 Anita C. Truttmann
Anita C. Truttmann Vanessa Ginet1,2†
Vanessa Ginet1,2† Julien Puyal
Julien Puyal
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 18 February 2020
Sec. Cell Death and Survival
Volume 8 - 2020 | https://doi.org/10.3389/fcell.2020.00027
Despite tremendous advances in neonatal intensive care over the past 20 years, prematurity carries a high burden of neurological morbidity lasting lifelong. The term encephalopathy of prematurity (EoP) coined by Volpe in 2009 encompasses all aspects of the now known effects of prematurity on the immature brain, including altered and disturbed development as well as specific lesional hallmarks. Understanding the way cells are damaged is crucial to design brain protective strategies, and in this purpose, preclinical models largely contribute to improve the comprehension of the cell death mechanisms. While neuronal cell death has been deeply investigated and characterized in (hypoxic–ischemic) encephalopathy of the newborn at term, little is known about the types of cell death occurring in preterm brain injury. Three main different morphological cell death types are observed in the immature brain, specifically in models of hypoxic–ischemic encephalopathy, namely, necrotic, apoptotic, and autophagic cell death. Features of all three types may be present in the same dying neuron. In preterm brain injury, description of cell death types is sparse, and cell loss primarily concerns immature oligodendrocytes and, infrequently, neurons. In the present review, we first shortly discuss the different main severe preterm brain injury conditions that have been reported to involve cell death, including periventricular leucomalacia (PVL), diffuse white matter injury (dWMI), and intraventricular hemorrhages, as well as potentially harmful iatrogenic conditions linked to premature birth (anesthesia and caffeine therapy). Then, we present an overview of current evidence concerning cell death in both clinical human tissue data and preclinical models by focusing on studies investigating the presence of cell death allowing discriminating between the types of cell death involved. We conclude that, to improve brain protective strategies, not only apoptosis but also other cell death (such as regulated necrotic and autophagic) pathways now need to be investigated together in order to consider all cell death mechanisms involved in the pathogenesis of preterm brain damage.
Neurodevelopmental deficits are frequent among infants born prematurely, particularly those born before 32 weeks of gestational age (very preterm infants, VPT). Around 5–10% VPT exhibit severe impairments such as cerebral palsy and cognitive delay, which are linked frequently to cystic periventricular leucomalacia (cPVL) and intraventricular and parenchymatous hemorrhages grades III and IV, corresponding to the most severe brain lesions. Around 10–15% of VPT exhibit more moderate impairments, such as limited cognitive functions and motoric deficits, linked morphologically to diffuse white and gray matter lesions. Finally, around 30–40% of VPT exhibit impaired academic achievement and behavioral disorders (Pierrat et al., 2017), associated presumably with altered brain development, dysmaturational disorders, and connectivity dysfunctions.
The major substrate of human preterm brain injury is the encephalopathy of prematurity (EoP) that is characterized by gray and white matter lesions reflecting combined acquired insults, altered developmental trajectories, and reparative phenomena (Kinney and Volpe, 2012). This term was quoted by Volpe in 2009 to characterize the multiple hit hypothesis and heterogeneous WM and GM impairments (including pre-oligodendrocytes injury, axonal injury, thalamic injury, subplate injury, and migrating GABAergic neuronal injury) affecting key developmental pathways and resulting in adverse clinical outcomes of VPT (Van’t Hooft et al., 2015; Schneider et al., 2016; Synnes and Hicks, 2018). On one hand, EoP covers severe brain lesions occurring after cell death such as in cPVL or intraventricular–parenchymatous hemorrhages (IVH). However, less severe brain lesions as in non-cystic and diffuse WM injury (detected by advanced MRI techniques and characterized by astrogliosis and microgliosis) are now more frequently observed with some features of cell death. On the other hand, subtler brain alterations can reflect disturbances of brain development or delay in brain maturation and affect functional connectivity with no apparent cell death (Ball et al., 2013; Gozdas et al., 2018).
The mostly studied anatomical structures involved in preterm brain injury are the periventricular white matter (PWM), the germinal matrix (source of most intraventricular hemorrhages), and the subventricular zone (SVZ, source of migrational and maturational disorders). Although there is growing evidence of the GM involvement (cortex, thalamus, nucleus caudatus, and deep GM injury), literature is sparser (Kinney and Volpe, 2012). Recent studies have shown that cortical interneurons development can be disrupted (reduced number and morphological complexity) in preterm infants with non-cystic WM injury or in inflammatory conditions (Panda et al., 2018; Stolp et al., 2019). Moreover, constant improvement in imaging techniques allows to study and detect microstructural alterations not only in WM but also in GM related to neurodevelopmental disorders (Chau et al., 2013; Nossin-Manor et al., 2013; Kersbergen et al., 2016).
Therefore, it is mandatory to understand the underlying pathophysiological cellular mechanisms contributing to WM and GM damage and/or organization defects. It is now widely recognized that events that induce an inflammatory context sensitize preterm brain to injury (Hagberg et al., 2015). Beside the inflammatory, vascular, and maturational factors implicated in the origin of preterm brain injury and some of those specific lesions, the etiology of the cellular factors and the cell death mechanisms encountered, either during altered brain development or in relation with specific lesions, are complex and interrelated. Focusing only on studies using cell death markers on pathological tissues, the present review proposes to do an overview of what has been described so far in human autopsy tissue and preclinical models regarding the different types of cell death involved in preterm brain injury. We here considered only studies using classical known and widely accepted cell death markers allowing the discrimination between cell death types (i.e., excluding studies reporting loss of cells without clarification of the type of cell death involved). Therefore, according to our selection criteria, this review will be focused mainly on the most severe cases of preterm brain injury involving “anatomical” evidence of brain damage.
Since EoP includes diverse causes that cannot be reproduced in one unique model, our investigations addressed the main pathological conditions related to EoP such as inflammation, hypoxia–ischemia (HI)/excitotoxicity, and other cytototoxic conditions related to current therapies such as hyperoxia, caffeine, and anesthetics. We will then discuss the need to investigate more in detail and improve our understanding of the different oligodendrocyte and neuronal death pathways that can play, even if not massive in some conditions, a role in neurological deficits developed by both preterm humans and animal models.
It is now globally accepted that three main morphological types of cell death exist as previously proposed by Peter Clarke in 1990 in physiological cell death conditions during development (Clarke, 1990): type 1 or apoptosis, type 2 or autophagic cell death, and type 3 or necrotic cell death (Figure 1). This classification is also valid for cell death involved in different pathologies, including brain injury. However, it has been described that, especially in the immature brain, hybrid morphological forms of cell death could occur in the same dying cell, reflecting the presence of interconnections between the molecular pathways controlling the execution of different cell death types (Portera-Cailliau et al., 1997; Puyal et al., 2013). The multiplicity of the cell death pathways and their positive/negative crosstalks that depend, moreover, on the cell type, their maturity, and the type of stress (intensity, duration, hypoxic, ischemic, inflammatory, etc.) represent then a major challenge for the design of neuroprotective strategies (Puyal et al., 2013). We will do here a very brief summary of the three main types of cell death (reviewed in Northington et al., 2011; Puyal et al., 2013).
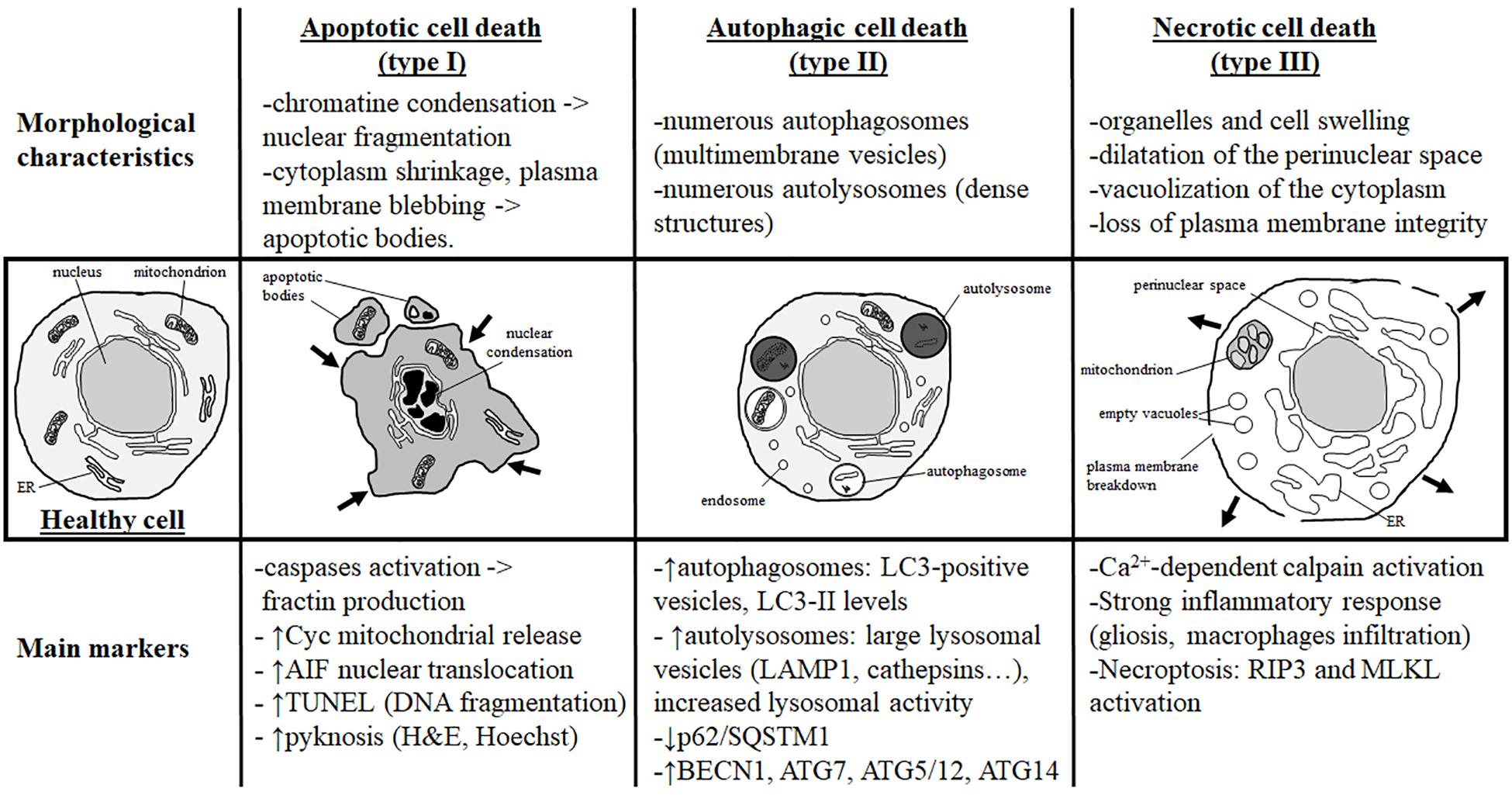
Figure 1. Morphological characteristics and most currently used markers of the three main types of cell death. Analysis of the dying cell ultrastructure is the most reliable method to identify the cell death type. The hallmark of apoptotic cell death (or type I) is a modification of the nuclear structure with presence of chromatin condensation and nuclear fragmentation. The cytoplasm shrinks and plasma membrane deforms until it forms apoptotic bodies that will be removed by phagocytic cells. Molecular markers of apoptosis are caspases (mainly caspase-3) and sometimes cytochrome c (Cyc) mitochondrial release in the cytosol. As a marker of caspase-independent apoptosis, nuclear translocation of AIF (Apoptosis-Inducing Factor) is mainly used. Pyknosis (condensation) of the nucleus shown by H&E (hematoxylin/eosin) or Hoechst stainings is also a main feature of apoptosis. TUNEL (Terminal deoxynucleotidyl transferase dUTP nick end labeling) is a staining revealing DNA fragmentation used as a marker of apoptosis with caution if not complemented with a cleaved-CASP3 immunostaining. Autophagic cell death (or type II) is mainly characterized by the presence of numerous autophagosomes (multimembrane vesicles containing intact cytoplasmic material such organelles) and autolysosomes (electron dense structure due to material at different stage of degradation). To conclude on autophagy flux with biochemical markers, it is necessary to study both autophagosome formation and autolysosome degradation. In autophagic cell death, both processes have to be enhanced. Autophagosome presence is reflected by the number of LC3-positive vesicles or by LC3-II level of expression on immunoblot. When autophagic degradation activity is enhanced, vesicles positive for lysosomal markers [such as LAMP1 (lysosomal-associated membrane protein 1) or cathepsins] are increased in number and size since autolysosomes are larger than primary lysosomes. The decrease in p62/SQSTM1 (Sequestosome-1), an autophagosome cargo protein degraded selectively by autophagy, reveals enhanced autophagy. In some cases, the level of autophagy-related (ATG) proteins such as BECN1 (BECLIN1/ATG6), ATG7, ATG5/ATG12, or ATG14 is increased. In necrotic cell death (or type III), ultrastructural changes are mainly cytoplasmic with organelles and cell swelling and presence of empty vacuoles. Perinuclear space is also dilated. Plasma membrane integrity can be finally lost, inducing a strong inflammatory response. Very few tools are available to identify necrotic cell death and mainly indirect markers are used such as strong inflammation response or Ca2+-dependent activation of calpains [mainly suggested by the production of a 145- to 150-kDa calpain-dependent cleavage of spectrin (fodrin)]. Necroptosis type of necrosis could be investigated by RIP3/RIPK3 (Receptor-interacting serine/threonine-protein kinase) or MLKL (mixed lineage kinase domain-like) expression and activation.
Since about 50 years, apoptosis is largely the best-known and characterized form of programmed cell death, which has been proposed to be the most important mechanism of delayed cell death occurring in different physiological and pathological conditions, including the developing brain. The most obvious morphological changes occurring during apoptosis concern the nucleus. Chromatin is compacted and the nucleus condensates, leading to the characteristic apoptotic feature of pyknosis and finally nuclear fragmentation. The cytoplasm is also shrunk and plasma membrane is distorted, forming blebbs that will finally detach and then form apoptotic bodies (Figure 1).
At the molecular level, apoptosis has been extensively characterized and can occur through two different but convergent pathways: the intrinsic and the extrinsic pathways (Galluzzi et al., 2018).
The intrinsic apoptotic pathway, also called the mitochondrial pathway of apoptosis, involves mitochondria, which represent a reservoir of apoptotic factors (stored in the intermembrane space of mitochondria). The balance of pro- and anti-apoptotic proteins from the BCL2 protein family controls mitochondrial membrane integrity. These proteins are responsive to intracellular stress coming from mitochondria, endoplasmic reticulum (ER), cytoskeleton, or nucleus. Pro-apoptotic proteins are directly (BAX and BAK) or indirectly (BAD, PUMA, BIM) involved in mitochondrial membrane permeabilization. As anti-apoptotic proteins, BCL2, BCL-xL, or MCL1 prevents BAX and BAK-mediated permeabilization of the outer mitochondrial membrane and then the release of apoptotic factors in the cytosol. Among these factors, some, such as cytochrome c, SMAC/Diablo, and Omi/HtrA2, activate caspase-dependent pathways and others, such as AIF and endoG, will trigger DNA fragmentation independently of caspases.
Caspases, which are cysteine-aspartic proteases, are the main common effectors of both intrinsic and extrinsic apoptotic pathways. Caspases activation is then widely considered as a gold standard marker of apoptosis in many studies including those on human tissues. Initiator caspases (CASP), CASP2, -8, -9, -10, and -12, activate the executor caspases (CASP3, -6, and -7) (Riedl and Shi, 2004), which have as substrates many essential proteins such as structural proteins, enzymes involved in DNA repair, or transcription factors resulting in the morphological changes induced by apoptosis (cell shrinkage, membrane blebbing, chromatin condensation, and DNA fragmentation).
Whereas CASP9 is involved in the intrinsic pathway of apoptosis, CASP8 and 10 are the initiators of the extrinsic pathway. CASP8 and -10 activations depend on the binding of extracellular death signals (such as TNFα, FasL, and TRAIL) on “death” receptors, resulting in the formation of a death-inducing signaling complex (DISC) used as a platform for CASP8 autoactivation. CASP8 can then directly activate the executor caspases (CASP3, -6, and -7). However, CASP8 can also interfere with the intrinsic pathway of apoptosis by mediating the cleavage of BID and then producing a truncated form of BID (tBID) able to induce BAX anchoring to the mitochondrial membrane and subsequent membrane permeabilization.
Finally, the executor caspases (CASP3, -6, and -7) could also be activated by CASP12 in some ER stress conditions (oxydative stress, unfolded protein response, etc.).
Autophagic cell death is morphologically characterized by the presence of numerous autophagosomes and autolysosomes in the cytoplasm of the dying cells, reflecting an excessive autophagy activity (Figures 1, 2).
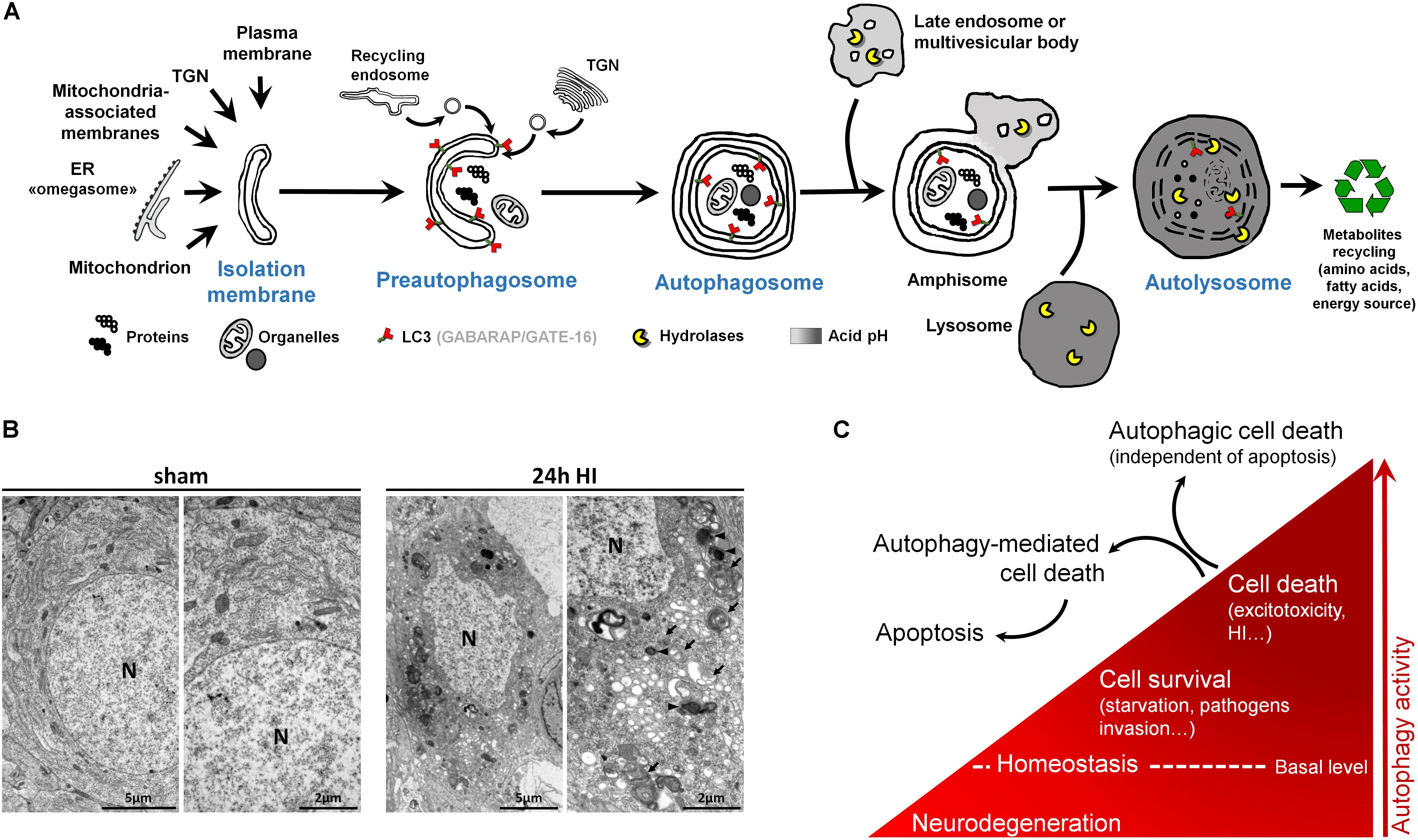
Figure 2. Autophagy and autophagic cell death. (A) Schematic illustration of (macro)autophagy degradation and recycling process showing the following: (1) isolation membrane formation from various possible intracellular membrane sources; (2) elongation and incurvation of the preautophagosome around the cytoplasmic material that has to be degraded (proteins, organelles); (3) closure end-to-end of the preautophagosome forming the autophagosome, a double-membrane vesicle containing undigested materials; (4) fusion of autophagosome with endosomes; (5) fusion of this amphisome with a lysosome to form a mature autophagosome able to degrade its content (autolysosome) thanks to lysosomal hydrolases. LC3 = Microtubule-associated protein 1A/1B-light chain 3 (and other family members). LC3 is the main marker of autophagosome. TGN = trans-Golgi network, ER = endoplasmic reticulum. (B) Electron micrographs showing CA3 neurons presenting features of autophagic cell death 24 h after perinatal hypoxia–ischemia (HI) in P7 rats (unilateral common carotid artery occlusion followed by 2 h of hypoxia at 8% of oxygen). Dying neurons displayed both numerous multimembrane vacuoles loaded with cytoplasmic material (autophagosomes, arrow) and electron dense vesicles containing digested material (autolysosomes, arrowhead). Note that nuclei (N) did not show chromatin condensation. Electron micrographs of CA3 neurons from sham-operated rat brain present healthy neurons. This study was carried out in accordance with the Swiss National Institutional Guidelines for Animal Experimentation. All experiments and methods were approved by the Veterinary Office of the Canton de Vaud. (C) Scheme illustrating the dual role of autophagy in neurons. At basal level, autophagy maintains cellular homeostasis. A reduction or impairment of autophagy can lead to neurodegeneration (such as for some proteinopathies). Autophagy can be activated, above its basal level, as a survival response to maintain energy level during a period of starvation, for example, or to eliminate invading bacteria or virus. However, in other stress conditions such as excitotoxicity, enhanced autophagy could lead to cell death as a mechanism of cell death by itself, independently of apoptosis or necrosis (autophagic cell death or type II), or as a mediator of another type of cell death, mainly apoptosis (autophagy-mediated cell death).
