiated into mature astrocytes, oligodendrocytes and neurons with normal karyotypes, although MS iPSC-derived neurons showed atypical electrophysiological characteristics (Song et al., 2012). In addition, iPSCs from PPMS patients of both sexes in the age range 50–62 years have been used to generate OPCs that achieved the efficient myelination of neurons in vivo and could be suitable for the development of autologous cell-replacement therapies for MS in the future (Douvaras et al., 2014). The advent of iPSC-derived OPCs allows researchers to study the impact of genetic variants on specific cell populations to facilitate the identification of disease subtypes and the functional evaluation of genetic variants and their role in MS (Di Ruscio et al., 2015). MS is a complex, multifactorial disease with multiple susceptibility phenotypes, many of which are related to the adaptive immune system (International Multiple Sclerosis Genetics Consortium [IMSGC] et al., 2011; Stys and Tsutsui, 2019). A broad panel of patients and controls is therefore required to generate a sufficient number of cell lines for the detection and validation of biologically relevant MS phenotypes (Song et al., 2012). The real limitation of MS organotypic models, such as iPSC-derived OPC cultures, is that they exclude the peripheral immune system, which is a key player in both the progression of MS and regeneration. It is unclear whether neuronal damage reflects the different phenotypes of autoreactive T lymphocytes. Therefore, it may be possible in the future to establish co-cultures of neurons and T cells derived from individual MS patients. This would allow both the detection of genetic variants in the T cells involved in neuronal damage and the screening of neuroprotective compounds blocking the detrimental effects of this T cell subpopulation, in order to improve our understanding of their role in the progression of MS.
Conclusion and Future Directions
Cellular models of neurodegenerative diseases range in complexity from conventional monolayers derived from immortalized cell lines to complex multicellular 3D tissue mimetics based on patient-derived iPSCs, which can replicate many disease hallmarks and in vivo physiological conditions, such as protein aggregation. Indeed, 3D cell culture systems can recapitulate the extracellular aggregation of Aβ and NFT typical of AD (Choi et al., 2016) and can spread the PrPSc responsible for prion diseases (Falsig and Aguzzi, 2008; Collins and Haigh, 2017). Furthermore, the aggregation of α-synuclein has been demonstrated in neurons differentiated from iPSCs derived from sporadic and familial PD patients, suggesting they can reproduce this hallmark of PD without the use of exogenous factors (Oh, 2019).
Traditional cell culture systems and animal models have improved our understanding of human neurodegenerative diseases, but models based on recent technological innovations in 3D culture systems can achieve the better characterization of pathological mechanisms, which also makes them more suitable for high-throughput drug screening. Indeed, the advent of new technologies involving the generation of organoids from iPSCs and the use of microfluidic devices could accelerate drug discovery compared to current approaches based on animal models. Despite these recent advances, we still lack a 3D model that recapitulates all the key aspects of neurodegenerative diseases and thus allows the detailed analysis of their pathophysiology.
Most in vitro cultures are composed primarily of neuronal cells, and their complexity (and therefore suitability as disease models) should be increased by adding microglial cells, astrocytes, oligodendrocytes, and pericytes to better mimic the structural and molecular complexity of the CNS environment in different diseases (Ormel et al., 2018; Song et al., 2019; Speicher et al., 2019). The genetic background of the cells used to establish the model may also influence drug responses, as recently shown for iPSCs isolated from PD patients and cultured in a microfluidic 3D platform (Bolognin et al., 2019). The unique genetic background of each individual can affect their susceptibility to neurodegenerative diseases. Patient-derived iPSCs could therefore reveal common pathways even in sporadic disease cases, allowing the development and testing of new therapeutic strategies. Interestingly, iPSC-derived neurons from patients with sporadic AD featured mutations that have not been identified by genome-wide association studies, and these mutations generated neuronal phenotypes in a manner analogous to familial AD, suggesting common pathogenetic mechanisms (Israel et al., 2012). However, previous experience with the handling of iPSCs has revealed the potential for in vitro reprogramming, which is a significant drawback. Polymorphisms affecting neuronal functions could overcome defects introduced during reprogramming process or generated by genomic instability due to the long in vitro culture durations. Furthermore, donor age, sex, and ancestry could influence the physiology of the iPSCs (Mackey et al., 2018). X-linked genes in cells generated from female patients with neurological diseases (including MS) tend to be inactivated, leading to a significant effect in the neurodegeneration models (Mekhoubad et al., 2012). Moreover, skin fibroblasts from older donors show very inefficient iPSC reprogramming (Ohmine et al., 2012), so we must also take into account the fact that neurodegenerative diseases are often aged related, or (in the case of MS) diagnosed later in life. Fibroblasts were recently shown to possess anatomic positional memory, which is clearly important for reprogramming strategies (Sacco et al., 2019). Moreover, in aged-associated diseases such as PD and AD, neuronal cells suffer epigenetic drift, mitochondrial dysfunction and the accumulation of damaged proteins, which can disrupt gene regulation and homeostasis leading to abnormal phenotypes. These features could be lost by cell rejuvenation during the reprogramming of iPSCs, although this could be addressed by the direct conversion of aging neurons (Mertens et al., 2018). It therefore follows that the variability, together with the inadequate purity of the reprogrammed cells and the lack of standardized methodologies still hamper the reproducibility of iPSCs-derived methodologies across different laboratories.
Brain function and homeostasis depend not only on neuronal cells but also on the regulation of cerebral blood flow, which supplies nutrients and oxygen while removing waste products. The BBB is essential in this context because it connects the CNS to both the peripheral circulation and the immune system (Zhao et al., 2015). The disruption of the BBB followed by the infiltration of peripheral immune cells is correlated with the pathogenesis of neurodegenerative diseases such as AD and PD (Schwartz and Shechter, 2010; Poewe et al., 2017; Zenaro et al., 2017). Complex in vitro models that include fluidic systems to mimic the BBB are therefore required to fully replicate neurodegenerative diseases affecting the CNS (Aucouturier et al., 2000; Zlokovic, 2008; Lopes Pinheiro et al., 2016). Recent studies have included vascularization in 3D models, highlighting the importance of this component (Lee et al., 2017; Mansour et al., 2018; Pham et al., 2018; Cakir et al., 2019; Worsdorfer et al., 2019). The most recent BBB model uses a chip to mimic the physiological interactions between brain endothelial cells, pericytes, and astrocytes, although the neuronal component and immune cells are still missing (Ahn et al., 2020). The role of immune dysfunction in neurodegeneration can only be understood in detail by the incorporation of immune cells into future 3D models (Figure 1). Indeed, an unlimited source of circulating leukocytes can be obtained from human blood, and these cells should therefore be considered in future models of neurodegenerative diseases. A recent study using a microfluidic system to mimic central–peripheral innate immunity in AD (Park et al., 2019) suggested that targeting the crosstalk between central and peripheral immune cells may reduce the immunological burden in other neuroinflammatory diseases of the CNS. Moreover, the BBB should be considered as a key component of 3D brain models because it is one of the major challenges in the development of drugs targeting the CNS (Pandit et al., 2019). The BBB prevents up to 98% of small-molecule drugs from reaching the brain (Pardridge, 2005; Neuwelt et al., 2008). The delivery of drugs to the CNS therefore requires new strategies that take advantage of BBB transporter systems, highlighting the need for more research into the physiology this barrier.
