 Joseph Sia
Joseph Sia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 13 February 2020
Sec. Cell Growth and Division
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00041
This article is part of the Research TopicProceedings from ACCM19: Cell Cycle, DNA Damage Response and TelomeresView all 12 articles
Radiation therapy (RT) is responsible for at least 40% of cancer cures, however treatment resistance remains a clinical problem. There have been recent advances in understanding the molecular mechanisms of radiation-induced cell death. The type of cell death after radiation depends on a number of factors including cell type, radiation dose and quality, oxygen tension, TP53 status, DNA repair capacity, cell cycle phase at time of radiation exposure, and the microenvironment. Mitotic catastrophe (a pathway preceding cell death that happens in mitosis or as a consequence of aberrant mitotic progression) is the primary context of radiation-induced cell death in solid cancers, although in a small subset of cancers such as haematopoietic malignancies, radiation results in immediate interphase apoptosis, occurring within hours after exposure. There is intense therapeutic interest in using stereotactic ablative body radiotherapy (SABR), a precise, high-dose form of RT given in a small number of fractions, to prime the immune system for cancer cell killing, but the optimal radiation dose and fractionation remain unclear. Additionally, promising novel radiosensitisers targeting the cell cycle and DNA repair pathways are being trialled. In the context of the increasing use of SABR and such novel agents in the clinic, we provide an updated primer on the major types of radiation-induced cell death, focussing on their molecular mechanisms, factors affecting their initiation, and their implications on immunogenicity.
Radiation therapy (RT) is a major cancer treatment modality and is responsible for at least 40% of cancer cures (Ringborg et al., 2003), yet treatment resistance remains a clinical problem. A primary reason for this is the capacity for cancer cells to evade radiation-induced cell death. Treatment paradigms have traditionally viewed cancer as a cell-autonomous problem of dysregulated proliferation while side-lining host-tumour interactions, but this dogma has undergone a remarkable revolution in the last few decades, with increasing appreciation of the tumour stroma and immune milieu in shaping tumour evolution. Trials of novel radiosensitisers targeting not only key cell death pathways but also the stromal and immune microenvironment, particularly together with stereotactic ablative body radiotherapy (SABR), have gained intense interest.
Here, we provide an updated primer on the major types of radiation-induced cell death, focussing on their molecular mechanisms, factors affecting their initiation, and their implications on immunogenicity. We conclude with discussing these aspects in the context of the increasing use of SABR and novel agents in the clinic. We point readers interested in further detail to the excellent referenced reviews.
The cell cycle is a highly regulated process occurring in two major phases: interphase (consisting of the G1, S, and G2 phases) and mitosis (cell division). During interphase, the cell grows its organelle counts (G1 phase), copies its DNA (S phase), and reorganises contents in preparation for division (G2 phase). Radiation-induced DNA damage is sensed by the kinases ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad3-related protein (ATR), which activate downstream proteins to initiate the DNA damage response (reviewed in Maier et al., 2016). This places the cell into cell cycle arrest, during which DNA double-strand breaks (DSBs) are repaired mainly by two pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). NHEJ is error-prone but active throughout the cell cycle, while HR is error-free but requires an undamaged sister chromatid as the repair template, and therefore is only available in the late S and G2 phases. If damage is too significant for repair, cell death will occur via one of the types described below, at varying time points after the initial irradiation event (interphase or mitotic). In classical radiobiology, cell death is defined by loss of replicative capacity (i.e., replicative or reproductive death) and is determined by clonogenic assays. While this has undoubtedly served the field tremendously, it ignores the types and effects of cell death, discussed in this review.
Mitotic catastrophe is a mechanism for the control of cells unable to complete mitosis, by the triggering of mitotic arrest and ultimately regulated cell death (RCD) or senescence. Mitotic death refers to RCD (usually intrinsic apoptosis) that is driven by mitotic catastrophe (Galluzzi et al., 2018). Because cells in mitotic catastrophe are almost always incapable of further replication, mitotic catastrophe is often confused as a bona fide cell death type. However, these cells eventually trigger one of the ‘other’ cell death pathways, or less commonly escape from this state (Gascoigne and Taylor, 2008; Vakifahmetoglu et al., 2008). Dysfunctional cell cycle checkpoints, a common hallmark of cancer cells, allow radiation-damaged cells to enter mitosis prematurely with misrepaired DNA, leading to mitotic catastrophe. The exact mechanisms for the initiation of mitotic catastrophe and the deciding of final cell fate are unclear. Several attempted divisions can occur before sufficient genetic damage is accumulated to trigger mitotic death, underlining why solid tumours often demonstrate delayed responses to RT.
Apoptosis is a highly regulated form of cell death with characteristic morphological and molecular features. The intrinsic (mitochondrial) apoptotic pathway is activated when the DNA damage repair machinery, with p53 as a central player, disrupts the balance between pro- and anti-apoptotic factors, resulting in the release of cytochrome c from the mitochondria into the cytoplasm to activate the intrinsic pathway-specific caspase 9 (Eriksson and Stigbrand, 2010). The extrinsic pathway, in contrast, is initiated by external signalling via binding of the tumour necrosis factor (TNF) family of ligands to plasma membrane death receptors, eventually causing downstream activation of the extrinsic pathway-specific caspase 8. Irradiated cells can upregulate death receptors, making them susceptible to death through this pathway (Wu et al., 1997; Chakraborty et al., 2003; Sheard et al., 2003). The third pathway, the ceramide pathway, is triggered by radiation-induced activation of acid sphingomyelinase in the plasma membrane, producing ceramide via hydrolysis of sphingomyelin. Radiation-induced DNA damage can also activate mitochondrial ceramide synthase for de novo synthesis of ceramide. Ceramide acts as a second messenger in initiating the apoptotic programme, but its signalling targets are complex (Pettus et al., 2002; Kolesnick and Fuks, 2003). The intrinsic, extrinsic and ceramide pathways converge in the activation of caspase 3 and 7, which sets off a cascade of controlled degradation of cellular components.
Necrosis is an unregulated, chaotic form of cell death, triggered by unfavourable conditions such as extreme changes in pH, energy loss, and ion imbalance within the cell and its microenvironment after irradiation (Kroemer et al., 2009). Traditionally identified in histological specimens, it is a diagnosis of exclusion and based on morphology, characterised by a gain in cellular volume, plasma membrane rupture and spill of intracellular contents.
