 Tatiana V. Denisenko1
Tatiana V. Denisenko1 Anna S. Gorbunova
Anna S. Gorbunova Boris Zhivotovsky
Boris Zhivotovsky- 1Faculty of Medicine, Lomonosov Moscow State University, Moscow, Russia
- 2Institute of Environmental Medicine, Division of Toxicology, Karolinska Institute, Stockholm, Sweden
Mitochondria in addition to be a main cellular power station, are involved in the regulation of many physiological processes, such as generation of reactive oxygen species, metabolite production and the maintenance of the intracellular Ca2+ homeostasis. Almost 100 years ago Otto Warburg presented evidence for the role of mitochondria in the development of cancer. During the past 20 years mitochondrial involvement in programmed cell death regulation has been clarified. Moreover, it has been shown that mitochondria may act as a switchboard between various cell death modalities. Recently, accumulated data have pointed to the role of mitochondria in the metastatic dissemination of cancer cells. Here we summarize the modern knowledge concerning the contribution of mitochondria to the invasion and dissemination of tumor cells and the possible mechanisms behind that and attempts to target metastatic cancers involving mitochondria.
Introduction
Mitochondria are intracellular organelles that produce the majority of the energy in the cells, providing synthesis of ATP by oxidative phosphorylation (OXPHOS) (Mitchell, 1961). Beyond energy production mitochondria have multiple functions including the generation of reactive oxygen species (ROS), metabolite production, the regulation of intracellular Ca2+ homeostasis and modulation of cell death pathways. Additionally, mitochondria contribute to the regulation of signaling pathways linked to the cell proliferation, differentiation, and many others (Porporato et al., 2018). The multiple functions of mitochondria allow cells to adapt to the changing of environment, including the availability of nutrients and oxygen, making them perfect stress sensors (Vyas et al., 2016). These functions also determine the crucial role of mitochondria in development and progression of cancer. Indeed, mitochondria may drive tumor progression through adaptation to changing metabolic demand, contributing to chemoresistance, and regulating cell death pathways (Gogvadze et al., 2008). Furthermore, mitochondria have been shown to be linked to the metastatic dissemination of cancer cells. Importantly, mitochondrial turnover, i.e., fission/fusion, is deeply involved in the regulation of different mitochondrial functions and metastatic cascade. However, the role of mitochondrial dynamics in cancer cell invasion and metastasis remains highly controversial. Here we took an attempt to summarize the present knowledge about the functions of mitochondria that contribute to the metastatic dissemination and invasion including mitochondrial dynamics, cell death, oxidative stress, metabolism and bioenergetics, Ca2+ signaling, and mtDNA (Figure 1). Additionally, we highlight the existing therapy approaches to target metastatic cancers involving mitochondria.
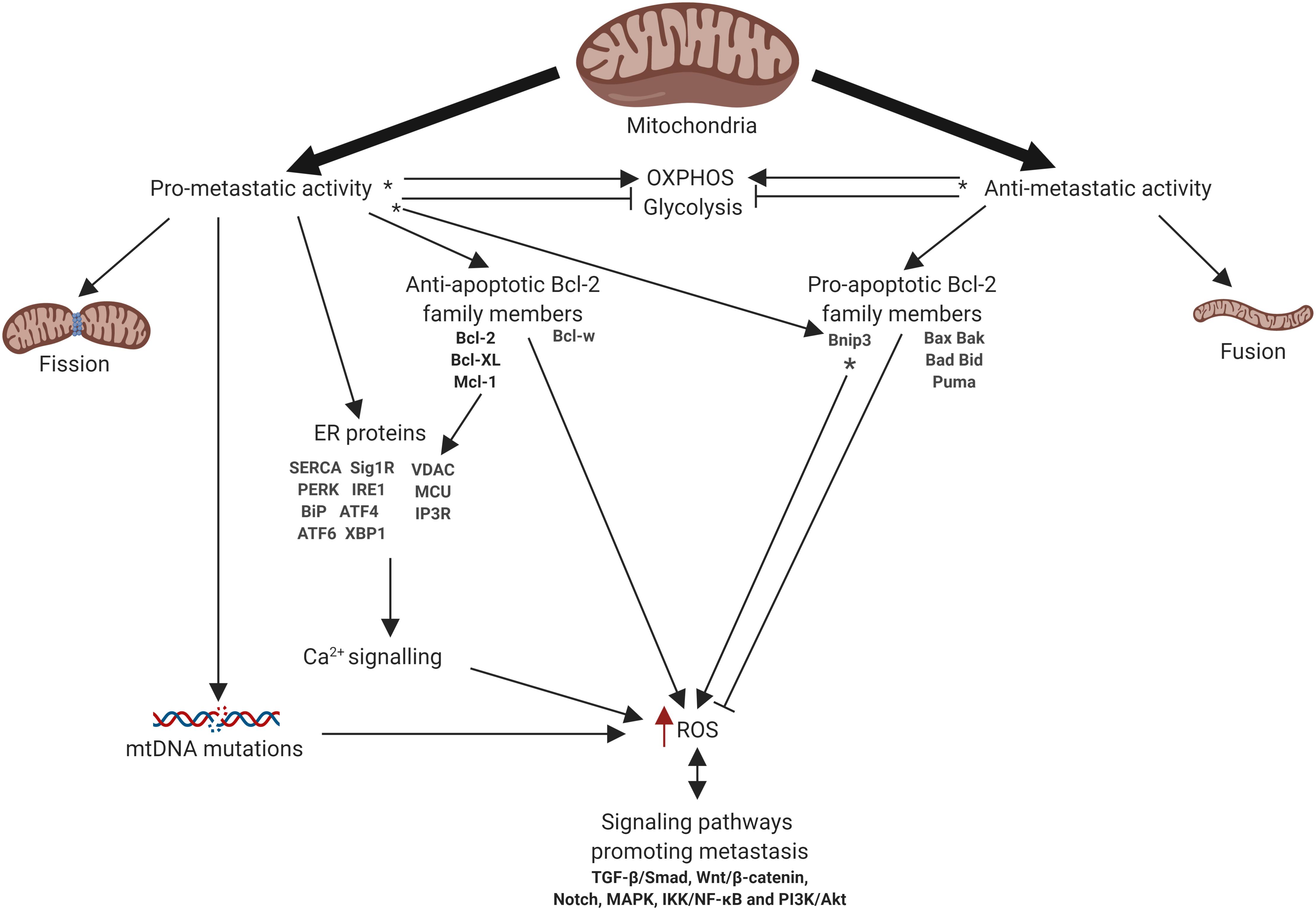
Figure 1. Schematic representation of mitochondrial involvement in metastasis. Arrows or blunt ends indicate activation or inhibition, respectively. Red arrow indicates increased level. ∗ - function depends on the tumor type. OXPHOS, oxidative phosphorylation; ER, endoplasmic reticulum; ROS, reactive oxygen species. For details, see text. Figure is created using BioRender.
Mitochondria and Migration
Metastasis is one of the main cause of cancer patients’ death. Metastatic dissemination is characterized by cell detachment from the primary tumor mass, further migration through blood and lymphatic vessels and colonization of different tissues. The metastatic cascade can be subdivided into different stages, including local invasion, intravasation, survival in the circulation, extravasation, survival at a second site and finally outgrowth at this site. The epithelial to mesenchymal transition (EMT) is a biological phenomenon occurring during embryonic development but also associated with cancer metastasis (Chaffer and Weinberg, 2011). During EMT cancer cells lose their epithelial features and temporally acquire mesenchymal characteristics which allow them to migrate from the original site in order to colonize different tissues.
Epithelial to mesenchymal transition is regulated by various molecular pathways including TGF-β/Smad, Wnt/β-catenin, Notch, mitogen-activated protein kinase (MAPK), IKK/NF-κB and PI3K/Akt (Lamouille et al., 2014), cytokines (e.g., TGF-β and EGF) and transcription factors Snail (Snai1), Slug (Snai2), Twist (helix-loop-helix factor), and ZEB1/2 (zinc-finger E-box-binding homeobox) (Lu and Kang, 2019), etc. Overexpression of EMT transcriptional factors leads to the downregulation of epithelial markers and Tight Junction proteins, such as E-cadherin, occludin, and claudins, which in turn results in the loose of apical cell polarity (Lu and Kang, 2019). On the other hand, EMT activation provides the upregulation of mesenchymal markers: N-cadherin, vimentin, and fibronectin. In addition, EMT is accompanied by increased expression of matrix metalloproteinases (MMPs) and urokinase plasminogen activator (uPA), which contribute to the degradation of the extracellular matrix (ECM) and the basal membrane of epithelial tissue (Lu and Kang, 2019). These events lead to the loss of cell-cell and cell matrix adhesion contacts and an increase in cell motility and cell migration, which are the hallmarks of metastasis (Chaffer and Weinberg, 2011).
Mitochondrial Dynamics and Migration
Mitochondria have been shown to contribute to carcinogenesis including metastatic dissemination and EMT by different mechanisms. Being extremely dynamic organelles mitochondria continuously change their morphology undergoing fission (fragmentation) and fusion (elongation). These processes are regulated by highly conserved guanosine triphosphatases (GTPases) (Senft and Ronai, 2016). Fission is controlled by cytosolic dynamin-related protein 1 (Drp1), which is recruited to mitochondria by adapter proteins, including mitochondrial fission factor (Mff) and mitochondrial dynamics proteins of 49 and 51 kDa (Mid49/51), where it forms oligomeric ring structures and executes mitochondrial fission. Fusion is mediated by two GTPases in the outer mitochondrial membrane (OMM), i.e., mitofusins 1 and 2 (Mfn1, 2), whereas the inner mitochondrial membrane (IMM) fusion is promoted by the cristae-shaping protein Opa1 (Losón et al., 2013). In cancer cells, mitochondrial fission/fusion is unbalanced due to the mitochondrial dysfunction (Srinivasan et al., 2017). Several studies have demonstrated that increased fission and/or reduced fusion are associated with malignant transformation in different types of cancer (Senft and Ronai, 2016). Furthermore, upregulation of mitochondrial fission and increased expression of Drp1 was shown to promote cancer metastasis (Zhao et al., 2013; Sun et al., 2018a). For example, overexpression of Drp1 was detected in breast cancer metastatic cells compared to the non-metastatic, whereas silencing of Drp1 or overexpression of Mfn1 resulted in mitochondrial elongation and significantly suppressed the metastatic properties of breast cancer cells (Zhao et al., 2013). Similarly, increased mitochondrial fission was observed in hepatocellular carcinoma (HCC) metastatic cells. Comparison of levels of Drp1 in tumor samples and in the normal tissues revealed its higher expression in the former, which is associated with the promotion of tumor cell survival and metastasis formation (Sun et al., 2018a). In addition, downregulation of Drp1 inhibits glioma cells invasive properties affecting cytoskeleton remodeling through the RhoA/ROCK1 pathway (Yin et al., 2016). Recent data have also suggested the existence of a link between mitochondrial fission and hypoxia-induced migration. The inhibition of Drp1 by Mdivi-1 leads to the decreased migration induced by hypoxia (Han et al., 2015). Taken together, these studies provide the evidence that mitochondrial fission is required for cancer cell migration and to support the metastatic potential of cancer cells. In migrating cancer cells, mitochondria localize at the leading edge along microtubules, where the energy demand is higher, providing necessary supply (Senft and Ronai, 2016). Unfortunately, the mechanism involving Drp1 in regulation of the process of cancer metastasis remains not fully understood. Different studies suggest that fission is required for efficient redistribution of mitochondria, and the upregulation/activation of Drp1 is associated with the migration of cancer cells (Zhao et al., 2013; Senft and Ronai, 2016). Another study has shown that inhibition of mitochondrial fusion may abolish invasion of syntaphilin-depleted prostate adenocarcinoma cells. Syntaphilin suppresses mitochondrial dynamics, cancer cell dissemination in vivo. Moreover, its downregulation correlates with poor outcome of cancer patients. Thereby, the silencing of both Mfn1, 2 and syntaphilin abolished mitochondrial trafficking and abrogated the migratory response (Caino et al., 2016). Thus, mitochondrial dynamics are linked to cancer cell migration, and the relative contribution of fission or fusion depends on tumor type and molecular context.
ROS Contributes to Migration and Metastasis
Reactive oxygen species constantly generated during the metabolic process and play a crucial role in the regulation of various cellular functions (Vyas et al., 2016). Mitochondrial electron transport chain (ETC) is the main source of ROS (Vyas et al., 2016). Complexes I and III are often regarded as the major sites of mitochondrial ROS (mtROS) production, but more recent studies indicate that at least ten other mitochondrial enzymes also contribute to ROS generation, including Complex II (Quinlan et al., 2013). The role of ROS in cancer remains highly controversial. First, in cancer cells a higher level of ROS is detected compared to their normal counterparts (Cannito et al., 2010; Vyas et al., 2016). A moderate increase of ROS level was shown to support cancer cell proliferation and migration and to activate different signaling pathways associated with cell survival, contributing to tumor growth and malignant transformation (Kumari et al., 2018). Indeed, the level of ROS has been shown to activate the PI3K pathway. The primary known ROS target in the PI3K pathway is phosphatase and tensin homolog (PTEN). ROS promote the inactivation of the tumor suppressor PTEN by oxidizing active-site cysteine residues, causing the formation of a disulfide bond, which prevents PTEN from inactivating the PI3K pathway (Lee et al., 2002; Sullivan and Chandel, 2014). Since ROS can inactivate protein tyrosine phosphatases through oxidation of cysteine residues, ROS may have many yet-to-be discovered effects on diverse, mitogen-activated pathways that are normally inhibited by phosphatases (Sullivan and Chandel, 2014). ROS can stimulate the phosphorylation of MAPK and extracellular signal-regulated kinase (ERK), cyclin D1 expression and JUN N-terminal kinase (JNK) activation, all of which are linked to tumor cell survival and growth (Gorrini et al., 2013). ROS may activate different processes associated with metastatic dissemination and invasion. They may be involved in cytoskeleton remodeling. The cell cytoskeleton is dynamic structure composed of microtubules and filaments. Cytoskeletal rearrangements are important for driving cell migration and invasion through the formation of different types of cellular protrusions including filopodia, lamellipodia, and invadopodia (Ridley, 2011). Recent studies have shown that Rac-mediated actin remodeling is attributed to increased O2– levels (Jiang et al., 2017). Specifically, Rho activation leads to the filopodia formation, while induction of Rac contributes to the formation of lamellipodia (Kozma et al., 1995; Narumiya et al., 2009; Galadari et al., 2017). Another mechanism by which ROS may promote tumor cell invasion is by stimulation of the proteolytic degradation of ECM components such as glycosaminoglycan (GAG), contributing to metastatic dissemination (Galadari et al., 2017).