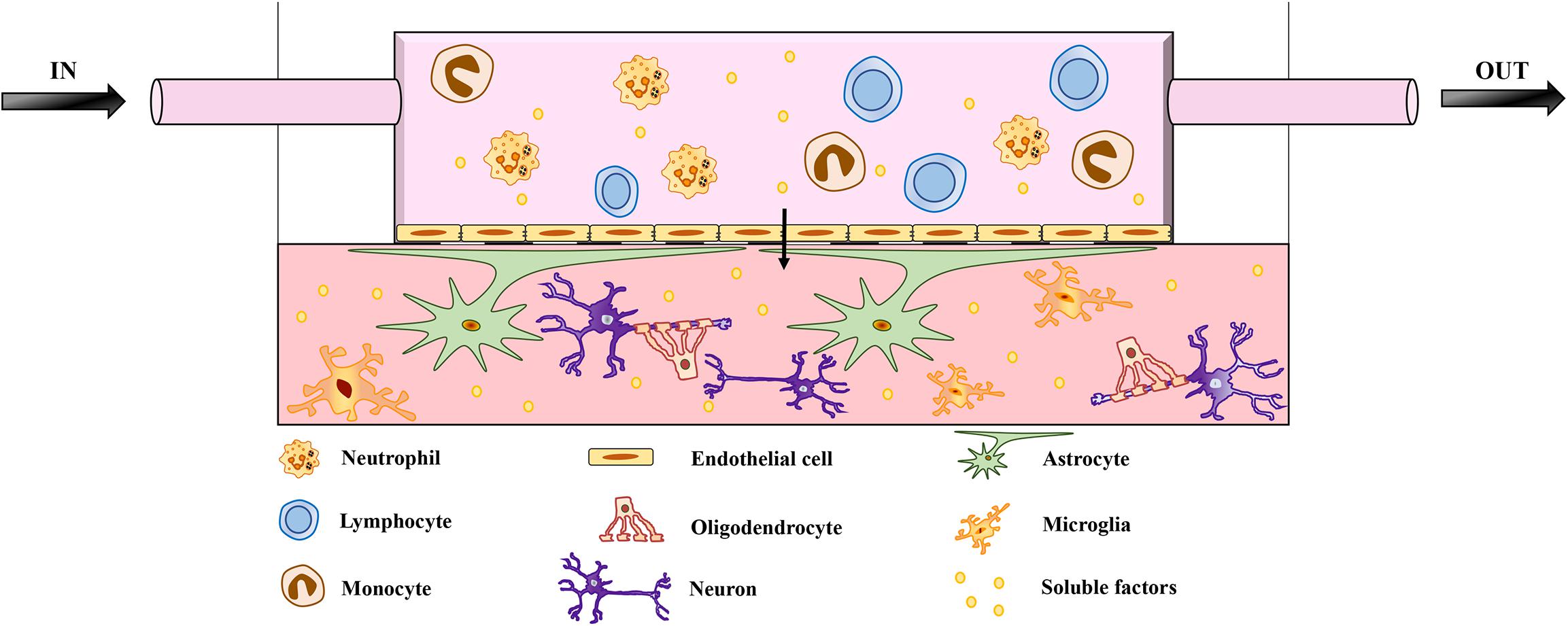
Figure 1. Schematic representation of microfluidic brain model. The model features neurons and glial cells embedded in a matrix. The architecture includes a flow of medium mimicking the BBB, enriched with soluble factors and peripheral immune cells, which are key players in neuroinflammation and neurodegeneration. The migration of peripheral immune cells through the BBB has been implicated in the pathogenesis of several neurodegenerative diseases. The role of infiltrating peripheral immune cells has been investigated in detail for MS, which involves the breakdown of the BBB and multifocal inflammation caused by the innate and adaptive immune systems. However, BBB impairment and the infiltration of peripheral immune cells also correlate with the pathogenesis of other neurodegenerative diseases, such as AD and PD. Adding a fluidic system to mimic the BBB is therefore necessary to investigate the pathological mechanisms of neurodegenerative diseases and eventually to study the transport of drugs across the BBB.
Although in vitro models of neurodegenerative diseases are still incomplete, the 3D culture methods discussed herein offer an important new strategy to characterize disease mechanisms, leading to the discovery of new therapies. Personalized treatments are not yet economically viable, but 3D models based on patient-derived cells will show how the genetic landscape of the human population contributes to the pathogenesis of neurodegenerative diseases, bringing the prospect of personalized medicine another step closer.
Author Contributions
AS and GI searched the literature, performed review design, designed the figure and tables, and wrote the manuscript. BR, EZ, and GC contributed to review conception and manuscript revision.
Funding
This work was supported in part by the European Research Council (ERC) grants (695714 IMMUNOALZHEIMER, 693606 IMPEDE, and 261079 NEUROTRAFFICKING), Alzheimer’s Drug Discovery Foundation (ADDF) United States, Fondazione Italiana Sclerosi Multipla (FISM), and by the University of Verona (to GC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Agholme, L., Lindstrom, T., Kagedal, K., Marcusson, J., and Hallbeck, M. (2010). An in vitro model for neuroscience: differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J. Alzheimers Dis. 20, 1069–1082. doi: 10.3233/JAD-2010-091363
Ahn, S. I., Sei, Y. J., Park, H. J., Kim, J., Ryu, Y., Choi, J. J., et al. (2020). Microengineered human blood-brain barrier platform for understanding nanoparticle transport mechanisms. Nat. Commun. 11:175. doi: 10.1038/s41467-019-13896-7
Almeida, S., Zhang, Z., Coppola, G., Mao, W., Futai, K., Karydas, A., et al. (2012). Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2, 789–798. doi: 10.1016/j.celrep.2012.09.007
An, M. C., Zhang, N., Scott, G., Montoro, D., Wittkop, T., Mooney, S., et al. (2012). Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11, 253–263. doi: 10.1016/j.stem.2012.04.026
Anderson, R. M., Hadjichrysanthou, C., Evans, S., and Wong, M. M. (2017). Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet 390, 2327–2329. doi: 10.1016/S0140-6736(17)32399-1
Aucouturier, P., Carp, R. I., Carnaud, C., and Wisniewski, T. (2000). Prion diseases and the immune system. Clin. Immunol. 96, 79–85.
Baik, S. H., Cha, M. Y., Hyun, Y. M., Cho, H., Hamza, B., Kim, D. K., et al. (2014). Migration of neutrophils targeting amyloid plaques in Alzheimer’s disease mouse model. Neurobiol. Aging 35, 1286–1292. doi: 10.1016/j.neurobiolaging.2014.01.003
Barateiro, A., and Fernandes, A. (2014). Temporal oligodendrocyte lineage progression: in vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta 1843, 1917–1929. doi: 10.1016/j.bbamcr.2014.04.018
Bartzokis, G. (2011). Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol. Aging 32, 1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007
Bechler, M. E., Byrne, L., and Ffrench-Constant, C. (2015). CNS myelin sheath lengths are an intrinsic property of oligodendrocytes. Curr. Biol. 25, 2411–2416. doi: 10.1016/j.cub.2015.07.056
Bekris, L. M., Yu, C. E., Bird, T. D., and Tsuang, D. W. (2010). Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 23, 213–227. doi: 10.1177/0891988710383571
Bidollari, E., Rotundo, G., Ferrari, D., Candido, O., Bernardini, L., Consoli, F., et al. (2018). Generation of induced pluripotent stem cell line, CSSi004-A (2962), from a patient diagnosed with Huntington’s disease at the presymptomatic stage. Stem Cell Res. 28, 145–148. doi: 10.1016/j.scr.2018.02.014
Birgbauer, E., Rao, T. S., and Webb, M. (2004). Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. J. Neurosci. Res. 78, 157–166. doi: 10.1002/jnr.20248
Bolam, J. P., and Pissadaki, E. K. (2012). Living on the edge with too many mouths to feed: why dopamine neurons die. Mov. Disord. 27, 1478–1483. doi: 10.1002/mds.25135
Bolognin, S., Fossepre, M., Qing, X., Jarazo, J., Scancar, J., Moreno, E. L., et al. (2019). 3D cultures of Parkinson’s disease-specific dopaminergic neurons for high content phenotyping and drug testing. Adv. Sci. 6:1800927. doi: 10.1002/advs.201800927
Bousquet, J., and Meunier, J. M. (1962). [Organotypic culture, on natural and artificial media, of fragments of the adult rat hypophysis]. C. R. Seances Soc. Biol. Fil. 156, 65–67.