Senescence refers to permanent cell cycle arrest. Radiation-induced senescence is triggered by DNA damage and the induction of the p53 and pRb pathways causing cell cycle block, but other factors including oxidative stress may also be relevant triggers in the irradiation context (Sabin and Anderson, 2011; Li et al., 2018). Importantly, senescent cells while “dead” in terms of clonogenicity continue to be viable and metabolically active, over time developing a specific expression pattern of immunomodulatory factors. This senescence-associated secretory phenotype is a result of the extrusion of cytoplasmic chromatin fragments in senescent cells, which act on the DNA sensing and effector cGAS-STING pathway to drive expression of interferon and NF-kB elements (Dou et al., 2017; Gluck et al., 2017).
Autophagy describes the process of sequestrating damaged or old cytoplasmic organelles within vesicles for lysosomal degradation in response to cellular stress. It is usually a cytoprotective process, but autophagic cell death occurs when the autophagic response is excessive (Kroemer and Levine, 2008). The autophagic machinery is a highly regulated pathway involving the ATG genes and is known to be triggered by irradiation, likely via the endoplasmic stress module and mTOR pathway, although the exact mechanisms are unclear (Tam et al., 2017). Autophagy observed after irradiation may be a mechanism of treatment resistance or of cell death, being likely context-dependent (Levy et al., 2017; Dikic and Elazar, 2018).
More recently, forms of RCD have been identified in response to irradiation, including necroptosis and ferroptosis, although the extent to which these occur in vivo are uncertain (Gong et al., 2019; Lang et al., 2019; Lei et al., 2019). Necroptosis has similar morphological appearances to necrosis, but is regulated. It can be activated by death receptors, but signalling is transduced via caspase-independent pathways converging on RIPK3 and MLKL. Ferroptosis is a type of cell death triggered by the accumulation of lipid peroxides as the lethal event due to decreased degradation by glutathione peroxidase (GPX4). With irradiation, this is shown to happen via ATM, which represses the cystene-glutamate antiporter system xc-, resulting in decreased glutathione, a co-factor for GPX4 (Lang et al., 2019).
The type of cell death after irradiation depends on a number of factors, with many remaining uncertainties. Cell type is a key determinant. In a subset of tumours such as haematopoietic malignancies (Radford et al., 1994; Jonathan et al., 1999), radiation results in immediate interphase apoptosis. In the majority of solid tumours however, mitotic catastrophe is the most frequent context of radiation-induced cell death (Eriksson and Stigbrand, 2010), whereas normal tissues commonly undergo senescence after irradiation (Nguyen et al., 2018).
The importance of cell type could be related to the cellular status and function of p53 and ATM. p53 is a key regulator of apoptosis and senescence. Intact p53 is required for interphase apoptosis (Fridman and Lowe, 2003), while disruptive TP53 mutations are associated with increased radioresistance via the inhibition of senescence (Skinner et al., 2012). As most tumour cells have lost normal p53 function or inactivated its downstream pathways, the G1/S checkpoint is impaired and DNA damage repair is consequently dependent on ATR/Chk1-mediated intra-S and G2/M arrest (Soussi and Beroud, 2001; Brady et al., 2011; Dillon et al., 2014). Thus, irradiated cells with inactivated p53 will enter mitosis with unrepaired DNA damage, resulting in mitotic catastrophe rather than immediate apoptosis or senescence (Ianzini et al., 2006). While manipulation of p21 or p53 levels may dramatically alter the kinetics of cell death after irradiation, it may not always correlate with the overall loss of replicative capacity (Wouters et al., 1997; Brown and Wouters, 1999). Similarly, ATM impacts cell death by regulating apoptosis, autophagy, and possibly necroptosis (Liang et al., 2013; Tripathi et al., 2013; Chen et al., 2015) (reviewed in Wu et al., 2017), but it is not clear yet how ATM may affect the decision between these types of cell death.
The phase of the cell cycle in which irradiation occurs also influences timing and type of cell death. Studies have demonstrated cells irradiated in G1 phase divide more times and survive longer before undergoing apoptosis compared to cells irradiated in G2 phase, while cells irradiated in mid-to-late S phase died without undergoing mitosis. Post-mitotic apoptosis however appeared to be stochastic and variable between cell lines (Forrester et al., 1999; Endlich et al., 2000).
It may be tempting to speculate that ablative doses incline cells to a different type of cell death compared to conventional doses per fraction, but whether this occurs as a direct effect of irradiation is uncertain. The complexities of establishing this link surround dissecting the effect of dose per fraction from biological effective dose (BED), and accounting for temporal confounding by repopulation and cell death dynamics. Worth noting here is that the induction of sufficient damage to non-nuclear cellular components to evoke upfront necrosis may require clinically-irrelevant high doses (Warters et al., 1978). Nonetheless, the rate of apoptotic events after irradiation can be proportional to the number of fractions given, in line with the reassortment principle (cells cycling inter-fraction into states predisposed for interphase apoptosis) (Verheij and Bartelink, 2000).
Radiation quality may also affect mode of cell death. High linear energy transfer (LET) radiation such as alpha particles has been shown to result in enhanced chromosome rearrangements and reproductive death (Franken et al., 2011), due to both the complexity and absolute number of DNA damage clusters (Pinto et al., 2005; Obe et al., 2010).
The above factors are strongly influenced by the cellular microenvironment. In brief, oxygen tension modulates cell death after irradiation, with a reduction in chromosomal aberrations and reproductive capacity of cells irradiated under hypoxic conditions. This is presumed due to kinetic competition between the oxygen ‘fixation’ of DNA damage and chemical repair processes (reviewed in Stewart et al., 2011). Under the hypoxic, nutrient-deprived conditions in vivo, cell death will be influenced both by the chemical properties of oxygen fixation and indirect effects of hypoxia on cellular processes including cell metabolism, DNA repair, and hypoxia inducible factor (HIF)-related survival mechanisms (Harris, 2002). An acidic microenvironment and serum deprivation also suppress the progression of cells to apoptosis and the formation of micronuclei after irradiation (Paglin et al., 1997; Park et al., 2000).