Increased ROS levels can activate different pathways that induce morphological changes associated with the EMT (Jiang et al., 2017). For example, increased ROS generation stimulates the acquisition of invasive properties by pancreatic cancer cells through the activation of NF-κB signaling. In turn, treatment with antioxidants leads to the suppression of EMT and attenuates metastasis (Shimojo et al., 2013). NF-κB signaling is strongly associated with the EMT process by promoting the expression of the main EMT-related transcription factors Snail, Slug, Twist1, and ZEB1/2, which is also involved in the disruption of the cell–cell junctions (Min et al., 2008; Jiang et al., 2017). Furthermore, NF-κB activation may contribute to the transcription of vimentin and MMPs such as MMP-2, MMP-9, to maintain the mesenchymal phenotype and promote tumor cell migration (Jiang et al., 2017). Another pathway involved in EMT and regulated by ROS is the transcription factor hypoxia inducible factor 1-alpha (HIF-1α) (Jiang et al., 2017), which is induced under hypoxic conditions and can stimulate cancer cell EMT by activating EMT-inducing transcription factors such as Twist, Snail and ZEB1/2 (Joseph et al., 2015; Zhang et al., 2015). Thus, NF-κB is activated under hypoxic conditions, and thereby, in the presence of hypoxia, may co-regulate many of EMT-linked transcription factors (D’Ignazio et al., 2017). Importantly, ROS accumulation leads to the stabilization of HIF-1 due to inhibition of the HIF-degrading enzyme prolyl hydroxylase (Comito et al., 2011).
There is a complex interplay between the level of ROS and the TGF-β signaling pathway exists, which is the one of the most important pathways involved in EMT regulation. It was reported that ROS mediate TGF-β-induced EMT in cancer (Corcoran and Cotter, 2013; Liu and Desai, 2015). ROS may affect the activation of TGF-β downstream effector Smad, while treatment with the ROS scavenger N-acetyl cysteine (NAC) abolishes Smad phosphorylation (Krstiæ et al., 2015). Additionally, ROS may regulate TGF-β activation through different signaling pathways as described above, including MAPK and NF-κB (Corcoran and Cotter, 2013). Conversely, TGF-β can induce ROS production by many alterations in mitochondrial functioning and antioxidant protection. For example, TGF-β affects ROS levels by blocking of ETC Complex IV and upregulation of NADPH oxidase 4 (NOX4) (Yoon et al., 2005). Further, it was revealed that TGF-β increases ROS levels inhibiting ETC Complex III (Jain et al., 2013). TGF-β also downregulates the synthesis of the antioxidant glutathione (GSH) and several antioxidant enzymes contributing to cellular redox misbalance and EMT-related processes, such as fibrosis (Liu and Desai, 2015). Using the mitochondria-targeted antioxidant SkQ1 was also shown that oxidative stress is implicated in EMT induced by TGF-β. In cervical carcinoma SiHa cells depletion of ROS leads to increase of E-cadherin and downregulation of Snail, the main negative regulator of E-cadherin (Shagieva et al., 2017). Similarly, pretreatment with the ROS scavenger carotenoid astaxanthin (AST) leads to the suppression of EMT and the production of inflammatory cytokines by mesothelial cells (Hara et al., 2017). Thus, TGF-β, as inducer of EMT, is likely to affect this process through the ROS production.
Reactive oxygen species accumulation may influence migration and metastasis of cancer cells through different mechanisms affecting cytoskeleton remodeling, ECM degradation and the activation of signaling pathways. However, in conditions of strong oxidative stress, ROS suppress metastatic dissemination, due to induction of cell death or cellular senescence (Piskounova et al., 2015). Furthermore, elevated ROS levels may activate antioxidant pathways (Vyas et al., 2016). Indeed, oncogenic K- Ras-, B- Raf-, and c-Myc-mediated pathways may downregulate ROS production through regulation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2), one of the main regulators of the antioxidant response (Vyas et al., 2016). Nrf2 provides a transcriptional activation of several genes involved in glutathione (GSH) synthesis. It promotes tumorigenesis contributing to the cancer cell protection against oxidative stress and chemotherapeutic agents (Rojo de la Vega et al., 2018). Recent data have demonstrated that Nrf2 activation can stimulate cancer cell migration and metastasis and Nrf2 deletion attenuates metastatic potential breast cancer cells suppressing RhoA GTPases activity (Zhang et al., 2016). Altogether, these observations demonstrate that the role of ROS in tumor progression and metastasis remains highly controversial. It has been suggested that tumors should maintain the ROS at a definite level in order to sustain their growth and metastasis without causing cytotoxicity (Vyas et al., 2016). Moreover, for tumor promotion it also necessary to provide the right balance between ROS production and antioxidants.
Mitochondrial DNA Mutations Contribute to the Migration and Metastasis
It is known that mtDNA mutations can contribute to tumor initiation and progression (Vyas et al., 2016). Variations in copy number of mtDNA are associated with tumorigenesis and depend on tumor type (Sun et al., 2018b). Thus, decreased copy number of mtDNA was detected in breast cancer, HCC, non-small cell lung cancer (NSCLC) and gastric cancer (Mambo et al., 2005). On the other hand, increased copy number of mtDNA was found in prostate, head and neck, and colorectal cancers (Sun et al., 2018b). Mutations and variations in mtDNA content might be associated with regulation of the metastatic properties of tumor cells (Ishikawa et al., 2008). Replacement of mtDNA from a highly metastatic to a poorly metastatic cell line led to an increase in the metastatic potential in the recipient cell line (Ishikawa et al., 2008). mtDNA mutations are also associated with EMT of cancer cells. Indeed, EMT induced by TGF-β leads to an increase of mtDNA copy number in NSCLC cells (Xu and Lu, 2015). Conversely, knockdown of mitochondrial transcription factor A (TFAM) leads to a decrease in mtDNA copy number, upregulation of E-cadherin expression, and suppression of cell migration rate in esophageal squamous cell carcinoma (Lin et al., 2012). Increased mtDNA content in this type of tumor is associated with the higher energy required for EMT. Furthermore, mtDNA mutations contribute to the acquisition of an aggressive phenotype in oncocytic thyroid tumors leading to their bioenergetic crisis (De Luise et al., 2017). As a consequence, mitochondrial dysfunction may lead to the activation of glycolysis (Smolková et al., 2011). Thus, oxygen deprivation may provide positive selective pressure for cancer cells carrying damaging mtDNA mutations. However, another study provided the evidence that EMT could also be induced in mtDNA-depleted cells. Indeed, it has been demonstrated that TGF-β–induced EMT occurs in mitochondria-depleted cell lines leading to the stimulation of invasive properties through activation of Raf/MAPK (Naito et al., 2008). These data are consistent with the observation that in human mammary epithelial cells (hMECs) a decrease in mtDNA copy number promotes calcineurin-mediated mitochondrial retrograde signaling, which initiates EMT (Guha et al., 2014). Likewise, reduction of mtDNA content by suppression of mitochondrial pyrimidine nucleotide carrier 1 (PNC1), which is responsible for mitochondrial DNA replication, leads to EMT induction in hMECs (Favre et al., 2010). A recent study has revealed that increased mtDNA copy number may sustain tumor progression and metastasis by upregulating OXPHOS function in cancer cells that rely on mitochondrial OXPHOS. On the other hand, in cancer cells that depend on glycolytic type of metabolism reduction of mtDNA was shown to promote proliferation and chemoresistance (Sun et al., 2018b). Summing up, mtDNA mutations and variations of mtDNA copy number are associated with EMT, increased invasiveness and metastasis in different types of cancer. The opposite role of mtDNA content in cancer progression and metastatic dissemination depends on metabolic pattern of different types of cancer.
Bcl-2 and Metastasis
B-cell lymphoma/leukemia gene 2 (Bcl-2) family proteins are considered to be regulators of the apoptotic mitochondrial pathway. This family includes both anti-apoptotic Bcl-2, Bcl-XL, Bcl-w, Mcl-1, Bcl-B, and pro-apoptotic multidomain Bax and Bak proteins. In addition, the pro-apoptotic subfamily includes so-called BH3-only domain proteins, such as Bim, Puma, Noxa, Bad, Bid, and Bnip3. The ratio between these proteins with opposite functions determines the success of apoptosis (Adams and Cory, 2018). Upregulation of anti-apoptotic and downregulation of pro-apoptotic proteins is a hallmark of cancer, and their misbalance is contributed to the chemo-, immune-, and radio-resistance of anticancer therapies (Opferman and Kothari, 2018). However, further evidence has demonstrated that the functions of Bcl-2 family proteins are not limited to cell death control and tumor resistance. It has been established, that Bcl-2 family proteins play crucial roles in the regulation of migration, invasion and metastasis (Um, 2016) (Figure 2).
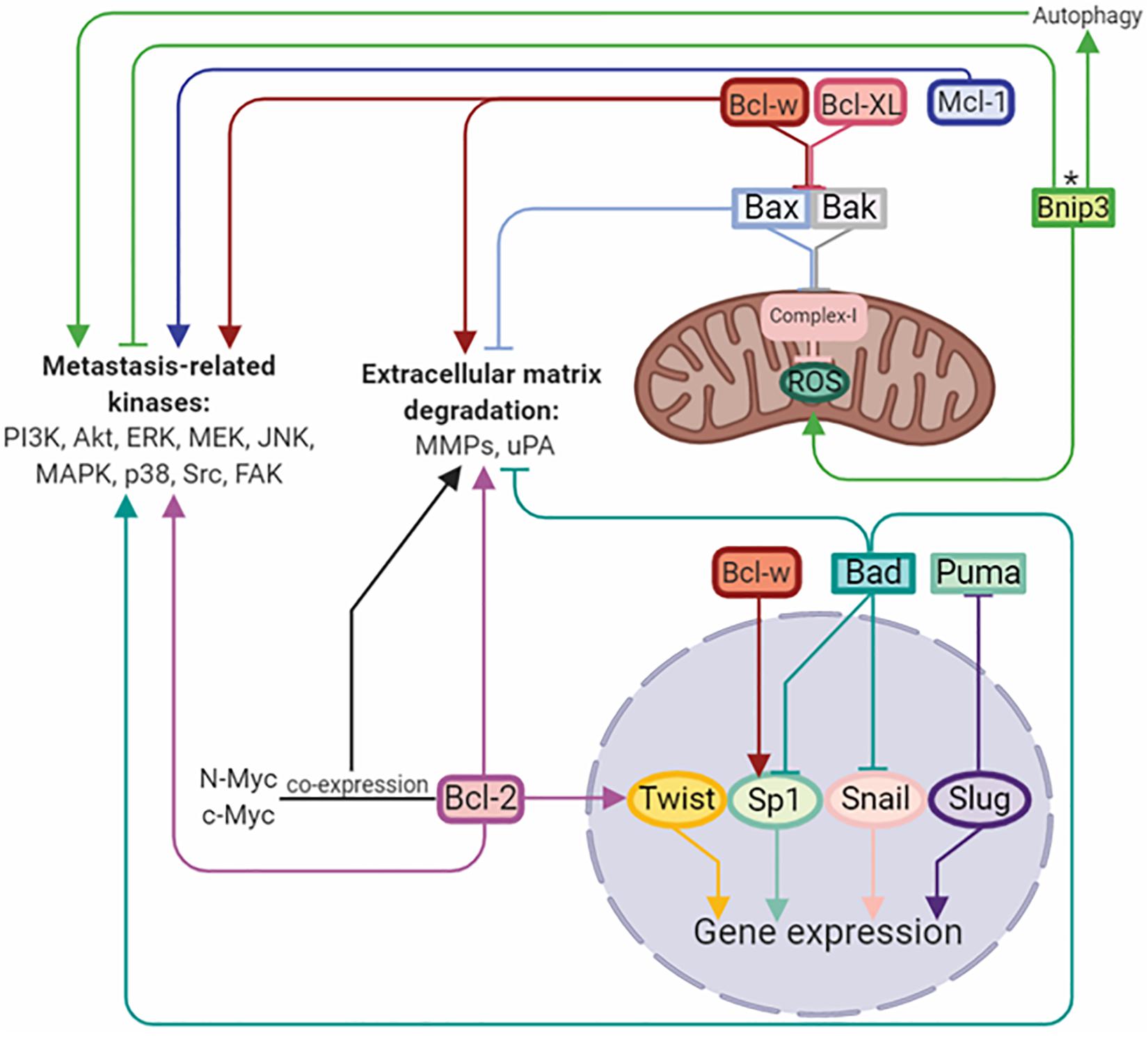
Figure 2. Bcl-2 family members regulate metastasis by activation/inhibition of signaling kinases, matrix-degrading enzymes, and transcriptional factors. Arrows or blunt ends indicate activation or inhibition, respectively. Colored lines corresponds to each protein. ∗ - function depends on the tumor type. Bcl-2, B-cell lymphoma 2; Mcl-1, myeloid cell leukemia 1; Bcl-XL, B-cell lymphoma-extra large; Bcl-w, Bcl-2-like protein 2; Bnip3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; Bax, Bcl-2-like protein 4; Bak, Bcl-2 homologous antagonist/killer; Bad, Bcl-2-associated death promoter; Puma, p53 upregulated modulator of apoptosis; Twist, class A basic helix-loop-helix protein 38; Sp1, specificity protein 1; Snail, zinc finger protein SNAI1; Slug, zinc finger protein SNAI2. For details, see text. Figure is created using BioRender.
Indeed, the overexpression of Bcl-2, Bcl-XL, Bcl-w, and Mcl-1 in different cancers, including glioma, neuroblastoma, melanoma, squamous carcinoma, and breast, lung, and colorectal cancer cells, leads to significant increase in their migratory and invasive properties (Sun et al., 2011; Lee et al., 2013; Um, 2016; Young et al., 2016; Trisciuoglio et al., 2017). On the other hand, downregulation of these proteins attenuates invasiveness without affecting apoptosis or tumor growth, indicating that their pro-survival functions are not linked to regulation of cell migration and invasion (Um, 2016). Importantly, several studies have demonstrated that overexpression of the Bcl-2 family proteins is not always sufficient to induce pro-invasive properties of cancer cells, and could require the co-expression of other proteins stimulating invasiveness, such as c-Myc (Lu and Hong, 2009), N-Myc (Noujaim et al., 2002), or Twist1 (Sun et al., 2011). In certain cases, the exposure to hypoxic conditions may also be essential (Trisciuoglio et al., 2005). It has been suggested that the pro-invasive activity of pro-survival Bcl-2 family members appears to vary depending on the cell type and environment (Um, 2016).