Macroautophagy, the main type of autophagy (beside microautophagy and chaperone-mediated autophagy) (Cuervo, 2004), is a physiological intracellular catabolic process using lysosomes for degradation. Macroautophagy (hereafter called autophagy) consists in the engulfment of some cytoplasmic material that has to be degraded (such as impaired long-lived proteins and organelles), in an intermediate compartment termed autophagosome. Autophagy can be divided into three major steps: (1) the induction phase, which consists in the isolation of a membrane termed the preautophagosome; (2) the autophagosome formation, which involves elongation, incurvation, and finally closure of the preautophagosome, resulting in multimembrane vesicles containing cytoplasmic material including organelles; and (3) the autophagosome maturation, which consists in the fusion of the autophagosome with a lysosome leading to the formation of the functional degradative compartment of autophagy, the autolysosome (Figure 2A).
Besides, multiple analyses are needed to characterize the presence of autophagy (Klionsky et al., 2016), and the analysis of the autophagosomal marker, microtubule-associated protein 1 light chain 3 (LC3), is an essential step (Figure 2A). This ATG protein (homologue of yeast ATG8) is subject to an important ubiquitylation-like modification resulting in LC3 conjugation to a phosphatidylethanolamine (PE), a membrane phospholipid that allows LC3 to be incorporated into the membrane of early and late autophagosomes. Despite the fact that other methods could be used (such as the gold standard electron microscopy), an increased number of LC3-positive dots combined with an increased lysosomal activity (e.g., monitored by p62/SQSTM1 degradation) is, in many cases, sufficient to conclude for enhanced autophagy.
The recent discovery (since the 1990s) of more than 30 evolutionarily conserved autophagy-related (ATG) genes has largely contributed to understand the roles of autophagy in cell survival and death (Levine and Kroemer, 2019). Due to the important roles of autophagy in maintaining cell homeostasis and in cell adaptation to stress conditions (such as starvation or pathogen invasion) (Marino et al., 2011), the existence of a type of cell death mediated exclusively by autophagy has long been controversial (Debnath et al., 2005; Kroemer and Levine, 2008; Clarke and Puyal, 2012). However, it is now accepted that in specific conditions, autophagic cell death can occur, independently of necrosis and apoptosis, such as during development (morphogenesis or metamorphosis) or in particular stressful conditions (Clarke, 1990; Liu et al., 2013; Ginet et al., 2014b) (Figures 2B,C). Despite this, pure autophagic cell death is rarely observed, and there is now growing evidence that autophagy-mediated cell death is more frequently involved, meaning situations where autophagy is acting upstream of another type of cell death (Kriel and Loos, 2019), especially apoptosis (Grishchuk et al., 2011; Puyal et al., 2013) (Figure 2C).
Necrosis (or type 3B cell death) (Clarke, 1990) is usually considered as a passive form of cell death (unregulated and energy-independent) occuring rapidly and irreversibly in conditions precluding apoptotic and autophagic mechanisms (Golstein and Kroemer, 2007). Necrosis is characterized morphologically by the presence of dilated organelles (primarily mitochondria and ER), the vacuolization of the cytoplasm (empty vacuoles), and the swelling of the perinuclear space. The nucleus is first preserved; however, it ends up also swelling as the cell rounds up until plasma membrane ruptures (Figure 1), leading to the release of cellular contents [including damage-associated molecular patterns (DAMPs)] that promote and exacerbate the inflammatory response.
More recently, programmed forms of necrotic cell deaths have been identified, demonstrating that, in some cases, necrotic cell death could be regulated. One of the best characterized programmed form of necrosis is necroptosis. Necroptosis is initiated by the activation of death receptors (Kitanaka and Kuchino, 1999; Galluzzi et al., 2017) and is dependent on the RIPK1/3-MLKL pathway leading to the translocation of MLKL to intracellular and plasma membranes, and then inducing membrane permeabilization and rupture.
As mentioned in the Introduction, EoP that covers the different injury patterns related to premature birth can affect either the WM or GM but, depending on the nature and the severity of the lesion, the extent of brain lesions could vary strongly between the individuals. With new emerging MRI techniques, better characterization of EoP now became possible and opens new understanding, such as functional connectivity, diffusion tensor imaging, fractional anisotropy measures and fiber tractography, and automated segmentation techniques for volume determination and growth (Ball et al., 2013; Gozdas et al., 2018; Schneider et al., 2018; Duerden et al., 2019; Jakab et al., 2019). The most prominent findings of EoP in VPT are (1) decreased global brain volume due to white (WM) and gray matter reduction [(GM); cortical GM as well as deep nuclear GM], (2) hydrocephalus ex vacuo (CSF), and (3) connectivity impairment (Dubois et al., 2008; Ball et al., 2013; Chau et al., 2013; Kidokoro et al., 2013; Friedrichs-Maeder et al., 2017; Gozdas et al., 2018; Duerden et al., 2019; Jakab et al., 2019).
This new understanding facilitates the development of new preclinical animal models, but still implementation of multiple features of EoP seems difficult (see review from Kinney and Volpe, 2012) and specific features of cell death in particular models, either neuronal or glial cell death, are lacking.
While it is widely accepted that cell death could happen in those conditions, cell death evidence in preterm brain injury are sparse and controversial, notably because few studies on human preterm brain tissue have characterized and determined either the type of cell death or, more importantly, the nature of the dying cell type (Tables 1, 2). Another point to mention is that in the studies claiming that cell death is not involved, not all the different types of cell death have been investigated (most often only apoptotic cell death markers were considered) and one can speculate that cell death types other than apoptosis might be involved in EoP. Finally, when comparing the different cohorts in Tables 1, 2, it is important to keep in mind that some infants have experienced a re-direction of care (most European countries) whereas some infants have died reaching their natural life span (US and Canada) and that the extent of cell death might have been influenced by different ethical practices. Moreover, as shown in Table 1, most of the studies were performed with cohorts from the Boston Children’s Hospital, meaning that some data could come from the same tissue collection.
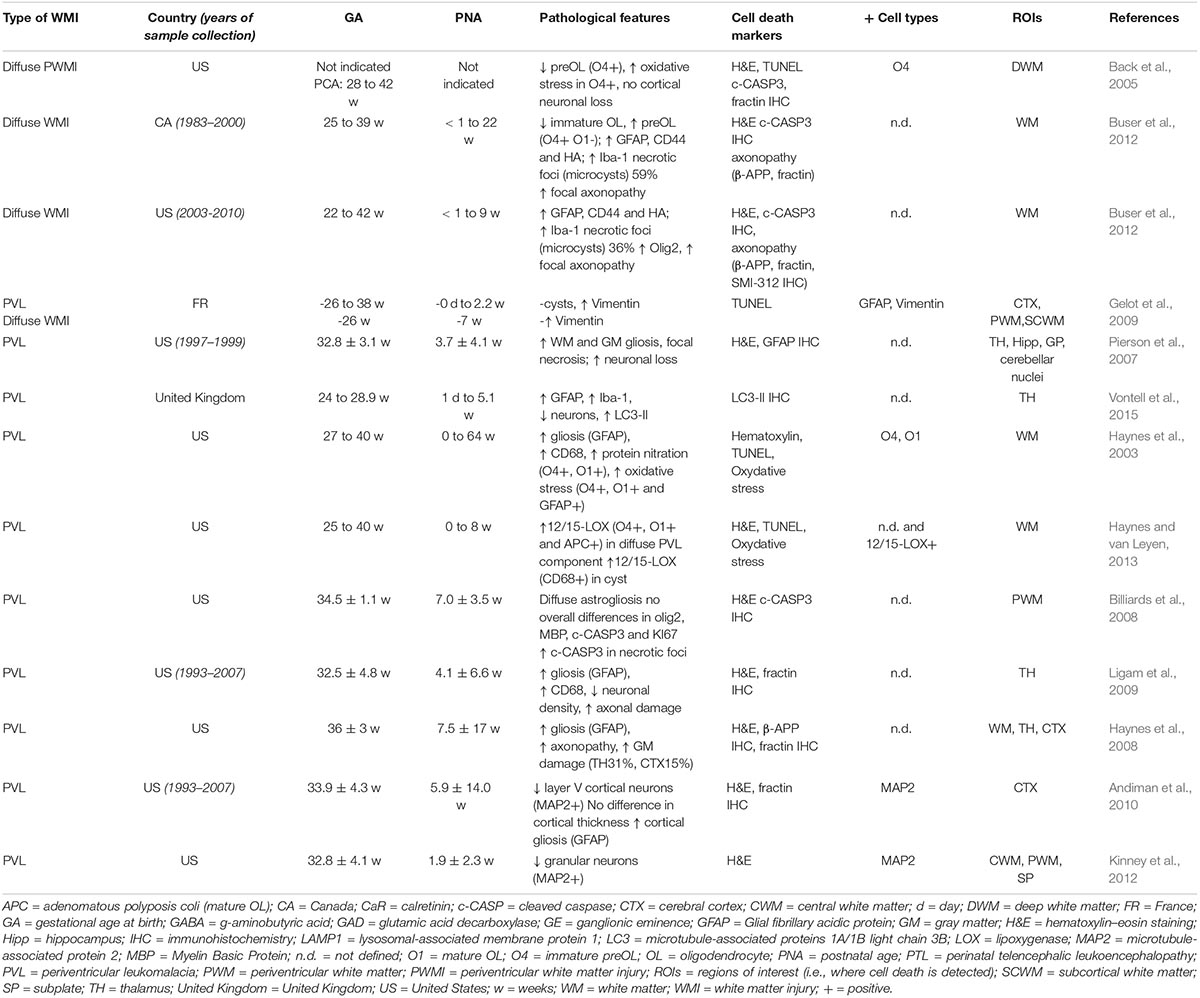
Table 1. Cell death after WMI in human preterm brain from autopsy.
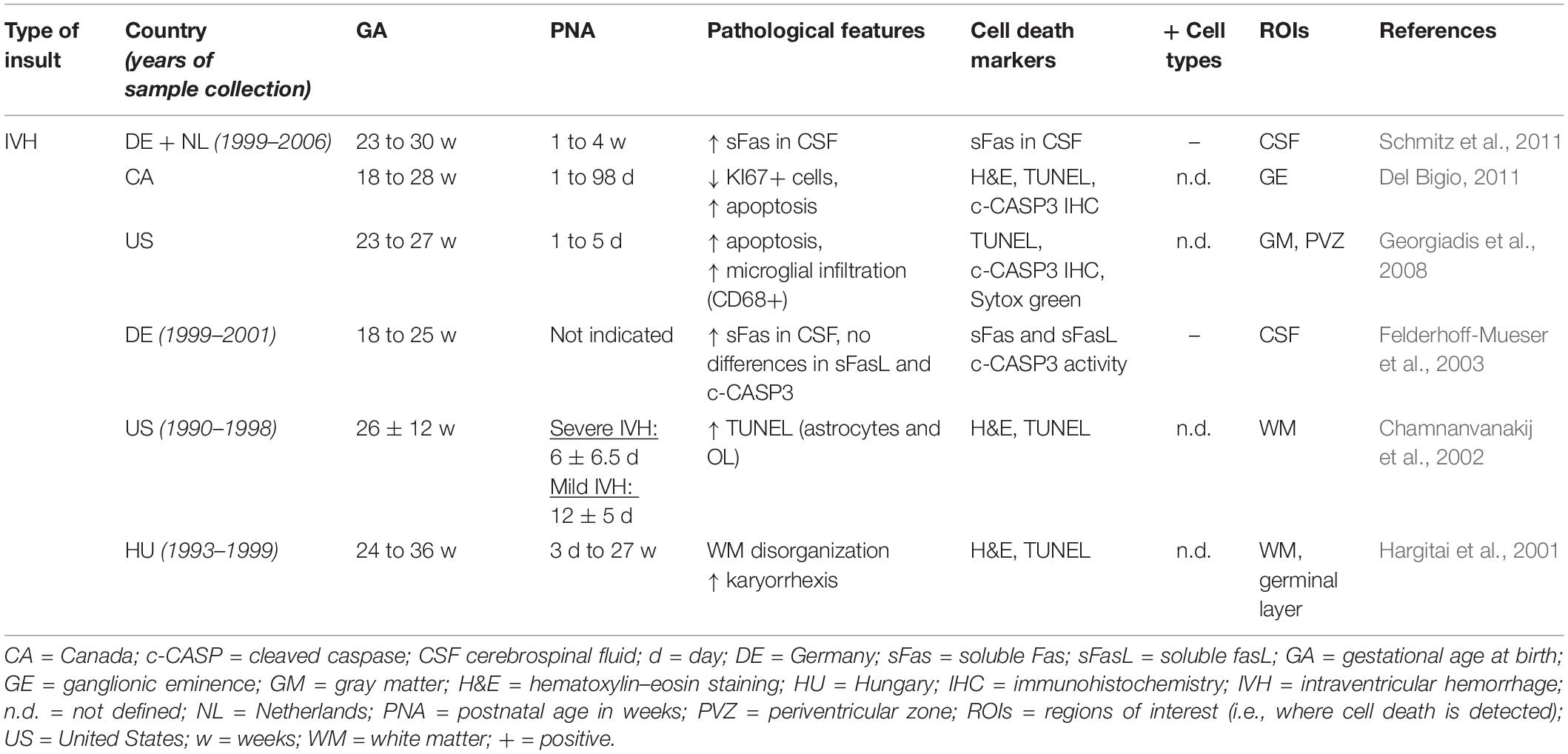
Table 2. Cell death after IVH in human preterm brains from autopsy.
The most important type of WM injury (WMI) in EoP is the PVL (malacia: infarction and leucos: white), first described as focal coagulative necrosis occurring in deep hemispheric WM (Banker and Larroche, 1962). The cPVL is the most severe form of PVL, whose incidence has decreased fivefold in the last decade and which carries the highest burden of neurological morbidity, especially cerebral palsy. Depending on the centers, the incidence of cPVL is around 4–8% for the babies born before 32 weeks of gestation and around 6 to 12% for babies born before 27 weeks of gestation (McGowan and Vohr, 2019).
cPVL is characterized by the presence of large focal periventricular WM necrotic lesions (necrotic foci), where all the cells are destroyed without specificity regarding the cell type (pan-cell death). These necrotic foci (evolving in cyst) are surrounded by reactive glial and microglial cells localized in the neighboring WM site where delayed cell death has been proposed to occur. Histological observations have reported that cPVL cases are characterized, in addition to the presence of necrotic foci, by a massive loss not only of WM (i.e., evidenced by the thinning of corpus callosum and subcortical WM) but also of GM (strong volume reduction of either cortical and deep nuclear structures). Several studies have reported increasing presence of apoptosis (mainly evidenced by TUNEL staining and c-CASP-3 immunohistochemistry) in cPVL in WM and GM, but few of these studies have identified the cell type of these apoptotic dying cells (Hargitai et al., 2001; Chamnanvanakij et al., 2002; Back et al., 2005; Robinson et al., 2006; Pierson et al., 2007; Billiards et al., 2008; Haynes et al., 2008; Ligam et al., 2009; Andiman et al., 2010; Zubiaurre-Elorza et al., 2011; Buser et al., 2012; Kinney et al., 2012; Haynes and van Leyen, 2013; Vontell et al., 2015) (see Table 1). However, apoptosis has been reported to mediate cell death of surrounding necrotic foci pre-oligodendrocytes (O4+ cells) (Haynes et al., 2003; Back et al., 2005; Robinson et al., 2006; Haynes and van Leyen, 2013), astrocytes (Gelot et al., 2009), but also of neurons in different locations including WM, subplate, cortex, basal ganglia, thalamus, and hippocampus (Robinson et al., 2006; Pierson et al., 2007; Ligam et al., 2009; Andiman et al., 2010; Kinney et al., 2012; Haynes and van Leyen, 2013; Vontell et al., 2015). Interestingly, the density of a type of subcortical neurons, granular neurons (GAD67/65+), is significantly reduced not only in the periventricular and central WM, but also in the subplate region in the presence of PVL, suggesting that this particular subtype of neurons is more selectively sensitive to preterm brain injury (Robinson et al., 2006). Then, diffuse axonal injury (distant from necrotic foci) is also observed in the WM and positive for the apoptotic marker fractin (Haynes et al., 2008; Andiman et al., 2010; Buser et al., 2012). This widespread axonopathy in PVL is reflecting either secondary degeneration to GM damage (such as thalamus) or primary alteration following a direct injury to the axon.
Focal or diffuse nondestructive lesions characterize a milder form of WMI with an incidence up to 20 to 25% in VPT infants (Schneider and Miller, 2019). This form is more frequently seen in preterm neonates in recent years and has been histologically characterized by the absence of focal necrotic lesions and the presence of diffuse reactive astrocytes (Billiards et al., 2008; Buser et al., 2012) and activated microglia (without macrophages infiltration) maintained in a pro-inflammatory state (Haynes et al., 2003; Buser et al., 2012). These observations are mostly confirmed by human imaging studies (Kidokoro et al., 2014; Van’t Hooft et al., 2015; Schneider et al., 2016; Synnes and Hicks, 2018), with microstructural alterations measured on advanced magnetic resonance (MR) techniques such as diffusion-weighted imaging (DWI) and apparent diffusion coefficients (ADC), T1 maps, and fractional anisotropy vectors (FA) and interesting longitudinal observations.
