 Ornella Moltedo1
Ornella Moltedo1 Paolo Remondelli
Paolo Remondelli Giuseppina Amodio
Giuseppina Amodio
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 21 August 2019
Sec. Molecular and Cellular Pathology
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00172
The recent discovery of interconnections between the endoplasmic reticulum (ER) membrane and those of almost all the cell compartments is providing novel perspectives for the understanding of the molecular events underlying cellular mechanisms in both physiological and pathological conditions. In particular, growing evidence strongly supports the idea that the molecular interactions occurring between ER and mitochondrial membranes, referred as the mitochondria (MT)–ER contacts (MERCs), may play a crucial role in aging and in the development of age-associated diseases. As emerged in the last decade, MERCs behave as signaling hubs composed by structural components that act as critical players in different age-associated disorders, such as neurodegenerative diseases and motor disorders, cancer, metabolic syndrome, as well as cardiovascular diseases. Age-associated disorders often derive from mitochondrial or ER dysfunction as consequences of oxidative stress, mitochondrial DNA mutations, accumulation of misfolded proteins, and defective organelle turnover. In this review, we discuss the recent advances associating MERCs to aging in the context of ER–MT crosstalk regulating redox signaling, ER-to MT lipid transfer, mitochondrial dynamics, and autophagy.
The existence of MT–ER contacts (MERCs) was demonstrated in the late 50s by electron microscopy (Copeland and Dalton, 1959). Biological functions, for instance modulation of phospholipid transfer and Ca2+ interchange were the first functions established for MERCs (Vance, 1990; Rizzuto et al., 1998), but more recently, additional roles, such as the regulation of mitochondrial dynamics (Friedman et al., 2011), inflammasome formation (Zhou et al., 2011), activation of autophagy (Hamasaki et al., 2013), and redox signaling control (Booth et al., 2016) have been charged to these structures. To better define the molecular composition of MERCs different methods, ranging from subcellular fractionations to proteomics and electron microscopy, have been deployed. As a result, a structural and functional equivalence has been established between MERCs and MT-associated membranes (MAMs) (Giacomello and Pellegrini, 2016). The first refers actually to the ultrastructural architecture that can be observed by electron microscopy, whereas the second hints at the molecular composition of fractions derived from ER–MT membranes isolated by subcellular fractionation, respectively. Recently, high-electron microscopy and super-resolution optical microscopy have hugely contributed to the uncovering of the architectural complexity of MERCs (Csordas et al., 2006; Sood et al., 2014; Giacomello and Pellegrini, 2016; Krols et al., 2016; Sezgin, 2017; Chakkarapani et al., 2018). By these methods, MERCs appear as site of parallel juxtaposition between MT and smooth or rough ER tubules at a distance ranging from 10 to 80 nm. In different tissues, the length and the thickness of the contact zones and the protein composition are variable and differently tuned by signaling pathways, which are under the control of apoptosis, ER stress response, or metabolic dysfunction (Rowland and Voeltz, 2012). In addition to that, the increasing number of proteomic studies and the new toolkits utilized have highlighted a novel group of proteins frequently involved in MERCs (Poston et al., 2013; Hung et al., 2017). Some of them retain the function of tethering factors that hold the two organelles in close proximity (Figure 1). The best established ones include mfn2 (de Brito and Scorrano, 2008; Cosson et al., 2012; Naon et al., 2016), the PhosphoAcidic Cluster Sorting protein 2 (PACS2) (Simmen et al., 2005), the complex formed by VAPB, the PTPIP51 (Gomez-Suaga et al., 2017), and the association between the subtype 3 of the 1,4,5-triphosphate receptor (IP3R3) and the mitochondrial VDAC1 (Szabadkai et al., 2006). However, although the growing effort to define the exact protein composition of MERCs, their high plasticity represents such a great challenge for modern biology that their identity in mammalian cells still remains debated (Rowland and Voeltz, 2012).