Buntinx, M., Vanderlocht, J., Hellings, N., Vandenabeele, F., Lambrichts, I., Raus, J., et al. (2003). Characterization of three human oligodendroglial cell lines as a model to study oligodendrocyte injury: morphology and oligodendrocyte-specific gene expression. J. Neurocytol. 32, 25–38. doi: 10.1023/a:1027324230923
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 355–364. doi: 10.1016/j.mcn.2013.07.007
Buttiglione, M., Vitiello, F., Sardella, E., Petrone, L., Nardulli, M., Favia, P., et al. (2007). Behaviour of SH-SY5Y neuroblastoma cell line grown in different media and on different chemically modified substrates. Biomaterials 28, 2932–2945. doi: 10.1016/j.biomaterials.2007.02.022
Cakir, B., Xiang, Y., Tanaka, Y., Kural, M. H., Parent, M., Kang, Y. J., et al. (2019). Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175. doi: 10.1038/s41592-019-0586-5
Camnasio, S., Delli Carri, A., Lombardo, A., Grad, I., Mariotti, C., Castucci, A., et al. (2012). The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 46, 41–51. doi: 10.1016/j.nbd.2011.12.042
Cavaliere, F., Vicente, E. S., and Matute, C. (2010). An organotypic culture model to study nigro-striatal degeneration. J. Neurosci. Methods 188, 205–212. doi: 10.1016/j.jneumeth.2010.02.008
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Charvin, D., Medori, R., Hauser, R. A., and Rascol, O. (2018). Therapeutic strategies for Parkinson disease: beyond dopaminergic drugs. Nat. Rev. Drug Discov. 17:844. doi: 10.1038/nrd.2018.184
Che, Y., Hou, L., Sun, F., Zhang, C., Liu, X., Piao, F., et al. (2018). Taurine protects dopaminergic neurons in a mouse Parkinson’s disease model through inhibition of microglial M1 polarization. Cell Death Dis. 9:435. doi: 10.1038/s41419-018-0468-2
Chen, H., Qian, K., Du, Z., Cao, J., Petersen, A., Liu, H., et al. (2014). Modeling ALS with iPSCs reveals that mutant SOD1 misregulates neurofilament balance in motor neurons. Cell Stem Cell 14, 796–809. doi: 10.1016/j.stem.2014.02.004
Chen, H., Zhang, Y., Yang, Z., and Zhang, H. (2013). Human umbilical cord Wharton’s jelly-derived oligodendrocyte precursor-like cells for axon and myelin sheath regeneration. Neural Regen. Res. 8, 890–899. doi: 10.3969/j.issn.1673-5374.2013.10.003
Chew, L. J., Deboy, C. A., and Senatorov, V. V. Jr. (2014). Finding degrees of separation: experimental approaches for astroglial and oligodendroglial cell isolation and genetic targeting. J. Neurosci. Methods 236, 125–147. doi: 10.1016/j.jneumeth.2014.08.017
Chlebanowska, P., Tejchman, A., Sulkowski, M., Skrzypek, K., and Majka, A. M. (2020). Use of 3D organoids as a model to study idiopathic form of Parkinson’s disease. Int. J. Mol. Sci. 21:694. doi: 10.3390/ijms21030694
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278. doi: 10.1038/nature13800
Choi, S. H., Kim, Y. H., Quinti, L., Tanzi, R. E., and Kim, D. Y. (2016). 3D culture models of Alzheimer’s disease: a road map to a “cure-in-a-dish”. Mol. Neurodegener. 11:75. doi: 10.1186/s13024-016-0139-7
Chuang, C. Y., Yang, C. C., Soong, B. W., Yu, C. Y., Chen, S. H., Huang, H. P., et al. (2019). Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology. Sci. Rep. 9:1166. doi: 10.1038/s41598-018-37774-2
Collins, S., Mclean, C. A., and Masters, C. L. (2001). Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J. Clin. Neurosci. 8, 387–397. doi: 10.1054/jocn.2001.0919
Collins, S. J., and Haigh, C. L. (2017). Simplified murine 3D neuronal cultures for investigating neuronal activity and neurodegeneration. Cell Biochem. Biophys. 75, 3–13. doi: 10.1007/s12013-016-0768-z
Compston, A., and Coles, A. (2008). Multiple sclerosis. Lancet 372, 1502–1517. doi: 10.1016/S0140-6736(08)61620-7
Costamagna, G., Andreoli, L., Corti, S., and Faravelli, I. (2019). iPSCs-based neural 3D systems: a multidimensional approach for disease modeling and drug discovery. Cells 8:1438. doi: 10.3390/cells8111438
Crain, S. M. (1966). Development of “organotypic” bioelectric activities in central nervous tissues during maturation in culture. Int. Rev. Neurobiol. 9, 1–43. doi: 10.1016/s0074-7742(08)60135-x
Cummings, J. L., Morstorf, T., and Zhong, K. (2014). Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res. Ther. 6:37. doi: 10.1186/alzrt269
D’Avanzo, C., Aronson, J., Kim, Y. H., Choi, S. H., Tanzi, R. E., and Kim, D. Y. (2015). Alzheimer’s in 3D culture: challenges and perspectives. Bioessays 37, 1139–1148. doi: 10.1002/bies.201500063
Daviaud, N., Garbayo, E., Lautram, N., Franconi, F., Lemaire, L., Perez-Pinzon, M., et al. (2014). Modeling nigrostriatal degeneration in organotypic cultures, a new ex vivo model of Parkinson’s disease. Neuroscience 256, 10–22. doi: 10.1016/j.neuroscience.2013.10.021
De Strooper, B. (2014). Lessons from a failed gamma-secretase Alzheimer trial. Cell 159, 721–726. doi: 10.1016/j.cell.2014.10.016
Desai, M. K., Mastrangelo, M. A., Ryan, D. A., Sudol, K. L., Narrow, W. C., and Bowers, W. J. (2010). Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am. J. Pathol. 177, 1422–1435. doi: 10.2353/ajpath.2010.100087
Devine, M. J., Ryten, M., Vodicka, P., Thomson, A. J., Burdon, T., Houlden, H., et al. (2011). Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat. Commun. 2:440. doi: 10.1038/ncomms1453
Devos, D., Lebouvier, T., Lardeux, B., Biraud, M., Rouaud, T., Pouclet, H., et al. (2013). Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 50, 42–48. doi: 10.1016/j.nbd.2012.09.007
Di Ruscio, A., Patti, F., Welner, R. S., Tenen, D. G., and Amabile, G. (2015). Multiple sclerosis: getting personal with induced pluripotent stem cells. Cell Death Dis. 6:e1806. doi: 10.1038/cddis.2015.179
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221. doi: 10.1126/science.1158799
Doussau, F., Dupont, J. L., Neel, D., Schneider, A., Poulain, B., and Bossu, J. L. (2017). Organotypic cultures of cerebellar slices as a model to investigate demyelinating disorders. Expert Opin. Drug Discov. 12, 1011–1022. doi: 10.1080/17460441.2017.1356285
Douvaras, P., Wang, J., Zimmer, M., Hanchuk, S., O’bara, M. A., Sadiq, S., et al. (2014). Efficient generation of myelinating oligodendrocytes from primary progressive multiple sclerosis patients by induced pluripotent stem cells. Stem Cell Rep. 3, 250–259. doi: 10.1016/j.stemcr.2014.06.012
Dugas, J. C., and Emery, B. (2013). Purification and culture of oligodendrocyte lineage cells. Cold Spring Harb. Protoc. 2013, 810–814. doi: 10.1101/pdb.top074898
Ebert, A. D., Yu, J., Rose, F. F. Jr., Mattis, V. B., Lorson, C. L., Thomson, J. A., et al. (2009). Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 457, 277–280. doi: 10.1038/nature07677
Efremova, L., Chovancova, P., Adam, M., Gutbier, S., Schildknecht, S., and Leist, M. (2017). Switching from astrocytic neuroprotection to neurodegeneration by cytokine stimulation. Arch. Toxicol. 91, 231–246. doi: 10.1007/s00204-016-1702-2
Ehrlich, M., Hallmann, A. L., Reinhardt, P., Arauzo-Bravo, M. J., Korr, S., Ropke, A., et al. (2015). Distinct neurodegenerative changes in an induced pluripotent stem cell model of frontotemporal dementia linked to mutant TAU protein. Stem Cell Rep. 5, 83–96. doi: 10.1016/j.stemcr.2015.06.001
Ehrlich, M., Mozafari, S., Glatza, M., Starost, L., Velychko, S., Hallmann, A. L., et al. (2017). Rapid and efficient generation of oligodendrocytes from human induced pluripotent stem cells using transcription factors. Proc. Natl. Acad. Sci. U.S.A. 114, E2243–E2252. doi: 10.1073/pnas.1614412114
Encinas, M., Iglesias, M., Liu, Y. H., Wang, H. Y., Muhaisen, A., Cena, V., et al. (2000). Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 75, 991–1003. doi: 10.1046/j.1471-4159.2000.0750991.x
Falkenburger, B. H., and Schulz, J. B. (2006). Limitations of cellular models in Parkinson’s disease research. J. Neural Transm. Suppl. 70, 261–268. doi: 10.1007/978-3-211-45295-0_40
Falsig, J., and Aguzzi, A. (2008). The prion organotypic slice culture assay–POSCA. Nat. Protoc. 3, 555–562. doi: 10.1038/nprot.2008.13
Fitzgerald, K. A., Malhotra, M., Curtin, C. M., O’ Brien, F. J., and O’Driscoll, C. M. (2015). Life in 3D is never flat: 3D models to optimise drug delivery. J. Control. Release 215, 39–54. doi: 10.1016/j.jconrel.2015.07.020
Freundt, E. C., Maynard, N., Clancy, E. K., Roy, S., Bousset, L., Sourigues, Y., et al. (2012). Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524. doi: 10.1002/ana.23747
Frohman, E. M., Racke, M. K., and Raine, C. S. (2006). Multiple sclerosis — the plaque and its pathogenesis. N. Engl. J. Med. 354, 942–955. doi: 10.1056/NEJMra052130
Fujimori, K., Ishikawa, M., Otomo, A., Atsuta, N., Nakamura, R., Akiyama, T., et al. (2018). Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 24, 1579–1589. doi: 10.1038/s41591-018-0140-5
Fuller, H. R., Mandefro, B., Shirran, S. L., Gross, A. R., Kaus, A. S., Botting, C. H., et al. (2015). Spinal muscular atrophy patient iPSC-derived motor neurons have reduced expression of proteins important in neuronal development. Front. Cell. Neurosci. 9:506. doi: 10.3389/fncel.2015.00506
Gaven, F., Marin, P., and Claeysen, S. (2014). Primary culture of mouse dopaminergic neurons. J. Vis. Exp. 91:e51751.