The mechanisms involved in Bcl-2 proteins-mediated regulation of invasiveness and metastasis remain incompletely understood. Anti-apoptotic Bcl-2 family proteins have been shown to activate different signaling pathways controlling migration, invasiveness and metastasis ability in cancer. Indeed, Bcl-2 may modulate the EMT program by direct interaction with Twist1 through the helix-loop-helix DNA binding domain of Twist1 and two domains of Bcl-2 in hepatocellular and oral squamous cells (Sun et al., 2011; Duan et al., 2017). In addition, almost all anti-apoptotic Bcl-2 family members regulate the PI3K pathway involved in metastasis progression (Um, 2016). Interplay between Bcl-2 and the p110α subunit of PI3K regulates human colorectal cancer cell migration through actin polymerization and filopodia formation (Wan et al., 2015). Likewise, in lung cancer cells, Bcl-XL increases PI3K and p38 MAPK activities, which subsequently stimulate MMP-2 expression via Akt (Ho et al., 2010). Specifically, Bcl-w was shown to affect migration and invasion pathways through regulation of PI3K, EGF, Src, MMP-2, uPA, and focal adhesion kinase (FAK) (Um, 2016). Furthermore, Bcl-w promotes migration and invasion of glioblastoma cells through β-catenin signaling via its translocation into the nucleus to act as transcription factor for MMP-2 and mesenchymal marker expression (Lee et al., 2013). Additionally, Mcl-1 supports breast cancer cell migration and invasion via Src family kinases (SFKs) and their targets, and also by alteration of the phosphorylation state of the cytoskeletal protein cofilin (Young et al., 2016).
It has been reported that Bcl-2 family members are capable of regulating the functioning of mitochondria, during cellular respiration and of stimulating ROS generation in the form of the superoxide anion radical O2– and H2O2 (Um, 2016). Thus, multidomain pro-apoptotic proteins Bax and Bak have been shown to bind to the ETC Complex-I resulting in decreased ROS production, whereas anti-apoptotic Bcl-w and Bcl-XL interact with Bax and Bak, and abolish their binding to the Complex-I, stimulating ROS production and PI3K-, Src-, and EGFR-dependent cell migration and invasion (Jung et al., 2018; Kim et al., 2018).
Analysis of patients’ clinical samples confirmed the involvement of pro-survival Bcl-2 family members in metastasis. Bcl-2 expression is associated with lymph node metastases of bladder (Kiss et al., 2015) and gastric (Geng et al., 2013) cancer, liver metastases of colorectal cancer (Ishijima et al., 1999) and lymphovascular invasion of patients with breast cancer (Neri et al., 2006). Upregulation of Bcl-XL was observed in lymph node metastases and venous permeation in colorectal cancer (Jin-Song et al., 2011), hematogenous metastases of osteosarcoma patients (Wang et al., 2010) and lymph node metastases in oral tongue squamous cell carcinoma (Zhang et al., 2014). Overexpression of Bcl-w is associated with the infiltrative morphotypes of gastric cancer (Lee et al., 2003) and is overexpressed in patients with lung and breast cancers (Kim et al., 2019). Mcl-1 is upregulated among III–IV a stage of esophageal squamous cell carcinoma patients with lymph node metastases (Xu et al., 2017).
Conversely, pro-apoptotic proteins of this family may suppress cancer cell invasion and metastatic dissemination. Thus, Bax and Bad expression is associated with downregulation of MMP-2, -9, and -10 (Lee et al., 2010; Cekanova et al., 2015). Interestingly, Bax and Bid may downregulate tumor cell invasiveness, indirectly repressing the gene expression of c-Jun, cyclin D1, β-catenin, and Sp1, which are known to stimulate invasive properties and metastasis in breast cancer (Cekanova et al., 2015). In addition, Bad, Bim, and Puma were shown to suppress EMT, inhibiting related transcription factors, including Snai1, Sp1, Snai2, and Slug, and subsequent upregulation of epithelial phenotype markers (Kim et al., 2014; Cekanova et al., 2015; Merino et al., 2015).
Another member of the Bcl-2 family implicated in cancer cell migration, invasion and metastasis is Bnip3 (Maes et al., 2014; Chourasia et al., 2015), which is considered to be a pro-apoptotic Bcl-2 protein. Bnip3 also plays an important role in autophagy and mitophagy regulation (Chourasia et al., 2015). However, its role in cancer progression and metastasis remains highly controversial. Thus, in human triple-negative breast cancer (TNBC) the lack of Bnip3 results in tumor progression and metastasis via storage of dysfunctional mitochondria and subsequent ROS accumulation; the events that, as was discussed earlier, lead to expression of HIF-inducible genes including metastasis-related angiogenesis genes (Chourasia et al., 2015). Conversely, in melanoma cells Bnip3 silencing reduces the formation of lamellipodia and filopodia as well as cell migration through the downregulation of integrin-associated glycoprotein CD47, Rac1 and Cdc42 (Maes et al., 2014). Bnip3 in HCC may suppress metastasis through the JNK/Bnip3/SERCA/CaMKII axis, leading further to the cofilin/F-actin/lamellipodia inhibition (Shi et al., 2018). As mentioned above, Bnip3 is involved in autophagy, the process, which is tightly linked to EMT, migration and metastasis. Specifically, Bnip3-dependent autophagy via hypoxia-induced ROS-modulated p38 MAPK and JNK activation contributes to keratinocytes migration (Zhang et al., 2019). In NSCLC Bnip3 supports metastasis via its modulation by aryl hydrocarbon receptor (AhR), which has an impact on Bnip3 proteasomal degradation and subsequent autophagy disturbance. These events result in decreased EMT progress (Tsai et al., 2017). Thus, Bnip3, depending on cancer cell type and hypoxic conditions, fulfills opposite functions in metastasis.
Analysis of clinical samples confirmed the participation of Bcl-2 pro-apoptotic members in metastasis formation. Increased expression of Bax, Bak and Puma is associated with a lack of vascular invasion in patients with oral squamous cell carcinoma (Coutinho-Camillo et al., 2010). Loss of Bax protein was demonstrated in retinoblastoma specimens with massive choroidal invasion (Singh et al., 2015). Downregulation of Bnip3 is characteristic of lymph node metastases in breast cancer (Koop et al., 2009). Conversely, in patients with renal cell carcinomas Bnip3 expression is correlated with lymph node metastasis (Macher-Goeppinger et al., 2017). Methylation of Bim and Bnip3 genes is associated with metastasis and the gene methylation rate is increased among colorectal and pancreatic cancer patients compared to healthy individuals (Shimizu et al., 2010; Mhaidat et al., 2017; Zhu et al., 2017). Interestingly, overexpression of pro-apoptotic Bcl-2 family members, except Bnip3, frequently correlates with decreased metastasis and favorable outcomes in patients with various cancer types (Chi et al., 2016).
Thus, anti-apoptotic members of the Bcl-2 family, including Bcl-2, Bcl-XL, Bcl-w, and Mcl-1, support the invasion and metastasis in various types of cancer. This positive influence is achieved through EMT, subsequent cytoskeleton rearrangement, overexpression of MMPs and uPA, and regulation of PI3K, p38 MAPK, Akt, and ERK. In contrast to anti-apoptotic proteins, almost all pro-apoptotic members, such as Bax, Bak, Bad, Bid, and Puma, were characterized by suppression of the metastatic potential of cancer cells. Despite Bnip3 being considered a pro-apoptotic member, its influence on metastasis remains highly controversial, likely due to its atypical BH3-domain contributing to autophagy-dependent processes (Mazure and Pouysségur, 2009). Bcl-2 family members play important role in the regulation of ROS production and activity of mitochondrial complexes leading to the activation of molecular pathways controlling invasion and metastasis. Moreover, the Bcl-2 family proteins affect cell migration of both malignant and normal tissues. Analysis of patient specimens with tumors confirms the participation of Bcl-2 family members in invasion and metastasis, which gives a reason to consider these proteins for target therapy.
ER-Mitochondria Network and Metastasis
The endoplasmic reticulum (ER) is a crucial cellular Ca2+ reservoir, that coordinates Ca2+ signaling, protein synthesis and folding and traffic of properly folded proteins to the Golgi apparatus. Accumulation of misfolded proteins in the ER lumen triggers unfolded protein response (UPR), which is an adaptive signaling pathway to restore protein homeostasis (proteostasis). If accumulation of misfolded proteins remains unresolved activation of UPR signaling may lead to the initiation of apoptotic cascades. Crosstalk between apoptosis and UPR is maintained by mitochondria functioning partly due to contact sites with ER. These so-called mitochondria-associated ER membranes (MAMs) are key for Ca2+ transport between the ER and mitochondria to maintain cellular homeostasis and regulate ER stress. Moreover, MAMs form functional networks essential in determining pro-survival/pro-death and inflammation signaling (Malhotra and Kaufman, 2011).
As mentioned above, ER is a multifunctional organelle, the main function of which is to control protein-folding quality. Numerous factors may affect proper protein folding in the ER, including oxidative stress, hypoxia, glucose deficiency, viral infections and other physical/chemical stresses. As a result, it leads to ER stress and subsequent UPR. In mammals the UPR is carried out by three distinct ER-related transmembrane proteins, including protein kinase RNA-like ER kinase (PERK), endoribonuclease inositol-requiring enzyme 1 alpha (IRE1α/IRE1), and activating transcription factor 6 (ATF6) (Malhotra and Kaufman, 2011). In unstressed cell, ER-related transmembrane proteins including PERK, IRE1α/IRE1, and ATF6 are bound to immunoglobulin heavy chain protein/glucose-regulated protein 78 (BiP/GRP78). Under ER stress BiP dissociates from these proteins to trigger signaling pathways that result in the reduction of global protein synthesis, degradation of unfolded proteins and increase of protein-folding capacity of the ER (Lee, 2005).
Endoplasmic reticulum plays an important role in mitochondrial calcium signaling via the contact sites between mitochondria and ER (MERCs). The portion of membranes involved in these interactions defines the MAMs, which, as mentioned above, provide Ca2+ traffic between these organelles (Martinvalet, 2018). The transport of extracellular Ca2+ into the cytosol occurs through the voltage-gated, ligand-gated, and store-operated Ca2+-channels (SOCCs) including Orai and/or transient receptor potential channels (TRPC). Orai1 and TRPC are activated through their binding to the stromal interaction molecule 1 (STIM1), which is the ER Ca2+-sensor. Ca2+ transport from the cytosol and its accumulation in the ER depends on the action of ATP-driven sarco/ER Ca2+-ATPase (SERCA) (Bower et al., 2017). Ca2+ is transported from the ER via 1,4,5-trisphosphate (IP3) and ryanodine receptors (IP3Rs, RyRs), after which Ca2+ invades the mitochondria through the voltage-dependent anion channels (VDACs) on the OMM (Báthori et al., 2006). Ca2+ then is transferred by the mitochondrial calcium uniporter (MCU) on the IMM (Martinvalet, 2018). Expression levels of these calcium-signaling proteins are frequently altered in numerous types of tumor cells (Singh et al., 2017) and most of all govern metastasis-related processes.
As mentioned above, ER is the main cellular Ca2+-store. Decreased Ca2+ level in the ER results in STIM1 oligomerization and its transfer from ER to the plasma membrane where it promotes Orai1-dependent store-operated Ca2+ entry (SOCE) (Yang, 2018). A growing body of evidence indicates the existence of an interplay between mitochondria and SOCE. Indeed, SOCE activation is accompanied by TRPC-modulated increase in cytosolic Na+ level that in turn promotes the activation of mitochondrial Na+/Ca2+ exchanger (NCLX) leading to mitochondrial Na+ influx and Ca2+ efflux. Therefore, NCLX tightly regulates mitochondrial Ca2+ level and prevents excessive Ca2+ accumulation in mitochondria that can lead to the increase of the mtROS level and subsequent SOCE suppression via oxidation of redox-sensitive Cys195 of Orai1 (Ben-Kasus Nissim et al., 2017). Besides NCLX, the activity of SOCE-related proteins is regulated by Bcl-2. The mutations in BH1 domain of Bcl-2 protein leads to STIM1, Orai1-3, TRPC1 overexpression and SOCE enhancement (Chiu et al., 2018). It has been established, that hyperactive SOCE induced by STIM1 and Orai1 overexpression correlates with increased metastasis in different types of cancer. Intensified SOCE supports tumor cell invasion and migration by cytoskeleton rearrangement, ECM degradation and tumor microenvironment remodeling (Yang, 2018).
Sarco/ER Ca2+-ATPase is a well-known regulator of Ca2+ stores in the ER and maintains the level of Ca2+ uptake and leak properties. Furthermore, SERCA inactivation associated with Yap deficiency has been shown to inhibit HCC metastasis through the cofilin/F-actin/lamellipodium pathway (Shi et al., 2018). Additionally, the downregulation of SERCA leads to a significant decrease in Ca2+ level in migrating cells that in turn inhibits cell migration and tracheogenesis (Bower et al., 2017). Other proteins localized to MAMs are VDAC and IP3R, the action of which is dependent on Bcl-2 family proteins and, in addition to their role in apoptosis, partly regulate Ca2+ signaling via their complex with MAM proteins (Monaco et al., 2015; Bittremieux et al., 2019). Anti-apoptotic family members Bcl-2, Bcl-XL, and Mcl-1 bind to VDACs and suppress mitochondrial Ca2+ transport that in turn supports cell migration and invasion (Huang et al., 2013, 2014; Fouqué et al., 2016). Both Bcl-2 and Bcl-XL interact with VDAC1 through BH4 domain; however, Bcl-XL BH4 is more effective than Bcl-2-BH4 in targeting VDAC1 activity (Monaco et al., 2015). Dissociation between these anti-apoptotic Bcl-2 family members and VDACs results in decreased migration of TNBC (Fouqué et al., 2016) and NSCLC cells (Huang et al., 2014). Bcl-2 family proteins could also interact with IP3R suppressing Ca2+-release. Like VDACs, Bcl-2 binds IP3R through its BH4 domain inhibiting its activity. Besides Bcl-2, other anti-apoptotic family members, including Bcl-XL and Mcl-1, are able to influence IP3R activity and Ca2+ signaling, but their role in mitochondria-associated ER membrane-related calcium signaling still remains controversial (Eckenrode et al., 2010; Monaco et al., 2012). Also like VDACs, IP3R may regulate cell migration separately from its complex with Bcl-2 family members. It has been revealed, that inhibition of ryanodine receptor subtype IP3R3 and subsequent decrease in Ca2+ release results in suppression of the invasion and migration of glioblastoma cell lines and metastasis in glioblastoma mouse model (Kang et al., 2010). Overexpression of IP3R3, but not of IP3R1 and IP3R2, leads to stimulation of the migration properties of breast cancer cells sustaining Ca2+ signaling (Mound et al., 2017). Thus, IP3R could regulate cancer cell migration and metastasis through modifying calcium ER level.