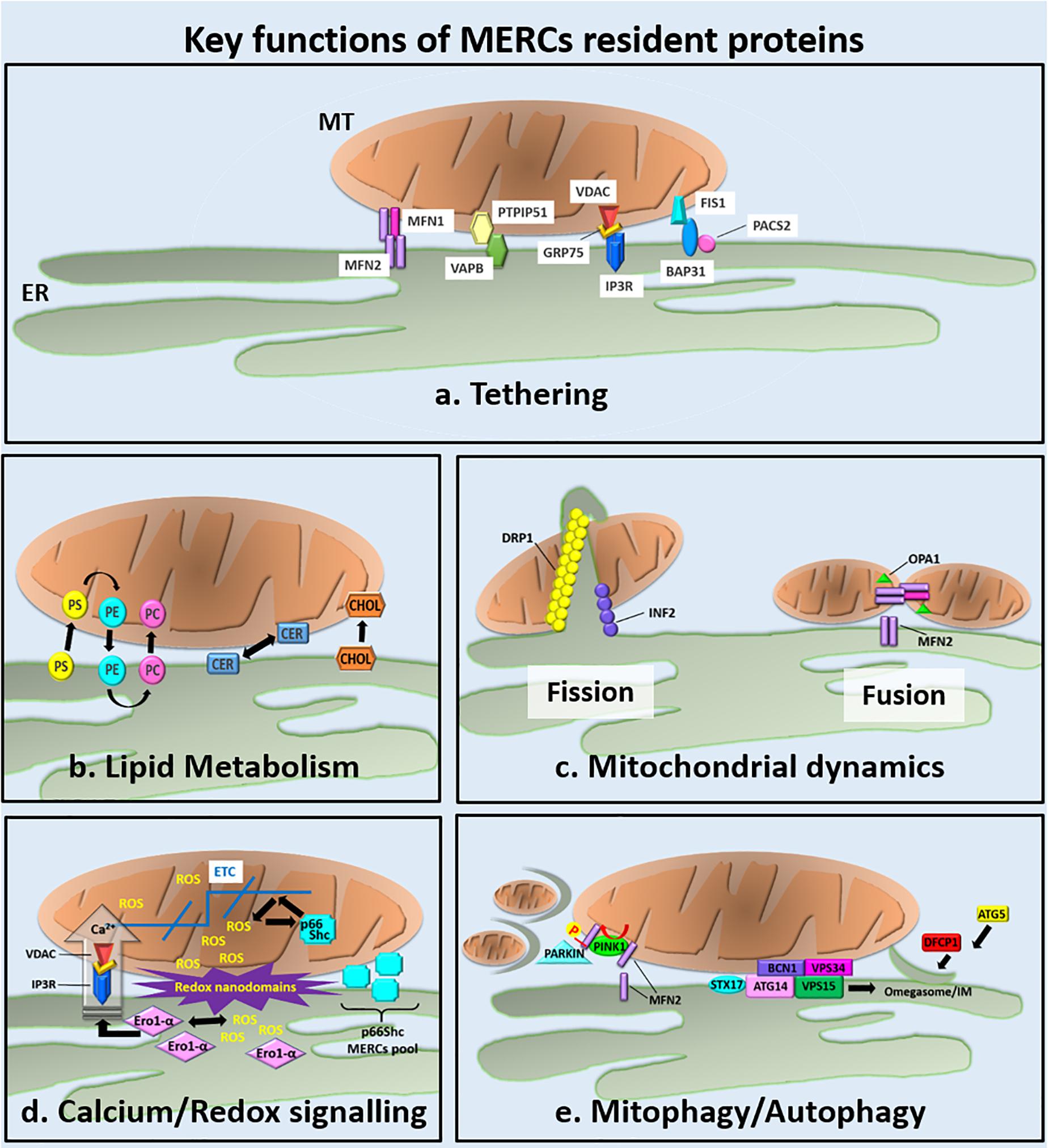
Figure 1. Overview of the key functions of the MERCs resident proteins. (a) The main ER–MT tethering factors are described from left to right. Although it is mainly localized at MT, a small amount of Mfn2 is found at ER, where it participates to the tethering of ER to MT through the formation of ER–Mfn2/MT–Mfn2 homodimers or ER–Mfn2/MT–Mfn1 heterodimers (de Brito and Scorrano, 2008; Cosson et al., 2012; Filadi et al., 2015; Naon et al., 2016). VAPB is an integral ER protein and binds the mitochondrial PTPIP51 protein. The reduced or increased expression of IP3R decreases or increases, respectively, the number of ER–MT contacts (Stoica et al., 2014; Gomez-Suaga et al., 2017). The ER-localized IP3R Ca2+ channel forms a tether with the mitochondrial Ca2+ channel VDAC. Their interaction is mediated by the mitochondrial chaperone 75 kDa Grp75 and modulates Ca2+ fluxes from the ER to the mitochondrial intermembrane space (Mendes et al., 2005; Szabadkai et al., 2006). The ER protein BAP31 interacts with the mitochondrial fission protein Fis1. The BAP31–Fis1 complex bridges the ER–MT interface and regulates the mitochondrial induction of apoptosis. The exact function of PACS2 at MERCS is still unknown but its depletion uncouples the ER from MT by inducing the cleavage of BAP31 and MT fragmentation (Simmen et al., 2005; Iwasawa et al., 2011). (b–e) Key cellular functions handled at MERCs (see the text for details). (b) MERCs are involved in lipid metabolism through the ER–MT exchange of PS, PE, PC, Cers, and Chol (Vance, 2014). (c) MERCs appear as sites promoting mitochondrial fission and fusion. During mitochondrial fission, INF2 recruits DRP1 at MERCs mediating the formation of the constriction ring around the mitochondrial outer membrane (Friedman et al., 2011; Chakrabarti et al., 2018). MFN2, the core component of mitochondrial fusion machinery, was found to localize at MERCs, were, together with OPA1, promotes the fusion of mitochondrial membranes. (d) ER–MT redox crosstalk occurs at MERCs where different mechanisms are responsible for ROS production: Ca2+ flux from the ER to MT through the IP3R/VADC Ca2+ channels, the oxidative folding activity of the ER chaperone Ero-1α, and the electron transport promoted by p66Shc at mitochondrial ETC. The high amount of ROS produced at MERCs generates redox nanodomains at ER–MT interface that modulates ER–MT apposition (Debattisti et al., 2017). (e) MERCs are emerged as important regulators of mitophagy/autophagy. During mitophagy, the MERCs localized Mfn2 is phosphorylated by PINK1. Phosphorylated Mfn2 recruits parkin that, in turn, mediates MFN2 ubiquitination leading to mitophagy initiation (Bockler and Westermann, 2014). Concomitantly, MERCs have been proposed as sites of autophagy initiation (Hamasaki et al., 2013; Bockler and Westermann, 2014). Upon starvation, the ER-resident protein STX17 recruits ATG14 to the MERCs. ATG14, along with other subunits of the Beclin 1/VPS34 complex, became enriched in the MAM and generates concentrated pools of PI3P necessary for IMs formation and expansion. The omegasome marker DFCP1 was also observed to translocate onto these PI3P-enriched regions with further associated markers like ATG5 (Axe et al., 2008; Hamasaki et al., 2013) contributing to the first steps of autophagosomes assembly.