Few studies on postmortem tissue of human preterm suggested the absence of neuronal and cortical injury in diffuse WMI (dWMI) (Back et al., 2005; Robinson et al., 2006; Pierson et al., 2007) (see Table 1). Moreover, the study of Verney et al. (2012) reported that there was no significant loss of the number of Olig2+ oligodendrocytes in diffuse lesions (near necrotic foci) in preterm non-cystic PWMI (Verney et al., 2012). Their conclusion was that axonal injury could occur without pre-oligodendrocytes death. Billiards et al. (2008) showed also no decrease in Olig2+ cell density. However, they observed Olig2+ cell proliferation and an increase in Olig2+ cell density in necrotic foci and they assumed that if some loss of pre-oligodendrocytes still occurs, it could be compensated by a subsequent proliferation of OL progenitors (Billiards et al., 2008). Anyway, even if pre-oligodendrocytes death did not occur, this would not necessarily indicate that there is no cell death in PWMI (especially in foci). Billiards et al. (2008) showed the presence of apoptotic cells (without identifying the cell type) in necrotic foci (microcysts) and reported also that Olig2+ cell (that “did not appear apoptotic”) density was not affected in periventricular necrotic foci or in the surrounding WM. The study of the group of Charriaut-Marlangue showed that the majority of astrocytes “in the penumbral WM” were apoptotic (TUNEL+) in human preterm cPVL and in one case of non-cystic PVL (Gelot et al., 2009). Regarding these two studies, one can hypothesize that astrocytes could be the apoptotic cells described by Billiards and colleagues (2008). Another hypothesis could be that pre-oligodendrocytes death, even if not massive, is occurring through an apoptotic-independent mechanism, i.e., an alternative cell death pathway.
Apart from few studies reporting the loss of pre-oligodendrocytes (preOL) (Haynes et al., 2003; Haynes and van Leyen, 2013), it appears that the main mechanism of the glial dysfunction in dWMI is not due to glial cell death, but more due to dysmaturation of the oligodendrocyte precursor cells (PreO4+) and the imbalance and implication of “bad” microglia (M1) and “good” (M2) microglia phenotypes (Billiards et al., 2008; Buser et al., 2012). This disequilibrium seems to be responsible for the dysmyelination and the gliosis observed for instance in dWMI. Evidence of increased oxidative stress has been reported in immature OLs in dWMI, suggesting that oxidative stress could be a key player of the dysmaturation of OLs supporting the formation of dWMI lesions (Haynes et al., 2003; Billiards et al., 2008; Buser et al., 2012; Haynes and van Leyen, 2013). Multiple preclinical and clinical studies have shown that preOL are less resistant to radical stress, certainly because of decreased expression and function of antioxidant enzymes (such as SOD and GN-oxidase) compared to more mature babies, such as term or even later age (Van’t Hooft et al., 2015).
In summary, cell death is rarely reported in human preterm WMI and PVL tissue from autopsy and cell death characterization (co-localization of cell death and cell type markers) is either missing or sparse.
Severe to moderate cerebral intraventricular hemorrhage (IVH grades III and IV or newly IPE) in preterm infants continues to be a major clinical problem and a neurological issue, occurring in about 5–10% of VPT. In contrast to cPVL, the incidence of IVH has remained stable over the last decade. Over 40% of surviving infants with IVH grade III develop post-hemorrhagic ventricular dilatation and about one third develop severe neurological impairment, such as cerebral palsy and mental retardation. To date, there is only little evidence for therapy to prevent infants from developing either hydrocephalus or serious neurological disability in this condition. There is one trial using a drainage and fibrinolytic approach (DRIFT Trial) that has shown some potential benefits on the neurological outcome, but data were inconclusive due to follow-up problems and other methodological issues (Luyt et al., 2019). There is also a management trial (ELVIS TRIAL) comparing early versus late approach in lumbar tapping and rickham insertion, which is underway (de Vries et al., 2019). It is known that blood rapidly accumulates within the ventricles following IVH, and this leads to disruption of normal cerebrospinal fluid flux and can cause obstruction and increased local tissue pressure as one of the potential etiological mechanisms (Klebe et al., 2020). As described in Table 2, reports about preterm human data, brain cell death, and IVH are rare, and consist in analyses of tissue from autopsy and clinical data in preterm infants, about apoptotic and necrotic markers detected in the cerebrospinal fluid (Felderhoff-Mueser et al., 2003; Schmitz et al., 2011), in the ganglionic eminence (Del Bigio, 2011), and in the periventricular zone (Hargitai et al., 2001; Chamnanvanakij et al., 2002; Georgiadis et al., 2008). However, to date, no study has systematically investigated the involvement of the different types of cell death and characterized the type of cell specifically affected by IVH.
Iatrogenic conditions involved during prematurity are contributory factors changing the normal developmental trajectory of the brain, notably the impact of nutrition on growth and maturation (Beauport et al., 2017; Schneider et al., 2018) and the use of supplemental oxygen for instance. Further, some drugs such as caffeine as well as anesthetic and sedative drugs might influence physiological cerebral development. In regard to cell death types and iatrogenic factors, there are no direct reports in human tissue associating them with cell death types and cells involved, and only suppositions can be made based on preclinical studies as detailed extensively further.
When leaving the hypoxic intrauterine milieu, the preterm infant will be exposed to either diminished or excessive oxygen saturation and variation in cerebral blood flow, which are potentially harmful for the retina, the lung, but also the brain and the neurodevelopmental outcome at long term [Safe Boosc (Askie et al., 2011; Panfoli et al., 2018)]. Therefore, targeting specific oxygen saturation, and regional cerebral oxygenation levels through different methods such as near-infrared spectroscopy (NIRS) are widespread in the neonatal intensive care units, and several trials are still ongoing to confirm the detrimental effect of hypo/hyperoxia and free radical stress in preterm brain injury (Klebe et al., 2020). These data are supported by a tremendous work of body on preclinical models, which are described later.
Caffeine therapy for the apnea of prematurity syndrome (by stimulating the immature respiration center) is the most widespread medication used in premature infants. Babies start to be treated with caffeine between day of life 1 until they reach 34 weeks of corrected age and doses are relatively high (between 5 and 10 mg/kg/day 1 × daily). Clinical studies such as the CAP Trial (Caffeine for Apnea of Prematurity) have shown a protective effect of caffeine administrated in VPT infants, in terms of neurodevelopmental outcome and mortality (Schmidt et al., 2012, 2017; Panfoli et al., 2018). While the short-term data at 18 months was slightly convincing and significant, the effect about the neurodevelopmental outcome was ephemeral at 5 years of follow-up. In fact, a pilot study using high-dose caffeine therapy in premature infants showed increased cerebellar injury and neurobehavioral deficits when compared to low dose (McPherson et al., 2015). This controversy between preclinical worrying and clinical reassuring data is not really rational and raises many questions and doubts. There is probably an effect of the dose. Therapeutical levels preventing apnea consequences in babies are low and harmless, whereas higher doses and/or the multiplicity of drugs combined are linked to neurotoxicity, but this has still to be confirmed.
When looking at clinical risk factors for neurodevelopmental outcome, surgery is a major and constant contributor for impaired neurological outcome, and these concerns are true for neonates at term and premature infants (Sun, 2010; Morriss et al., 2014; Sinner et al., 2014; Stolwijk et al., 2016; Walker et al., 2018). Premature infants may undergo several surgical procedures such as ductus ligation, laparotomies, and thoracotomies defined as major surgeries and herniotomies or endoscopic laryngoscopies defined as minor surgeries. It is still unclear whether the surgical stress, the pain perception, or the administrated drugs for anesthesia are neurotoxic (Broad et al., 2017; Walker et al., 2018), but the combination of both seems to play a major role. This also explains the development of several preclinical models in rodent and primate animals in order to answer the unsolved questions (Baud and Saint-Faust, 2019).
There are only very few clinical data on humans and associated secondary effects with anesthetics/or surgery. The group of Buonocuore measured oxidative stress (Stolwijk et al., 2017), using non-protein bound iron (NPBI) in neonates who underwent noncardiac surgery. These biomarkers were found to be predictive for brain injury post-operatively. Walker et al. (2018) in a cohort of United Kingdom Epicure in ELBW infants found an association of neonatal surgery in the volume of the amygdala and the somatosensory response and pain response in young adults ex-preterm. Filan et al. (2012) analyzed post hoc 30 babies exposed to surgery who had more evidence of WMI and smaller brain volumes, particularly in the deep GM. They could not find any difference in the neurodevelopmental outcome.
The most used anesthetic drugs in premature infants are GABA agonists such as thiopental and propofol, and NMDA antagonists (either ketamine or anesthetic gazes such as isoflurane or sevoflurane). Potential effects of anesthetic drugs on the brain development are driven from the multiplicity of preclinical studies, but very few are described on human data (Sun, 2010; Sinner et al., 2014).
Due to the multifactorial etiologies and the heterogeneity of the brain pathologies involved in EoP, the challenge was, over the last decades, to develop preclinical models reproducing as best as possible the pathophysiology of brain injury and/or brain development impairment observed in the premature human brain. However, the use of a single preclinical model of preterm brain injury cannot reproduce exactly the human situation, and in conclusion, each model presents advantages/disadvantages as reviewed by others (Back et al., 2012; Kinney and Volpe, 2012; Jantzie and Robinson, 2015).
Cell death has been investigated in preterm models in three main species: rodent (rat and mice), sheep, and non-human primate (macaque, baboon) (Tables 3–9). Few studies were undertaken in rabbit and pig. Human preterm neonates are highly exposed to WM injury between 23 and 32 weeks of gestation that corresponds in rodents to the postnatal period before 7 days (P1–P6), in sheep to gestational age around 95 days (90–120 days, term at 145 days), and in macaques to gestational age 125–145 days (term at around 160 days). Whereas substantial cell death can occur in severe models such as those of cPVL in rodents (Table 3), the identification of the presence of cell death frequently showed sparse cell death in specific sites both in WM and GM affecting mainly oligodendrocytes (OL) but also neurons. Animal models have contributed to hypothesize that, in preterm brain injury, a primary OL loss during the “acute” phase is followed by a regenerative process producing immature OL that leads to impaired myelination (Rousset et al., 2006; Segovia et al., 2008). Neuronal injury, including not only neuronal death but also deficits in dendrite and spine maturation (Balakrishnan et al., 2013), could be a direct consequence of primary GM injury as well as secondary through axonal deleterious factors linked to primary WM injury (Rousset et al., 2006; Segovia et al., 2008; Back and Volpe, 2018).
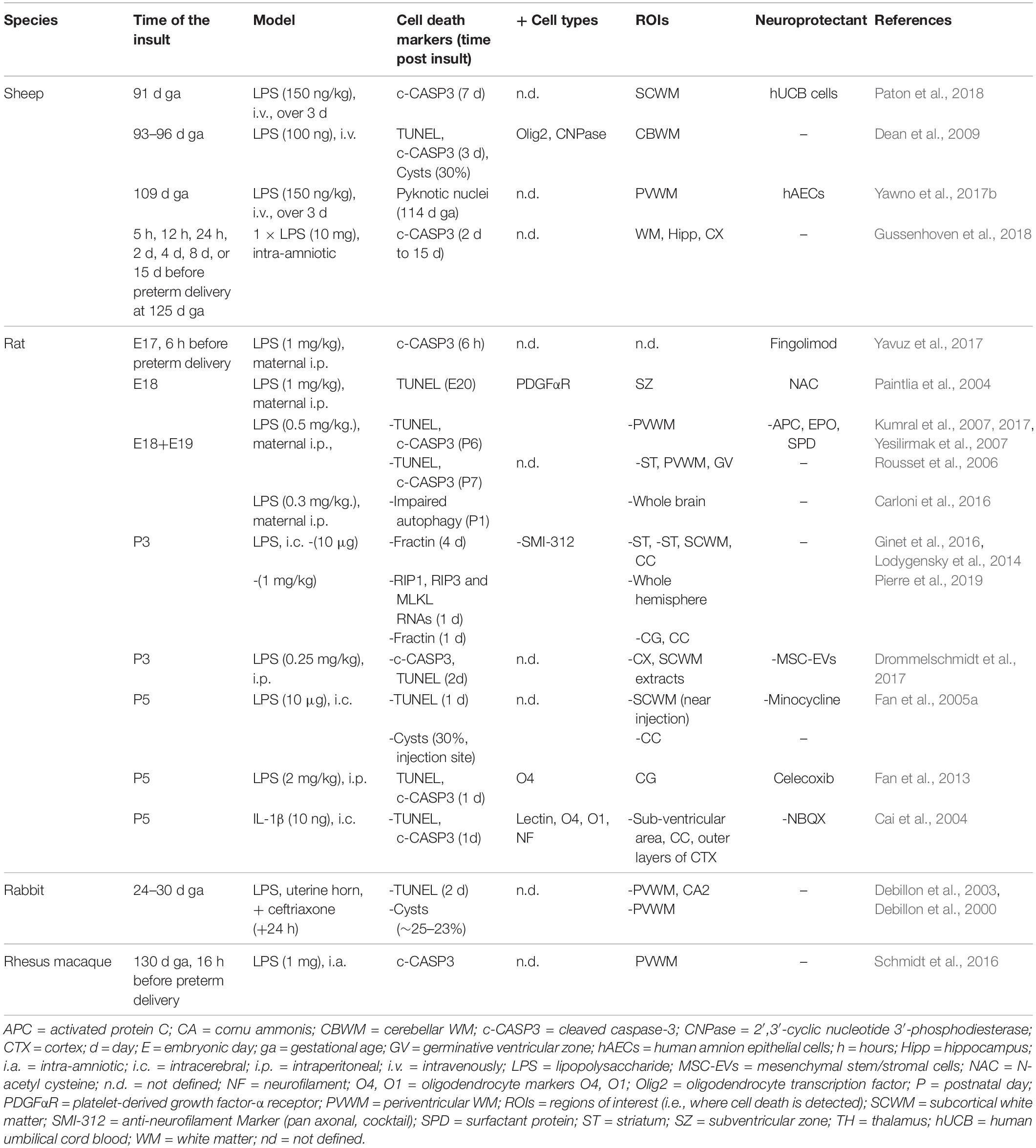
Table 3. Cell death in preclinical models of preterm white matter injury following inflammation.
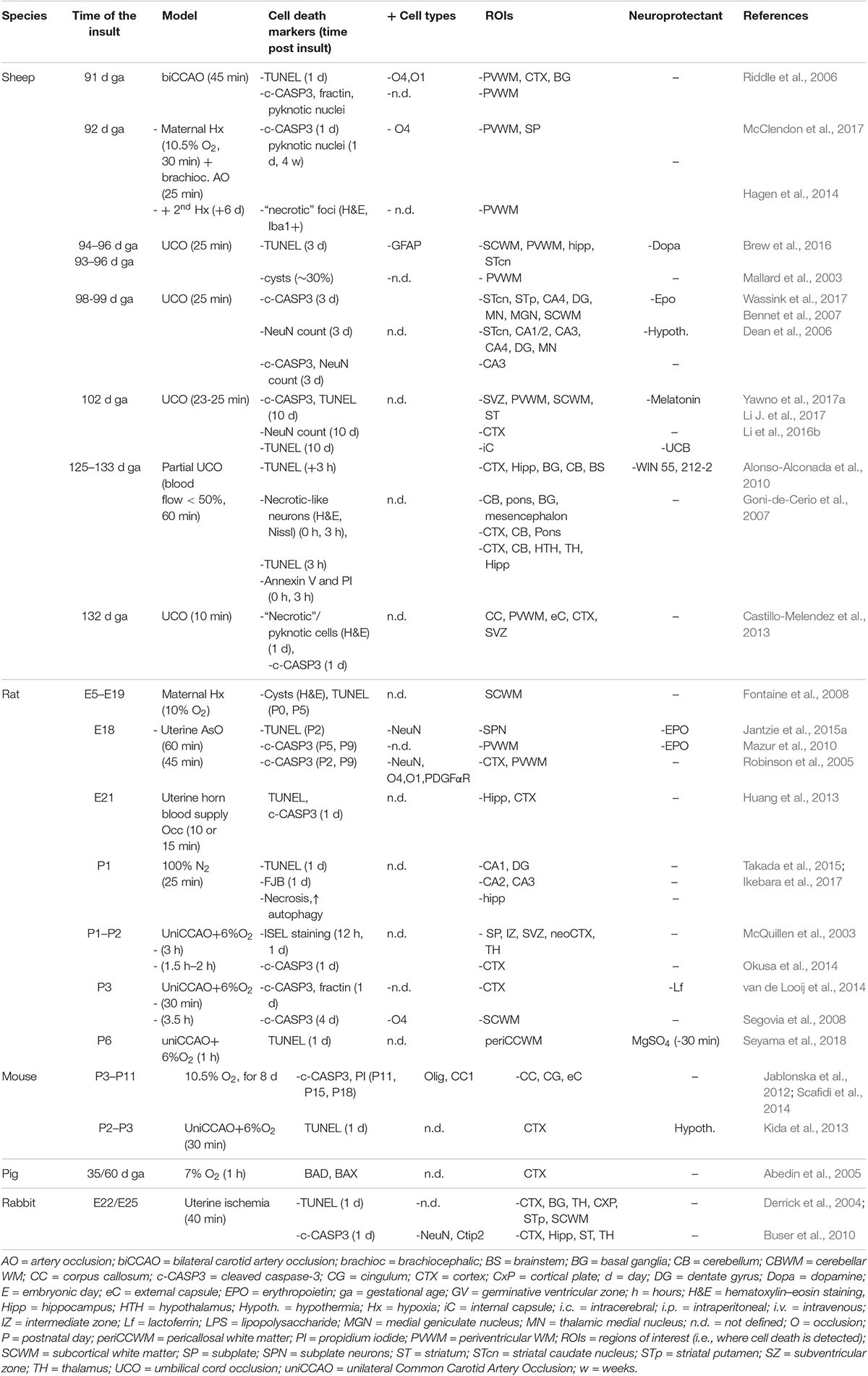
Table 4. Cell death in preclinical models of preterm brain injury following hypoxia/ischemia.

Table 5. Cell death in preclinical models of preterm LPS-sensitized HI brain injury.

Table 6. Cell death in preclinical models of excitotoxin-induced preterm brain injury.