Another MAM-related protein is Sig1R (stress-activated chaperone sigma-1 receptor). When ER stress is not activated, Sig1R cooperates with MAMs chaperone BiP/GRP78, whereas under activation of IP3Rs Sig1R dissociates from chaperone BiP and binds to IP3R3, leading to its stabilization at the MAM and increasing Ca2+ flux to the mitochondria (De Pinto and Palmieri, 1992; Naon and Scorrano, 2014). The expression level of MAM-associated Sig1R is increased in metastatic breast and colorectal cancer cells as compared to normal tissues (Gueguinou et al., 2017). Consistently with the above-mentioned MAM-related proteins, MCU also affects migration, invasion and metastasis. Silencing of this uniporter results in decreased mitochondrial Ca2+ level and ROS production, as well as migratory and invasiveness capacities. These findings are in good agreement with in vivo experiment. MCU gene deletion reduces tumor metastasis in TNBC MDA-MB-231 xenografts via HIF-1-dependent gene expression (Tosatto et al., 2016). Yu et al. (2017) demonstrated similar results in breast cancer MCF-7 cells by MCU overexpression, which leads to enhanced migratory and invasiveness potential in vitro and lung metastasis mouse model in vivo. Furthermore, overexpressed MCU was found in specimens from breast cancer patients with metastases. Thus, MCU expression correlates with migration and invasion of cancer cells, as well as with tumor metastasis, which has been proved by both in vitro and in vivo studies. Besides MAM-anchored proteins, UPR regulators, including PERK, IRE1 and BiP/GRP78, all influence the migration and metastasis. PERK as a key UPR sensor, also participates in MAM signaling (Verfaillie et al., 2012). A growing amount of evidence proves that UPR signaling and EMT reprogramming mutually activate each other. In gastric cancer cells knockdown of UPR-related proteins such as PERK, ATF4, and ATF6, decrease TGF-β expression and abrogates EMT under severe hypoxia (Shen et al., 2015). The inverse pattern in this UPR-EMT axis has been demonstrated in both in vivo and in vitro models of breast cancer: cells undergoing EMT have a branched ER structure and activated PERK–eIF2α link of the UPR, which helps cells to metastasize. Analysis of specimens from patients with breast, gastric, colon and lung metastatic tumors revealed correlations between expression of EMT and PERK–eIF2α genes (Feng et al., 2014). Additionally, ATF4, ATF6, another ER-transmembrane protein IRE1 and its-related X-box binding protein-1 (XBP1) play a role in metastatic progression. IRE1α regulates actin cytoskeleton rearrangement and influences the cell migration via filamin A in MEFs, fly, and zebrafish models (Urra et al., 2018). IRE1-XBP1 pathway is regulated by lysyl oxidase-like 2 (LOXL2) overexpression, and activates EMT via Snai1/2, ZEB2 and TCF3 transcription factors in breast carcinoma cells (Cuevas et al., 2017). Notably, XBP1 expression is significantly upregulated in tumor and lymph node metastases compared to normal tissues from patients with oral squamous cell carcinoma, and downregulated XBP1 expression results in decreased cell invasion capacity (Sun et al., 2018c). Thus, proteins of three distinct UPR branches, including PERK, ATF4, ATF6, IRE1, XBP1, and BiP/GRP78, contribute to cancer cell invasive properties and metastatic dissemination regulating MAM signaling (Figure 3).
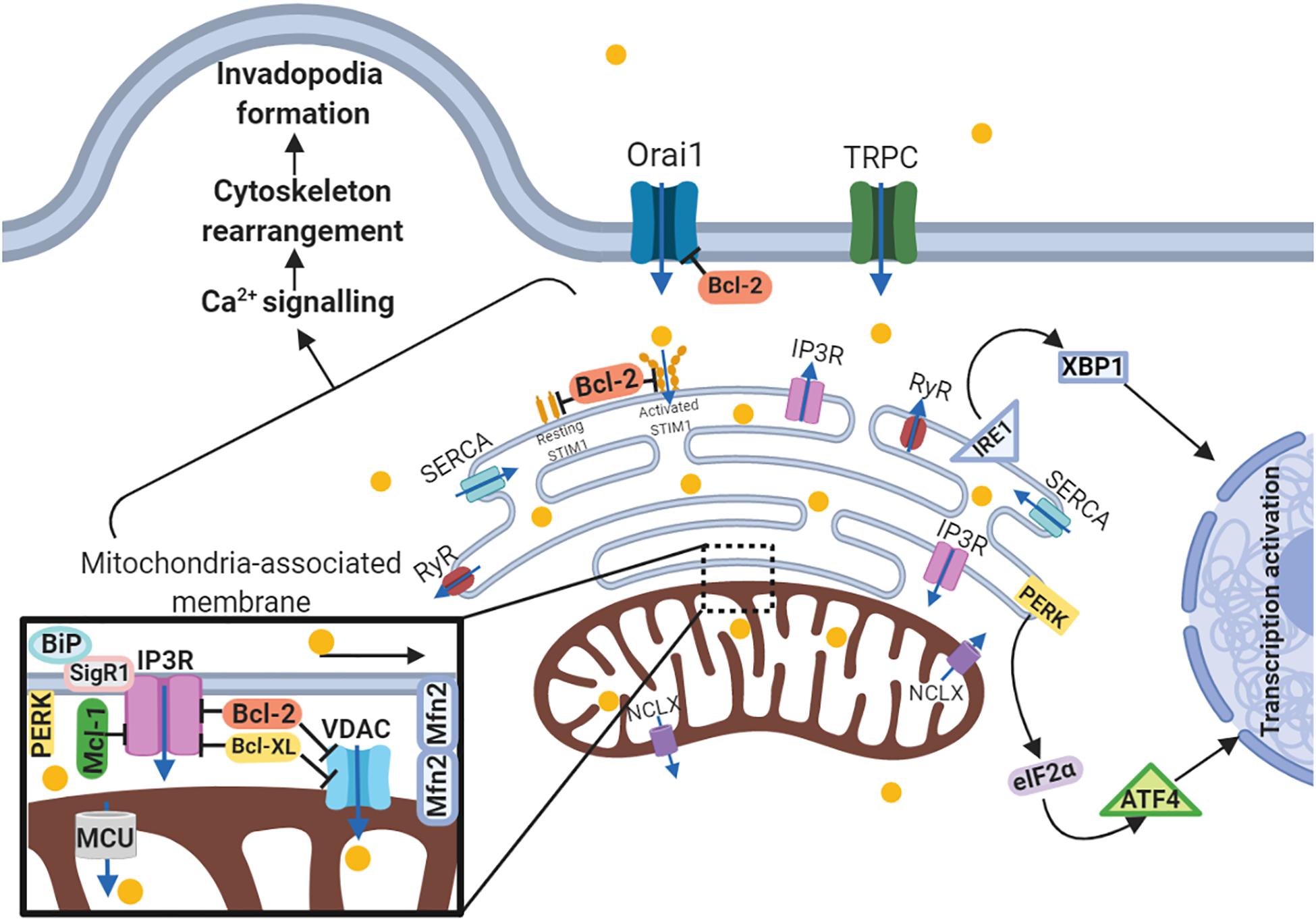
Figure 3. Schematic representation of the link between ER-mitochondria network and motility of cancer cells. Arrows or blunt ends indicate activation or inhibition, respectively. Blue arrows indicate direction of Ca2+ current. Yellow circles – Ca2+. MAM, mitochondria-associated ER membrane; Orai1, calcium release-activated calcium channel protein 1; TRPC, transient receptor potential cation channel; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; STIM1, stromal interaction molecule 1; IP3R, inositol trisphosphate receptor; RyR, ryanodine receptor; IRE1, serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1; XBP1, X-box binding protein 1; PERK, protein kinase RNA-like endoplasmic reticulum kinase; eIF2α, eukaryotic translation initiation factor 2; ATF4, activating transcription factor 4; NCLX, mitochondrial Na+/Ca2+ exchanger; BiP, binding immunoglobulin protein; SigR1, sigma receptor 1; Mfn2, mitofusin2; VDAC, voltage-dependent anion-selective channel; MCU, mitochondrial calcium uniporter. For details, see text. Figure is created using BioRender.
Metabolism and Metastasis
Altered metabolic activity is one of the hallmarks of cancer. Cancer cells change their metabolism in order to satisfy increasing of bioenergetic and biosynthetic demand and maintain tumor growth (DeBerardinis and Chandel, 2016). Unlike normal cells, which generate much of their ATP via mitochondrial-dependent OXPHOS, cancer cells often demonstrate upregulation of glycolysis even under conditions when oxygen concentration is not limited (Lehuédé et al., 2016; Teoh and Lunt, 2018). This phenomenon was observed in the 1920s by Otto Warburg, who demonstrated that tumor tissues metabolize approximately ten-fold more glucose to lactate in a given time than do normal tissues, which led him to conclude that cancer cells rely on glycolysis more than do healthy cells. Enhanced aerobic glycolysis has been detected in many types of cancer and is correlated with worse clinical outcome (Yu et al., 2019). However, further studies have demonstrated that cancer cells may also engage mitochondrial respiration in addition to glycolysis (Jia et al., 2018). Indeed, breast cancer cells produce most of their ATP through mitochondrial oxidation (Park et al., 2016). Similarly, glioma cell lines are strongly dependent on mitochondrial OXPHOS for ATP production (Griguer et al., 2005). Moreover, cancer cells may display distinct metabolic characteristics depending on the tissue of origin (Elia et al., 2015). Thus, lung, liver and colorectal cancers, and leukemias depend on glycolysis, whereas, melanomas, lymphomas, and glioblastomas are characterized as oxidative tumors (Elia et al., 2015; Obre and Rossignol, 2015; Lehuédé et al., 2016). Tumor cells can also switch from one type of metabolism to another under glucose-limiting conditions as observed in cervical cancer, breast carcinoma, hepatoma and pancreatic cancer cells (Rossignol et al., 2004; Beckner et al., 2005; Jose et al., 2011; Smolková et al., 2010). A growing number of studies provide the evidence that cancer cell migration is associated with significant metabolic alterations supporting metastatic dissemination (Morandi et al., 2017; Teoh and Lunt, 2018). Thus, it was reported that increased motility of cancer cells requires the shift toward utilization of glycolytic pathways (Shiraishi et al., 2015). Glycolytic genes activation has been detected in different tumors and is often associated with malignant and aggressive phenotypes (Jose et al., 2011). For example, expression of hexokinase 2 (HK2), the embryonic isoform of hexokinase, the enzyme which defines the start of glycolysis, is associated with increased risk of recurrence, and adverse clinical outcome for breast cancer, pancreatic cancer, and neuroblastoma patients (Teoh and Lunt, 2018). Further studies have demonstrated that glycolytic enzymes also contribute to the metastatic progression of cancer cells (Teoh and Lunt, 2018). For example, pyruvate kinase isozyme M2 (PKM2), which mediates the final rate-limiting step of glycolysis, promotes aggressive phenotype and metastasis in different types of tumors (Zhou et al., 2012; Yu et al., 2015; Teoh and Lunt, 2018). This enzyme also acts as a transcriptional coactivator of HIF-1α in cancer cells, thus promoting glycolysis and inducing EMT (Xu et al., 2012; Morandi et al., 2017). Furthermore, EMT stimulation induced by TGF-β leads to the nuclear translocation of PKM2 in colon cancer cells, where it interacts with TGIF2 and other transcription factors, promoting EMT and supporting the malignant properties of tumor cells (Hamabe et al., 2014). Phosphohexose isomerase (PHI) is another glycolytic enzyme that involved in stimulation of invasion and metastatic dissemination through extracellular autocrine motility factor (AMF) (Watanabe et al., 1996). The overexpression of PHI leads to the increased invasion and metastasis of colon cancer cells (Tsutsumi, 2009). Additionally, PHI/AMF overexpression has been reported to promote the EMT activation through the NF-κB pathway and increased expression of EMT markers such as Snai1 and ZEB1/2 (Ahmad et al., 2011). In keeping with these observations, high PHI levels in the serum correlate positively with metastases of colorectal and esophageal squamous cells, and lung tumors (Nakamori et al., 1994; Takanami et al., 1998). Conversely, downregulation of glycolytic enzymes including PKM2 and PHI, inhibit the proliferation and migration of cancer cells (Zhou et al., 2012; Teoh and Lunt, 2018). Inhibition of glycolysis attenuates cell motility even while mitochondrial ATP synthesis remains intact, and inhibition of mitochondrial respiration reduces cell motility only minimally compared to inhibition of glycolysis (Shiraishi et al., 2015).
Glycolysis regulates different stages of metastatic dissemination, contributing to the different stages of the metastatic cascade. Thus, prostate cancer cells undergoing EMT and acquiring mesenchymal features exhibit higher glycolytic activity than their epithelial counterparts. High glycolysis rate is associated with increased cytoskeletal rearrangement and cell migration. In turn, inhibition of glycolysis suppresses the migration properties of prostate cancer cells (Shiraishi et al., 2015). In addition, an interrelation between EMT induced by TGF-β, activation of the glycolytic pathway, and repression of mitochondrial function was demonstrated (Morandi et al., 2017). In breast cancer, loss of fructose-1,6-bisphosphatase together with the loss of E-cadherin promotes cancer stem cell (CSC)-like features and cancer cell dissemination by enhancing β-catenin signaling and the EMT program. These events are concomitant with the induction of glycolysis, increase in glucose uptake, and inhibition of oxygen consumption (Dong et al., 2013).
Although it is known that metastasis requires activation of the glycolytic program, recent studies demonstrate an equal importance of OXPHOS for metastatic dissemination (Porporato et al., 2018). For example, the strong correlation between expression of PGC-1a (peroxisome proliferator-activated receptor gamma coactivator-1a), a key regulator of mitochondrial biogenesis and OXPHOS, and invasive properties was observed in breast cancer cells (LeBleu et al., 2014). PGC-1a supports migration of cancer cells, stimulating mitochondrial biogenesis and respiration, whereas downregulation of PGC-1a decreases the frequency of metastasis. Elevated OXPHOS activity is also linked to the high metastatic potential in mouse melanoma and human cervical cancer cells (Porporato et al., 2014). In turn, prostate cancer cells exhibit a mixed phenotype, where both glycolysis and OXPHOS are required for energy metabolism at different stages of disease progression (Costello and Franklin, 2005). This hybrid metabolic state, also called metabolic plasticity, can sustain tumor cell survival under different micro-environmental conditions, while at the same time supporting tumor metastasis and therapy-resistance. Thus, a hybrid metabolic phenotype, characterized by high HIF-1/AMPK activities and high glycolysis/OXPHOS (glucose oxidation and FAO) activities, allows cancer cells to acquire metabolic plasticity and utilize different types of nutrients (Jia et al., 2019). Furthermore, it permits the cells to produce energy efficiently through multiple metabolic pathways and meanwhile synthesize biomass for rapid proliferation using by-products from glycolysis. A hybrid metabolic phenotype maintains the cellular ROS at a moderate level so that cancer cells can benefit from ROS signaling and avoid DNA damage due to excessive ROS (Vyas et al., 2016).