Abnormal production of ROS, leading to oxidative damage to DNA, proteins, and lipids, is a key event contributing to aging and age-associated disorders such as neurodegenerative and motor disorders (Krols et al., 2016; Bernard-Marissal et al., 2018; Fasano et al., 2018), cancer (Danese et al., 2017; De Marco et al., 2018; Iorio et al., 2018; Morciano et al., 2018), metabolic syndromes (MetSs) (Theurey and Rieusset, 2017), as well as cardiovascular diseases (CVDs) (Barja, 2014; Holmström and Finkel, 2014; Wang et al., 2018; De la Fuente and Sheu, 2019). According to recent discoveries, almost all intracellular compartments produce ROS, as side effects of metabolic pathways and in a manner depending on the cell type and/or on the pathophysiological state (Brown and Borutaite, 2012; Holmström and Finkel, 2014). When under control, ROS level is important to modulate many physiological events. Instead, an excessive ROS production can affect molecular structures and function of intracellular compartments, ultimately leading to cellular senescence.
Both MT and ER are sites of ROS production, therefore the communication at MERCs participates to the diffusion of the harmful effects of ROS production inside the cell.
The involvement of MT in age-related oxidative stress has been historically assessed by the mitochondrial free radical theory of aging (Barja, 2014). At mitochondrial level, the respiratory chain and, in particular, Complexes I and III components are the main source of superoxide radical anion (). Together with the ETC Complexes I and III, other mitochondrial enzymes are known sources of ROS, including cytochrome b5 reductase, monoamine oxidase, α-ketoglutarate dehydrogenase, pyruvate dehydrogenase, and the flavoprotein–ubiquinone oxidoreductase (Balaban et al., 2005). Proteomic studies allowed the quantification of carbonylated proteins that are produced by oxidative stress in relation to aging. According to these analyses, during lifespan, mitochondrial proteins result the over represented ones and also showed the greatest increase in carbonylation (Cabiscol et al., 2014).
More recently, several evidences reveal that also the ER is an important source of ROS even though its impact on intracellular oxidative stress is less prominent compared to the MT (Amodio et al., 2011, 2018; Cao and Kaufman, 2014). Within the ER, ROS is produced predominantly by members of cytochrome P450, NADPH oxidase 4 (Nox4), and by the process of oxidative protein folding mediated by the ER oxidoreductin (Ero1) α and β (Amodio et al., 2018). Little is known about the age-associated modulation of ROS production in the ER. Nevertheless, as revealed by some studies looking at the hepatic tissues of aged mice, there are evidences that ER-resident proteins, such as the molecular chaperones PDI and Bip/Grp78, undergo oxidative damage and progressive dysfunction during senescence (Rabek et al., 2003; Nuss et al., 2008). In any case it is important to point out that the transmembrane protein protein kinase RNA-like ER kinase (PERK), a component of the ER stress/unfolded protein response (UPR) machinery (Cao and Kaufman, 2014; Amodio et al., 2018), is found abundant in MAMs (Verfaillie et al., 2012). Curiously, in this contest, PERK would play the role of tethering factor by making tighter the ER–MT membrane contacts during ER stress to ease Ca2+ influx and, as a consequence, the ROS-dependent mitochondrial apoptosis. Thus, this is a further important example, which suggests that MERCs are decisive to mediate inter-organelle signaling leading to decision between cell death or surviving that could take an important part during aging.
Definitely, a large amount of scientific literature demonstrates the involvement of the ER–MT interplay in redox signaling and aging, but the role of MERCs in such a mutual dependency has emerged only in the past years. One example is the redox crosstalk between the ER and MT described for the regulation of Ca2+ signaling. In particular, the work by Booth et al. (2016) demonstrated that Ca2+ fluxes from ER evoke the production of large amount of ROS from MT cristae that are responsible for the generation of redox nanodomains at ER–MT interface. Such a strongly oxidizing environment, found around MERCs, modulates ER–MT apposition through the mitogen-activated protein kinase (MAPK)-dependent control of mitochondrial mobility (Debattisti et al., 2017) and, ultimately, can affect the function of proteins involved in MERCs.