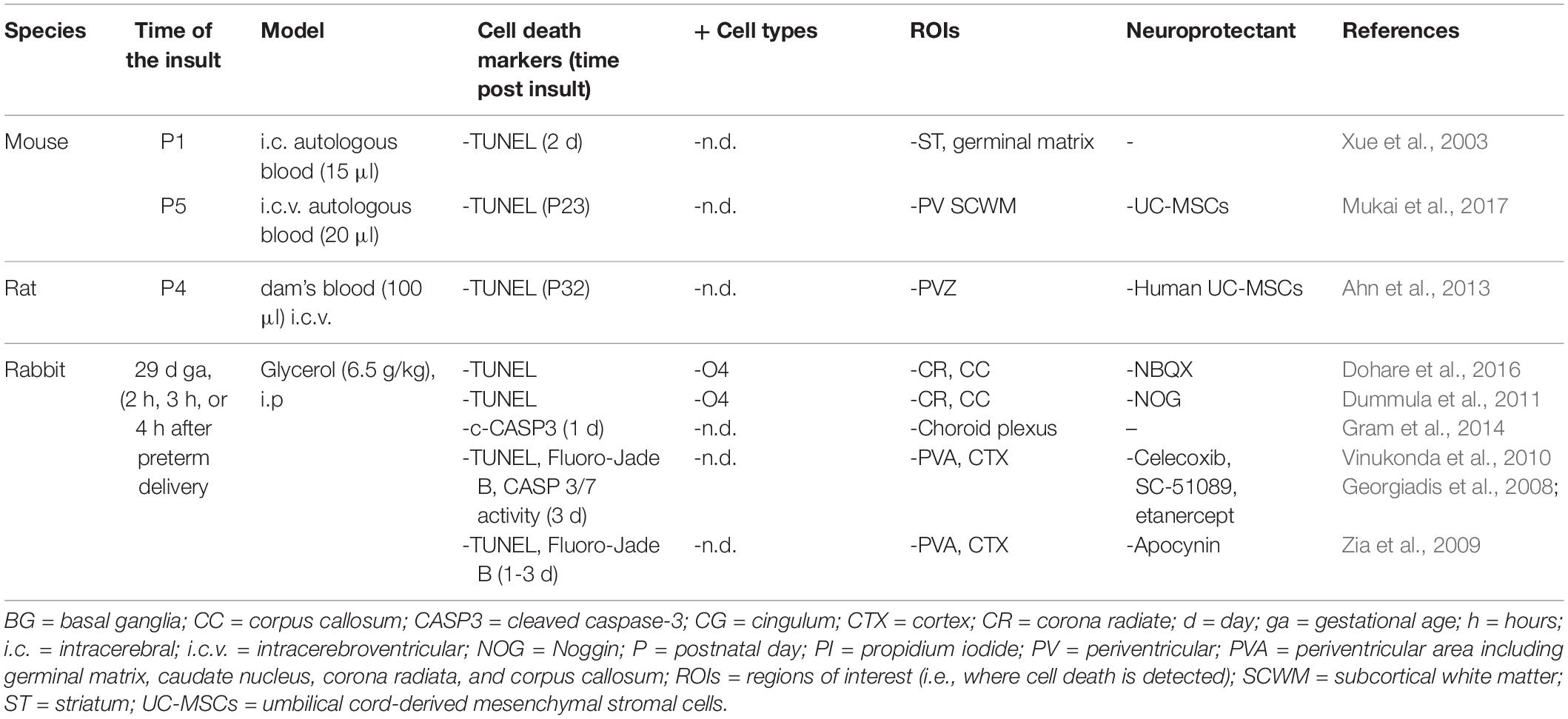
Table 7. Cell death in preclinical models of preterm brain injury following intracerebral hemorrhage.
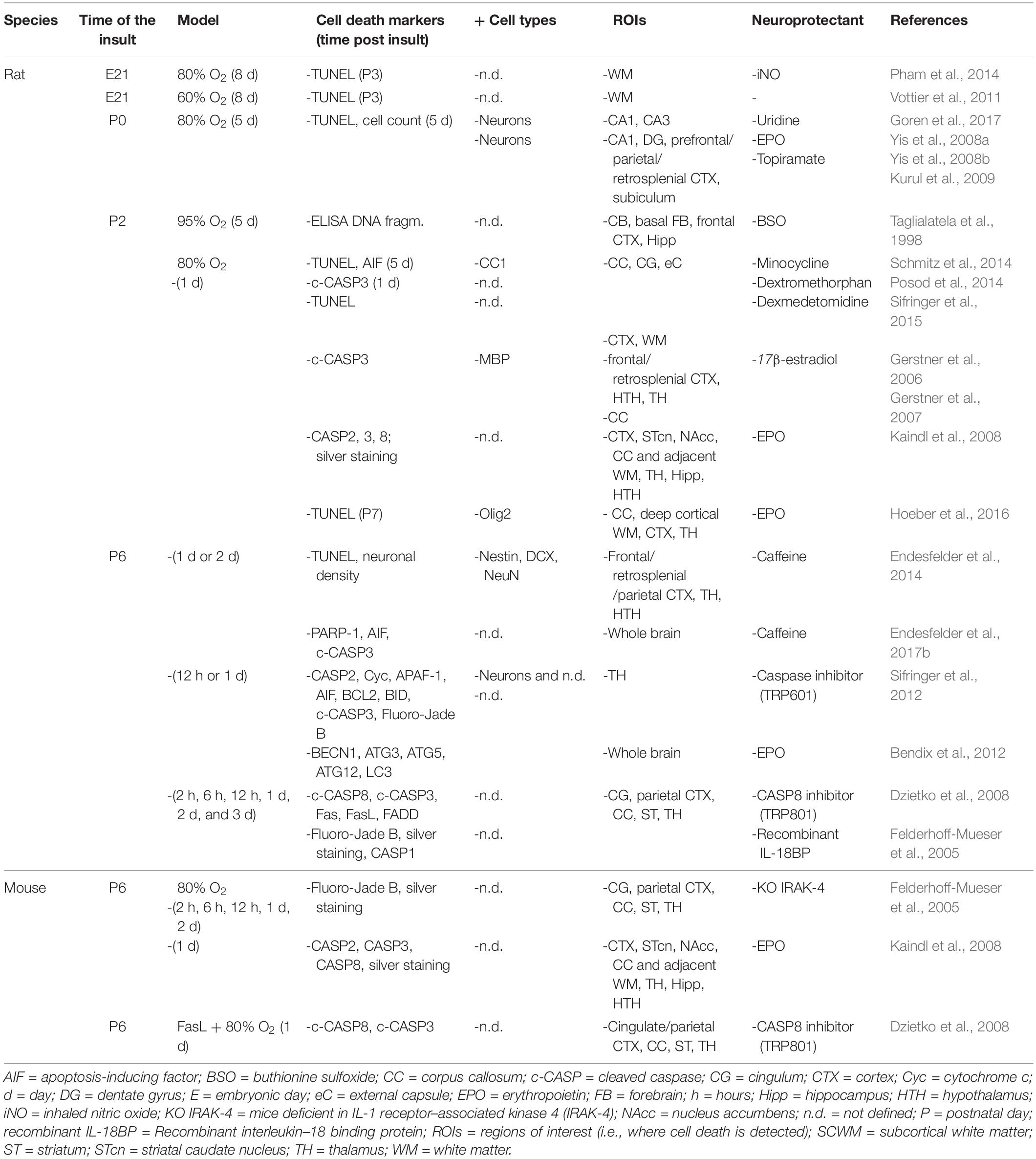
Table 8. Cell death in preclinical models of iatrogenic preterm brain injury: hyperoxia.
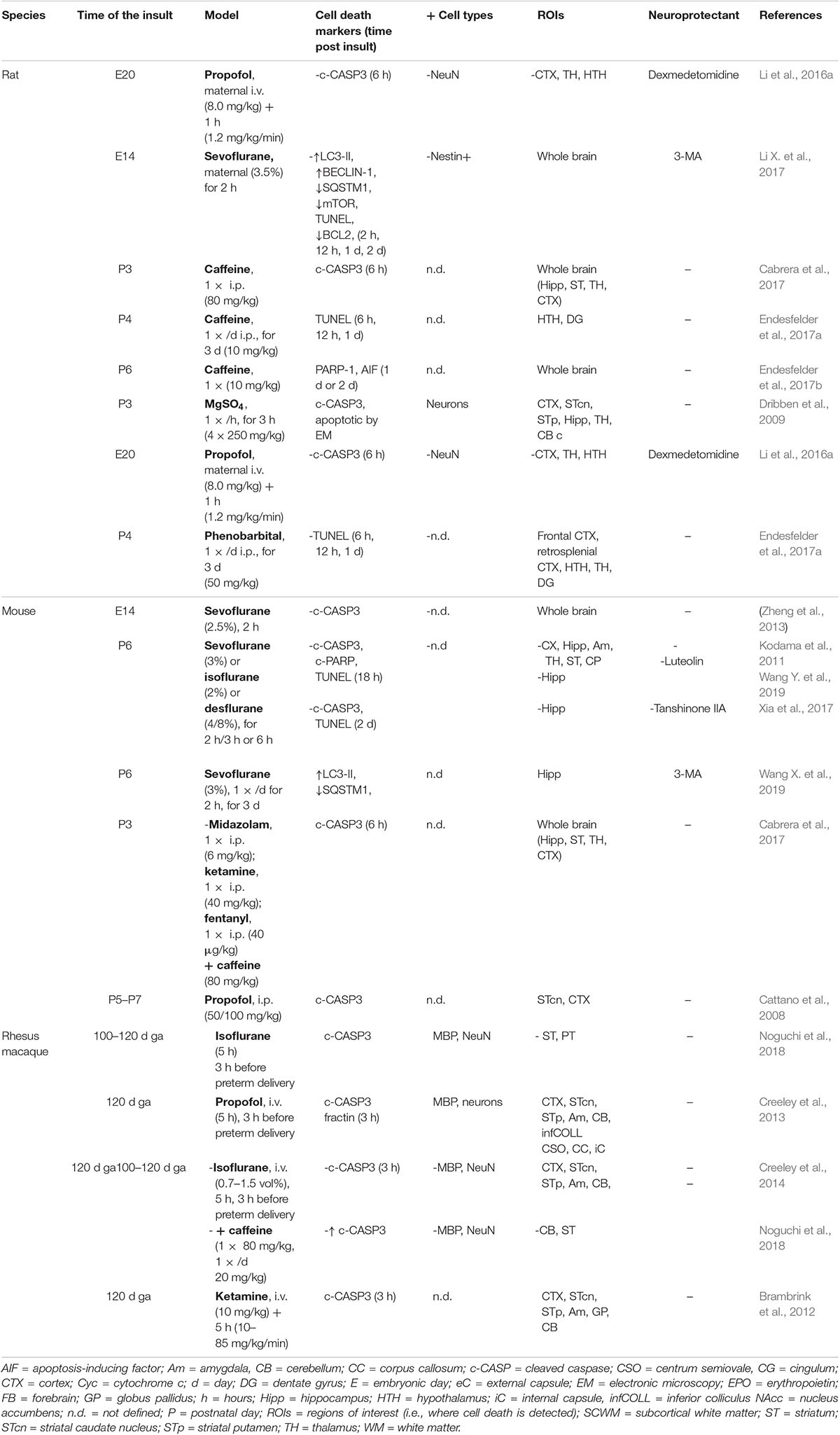
Table 9. Cell death in preclinical models of iatrogenic preterm brain injury: caffeine and anesthetics.
The need to better understand preterm brain damage at the molecular level requires selecting the most appropriate species, age (at which the insult is done), and type of injury, as well as the severity of the insult according to the precise addressed question. Preterm birth is by itself a high-risk factor for brain injury but different deleterious events (alone or combined) can occur and aggravate brain development such as inflammation, hypoxia, ischemia, or hemorrhage. Preclinical models were developed to reproduce one or two of these factors. In the present review, we mainly focused on studies revealing the presence of a type of cell death in the different panels of preterm brain injury models using cell death markers (i.e., studies using more than classical histological observations of pyknosis and karyorrhexis or cell counting).
Even if, thanks to the important progress in neonatal care, the incidence of cPVL has considerably decreased over the past decades, cPVL remains a major cause of cerebral palsy (Hielkema and Hadders-Algra, 2016; Pierrat et al., 2017). Some models were developed and found to reproduce the formation of cysts (or necrotic foci), i.e., macroscopic areas of massive cell death surrounded by activated microglia suggesting rapid necrotic mechanisms (Debillon et al., 2003).
In a rat inflammatory model, intracerebral injection of LPS formed cysts at the injection site in the corpus callosum with TUNEL+ cells surrounding the necrotic foci (Fan et al., 2013). Models of intrauterine infection in rabbit and sheep have also been shown to induce the formation of WM cysts (25–30% of survivors) accompanied by apoptotic cell death of WM oligodendrocytes (c-CASP3+) (Dean et al., 2009) and, in rabbit, cell death in the CA2 region of the hippocampus (TUNEL+) (Debillon et al., 2003) (Table 3). These models showed not only that a local strong inflammation can lead to cyst formation but also that fetal systemic inflammation can, for some treated animals, induce necrotic foci formation in the WM and/or apoptotic cell death in both oligodendrocytes and neurons. Many in vitro studies strongly suggested that activated microglia (M1 phenotype) directly contribute to oligodendrocyte and neuron apoptotic death by releasing reactive oxygen species or TNF-α (Dean et al., 2010; Baburamani et al., 2014).
Beside inflammation, excitotoxicity is one of the most common deleterious mechanisms involved in the formation of cysts. Excitotoxicity consists in a massive intracellular increase of calcium initiated by overactivation of excitatory amino acids receptors (mainly glutamate) (Puyal et al., 2013). In the human preterm brain, the expression of N-methyl-D-aspartate (NMDA) glutamate receptors peaks in pre-oligodendrocytes, allowing the WM to be particularly vulnerable to excitotoxic insult (Jantzie et al., 2015b). Then, the model using intracerebral injection of the glutamate receptor agonist ibotenate (Table 6) has been widely used to reproduce some aspects of human preterm PVL, including WM cyst formation, ventriculomegaly, reduction in brain structure volume, and decrease of WM thickness and myelination (Descloux et al., 2018). Previous studies showed that ibotenate-induced excitotoxicity triggers an important inflammatory response involved in cyst formation (Tahraoui et al., 2001; Dommergues et al., 2003). Concerning cell death type, compiling evidence suggested that apoptosis is importantly involved in this model. One study mentioned the presence of c-CASP3+-positive cells in WM (Neubauer et al., 2016). Neurodegeneration of cortical neurons has also been demonstrated by Fluoro-Jade staining and evidence suggested that caspase-dependent (CASP3) and -independent apoptotic mechanisms (AIF nuclear translocation) were implicated in cortical damage without performing, in most of the studies, an identification of the type of dying cells. Astrocytic cell death has been reported to occur in the site of ibotenate injection (Tahraoui et al., 2001). More recently, we have shown that cortical neurons [(RBFOX3)/NeuN+] presented both activation of CASP3 and nucleus fragmentation (Descloux et al., 2018). Moreover, we have clearly demonstrated that apoptotic neurons displayed mixed morphological features of apoptosis (chromatin condensation) and intense autophagy (increased presence of autophagosomes and autolysosomes). Ibotenate increased both the number of autophagosomes (increased LC3-II and LC3+ dots) and autophagic degradation [p62/SQSTM1 reduction and increased number of autolysosomes (LAMP1- and CATHEPSIN B+ vesicles)]. Inhibition of autophagy with 3-methyladenin (3-MA) prevented CASP3 activation, suggesting a role of autophagy upstream of apoptosis as previously shown in other models (Puyal et al., 2009; Grishchuk et al., 2011; Xie et al., 2016). Interestingly, 3-MA afforded strong protection by reducing not only cortical lesion but also WM deficits, indicating that preventing primary neuronal death by autophagy inhibition could reduce WM damage (Descloux et al., 2018).
Lesions mimicking human PVL were also observed in a sheep model of severe hypoxia–ischemia (Table 4) involving a double hit protocol and producing WM necrotic foci related to GM (CX and TH) damage (transient maternal hypoxia and brachiocephalic artery occlusion followed by a second hypoxia 6 h later) (Hagen et al., 2014).
While hypoxia/ischemia represents one of the major risk factors for neonatal encephalopathy in term babies, for preterm, however, inflammation has been of, in the last decade, growing importance in the field. The role of a hypoxic event in preterm brain injury has, indeed, no clear evidence. Argument that is more rigorous is needed today before claiming hypoxic–ischemic encephalopathy such as measurement of hypoxemia and metabolic acidosis (Graham et al., 2004; Gilles et al., 2017; Paneth, 2018). It is now proposed that inflammation could be a leading cause of preterm brain damage (Gilles et al., 2017). In human preterm babies, chorioamnionitis and especially postnatal sepsis are associated with WM injury and adverse neurodevelopmental outcomes (Shah et al., 2008; Anblagan et al., 2016; Bierstone et al., 2018; Heo et al., 2018). Moreover, inflammation is a common process in different preterm brain injuries either directly as in intrauterine infection/chorioamnionitis or secondarily because of a primary insult such as hemorrhage or hypoxia/ischemia. Despite the current questioning of the role of hypoxia in preterm brain damage, most of the studies investigating cell death mechanisms in diffuse WM injury have been performed in models involving both stimuli: inflammation (Table 3) and hypoxia/ischemia (HI) (Tables 4, 5) (Jantzie and Robinson, 2015).
Direct inflammatory lesions are induced in most of the preclinical models by an acute or chronic exposure to either gram-negative bacteria lipopolysaccharide (LPS) endotoxin or more recently by injection of interleukin-1β that leads to a persistent inflammatory response, astrogliosis, and myelination deficits (Dean et al., 2015). It has been shown that exposure to inflammation during the perinatal period could alter transiently or permanently neurodevelopmental outcome (learning and motor difficulties and development of psychiatric disease such as schizophrenia or autism) (Fan et al., 2005b; Boksa, 2010; Rousset et al., 2013; Schaafsma et al., 2017; Straley et al., 2017; Zhang et al., 2018). Cell death has been mostly investigated in models of fetal infection in rat, sheep, and macaques in which apoptotic cell death (c-CASP3) was found to be scattered, located essentially in WM, and observed mainly after days following inflammatory insult (Table 3). When investigated, cell type identification indicated that CASP3 activation occurred in oligodendrocyte lineage (Olig2, CNPase, and PDGFαR). However, injury of the GM was also described. A study in sheep involving intra-amniotic LPS injection also described CASP3 activation in the cortex and the hippocampus 8 days after LPS exposure (Gussenhoven et al., 2018). Another study involving i.p. LPS injections in rat dam (at E18 and E19) showed the presence of TUNEL and c-CASP3-positive cells in the periventricular striatum of pups 7 days after birth (Rousset et al., 2006).
An intracerebral injection of LPS consists of much more severe inflammatory models by mimicking late conditions of pathogen infiltration after the breakdown of the blood–brain barrier. In a model of LPS exposure in postnatal rat (P3), the production of the CASP-dependent fragment of β-actin, fractin, was observed in the subcortical WM (SCWM) as well as in axonal fascicles of striatum (Lodygensky et al., 2014; Ginet et al., 2016), suggesting a LPS-induced axonopathy (Sokolowski et al., 2014). The mechanisms involved in apoptosis induction could depend on the cell type. In fact, in a model of intracerebral injection of IL-1β in P5 rats inducing a global CASP3 activation, TUNEL staining was observed in Lectin+ microglia, in O4+ and O1+ oligodendrocyte precursors, and in some NF+ neurons 1 day after the insult (Cai et al., 2004). Inflammation-related cell death was partially associated with excitotoxicity since the AMPA/kainate receptors antagonist NBQX reduced the global number of TUNEL+ cells without protecting oligodendrocyte precursors. This could be explained by an age-dependent sensitivity of WM pre-oligodendrocytes to AMPA toxicity that occurs later at P7 (Follett et al., 2000).
A single study investigated autophagy in an inflammatory model (Carloni et al., 2016), showing that, when LPS was injected in the rat dam (at E18 and E19), autophagy appeared impaired (LC3-II and p62/SQSTM1 levels were increased in P1 brain rat pups). However, this study evaluated the overall autophagy response on whole brain extracts, a method that limits the conclusions concerning the effect on autophagy according to either WM, GM, or the cell type.
Very recently, an involvement of necroptosis has been proposed to occur after LPS-induced brain inflammation in P3 old rats by showing an increase in necroptotic gene expressions (RIPK1, RIPK3, and MLKL) 24 h after the insult (Pierre et al., 2019).
Even if the role of HI has become a subject of debate (Gilles et al., 2017), it remains one of the most used models for studying preterm brain injury that induces both severe WM and GM injury and are then used mainly as a model of cPVL. Cell death has been investigated mainly in sheep and rodent (Table 4).