Ghasemi, N., Razavi, S., and Nikzad, E. (2017). Multiple sclerosis: pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 19, 1–10. doi: 10.22074/cellj.2016.4867
Giri, M., Zhang, M., and Lu, Y. (2016). Genes associated with Alzheimer’s disease: an overview and current status. Clin. Interv. Aging 11, 665–681. doi: 10.2147/CIA.S105769
Gogolla, N., Galimberti, I., Depaola, V., and Caroni, P. (2006). Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat. Protoc. 1, 1165–1171. doi: 10.1038/nprot.2006.168
Gonzalez, C., Armijo, E., Bravo-Alegria, J., Becerra-Calixto, A., Mays, C. E., and Soto, C. (2018). Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 23, 2363–2374. doi: 10.1038/s41380-018-0229-8
Gonzalez-Reyes, R. E., Nava-Mesa, M. O., Vargas-Sanchez, K., Ariza-Salamanca, D., and Mora-Munoz, L. (2017). Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 10:427. doi: 10.3389/fnmol.2017.00427
Gordon, K., Clouaire, T., Bao, X. X., Kemp, S. E., Xenophontos, M., De Las Heras, J. I., et al. (2014). Immortality, but not oncogenic transformation, of primary human cells leads to epigenetic reprogramming of DNA methylation and gene expression. Nucleic Acids Res. 42, 3529–3541. doi: 10.1093/nar/gkt1351
Groveman, B. R., Foliaki, S. T., Orru, C. D., Zanusso, G., Carroll, J. A., Race, B., et al. (2019). Sporadic Creutzfeldt-Jakob disease prion infection of human cerebral organoids. Acta Neuropathol. Commun. 7:90. doi: 10.1186/s40478-019-0742-2
Harischandra, D. S., Rokad, D., Ghaisas, S., Verma, S., Robertson, A., Jin, H., et al. (2019). Enhanced differentiation of human dopaminergic neuronal cell model for preclinical translational research in Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1866:165533. doi: 10.1016/j.bbadis.2019.165533
Hemmer, B., Kerschensteiner, M., and Korn, T. (2015). Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 14, 406–419. doi: 10.1016/S1474-4422(14)70305-9
Henderson, A. P., Barnett, M. H., Parratt, J. D., and Prineas, J. W. (2009). Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 66, 739–753. doi: 10.1002/ana.21800
Heneka, M. T. (2020). An immune-cell signature marks the brain in Alzheimer’s disease. Nature 577, 322–323. doi: 10.1038/d41586-019-03892-8
Henley, D. B., Sundell, K. L., Sethuraman, G., Dowsett, S. A., and May, P. C. (2014). Safety profile of semagacestat, a gamma-secretase inhibitor: identity trial findings. Curr. Med. Res. Opin. 30, 2021–2032. doi: 10.1185/03007995.2014.939167
Hirsch, E. C., and Hunot, S. (2009). Neuroinflammation in Parkinson’s disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. doi: 10.1016/S1474-4422(09)70062-6
Honegger, P. (2001). Overview of cell and tissue culture techniques. Curr. Protoc. Pharmacol. Chapter 12:Unit1211. doi: 10.1002/0471141755.ph1201s04
Hor, J. H., Soh, E. S., Tan, L. Y., Lim, V. J. W., Santosa, M. M., and Winanto, et al. (2018). Cell cycle inhibitors protect motor neurons in an organoid model of Spinal Muscular Atrophy. Cell Death Dis. 9:1100. doi: 10.1038/s41419-018-1081-0
Hu, B. Y., Du, Z. W., and Zhang, S. C. (2009). Differentiation of human oligodendrocytes from pluripotent stem cells. Nat. Protoc. 4, 1614–1622. doi: 10.1038/nprot.2009.186
Hu, W., Qiu, B., Guan, W., Wang, Q., Wang, M., Li, W., et al. (2015). Direct conversion of normal and Alzheimer’s disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 17, 204–212. doi: 10.1016/j.stem.2015.07.006
Huang, W. J., Chen, W. W., and Zhang, X. (2017). Multiple sclerosis: pathology, diagnosis and treatments. Exp. Ther. Med. 13, 3163–3166. doi: 10.3892/etm.2017.4410
Hubler, Z., Allimuthu, D., Bederman, I., Elitt, M. S., Madhavan, M., Allan, K. C., et al. (2018). Accumulation of 8,9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 560, 372–376. doi: 10.1038/s41586-018-0360-3
Huch, M., Knoblich, J. A., Lutolf, M. P., and Martinez-Arias, A. (2017). The hope and the hype of organoid research. Development 144, 938–941. doi: 10.1242/dev.150201
International Multiple Sclerosis Genetics Consortium [IMSGC], Wellcome Trust Case Control Consortium 2, Sawcer, S., Hellenthal, G., Pirinen, M., Spencer, C. C., et al. (2011). Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219. doi: 10.1038/nature10251
Iqbal, K., and Grundke-Iqbal, I. (2010). Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement. 6, 420–424. doi: 10.1016/j.jalz.2010.04.006
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Jang, J., Yoo, J. E., Lee, J. A., Lee, D. R., Kim, J. Y., Huh, Y. J., et al. (2012). Disease-specific induced pluripotent stem cells: a platform for human disease modeling and drug discovery. Exp. Mol. Med. 44, 202–213. doi: 10.3858/emm.2012.44.3.015
Jorfi, M., D’avanzo, C., Tanzi, R. E., Kim, D. Y., and Irimia, D. (2018). Human neurospheroid arrays for in vitro studies of Alzheimer’s Disease. Sci. Rep. 8:2450. doi: 10.1038/s41598-018-20436-8
Juopperi, T. A., Kim, W. R., Chiang, C. H., Yu, H., Margolis, R. L., Ross, C. A., et al. (2012). Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 5:17. doi: 10.1186/1756-6606-5-17
Kane, K. I. W., Moreno, E. L., Hachi, S., Walter, M., Jarazo, J., Oliveira, M. A. P., et al. (2019). Automated microfluidic cell culture of stem cell derived dopaminergic neurons. Sci. Rep. 9:1796. doi: 10.1038/s41598-018-34828-3
Kearns, S. M., Scheffler, B., Goetz, A. K., Lin, D. D., Baker, H. D., Roper, S. N., et al. (2006). A method for a more complete in vitro Parkinson’s model: slice culture bioassay for modeling maintenance and repair of the nigrostriatal circuit. J. Neurosci. Methods 157, 1–9. doi: 10.1016/j.jneumeth.2006.03.020
Kelava, I., and Lancaster, M. A. (2016). Stem cell models of human brain development. Cell Stem Cell 18, 736–748. doi: 10.1016/j.stem.2016.05.022
Kim, H., Park, H. J., Choi, H., Chang, Y., Park, H., Shin, J., et al. (2019). Modeling G2019S-LRRK2 sporadic Parkinson’s disease in 3D midbrain organoids. Stem Cell Rep. 12, 518–531. doi: 10.1016/j.stemcr.2019.01.020
Knight, R. (2008). Clinical features and diagnosis of human prion diseases. Future Neurol. 3, 473–481.