In our opinion, given the importance of oxidative damage in senescence, the ER–MT redox crosstalk cannot be separated by the age-dependent deterioration of proteins. One example comes from the regulation of ryanodine receptors (RyRs) activity. RyR is the ER Ca2+ release channel required for skeletal muscle contraction. Although the localization at MERCs of the RyR has not been defined yet, in the skeletal muscle of aged mice it has been reported how the increased carbonylation and oxidation of RyRs is associated to Ca2+ leak, ROS production, and muscle weakness (Andersson et al., 2011). Interestingly, the forced expression of catalase into the MT rescues RyR oxidation and prevents Ca2+ leak (Umanskaya et al., 2014). These data suggest the existence of bi-directional communication between the ER-localized RyR and MT that, not surprisingly, occur at MERCs.
Many other proteins known to be part of the MERCs structure are directly involved in the ER/MT redox crosstalk in aging and age-associated diseases.
As an example, a well-established case of MERCs-localized protein involved in redox signaling and aging is the 66-kilodalton (66-KDa) isoform of the growth factor adapter Shc (p66Shc) protein. It is well-known that the 66-KDa Shc isoform, a negative regulator of the epidermal growth factor (EGF)-stimulated MAPK pathway, controls oxidative stress and life span in mammals (Okada et al., 1997; Migliaccio et al., 1999). In addition, p66Shc catalyses the electron transfer from cytochrome c to oxygen in the mitochondrial intermembrane space inducing the formation of H2O2, which in turn triggers the activation of the mitochondrial dependent apoptotic pathway (Giorgio et al., 2005). The localization of p66Shc at MT can be induced by oxidative stress insults that exert its critical phosphorylation to Ser36 residue and therefore its association to the MT (Migliaccio et al., 1999; Pinton et al., 2007). Thus, one can envisage a feedback loop regulation, where p66Shc is firstly activated by oxidative stress and, in turn, induces mitochondrial oxidative stress and apoptosis. In this context, the capacity to activate apoptosis in response to ROS renders p66Shc a potential life-span determinant. This hypothesis is supported by a large amount of reports. For example, it is known that the enrichment of Ero1-α at MAM fractions is modulated by the redox state of MERCs (Gilady et al., 2010). This event, in turn, can potentiate Ca2+ signaling at MERCs through the Ero1-α-dependent production of H2O2 and the consequent oxidation of the IP3R (Li et al., 2009; Anelli et al., 2012). p66Shc has attracted considerable attention since it was found that p66Shc-knockout mice exhibited extended lifespan, increased resistance to oxidative and hypoxic stress, and a reduced amount of atherosclerotic and ischemic lesions (Trinei et al., 2002; Napoli et al., 2003; Zaccagnini et al., 2004). Additionally, it was found that primary fibroblasts obtained from centenarians, as well as liver, heart, lungs, skin, and diaphragm from adult mice, express higher levels of p66Shc compared to the younger counterparts (Pandolfi et al., 2005; Lebiedzinska et al., 2009). Likewise, work by Pinton et al. (2007) reported that the induced mitochondrial Ca2+ uptake inversely correlates with the number of passages of cultured mouse embryonic fibroblasts (MEFs) cells, whereas this was not observed in p66Shc-deficient cells. On the same line, oxidative stress induced mitochondrial fragmentation in wild-type MEFs but not in the p66Shc-deficient cells or in MEFs treated with a blocker of p66Shc phosphorylation at Ser36 (Pinton et al., 2007).
All together, these data suggest that p66Shc is a key player in preserving the mitochondrial fitness and, as a consequence, in regulating cellular physiology and senescence. Nevertheless, it is important to point out that its exact localization within the cell is still undefined. Indeed, p66Shc was previously thought to be a cytosolic protein but, recently, it was found also in different MT compartments, in the MAM fraction and in the plasma membrane-associated membranes (PAMs) (Giorgio et al., 2005; Lebiedzinska et al., 2009). Interestingly, its localization at MAMs is considered as the origin of the mitochondrial p66Shc pool (Lebiedzinska et al., 2009). Moreover, the level of p66Shc in the PAM or in the MAM fractions is a function of the age as reported in animal models, where higher levels of p66Shc are detected in the MAM fraction of older animals, indicating a role for this protein in the “induction” of mitochondrial oxidative stress correlated with aging and senescence (Lebiedzinska et al., 2009).
All together, these data confirm the detrimental outcome of redox signaling in aging and strongly indicate that MERCs take a major part in the accumulation of ROS-induced damages during senescence.