Sheep models consist in inducing a hypoxic–ischemic event in the fetus by either umbilical cord occlusion or by brachiocephalic arteries occlusion combined with maternal hypoxia. CASP3 activation, pyknotic nuclei, as well as TUNEL staining were described mainly in PVWM (periventricular and subcortical WM) and SCWM but also in cortex, basal ganglia (caudate nucleus and putamen), and less frequently in thalamus, hypothalamus, and hippocampus. One study investigating cell death within the first 3 h following HI showed that necrotic features are detected early, from time zero onwards, without signs of apoptosis in different cerebral regions (mesencephalon, cerebellum, pons, basal ganglia, cortex, hippocampus, thalamus, and hypothalamus) (Goni-de-Cerio et al., 2007). From 3 h onwards, apoptotic dying cells are detected and not restricted to cerebral regions where necrotic neurons were histologically observed at 0 h (mesencephalon, cerebellum, pons, and basal ganglia). Interestingly, some early necrotic regions will then become strongly apoptotic later (especially cortex, cerebellum, and pons), suggesting that different cell death processes are involved sequentially in the same damaged cerebral region. However, since all these observations are focused on unregulated necrosis, it could be speculated that regions displaying either apoptotic or necrotic features at later time points are regions where delayed cell death is prominent, including potentially not only apoptosis but also a regulated form of necrosis such as necroptosis.
The subplate zone is a transient layer of the developing cerebral cortex that plays a highly important role in the structural brain development and its plasticity. It contains different neuronal cells and especially early-generated subplate neurons whose cell bodies are located in developing WM. Subplate injury has been since years discussed in EoP and is becoming more and more important over the last decade. When sheep maternal hypoxia and fetal ischemia were induced early (92d GA), CASP3 activation was also present in the subplate but essentially in oligodendrocytes, suggesting a more important resistance of subplate neurons in sheep (McClendon et al., 2017). On the contrary, selective vulnerability of subplate neurons was described in the most widely used model of HI in P2–P3 rodents (uniCCAO followed by systemic hypoxia) (McQuillen et al., 2003; Mikhailova et al., 2017). However, another study could not confirm a specific vulnerability of subplate neurons compared to other deep layers or to the WM (Okusa et al., 2014). It is important to point out that these studies have evaluated only the density of neurons in the subplate and did not perform co-labeling with cell death markers, which is not sufficient to conclude for a specific neuron death in the subplate since an impairment in neuronal differentiation could also be involved. In a different rat model consisting in placental transient hypoxia–ischemia at E18 (uterine arteries occlusion), however, the presence of TUNEL+ neurons has been reported in the subplate at P2 (Jantzie et al., 2015a).
Even if widespread CASP3 activation or TUNEL labeling were shown in models of HI in P1 to P6 rodents in both GM (cortex, thalamus) and WM (Segovia et al., 2008; Jablonska et al., 2012; Scafidi et al., 2014; Seyama et al., 2018), few studies characterized the type of dying cells. In a mouse model of chronic hypoxia (8 days at 10.5% O2 from P3), oligodendrocytes were shown to undergo CASP3-dependent apoptosis even 1 week after the end of hypoxic treatment (Jablonska et al., 2012). In the model of HI in P3 rats pups, Segovia and colleagues (Segovia et al., 2008) showed that, in addition to pre-oligodendrocyte maturation arrest, O4+ cells underwent mainly caspase-independent cell death mechanisms (c-CASP3 negative but morphological characteristics of degeneration) 24 h after the insult, whereas 4 days later, they died mostly through caspase-dependent pathways, suggesting that different forms of cell death are involved in acute or delayed cell death of oligodendrocytes (Segovia et al., 2008).
In a model of anoxia in P1 rat pups, damaged hippocampus showed cell death with EM mixed morphological features of apoptosis, necrosis, and enhanced autophagy (Takada et al., 2015), suggesting that more than one cell death type could be involved in hypoxic–ischemic brain damage with a continuum of cell death that could include the three cell death types as it has been proposed in models of HI in more mature P7 brain (Portera-Cailliau et al., 1997; Ginet et al., 2009a; Northington et al., 2011). Interestingly, this study showed that whereas CA2/CA3 presented a neurodegeneration revealed by both Fluoro-Jade B+ staining and EM ultrastructure study, this hippocampal region was TUNEL negative and presented no more CASP3 activation than in control. This result points out the necessity to study cell death with alternative markers of cell death, different from CASP3 activation (that is moreover developmentally expressed in the immature brain) and TUNEL staining.
In summary, in inflammatory and/or HI premature models, cell death has been investigated in different models and species, and is present in cortical and deep GM neurons and in different glial cells (OL and microglia) with different cell death features reported (necrosis, apoptosis, and autophagic cell death).
Very few studies have investigated cell death occurring after IVH in preclinical preterm models (Table 7). Cell death has been studied in two main models reproducing the conditions of germinal matrix hemorrhage (GMH) (germinal matrix ruptures through the ependyma into the lateral ventricle) leading to IVH in preterm human brains. The first approach consists of applying periventricular or intracerebroventricular injections of blood (from dam or autologous) in rodent pups (P1 to P4). The second approach is to perform an i.p. injection of glycerol in rabbit fetus (29 days GA) that produces an intracranial pressure through profound osmotic changes followed by a reperfusion leading to GMH that can extend to the lateral ventricle (Table 7). In both models, pups developed severe IVH associated with progressive post-hemorrhagic hydrocephalus (PHH) compatible with grade 3 or grade 4 IVH in humans, even if the percentage of induced PHH is variable. The IVH pups displayed impaired sensorimotor functions, inflammation, defects in myelination, and increased levels of inflammatory cytokines in the CSF.
Increased cell death and apoptosis have been evidenced by TUNEL staining and CASP3 activation in the periventricular WM (Dummula et al., 2011; Ahn et al., 2013; Gram et al., 2014; Dohare et al., 2016; Mukai et al., 2017) and adjacent GM [striatum (Xue et al., 2003) or cortex (Georgiadis et al., 2008; Zia et al., 2009; Vinukonda et al., 2010)]. However, the identification of the dying cells has not been determined except in some studies showing that cell death (Fluoro-Jade+ cells) could affect O4+ pre-oligodendrocytes or neurons.
The involvement of cell death and apoptosis in preterm brain injury is well documented in preterm models of hyperoxia in postnatal rodent (from P0 to P6) (Table 8). Hyperoxia was shown in those models to affect both WM and GM (cortex, striatum, thalamus, and hippocampus) by promoting cell death of oligodendrocytes (Gerstner et al., 2006, 2007; Schmitz et al., 2014; Hoeber et al., 2016) and neurons (Yis et al., 2008a, b; Endesfelder et al., 2014). The involvement of apoptosis as a major cell death process was demonstrated by the implication of not only CASP3 but also numerous molecular markers of both intrinsic (Cyc, AIF, and APAF-1) and extrinsic (Fas, FasL, FADD, CASP8, and BID) apoptotic pathways inducing caspase-dependent (CASP1, 2, 3, and 8) and independent (AIF) mechanisms.
Furthermore, Bendix and colleagues (Bendix et al., 2012) showed an effect of hyperoxia on autophagy during the first 24 h of hyperoxia in the developing rat preterm brain. Some important autophagy-related genes (atg) for autophagosome formation were upregulated (BECLIN1, ATG3, ATG5, ATG12, and LC3-II) (Bendix et al., 2012) at 12 h after hyperoxia treatment. However, we cannot conclude on the effect of hyperoxia on autophagic flux (active autophagic process of degradation) since efficiency of the degradative part of autophagy was not investigated. Increased autophagosome presence could result from enhanced autophagic flux as well as impaired lysosomal degradation (Puyal et al., 2013; Klionsky et al., 2016). While this study showed that autophagy is certainly affected by hyperoxia, more investigations (assessing the state of the flux and its function) are needed before to conclude on the role of autophagy in hyperoxia-mediated cell death.
It has been shown that caffeine treatment (10 to 80 mg/kg, i.p.) activated apoptosis (CASP3, AIF) and increased TUNEL staining in the brain of rat pups (P3, P6) (Table 9) (Cabrera et al., 2017; Endesfelder et al., 2017a, b). Recently, Noguchi and colleagues showed that caffeine treatment in fetal macaque (110–120 days GA) anesthetized with isoflurane strongly sensitized brain (CX, ST, and CB) against isoflurane-induced apoptosis, especially NeuN-positive neurons, more than MBP-positive oligodendrocytes (Noguchi et al., 2018).
Different studies demonstrated pro-apoptotic effects of anesthetics [especially propofol, ketamine, and ethers (isoflurane, sevoflurane)] on immature brain of rodents and macaques (Table 9). Combination of anesthetic with caffeine is a powerful aggravating factor (Noguchi et al., 2018). These anesthetics have been shown to trigger widespread CASP3 activation or to increase TUNEL labeling in neurons and oligodendrocytes (Cattano et al., 2008; Dribben et al., 2009; Brambrink et al., 2010, 2012; Creeley et al., 2013; Li et al., 2016a; Xiong et al., 2016) associated with some cognitive deficits. In two studies using sevoflurane, autophagy was shown to be enhanced, in addition to apoptosis, in immature brains, and autophagy inhibition using 3-MA was both anti-apoptotic and neuroprotective (Li X. et al., 2017; Wang X. et al., 2019).
In conclusion, common and converging observations from animal models and human brains from autopsy suggested that cell death mechanisms in preterm brain injuries mainly involve apoptotic mechanisms. Although most of the studies missed to identify the type of cells subjected to cell death, some suggested that different cell types are affected depending not only on the severity and the type of the insult but also on the time point investigated. In mild preterm injuries, the cell death appeared to be diffuse and restricted (when detected) to preOL, whereas in more severe cases, cell death could affect almost all cell types including neurons.
Nevertheless, it appears that, although cell death is an ineluctable event occurring in severe preterm brain injuries such as cPVL and IVH, but not necessarily in dWMI, cell death has not been deeply investigated and characterized either in preclinical models or in tissues from human preterm neonates. The importance of apoptosis versus other cell death pathways (such as regulated necrotic and autophagic) is certainly overestimated since almost all the studies used only apoptotic (such as c-CASP3 and TUNEL) markers as indicators of cell death. Moreover, by using only c-CASP3 and TUNEL markers, cell death occurrence can be simply underestimated or wrongly declared absent since cell death types independent of CASP3 and not inducing (at least in an early stage) DNA fragmentation exist. In the model of anoxia in P1 rat pups, damaged hippocampal CA2/CA3 showed, for example, cell death mechanisms mainly independent of CASP3, not positive for TUNEL and with EM morphological features of necrosis and enhanced autophagy (Takada et al., 2015). We have previously clearly demonstrated the presence and the prodeath role of enhanced neuronal autophagy in models of both term (Ginet et al., 2009b; Puyal et al., 2009; Ginet et al., 2014a; Xie et al., 2016) and more recently preterm (Descloux et al., 2018) brain injury. We showed that, after neonatal cerebral HI, rat hippocampal dying CA3 neurons displayed high autophagic features without apoptosis activation, i.e., TUNEL and CASP3 negative (Ginet et al., 2009b). In addition, the discovery of programmed forms of necrosis and some of its molecular actors will allow one to investigate the implication of regulated necrosis. Necroptosis is especially concerned since this type of cell death is known to be activated by inflammatory signals.
Since “we find what we are looking for”, one could speculate that most of the studies missed to identify cell death mechanisms involved in the pathogenesis of the studied preterm brain damage. The increasing advances made to understand the biochemical mechanisms of the different cell death pathways and the recognition of regulated necrosis and autophagic cell death lead to switch, over the last 10 years, in the identification of the cell death type from a purely morphological to a molecular analysis (Galluzzi et al., 2012). Today, many markers of the different types of cell death could be used in human samples to systematically evaluate the presence not only of apoptosis but also of other forms of cell death including regulated necrosis and autophagic or autophagy-mediated cell death. Expression and activation of RIPK3 and MLKL could be investigated as markers for necroptosis. Concerning autophagic or autophagy-mediated cell death, an increase in autophagosome presence alone is not enough to demonstrate an increased autophagic flux since it could result from a lysosomal degradation failure. Then, an increase in punctate LC3 immunolabeling (marker of autophagosomes) together with an increase lysosomal activity, evaluated by either a decrease in p62/SQSTM1 labeling (selectively degraded by autophagy) or an increase in the size and number of vesicles labeled by lysosomal markers such as LAMP1 or different cathepsins (presumably autolysosomes), should be used to detect enhanced autophagy. Moreover, since increased autophagy could be either a prosurvival or a prodeath mechanism, conclusions on the role of enhanced autophagy in cell death has to be taken with caution due to the duality of autophagy in cell survival (protective vs. destructive) depending the conditions and cell type.
Future studies designed to provide a more precise characterization of the molecular cell death mechanisms and also identification of dying cell types will allow a better understanding of the preterm brain pathogenesis and will certainly participate to develop new and efficient protective strategies to prevent or treat injuries of the preterm human brain.
AT, VG, and JP conceived, wrote the manuscript, drew the tables and figures, and critically revised the manuscript.
This work was supported by the grants from the Swiss National Science Foundation (310030-163064 and 310030-182332) and by the Fondation Paralysie Cérébrale.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
3-MA, 3-methyladenin; ADC, apparent diffusion coefficients; ATG, autophagy-related gene; CASP, caspase; c-CASP, cleaved caspase; cPVL, cystic periventricular leucomalacia; CSF, cerebrospinal fluid; dWMI, diffuse white matter injury; EM, electronic microscopy; EoP, encephalopathy of prematurity; ER, endoplasmic reticulum; FA, fractional anisotropy; GA, gestational age; GM, gray matter reduction; GMH, germinal matrix hemorrhage; HI, hypoxia–ischemia; IVH, intraventricular hemorrhage; LC3, microtubule-associated protein 1 light chain 3; LPS, lipopolysaccharide; MRI, magnetic resonance imaging; OL, oligodendrocytes; PE, phosphatidylethanolamine; PHH, post-hemorrhagic hydrocephalus; preOL, pre-oligodendrocyte; PVL, periventricular leucomalacia; PWM, periventricular white matter; SCWM, subcortical WM; SVZ, subventricular zone; VPT, very preterm infants; WM, white matter; WMI, white matter injury.
Abedin, N., Ashraf, Q., Mishra, O. P., and Delivoria-Papadopoulos, M. (2005). Effect of hypoxia on the expression of pro- and anti-apoptotic proteins in neuronal nuclei of the guinea pig fetus during gestation. Brain Res. Dev. Brain Res. 156, 32–37. doi: 10.1016/j.devbrainres.2005.01.006
Ahn, S. Y., Chang, Y. S., Sung, D. K., Sung, S. I., Yoo, H. S., Lee, J. H., et al. (2013). Mesenchymal stem cells prevent hydrocephalus after severe intraventricular hemorrhage. Stroke 44, 497–504. doi: 10.1161/STROKEAHA.112.679092
Alonso-Alconada, D., Alvarez, F. J., Alvarez, A., Mielgo, V. E., Goni-de-Cerio, F., Rey-Santano, M. C., et al. (2010). The cannabinoid receptor agonist WIN 55,212-2 reduces the initial cerebral damage after hypoxic-ischemic injury in fetal lambs. Brain Res. 1362, 150–159. doi: 10.1016/j.brainres.2010.09.050
Anblagan, D., Pataky, R., Evans, M. J., Telford, E. J., Serag, A., Sparrow, S., et al. (2016). Association between preterm brain injury and exposure to chorioamnionitis during fetal life. Sci. Rep. 6:37932. doi: 10.1038/srep37932
Andiman, S. E., Haynes, R. L., Trachtenberg, F. L., Billiards, S. S., Folkerth, R. D., Volpe, J. J., et al. (2010). The cerebral cortex overlying periventricular leukomalacia: analysis of pyramidal neurons. Brain Pathol. 20, 803–814. doi: 10.1111/j.1750-3639.2010.00380.x
Askie, L. M., Brocklehurst, P., Darlow, B. A., Finer, N., Schmidt, B., Tarnow-Mordi, W., et al. (2011). NeOProM: neonatal oxygenation prospective meta-analysis collaboration study protocol. BMC Pediatr. 11:6. doi: 10.1186/1471-2431-11-6
Baburamani, A. A., Supramaniam, V. G., Hagberg, H., and Mallard, C. (2014). Microglia toxicity in preterm brain injury. Reprod. Toxicol. 48, 106–112. doi: 10.1016/j.reprotox.2014.04.002
Back, S. A., Luo, N. L., Mallinson, R. A., O’Malley, J. P., Wallen, L. D., Frei, B., et al. (2005). Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann. Neurol. 58, 108–120. doi: 10.1002/ana.20530
Back, S. A., Riddle, A., Dean, J., and Hohimer, A. R. (2012). The instrumented fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics 9, 359–370. doi: 10.1007/s13311-012-0108-y
Back, S. A., and Volpe, J. J. (2018). “Encephalopathy of prematurity: pathophysiology. pathophysiology,” in Volpe’s Neurology of the Newborn, (Amsterdam: Elsevier), 405.e8–424.e8.
Balakrishnan, B., Dai, H., Janisse, J., Romero, R., and Kannan, S. (2013). Maternal endotoxin exposure results in abnormal neuronal architecture in the newborn rabbit. Dev. Neurosci. 35, 396–405. doi: 10.1159/000353156
Ball, G., Boardman, J. P., Aljabar, P., Pandit, A., Arichi, T., Merchant, N., et al. (2013). The influence of preterm birth on the developing thalamocortical connectome. Cortex 49, 1711–1721. doi: 10.1016/j.cortex.2012.07.006
Banker, B. Q., and Larroche, J. C. (1962). Periventricular leukomalacia of infancy. A form of neonatal anoxic encephalopathy. Arch. Neurol. 7, 386–410.