The observation that an intact host immune system improves the tumour control probability of RT, first made in 1979 (Stone et al., 1979), suggests that the anti-tumour effects of RT must extend beyond its direct effects on cancer cells. Over the last decade, the concept of immunogenic cell death (ICD) has emerged to redefine cancer cell death from an immune-functional aspect, whereby the dying cell through peri-mortem processes is able to evoke anti-tumour adaptive immune responses (Ma et al., 2010; Kepp et al., 2014).
Historically, necrosis has been traditionally thought to be immunogenic (Scaffidi et al., 2002; Sancho et al., 2009), while apoptosis has been viewed as immunologically silent or even tolerogenic (Voll et al., 1997; Krysko and Vandenabeele, 2010). This relationship is an oversimplification. Apoptosis (Casares et al., 2005; Obeid et al., 2007b), necrosis (Scaffidi et al., 2002; Sancho et al., 2009), and autophagy (Michaud et al., 2011; Ko et al., 2014), have all been demonstrated to be capable of inducing immunogenic responses, while necrosis can in fact contribute to a chronic inflammatory microenvironment that is protumourigenic and immunosuppressive (Vakkila and Lotze, 2004).
The current framework for understanding ICD disengages from the traditional forms of cell death and requires two components: the exposure of danger signals, termed damage-associated molecular patterns (DAMPs), from the dying cell to alert antigen presenting cells (APCs), which include dendritic cells (DCs); and an antigenic epitope to be cross-presented by APCs for training of T cells (Galluzzi et al., 2017).
Bona fide ICD induced by RT has been directly demonstrated with in vivo vaccination assays in mouse models (Apetoh et al., 2007a; Obeid et al., 2007a; Gorin et al., 2014). A strong body of work now corroborate these landmark findings across different murine and human cell lines using surrogate measures (Deng et al., 2014; Gameiro et al., 2014; Golden et al., 2014; Ko et al., 2014; Lim et al., 2014). Radiation-induced ICD can also be implied from a growing number of pre-clinical and clinical studies combining RT with immunotherapy showing improved systemic tumour responses (reviewed in Deloch et al., 2016; Grassberger et al., 2019). Although the link is indirect, the release of DAMPs and antigenic determinants to evoke cancer-specific immunity in these studies are thought to be at least partially mediated through radiation-induced ICD. Other important trials testing RT for this role have yielded negative results for yet-uncertain reasons (Kwon et al., 2014; Voorwerk et al., 2019), suggesting that while the phenomenon of radiation-induced ICD is real, its desired clinical impact is a highly selective event.
Our understanding of the molecular mechanisms of radiation-induced ICD and the determinants for their efficient triggering are in its nascency. Calreticulin, HMGB1, and ATP, the classically described DAMPs, act on CD91, TLR4, and purinergic receptors on DCs respectively, to attract, activate, and promote phagocytosis of cellular corpses by DCs (Ma et al., 2010). Calreticulin, an endoplasmic reticulum chaperone, is translocated to the cell surface as part of the unfolded protein response in stressed cells as a pre-apoptotic event (Obeid et al., 2007b). HMGB1 is a nuclear chromatin-binding protein that is released on necrosis (Scaffidi et al., 2002; Apetoh et al., 2007b). Unlike HMGB1 where passive release can occur, optimal secretion of ATP in radiation-induced ICD is active and dependent on intact pre-mortem autophagic machinery for the accumulation of ATP within autolysosomes (Michaud et al., 2011; Ko et al., 2014). A host of other DAMPs have since been identified, including cytosolic double stranded DNA (dsDNA), the heat shock proteins 70 kDa (HSP70) and 90 kDa (HSP90), F-actin, and interleukin-33 (reviewed in Krysko et al., 2012; Galluzzi et al., 2017).
Cytosolic dsDNA and the cGAS-STING axis have recently emerged as subjects of intense therapeutic interest. dsDNA is released from either the mitochondria and/or nucleus of irradiated cells into the cytoplasm and binds to its sensor cGAS, producing the cyclic dinucleotide cGAMP. Micronuclei that accumulate when irradiated cells progress through mitosis with radiation-induced DSBs also constitute a source of dsDNA (Harding et al., 2017). cGAMP ligates with the adaptor protein STING to ultimately upregulate the expression of type I interferon, an important immunomodulatory cytokine (Burdette and Vance, 2013; Paludan and Bowie, 2013). Importantly, dsDNA does not need to activate STING in a cell-intrinsic fashion, but dsDNA or cGAMP can be released upon ICD and taken up by myeloid cells, including DCs, to activate STING within those cells (Deng et al., 2014; Diamond et al., 2018; Schadt et al., 2019). This axis can be shut down by ablative radiation doses due to the simultaneous induction of the exonuclease TREX1, which degrades cytosolic dsDNA (Vanpouille-Box et al., 2017). While this mechanism explains the negative impact of ablative radiation doses on abscopal responses in a murine mammary carcinoma model (Dewan et al., 2009), the observation is not consistently held up in non-abscopal readouts of tumour immunity, suggesting that other factors may be involved in this regulation (Burnette et al., 2011; Deng et al., 2014; Schadt et al., 2019). On the other hand, generation of DAMPs such as ATP and HMGB1 appears to be proportional to radiation doses of up to 100 Gy in vitro (Gameiro et al., 2014), but whether this translates to increased immunogenicity in vivo is unknown.
On the antigenicity side, RT increases the processing and MHC-I-restricted presentation of surface antigens in a cell-autonomous manner, at least partially via the mTOR pathway (Santin et al., 1997; Verbrugge et al., 2014). More provocatively, as a result of altered transcriptional activity and antigen processing, irradiated tumour cells in vitro demonstrate a diversification of epitope repertoire presented on MHC-I (Reits et al., 2006). A recent clinical trial combining RT with immune checkpoint blockade reported a patient case with expansion of T cell clones against a radiation-induced neoantigen, serving as the first in-human proof of this concept (Formenti et al., 2018).