Knopfel, T., Rietschin, L., and Gahwiler, B. H. (1989). Organotypic co-cultures of rat locus coeruleus and hippocampus. Eur. J. Neurosci. 1, 678–689. doi: 10.1111/j.1460-9568.1989.tb00374.x
Kondo, T., Asai, M., Tsukita, K., Kutoku, Y., Ohsawa, Y., Sunada, Y., et al. (2013). Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12, 487–496. doi: 10.1016/j.stem.2013.01.009
Korecka, J. A., Van Kesteren, R. E., Blaas, E., Spitzer, S. O., Kamstra, J. H., Smit, A. B., et al. (2013). Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PLoS One 8:e63862. doi: 10.1371/journal.pone.0063862
Krejciova, Z., Alibhai, J., Zhao, C., Krencik, R., Rzechorzek, N. M., Ullian, E. M., et al. (2017). Human stem cell-derived astrocytes replicate human prions in a PRNP genotype-dependent manner. J. Exp. Med. 214, 3481–3495. doi: 10.1084/jem.20161547
Krencik, R., Seo, K., Van Asperen, J. V., Basu, N., Cvetkovic, C., Barlas, S., et al. (2017). Systematic three-dimensional coculture rapidly recapitulates interactions between human neurons and astrocytes. Stem Cell Rep. 9, 1745–1753. doi: 10.1016/j.stemcr.2017.10.026
Kumar, D., Hussain, A., Srivastava, A. K., Mukerji, M., Mukherjee, O., and Faruq, M. (2018). Generation of three spinocerebellar ataxia type-12 patients derived induced pluripotent stem cell lines (IGIBi002-A, IGIBi003-A and IGIBi004-A). Stem Cell Res. 31, 216–221. doi: 10.1016/j.scr.2018.08.008
Kumar, S., Blangero, J., and Curran, J. E. (2018). Induced pluripotent stem cells in disease modeling and gene identification. Methods Mol. Biol. 1706, 17–38. doi: 10.1007/978-1-4939-7471-9_2
Laferla, F. M., and Green, K. N. (2012). Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006320.
Lancaster, M. A., Renner, M., Martin, C. A., Wenzel, D., Bicknell, L. S., Hurles, M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. doi: 10.1038/nature12517
Lavail, J. H., and Wolf, M. K. (1973). Postnatal development of the mouse dentate gyrus in organotypic cultures of the hippocampal formation. Am. J. Anat. 137, 47–65. doi: 10.1002/aja.1001370105
Lazaro, D. F., Dias, M. C., Carija, A., Navarro, S., Madaleno, C. S., Tenreiro, S., et al. (2016). The effects of the novel A53E alpha-synuclein mutation on its oligomerization and aggregation. Acta Neuropathol. Commun. 4:128. doi: 10.1186/s40478-016-0402-8
Lazaro, D. F., Pavlou, M. A. S., and Outeiro, T. F. (2017). Cellular models as tools for the study of the role of alpha-synuclein in Parkinson’s disease. Exp. Neurol. 298, 162–171. doi: 10.1016/j.expneurol.2017.05.007
Lazaro, D. F., Rodrigues, E. F., Langohr, R., Shahpasandzadeh, H., Ribeiro, T., Guerreiro, P., et al. (2014). Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 10:e1004741. doi: 10.1371/journal.pgen.1004741
Lee, H. K., Velazquez Sanchez, C., Chen, M., Morin, P. J., Wells, J. M., Hanlon, E. B., et al. (2016). Three dimensional human neuro-spheroid model of Alzheimer’s disease based on differentiated induced pluripotent stem cells. PLoS One 11:e0163072. doi: 10.1371/journal.pone.0163072
Lee, S., Park, C., Han, J. W., Kim, J. Y., Cho, K., Kim, E. J., et al. (2017). Direct reprogramming of human dermal fibroblasts into endothelial cells using ER71/ETV2. Circ. Res. 120, 848–861. doi: 10.1161/CIRCRESAHA.116.309833
Leite, C., Silva, N. T., Mendes, S., Ribeiro, A., De Faria, J. P., Lourenco, T., et al. (2014). Differentiation of human umbilical cord matrix mesenchymal stem cells into neural-like progenitor cells and maturation into an oligodendroglial-like lineage. PLoS One 9:e111059. doi: 10.1371/journal.pone.0111059
Li, T., Pires, C., Nielsen, T. T., Waldemar, G., Hjermind, L. E., Nielsen, J. E., et al. (2016). Generation of induced pluripotent stem cells (iPSCs) from an Alzheimer’s disease patient carrying an A79V mutation in PSEN1. Stem Cell Res. 16, 229–232. doi: 10.1016/j.scr.2016.01.002
Liebner, S., Dijkhuizen, R. M., Reiss, Y., Plate, K. H., Agalliu, D., and Constantin, G. (2018). Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 135, 311–336. doi: 10.1007/s00401-018-1815-1
Logan, S., Arzua, T., Canfield, S. G., Seminary, E. R., Sison, S. L., Ebert, A. D., et al. (2019). Studying human neurological disorders using induced pluripotent stem cells: from 2D monolayer to 3D organoid and blood brain barrier models. Compr. Physiol. 9, 565–611. doi: 10.1002/cphy.c180025
Lopes, F. M., Bristot, I. J., Da Motta, L. L., Parsons, R. B., and Klamt, F. (2017). Mimicking Parkinson’s disease in a dish: merits and pitfalls of the most commonly used dopaminergic in vitro models. Neuromol. Med. 19, 241–255. doi: 10.1007/s12017-017-8454-x
Lopes Pinheiro, M. A., Kooij, G., Mizee, M. R., Kamermans, A., Enzmann, G., Lyck, R., et al. (2016). Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta 1862, 461–471. doi: 10.1016/j.bbadis.2015.10.018
Lotharius, J., Falsig, J., Van Beek, J., Payne, S., Dringen, R., Brundin, P., et al. (2005). Progressive degeneration of human mesencephalic neuron-derived cells triggered by dopamine-dependent oxidative stress is dependent on the mixed-lineage kinase pathway. J. Neurosci. 25, 6329–6342. doi: 10.1523/JNEUROSCI.1746-05.2005
Lucchinetti, C., Bruck, W., Parisi, J., Scheithauer, B., Rodriguez, M., and Lassmann, H. (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707–717. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q
Mackey, L. C., Annab, L. A., Yang, J., Rao, B., Kissling, G. E., Schurman, S. H., et al. (2018). Epigenetic enzymes, age, and ancestry regulate the efficiency of human iPSC reprogramming. Stem Cells 36, 1697–1708. doi: 10.1002/stem.2899
Madhavan, M., Nevin, Z. S., Shick, H. E., Garrison, E., Clarkson-Paredes, C., Karl, M., et al. (2018). Induction of myelinating oligodendrocytes in human cortical spheroids. Nat. Methods 15, 700–706. doi: 10.1038/s41592-018-0081-4
Maguire, J. A., Gagne, A. L., Gonzalez-Alegre, P., Davidson, B. L., Shakkottai, V., Gadue, P., et al. (2019). Generation of Spinocerebellar Ataxia Type 2 induced pluripotent stem cell lines, CHOPi002-A and CHOPi003-A, from patients with abnormal CAG repeats in the coding region of the ATXN2 gene. Stem Cell Res. 34:101361. doi: 10.1016/j.scr.2018.101361
Mahad, D. H., Trapp, B. D., and Lassmann, H. (2015). Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 14, 183–193. doi: 10.1016/S1474-4422(14)70256-X
Mansour, A. A., Goncalves, J. T., Bloyd, C. W., Li, H., Fernandes, S., Quang, D., et al. (2018). An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 432–441. doi: 10.1038/nbt0818-772e
Marella, M., and Chabry, J. (2004). Neurons and astrocytes respond to prion infection by inducing microglia recruitment. J. Neurosci. 24, 620–627. doi: 10.1523/JNEUROSCI.4303-03.2004
Marotta, N., Kim, S., and Krainc, D. (2020). Organoid and pluripotent stem cells in Parkinson’s disease modeling: an expert view on their value to drug discovery. Expert Opin. Drug Discov. 15, 427–441. doi: 10.1080/17460441.2020.1703671
Marthaler, A. G., Schmid, B., Tubsuwan, A., Poulsen, U. B., Engelbrecht, A. F., Mau-Holzmann, U. A., et al. (2016). Generation of an isogenic, gene-corrected control cell line of the spinocerebellar ataxia type 2 patient-derived iPSC line H271. Stem Cell Res. 16, 180–183. doi: 10.1016/j.scr.2015.12.028
Marton, R. M., Miura, Y., Sloan, S. A., Li, Q., Revah, O., Levy, R. J., et al. (2019). Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 22, 484–491. doi: 10.1038/s41593-018-0316-9
Matamoros-Angles, A., Gayosso, L. M., Richaud-Patin, Y., Di Domenico, A., Vergara, C., Hervera, A., et al. (2018). iPS cell cultures from a Gerstmann-Straussler-Scheinker Patient with the Y218N PRNP mutation recapitulate tau pathology. Mol. Neurobiol. 55, 3033–3048. doi: 10.1007/s12035-017-0506-6
McManus, R. M., and Heneka, M. T. (2020). T cells in Alzheimer’s disease: space invaders. Lancet Neurol. 19, 285–287. doi: 10.1016/S1474-4422(20)30076-4
Mehta, S. R., Tom, C. M., Wang, Y., Bresee, C., Rushton, D., Mathkar, P. P., et al. (2018). Human Huntington’s disease iPSC-derived cortical neurons display altered transcriptomics, morphology, and maturation. Cell Rep. 25, 1081–1096.e6. doi: 10.1016/j.celrep.2018.09.076
Mekhoubad, S., Bock, C., De Boer, A. S., Kiskinis, E., Meissner, A., and Eggan, K. (2012). Erosion of dosage compensation impacts human iPSC disease modeling. Cell Stem Cell 10, 595–609. doi: 10.1016/j.stem.2012.02.014