Historically, lipid transfer from ER to MT was the first function ascribed to MERCs (Vance, 1990). In mammalian cells, the key event in this biological process is the transfer of PS from ER, where it is synthetized by the PS synthase 1 and 2 (PSS1 and PSS2), to the outer surface of the inner mitochondrial membrane. Here, PS-decarboxylase converts PS in PE, which is transferred back to the ER, where it is methylated to produce PC, the most abundant phospholipid in cellular membranes (Flis and Daum, 2013; Vance, 2014). The transfer of PS from ER to MT, which occurs at MERCs, is the rate-limiting step and becomes essential in condition of ethanolamine restriction. Interestingly, new components involved in the direct transfer of phospholipids between ER and MT have been recently described in yeast and defined as ER–MT encounter structures (ERMESs) and mitochondrial contact site and cristae organizing systems (MICOSs) (Aaltonen et al., 2016; Kojima et al., 2016). ERMESs were proposed to facilitate phospholipid transfer between the ER and MT, while MICOSs appeared to be involved in the beginning of close contacts between mitochondrial outer and inner membranes to facilitate mitochondrial PS import and decarboxylation. Remarkably, homologs of ERMESs and MICOSs have been identified also in mammalian cells. However, their role in the lipid transfer at MERCs in mammals is still under definition.
In addition to phospholipids synthesis, MERCs are involved in the metabolism of Chol and Cers (Bionda et al., 2004; Fujimoto et al., 2012). In particular, steroid hormones are produced from Chol in the MT, where the P450 side chain cleavage enzyme (CYP11A1) converts it to pregnenolone, the steroid precursor (Issop et al., 2015). The rate-limiting step in steroid biosynthesis is the availability of Chol, which is synthetized at the ER level and then transferred to MT. As such, the MERCs resident proteins play a pivotal role in dictating and promoting the Chol efflux to MT.
In this context, very attractive is the observation that caveolin-1 (CAV1), a protein involved in Chol intracellular transport and plasma-membrane organization, was found, by mass spectrometry, as a specific component in MAM fractions. At this level, CAV1 controls Chol levels (Bosch et al., 2011; Sala-Vila et al., 2016). Therefore, taking also in consideration the massive presence of Chol at the ER–MT interface, CAV1 plays a fundamental role. Indeed, the amount of Chol present in MAMs is particularly elevated compared to the ER and MT content, so that lipid rafts-like microdomains are present in MAM (Hayashi and Fujimoto, 2010). Moreover, the level of Chol at ER–MT interface seems to be critical for the integrity and function of MERCs. In this regard, it was found that aberrant Chol accumulation at these ER subdomains, due to CAV1 genetic deficiency, leads to reduced MERCs physical extension (Bosch et al., 2011; Sala-Vila et al., 2016). In agreement with previous data, these observations show that Chol depletion strengthen ER and MT association and reduce de novo synthesis of PS in association to the increase of PE (Fujimoto et al., 2012). Additionally, MERCs also accommodate enzymes needed for synthesizing Cer (Bionda et al., 2004; Stiban et al., 2008). Since increased mitochondrial Cer levels are associated with the permeabilization of the outer mitochondrial membrane and the initiation of apoptosis (Stiban et al., 2008), MERCs represent again crucial modulators of cellular lifespan (Stiban et al., 2008).
Mitochondria–endoplasmic reticulum contacts coordinate lipid membrane composition that, in turn, is a determining factor for cellular lifespan and aging as assessed since when the “membrane theory of aging” was postulated (Zs-Nagy, 1997). In few words, according to this theory aging is directly correlated to the membrane level of unsaturated fatty acids that are more sensitive to peroxidative damage (Pamplona et al., 2002). Indeed, accumulating evidences corroborate this theory and show that decreasing lipid unsaturation contribute positively to lifespan (Puca et al., 2008).
Another effective strategy to control aging and age-related diseases is the caloric restriction leading to a reduced peroxidation index of membrane fatty acids (Lambert et al., 2004; Lopez-Lluch and Navas, 2016).
The best-established example of association between fatty acids unsaturation and lifespan regulation is provided by cardiolipin (CL). CL is predominantly present in the inner mitochondrial membrane, where it governs crucial mitochondrial functions, including the activity and organization of respiratory chain, the regulation of mitochondrial dynamics and apoptosis, through the retention of cytochrome c (Claypool and Koehler, 2012; Hsu and Shi, 2017). CL is a dimeric phospholipid consisting of four acyl chains characterized by mono- or di-unsaturated chains bearing 16–18 carbons, which predispose CL to be highly susceptible to oxidative damage (Schlame et al., 2005; Hsu and Shi, 2017). Indeed, depletion of CL and remodeling of its fatty acids have been associated to aging. In particular, a significant increase in more unsaturated CL fatty acids, predominantly arachidonic and docosahexaenoic acid, was found in the heart of aged, compared to younger mice (Lee et al., 2006). Interestingly, one of the CL remodeling enzymes is the MAM-enriched enzyme acyl-Coa:lysocardiolipin acyltransferase 1 (ALCAT1), which catalyses the “bad” remodeling of CL, since ALCAT1 incorporates CoA loaded with long-chain highly unsaturated fatty acyl chains (Cao et al., 2004). This ALCAT1-mediated pathological remodeling has a broad impact of mitochondrial function, autophagy, and MAM structure, and has been implicated in aging and age-related diseases (Lee et al., 2006; Petrosillo et al., 2008; He and Han, 2014; Hsu and Shi, 2017). Accordingly, ALCAT1-knockout mice have reduced susceptibility to the onset of age-related diseases including obesity, diabetes, hepatosteatosis, and brain dysfunction (Hsu and Shi, 2017). Another MAM-enriched enzyme, the stearoyl-CoA desaturase 1 (SCD1) is involved in the regulation of membrane saturated/mono-unsaturated fatty acids levels and seems to contribute to aging. In particular, its ER-colocalizing partner diacylglycerol O-acyltransferase (DGAT2) was found overexpressed in the skin of aged individuals (Mitchell and Thompson, 1990; Man et al., 2006). In addition, inhibition of SCD1 was associated to reduced accumulation, composition, and saturation of cellular membrane phospholipids, leading to impaired autophagy and autophagosome formation (Ogasawara et al., 2014; Janikiewicz et al., 2015).