Finally, different metabolic profiles may dictate metastatic fitness to distinct organ sites (Lehuédé et al., 2016). It has been shown that metastatic breast cancer cells may display different metabolic pathways depending on the site of metastasis. Hence, breast cancer cells obtained from bone and lung metastases rely on OXPHOS, whereas liver-metastatic breast cancer cells engage a glycolytic type of metabolism (Dupuy et al., 2015).
In consequence, metabolic pathways of migrating cancer cells appear to be inter-connected and characterized by plasticity depending on different factors, i.e., tumor type, microenvironment, site of metastasis formation, etc. A better understanding of this metabolic plasticity will permit the design of specific therapy approaches in order to target metastatic cancer cells more efficiently.
Therapeutic Targeting Metastasis
Metastasis is associated with poor outcome of cancer patients (Porporato et al., 2014). Existing therapeutic approaches are often ineffective or provide limited clinical benefit. Hence, mitochondria play an important role in metastatic dissemination, the targeting mitochondria might represent an attractive approach for the development of new strategies for treatment of metastatic cancers.
Metastatic tumors have been shown to reprogram their metabolism in order to successfully metastasize (Lehuédé et al., 2016). Accordingly, significant efforts have been made to target cancer cell metabolism in different tumors for the prevention of metastasis progression. For example, the anti-diabetic drug metformin has been shown to possess anticancer properties in different types of cancer (Rattan et al., 2011; Schexnayder et al., 2018; Thakur et al., 2018). Metformin is a Complex I inhibitor, providing cancer metabolism suppression through downregulation of mitochondrial glycerophosphate dehydrogenase (mGPDH) and OXPHOS inhibition, leading to decreased metastasis levels in a thyroid cancer mouse model (Thakur et al., 2018). Metformin also attenuates the growth of lung metastatic nodules in an ovarian cancer mouse model by inhibiting the mTOR signaling pathway (Rattan et al., 2011). At low concentrations metformin inhibits breast cancer invasion and metastasis by suppressing ROS production, suggesting the use of metformin as a chemopreventive agent to block cancer cell invasiveness (Schexnayder et al., 2018). Although, the precise mechanisms of action of metformin are still debated, a number of clinical studies have confirmed its antitumor properties (Pollak, 2012). At present metformin is used during the treatment of different cancer types in order compound to inhibit hypoglycemia non-target effect of various chemotherapy drugs (da Veiga Moreira et al., 2019). Further clinical studies are required to clarify its anti-metastatic properties.
Glycolysis inhibition has been shown to suppress metastasis in several types of cancer (Caino and Altieri, 2016). For example, HKII inhibitor lonidamine (TH-070), a derivative of indazole-3-carboxylic acid, provided significant effectiveness in preclinical studies when the drug was administered in combination with paclitaxel and cisplatin (Sborov et al., 2015; Caino and Altieri, 2016). However, despite promising early stage results, further phase II and phase III trials targeting lung cancer with lonidamine have shown its limited efficacy and hepatic toxicity (Cervantes-Madrid et al., 2015). Importantly, a more recent study demonstrated that the use of modified lonidamine is significantly more efficacious in inhibiting mitochondrial bioenergetics in lung cancer cells, leading to suppression of lung cancer progression and metastasis. Mitochondrial-lonidamine activates the generation of ROS in lung cancer cells, which leads to the inactivation of the Akt/mTOR/p70S6K signaling pathways and autophagic cell death (Cheng et al., 2019). Glycolysis can be targeted by 2-deoxyglucose (2-DG), a non-metabolizable glucose analog, which is also pursued in the clinic. However, dose-escalation phase I trials in patients with castrate-resistant prostate cancer and other advanced solid tumors resulted in asymptomatic QTc prolongation that limited further drug evaluation (Sborov et al., 2015). Since tumors may shift from glycolysis to OXPHOS, or even engage hybrid metabolisms, several studies have proposed the dual inhibition of cancer metabolism using metformin and 2-DG (Cheong et al., 2011; Jia et al., 2019). Indeed, combined treatment with metformin and 2-DG led to the significant suppression of tumor growth and metastasis in preclinical models (Cheong et al., 2011).
Mitochondrial ROS have been reported to function as signaling molecules implicated in the regulation of tumor growth and metastasis (Porporato et al., 2014). The different mechanisms by which ROS contribute to tumor growth and metastatic dissemination were discussed above. Thus, targeting mtROS seems to be an attractive approach for cancer therapy. However, contrary to the expected results, the use of antioxidants for anticancer treatment led to increased risk of cancer (Klein et al., 2011; Sullivan and Chandel, 2014). Furthermore, the treatment with NAC was shown to enhance the metastatic dissemination of human melanoma cells, providing evidence that oxidative stress may, in certain circumstances, stimulate metastasis (Piskounova et al., 2015). The cause of the failure of treatment with antioxidants could be their lack of specificity. They also may regulate many different processes involved in tumor growth and metastasis (Sullivan and Chandel, 2014). On the other hand, it has been shown that inhibition of ROS with antioxidants that target precisely mitochondrial oxidative stress may stop metastatic spread. Scavenging with MitoTempo, specific mitochondrial antioxidant, significantly reduced cancer cell invasion and prevented metastasis (Porporato et al., 2014). Thus, ROS targeting appears to be more complex than believed before and requires further detailed investigation.
Another therapeutic agent that has shown promising results in preclinical studies is an inhibitor of mitochondrial heat shock protein 90 (Hsp90) Gamitrinib (Kang et al., 2011). This compound induces mitochondrial dysfunction, providing depolarization of inner membrane potential that in turn regulates the release of cytochrome c. In mouse model of prostate cancer, Gamitrinib administration inhibited tumor growth and metastasis affecting mitochondria (Kang et al., 2011). Furthermore, targeting Hsp90 with Gamitrinib suppresses cancer cell migration and metastasis preventing metabolic reprogramming and increasing AMPK phosphorylation (Caino et al., 2013), Gamitrinib is also effective in combination therapies with inhibitors of both TRAIL and PI3K (Siegelin et al., 2011; Ghosh et al., 2015).
Since the expression of Bcl-2 family proteins has been detected in metastases of different tumors, another possible approach for cancer therapy may be focused on targeting Bcl-2 family members. BH3-mimetics are promising therapeutic drugs that mimic endogenous Bcl-2 family member antagonists, thereby target some of them and abrogating their anti-apoptotic functions. Initially, BH3-mimetics displayed encouraged results in hematological malignancies including lymphoma lymphocytic leukemia, acute myeloid leukemia, small lymphocytic lymphoma and mantle-cell lymphoma (Cang et al., 2015; Mullard, 2016; DiNardo et al., 2018; Tam et al., 2018). Thus, first-generation BH3-mimetics such as ABT-737 and its orally available derivative navitoclax (ABT-263), which are inhibitors of Bcl-2 and Bcl-W, have shown clinical efficacy (Billard, 2013). However, in several cases the treatment with these agents was limited by severe thrombocytopenia (Mullard, 2016). Clinical studies of ABT-199 in chronic lymphocytic leukemia and non-Hodgkin’s lymphoma have shown impressive antitumor efficacy, with higher response rates than navitoclax and without thrombocytopenia (Besbes et al., 2015). Several clinical trials have also demonstrated the efficacy of BH3-mimetics in solid tumors (Boisvert-Adamo et al., 2009; McKee et al., 2013; Mukherjee et al., 2018). In particular Mcl-1 has emerged as a promising target for the treatment of melanoma (Boisvert-Adamo et al., 2009; McKee et al., 2013). Additionally, a novel gossypol derivative and BH3-mimetic ch282-5 (2-aminoethanesulfonic acid sodium-gossypolone) induced colon cancer cell death in vitro and in vivo. Ch282-5 treatment activated mitochondria-dependent apoptotic pathway accompanied by mitophagy disruption and mTOR pathway activation. Furthermore, Ch282-5 provided suppression of colon cancer cell migration, invasion and liver metastasis (Wang et al., 2016). Notably, Bcl-2 family members were shown to interact with Drp1 and treatment with BH3-mimetic A-1210477 led to Drp1-dependent mitochondria fragmentation, whereas Drp1 silencing significantly reduced apoptosis induced by BH3-mimetic in lung, cervical, and breast cancer cell lines (Milani et al., 2018). Conversely, inhibition of Drp1 in combination with BH3-mimetic treatment significantly enhanced apoptotic response in melanoma cells (Mukherjee et al., 2018). Additionally, inhibition of Drp1 by Mdivi-1 increased the cytotoxic effect of combination treatment with A-1210477 and ABT-263 in different melanoma cell lines (Mukherjee et al., 2018).
Another interesting approach to target metastatic cancers is the regulation of mitochondrial K+/H+ exchange. Salinomycin is an antibiotic from the polyether ionophores group widely used in agriculture (Managò et al., 2015). Recently, it has been revealed that it possesses anticancer properties in different types of cancer (Klose et al., 2019; Tang et al., 2011). Salinomycin may target chemoresistant tumor cells, inhibiting Wnt/β-catenin and Sonic Hedgehog signaling pathways (Managò et al., 2015). Furthermore, it suppresses the migration of colorectal, breast, lung and colon cancer cell lines as well the invasion of nasopharyngeal carcinoma and bladder cancer cells in vitro (Kopp et al., 2014; Wu et al., 2014; Qu et al., 2015; Klose et al., 2016). Consistently, in vivo studies proved that salinomycin may reduce metastasis formation in mammary tumor mouse model, bladder tumor rat model and intravenous mouse tumor model (Gupta et al., 2009; Kopp et al., 2014; Qu et al., 2015). Importantly, salinomycin is able to suppress the late stages of autophagy contributing to the ROS generation and mitochondria dysfunction (Klose et al., 2019). This might explain the mechanism by which salinomycin targets mitochondrial K+/H+ exchange and prevents migration, invasion and metastasis.
Summarizing, mitochondria contribute to tumor progression and metastasis through different mechanisms including redox signaling, mitochondrial biogenesis, regulating Bcl-2 family members, metabolic reprogramming and mitochondrial K+/H+ exchange. The better understanding of these mechanisms and the possible interplay between them may provide new therapeutic approaches to target metastatic diseases.
Conclusion
Mitochondria are very important and complex organelles that affect tumorigenesis and metastatic dissemination through different mechanisms including regulation of metabolism, redox status, signaling and cell death pathways. Recent evidence has demonstrated the existence of complex interplay between mitochondria-related functions and mitochondrial dynamics. Thus, dysregulated mitochondrial turnover contributes to tumorigenesis and metastases. However, the mechanisms connecting mitochondrial dynamics to the development of metastasis remain poorly understood. In addition, the flexibility of mitochondria that allow cancer cells to adapt to the changing microenvironment and stresses should be considered in order to combat cancer successfully. Consequently, a better understanding of the processes regulated by mitochondria and their complex interplay with mitochondrial biogenesis may offer new promising therapeutic strategies for cancer treatment.
Author Contributions
All authors discussed the outline of the manuscript and involved in the preparation of the final version of the manuscript. TD and AG prepared the first draft, figure, and table.