Increasingly, our appreciation of the anti-tumour effects of RT extends beyond a DNA and cancer cell-centric perspective (Figure 1). The need to better understand the molecular pathways of radiation-induced cell death is even more important in light of recent technological advances in the safe delivery of RT and the increasing use of novel agents in clinic. SABR is progressively adopted into routine clinical practice with a growing number of studies backing its remarkable efficacy, greater than predicted by a simple extrapolation from lower doses per fraction. For example, it has been shown to double median survival for oligometastatic disease (Palma et al., 2019) and more than double the response rate to immunotherapy (Formenti et al., 2018; Theelen et al., 2019), leading to the hypothesis that the biology of tumour response to irradiation is different when given high dose per fraction. Theoretically, SABR would induce DNA damage that was more difficult to repair and decrease tumour cell repopulation due to reduced overall treatment time, albeit with a cost from decreased inter-fraction cellular reassortment and reoxygenation. There is currently insufficient data to confidently propose a model by which SABR achieves such high efficacy (Moding et al., 2016; Song et al., 2019). Indeed, some authors argue that the improved local control seen with SABR purely results from a higher dose (Brown et al., 2013, 2014).
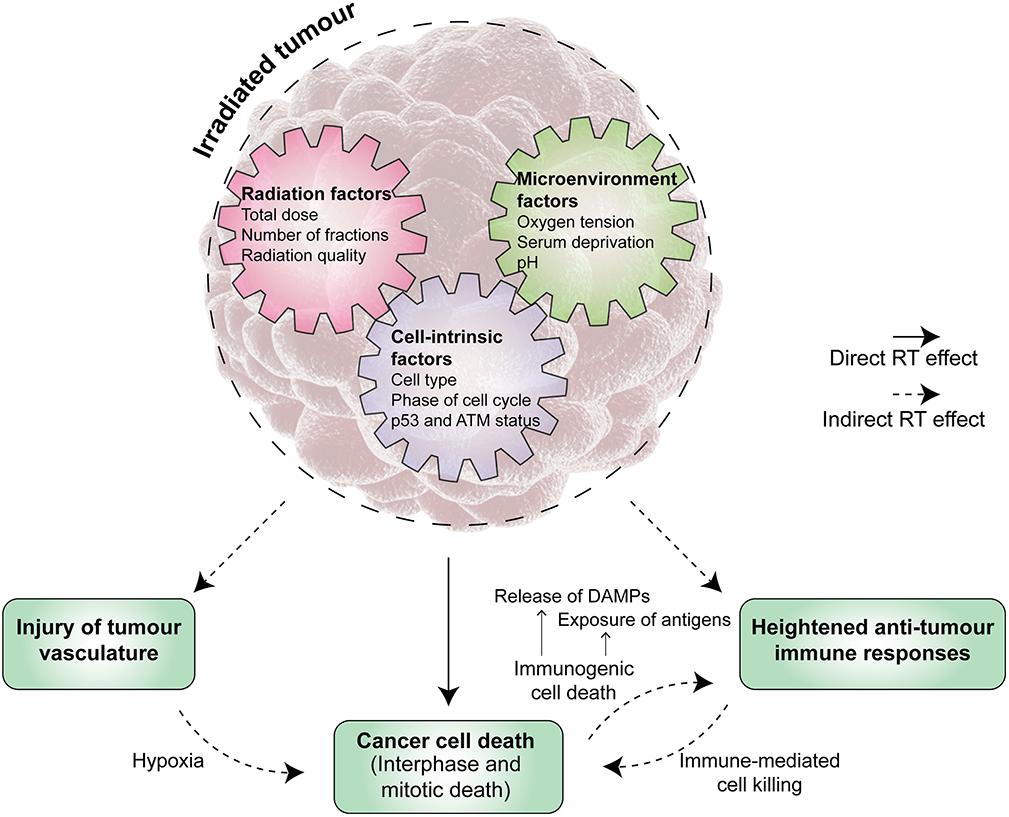
Figure 1. The anti-tumour activity of radiation therapy (RT) is multi-faceted. Tumour irradiation induces both direct and indirect effects in controlling the tumour. Direct effects are the result of significant radiation-induced DNA damage in cancer cells leading to their death, which may occur at various time points after the initial irradiation event. Radiation therapy (RT) also exerts anti-tumour activity via indirect effects, which include, but are not limited to, injury of tumour vasculature and priming of host anti-tumour immune responses. A range of factors influence the triggering and magnitude of these effects, broadly falling into cell-intrinsic, radiation, and microenvironment factors. A deeper knowledge of the underpinning mechanisms and their interplay will reveal opportunities for enhancing the overall anti-tumour activity of RT. ATM, ataxia-telangiectasia mutated. DAMP, danger associated-molecular pattern.
Others posit that SABR has additional indirect effects on cancer cells. Significant tumour vascular injury has been shown to result from SABR, leading to hypoxic cancer cell death several days after irradiation (Song et al., 2019). Interestingly, the ceramide apoptotic pathway in the tumour vascular endothelium, activated with ablative radiation doses, is thought to be especially important (Garcia-Barros et al., 2003; Rao et al., 2014). These results, however, have been directly challenged in other models (Moding et al., 2015), while others contend that hypoxia induced by SABR in fact inhibits its efficacy (Carlson et al., 2011).
The other reason measurements of radiation-induced cancer cell death in vitro fail to represent the extent of clinically observed responses is due to host immune responses (Golden et al., 2012; Haikerwal et al., 2015). For example, inhibition of the pro-survival autophagy machinery radiosensitises cell kill in vitro, but in fact impairs radiation tumour control in immunocompetent mice due to loss of autophagy-dependent ATP release as an immune adjuvant (Ko et al., 2014). As such, a pressing question is the impact of radiation dose-fractionation on cell death pathways, especially those related to ICD. Studying fractionated regimens in the pre-clinical setting is complicated because of the physiologically artificial context and logistical difficulties of irradiating animals in vivo over an extended number of fractions. Pre-clinical studies therefore typically employ moderately hypofractionated or ablative regimens (Demaria and Formenti, 2012; Deloch et al., 2016), potentially biassing the commonly-held hypothesis that such regimens are better inducers of ICD.
DEFINITIONS
Radiation therapy terms:
Fraction – single treatment session into which the overall radiation therapy course is broken down.
Stereotactic ablative body radiotherapy/stereotactic body radiation therapy (SABR/SBRT) – precise, high-dose radiation therapy given in a small number of fractions.
Abscopal effect – regression of a tumour lesion distant to the radiation field.
Oligometastatic – Stage IV (distant spread) but with a limited number of metastases (usually less than five).
Linear energy transfer (LET) – measure of radiation quality, describing the pattern of energy deposition by the radiation.
Biological effective dose (BED) – an expression of total dose for comparison of biological effects between radiation dose-fractionation regimens.