Moreover, as detailed later on in this review, MERCs can contribute with an additional mechanism to lifespan determination, which is the regulation of lipid composition (essential) for the autophagy initiation.
Biological aging is a multifactorial process defined as the time-dependent breakdown in the ability to efficiently regenerate tissues and organs. Such a progressive transformation drives to hypofunctional capacity to counteract cell stress (known as homeostenosis) consequent to the exposure to environmental or endogenous agents. Genomic instability, telomere shortening, epigenetic alterations, impairment of proteostasis, altered nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cells depletion, and altered cell-to-cell communication have been proposed as molecular hallmarks of aging (Lopez-Otin et al., 2013). All these events are under the influence of genetic, epigenetic, and environmental factors thus explaining different course of age-related decline of individuals having the same chronological age. Hence, mitochondrial dysfunction is among the acknowledged hallmarks of aging (Lopez-Otin et al., 2013) and the modulation of mitochondrial fission and fusion dynamics is crucial mechanisms engaged by the cell to cope with the decline in mitochondrial activity associated to mitochondrial DNA damage, accumulation of misfolded protein aggregates, or exposure to stress (Sun et al., 2016). Recently, a wide area of research highlights the participation of MERCs to the regulation of MT dynamics and biogenesis (Friedman et al., 2011; Westermann, 2011; Korobova et al., 2013), pointing up to an additional role of MERCs in the biogenesis of aging.
Mitochondria form a highly dynamic network with morphology varying from fragmented to filamentous as the result of the combination of fusion and fission events (Westermann, 2010). Under physiological conditions, the balance between fusion and fission is necessary to optimize mitochondrial function and quality control. However, when the mitochondrial bioenergetic state became critical, fusion or fission events must be modulated in order to isolate damaged MT or to maximize mitochondrial function and, therefore, to prevent cell degeneration and senescence (Twig et al., 2008; Westermann, 2010). Interestingly, proteins involved in mitochondrial fission and fusion were found significantly enriched in MAM fractions (Friedman et al., 2011; Schon and Area-Gomez, 2013).
Indeed, the Drp1, a mitochondrial adaptor involved in the recruitment of the key fission proteins, was found at MERCs (Arasaki et al., 2015; Elgass et al., 2015). At the molecular level, Drp1 is the crucial protein promoting mitochondrial fission, together with its two adaptor proteins, namely the mitochondrial fission factor (Mff) and the mitochondrial Fis1 (Frank et al., 2001; Westermann, 2010). Interestingly, MERCs participate directly in the early steps of mitochondrial fission, by wrapping around the MT and initiating the mitochondrial constriction required for fission and facilitating the recruitment of Drp1 on the mitochondrial outer membrane (Westermann, 2010). In addition, later studies have showed that an ER-bound protein, the INF2 is required for the recruitment of Drp1 (Korobova et al., 2013; Chakrabarti et al., 2018; Steffen and Koehler, 2018). This event requires two steps: the polymerization of actin at ER–MT microdomains and the amplification of Ca2+ efflux from the ER to MT. Both mechanisms are crucial to mediate both the formation of the constriction ring around the mitochondrial outer membrane and to increase the ER–MT contact area, which, in turn, is essential to ensure the execution of mitochondrial fission.
Besides, these observations highlight the existence of a positive feedback between MERCs and MT during fission and this crosstalk is essential in critical situations that require the elimination of MT by mitophagy, as discussed in the next section.
On the other hand, MERCs are equally important for mitochondrial membrane fusion. The core components of the mitochondrial fusion machinery are Mfn1 and Mfn2, along with the OPA1. MFNs are GTPase embedded on the outer mitochondrial membrane and required for the tethering and subsequent fusion of the two lipid bilayers constituting the outer mitochondrial membranes. Following to the realization of the outer membrane fusion, OPA1 mediates the fusion of the inner mitochondrial membranes (Chan, 2006; Westermann, 2010). Interestingly, Mfn2 was found to localize not only at the outer mitochondrial membrane but also at the ER membrane and at MERCs (Koshiba et al., 2004).