Funding
This work was supported by the Grant from the Russian Science Foundation (19-15-00125). The work in the authors’ laboratories is also supported by the Grant from the Russian Foundation for Basic Research (19-015-00332\19), the Swedish (190345) and Stockholm (181301) Cancer Societies. We apologize to those authors whose primary works could not be cited owing to space limitations.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, J. M., and Cory, S. (2018). The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 25, 27–36. doi: 10.1038/cdd.2017.161
Ahmad, A., Aboukameel, A., Kong, D., Wang, Z., Sethi, S., Chen, W., et al. (2011). Phosphoglucose Isomerase/Autocrine Motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res. 71, 3400–3409. doi: 10.1158/0008-5472.CAN-10-0965
Báthori, G., Csordás, G., Garcia-Perez, C., Davies, E., and Hajnóczky, G. (2006). Ca2+ -dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 281, 17347–17358. doi: 10.1074/jbc.M600906200
Beckner, M. E., Gobbel, G. T., Abounader, R., Burovic, F., Agostino, N. R., Laterra, J., et al. (2005). Glycolytic glioma cells with active glycogen synthase are sensitive to PTEN and inhibitors of PI3K and gluconeogenesis. Lab. Invest. 85, 1457–1470. doi: 10.1038/labinvest.3700355
Ben-Kasus Nissim, T., Zhang, X., Elazar, A., Roy, S., Stolwijk, J. A., Zhou, Y., et al. (2017). Mitochondria control store-operated Ca2+ entry through Na + and redox signals. EMBO J. 36, 797–815. doi: 10.15252/embj.201592481
Besbes, S., Mirshahi, M., Pocard, M., and Billard, C. (2015). New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget 6, 12862–12871. doi: 10.18632/oncotarget.3868
Billard, C. (2013). BH3 mimetics: status of the field and new developments. Mol. Cancer Ther. 12, 1691–1700. doi: 10.1158/1535-7163.MCT-13-0058
Bittremieux, M., La Rovere, R. M., Akl, H., Martines, C., Welkenhuyzen, K., Dubron, K., et al. (2019). Constitutive IP3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP3 receptor disruptor BIRD-2. Cell Death Differ. 26, 531–547. doi: 10.1038/s41418-018-0142-3
Boisvert-Adamo, K., Longmate, W., Abel, E. V., and Aplin, A. E. (2009). Mcl-1 is required for melanoma cell resistance to anoikis. Mol. Cancer Res. 7, 549–556. doi: 10.1158/1541-7786.MCR-08-0358
Bower, D. V., Lansdale, N., Navarro, S., Truong, T. V., Bower, D. J., Featherstone, N. C., et al. (2017). SERCA directs cell migration and branching across species and germ layers. Biol. Open 6, 1458–1471. doi: 10.1242/bio.026039
Caino, M. C., and Altieri, D. C. (2016). Molecular pathways: mitochondrial reprogramming in tumor progression and therapy. Clin. Cancer Res. 22, 540–545. doi: 10.1158/1078-0432.CCR-15-0460
Caino, M. C., Chae, Y. C., Vaira, V., Ferrero, S., Nosotti, M., Martin, N. M., et al. (2013). Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J. Clin. Invest. 123, 2907–2920. doi: 10.1172/JCI67841
Caino, M. C., Seo, J. H., Aguinaldo, A., Wait, E., Bryant, K. G., Kossenkov, A. V., et al. (2016). A neuronal network of mitochondrial dynamics regulates metastasis. Nat. Commun. 7:13730. doi: 10.1038/ncomms13730
Cang, S., Iragavarapu, C., Savooji, J., Song, Y., and Liu, D. (2015). ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J. Hematol. Oncol. 8:129. doi: 10.1186/s13045-015-0224-3
Cannito, S., Novo, E., di Bonzo, L. V., Busletta, C., Colombatto, S., and Parola, M. (2010). Epithelial–mesenchymal transition: from molecular mechanisms. Redox regulation to implications in human health and disease. Antioxid. Redox Signal. 12, 1383–1430. doi: 10.1089/ars.2009.2737
Cekanova, M., Fernando, R. I, Siriwardhana, N., Sukhthankar, M., De la Parra, C., Woraratphoka, J., et al. (2015). BCL-2 family protein, BAD is down-regulated in breast cancer and inhibits cell invasion. Exp. Cell Res 331, 1–10. doi: 10.1016/j.yexcr.2014.11.016
Cervantes-Madrid, D., Romero, Y., and Dueñas-González, A. (2015). Reviving lonidamine and 6-Diazo-5-oxo-L-norleucine to be used in combination for metabolic cancer therapy. Biomed. Res. Int. 2015, 1–13. doi: 10.1155/2015/690492
Chaffer, C. L., and Weinberg, R. A. (2011). A perspective on cancer cell metastasis. Science 331, 1559–1564. doi: 10.1126/science.1203543
Cheng, G., Zhang, Q., Pan, J., Lee, Y., Ouari, O., Hardy, M., et al. (2019). Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat. Commun. 10:2205. doi: 10.1038/s41467-019-10042-1
Cheong, J.-H., Park, E. S., Liang, J., Dennison, J. B., Tsavachidou, D., Nguyen-Charles, C., et al. (2011). Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol. Cancer Ther. 10, 2350–2362. doi: 10.1158/1535-7163.MCT-11-0497
Chi, H.-C., Chen, S.-L., Cheng, Y.-H., Lin, T.-K., Tsai, C.-Y., Tsai, M.-M., et al. (2016). Chemotherapy resistance and metastasis-promoting effects of thyroid hormone in hepatocarcinoma cells are mediated by suppression of FoxO1 and Bim pathway. Cell Death Dis. 7:e2324. doi: 10.1038/cddis.2016.227
Chiu, W.-T., Chang, H.-A., Lin, Y.-H., Lin, Y.-S., Chang, H.-T., Lin, H.-H., et al. (2018). Bcl-2 regulates store-operated Ca2+ entry to modulate ER stress-induced apoptosis. Cell Death Discov. 4:37. doi: 10.1038/s41420-018-0039-4
Chourasia, A. H., Tracy, K., Frankenberger, C., Boland, M. L., Sharifi, M. N., Drake, L. E., et al. (2015). Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 16, 1145–1163. doi: 10.15252/embr.201540759
Comito, G., Calvani, M., Giannoni, E., Bianchini, F., Calorini, L., Torre, E., et al. (2011). HIF-1α stabilization by mitochondrial ROS promotes Met-dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic. Biol. Med. 51, 893–904. doi: 10.1016/j.freeradbiomed.2011.05.042
Corcoran, A., and Cotter, T. G. (2013). Redox regulation of protein kinases. FEBS J. 280, 1944–1965. doi: 10.1111/febs.12224
Costello, L. C., and Franklin, R. B. (2005). Why do tumour cells glycolyse?”: from glycolysis through citrate to lipogenesis. Mol. Cell. Biochem. 280, 1–8. doi: 10.1007/s11010-005-8841-8
Coutinho-Camillo, C. M., Lourenço, S. V., Nishimoto, I. N., Kowalski, L. P., and Soares, F. A. (2010). Expression of Bcl-2 family proteins and association with clinicopathological characteristics of oral squamous cell carcinoma. Histopathology 57, 304–316. doi: 10.1111/j.1365-2559.2010.03621.x
Cuevas, E. P., Eraso, P., Mazón, M. J., Santos, V., Moreno-Bueno, G., Cano, A., et al. (2017). LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci. Rep. 7:44988. doi: 10.1038/srep44988
da Veiga Moreira, J., Hamraz, M., Abolhassani, M., Schwartz, L., Jolicoeur, M., et al. (2019). Metabolic therapies inhibit tumor growth in vivo and in silico. Sci. Rep. 9:3153. doi: 10.1038/s41598-019-39109-1
De Luise, M., Girolimetti, G., Okere, B., Porcelli, A. M., Kurelac, I., and Gasparre, G. (2017). Molecular and metabolic features of oncocytomas: seeking the blueprints of indolent cancers. Biochim. Biophys. Acta Bioenerg. 1858, 591–601. doi: 10.1016/j.bbabio.2017.01.009
De Pinto, V. D., and Palmieri, F. (1992). Transmembrane arrangement of mitochondrial porin or voltage-dependent anion channel (VDAC). J. Bioenerg. Biomembr. 24, 21–26. doi: 10.1007/bf00769526
DeBerardinis, R. J., and Chandel, N. S. (2016). Fundamentals of cancer metabolism. Sci. Adv. 2:e1600200. doi: 10.1126/sciadv.1600200
D’Ignazio, L., Batie, M., and Rocha, S. (2017). Hypoxia and inflammation in cancer, focus on HIF and NF-κB. Biomedicines 5:21. doi: 10.3390/biomedicines5020021
DiNardo, C. D., Pratz, K. W., Letai, A., Jonas, B. A., Wei, A. H., Thirman, M., et al. (2018). Safety and preliminary efficacy of venetoclax with decitabine or azacitidine in elderly patients with previously untreated acute myeloid leukaemia: a non-randomised, open-label, phase 1b study. Lancet Oncol. 19, 216–228. doi: 10.1016/S1470-2045(18)30010-X
Dong, C., Yuan, T., Wu, Y., Wang, Y., Fan, T. W. M., Miriyala, S., et al. (2013). Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23, 316–331. doi: 10.1016/j.ccr.2013.01.022
Duan, Y., He, Q., Yue, K., Si, H., Wang, J., Zhou, X., et al. (2017). Hypoxia induced Bcl-2/Twist1 complex promotes tumor cell invasion in oral squamous cell carcinoma. Oncotarget 8, 7729–7739. doi: 10.18632/oncotarget.13890
Dupuy, F., Tabariès, S., Andrzejewski, S., Dong, Z., Blagih, J., Annis, M. G., et al. (2015). PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 22, 577–589. doi: 10.1016/j.cmet.2015.08.007
Eckenrode, E. F., Yang, J., Velmurugan, G. V., Foskett, J. K., and White, C. (2010). Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol. Chem. 285, 13678–13684. doi: 10.1074/jbc.M109.096040
Elia, I., Schmieder, R., Christen, S., and Fendt, S.-M. (2015). Organ-specific cancer metabolism and its potential for therapy. Hand. Exp. Pharmacol. 233, 321–353. doi: 10.1007/164_2015_10
Favre, C., Zhdanov, A., Leahy, M., Papkovsky, D., and O’Connor, R. (2010). Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene 29, 3964–3976. doi: 10.1038/onc.2010.146
Feng, Y.-X., Sokol, E. S., Del Vecchio, C. A., Sanduja, S., Claessen, J. H. L., Proia, T. A., et al. (2014). Epithelial-to-mesenchymal transition activates PERK-eIF2 and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 4, 702–715. doi: 10.1158/2159-8290.CD-13-0945
Fouqué, A., Lepvrier, E., Debure, L., Gouriou, Y., Malleter, M., Delcroix, V., et al. (2016). The apoptotic members CD95. BclxL and Bcl-2 cooperate to promote cell migration by inducing Ca2+ flux from the endoplasmic reticulum to mitochondria. Cell Death Differ. 23, 1702–1716. doi: 10.1038/cdd.2016.61
Galadari, S., Rahman, A., Pallichankandy, S., and Thayyullathil, F. (2017). Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic. Biol. Med. 104, 144–164. doi: 10.1016/j.freeradbiomed.2017.01.004
Geng, M., Wang, L., and Li, P. (2013). Correlation between chemosensitivity to anticancer drugs and Bcl-2 expression in gastric cancer. Int. J. Clin. Exp. Pathol. 6, 2554–2559.
Ghosh, J. C., Siegelin, M. D., Vaira, V., Faversani, A., Tavecchio, M., Chae, Y. C., et al. (2015). adaptive mitochondrial reprogramming and resistance to PI3K therapy. JNCI J. Natl. Cancer Inst. 107, doi: 10.1093/jnci/dju502
Gogvadze, V., Orrenius, S., and Zhivotovsky, B. (2008). Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 18, 165–173. doi: 10.1016/j.tcb.2008.01.006
Gorrini, C., Harris, I. S., and Mak, T. W. (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. doi: 10.1038/nrd4002
Griguer, C. E., Oliva, C. R., and Gillespie, G. Y. (2005). Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J. Neurooncol. 74, 123–133. doi: 10.1007/s11060-004-6404-6
Gueguinou, M., Crottès, D., Chantôme, A., Rapetti-Mauss, R., Potier-Cartereau, M., Clarysse, L., et al. (2017). The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca2+ homeostasis. Oncogene 36, 3640–3647. doi: 10.1038/onc.2016.501
Guha, M., Srinivasan, S., Ruthel, G., Kashina, A. K., Carstens, R. P., Mendoza, A., et al. (2014). Mitochondrial retrograde signaling induces epithelial–mesenchymal transition and generates breast cancer stem cells. Oncogene 33, 5238–5250. doi: 10.1038/onc.2013.467
Gupta, P. B., Onder, T. T., Jiang, G., Tao, K., Kuperwasser, C., Weinberg, R. A., et al. (2009). Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 138, 645–659. doi: 10.1016/j.cell.2009.06.034
Hamabe, A., Konno, M., Tanuma, N., Shima, H., Tsunekuni, K., Kawamoto, K., et al. (2014). Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. U.S.A. 111, 15526–15531. doi: 10.1073/pnas.1407717111
Han, X.-J., Yang, Z.-J., Jiang, L.-P., Wei, Y.-F., Liao, M.-F., Qian, Y., et al. (2015). Mitochondrial dynamics regulates hypoxia-induced migration and antineoplastic activity of cisplatin in breast cancer cells. Int. J. Oncol. 46, 691–700. doi: 10.3892/ijo.2014.2781
Hara, K., Hamada, C., Wakabayashi, K., Kanda, R., Kaneko, K., Horikoshi, S., et al. (2017). Scavenging of reactive oxygen species by astaxanthin inhibits epithelial–mesenchymal transition in high glucose-stimulated mesothelial cells. PLoS One 12:e0184332. doi: 10.1371/journal.pone.0184332
Ho, J.-N., Kang, G. Y., Lee, S.-S., Kim, J., Bae, I. H., Hwang, S.-G., et al. (2010). Bcl-XL and STAT3 mediate malignant actions of γ-irradiation in lung cancer cells. Cancer Sci. 101, 1417–1423. doi: 10.1111/j.1349-7006.2010.01552.x
Huang, H., Hu, X., Eno, C. O., Zhao, G., Li, C., and White, C. (2013). An interaction between Bcl-x L and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J. Biol. Chem. 288, 19870–19881. doi: 10.1074/jbc.M112.448290
Huang, H., Shah, K., Bradbury, N. A., Li, C., and White, C. (2014). Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 5:e1482. doi: 10.1038/cddis.2014.419
Ishijima, N., Miki, C., Ishida, T., Kinoshita, T., and Suzuki, H. (1999). The immunohistochemical expression of BCL-2 oncoprotein in colorectal adenocarcinoma. Surg. Today 29, 682–684. doi: 10.1007/BF02483002
Ishikawa, K., Takenaga, K., Akimoto, M., Koshikawa, N., Yamaguchi, A., Imanishi, H., et al. (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320, 661–664. doi: 10.1126/science.1156906
Jain, M., Rivera, S., Monclus, E. A., Synenki, L., Zirk, A., Eisenbart, J., et al. (2013). Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 288, 770–777. doi: 10.1074/jbc.M112.431973
Jia, D., Lu, M., Jung, K. H., Park, J. H., Yu, L., Onuchic, J. N., et al. (2019). Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. U.S.A. 116, 3909–3918. doi: 10.1073/pnas.1816391116
Jia, L., Huang, S., Yin, X., Zan, Y., Guo, Y., and Han, L. (2018). Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 208, 123–130. doi: 10.1016/j.lfs.2018.07.027
Jiang, J., Wang, K., Chen, Y., Chen, H., Nice, E. C., and Huang, C. (2017). Redox regulation in tumor cell epithelial–mesenchymal transition: molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2:17036. doi: 10.1038/sigtrans.2017.36
Jin-Song, Y., Zhao-Xia, W., Cheng-Yu, L., Xiao-Di, L., Ming, S., Yuan-Yuan, G., et al. (2011). Prognostic significance of Bcl-xL gene expression in human colorectal cancer. Acta Histochem. 113, 810–814. doi: 10.1016/j.acthis.2011.01.002
Jose, C., Bellance, N., and Rossignol, R. (2011). Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim. Biophys. Acta - Bioenerg. 1807, 552–561. doi: 10.1016/j.bbabio.2010.10.012
Joseph, J. V., Conroy, S., Pavlov, K., Sontakke, P., Tomar, T., Eggens-Meijer, E., et al. (2015). Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α–ZEB1 axis. Cancer Lett. 359, 107–116. doi: 10.1016/j.canlet.2015.01.010
Jung, C.-H., Kim, E. M., Um, H.-D., Song, J.-Y., and Park, J. K. (2018). Pro-apoptotic Bax promotes mesenchymal-epithelial transition by binding to respiratory complex-I and antagonizing the malignant actions of pro-survival Bcl-2 proteins. Cancer Lett. 424, 127–135. doi: 10.1016/j.canlet.2018.03.033
Kang, B. H., Tavecchio, M., Goel, H. L., Hsieh, C.-C., Garlick, D. S., Raskett, C. M., et al. (2011). Targeted inhibition of mitochondrial Hsp90 suppresses localised and metastatic prostate cancer growth in a genetic mouse model of disease. Br. J. Cancer 104, 629–634. doi: 10.1038/bjc.2011.9
Kang, S. S., Han, K.-S., Ku, B. M., Lee, Y. K., Hong, J., Shin, H. Y., et al. (2010). Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 70, 1173–1183. doi: 10.1158/0008-5472.CAN-09-2886
Kim, E. M., Jung, C.-H., Song, J.-Y., Park, J. K., and Um, H.-D. (2018). Pro-apoptotic Bax promotes mesenchymal-epithelial transition by binding to respiratory complex-I and antagonizing the malignant actions of pro-survival Bcl-2 proteins. Cancer Lett. 424, 127–135. doi: 10.1016/j.canlet.2018.03.033
Kim, E. S., Choi, J. Y., Hwang, S. J., and Bae, I. H. (2019). Hypermethylation of miR-205-5p by IR governs aggressiveness and metastasis via regulating Bcl-w and Src. Mol. Ther. Nucleic Acids 14, 450–464. doi: 10.1016/j.omtn.2018.12.013
Kim, S., Yao, J., Suyama, K., Qian, X., Qian, B.-Z., Bandyopadhyay, S., et al. (2014). Slug promotes survival during metastasis through suppression of Puma-mediated apoptosis. Cancer Res. 74, 3695–3706. doi: 10.1158/0008-5472.CAN-13-2591
Kiss, B., Skuginna, V., Fleischmann, A., Bell, R. H., Collins, C., Thalmann, G. N., et al. (2015). Bcl-2 predicts response to neoadjuvant chemotherapy and is overexpressed in lymph node metastases of urothelial cancer of the bladder. Urol. Oncol. Semin. Orig. Investig 33:166.e1-8. doi: 10.1016/j.urolonc.2014.12.005
Klein, E. A., Thompson, I. M., Tangen, C. M., Crowley, J. J., Lucia, M. S., Goodman, P. J., et al. (2011). Vitamin E and the risk of prostate cancer. JAMA 306:1549. doi: 10.1001/jama.2011.1437
Klose, J., Eissele, J., Volz, C., Schmitt, S., Ritter, A., Ying, S., et al. (2016). Salinomycin inhibits metastatic colorectal cancer growth and interferes with Wnt/β-catenin signaling in CD133+ human colorectal cancer cells. BMC Cancer 16:896. doi: 10.1186/s12885-016-2879-8
Klose, J., Trefz, S., Wagner, T., Steffen, L., Preißendörfer Charrier, A., Radhakrishnan, P., et al. (2019). Salinomycin: anti-tumor activity in a pre-clinical colorectal cancer model. PLoS One 14:e0211916. doi: 10.1371/journal.pone.0211916
Koop, E. A., van Laar, T., van Wichen, D. F., de Weger, R. A., Wall, E., and van der, et al. (2009). Expression of BNIP3 in invasive breast cancer: correlations with the hypoxic response and clinicopathological features. BMC Cancer 9:175. doi: 10.1186/1471-2407-9-175
Kopp, F., Hermawan, A., Oak, P. S., Herrmann, A., Wagner, E., and Roidl, A. (2014). Salinomycin treatment reduces metastatic tumor burden by hampering cancer cell migration. Mol. Cancer 13:16. doi: 10.1186/1476-4598-13-16
Kozma, R., Ahmed, S., Best, A., and Lim, L. (1995). The Ras-related protein Cdc42Hs and bradykinin promote formation of peripheral actin microspikes and filopodia in Swiss 3T3 fibroblasts. Mol. Cell. Biol. 15, 1942–1952. doi: 10.1128/MCB.15.4.1942
Krstić, J., Trivanović, D., Mojsilović, S., and Santibanez, J. F. (2015). Transforming growth factor-beta and oxidative stress interplay: implications in tumorigenesis and cancer progression. Oxid. Med. Cell. Longev. 2015, 1–15. doi: 10.1155/2015/654594
Kumari, S., Badana, A. K., G, M. M., G, S., and Malla, R. (2018). Reactive oxygen species: a key constituent in cancer survival. Biomark. Insights 13:117727191875539. doi: 10.1177/1177271918755391
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. doi: 10.1038/nrm3758
LeBleu, V. S., O’Connell, J. T., Gonzalez Herrera, K. N., Wikman, H., Pantel, K., Haigis, M. C., et al. (2014). PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003. doi: 10.1038/ncb3039
Lee, A. S. (2005). The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 35, 373–381. doi: 10.1016/j.ymeth.2004.10.010
Lee, G.-H., Yan, C., Shin, S.-J., Hong, S.-C., Ahn, T., Moon, A., et al. (2010). BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene 29, 2130–2141. doi: 10.1038/onc.2009.491
Lee, H. W., Lee, S.-S., Lee, S. J., and Um, H.-D. (2003). Bcl-w is expressed in a majority of infiltrative gastric adenocarcinomas and suppresses the cancer cell death by blocking stress-activated protein kinase/c-Jun NH2-terminal kinase activation. Cancer Res 63, 1093–1100.