Mertens, J., Reid, D., Lau, S., Kim, Y., and Gage, F. H. (2018). Aging in a Dish: iPSC-derived and directly induced neurons for studying brain aging and age-related neurodegenerative diseases. Annu. Rev. Genet. 52, 271–293. doi: 10.1146/annurev-genet-120417-031534
Mohamet, L., Miazga, N. J., and Ward, C. M. (2014). Familial Alzheimer’s disease modelling using induced pluripotent stem cell technology. World J. Stem Cells 6, 239–247. doi: 10.4252/wjsc.v6.i2.239
Moore, S., Evans, L. D., Andersson, T., Portelius, E., Smith, J., Dias, T. B., et al. (2015). APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 11, 689–696. doi: 10.1016/j.celrep.2015.03.068
Moreno, E. L., Hachi, S., Hemmer, K., Trietsch, S. J., Baumuratov, A. S., Hankemeier, T., et al. (2015). Differentiation of neuroepithelial stem cells into functional dopaminergic neurons in 3D microfluidic cell culture. Lab Chip 15, 2419–2428. doi: 10.1039/c5lc00180c
Muratore, C. R., Rice, H. C., Srikanth, P., Callahan, D. G., Shin, T., Benjamin, L. N., et al. (2014). The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 23, 3523–3536. doi: 10.1093/hmg/ddu064
Nagele, R. G., Wegiel, J., Venkataraman, V., Imaki, H., Wang, K. C., and Wegiel, J. (2004). Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol. Aging 25, 663–674. doi: 10.1016/j.neurobiolaging.2004.01.007
Nakamura, M., Shiozawa, S., Tsuboi, D., Amano, M., Watanabe, H., Maeda, S., et al. (2019). Pathological progression induced by the frontotemporal dementia-associated R406W Tau mutation in patient-derived iPSCs. Stem Cell Rep. 13, 684–699. doi: 10.1016/j.stemcr.2019.08.011
Nayler, S. P., Powell, J. E., Vanichkina, D. P., Korn, O., Wells, C. A., Kanjhan, R., et al. (2017). Human iPSC-derived cerebellar neurons from a patient with ataxia-telangiectasia reveal disrupted gene regulatory networks. Front. Cell. Neurosci. 11:321. doi: 10.3389/fncel.2017.00321
Nekrasov, E. D., Vigont, V. A., Klyushnikov, S. A., Lebedeva, O. S., Vassina, E. M., Bogomazova, A. N., et al. (2016). Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 11:27. doi: 10.1186/s13024-016-0092-5
Neuwelt, E., Abbott, N. J., Abrey, L., Banks, W. A., Blakley, B., Davis, T., et al. (2008). Strategies to advance translational research into brain barriers. Lancet Neurol. 7, 84–96. doi: 10.1016/S1474-4422(07)70326-5
Nguyen, H. N., Byers, B., Cord, B., Shcheglovitov, A., Byrne, J., Gujar, P., et al. (2011). LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8, 267–280. doi: 10.1016/j.stem.2011.01.013
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 16, 653–661. doi: 10.1038/nm.2165
Oh, Y. (2019). Patient-specific pluripotent stem cell-based Parkinson’s disease models showing endogenous alpha-synuclein aggregation. BMB Rep. 52, 349–359. doi: 10.5483/BMBRep.2019.52.6.142
Ohmine, S., Squillace, K. A., Hartjes, K. A., Deeds, M. C., Armstrong, A. S., Thatava, T., et al. (2012). Reprogrammed keratinocytes from elderly type 2 diabetes patients suppress senescence genes to acquire induced pluripotency. Aging 4, 60–73. doi: 10.18632/aging.100428
Olanow, C. W., Kieburtz, K., and Schapira, A. H. (2008). Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann. Neurol. 64(Suppl. 2), S101–S110. doi: 10.1002/ana.21461
Ormel, P. R., Vieira De Sa, R., Van Bodegraven, E. J., Karst, H., Harschnitz, O., Sneeboer, M. A. M., et al. (2018). Microglia innately develop within cerebral organoids. Nat. Commun. 9:4167. doi: 10.1038/s41467-018-06684-2
Osaki, T., Uzel, S. G. M., and Kamm, R. D. (2018). Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4:eaat5847. doi: 10.1126/sciadv.aat5847
Ostergaard, K., Finsen, B., and Zimmer, J. (1995). Organotypic slice cultures of the rat striatum: an immunocytochemical, histochemical and in situ hybridization study of somatostatin, neuropeptide Y, nicotinamide adenine dinucleotide phosphate-diaphorase, and enkephalin. Exp. Brain Res. 103, 70–84. doi: 10.1007/bf00241966
Pandit, R., Chen, L., and Gotz, J. (2019). The blood-brain barrier: physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. (in press). doi: 10.1016/j.addr.2019.11.009
Pansarasa, O., Bordoni, M., Drufuca, L., Diamanti, L., Sproviero, D., Trotti, R., et al. (2018). Lymphoblastoid cell lines as a model to understand amyotrophic lateral sclerosis disease mechanisms. Dis. Model. Mech. 11:dmm031625. doi: 10.1242/dmm.031625
Paquet, D., Kwart, D., Chen, A., Sproul, A., Jacob, S., Teo, S., et al. (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129. doi: 10.1038/nature17664
Pardridge, W. M. (2005). The blood-brain barrier: bottleneck in brain drug development. NeuroRx 2, 3–14. doi: 10.1602/neurorx.2.1.3
Park, I. H., Arora, N., Huo, H., Maherali, N., Ahfeldt, T., Shimamura, A., et al. (2008). Disease-specific induced pluripotent stem cells. Cell 134, 877–886. doi: 10.1016/j.cell.2008.07.041
Park, J., Baik, S. H., Mook-Jung, I., Irimia, D., and Cho, H. (2019). Mimicry of central-peripheral immunity in Alzheimer’s disease and discovery of neurodegenerative roles in neutrophil. Front. Immunol. 10:2231. doi: 10.3389/fimmu.2019.02231
Park, J., Wetzel, I., Marriott, I., Dreau, D., D’avanzo, C., Kim, D. Y., et al. (2018). A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 21, 941–951. doi: 10.1038/s41593-018-0175-4
Pfeuffer, S., Ruck, T., Kleinschnitz, C., Wiendl, H., and Meuth, S. G. (2016). Failed, interrupted and inconclusive trials on relapsing multiple sclerosis treatment: update 2010-2015. Expert Rev. Neurother. 16, 689–700. doi: 10.1080/14737175.2016.1176531
Pham, M. T., Pollock, K. M., Rose, M. D., Cary, W. A., Stewart, H. R., Zhou, P., et al. (2018). Generation of human vascularized brain organoids. Neuroreport 29, 588–593. doi: 10.1097/WNR.0000000000001014
Pietronigro, E., Zenaro, E., Bianca, V. D., Dusi, S., Terrabuio, E., Iannoto, G., et al. (2019). Blockade of alpha4 integrins reduces leukocyte-endothelial interactions in cerebral vessels and improves memory in a mouse model of Alzheimer’s disease. Sci. Rep. 9:12055. doi: 10.1038/s41598-019-48538-x
Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., et al. (2017). Parkinson disease. Nat. Rev. Dis. Primers 3:17013. doi: 10.1038/nrdp.2017.13
Raja, W. K., Mungenast, A. E., Lin, Y. T., Ko, T., Abdurrob, F., Seo, J., et al. (2016). Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One 11:e0161969. doi: 10.1371/journal.pone.0161969
Reinhardt, P., Glatza, M., Hemmer, K., Tsytsyura, Y., Thiel, C. S., Hoing, S., et al. (2013). Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One 8:e59252. doi: 10.1371/journal.pone.0059252
Reitz, C. (2012). Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int. J. Alzheimers Dis. 2012:369808. doi: 10.1155/2012/369808
Renner, M., Lancaster, M. A., Bian, S., Choi, H., Ku, T., Peer, A., et al. (2017). Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 36, 1316–1329. doi: 10.15252/embj.201694700
Robertson, R. T., Baratta, J., Kageyama, G. H., Ha, D. H., and Yu, J. (1997). Specificity of attachment and neurite outgrowth of dissociated basal forebrain cholinergic neurons seeded on to organotypic slice cultures of forebrain. Neuroscience 80, 741–752. doi: 10.1016/s0306-4522(97)00067-5
Ryan, S. D., Dolatabadi, N., Chan, S. F., Zhang, X., Akhtar, M. W., Parker, J., et al. (2013). Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 155, 1351–1364. doi: 10.1016/j.cell.2013.11.009
Sacco, A. M., Belviso, I., Romano, V., Carfora, A., Schonauer, F., Nurzynska, D., et al. (2019). Diversity of dermal fibroblasts as major determinant of variability in cell reprogramming. J. Cell Mol. Med. 23, 4256–4268. doi: 10.1111/jcmm.14316
Sareen, D., O’rourke, J. G., Meera, P., Muhammad, A. K., Grant, S., Simpkinson, M., et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5:208ra149. doi: 10.1126/scitranslmed.3007529
Savica, R., Rocca, W. A., and Ahlskong, E. (2010). When does Parkinson disease start? Arch. Neurol. 67, 798–801. doi: 10.1001/archneurol.2010.135
Scheckel, C., and Aguzzi, A. (2018). Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 19, 405–418. doi: 10.1038/s41576-018-0011-4
Scheltens, P., Blennow, K., Breteler, M. M. B., De Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517.