From the molecular standpoint, the localization of Mfn2 at MERCs generates multiple effects. As mentioned above, the MERCs-localized Mfn2 seems to be required for tethering MT to the ER and to stabilize MERCs structure through the formation of tight Mfn2–Mfn1 multimer (de Brito and Scorrano, 2008). In support to this notion, the ablation or silencing of Mfn2 expression in mammalian cells disrupts ER morphology and loosens ER–MT interactions and, as a consequence, mitochondrial Ca2+ uptake (de Brito and Scorrano, 2008; Naon et al., 2016). However, there are contrasting reports showing that ablation of Mfn2 increases ER-to-MT Ca2+ transport (Cosson et al., 2012; Filadi et al., 2015). These discrepancies are probably due to the multiple roles played by Mfn2 at MERCs in different cell types and circumstances and/or to the different methodologies deployed to analyze ER–MT juxtaposition (Filadi et al., 2018).
Autophagy exerts a protective role against cellular senescence through the elimination of damaged organelles and intracellular protein aggregates (Rubinsztein et al., 2011; Ranieri et al., 2018). Interestingly, MERCs have been proposed recently as platforms for autophagy initiation and function (Hamasaki et al., 2013; Bockler and Westermann, 2014) on the basis of their role in modulating lipid composition of ER–MT interface. In particular, the artificial increase of PE was found to regulate positively the autophagic flux and, thus, to extend significantly the lifespan in yeast, mammalian cells, and flies (Rockenfeller et al., 2015). In addition, MAM lipid-rafts microdomains and the GD3 ganglioside were reported to participate in the initial organelle scrambling activity that finally leads to the formation of autophagosome (Garofalo et al., 2016).
Besides its role as tethering factor, Mfn2 has emerged as an important regulator of mitophagy, the selective degradation of MT during autophagy activation (Chen and Dorn, 2013; Bockler and Westermann, 2014). In particular, during mitophagy PINK1-phosphorylated Mfn2 functions as a receptor for parkin that, in turn, mediates MFN2 ubiquitination, as a signal to mark damaged MT recruitment and ubiquitination leading to mitophagy initiation. Thus, giving that Mfn2 localizes at MERCs, it is conceivable to speculate that MERCs is primarily involved and participate to mitophagy, rather than to fission and fusion processes.
Studies performed in yeast have provided an interesting model for the role of MERCs in the removal of aged MT from the cell. Indeed, during yeast cells mitosis, tethering activity of MERCs is essential to segregate maternal MT and accumulate toxic protein aggregates, separating those from the MT acquired by the bud, which are largely free of aggregates (Mogk and Bukau, 2014; Zhou et al., 2014). Interestingly, this mechanism, which account for a strategy to rejuvenate cellular environment, involves MERCs and is gradually lost by cells with advanced replicative age, suggesting the participation of MERCs in the mitochondrial quality control (Zhou et al., 2014). More interestingly, in human mammary epithelial cells, a similar event was observed following cellular division. In mammary cells, fine-tuned fission events allow daughter cells, which must maintain stemness properties, to receive newly synthetized MT, while the daughter cells, undergoing to differentiation, receive aged MT (Katajisto et al., 2015). As a consequence, this mechanism is settled to preserve the regenerative capacity of the tissue and prevent senescence. Similarly, recent works have revealed how MERCs were spatially linked to the mitochondrial DNA synthesis both in human and yeast (Murley et al., 2013; Lewis et al., 2016). In other words, it was found that MERCs coordinate replication of mitochondrial nucleoids with mitochondrial fission in order to distribute the proper nucleoids into the daughter MT. Altogether, these evidences confirm the involvement of MERCs in the modulation of mitochondrial fission as a strategy to cope with the establishment of cellular aging.
Giving the crucial role of Mfn2 in ER–MT tethering, mitochondrial fusion, and mitophagy, it is not surprising the increase of experimental evidences and studies linking Mfn2 to aging and age-related diseases. Notably, all the processes related to mitochondrial dynamics ascribed to Mfn2 are important to optimize mitochondrial function and avoid senescence and degeneration. Indeed, Mfn2-knockout in MEFs, cardiomyocyte, and neurons, impairs mitophagy leading to damaged MT accumulation, cell death, and tissue degeneration. Remarkably, these pathological mechanisms involving Mfn2 dysfunction are found in numerous age-related diseases, such as Alzheimer, Parkinson, diabetes, and cardiomyopathies (Filadi et al., 2018).
Not by coincidence, a progressive reduction of Mfn2 that caused impaired mitophagy and accumulation of dysfunctional MT has been reported in the skeletal muscles of aging mice, linked to sarcopenia (Sebastian et al., 2016). In the PolG mice model of premature aging, obtained by expressing a proofreading-deficient version of mtDNA polymerase gamma (PolG mice), mouse cells displayed higher level of the mitochondrial fission protein Fis1, in parallel with increased mitophagy, which likely contributes to the sarcopenic phenotype observed in premature aging (Joseph et al., 2013). In contrast, wild-type aged mice were characterized by higher level of Mfn2 and Mfn1 and reduced levels of Fis1, suggesting increased mitochondrial fusion and reduced mitophagy, probably in response to the physiological accumulation of mitochondrial DNA mutations in the aged muscles. Similarly, increased mitochondrial fusion was also found in senescent mesenchymal stromal stem cells that showed higher levels of Mfn1 and OPA1, together with increased mitochondrial mass and ROS, compared to younger cells at lower passages (Stab et al., 2016).