Reassortment – the redistribution of cells previously in radioresistant phases into more radiosensitive phases after each radiation fraction.
Oxygen fixation – the making permanent of DNA damage induced by radiation, via the formation of irreparable peroxyl radicals.
Finally, greater understanding of the mechanisms of cell death will enable the application of novel radiosensitisers (Moding et al., 2016). For example, the dependence of many cancer cells specifically on the G2/M checkpoint has led to the development of agents that target this checkpoint, particularly Chk1, Wee1, ATM, and ATR (Dillon et al., 2014). Hypoxia-modifying therapy might prevent HIF-mediated revascularisation, recurrence, and metastasis (Moding et al., 2016). Additionally, targeting previously under-appreciated cell death types such as ferroptosis may support the combination of RT and immunotherapy (Lang et al., 2019). Alongside these developments, biomarker driven studies must not be neglected to help guide patient selection.
Cancer cells undergo a range of interphase and mitotic death after irradiation via direct and indirect effects of RT. An increased understanding of the molecular mechanisms of radiation-induced cell death will reveal novel opportunities for improving the overall anti-tumour efficacy of RT.
JS, RS, EH, and HG conceived and designed the study, revised the manuscript, and read and approved the submitted version. JS wrote the first draft of the manuscript. HG wrote sections of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Apetoh, L., Ghiringhelli, F., Tesniere, A., Criollo, A., Ortiz, C., Lidereau, R., et al. (2007a). The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 220, 47–59. doi: 10.1111/j.1600-065X.2007.00573.x
Apetoh, L., Ghiringhelli, F., Tesniere, A., Obeid, M., Ortiz, C., Criollo, A., et al. (2007b). Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059. doi: 10.1038/nm1622
Brady, C. A., Jiang, D., Mello, S. S., Johnson, T. M., Jarvis, L. A., Kozak, M. M., et al. (2011). Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145, 571–583. doi: 10.1016/j.cell.2011.03.035
Brown, J. M., Brenner, D. J., and Carlson, D. J. (2013). Dose escalation, not “new biology,” can account for the efficacy of stereotactic body radiation therapy with non-small cell lung cancer. Int. J. Radiat. Oncol. Biol. Phys. 85, 1159–1160. doi: 10.1016/j.ijrobp.2012.11.003
Brown, J. M., Carlson, D. J., and Brenner, D. J. (2014). The tumor radiobiology of SRS and SBRT: are more than the 5 Rs involved? Int. J. Radiat. Oncol. Biol. Phys. 88, 254–262. doi: 10.1016/j.ijrobp.2013.07.022
Brown, J. M., and Wouters, B. G. (1999). Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 59, 1391–1399.
Burdette, D. L., and Vance, R. E. (2013). STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 14, 19–26. doi: 10.1038/ni.2491
Burnette, B. C., Liang, H., Lee, Y., Chlewicki, L., Khodarev, N. N., Weichselbaum, R. R., et al. (2011). The efficacy of radiotherapy relies upon induction of type i interferon-dependent innate and adaptive immunity. Cancer Res. 71, 2488–2496. doi: 10.1158/0008-5472.CAN-10-2820
Carlson, D. J., Keall, P. J., Loo, B. W. Jr., Chen, Z. J., and Brown, J. M. (2011). Hypofractionation results in reduced tumor cell kill compared to conventional fractionation for tumors with regions of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 79, 1188–1195. doi: 10.1016/j.ijrobp.2010.10.007
Casares, N., Pequignot, M. O., Tesniere, A., Ghiringhelli, F., Roux, S., Chaput, N., et al. (2005). Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701. doi: 10.1084/jem.20050915
Chakraborty, M., Abrams, S. I., Camphausen, K., Liu, K., Scott, T., Coleman, C. N., et al. (2003). Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 170, 6338–6347. doi: 10.4049/jimmunol.170.12.6338
Chen, J. H., Zhang, P., Chen, W. D., Li, D. D., Wu, X. Q., Deng, R., et al. (2015). ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 11, 239–252. doi: 10.1080/15548627.2015.1009767
Deloch, L., Derer, A., Hartmann, J., Frey, B., Fietkau, R., and Gaipl, U. S. (2016). Modern radiotherapy concepts and the impact of radiation on immune activation. Front. Oncol. 6:141. doi: 10.3389/fonc.2016.00141
Demaria, S., and Formenti, S. C. (2012). Radiation as an immunological adjuvant: current evidence on dose and fractionation. Front. Oncol. 2:153. doi: 10.3389/fonc.2012.00153
Deng, L., Liang, H., Xu, M., Yang, X., Burnette, B., Arina, A., et al. (2014). STING-Dependent Cytosolic DNA sensing promotes radiation-induced Type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852. doi: 10.1016/j.immuni.2014.10.019
Dewan, M. Z., Galloway, A. E., Kawashima, N., Dewyngaert, J. K., Babb, J. S., Formenti, S. C., et al. (2009). Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 15, 5379–5388. doi: 10.1158/1078-0432.ccr-09-0265
Diamond, J. M., Vanpouille-Box, C., Spada, S., Rudqvist, N. P., Chapman, J. R., Ueberheide, B. M., et al. (2018). Exosomes shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res. 6, 910–920. doi: 10.1158/2326-6066.CIR-17-0581
Dikic, I., and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19, 349–364. doi: 10.1038/s41580-018-0003-4
Dillon, M. T., Good, J. S., and Harrington, K. J. (2014). Selective targeting of the G2/M cell cycle checkpoint to improve the therapeutic index of radiotherapy. Clin. Oncol. 26, 257–265. doi: 10.1016/j.clon.2014.01.009
Dou, Z., Ghosh, K., Vizioli, M. G., Zhu, J., Sen, P., Wangensteen, K. J., et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. doi: 10.1038/nature24050
Endlich, B., Radford, I. R., Forrester, H. B., and Dewey, W. C. (2000). Computerized video time-lapse microscopy studies of ionizing radiation-induced rapid-interphase and mitosis-related apoptosis in lymphoid cells. Radiat. Res. 153, 36–48. doi: 10.1667/0033-7587(2000)153%5B0036:cvtlms%5D2.0.co;2
Eriksson, D., and Stigbrand, T. (2010). Radiation-induced cell death mechanisms. Tumour. Biol. 31, 363–372. doi: 10.1007/s13277-010-0042-8
Formenti, S. C., Rudqvist, N. P., Golden, E., Cooper, B., Wennerberg, E., Lhuillier, C., et al. (2018). Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat. Med. 24, 1845–1851. doi: 10.1038/s41591-018-0232-2
Forrester, H. B., Vidair, C. A., Albright, N., Ling, C. C., and Dewey, W. C. (1999). Using computerized video time lapse for quantifying cell death of X-irradiated rat embryo cells transfected with c-myc or c-Ha-ras. Cancer Res. 59, 931–939.