Baud, O., and Saint-Faust, M. (2019). Neuroinflammation in the developing brain: risk factors. involvement of microglial cells, and implication for early anesthesia. Anesth. Analg. 128, 718–725. doi: 10.1213/ANE.0000000000004032
Beauport, L., Schneider, J., Faouzi, M., Hagmann, P., Huppi, P. S., Tolsa, J. F., et al. (2017). Impact of early nutritional intake on preterm brain: a magnetic resonance imaging study. J. Pediatr. 181:e21. doi: 10.1016/j.jpeds.2016.09.073
Bendix, I., Schulze, C., Haefen, C., Gellhaus, A., Endesfelder, S., Heumann, R., et al. (2012). Erythropoietin modulates autophagy signaling in the developing rat brain in an in vivo model of oxygen-toxicity. Int. J. Mol. Sci. 13, 12939–12951. doi: 10.3390/ijms131012939
Bennet, L., Roelfsema, V., George, S., Dean, J. M., Emerald, B. S., and Gunn, A. J. (2007). The effect of cerebral hypothermia on white and grey matter injury induced by severe hypoxia in preterm fetal sheep. J. Physiol. 578(Pt 2), 491–506. doi: 10.1113/jphysiol.2006.119602
Bierstone, D., Wagenaar, N., Gano, D. L., Guo, T., Georgio, G., Groenendaal, F., et al. (2018). Association of histologic chorioamnionitis with perinatal brain injury and early childhood neurodevelopmental outcomes among preterm neonates. JAMA Pediatr. 172, 534–541. doi: 10.1001/jamapediatrics.2018.0102
Billiards, S. S., Haynes, R. L., Folkerth, R. D., Borenstein, N. S., Trachtenberg, F. L., Rowitch, D. H., et al. (2008). Myelin abnormalities without oligodendrocyte loss in periventricular leukomalacia. Brain Pathol. 18, 153–163. doi: 10.1111/j.1750-3639.2007.00107.x
Boksa, P. (2010). Effects of prenatal infection on brain development and behavior: a review of findings from animal models. Brain Behav. Immun. 24, 881–897. doi: 10.1016/j.bbi.2010.03.005
Brambrink, A. M., Evers, A. S., Avidan, M. S., Farber, N. B., Smith, D. J., Martin, L. D., et al. (2012). Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology 116, 372–384. doi: 10.1097/ALN.0b013e318242b2cd
Brambrink, A. M., Evers, A. S., Avidan, M. S., Farber, N. B., Smith, D. J., Zhang, X., et al. (2010). Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology 112, 834–841. doi: 10.1097/ALN.0b013e3181d049cd
Brew, N., Azhan, A., den Heijer, I., Boomgardt, M., Davies, G. I., Nitsos, I., et al. (2016). Dopamine treatment during acute hypoxia is neuroprotective in the developing sheep brain. Neuroscience 316, 82–93. doi: 10.1016/j.neuroscience.2015.12.022
Broad, K. D., Kawano, G., Fierens, I., Rocha-Ferreira, E., Hristova, M., Ezzati, M., et al. (2017). Surgery increases cell death and induces changes in gene expression compared with anesthesia alone in the developing piglet brain. PLoS One 12:e0173413. doi: 10.1371/journal.pone.0173413
Buser, J. R., Maire, J., Riddle, A., Gong, X., Nguyen, T., Nelson, K., et al. (2012). Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann. Neurol. 71, 93–109. doi: 10.1002/ana.22627
Buser, J. R., Segovia, K. N., Dean, J. M., Nelson, K., Beardsley, D., Gong, X., et al. (2010). Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of preterm rabbit cerebral white matter to hypoxia-ischemia. J. Cereb. Blood Flow Metab. 30, 1053–1065. doi: 10.1038/jcbfm.2009.286
Cabrera, O. H., O’Connor, S. D., Swiney, B. S., Salinas-Contreras, P., Manzella, F. M., Taylor, G. T., et al. (2017). Caffeine combined with sedative/anesthetic drugs triggers widespread neuroapoptosis in a mouse model of prematurity. J. Matern. Fetal. Neonatal. Med. 30, 2734–2741. doi: 10.1080/14767058.2016.1261400
Cai, Z., Lin, S., Pang, Y., and Rhodes, P. G. (2004). Brain injury induced by intracerebral injection of interleukin-1beta and tumor necrosis factor-alpha in the neonatal rat. Pediatr. Res. 56, 377–384. doi: 10.1203/01.PDR.0000134249.92944.14
Carloni, S., Favrais, G., Saliba, E., Albertini, M. C., Chalon, S., Longini, M., et al. (2016). Melatonin modulates neonatal brain inflammation through endoplasmic reticulum stress, autophagy, and miR-34a/silent information regulator 1 pathway. J. Pineal. Res. 61, 370–380. doi: 10.1111/jpi.12354
Carlsson, Y., Schwendimann, L., Vontell, R., Rousset, C. I., Wang, X., Lebon, S., et al. (2011). Genetic inhibition of caspase-2 reduces hypoxic-ischemic and excitotoxic neonatal brain injury. Ann. Neurol. 70, 781–789. doi: 10.1002/ana.22431
Castillo-Melendez, M., Baburamani, A. A., Cabalag, C., Yawno, T., Witjaksono, A., Miller, S. L., et al. (2013). Experimental modelling of the consequences of brief late gestation asphyxia on newborn lamb behaviour and brain structure. PLoS One 8:e77377. doi: 10.1371/journal.pone.0077377
Cattano, D., Young, C., Straiko, M. M., and Olney, J. W. (2008). Subanesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth. Analg. 106, 1712–1714. doi: 10.1213/ane.0b013e318172ba0a
Chamnanvanakij, S., Margraf, L. R., Burns, D., and Perlman, J. M. (2002). Apoptosis and white matter injury in preterm infants. Pediatr. Dev. Pathol. 5, 184–189. doi: 10.1007/s10024-001-0205-0
Chau, V., Synnes, A., Grunau, R. E., Poskitt, K. J., Brant, R., and Miller, S. P. (2013). Abnormal brain maturation in preterm neonates associated with adverse developmental outcomes. Neurology 81, 2082–2089. doi: 10.1212/01.wnl.0000437298.43688.b9
Clarke, P. G. (1990). Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. 181, 195–213.
Clarke, P. G., and Puyal, J. (2012). Autophagic cell death exists. Autophagy 8, 867–869. doi: 10.4161/auto.20380
Creeley, C., Dikranian, K., Dissen, G., Martin, L., Olney, J., and Brambrink, A. (2013). Propofol-induced apoptosis of neurones and oligodendrocytes in fetal and neonatal rhesus macaque brain. Br. J. Anaesth. 110(Suppl. 1), i29–i38. doi: 10.1093/bja/aet173
Creeley, C. E., Dikranian, K. T., Dissen, G. A., Back, S. A., Olney, J. W., and Brambrink, A. M. (2014). Isoflurane-induced apoptosis of neurons and oligodendrocytes in the fetal rhesus macaque brain. Anesthesiology 120, 626–638. doi: 10.1097/ALN.0000000000000037
Cuervo, A. M. (2004). Autophagy: many paths to the same end. Mol. Cell Biochem. 263, 55–72. doi: 10.1023/B:MCBI.0000041848.57020.57
de Vries, L. S., Groenendaal, F., Liem, K. D., Heep, A., Brouwer, A. J., van ’t Verlaat, E., et al. (2019). Treatment thresholds for intervention in posthaemorrhagic ventricular dilation: a randomised controlled trial. Arch. Dis. Child Fetal. Neonatal. Ed. 104, F70–F75. doi: 10.1136/archdischild-2017-314206
Dean, J. M., Farrag, D., Zahkouk, S. A., El Zawahry, E. Y., Hagberg, H., Kjellmer, I., et al. (2009). Cerebellar white matter injury following systemic endotoxemia in preterm fetal sheep. Neuroscience 160, 606–615. doi: 10.1016/j.neuroscience.2009.02.071
Dean, J. M., Gunn, A. J., Wassink, G., George, S., and Bennet, L. (2006). Endogenous alpha2-adrenergic receptor-mediated neuroprotection after severe hypoxia in preterm fetal sheep. Neuroscience 142, 615–628. doi: 10.1016/j.neuroscience.2006.06.066
Dean, J. M., Shi, Z., Fleiss, B., Gunn, K. C., Groenendaal, F., van Bel, F., et al. (2015). A Critical Review of Models of Perinatal Infection. Dev. Neurosci. 37, 289–304. doi: 10.1159/000370309
Dean, J. M., Wang, X., Kaindl, A. M., Gressens, P., Fleiss, B., Hagberg, H., et al. (2010). Microglial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microglia in vitro. Brain Behav. Immun. 24, 776–783. doi: 10.1016/j.bbi.2009.10.018
Debillon, T., Gras-Leguen, C., Leroy, S., Caillon, J., Rozé, J. C., and Gressens, P. (2003). Patterns of cerebral inflammatory response in a rabbit model of intrauterine infection-mediated brain lesion. Dev. Brain Res. 145, 39–48. doi: 10.1016/s0165-3806(03)00193-7
Debillon, T., Gras-Leguen, C., Verielle, V., Winer, N., Caillon, J., Roze, J. C., et al. (2000). Intrauterine infection induces programmed cell death in rabbit periventricular white matter. Pediatr. Res. 47, 736–742. doi: 10.1203/00006450-200006000-00009
Debnath, J., Baehrecke, E. H., and Kroemer, G. (2005). Does autophagy contribute to cell death? Autophagy 1, 66–74. doi: 10.4161/auto.1.2.1738
Del Bigio, M. R. (2011). Cell proliferation in human ganglionic eminence and suppression after prematurity-associated haemorrhage. Brain 134(Pt 5), 1344–1361. doi: 10.1093/brain/awr052
Derrick, M., Luo, N. L., Bregman, J. C., Jilling, T., Ji, X., Fisher, K., et al. (2004). Preterm fetal hypoxia-ischemia causes hypertonia and motor deficits in the neonatal rabbit: a model for human cerebral palsy? J. Neurosci. 24, 24–34. doi: 10.1523/JNEUROSCI.2816-03.2004
Descloux, C., Ginet, V., Rummel, C., Truttmann, A. C., and Puyal, J. (2018). Enhanced autophagy contributes to excitotoxic lesions in a rat model of preterm brain injury. Cell Death Dis. 9:853. doi: 10.1038/s41419-018-0916-z
Destot-Wong, K. D., Liang, K., Gupta, S. K., Favrais, G., Schwendimann, L., Pansiot, J., et al. (2009). The AMPA receptor positive allosteric modulator, S18986, is neuroprotective against neonatal excitotoxic and inflammatory brain damage through BDNF synthesis. Neuropharmacology 57, 277–286. doi: 10.1016/j.neuropharm.2009.05.010
Dohare, P., Zia, M. T., Ahmed, E., Ahmed, A., Yadala, V., Schober, A. L., et al. (2016). AMPA-kainate receptor inhibition promotes neurologic recovery in premature rabbits with intraventricular hemorrhage. J. Neurosci. 36, 3363–3377. doi: 10.1523/JNEUROSCI.4329-15.2016
Dommergues, M. A., Plaisant, F., Verney, C., and Gressens, P. (2003). Early microglial activation following neonatal excitotoxic brain damage in mice: a potential target for neuroprotection. Neuroscience 121, 619–628. doi: 10.1016/s0306-4522(03)00558-x
Dribben, W. H., Creeley, C. E., Wang, H. H., Smith, D. J., Farber, N. B., and Olney, J. W. (2009). High dose magnesium sulfate exposure induces apoptotic cell death in the developing neonatal mouse brain. Neonatology 96, 23–32. doi: 10.1159/000201327
Drommelschmidt, K., Serdar, M., Bendix, I., Herz, J., Bertling, F., Prager, S., et al. (2017). Mesenchymal stem cell-derived extracellular vesicles ameliorate inflammation-induced preterm brain injury. Brain Behav. Immun. 60, 220–232. doi: 10.1016/j.bbi.2016.11.011
Dubois, J., Benders, M., Borradori-Tolsa, C., Cachia, A., Lazeyras, F., Ha-Vinh Leuchter, R., et al. (2008). Primary cortical folding in the human newborn: an early marker of later functional development. Brain 131(Pt 8), 2028–2041. doi: 10.1093/brain/awn137
Duerden, E. G., Halani, S., Ng, K., Guo, T., Foong, J., Glass, T. J. A., et al. (2019). White matter injury predicts disrupted functional connectivity and microstructure in very preterm born neonates. Neuroimage Clin. 21:101596. doi: 10.1016/j.nicl.2018.11.006
Dummula, K., Vinukonda, G., Chu, P., Xing, Y., Hu, F., Mailk, S., et al. (2011). Bone morphogenetic protein inhibition promotes neurological recovery after intraventricular hemorrhage. J. Neurosci. 31, 12068–12082. doi: 10.1523/JNEUROSCI.0013-11.2011
Dzietko, M., Boos, V., Sifringer, M., Polley, O., Gerstner, B., Genz, K., et al. (2008). A critical role for Fas/CD-95 dependent signaling pathways in the pathogenesis of hyperoxia-induced brain injury. Ann. Neurol. 64, 664–673. doi: 10.1002/ana.21516
Endesfelder, S., Weichelt, U., Schiller, C., Sifringer, M., Bendix, I., and Buhrer, C. (2017a). Caffeine protects against anticonvulsant-induced neurotoxicity in the developing rat brain. Neurotox. Res. 32, 460–472. doi: 10.1007/s12640-017-9768-z
Endesfelder, S., Weichelt, U., Strauss, E., Schlor, A., Sifringer, M., Scheuer, T., et al. (2017b). Neuroprotection by caffeine in hyperoxia-induced neonatal brain injury. Int. J. Mol. Sci. 18, E187. doi: 10.3390/ijms18010187
Endesfelder, S., Zaak, I., Weichelt, U., Buhrer, C., and Schmitz, T. (2014). Caffeine protects neuronal cells against injury caused by hyperoxia in the immature brain. Free Radic. Biol. Med. 67, 221–234. doi: 10.1016/j.freeradbiomed.2013.09.026
Fan, L. W., Kaizaki, A., Tien, L. T., Pang, Y., Tanaka, S., Numazawa, S., et al. (2013). Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience 240, 27–38. doi: 10.1016/j.neuroscience.2013.02.041
Fan, L. W., Pang, Y., Lin, S., Rhodes, P. G., and Cai, Z. (2005a). Minocycline attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neurosci 133, 159–168. doi: 10.1111/j.1460-9568.2006.04918.x
Fan, L. W., Pang, Y., Lin, S., Tien, L. T., Ma, T., Rhodes, P. G., et al. (2005b). Minocycline reduces lipopolysaccharide-induced neurological dysfunction and brain injury in the neonatal rat. J. Neurosci. Res. 82, 71–82. doi: 10.1002/jnr.20623
Felderhoff-Mueser, U., Buhrer, C., Groneck, P., Obladen, M., Bartmann, P., and Heep, A. (2003). Soluble Fas (CD95/Apo-1), soluble Fas ligand, and activated caspase 3 in the cerebrospinal fluid of infants with posthemorrhagic and nonhemorrhagic hydrocephalus. Pediatr. Res. 54, 659–664. doi: 10.1203/01.PDR.0000084114.83724.65
Felderhoff-Mueser, U., Sifringer, M., Polley, O., Dzietko, M., Leineweber, B., Mahler, L., et al. (2005). Caspase-1-processed interleukins in hyperoxia-induced cell death in the developing brain. Ann. Neurol. 57, 50–59. doi: 10.1002/ana.20322
Filan, P. M., Hunt, R. W., Anderson, P. J., Doyle, L. W., and Inder, T. E. (2012). Neurologic outcomes in very preterm infants undergoing surgery. J. Pediatr. 160, 409–414. doi: 10.1016/j.jpeds.2011.09.009
Follett, P. L., Rosenberg, P. A., Volpe, J. J., and Jensen, F. E. (2000). NBQX attenuates excitotoxic injury in developing white matter. J. Neurosci. 20, 9235–9241. doi: 10.1523/jneurosci.20-24-09235.2000
Fontaine, R. H., Olivier, P., Massonneau, V., Leroux, P., Degos, V., Lebon, S., et al. (2008). Vulnerability of white matter towards antenatal hypoxia is linked to a species-dependent regulation of glutamate receptor subunits. Proc. Natl. Acad. Sci. U.S.A. 105, 16779–16784. doi: 10.1073/pnas.0803004105
Friedrichs-Maeder, C. L., Griffa, A., Schneider, J., Huppi, P. S., Truttmann, A., and Hagmann, P. (2017). Exploring the role of white matter connectivity in cortex maturation. PLoS One 12:e0177466. doi: 10.1371/journal.pone.0177466
Galluzzi, L., Kepp, O., Chan, F. K., and Kroemer, G. (2017). Necroptosis: mechanisms and relevance to disease. Annu. Rev. Pathol. 12, 103–130. doi: 10.1146/annurev-pathol-052016-100247
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., et al. (2012). Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 19, 107–120. doi: 10.1038/cdd.2011.96
Gelot, A., Villapol, S., Billette, de Villemeur, T., Renolleau, S., and Charriaut-Marlangue, C. (2009). Astrocytic demise in the developing rat and human brain after hypoxic-ischemic damage. Dev. Neurosci. 31, 459–470. doi: 10.1159/000232564
Georgiadis, P., Xu, H., Chua, C., Hu, F., Collins, L., Huynh, C., et al. (2008). Characterization of acute brain injuries and neurobehavioral profiles in a rabbit model of germinal matrix hemorrhage. Stroke 39, 3378–3388. doi: 10.1161/STROKEAHA.107.510883
Gerstner, B., Buhrer, C., Rheinlander, C., Polley, O., Schuller, A., Berns, M., et al. (2006). Maturation-dependent oligodendrocyte apoptosis caused by hyperoxia. J. Neurosci. Res. 84, 306–315. doi: 10.1002/jnr.20880
Gerstner, B., Sifringer, M., Dzietko, M., Schuller, A., Lee, J., Simons, S., et al. (2007). Estradiol attenuates hyperoxia-induced cell death in the developing white matter. Ann. Neurol. 61, 562–573. doi: 10.1002/ana.21118
Gilles, F., Gressens, P., Dammann, O., and Leviton, A. (2017). Hypoxia-ischemia is not an antecedent of most preterm brain damage: the illusion of validity. Dev. Med. Child Neurol. 60, 120–125. doi: 10.1111/dmcn.13483
Ginet, V., Pittet, M. P., Rummel, C., Osterheld, M. C., Meuli, R., Clarke, P. G., et al. (2014a). Dying neurons in thalamus of asphyxiated term newborns and rats are autophagic. Ann. Neurol. 76, 695–711. doi: 10.1002/ana.24257
Ginet, V., Puyal, J., Clarke, P. G., and Truttmann, A. C. (2009a). Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am. J. Pathol. 175, 1962–1974. doi: 10.2353/ajpath.2009.090463
Ginet, V., Puyal, J., Magnin, G., Clarke, P. G., and Truttmann, A. C. (2009b). Limited role of the c-Jun N-terminal kinase pathway in a neonatal rat model of cerebral hypoxia-ischemia. J. Neurochem. 108, 552–562. doi: 10.1111/j.1471-4159.2008.05797.x
Ginet, V., Spiehlmann, A., Rummel, C., Rudinskiy, N., Grishchuk, Y., Luthi-Carter, R., et al. (2014b). Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 10, 846–860. doi: 10.4161/auto.28264
Ginet, V., van de Looij, Y., Petrenko, V., Toulotte, A., Kiss, J., Huppi, P. S., et al. (2016). Lactoferrin during lactation reduces lipopolysaccharide-induced brain injury. Biofactors 42, 323–336. doi: 10.1002/biof.1278
Golstein, P., and Kroemer, G. (2007). Cell death by necrosis: towards a molecular definition. Trends Biochem. Sci. 32, 37–43. doi: 10.1016/j.tibs.2006.11.001
Goni-de-Cerio, F., Alvarez, A., Caballero, A., Mielgo, V. E., Alvarez, F. J., Rey-Santano, M. C., et al. (2007). Early cell death in the brain of fetal preterm lambs after hypoxic-ischemic injury. Brain Res. 1151, 161–171. doi: 10.1016/j.brainres.2007.03.013
Goren, B., Cakir, A., Sevinc, C., Serter Kocoglu, S., Ocalan, B., Oy, C., et al. (2017). Uridine treatment protects against neonatal brain damage and long-term cognitive deficits caused by hyperoxia. Brain Res. 1676, 57–68. doi: 10.1016/j.brainres.2017.09.010
Gozdas, E., Parikh, N. A., Merhar, S. L., Tkach, J. A., He, L., and Holland, S. K. (2018). Altered functional network connectivity in preterm infants: antecedents of cognitive and motor impairments? Brain Struct. Funct. 223, 3665–3680. doi: 10.1007/s00429-018-1707-0