Schnadelbach, O., Ozen, I., Blaschuk, O. W., Meyer, R. L., and Fawcett, J. W. (2001). N-cadherin is involved in axon-oligodendrocyte contact and myelination. Mol. Cell. Neurosci. 17, 1084–1093. doi: 10.1006/mcne.2001.0961
Schneider, L. S., Mangialasche, F., Andreasen, N., Feldman, H., Giacobini, E., Jones, R., et al. (2014). Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J. Intern. Med. 275, 251–283. doi: 10.1111/joim.12191
Schwartz, M., and Shechter, R. (2010). Systemic inflammatory cells fight off neurodegenerative disease. Nat. Rev. Neurol. 6, 405–410. doi: 10.1038/nrneurol.2010.71
Seo, J., Kritskiy, O., Watson, L. A., Barker, S. J., Dey, D., Raja, W. K., et al. (2017). Inhibition of p25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J. Neurosci. 37, 9917–9924. doi: 10.1523/JNEUROSCI.0621-17.2017
Shi, Y., Kirwan, P., Smith, J., Maclean, G., Orkin, S. H., and Livesey, F. J. (2012). A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl. Med. 4:124ra29. doi: 10.1126/scitranslmed.3003771
Shin, Y., Choi, S. H., Kim, E., Bylykbashi, E., Kim, J. A., Chung, S., et al. (2019). Blood-brain barrier dysfunction in a 3D in vitro model of Alzheimer’s disease. Adv. Sci. 6:1900962. doi: 10.1002/advs.201900962
Shoji, E., Woltjen, K., and Sakurai, H. (2016). Directed myogenic differentiation of human induced pluripotent stem cells. Methods Mol. Biol. 1353, 89–99. doi: 10.1007/7651_2015_257
Silva, M. C., Cheng, C., Mair, W., Almeida, S., Fong, H., Biswas, M. H. U., et al. (2016). Human iPSC-derived neuronal model of Tau-A152T frontotemporal dementia reveals Tau-mediated mechanisms of neuronal vulnerability. Stem Cell Rep. 7, 325–340. doi: 10.1016/j.stemcr.2016.08.001
Smirnova, L., Harris, G., Delp, J., Valadares, M., Pamies, D., Hogberg, H. T., et al. (2016). A LUHMES 3D dopaminergic neuronal model for neurotoxicity testing allowing long-term exposure and cellular resilience analysis. Arch. Toxicol. 90, 2725–2743. doi: 10.1007/s00204-015-1637-z
Smith, I., Haag, M., Ugbode, C., Tams, D., Rattray, M., Przyborski, S., et al. (2015). Neuronal-glial populations form functional networks in a biocompatible 3D scaffold. Neurosci. Lett. 609, 198–202. doi: 10.1016/j.neulet.2015.10.044
Smits, L. M., Reinhardt, L., Reinhardt, P., Glatza, M., Monzel, A. S., Stanslowsky, N., et al. (2019). Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 5:5. doi: 10.1038/s41531-019-0078-4
Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G. W., Cook, E. G., et al. (2009). Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977. doi: 10.1016/j.cell.2009.02.013
Son, M. Y., Sim, H., Son, Y. S., Jung, K. B., Lee, M. O., Oh, J. H., et al. (2017). Distinctive genomic signature of neural and intestinal organoids from familial Parkinson’s disease patient-derived induced pluripotent stem cells. Neuropathol. Appl. Neurobiol. 43, 584–603. doi: 10.1111/nan.12396
Song, B., Sun, G., Herszfeld, D., Sylvain, A., Campanale, N. V., Hirst, C. E., et al. (2012). Neural differentiation of patient specific iPS cells as a novel approach to study the pathophysiology of multiple sclerosis. Stem Cell Res. 8, 259–273. doi: 10.1016/j.scr.2011.12.001
Song, L., Yuan, X., Jones, Z., Griffin, K., Zhou, Y., Ma, T., et al. (2019). Assembly of human stem cell-derived cortical spheroids and vascular spheroids to model 3-D brain-like tissues. Sci. Rep. 9:5977. doi: 10.1038/s41598-019-42439-9
Speicher, A. M., Wiendl, H., Meuth, S. G., and Pawlowski, M. (2019). Generating microglia from human pluripotent stem cells: novel in vitro models for the study of neurodegeneration. Mol. Neurodegener. 14:46. doi: 10.1186/s13024-019-0347-z
Spillantini, M. G., Schmidt, M. L., Lee, V. M.-Y., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). Alpha-synuclein in lewy bodies. Nature 388, 839–840.
Sproul, A. A., Jacob, S., Pre, D., Kim, S. H., Nestor, M. W., Navarro-Sobrino, M., et al. (2014). Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One 9:e84547. doi: 10.1371/journal.pone.0084547
Stahl, K., Skare, O., and Torp, R. (2009). Organotypic cultures as a model of Parkinson s disease. A twist to an old model. ScientificWorldJournal 9, 811–821. doi: 10.1100/tsw.2009.68
Stys, P. K., and Tsutsui, S. (2019). Recent advances in understanding multiple sclerosis. F1000Res. 8:F1000 Faculty Rev-2100. doi: 10.12688/f1000research.20906.1
Sulzer, D., Alcalay, R. N., Garretti, F., Cote, L., Kanter, E., Agin-Liebes, J., et al. (2017). T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546, 656–661. doi: 10.1038/nature22815
Sun, X., Song, J., Huang, H., Chen, H., and Qian, K. (2018). Modeling hallmark pathology using motor neurons derived from the family and sporadic amyotrophic lateral sclerosis patient-specific iPS cells. Stem Cell Res. Ther. 9:315. doi: 10.1186/s13287-018-1048-1
Szlachcic, W. J., Wiatr, K., Trzeciak, M., Figlerowicz, M., and Figiel, M. (2017). The generation of mouse and human Huntington disease iPS cells suitable for in vitro studies on Huntingtin function. Front. Mol. Neurosci. 10:253. doi: 10.3389/fnmol.2017.00253
t Hart, B. A., Gran, B., and Weissert, R. (2011). EAE: imperfect but useful models of multiple sclerosis. Trends Mol. Med. 17, 119–125. doi: 10.1016/j.molmed.2010.11.006
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Tan, G. A., Furber, K. L., Thangaraj, M. P., Sobchishin, L., Doucette, J. R., and Nazarali, A. J. (2018). Organotypic cultures from the adult CNS: a novel model to study demyelination and remyelination ex vivo. Cell. Mol. Neurobiol. 38, 317–328. doi: 10.1007/s10571-017-0529-6
Taylor-Whiteley, T. R., Le Maitre, C. L., Duce, J. A., Dalton, C. F., and Smith, D. P. (2019). Recapitulating Parkinson’s disease pathology in a three-dimensional human neural cell culture model. Dis. Model. Mech. 12:dmm038042. doi: 10.1242/dmm.038042
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. doi: 10.1101/sqb.2008.73.038
Tong, Z. B., Hogberg, H., Kuo, D., Sakamuru, S., Xia, M., Smirnova, L., et al. (2017). Characterization of three human cell line models for high-throughput neuronal cytotoxicity screening. J. Appl. Toxicol. 37, 167–180. doi: 10.1002/jat.3334
Town, T., Tan, J., Flavell, R. A., and Mullan, M. (2005). T-cells in Alzheimer’s disease. Neuromol. Med. 7, 255–264.