Franken, N. A., ten Cate, R., Krawczyk, P. M., Stap, J., Haveman, J., Aten, J., et al. (2011). Comparison of RBE values of high-LET alpha-particles for the induction of DNA-DSBs, chromosome aberrations and cell reproductive death. Radiat. Oncol. 6:64. doi: 10.1186/1748-717X-6-64
Fridman, J. S., and Lowe, S. W. (2003). Control of apoptosis by p53. Oncogene 22, 9030–9040. doi: 10.1038/sj.onc.1207116
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L., and Kroemer, G. (2017). Immunogenic cell death in cancer and infectious disease. Nat. Re.v Immunol. 17, 97–111. doi: 10.1038/nri.2016.107
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on Cell Death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Gameiro, S. R., Jammeh, M. L., Wattenberg, M. M., Tsang, K. Y., Ferrone, S., and Hodge, J. W. (2014). Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 5, 403–416. doi: 10.18632/oncotarget.1719
Garcia-Barros, M., Paris, F., Cordon-Cardo, C., Lyden, D., Rafii, S., Haimovitz-Friedman, A., et al. (2003). Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 300, 1155–1159. doi: 10.1126/science.1082504
Gascoigne, K. E., and Taylor, S. S. (2008). Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122. doi: 10.1016/j.ccr.2008.07.002
Gluck, S., Guey, B., Gulen, M. F., Wolter, K., Kang, T. W., Schmacke, N. A., et al. (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. 19, 1061–1070. doi: 10.1038/ncb3586
Golden, E. B., Frances, D., Pellicciotta, I., Demaria, S., Helen Barcellos-Hoff, M., and Formenti, S. C. (2014). Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology 3:e28518. doi: 10.4161/onci.28518
Golden, E. B., Pellicciotta, I., Demaria, S., Barcellos-Hoff, M. H., and Formenti, S. C. (2012). The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol. 2:88. doi: 10.3389/fonc.2012.00088
Gong, Y., Fan, Z., Luo, G., Yang, C., Huang, Q., Fan, K., et al. (2019). The role of necroptosis in cancer biology and therapy. Mol. Cancer 18:100. doi: 10.1186/s12943-019-1029-8
Gorin, J. B., Menager, J., Gouard, S., Maurel, C., Guilloux, Y., Faivre-Chauvet, A., et al. (2014). Antitumor immunity induced after alpha irradiation. Neoplasia 16, 319–328. doi: 10.1016/j.neo.2014.04.002
Grassberger, C., Ellsworth, S. G., Wilks, M. Q., Keane, F. K., and Loeffler, J. S. (2019). Assessing the interactions between radiotherapy and antitumour immunity. Nat. Rev. Clin. Oncol. 16, 729–745. doi: 10.1038/s41571-019-0238-9
Haikerwal, S. J., Hagekyriakou, J., MacManus, M., Martin, O. A., and Haynes, N. M. (2015). Building immunity to cancer with radiation therapy. Cancer Lett. 368, 198–208. doi: 10.1016/j.canlet.2015.01.009
Harding, S. M., Benci, J. L., Irianto, J., Discher, D. E., Minn, A. J., and Greenberg, R. A. (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. doi: 10.1038/nature23470
Harris, A. (2002). Hypoxia–a key regulatory factor in tumour growth. Nat. Rev. Cancer 2, 38–47. doi: 10.1038/nrc704
Ianzini, F., Bertoldo, A., Kosmacek, E. A., Phillips, S. L., and Mackey, M. A. (2006). Lack of p53 function promotes radiation-induced mitotic catastrophe in mouse embryonic fibroblast cells. Cancer Cell Int. 6:11. doi: 10.1186/1475-2867-6-11
Jonathan, E. C., Bernhard, E. J., and McKenna, W. G. (1999). How does radiation kill cells? Curr. Opin. Chem. Biol. 3, 77–83. doi: 10.1016/s1367-5931(99)80014-3
Kepp, O., Senovilla, L., Vitale, I., Vacchelli, E., Adjemian, S., Agostinis, P., et al. (2014). Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3:e955691. doi: 10.4161/21624011.2014.955691
Ko, A., Kanehisa, A., Martins, I., Senovilla, L., Chargari, C., Dugue, D., et al. (2014). Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ. 21, 92–99. doi: 10.1038/cdd.2013.124
Kolesnick, R., and Fuks, Z. (2003). Radiation and ceramide-induced apoptosis. Oncogene 22, 5897–5906. doi: 10.1038/sj.onc.1206702
Kroemer, G., Galluzzi, L., Vandenabeele, P., Abrams, J., Alnemri, E. S., Baehrecke, E. H., et al. (2009). Classification of cell death: recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 16, 3–11. doi: 10.1038/cdd.2008.150
Kroemer, G., and Levine, B. (2008). Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010. doi: 10.1038/nrm2529
Krysko, D. V., Garg, A. D., Kaczmarek, A., Krysko, O., Agostinis, P., and Vandenabeele, P. (2012). Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 12, 860–875. doi: 10.1038/nrc3380
Krysko, D. V., and Vandenabeele, P. (2010). Clearance of dead cells: mechanisms, immune responses and implication in the development of diseases. Apoptosis 15, 995–997. doi: 10.1007/s10495-010-0524-6
Kwon, E. D., Drake, C. G., Scher, H. I., Fizazi, K., Bossi, A., van den Eertwegh, A. J., et al. (2014). Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 15, 700–712. doi: 10.1016/S1470-2045(14)70189-5
Lang, X., Green, M. D., Wang, W., Yu, J., Choi, J. E., Jiang, L., et al. (2019). Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 9, 1673–1685. doi: 10.1158/2159-8290.CD-19-0338
Lei, P., Bai, T., and Sun, Y. (2019). Mechanisms of ferroptosis and relations with regulated cell death: a review. Front. Physiol. 10:139. doi: 10.3389/fphys.2019.00139
Levy, J. M. M., Towers, C. G., and Thorburn, A. (2017). Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542. doi: 10.1038/nrc.2017.53
Li, M., You, L., Xue, J., and Lu, Y. (2018). Ionizing radiation-induced cellular senescence in normal, non-transformed cells and the involved DNA damage response: a mini review. Front. Pharmacol. 9:522. doi: 10.3389/fphar.2018.00522
Liang, N., Jia, L., Liu, Y., Liang, B., Kong, D., Yan, M., et al. (2013). ATM pathway is essential for ionizing radiation-induced autophagy. Cell Signal. 25, 2530–2539. doi: 10.1016/j.cellsig.2013.08.010