Lee, S.-R., Yang, K.-S., Kwon, J., Lee, C., Jeong, W., and Rhee, S. G. (2002). Reversible inactivation of the tumor suppressor PTEN by H 2 O 2. J. Biol. Chem. 277, 20336–20342. doi: 10.1074/jbc.M111899200
Lee, W. S., Woo, E. Y., Kwon, J., Park, M.-J., Lee, J.-S., Han, Y.-H., et al. (2013). Bcl-w enhances mesenchymal changes and invasiveness of glioblastoma cells by inducing nuclear accumulation of β-Catenin. PLoS One 8:e68030. doi: 10.1371/journal.pone.0068030
Lehuédé, C., Dupuy, F., Rabinovitch, R., Jones, R. G., and Siegel, P. M. (2016). Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 76, 5201–5208. doi: 10.1158/0008-5472.CAN-16-0266
Lin, C.-S., Lee, H.-T., Lee, S.-Y., Shen, Y.-A., Wang, L.-S., Chen, Y.-J., et al. (2012). High mitochondrial DNA copy number and bioenergetic function are associated with tumor invasion of esophageal squamous cell carcinoma cell lines. Int. J. Mol. Sci. 13, 11228–11246. doi: 10.3390/ijms130911228
Liu, R.-M., and Desai, L. P. (2015). Reciprocal regulation of TGF-β and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 6, 565–577. doi: 10.1016/j.redox.2015.09.009
Losón, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1. Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.e12-10-0721
Lu, Q., and Hong, W. (2009). Bcl2 enhances c-Myc-mediated MMP-2 expression of vascular smooth muscle cells. Cell. Signal. 21, 1054–1059. doi: 10.1016/j.cellsig.2009.02.020
Lu, W., and Kang, Y. (2019). Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 49, 361–374. doi: 10.1016/j.devcel.2019.04.010
Macher-Goeppinger, S., Keith, M., Hatiboglu, G., Hohenfellner, M., Schirmacher, P., Roth, W., et al. (2017). Expression and functional characterization of the BNIP3 protein in renal cell carcinomas. Transl. Oncol. 10, 869–875. doi: 10.1016/j.tranon.2017.08.008
Maes, H., Van Eygen, S., Krysko, D. V., Vandenabeele, P., Nys, K., Rillaerts, K., et al. (2014). BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 5, e1127. doi: 10.1038/cddis.2014.94
Malhotra, J. D., and Kaufman, R. J. (2011). ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb. Perspect. Biol. 3:a004424. doi: 10.1101/cshperspect.a004424
Mambo, E., Chatterjee, A., Xing, M., Tallini, G., Haugen, B. R., Yeung, S.-C. J., et al. (2005). Tumor-specific changes in mtDNA content in human cancer. Int. J. Cancer 116, 920–924. doi: 10.1002/ijc.21110
Managò, A., Leanza, L., Carraretto, L., Sassi, N., Grancara, S., Quintana-Cabrera, R., et al. (2015). Early effects of the antineoplastic agent salinomycin on mitochondrial function. Cell Death Dis. 6:e1930. doi: 10.1038/cddis.2015.263
Martinvalet, D. (2018). The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death Dis. 9:336. doi: 10.1038/s41419-017-0237-7
Mazure, N. M., and Pouysségur, J. (2009). Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy 5, 868–869. doi: 10.4161/auto.9042
McKee, C. S., Hill, D. S., Redfern, C. P. F., Armstrong, J. L., and Lovat, P. E. (2013). Oncogenic BRAF signalling increases Mcl-1 expression in cutaneous metastatic melanoma. Exp. Dermatol. 22, 767–769. doi: 10.1111/exd.12254
Merino, D., Best, S. A., Asselin-Labat, M.-L., Vaillant, F., Pal, B., Dickins, R. A., et al. (2015). Pro-apoptotic Bim suppresses breast tumor cell metastasis and is a target gene of SNAI2. Oncogene 34, 3926–3934. doi: 10.1038/onc.2014.313
Mhaidat, N. M., Al Sheyab, F. M., Zyoud, A. M., Alzoubi, K. H., Khabour, O. F., Al-Quraan, N. A., et al. (2017). Study the epigenetic down-regulation of Bim on colorectal cancer chemotherapy response. J. King Saud Univ. Sci. 31, 308–313. doi: 10.1016/j.jksus.2017.09.012
Milani, M., Byrne, D. P., Greaves, G., Butterworth, M., Cohen, G. M., Eyers, P. A., et al. (2018). DRP-1 is required for BH3 mimetic-mediated mitochondrial fragmentation and apoptosis. Cell Death Dis. 8:e2552. doi: 10.1038/cddis.2016.485
Min, C., Eddy, S. F., Sherr, D. H., and Sonenshein, G. E. (2008). NF-κB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 104, 733–744. doi: 10.1002/jcb.21695
Mitchell, P. (1961). Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191, 144–148. doi: 10.1038/191144a0
Monaco, G., Beckers, M., Ivanova, H., Missiaen, L., Parys, J. B., De Smedt, H., et al. (2012). Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem. Biophys. Res. Commun. 428, 31–35. doi: 10.1016/j.bbrc.2012.10.002
Monaco, G., Decrock, E., Arbel, N., van Vliet, A. R., La Rovere, R. M., De Smedt, H., et al. (2015). The BH4 Domain of Anti-apoptotic Bcl-XL, but Not That of the Related Bcl-2. Limits the Voltage-dependent Anion Channel 1 (VDAC1)-mediated Transfer of Pro-apoptotic Ca 2+ Signals to Mitochondria. J. Biol. Chem. 290, 9150–9161. doi: 10.1074/jbc.M114.622514
Morandi, A., Taddei, M. L., Chiarugi, P., and Giannoni, E. (2017). Targeting the metabolic reprogramming that controls epithelial-to-mesenchymal transition in aggressive tumors. Front. Oncol. 7:40. doi: 10.3389/fonc.2017.00040
Mound, A., Vautrin-Glabik, A., Foulon, A., Botia, B., Hague, F., Parys, J. B., et al. (2017). Downregulation of type 3 inositol (1,4,5)-trisphosphate receptor decreases breast cancer cell migration through an oscillatory Ca2+ signal. Oncotarget 8, 72324–72341. doi: 10.18632/oncotarget.20327
Mukherjee, N., Strosnider, A., Vagher, B., Lambert, K. A., Slaven, S., Robinson, W. A., et al. (2018). BH3 mimetics induce apoptosis independent of DRP-1 in melanoma. Cell Death Dis. 9:907. doi: 10.1038/s41419-018-0932-z
Mullard, A. (2016). Pioneering apoptosis-targeted cancer drug poised for FDA approval. Nat. Rev. Drug Discov. 15, 147–149. doi: 10.1038/nrd.2016.23
Naito, A., Cook, C. C., Mizumachi, T., Wang, M., Xie, C., Evans, T. T., et al. (2008). Progressive tumor features accompany epithelial-mesenchymal transition induced in mitochondrial DNA-depleted cells. Cancer Sci. 99, 1584–1588. doi: 10.1111/j.1349-7006.2008.00879.x
Nakamori, S., Watanabe, H., Kameyama, M., Imaoka, S., Furukawa, H., Ishikawa, O., et al. (1994). Expression of autocrine motility factor receptor in colorectal cancer as a predictor for disease recurrence. Cancer 74, 1855–1862. doi: 10.1002/1097-0142(19941001)74:7<1855
Naon, D., and Scorrano, L. (2014). At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim. Biophys. Acta Mol. Cell Res. 1843, 2184–2194. doi: 10.1016/j.bbamcr.2014.05.011
Narumiya, S., Tanji, M., and Ishizaki, T. (2009). Rho signaling. ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28, 65–76. doi: 10.1007/s10555-008-9170-7
Neri, A., Marrelli, D., Roviello, F., De Marco, G., Mariani, F., De Stefano, A., et al. (2006). Bcl-2 expression correlates with lymphovascular invasion and long-term prognosis in breast cancer. Breast Cancer Res. Treat. 99, 77–83. doi: 10.1007/s10549-006-9183-2
Noujaim, D., van Golen, C. M., van Golen, K. L., Grauman, A., and Feldman, E. L. (2002). N-Myc and Bcl-2 coexpression induces MMP-2 secretion and activation in human neuroblastoma cells. Oncogene 21, 4549–4557. doi: 10.1038/sj.onc.1205552
Obre, E., and Rossignol, R. (2015). Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int. J. Biochem. Cell Biol. 59, 167–181. doi: 10.1016/j.biocel.2014.12.008
Opferman, J. T., and Kothari, A. (2018). Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 25, 37–45. doi: 10.1038/cdd.2017.170
Park, J. H., Vithayathil, S., Kumar, S., Sung, P.-L., Dobrolecki, L. E., Putluri, V., et al. (2016). Fatty acid oxidation-driven src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 14, 2154–2165. doi: 10.1016/j.celrep.2016.02.004
Piskounova, E., Agathocleous, M., Murphy, M. M., Hu, Z., Huddlestun, S. E., Zhao, Z., et al. (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. doi: 10.1038/nature15726
Pollak, M. N. (2012). Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2, 778–790. doi: 10.1158/2159-8290.CD-12-0263
Porporato, P. E., Filigheddu, N., Pedro, J. M. B.-S., Kroemer, G., and Galluzzi, L. (2018). Mitochondrial metabolism and cancer. Cell Res. 28, 265–280. doi: 10.1038/cr.2017.155
Porporato, P. E., Payen, V. L., Pérez-Escuredo, J., De Saedeleer, C. J., Danhier, P., Copetti, T., et al. (2014). A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766. doi: 10.1016/j.celrep.2014.06.043
Qu, H., Ma, B., Yuan, H.-F., Wang, Z.-Y., Guo, S.-J., and Zhang, J. (2015). Effect of salinomycin on metastasis and invasion of bladder cancer cell line T24. Asian Pac. J. Trop. Med. 8, 578–582. doi: 10.1016/j.apjtm.2015.06.004
Quinlan, C. L., Perevoshchikova, I. V., Hey-Mogensen, M., Orr, A. L., and Brand, M. D. (2013). Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312. doi: 10.1016/j.redox.2013.04.005
Rattan, R., Graham, R. P., Maguire, J. L., Giri, S., and Shridhar, V. (2011). Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 13, 483–491.