Graham, E. M., Holcroft, C. J., Rai, K. K., Donohue, P. K., and Allen, M. C. (2004). Neonatal cerebral white matter injury in preterm infants is associated with culture positive infections and only rarely with metabolic acidosis. Am. J. Obstet. Gynecol. 191, 1305–1310. doi: 10.1016/j.ajog.2004.06.058
Gram, M., Sveinsdottir, S., Cinthio, M., Sveinsdottir, K., Hansson, S. R., Morgelin, M., et al. (2014). Extracellular hemoglobin - mediator of inflammation and cell death in the choroid plexus following preterm intraventricular hemorrhage. J. Neuroinflammation 11:200. doi: 10.1186/s12974-014-0200-9
Griesmaier, E., Schlager, G., Wegleiter, K., Hermann, M., Urbanek, M., Simbruner, G., et al. (2010). Role of p75NTR in NMDAR-mediated excitotoxic brain injury in neonatal mice. Brain Res. 1355, 31–40. doi: 10.1016/j.brainres.2010.07.095
Grishchuk, Y., Ginet, V., Truttmann, A. C., Clarke, P. G., and Puyal, J. (2011). Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 7, 1115–1131. doi: 10.4161/auto.7.10.16608
Gussenhoven, R., Westerlaken, R. J. J., Ophelders, D., Jobe, A. H., Kemp, M. W., Kallapur, S. G., et al. (2018). Chorioamnionitis, neuroinflammation, and injury: timing is key in the preterm ovine fetus. J. Neuroinflammation 15:113. doi: 10.1186/s12974-018-1149-x
Hagberg, H., Mallard, C., Ferriero, D. M., Vannucci, S. J., Levison, S. W., Vexler, Z. S., et al. (2015). The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208. doi: 10.1038/nrneurol.2015.13
Hagen, M. W., Riddle, A., McClendon, E., Gong, X., Shaver, D., Srivastava, T., et al. (2014). Role of recurrent hypoxia-ischemia in preterm white matter injury severity. PLoS One 9:e112800. doi: 10.1371/journal.pone.0112800
Haldipur, P., Dupuis, N., Degos, V., Moniaux, N., Chhor, V., Rasika, S., et al. (2014). HIP/PAP prevents excitotoxic neuronal death and promotes plasticity. Ann. Clin. Transl. Neurol. 1, 739–754. doi: 10.1002/acn3.127
Hargitai, B., Szabo, V., Hajdu, J., Harmath, A., Pataki, M., Farid, P., et al. (2001). Apoptosis in various organs of preterm infants: histopathologic study of lung, kidney, liver, and brain of ventilated infants. Pediatr. Res. 50, 110–114. doi: 10.1203/00006450-200107000-00020
Haynes, R. L., Billiards, S. S., Borenstein, N. S., Volpe, J. J., and Kinney, H. C. (2008). Diffuse axonal injury in periventricular leukomalacia as determined by apoptotic marker fractin. Pediatr. Res. 63, 656–661. doi: 10.1203/PDR.0b013e31816c825c
Haynes, R. L., Folkerth, R. D., Keefe, R. J., Sung, I., Swzeda, L. I., Rosenberg, P. A., et al. (2003). Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J. Neuropathol. Exp. Neurol. 62, 441–450. doi: 10.1093/jnen/62.5.441
Haynes, R. L., and van Leyen, K. (2013). 12/15-lipoxygenase expression is increased in oligodendrocytes and microglia of periventricular leukomalacia. Dev. Neurosci. 35, 140–154. doi: 10.1159/000350230
Heo, J. S., Kim, E. K., Choi, Y. H., Shin, S. H., Sohn, J. A., Cheon, J. E., et al. (2018). Timing of sepsis is an important risk factor for white matter abnormality in extremely premature infants with sepsis. Pediatr. Neonatol. 59, 77–84. doi: 10.1016/j.pedneo.2017.07.008
Hielkema, T., and Hadders-Algra, M. (2016). Motor and cognitive outcome after specific early lesions of the brain - a systematic review. Dev. Med. Child Neurol. 58(Suppl. 4), 46–52. doi: 10.1111/dmcn.13047
Hoeber, D., Sifringer, M., van de Looij, Y., Herz, J., Sizonenko, S. V., Kempe, K., et al. (2016). Erythropoietin restores long-term neurocognitive function involving mechanisms of neuronal plasticity in a model of hyperoxia-induced preterm brain injury. Oxid. Med. Cell Longev. 2016:9247493. doi: 10.1155/2016/9247493
Huang, Y., Lai, H., Xu, H., Wu, W., Lai, X., Ho, G., et al. (2013). Impact of perinatal systemic hypoxic-ischemic injury on the brain of male offspring rats: an improved model of neonatal hypoxic-ischemic encephalopathy in early preterm newborns. PLoS One 8:e82502. doi: 10.1371/journal.pone.0082502
Husson, I., Mesplès, B., Bac, P., Vamecq, J., Evrard, P., and Gressens, P. (2002). Melatoninergic neuroprotection of the murine periventricular white matter against neonatal excitotoxic challenge. Ann. Neurol. 51, 82–92. doi: 10.1002/ana.10072
Husson, I., Rangon, C. M., Lelievre, V., Bemelmans, A. P., Sachs, P., Mallet, J., et al. (2005). BDNF-induced white matter neuroprotection and stage-dependent neuronal survival following a neonatal excitotoxic challenge. Cereb. Cortex 15, 250–261. doi: 10.1093/cercor/bhh127
Ikebara, J. M., Takada, S. H., Cardoso, D. S., Dias, N. M., de Campos, B. C., Bretherick, T. A., et al. (2017). Functional role of intracellular calcium receptor inositol 1,4,5-trisphosphate type 1 in rat hippocampus after neonatal anoxia. PLoS One 12:e0169861. doi: 10.1371/journal.pone.0169861
Jablonska, B., Scafidi, J., Aguirre, A., Vaccarino, F., Nguyen, V., Borok, E., et al. (2012). Oligodendrocyte regeneration after neonatal hypoxia requires FoxO1-mediated p27Kip1 expression. J. Neurosci. 32, 14775–14793. doi: 10.1523/JNEUROSCI.2060-12.2012
Jakab, A., Ruegger, C., Bucher, H. U., Makki, M., Huppi, P. S., Tuura, R., et al. (2019). Network based statistics reveals trophic and neuroprotective effect of early high dose erythropoetin on brain connectivity in very preterm infants. Neuroimage Clin. 22:101806. doi: 10.1016/j.nicl.2019.101806
Jantzie, L. L., Corbett, C. J., Firl, D. J., and Robinson, S. (2015a). Postnatal erythropoietin mitigates impaired cerebral cortical development following subplate loss from prenatal hypoxia-ischemia. Cereb. Cortex 25, 2683–2695. doi: 10.1093/cercor/bhu066
Jantzie, L. L., and Robinson, S. (2015). Preclinical models of encephalopathy of prematurity. Dev. Neurosci. 37, 277–288. doi: 10.1159/000371721
Jantzie, L. L., Talos, D. M., Jackson, M. C., Park, H. K., Graham, D. A., Lechpammer, M., et al. (2015b). Developmental expression of N-methyl-D-aspartate (n.d.) receptor subunits in human white and gray matter: potential mechanism of increased vulnerability in the immature brain. Cereb. Cortex 25, 482–495. doi: 10.1093/cercor/bht246
Kaindl, A. M., Sifringer, M., Koppelstaetter, A., Genz, K., Loeber, R., Boerner, C., et al. (2008). Erythropoietin protects the developing brain from hyperoxia-induced cell death and proteome changes. Ann. Neurol. 64, 523–534. doi: 10.1002/ana.21471
Kersbergen, K. J., Leroy, F., Isgum, I., Groenendaal, F., de Vries, L. S., Claessens, N. H. P., et al. (2016). Relation between clinical risk factors, early cortical changes, and neurodevelopmental outcome in preterm infants. Neuroimage 142, 301–310. doi: 10.1016/j.neuroimage.2016.07.010
Kida, H., Nomura, S., Shinoyama, M., Ideguchi, M., Owada, Y., and Suzuki, M. (2013). The effect of hypothermia therapy on cortical laminar disruption following ischemic injury in neonatal mice. PLoS One 8:e68877. doi: 10.1371/journal.pone.0068877
Kidokoro, H., Anderson, P. J., Doyle, L. W., Woodward, L. J., Neil, J. J., and Inder, T. E. (2014). Brain injury and altered brain growth in preterm infants: predictors and prognosis. Pediatrics 134, e444–e453. doi: 10.1542/peds.2013-2336
Kidokoro, H., Neil, J. J., and Inder, T. E. (2013). New MR imaging assessment tool to define brain abnormalities in very preterm infants at term. AJNR Am. J. Neuroradiol. 34, 2208–2214. doi: 10.3174/ajnr.A3521
Kinney, H. C., Haynes, R. L., Xu, G., Andiman, S. E., Folkerth, R. D., Sleeper, L. A., et al. (2012). Neuron deficit in the white matter and subplate in periventricular leukomalacia. Ann. Neurol. 71, 397–406. doi: 10.1002/ana.22612
Kinney, H. C., and Volpe, J. J. (2012). Modeling the encephalopathy of prematurity in animals: the important role of translational research. Neurol. Res. Int. 2012:295389. doi: 10.1155/2012/295389
Kitanaka, C., and Kuchino, Y. (1999). Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 6, 508–515. doi: 10.1038/sj.cdd.4400526
Klebe, D., McBride, D., Krafft, P. R., Flores, J. J., Tang, J., and Zhang, J. H. (2020). Posthemorrhagic hydrocephalus development after germinal matrix hemorrhage: established mechanisms and proposed pathways. J. Neurosci. Res. 98, 105–120. doi: 10.1002/jnr.24394
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. doi: 10.1080/15548627.2015.1100356
Kodama, M., Satoh, Y., Otsubo, Y., Araki, Y., Yonamine, R., Masui, K., et al. (2011). Neonatal desflurane exposure induces more robust neuroapoptosis than do isoflurane and sevoflurane and impairs working memory. Anesthesiology 115, 979–991. doi: 10.1097/ALN.0b013e318234228b
Kriel, J., and Loos, B. (2019). The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 26, 640–652. doi: 10.1038/s41418-018-0267-4
Kroemer, G., and Levine, B. (2008). Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010. doi: 10.1038/nrm2529
Kumral, A., Baskin, H., Yesilirmak, D. C., Ergur, B. U., Aykan, S., Genc, S., et al. (2007). Erythropoietin attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neonatology 92, 269–278. doi: 10.1159/000105493
Kumral, A., Iscan, B., Engur, D., Tuzun, F., Ozbal, S., Ergur, B. U., et al. (2017). Intranasal surfactant protein D as neuroprotective rescue in a neonatal rat model of periventricular leukomalacia. J. Matern. Fetal. Neonatal. Med. 30, 446–451. doi: 10.1080/14767058.2016.1174996
Kurul, S. H., Yis, U., Kumral, A., Tugyan, K., Cilaker, S., Kolatan, E., et al. (2009). Protective effects of topiramate against hyperoxic brain injury in the developing brain. Neuropediatrics 40, 22–27. doi: 10.1055/s-0029-1224101
Levine, B., and Kroemer, G. (2019). Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. doi: 10.1016/j.cell.2018.09.048
Li, J., Xiong, M., Nadavaluru, P. R., Zuo, W., Ye, J. H., Eloy, J. D., et al. (2016a). Dexmedetomidine attenuates neurotoxicity induced by prenatal propofol exposure. J. Neurosurg. Anesthesiol. 28, 51–64. doi: 10.1097/ANA.0000000000000181
Li, J., Yawno, T., Sutherland, A., Loose, J., Nitsos, I., Allison, B. J., et al. (2017). Term vs. preterm cord blood cells for the prevention of preterm brain injury. Pediatr. Res. 82, 1030–1038. doi: 10.1038/pr.2017.170
Li, J., Yawno, T., Sutherland, A., Loose, J., Nitsos, I., Bischof, R., et al. (2016b). Preterm white matter brain injury is prevented by early administration of umbilical cord blood cells. Exp. Neurol. 283(Pt A), 179–187. doi: 10.1016/j.expneurol.2016.06.017
Li, X., Wu, Z., Zhang, Y., Xu, Y., Han, G., and Zhao, P. (2017). Activation of autophagy contributes to sevoflurane-induced neurotoxicity in fetal rats. Front. Mol. Neurosci. 10:432. doi: 10.3389/fnmol.2017.00432
Ligam, P., Haynes, R. L., Folkerth, R. D., Liu, L., Yang, M., Volpe, J. J., et al. (2009). Thalamic damage in periventricular leukomalacia: novel pathologic observations relevant to cognitive deficits in survivors of prematurity. Pediatr. Res. 65, 524–529. doi: 10.1203/PDR.0b013e3181998baf
Liu, Y., Shoji-Kawata, S., Sumpter, R. M. Jr., Wei, Y., Ginet, V., Zhang, L., et al. (2013). Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. U.S.A. 110, 20364–20371. doi: 10.1073/pnas.1319661110
Lodygensky, G. A., Kunz, N., Perroud, E., Somm, E., Mlynarik, V., Huppi, P. S., et al. (2014). Definition and quantification of acute inflammatory white matter injury in the immature brain by MRI/MRS at high magnetic field. Pediatr. Res. 75, 415–423. doi: 10.1038/pr.2013.242
Luyt, K., Jary, S., Lea, C., Young, G. J., Odd, D., Miller, H., et al. (2019). Ten-year follow-up of a randomised trial of drainage, irrigation and fibrinolytic therapy (DRIFT) in infants with post-haemorrhagic ventricular dilatation. Health Technol. Assess. 23, 1–116. doi: 10.3310/hta23040
Mallard, C., Welin, A. K., Peebles, D., Hagberg, H., and Kjellmer, I. (2003). White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochem. Res. 28, 215–223. doi: 10.1023/a:1022368915400
Marino, G., Madeo, F., and Kroemer, G. (2011). Autophagy for tissue homeostasis and neuroprotection. Curr. Opin. Cell Biol. 23, 198–206. doi: 10.1016/j.ceb.2010.10.001
Mazur, M., Miller, R. H., and Robinson, S. (2010). Postnatal erythropoietin treatment mitigates neural cell loss after systemic prenatal hypoxic-ischemic injury. J. Neurosurg. Pediatr. 6, 206–221. doi: 10.3171/2010.5.PEDS1032
McClendon, E., Shaver, D. C., Degener-O’Brien, K., Gong, X., Nguyen, T., Hoerder-Suabedissen, A., et al. (2017). Transient hypoxemia chronically disrupts maturation of preterm fetal ovine subplate neuron arborization and activity. J. Neurosci. 37, 11912–11929. doi: 10.1523/JNEUROSCI.2396-17.2017
McGowan, E. C., and Vohr, B. R. (2019). Neurodevelopmental follow-up of preterm infants: what is new? Pediatr. Clin. North Am. 66, 509–523. doi: 10.1016/j.pcl.2018.12.015
McPherson, C., Neil, J. J., Tjoeng, T. H., Pineda, R., and Inder, T. E. (2015). A pilot randomized trial of high-dose caffeine therapy in preterm infants. Pediatr. Res. 78, 198–204. doi: 10.1038/pr.2015.72
McQuillen, P. S., Sheldon, R. A., Shatz, C. J., and Ferriero, D. M. (2003). Selective vulnerability of subplate neurons after early neonatal hypoxia-ischemia. J. Neurosci. 23, 3308–3315. doi: 10.1523/jneurosci.23-08-03308.2003
Medja, F., Lelievre, V., Fontaine, R. H., Lebas, F., Leroux, P., Ouimet, T., et al. (2006). Thiorphan, a neutral endopeptidase inhibitor used for diarrhoea, is neuroprotective in newborn mice. Brain 129(Pt 12), 3209–3223. doi: 10.1093/brain/awl239
Mikhailova, A., Sunkara, N., and McQuillen, P. S. (2017). Unbiased quantification of subplate neuron loss following neonatal hypoxia-ischemia in a rat model. Dev. Neurosci. 39, 171–181. doi: 10.1159/000460815
Morriss, F. H. Jr., Saha, S., Bell, E. F., Colaizy, T. T., Stoll, B. J., Hintz, S. R., et al. (2014). Surgery and neurodevelopmental outcome of very low-birth-weight infants. JAMA Pediatr. 168, 746–754. doi: 10.1001/jamapediatrics.2014.307
Mukai, T., Mori, Y., Shimazu, T., Takahashi, A., Tsunoda, H., Yamaguchi, S., et al. (2017). Intravenous injection of umbilical cord-derived mesenchymal stromal cells attenuates reactive gliosis and hypomyelination in a neonatal intraventricular hemorrhage model. Neuroscience 355, 175–187. doi: 10.1016/j.neuroscience.2017.05.006
Neubauer, V., Wegleiter, K., Posod, A., Urbanek, M., Wechselberger, K., Kiechl-Kohlendorfer, U., et al. (2016). Delayed application of the haematopoietic growth factors G-CSF/SCF and FL reduces neonatal excitotoxic brain injury. Brain Res. 1634, 94–103. doi: 10.1016/j.brainres.2015.12.058
Noguchi, K. K., Johnson, S. A., Manzella, F. M., Masuoka, K. L., Williams, S. L., Martin, L. D., et al. (2018). Caffeine augments anesthesia neurotoxicity in the fetal macaque brain. Sci. Rep. 8:5302. doi: 10.1038/s41598-018-23560-7
Northington, F. J., Chavez-Valdez, R., and Martin, L. J. (2011). Neuronal cell death in neonatal hypoxia-ischemia. Ann. Neurol. 69, 743–758. doi: 10.1002/ana.22419
Nossin-Manor, R., Card, D., Morris, D., Noormohamed, S., Shroff, M. M., Whyte, H. E., et al. (2013). Quantitative MRI in the very preterm brain: assessing tissue organization and myelination using magnetization transfer, diffusion tensor and T(1) imaging. Neuroimage 64, 505–516. doi: 10.1016/j.neuroimage.2012.08.086
Okusa, C., Oeschger, F., Ginet, V., Wang, W. Z., Hoerder-Suabedissen, A., Matsuyama, T., et al. (2014). Subplate in a rat model of preterm hypoxia-ischemia. Ann. Clin. Transl. Neurol. 1, 679–691. doi: 10.1002/acn3.97
Paintlia, M. K., Paintlia, A. S., Barbosa, E., Singh, I., and Singh, A. K. (2004). N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J. Neurosci. Res. 78, 347–361. doi: 10.1002/jnr.20261
Panda, S., Dohare, P., Jain, S., Parikh, N., Singla, P., Mehdizadeh, R., et al. (2018). Estrogen treatment reverses prematurity-induced disruption in cortical interneuron population. J. Neurosci. 38, 7378–7391. doi: 10.1523/JNEUROSCI.0478-18.2018
Paneth, N. (2018). Hypoxia-ischemia and brain injury in infants born preterm. Dev. Med. Child Neurol. 60:115. doi: 10.1111/dmcn.13642
Panfoli, I., Candiano, G., Malova, M., De Angelis, L., Cardiello, V., Buonocore, G., et al. (2018). Oxidative stress as a primary risk factor for brain damage in preterm newborns. Front. Pediatr. 6:369. doi: 10.3389/fped.2018.00369
Paton, M. C. B., Allison, B. J., Li, J., Fahey, M. C., Sutherland, A. E., Nitsos, I., et al. (2018). Human umbilical cord blood therapy protects cerebral white matter from systemic LPS exposure in preterm fetal sheep. Dev. Neurosci. 40, 258–270. doi: 10.1159/000490943
Pham, H., Vottier, G., Pansiot, J., Duong-Quy, S., Bollen, B., Dalous, J., et al. (2014). Inhaled NO prevents hyperoxia-induced white matter damage in neonatal rats. Exp. Neurol. 252, 114–123. doi: 10.1016/j.expneurol.2013.11.025
Pierrat, V., Marchand-Martin, L., Arnaud, C., Kaminski, M., Resche-Rigon, M., Lebeaux, C., et al. (2017). Neurodevelopmental outcome at 2 years for preterm children born at 22 to 34 weeks’ gestation in France in 2011: EPIPAGE-2 cohort study. BMJ 358:j3448. doi: 10.1136/bmj.j3448
Pierre, W. C., Akakpo, L., Londono, I., Pouliot, P., Chemtob, S., Lesage, F., et al. (2019). Assessing therapeutic response non-invasively in a neonatal rat model of acute inflammatory white matter injury using high-field MRI. Brain Behav. Immun. 81, 348–360. doi: 10.1016/j.bbi.2019.06.032
Pierson, C. R., Folkerth, R. D., Billiards, S. S., Trachtenberg, F. L., Drinkwater, M. E., Volpe, J. J., et al. (2007). Gray matter injury associated with periventricular leukomalacia in the premature infant. Acta Neuropathol. 114, 619–631. doi: 10.1007/s00401-007-0295-5
Portera-Cailliau, C., Price, D. L., and Martin, L. J. (1997). Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J. Comp. Neurol. 378, 70–87.