Lim, J. Y., Gerber, S. A., Murphy, S. P., and Lord, E. M. (2014). Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8(+) T cells. Cancer Immunol. Immunother. 63, 259–271. doi: 10.1007/s00262-013-1506-7
Ma, Y., Kepp, O., Ghiringhelli, F., Apetoh, L., Aymeric, L., Locher, C., et al. (2010). Chemotherapy and radiotherapy: cryptic anticancer vaccines. Semin. Immunol. 22, 113–124. doi: 10.1016/j.smim.2010.03.001
Maier, P., Hartmann, L., Wenz, F., and Herskind, C. (2016). Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int. J. Mol. Sci. 17:E102. doi: 10.3390/ijms17010102
Michaud, M., Martins, I., Sukkurwala, A. Q., Adjemian, S., Ma, Y., Pellegatti, P., et al. (2011). Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334, 1573–1577. doi: 10.1126/science.1208347
Moding, E. J., Castle, K. D., Perez, B. A., Oh, P., Min, H. D., Norris, H., et al. (2015). Tumor cells, but not endothelial cells, mediate eradication of primary sarcomas by stereotactic body radiation therapy. Sci. Transl. Med. 7:278ra234. doi: 10.1126/scitranslmed.aaa4214
Moding, E. J., Mowery, Y. M., and Kirsch, D. G. (2016). Opportunities for radiosensitization in the stereotactic body radiation therapy (SBRT) Era. Cancer J. 22, 267–273. doi: 10.1097/PPO.0000000000000203
Nguyen, H. Q., To, N. H., Zadigue, P., Kerbrat, S., De La Taille, A., Le Gouvello, S., et al. (2018). Ionizing radiation-induced cellular senescence promotes tissue fibrosis after radiotherapy. A review. Crit. Rev. Oncol. Hematol. 129, 13–26. doi: 10.1016/j.critrevonc.2018.06.012
Obe, G., Johannes, C., and Ritter, S. (2010). The number and not the molecular structure of DNA double-strand breaks is more important for the formation of chromosomal aberrations: a hypothesis. Mutat. Res. 701, 3–11. doi: 10.1016/j.mrgentox.2010.05.010
Obeid, M., Panaretakis, T., Joza, N., Tufi, R., Tesniere, A., van Endert, P., et al. (2007a). Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 14, 1848–1850. doi: 10.1038/sj.cdd.4402201
Obeid, M., Tesniere, A., Ghiringhelli, F., Fimia, G. M., Apetoh, L., Perfettini, J. L., et al. (2007b). Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61. doi: 10.1038/nm1523
Paglin, S., Delohery, T., Erlandson, R., and Yahalom, J. (1997). Radiation-induced micronuclei formation in human breast cancer cells: dependence on serum and cell cycle distribution. Biochem. Biophys. Res. Commun. 237, 678–684. doi: 10.1006/bbrc.1997.7117
Palma, D. A., Olson, R., Harrow, S., Gaede, S., Louie, A. V., Haasbeek, C., et al. (2019). Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (SABR-COMET): a randomised, phase 2, open-label trial. Lancet 393, 2051–2058. doi: 10.1016/S0140-6736(18)32487-5
Paludan, S. R., and Bowie, A. G. (2013). Immune sensing of DNA. Immunity 38, 870–880. doi: 10.1016/j.immuni.2013.05.004
Park, H. J., Lyons, J. C., Ohtsubo, T., and Song, C. W. (2000). Cell cycle progression and apoptosis after irradiation in an acidic environment. Cell Death Differ. 7, 729–738. doi: 10.1038/sj.cdd.4400702
Pettus, B. J., Chalfant, C. E., and Hannun, Y. A. (2002). Ceramide in apoptosis: an overview and current perspectives. Biochim. Biophys. Acta 1585, 114–125. doi: 10.1016/s1388-1981(02)00331-1
Pinto, M., Prise, K. M., and Michael, B. D. (2005). Evidence for complexity at the nanometer scale of radiation-induced DNA DSBs as a determinant of rejoining kinetics. Radiat. Res. 164, 73–85. doi: 10.1667/rr3394
Radford, I. R., Murphy, T. K., Radley, J. M., and Ellis, S. L. (1994). Radiation response of mouse lymphoid and myeloid cell lines. Part II. Apoptotic death is shown by all lines examined. Int. J. Radiat. Biol. 65, 217–227. doi: 10.1080/09553009414550251
Rao, S. S., Thompson, C., Cheng, J., Haimovitz-Friedman, A., Powell, S. N., Fuks, Z., et al. (2014). Axitinib sensitization of high Single Dose Radiotherapy. Radiother. Oncol. 111, 88–93. doi: 10.1016/j.radonc.2014.02.010
Reits, E. A., Hodge, J. W., Herberts, C. A., Groothuis, T. A., Chakraborty, M., Wansley, E. K., et al. (2006). Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 203, 1259–1271. doi: 10.1084/jem.20052494
Ringborg, U., Bergqvist, D., Brorsson, B., Cavallin-Stahl, E., Ceberg, J., Einhorn, N., et al. (2003). The swedish council on technology assessment in health care (SBU) systematic overview of radiotherapy for cancer including a prospective survey of radiotherapy practice in Sweden 2001–summary and conclusions. Acta Oncol. 42, 357–365. doi: 10.1080/02841860310010826
Sabin, R. J., and Anderson, R. M. (2011). Cellular Senescence - its role in cancer and the response to ionizing radiation. Genome Integr. 2:7. doi: 10.1186/2041-9414-2-7
Sancho, D., Joffre, O. P., Keller, A. M., Rogers, N. C., Martinez, D., Hernanz-Falcon, P., et al. (2009). Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903. doi: 10.1038/nature07750
Santin, A. D., Hermonat, P. L., Hiserodt, J. C., Chiriva-Internati, M., Woodliff, J., Theus, J. W., et al. (1997). Effects of irradiation on the expression of major histocompatibility complex class I antigen and adhesion costimulation molecules ICAM-1 in human cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 39, 737–742. doi: 10.1016/s0360-3016(97)00372-6
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi: 10.1038/nature00858
Schadt, L., Sparano, C., Schweiger, N. A., Silina, K., Cecconi, V., Lucchiari, G., et al. (2019). Cancer-Cell-Intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep 29, 1236.e7–1248.e7. doi: 10.1016/j.celrep.2019.09.065
Sheard, M. A., Uldrijan, S., and Vojtesek, B. (2003). Role of p53 in regulating constitutive and X-radiation-inducible CD95 expression and function in carcinoma cells. Cancer Res. 63, 7176–7184.