Ridley, A. J. (2011). Life at the leading edge. Cell 145, 1012–1022. doi: 10.1016/j.cell.2011.06.010
Rojo de la Vega, M., Chapman, E., and Zhang, D. D. (2018). NRF2 and the Hallmarks of Cancer. Cancer Cell 34, 21–43. doi: 10.1016/j.ccell.2018.03.022
Rossignol, R., Gilkerson, R., Aggeler, R., Yamagata, K., Remington, S. J., and Capaldi, R. A. (2004). Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 64, 985–993. doi: 10.1158/0008-5472.can-03-1101
Sborov, D. W., Haverkos, B. M., and Harris, P. J. (2015). Investigational cancer drugs targeting cell metabolism in clinical development. Expert Opin. Investig. Drugs 24, 79–94. doi: 10.1517/13543784.2015.960077
Schexnayder, C., Broussard, K., Onuaguluchi, D., Poché, A., Ismail, M., McAtee, L., et al. (2018). Metformin inhibits migration and invasion by suppressing ROS production and COX2 expression in MDA-MB-231 breast cancer cells. Int. J. Mol. Sci. 19:3692. doi: 10.3390/ijms19113692
Senft, D., and Ronai, Z. A. (2016). Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell Biol. 39, 43–52. doi: 10.1016/j.ceb.2016.02.001
Shagieva, G., Domnina, L., Makarevich, O., Chernyak, B., Skulachev, V., and Dugina, V. (2017). Depletion of mitochondrial reactive oxygen species downregulates epithelial-to-mesenchymal transition in cervical cancer cells. Oncotarget 8, 4901–4913. doi: 10.18632/oncotarget.13612
Shen, X., Xue, Y., Si, Y., Wang, Q., Wang, Z., Yuan, J., et al. (2015). The unfolded protein response potentiates epithelial-to-mesenchymal transition (EMT) of gastric cancer cells under severe hypoxic conditions. Med. Oncol. 32:447. doi: 10.1007/s12032-014-0447-0
Shi, C., Cai, Y., Li, Y., Li, Y., Hu, N., Ma, S., et al. (2018). Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 14, 59–71. doi: 10.1016/j.redox.2017.08.013
Shimizu, S., Iida, S., Ishiguro, M., Uetake, H., Ishikawa, T., Takagi, Y., et al. (2010). Methylated BNIP3 gene in colorectal cancer prognosis. Oncol. Lett. 1, 865–872. doi: 10.3892/ol_00000153
Shimojo, Y., Akimoto, M., Hisanaga, T., Tanaka, T., Tajima, Y., Honma, Y., et al. (2013). Attenuation of reactive oxygen species by antioxidants suppresses hypoxia-induced epithelial-mesenchymal transition and metastasis of pancreatic cancer cells. Clin. Exp. Metastasis 30, 143–154. doi: 10.1007/s10585-012-9519-8
Shiraishi, T., Verdone, J. E., Huang, J., Kahlert, U. D., Hernandez, J. R., Torga, G., et al. (2015). Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 6, 130–143. doi: 10.18632/oncotarget.2766
Siegelin, M. D., Dohi, T., Raskett, C. M., Orlowski, G. M., Powers, C. M., Gilbert, C. A., et al. (2011). Exploiting the mitochondrial unfolded protein response for cancer therapy in mice and human cells. J. Clin. Invest. 121, 1349–1360. doi: 10.1172/JCI44855
Singh, A., Sharma, R. K., Chagtoo, M., Agarwal, G., George, N., Sinha, N., et al. (2017). 1H NMR metabolomics reveals association of high expression of inositol 1, 4, 5 trisphosphate receptor and metabolites in breast cancer patients. PLoS One 12:e0169330. doi: 10.1371/journal.pone.0169330
Singh, L., Pushker, N., Saini, N., Sen, S., Sharma, A., Bakhshi, S., et al. (2015). Expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins in human retinoblastoma. Clin. Exp. Ophthalmol. 43, 259–267. doi: 10.1111/ceo.12397
Smolková, K., Bellance, N., Scandurra, F., Génot, E., Gnaiger, E., Plecitá-Hlavatá, L., et al. (2010). Mitochondrial bioenergetic adaptations of breast cancer cells to aglycemia and hypoxia. J. Bioenerg. Biomembr. 42, 55–67. doi: 10.1007/s10863-009-9267-x
Smolková, K., Plecitá-Hlavatá, L., Bellance, N., Benard, G., Rossignol, R., and Ježek, P. (2011). Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 43, 950–968. doi: 10.1016/j.biocel.2010.05.003
Srinivasan, S., Guha, M., Kashina, A., and Avadhani, N. G. (2017). Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta - Bioenerg. 1858, 602–614. doi: 10.1016/j.bbabio.2017.01.004
Sullivan, L. B., and Chandel, N. S. (2014). Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2:17. doi: 10.1186/2049-3002-2-17
Sun, T., Sun, B., Zhao, X., Zhao, N., Dong, X., Che, N., et al. (2011). Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: a study of hepatocellular carcinoma. Hepatology 54, 1690–1706. doi: 10.1002/hep.24543
Sun, X., Cao, H., Zhan, L., Yin, C., Wang, G., Liang, P., et al. (2018a). Mitochondrial fission promotes cell migration by Ca2+ /CaMKII/ERK/FAK pathway in hepatocellular carcinoma. Liver Int. 38, 1263–1272. doi: 10.1111/liv.13660
Sun, X., Zhan, L., Chen, Y., Wang, G., He, L., Wang, Q., et al. (2018b). Increased mtDNA copy number promotes cancer progression by enhancing mitochondrial oxidative phosphorylation in microsatellite-stable colorectal cancer. Signal Transduct. Target. Ther. 3:8. doi: 10.1038/s41392-018-0011-z
Sun, Y., Jiang, F., Pan, Y., Chen, X., Chen, J., Wang, Y., et al. (2018c). XBP1 promotes tumor invasion and is associated with poor prognosis in oral squamous cell carcinoma. Oncol. Rep. 40, 988–998. doi: 10.3892/or.2018.6498
Takanami, I., Takeuchi, K., Naruke, M., Kodaira, S., Tanaka, F., Watanabe, H., et al. (1998). Autocrine motility factor in pulmonary adenocarcinomas: results of an immunohistochemical study. Tumor Biol. 19, 384–389. doi: 10.1159/000030031
Tam, C. S., Anderson, M. A., Pott, C., Agarwal, R., Handunnetti, S., Hicks, R. J., et al. (2018). Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N. Engl. J. Med. 378, 1211–1223. doi: 10.1056/NEJMoa1715519
Tang, Q.-L., Zhao, Z.-Q., Li, J.-C., Liang, Y., Yin, J.-Q., Zou, C.-Y., et al. (2011). Salinomycin inhibits osteosarcoma by targeting its tumor stem cells. Cancer Lett. 311, 113–121. doi: 10.1016/j.canlet.2011.07.016
Teoh, S. T., and Lunt, S. Y. (2018). Metabolism in cancer metastasis: bioenergetics, biosynthesis, and beyond. Wiley Interdiscip. Rev. Syst. Biol. Med. 10:e1406. doi: 10.1002/wsbm.1406
Thakur, S., Daley, B., Gaskins, K., Vasko, V. V., Boufraqech, M., Patel, D., et al. (2018). Metformin targets mitochondrial glycerophosphate dehydrogenase to control rate of oxidative phosphorylation and growth of thyroid cancer in vitro and in vivo. Clin. Cancer Res. 24, 4030–4043. doi: 10.1158/1078-0432.CCR-17-3167
Tosatto, A., Sommaggio, R., Kummerow, C., Bentham, R. B., Blacker, T. S., Berecz, T., et al. (2016). The mitochondrial calcium uniporter regulates breast cancer progression via HIF−1α. EMBO Mol. Med. 8, 569–585. doi: 10.15252/emmm.201606255
Trisciuoglio, D., Desideri, M., Ciuffreda, L., Mottolese, M., Ribatti, D., Vacca, A., et al. (2005). Bcl-2 overexpression in melanoma cells increases tumor progression-associated properties and in vivo tumor growth. J. Cell. Physiol. 205, 414–421. doi: 10.1002/jcp.20413
Trisciuoglio, D., Tupone, M. G., Desideri, M., Di Martile, M., Gabellini, C., Buglioni, S., et al. (2017). BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis. 8, 3216. doi: 10.1038/s41419-017-0055-y
Tsai, C.-H., Li, C.-H., Cheng, Y.-W., Lee, C.-C., Liao, P.-L., Lin, C.-H., et al. (2017). The inhibition of lung cancer cell migration by AhR-regulated autophagy. Sci. Rep. 7:41927. doi: 10.1038/srep41927
Tsutsumi. (2009). Phosphoglucose isomerase enhances colorectal cancer metastasis. Int. J. Oncol. 35, 1117–1121. doi: 10.3892/ijo_00000427
Um, H.-D. (2016). Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: a review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget 7, 5193–5203. doi: 10.18632/oncotarget.6405
Urra, H., Henriquez, D. R., Cánovas, J., Villarroel-Campos, D., Carreras-Sureda, A., Pulgar, E., et al. (2018). IRE1α governs cytoskeleton remodelling and cell migration through a direct interaction with filamin A. Nat. Cell Biol. 20, 942–953. doi: 10.1038/s41556-018-0141-0
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J.-P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891. doi: 10.1038/cdd.2012.74
Vyas, S., Zaganjor, E., and Haigis, M. C. (2016). Mitochondria and cancer. Cell 166, 555–566. doi: 10.1016/j.cell.2016.07.002
Wan, G., Mahajan, A., Lidke, D., and Rajput, A. (2015). Bcl-2 together with PI3K p110α regulates cell morphology and cell migration. Cell Death Dis. 6:e2006. doi: 10.1038/cddis.2015.345
Wang, X., Zhang, C., Yan, X., Lan, B., Wang, J., Wei, C., et al. (2016). A novel bioavailable BH3 mimetic efficiently inhibits colon cancer via cascade effects of mitochondria. Clin. Cancer Res. 22, 1445–1458. doi: 10.1158/1078-0432.CCR-15-0732
Wang, Z.-X., Yang, J.-S., Pan, X., Wang, J.-R., Li, J., Yin, Y.-M., et al. (2010). Functional and biological analysis of Bcl-xL expression in human osteosarcoma. Bone 47, 445–454. doi: 10.1016/j.bone.2010.05.027
Watanabe, H., Takehana, K., Date, M., Shinozaki, T., and Raz, A. (1996). Tumor cell autocrine motility factor is the neuroleukin/phosphohexose isomerase polypeptide. Cancer Res. 56, 2960–2963.
Wu, D., Zhang, Y., Huang, J., Fan, Z., Shi, F., and Wang, S. (2014). Salinomycin inhibits proliferation and induces apoptosis of human nasopharyngeal carcinoma cell in vitro and suppresses tumor growth in vivo. Biochem. Biophys. Res. Commun. 443, 712–717. doi: 10.1016/j.bbrc.2013.12.032
Xu, C., Liu, Y., Huang, J., Wang, H., Tan, L., Xu, Y., et al. (2017). The prognostic significance of MCL1 copy number gain in esophageal squamous cell carcinoma. Oncotarget 8, 87699–87709. doi: 10.18632/oncotarget.21181
Xu, Y., and Lu, S. (2015). Transforming growth factor-β1-induced epithelial to mesenchymal transition increases mitochondrial content in the A549 non-small cell lung cancer cell line. Mol. Med. Rep. 11, 417–421. doi: 10.3892/mmr.2014.2678
Xu, Y., Zhao, F., Wang, Z., Song, Y., Luo, Y., Zhang, X., et al. (2012). MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene 31, 1398–1407. doi: 10.1038/onc.2011.340
Yang, S. (2018). The store-operated calcium channels in cancer metastasis from cell migration invasion to metastatic colonization. Front. Biosci. 23:4641. doi: 10.2741/4641
Yin, M., Lu, Q., Liu, X., Wang, T., Liu, Y., and Chen, L. (2016). Silencing Drp1 inhibits glioma cells proliferation and invasion by RHOA/ROCK1 pathway. Biochem. Biophys. Res. Commun. 478, 663–668. doi: 10.1016/j.bbrc.2016.08.003
Yoon, Y.-S., Lee, J.-H., Hwang, S.-C., Choi, K. S., and Yoon, G. (2005). TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 24, 1895–1903. doi: 10.1038/sj.onc.1208262
Young, A. I. J., Law, A. M. K., Castillo, L., Chong, S., Cullen, H. D., Koehler, M., et al. (2016). MCL-1 inhibition provides a new way to suppress breast cancer metastasis and increase sensitivity to dasatinib. Breast Cancer Res. 18:125. doi: 10.1186/s13058-016-0781-6
Yu, C., Wang, Y., Peng, J., Shen, Q., Chen, M., Tang, W., et al. (2017). Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 8, 83831–83844. doi: 10.18632/oncotarget.19747
Yu, G., Yu, W., Jin, G., Xu, D., Chen, Y., Xia, T., et al. (2015). PKM2 regulates neural invasion of and predicts poor prognosis for human hilar cholangiocarcinoma. Mol. Cancer 14:193. doi: 10.1186/s12943-015-0462-6
Yu, M., Chen, S., Hong, W., Gu, Y., Huang, B., Lin, Y., et al. (2019). Prognostic role of glycolysis for cancer outcome: evidence from 86 studies. J. Cancer Res. Clin. Oncol. 145, 967–999. doi: 10.1007/s00432-019-02847-w
Zhang, C., Wang, H.-J., Bao, Q.-C., Wang, L., Guo, T.-K., Chen, W.-L., et al. (2016). NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 7, 73593–73606. doi: 10.18632/oncotarget.12435
Zhang, J., Zhang, C., Jiang, X., Li, L., Zhang, D., Tang, D., et al. (2019). Involvement of autophagy in hypoxia-BNIP3 signaling to promote epidermal keratinocyte migration. Cell Death Dis. 10:234. doi: 10.1038/s41419-019-1473-9
Zhang, K., Jiao, K., Xing, Z., Zhang, L., Yang, J., Xie, X., et al. (2014). Bcl-xL overexpression and its association with the progress of tongue carcinoma. Int. J. Clin. Exp. Pathol. 7, 7360–7377.
Zhang, W., Shi, X., Peng, Y., Wu, M., Zhang, P., Xie, R., et al. (2015). HIF-1α promotes epithelial-mesenchymal transition and metastasis through direct regulation of ZEB1 in colorectal cancer. PLoS One 10:e0129603. doi: 10.1371/journal.pone.0129603
Zhao, J., Zhang, J., Yu, M., Xie, Y., Huang, Y., Wolff, D. W., et al. (2013). Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814–4824. doi: 10.1038/onc.2012.494
Zhou, C.-F., Li, X.-B., Sun, H., Zhang, B., Han, Y.-S., Jiang, Y., et al. (2012). Pyruvate kinase type M2 is upregulated in colorectal cancer and promotes proliferation and migration of colon cancer cells. IUBMB Life 64, 775–782. doi: 10.1002/iub.1066
Keywords: cell death, invasion, metastasis, migration, mitochondria
Citation: Denisenko TV, Gorbunova AS and Zhivotovsky B (2019) Mitochondrial Involvement in Migration, Invasion and Metastasis. Front. Cell Dev. Biol. 7:355. doi: 10.3389/fcell.2019.00355
Received: 20 August 2019; Accepted: 05 December 2019;
Published: 20 December 2019.
Edited by:
Satyendra Chandra Tripathi, All India Institute of Medical Sciences Nagpur, IndiaReviewed by:
Ildikò Szabò, University of Padua, ItalyCarlos Villalobos, Spanish National Research Council (CSIC), Spain
Copyright © 2019 Denisenko, Gorbunova and Zhivotovsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boris Zhivotovsky, Qm9yaXMuWmhpdm90b3Zza3lAa2kuc2U=