Posod, A., Pinzer, K., Urbanek, M., Wegleiter, K., Keller, M., Kiechl-Kohlendorfer, U., et al. (2014). The common antitussive agent dextromethorphan protects against hyperoxia-induced cell death in established in vivo and in vitro models of neonatal brain injury. Neuroscience 274, 260–272. doi: 10.1016/j.neuroscience.2014.05.059
Puyal, J., Ginet, V., and Clarke, P. G. (2013). Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog. Neurobiol. 105, 24–48. doi: 10.1016/j.pneurobio.2013.03.002
Puyal, J., Vaslin, A., Mottier, V., and Clarke, P. G. (2009). Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann. Neurol. 66, 378–389. doi: 10.1002/ana.21714
Riddle, A., Luo, N. L., Manese, M., Beardsley, D. J., Green, L., Rorvik, D. A., et al. (2006). Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J. Neurosci. 26, 3045–3055. doi: 10.1523/JNEUROSCI.5200-05.2006
Riedl, S. J., and Shi, Y. (2004). Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 5, 897–907. doi: 10.1038/nrm1496
Robinson, S., Li, Q., Dechant, A., and Cohen, M. L. (2006). Neonatal loss of gamma-aminobutyric acid pathway expression after human perinatal brain injury. J. Neurosurg/ 104 6(Suppl.), 396–408. doi: 10.3171/ped.2006.104.6.396
Robinson, S., Petelenz, K., Li, Q., Cohen, M. L., Dechant, A., Tabrizi, N., et al. (2005). Developmental changes induced by graded prenatal systemic hypoxic-ischemic insults in rats. Neurobiol. Dis. 18, 568–581. doi: 10.1016/j.nbd.2004.10.024
Rogido, M., Husson, I., Bonnier, C., Lallemand, M. C., Merienne, C., Gregory, G. A., et al. (2003). Fructose-1,6-biphosphate prevents excitotoxic neuronal cell death in the neonatal mouse brain. Brain Res. Dev. Brain Res. 140, 287–297. doi: 10.1016/s0165-3806(02)00615-6
Rousset, C. I., Chalon, S., Cantagrel, S., Bodard, S., Andres, C., Gressens, P., et al. (2006). Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatr. Res. 59, 428–433. doi: 10.1203/01.pdr.0000199905.08848.55
Rousset, C. I., Kassem, J., Aubert, A., Planchenault, D., Gressens, P., Chalon, S., et al. (2013). Maternal exposure to lipopolysaccharide leads to transient motor dysfunction in neonatal rats. Dev. Neurosci. 35, 172–181. doi: 10.1159/000346579
Scafidi, J., Hammond, T. R., Scafidi, S., Ritter, J., Jablonska, B., Roncal, M., et al. (2014). Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature 506, 230–234. doi: 10.1038/nature12880
Schaafsma, W., Basterra, L. B., Jacobs, S., Brouwer, N., Meerlo, P., Schaafsma, A., et al. (2017). Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol. Dis. 106, 291–300. doi: 10.1016/j.nbd.2017.07.017
Schmidt, A. F., Kannan, P. S., Chougnet, C. A., Danzer, S. C., Miller, L. A., Jobe, A. H., et al. (2016). Intra-amniotic LPS causes acute neuroinflammation in preterm rhesus macaques. J. Neuroinflammation 13:238. doi: 10.1186/s12974-016-0706-4
Schmidt, B., Anderson, P. J., Doyle, L. W., Dewey, D., Grunau, R. E., Asztalos, E. V., et al. (2012). Survival without disability to age 5 years after neonatal caffeine therapy for apnea of prematurity. JAMA 307, 275–282. doi: 10.1001/jama.2011.2024
Schmidt, B., Roberts, R. S., Anderson, P. J., Asztalos, E. V., Costantini, L., Davis, P. G., et al. (2017). Academic performance, motor function, and behavior 11 years after neonatal caffeine citrate therapy for apnea of prematurity: an 11-year follow-up of the CAP randomized clinical trial. JAMA Pediatr. 171, 564–572. doi: 10.1001/jamapediatrics.2017.0238
Schmitz, T., Felderhoff-Mueser, U., Sifringer, M., Groenendaal, F., Kampmann, S., and Heep, A. (2011). Expression of soluble Fas in the cerebrospinal fluid of preterm infants with posthemorrhagic hydrocephalus and cystic white matter damage. J. Perinat. Med. 39, 83–88. doi: 10.1515/JPM.2010.125
Schmitz, T., Krabbe, G., Weikert, G., Scheuer, T., Matheus, F., Wang, Y., et al. (2014). Minocycline protects the immature white matter against hyperoxia. Exp. Neurol. 254, 153–165. doi: 10.1016/j.expneurol.2014.01.017
Schneider, J., Fischer Fumeaux, C. J., Duerden, E. G., Guo, T., Foong, J., Graz, M. B., et al. (2018). Nutrient Intake in the first two weeks of life and brain growth in preterm neonates. Pediatrics 141, e20172169. doi: 10.1542/peds.2017-2169
Schneider, J., Kober, T., Bickle Graz, M., Meuli, R., Huppi, P. S., Hagmann, P., et al. (2016). Evolution of T1 relaxation, ADC, and fractional anisotropy during early brain maturation: a serial imaging study on preterm infants. AJNR Am. J. Neuroradiol. 37, 155–162. doi: 10.3174/ajnr.A4510
Schneider, J., and Miller, S. P. (2019). Preterm brain Injury: white matter injury. Handb. Clin. Neurol. 162, 155–172. doi: 10.1016/B978-0-444-64029-1.00007-2
Segovia, K. N., McClure, M., Moravec, M., Luo, N. L., Wan, Y., Gong, X., et al. (2008). Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 63, 520–530. doi: 10.1002/ana.21359
Seyama, T., Kamei, Y., Iriyama, T., Imada, S., Ichinose, M., Toshimitsu, M., et al. (2018). Pretreatment with magnesium sulfate attenuates white matter damage by preventing cell death of developing oligodendrocytes. J. Obstet. Gynaecol. Res. 44, 601–607. doi: 10.1111/jog.13568
Shah, D. K., Doyle, L. W., Anderson, P. J., Bear, M., Daley, A. J., Hunt, R. W., et al. (2008). Adverse neurodevelopment in preterm infants with postnatal sepsis or necrotizing enterocolitis is mediated by white matter abnormalities on magnetic resonance imaging at term. J. Pediatr. 153:175.e1–171.e1. doi: 10.1016/j.jpeds.2008.02.033
Sifringer, M., Bendix, I., Borner, C., Endesfelder, S., von Haefen, C., Kalb, A., et al. (2012). Prevention of neonatal oxygen-induced brain damage by reduction of intrinsic apoptosis. Cell Death Dis. 3:e250. doi: 10.1038/cddis.2011.133
Sifringer, M., von Haefen, C., Krain, M., Paeschke, N., Bendix, I., Buhrer, C., et al. (2015). Neuroprotective effect of dexmedetomidine on hyperoxia-induced toxicity in the neonatal rat brain. Oxid. Med. Cell Longev. 2015:530371. doi: 10.1155/2015/530371
Sinner, B., Becke, K., and Engelhard, K. (2014). General anaesthetics and the developing brain: an overview. Anaesthesia 69, 1009–1022. doi: 10.1111/anae.12637
Sokolowska, P., Passemard, S., Mok, A., Schwendimann, L., Gozes, I., and Gressens, P. (2011). Neuroprotective effects of NAP against excitotoxic brain damage in the newborn mice: implications for cerebral palsy. Neuroscience 173, 156–168. doi: 10.1016/j.neuroscience.2010.10.074
Sokolowski, J. D., Gamage, K. K., Heffron, D. S., Leblanc, A. C., Deppmann, C. D., and Mandell, J. W. (2014). Caspase-mediated cleavage of actin and tubulin is a common feature and sensitive marker of axonal degeneration in neural development and injury. Acta Neuropathol. Commun. 2:16. doi: 10.1186/2051-5960-2-16
Stolp, H. B., Fleiss, B., Arai, Y., Supramaniam, V., Vontell, R., Birtles, S., et al. (2019). Interneuron development is disrupted in preterm brains with diffuse white matter injury: observations in mouse and human. Front. Physiol. 10:955. doi: 10.3389/fphys.2019.00955
Stolwijk, L. J., Lemmers, P. M., Harmsen, M., Groenendaal, F., de Vries, L. S., van der Zee, D. C., et al. (2016). Neurodevelopmental outcomes after neonatal surgery for major noncardiac anomalies. Pediatrics 137:e20151728. doi: 10.1542/peds.2015-1728
Stolwijk, L. J., Lemmers, P. M. A., van Herwaarden, M. Y. A., van der Zee, D. C., van Bel, F., Groenendaal, F., et al. (2017). Predictive role of F2-isoprostanes as biomarkers for brain damage after neonatal surgery. Dis. Markers 2017:2728103. doi: 10.1155/2017/2728103
Straley, M. E., Van Oeffelen, W., Theze, S., Sullivan, A. M., O’Mahony, S. M., Cryan, J. F., et al. (2017). Distinct alterations in motor & reward seeking behavior are dependent on the gestational age of exposure to LPS-induced maternal immune activation. Brain Behav. Immun. 63, 21–34. doi: 10.1016/j.bbi.2016.06.002
Sun, L. (2010). Early childhood general anaesthesia exposure and neurocognitive development. Br. J. Anaesth. 105(Suppl. 1), i61–i68. doi: 10.1093/bja/aeq302
Synnes, A., and Hicks, M. (2018). Neurodevelopmental outcomes of preterm children at school age and beyond. Clin. Perinatol. 45, 393–408. doi: 10.1016/j.clp.2018.05.002
Taglialatela, G., Perez-Polo, J. R., and Rassin, D. K. (1998). Induction of apoptosis in the CNS during development by the combination of hyperoxia and inhibition of glutathione synthesis. Free Radic Biol. Med 25, 936–942. doi: 10.1016/s0891-5849(98)00131-2
Tahraoui, S. L., Marret, S., Bodenant, C., Leroux, P., Dommergues, M. A., Evrard, P., et al. (2001). Central role of microglia in neonatal excitotoxic lesions of the murine periventricular white matter. Brain Pathol. 11, 56–71. doi: 10.1111/j.1750-3639.2001.tb00381.x
Takada, S. H., dos Santos Haemmerle, C. A., Motta-Teixeira, L. C., Machado-Nils, A. V., Lee, V. Y., Takase, L. F., et al. (2015). Neonatal anoxia in rats: hippocampal cellular and subcellular changes related to cell death and spatial memory. Neuroscience 284, 247–259. doi: 10.1016/j.neuroscience.2014.08.054
van de Looij, Y., Ginet, V., Chatagner, A., Toulotte, A., Somm, E., Huppi, P. S., et al. (2014). Lactoferrin during lactation protects the immature hypoxic-ischemic rat brain. Ann. Clin. Transl. Neurol. 1, 955–967. doi: 10.1002/acn3.138
Van’t Hooft, J., van der Lee, J. H., Opmeer, B. C., Aarnoudse-Moens, C. S., Leenders, A. G., Mol, B. W., et al. (2015). Predicting developmental outcomes in premature infants by term equivalent MRI: systematic review and meta-analysis. Syst. Rev. 4:71. doi: 10.1186/s13643-015-0058-7
Verney, C., Pogledic, I., Biran, V., Adle-Biassette, H., Fallet-Bianco, C., and Gressens, P. (2012). Microglial reaction in axonal crossroads is a hallmark of noncystic periventricular white matter injury in very preterm infants. J. Neuropathol. Exp. Neurol. 71, 251–264. doi: 10.1097/NEN.0b013e3182496429
Vinukonda, G., Csiszar, A., Hu, F., Dummula, K., Pandey, N. K., Zia, M. T., et al. (2010). Neuroprotection in a rabbit model of intraventricular haemorrhage by cyclooxygenase-2, prostanoid receptor-1 or tumour necrosis factor-alpha inhibition. Brain 133(Pt 8), 2264–2280. doi: 10.1093/brain/awq107
Vontell, R., Supramaniam, V., Wyatt-Ashmead, J., Gressens, P., Rutherford, M., Hagberg, H., et al. (2015). Cellular mechanisms of toll-like receptor-3 activation in the thalamus are associated with white matter injury in the developing brain. J. Neuropathol. Exp. Neurol. 74, 273–285. doi: 10.1097/NEN.0000000000000172
Vottier, G., Pham, H., Pansiot, J., Biran, V., Gressens, P., Charriaut-Marlangue, C., et al. (2011). Deleterious effect of hyperoxia at birth on white matter damage in the newborn rat. Dev. Neurosci. 33, 261–269. doi: 10.1159/000327245)
Walker, S. M., Melbourne, A., O’Reilly, H., Beckmann, J., Eaton-Rosen, Z., Ourselin, S., et al. (2018). Somatosensory function and pain in extremely preterm young adults from the UK EPICure cohort: sex-dependent differences and impact of neonatal surgery. Br. J. Anaesth. 121, 623–635. doi: 10.1016/j.bja.2018.03.035
Wang, L. W., Chang, Y. C., Lin, C. Y., Hong, J. S., and Huang, C. C. (2010). Low-dose lipopolysaccharide selectively sensitizes hypoxic ischemia-induced white matter injury in the immature brain. Pediatr. Res. 68, 41–47. doi: 10.1203/00006450-201011001-00076
Wang, L. W., Tu, Y. F., Huang, C. C., and Ho, C. J. (2012). JNK signaling is the shared pathway linking neuroinflammation, blood-brain barrier disruption, and oligodendroglial apoptosis in the white matter injury of the immature brain. J. Neuroinflammation 9:175. doi: 10.1186/1742-2094-9-175
Wang, X., Dong, Y., Zhang, Y., Li, T., and Xie, Z. (2019). Sevoflurane induces cognitive impairment in young mice via autophagy. PLoS One 14:e0216372. doi: 10.1371/journal.pone.0216372
Wang, Y., Wang, C., Zhang, Y., Wang, G., and Yang, H. (2019). Pre-administration of luteoline attenuates neonatal sevoflurane-induced neurotoxicity in mice. Acta Histochem. 121, 500–507. doi: 10.1016/j.acthis.2019.04.004
Wassink, G., Davidson, J. O., Dhillon, S. K., Fraser, M., Galinsky, R., Bennet, L., et al. (2017). Partial white and grey matter protection with prolonged infusion of recombinant human erythropoietin after asphyxia in preterm fetal sheep. J. Cereb. Blood Flow Metab. 37, 1080–1094. doi: 10.1177/0271678X16650455
Wegleiter, K., Hermann, M., Posod, A., Wechselberger, K., Stanika, R. I., Obermair, G. J., et al. (2014). The sigma-1 receptor agonist 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) protects against newborn excitotoxic brain injury by stabilizing the mitochondrial membrane potential in vitro and inhibiting microglial activation in vivo. Exp. Neurol. 261, 501–509. doi: 10.1016/j.expneurol.2014.07.022
Xia, Y., Xu, H., Jia, C., Hu, X., Kang, Y., Yang, X., et al. (2017). Tanshinone IIA Attenuates Sevoflurane Neurotoxicity in Neonatal Mice. Anesth. Analg. 124, 1244–1252. doi: 10.1213/ANE.0000000000001942
Xie, C., Ginet, V., Sun, Y., Koike, M., Zhou, K., Li, T., et al. (2016). Neuroprotection by selective neuronal deletion of Atg7 in neonatal brain injury. Autophagy 12, 410–423. doi: 10.1080/15548627.2015.1132134
Xiong, M., Zhang, L., Li, J., Eloy, J., Ye, J. H., and Bekker, A. (2016). Propofol-induced neurotoxicity in the fetal animal brain and developments in modifying these effects-an updated review of propofol fetal exposure in laboratory animal studies. Brain Sci. 6:E11. doi: 10.3390/brainsci6020011
Xue, M., Balasubramaniam, J., Buist, R. J., Peeling, J., and Del Bigio, M. R. (2003). Periventricular/intraventricular hemorrhage in neonatal mouse cerebrum. J. Neuropathol. Exp. Neurol. 62, 1154–1165. doi: 10.1093/jnen/62.11.1154
Yavuz, A., Sezik, M., Ozmen, O., and Asci, H. (2017). Fingolimod against endotoxin-induced fetal brain injury in a rat model. J. Obstet. Gynaecol. Res. 43, 1708–1713. doi: 10.1111/jog.13444
Yawno, T., Mahen, M., Li, J., Fahey, M. C., Jenkin, G., and Miller, S. L. (2017a). The beneficial effects of melatonin administration following hypoxia-ischemia in preterm fetal sheep. Front. Cell Neurosci. 11:296. doi: 10.3389/fncel.2017.00296
Yawno, T., Sabaretnam, T., Li, J., McDonald, C., Lim, R., Jenkin, G., et al. (2017b). Human amnion epithelial cells protect against white matter brain injury after repeated endotoxin exposure in the preterm ovine fetus. Cell Transplant. 26, 541–553. doi: 10.3727/096368916X693572
Yesilirmak, D. C., Kumral, A., Baskin, H., Ergur, B. U., Aykan, S., Genc, S., et al. (2007). Activated protein C reduces endotoxin-induced white matter injury in the developing rat brain. Brain Res. 1164, 14–23. doi: 10.1016/j.brainres.2007.04.083
Yis, U., Kurul, S. H., Kumral, A., Cilaker, S., Tugyan, K., Genc, S., et al. (2008a). Hyperoxic exposure leads to cell death in the developing brain. Brain Dev. 30, 556–562. doi: 10.1016/j.braindev.2008.01.010
Yis, U., Kurul, S. H., Kumral, A., Tugyan, K., Cilaker, S., Yilmaz, O., et al. (2008b). Effect of erythropoietin on oxygen-induced brain injury in the newborn rat. Neurosci. Lett. 448, 245–249. doi: 10.1016/j.neulet.2008.10.060
Zheng, H., Dong, Y., Xu, Z., Crosby, G., Culley, D.J., Zhang, Y., et al. (2013). Sevoflurane anesthesia in pregnant mice induces neurotoxicity in fetal and offspring mice. Anesthesiology 118, 516–526. doi: 10.1097/ALN.0b013e3182834d5d
Zhang, Z., Narayan, S., Su, L., Al-Alawyat, H., Liu, J., and Kannan, S. (2018). Cerebellar injury and impaired function in a rabbit model of maternal inflammation induced neonatal brain injury. Neurobiol. Learn. Mem. 165:106901. doi: 10.1016/j.nlm.2018.07.005
Zhu, J., Qu, Y., Lin, Z., Zhao, F., Zhang, L., Huang, Y., et al. (2016). Loss of PINK1 inhibits apoptosis by upregulating alpha-synuclein in inflammation-sensitized hypoxic-ischemic injury in the immature brains. Brain Res. 1653, 14–22. doi: 10.1016/j.brainres.2016.10.009
Zia, M. T., Csiszar, A., Labinskyy, N., Hu, F., Vinukonda, G., LaGamma, E. F., et al. (2009). Oxidative-nitrosative stress in a rabbit pup model of germinal matrix hemorrhage: role of NAD(P)H oxidase. Stroke 40, 2191–2198. doi: 10.1161/STROKEAHA.108.544759
Keywords: autophagy, apoptosis, necrosis, neonatal, neuroprotection, autophagic cell death, periventricular leucomalacia
Citation: Truttmann AC, Ginet V and Puyal J (2020) Current Evidence on Cell Death in Preterm Brain Injury in Human and Preclinical Models. Front. Cell Dev. Biol. 8:27. doi: 10.3389/fcell.2020.00027
Received: 13 September 2019; Accepted: 14 January 2020;
Published: 18 February 2020.
Edited by:
Ivan Lilyanov Dzhagalov, National Yang-Ming University, TaiwanReviewed by:
Lauren Jantzie, Johns Hopkins University, United StatesCopyright © 2020 Truttmann, Ginet and Puyal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anita C. Truttmann, QW5pdGEuVHJ1dHRtYW5uQGNodXYuY2g=; Julien Puyal, SnVsaWVuUGllcnJlLlB1eWFsQHVuaWwuY2g=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.