Skinner, H. D., Sandulache, V. C., Ow, T. J., Meyn, R. E., Yordy, J. S., Beadle, B. M., et al. (2012). TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin. Cancer Res. 18, 290–300. doi: 10.1158/1078-0432.CCR-11-2260
Song, C. W., Glatstein, E., Marks, L. B., Emami, B., Grimm, J., Sperduto, P. W., et al. (2019). Biological principles of stereotactic body radiation therapy (SBRT) and stereotactic radiation surgery (SRS): indirect cell death. Int. J. Radiat. Oncol. Biol. Phys. S0360-3016, 30291–30293. doi: 10.1016/j.ijrobp.2019.02.047
Soussi, T., and Beroud, C. (2001). Assessing TP53 status in human tumours to evaluate clinical outcome. Nat. Rev. Cancer 1, 233–240. doi: 10.1038/35106009
Stewart, R. D., Yu, V. K., Georgakilas, A. G., Koumenis, C., Park, J. H., and Carlson, D. J. (2011). Effects of radiation quality and oxygen on clustered DNA lesions and cell death. Radiat. Res. 176, 587–602. doi: 10.1667/rr2663.1
Stone, H. B., Peters, L. J., and Milas, L. (1979). Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J. Natl. Cancer Inst. 63, 1229–1235.
Tam, S. Y., Wu, V. W., and Law, H. K. (2017). Influence of autophagy on the efficacy of radiotherapy. Radiat. Oncol. 12, 57. doi: 10.1186/s13014-017-0795-y
Theelen, W., Peulen, H. M. U., Lalezari, F., van der Noort, V., de Vries, J. F., Aerts, J., et al. (2019). Effect of pembrolizumab after stereotactic body radiotherapy vs pembrolizumab alone on tumor response in patients with advanced non-small cell lung cancer: results of the PEMBRO-RT Phase 2 randomized clinical trial. JAMA Oncol. doi: 10.1001/jamaoncol.2019.1478 [Epub ahead of print].,
Tripathi, D. N., Chowdhury, R., Trudel, L. J., Tee, A. R., Slack, R. S., Walker, C. L., et al. (2013). Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc. Natl. Acad. Sci. U.S.A. 110, E2950–E2957. doi: 10.1073/pnas.1307736110
Vakifahmetoglu, H., Olsson, M., and Zhivotovsky, B. (2008). Death through a tragedy: mitotic catastrophe. Cell Death Differ. 15, 1153–1162. doi: 10.1038/cdd.2008.47
Vakkila, J., and Lotze, M. T. (2004). Inflammation and necrosis promote tumour growth. Nat. Rev. Immunol. 4, 641–648. doi: 10.1038/nri1415
Vanpouille-Box, C., Formenti, S. C., and Demaria, S. (2017). TREX1 dictates the immune fate of irradiated cancer cells. Oncoimmunology 6, e1339857. doi: 10.1080/2162402X.2017.1339857
Verbrugge, I., Gasparini, A., Haynes, N. M., Hagekyriakou, J., Galli, M., Stewart, T. J., et al. (2014). The curative outcome of radioimmunotherapy in a mouse breast cancer model relies on mTOR signaling. Radiat. Res. 182, 219–229. doi: 10.1667/RR13511.1
Verheij, M., and Bartelink, H. (2000). Radiation-induced apoptosis. Cell Tissue Res. 301, 133–142. doi: 10.1007/s004410000188
Voll, R. E., Herrmann, M., Roth, E. A., Stach, C., Kalden, J. R., and Girkontaite, I. (1997). Immunosuppressive effects of apoptotic cells. Nature 390, 350–351. doi: 10.1038/37022
Voorwerk, L., Slagter, M., Horlings, H. M., Sikorska, K., van de Vijver, K. K., de Maaker, M., et al. (2019). Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat. Med. 25, 920–928. doi: 10.1038/s41591-019-0432-4
Warters, R. L., Hofer, K. G., Harris, C. R., and Smith, J. M. (1978). Radionuclide toxicity in cultured mammalian cells: elucidation of the primary site of radiation damage. Curr. Top. Radiat. Res. Q. 12, 389–407.
Wouters, B. G., Giaccia, A. J., Denko, N. C., and Brown, J. M. (1997). Loss of p21Waf1/Cip1 sensitizes tumors to radiation by an apoptosis-independent mechanism. Cancer Res. 57, 4703–4706.
Wu, G. S., Burns, T. F., McDonald, E. R. III, Jiang, W., Meng, R., Krantz, I. D., et al. (1997). KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17, 141–143. doi: 10.1038/ng1097-141
Keywords: radiotherapy, radiation therapy, stereotactic ablative radiotherapy, cell death, immunogenic cell death, abscopal effect
Citation: Sia J, Szmyd R, Hau E and Gee HE (2020) Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 8:41. doi: 10.3389/fcell.2020.00041
Received: 14 November 2019; Accepted: 17 January 2020;
Published: 13 February 2020.
Edited by:
Andrew Burgess, Anzac Research Institute, AustraliaReviewed by:
Brian Gabrielli, The University of Queensland, AustraliaCopyright © 2020 Sia, Szmyd, Hau and Gee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harriet E. Gee, harriet.gee@sydney.edu.au; Harriet.Gee@health.nsw.gov.au
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.