Valetdinova, K. R., Maretina, M. A., Kuranova, M. L., Grigor’eva, E. V., Minina, Y. M., Kizilova, E. A., et al. (2019). Generation of two spinal muscular atrophy (SMA) type I patient-derived induced pluripotent stem cell (iPSC) lines and two SMA type II patient-derived iPSC lines. Stem Cell Res. 34:101376. doi: 10.1016/j.scr.2018.101376
Van Den Eeden, S. K., Tanner, C. M., Bernstein, A. L., Fross, R. D., Leimpeter, A., Bloch, D. A., et al. (2003). Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 157, 1015–1022. doi: 10.1093/aje/kwg068
Van Vliet, E., Morath, S., Eskes, C., Linge, J., Rappsilber, J., Honegger, P., et al. (2008). A novel in vitro metabolomics approach for neurotoxicity testing, proof of principle for methyl mercury chloride and caffeine. Neurotoxicology 29, 1–12. doi: 10.1016/j.neuro.2007.09.007
Vigont, V., Nekrasov, E., Shalygin, A., Gusev, K., Klushnikov, S., Illarioshkin, S., et al. (2018). Patient-Specific iPSC-Based Models of Huntington’s disease as a tool to study store-operated calcium entry drug targeting. Front. Pharmacol. 9:696. doi: 10.3389/fphar.2018.00696
Virlogeux, A., Moutaux, E., Christaller, W., Genoux, A., Bruyere, J., Fino, E., et al. (2018). Reconstituting corticostriatal network on-a-chip reveals the contribution of the presynaptic compartment to Huntington’s disease. Cell Rep. 22, 110–122. doi: 10.1016/j.celrep.2017.12.013
Walsh, K., Megyesi, J., and Hammond, R. (2005). Human central nervous system tissue culture: a historical review and examination of recent advances. Neurobiol. Dis. 18, 2–18. doi: 10.1016/j.nbd.2004.09.002
Wang, H. (2018). Modeling neurological diseases with human brain organoids. Front. Synaptic Neurosci. 10:15. doi: 10.3389/fnsyn.2018.00015
Wang, S., Wang, B., Pan, N., Fu, L., Wang, C., Song, G., et al. (2015). Differentiation of human induced pluripotent stem cells to mature functional Purkinje neurons. Sci. Rep. 5:9232. doi: 10.1038/srep09232
Weinert, M., Selvakumar, T., Tierney, T. S., and Alavian, K. N. (2015). Isolation, culture and long-term maintenance of primary mesencephalic dopaminergic neurons from embryonic rodent brains. J. Vis. Exp. 96:52475. doi: 10.3791/52475
Whetsell, W. O., and Schwarcz, R. (1983). The organotypic tissue culture model of corticostriatal system used for examining amino acid neurotoxicity and its antagonism: studies on kainic acid, quinolinic acid and (-) 2-amino-7-phosphonoheptanoic acid. J. Neural Transm. Suppl. 19, 53–63.
Worsdorfer, P., Dalda, N., Kern, A., Kruger, S., Wagner, N., Kwok, C. K., et al. (2019). Generation of complex human organoid models including vascular networks by incorporation of mesodermal progenitor cells. Sci. Rep. 9:15663. doi: 10.1038/s41598-019-52204-7
Xia, G., Santostefano, K., Hamazaki, T., Liu, J., Subramony, S. H., Terada, N., et al. (2013). Generation of human-induced pluripotent stem cells to model spinocerebellar ataxia type 2 in vitro. J. Mol. Neurosci. 51, 237–248. doi: 10.1007/s12031-012-9930-2
Xicoy, H., Wieringa, B., and Martens, G. J. (2017). The SH-SY5Y cell line in Parkinson’s disease research: a systematic review. Mol. Neurodegener. 12:10. doi: 10.1186/s13024-017-0149-0
Yagi, T., Ito, D., Okada, Y., Akamatsu, W., Nihei, Y., Yoshizaki, T., et al. (2011). Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 20, 4530–4539. doi: 10.1093/hmg/ddr394
Yang, M., Liu, M., Vajda, A., O’brien, T., Henshall, D., Hardiman, O., et al. (2019). Generation of six induced pluripotent stem cell (iPSC) lines from two patients with amyotrophic lateral sclerosis (NUIGi043-A, NUIGi043-B, NUIGi043-C, NUIGi044-A, NUIGi044-B, NUIGi044-C). Stem Cell Res. 40:101558. doi: 10.1016/j.scr.2019.101558
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 107, 41–56. doi: 10.1016/j.nbd.2016.07.007
Zenaro, E., Pietronigro, E., Della Bianca, V., Piacentino, G., Marongiu, L., Budui, S., et al. (2015). Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. doi: 10.1038/nm.3913
Zhang, H., Jarjour, A. A., Boyd, A., and Williams, A. (2011). Central nervous system remyelination in culture–a tool for multiple sclerosis research. Exp. Neurol. 230, 138–148. doi: 10.1016/j.expneurol.2011.04.009
Zhang, J., Wilson, G. F., Soerens, A. G., Koonce, C. H., Yu, J., Palecek, S. P., et al. (2009). Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 104, e30–e41. doi: 10.1161/CIRCRESAHA.108.192237
Zhang, Q. J., Li, J. J., Lin, X., Lu, Y. Q., Guo, X. X., Dong, E. L., et al. (2017a). Modeling the phenotype of spinal muscular atrophy by the direct conversion of human fibroblasts to motor neurons. Oncotarget 8, 10945–10953. doi: 10.18632/oncotarget.14641
Zhang, Y., Li, X., Ciric, B., Ma, C. G., Gran, B., Rostami, A., et al. (2017b). Effect of fingolimod on neural stem cells: a novel mechanism and broadened application for neural repair. Mol. Ther. 25, 401–415. doi: 10.1016/j.ymthe.2016.12.008
Zhang, Y., Schmid, B., Nikolaisen, N. K., Rasmussen, M. A., Aldana, B. I., Agger, M., et al. (2017c). Patient iPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Rep. 8, 648–658. doi: 10.1016/j.stemcr.2017.01.012
Zhang, X. M., Yin, M., and Zhang, M. H. (2014). Cell-based assays for Parkinson’s disease using differentiated human LUHMES cells. Acta Pharmacol. Sin. 35, 945–956. doi: 10.1038/aps.2014.36
Zhang, Z., Almeida, S., Lu, Y., Nishimura, A. L., Peng, L., Sun, D., et al. (2013). Downregulation of microRNA-9 in iPSC-derived neurons of FTD/ALS patients with TDP-43 mutations. PLoS One 8:e76055. doi: 10.1371/journal.pone.0076055
Zhao, Z., Nelson, A. R., Betsholtz, C., and Zlokovic, B. V. (2015). Establishment and dysfunction of the blood-brain barrier. Cell 163, 1064–1078. doi: 10.1016/j.cell.2015.10.067
Zlokovic, B. V. (2008). The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57, 178–201. doi: 10.1016/j.neuron.2008.01.003
Keywords: neurodegenerative diseases, in vitro models, three-dimensional culture, induced pluripotent stem cells, organoids
Citation: Slanzi A, Iannoto G, Rossi B, Zenaro E and Constantin G (2020) In vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 8:328. doi: 10.3389/fcell.2020.00328
Received: 11 February 2020; Accepted: 16 April 2020;
Published: 13 May 2020.
Edited by:
Mariano Ponz-Sarvise, University Clinic of Navarra, SpainReviewed by:
Perez-Mediavilla Alberto, University of Navarra, SpainStefan Hauser, German Center for Neurodegenerative Diseases (DZNE), Germany
Copyright © 2020 Slanzi, Iannoto, Rossi, Zenaro and Constantin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriela Constantin, Z2FicmllbGEuY29uc3RhbnRpbkB1bml2ci5pdA==
†These authors have contributed equally to this work