To summarize, stimulating autophagy in animal models can certainly ameliorate several aging-associated phenotypes. Collectively, these data indicate that the beneficial effects derived from lifespan extension regimens can (at least in part) be explained by the induction of mitophagy. Future studies should provide further insights into how these mechanisms intersect with the mitophagy pathway in order to maintain mitochondrial fitness in vivo.
Aging favors the development of different kind of disorders such as neurodegenerative (Krols et al., 2016; Bernard-Marissal et al., 2018), MetS (Theurey and Rieusset, 2017), as well as CVDs (Barja, 2014; Holmström and Finkel, 2014; Wang et al., 2018; De la Fuente and Sheu, 2019) and cancer (Danese et al., 2017; Morciano et al., 2018).
Among neurodegenerative diseases, late onset Alzheimer and Parkinson’s diseases are the most recurrent age-related disorders. Deranged ER–MT interplay is a common hallmark of neurodegenerative disorders and several studies demonstrate that such pathologies correlate with structural and functional alterations of MERCs (Area-Gomez et al., 2009; Zampese et al., 2011; Cali et al., 2012, 2013). More interestingly, many proteins associated to neurodegenerative disorders are found in the MAMs fractions, although the significance of their presence in MERCs is still under investigation (Area-Gomez et al., 2009; Guardia-Laguarta et al., 2014).
Being the site of β-amyloid peptide (Aβ) production (Schreiner et al., 2015), MERCs play important role in AD pathogenesis. The release of Aβ occurs at MERCs throughout the processing of the amyloid precursor protein (APP) by the γ-secretase complex, composed by the Presenilin 1 and Presenilin 2. In genetic types of AD, mutated Presenilin 2 proteins affect ER–MT connections and their related functions (Zampese et al., 2011). Similar perturbations have been observed also in APP transgenic mouse models as well as in neuronal cells treated with Abβ (Hedskog et al., 2013). In truth, the presence AD-related proteins at MERCs produces quite self-contradicting effect. As a matter of fact, in some cases mutant AD proteins generate a significant up-regulation of MERCs functionality and number (Cali et al., 2012; Ottolini et al., 2013). Intriguingly, even the e4 allele of apolipoprotein E (ApoE4), the most common risk factor for AD-related senile dementia (Liu et al., 2013), has been shown to upregulate the activity of MERCs (Tambini et al., 2016). Instead, in other cases AD proteins generate a decrease of ER–MT tethering (Hedskog et al., 2013).
Analogously, it has been shown that PD-related proteins, such as α-synuclein, Parkin, and protein deglycase (DJ-1), promote ER–MT connections (Cali et al., 2012, 2013; Ottolini et al., 2013; Guardia-Laguarta et al., 2014). In particular, α-synuclein is among the protein found in MAM fractions, where it binds very stably to lipid rafts domain of mitochondrial membranes (Guardia-Laguarta et al., 2014). Likewise, the effect of a-synuclein mutations on the MERCs structure is contradictory. In fact, at least one report showed that expression of mutant forms of α-synuclein decreased ER–MT contacts, whereas another one the result was an increased number of MERCs in PD cells (Cali et al., 2012).
In synthesis, the variable response of MERCs to either AD- or PD-related proteins suggests the requirement of more work to better define the molecular events underlying MERCs function in the different pathological circumstances.
Many evidences show that ER stress is a common hallmark in neuronal diseases such as AD and PD, but also in Huntington’s disease (HD) and amyotrophic lateral sclerosis (ALS) (Remondelli and Renna, 2017). Different conditions can induce the accumulation of unfolded proteins within the ER lumen leading to ER stress (Amodio et al., 2011; Wang and Kaufman, 2016). This state initiates an adaptive response, known as the UPR composed by three integrated pathways: the PERK, the inositol requiring enzyme 1 (IRE1), and the activating transcription factor 6 (ATF6) pathways of the UPR (Walter and Ron, 2011). UPR pathways get started to re-establish proteostasis or, in the case of an unsuccessful recovery, to induce cell death. Several evidences demonstrate a tight link between the ER stress response and neurodegenerative disorders and indicate the UPR pathways as true therapeutic target for such diseases (Remondelli and Renna, 2017).
Remarkably, many reports show that MERCs are closely linked to ER stress and UPR. Indeed, this was shown for MERCs tethering factors such as MFN-2 and for the VAPB (Kanekura et al., 2006; de Brito and Scorrano, 2008; Gkogkas et al., 2008; De Vos et al., 2012; Muñoz et al., 2013; Stoica et al., 2014). In addition, MERCs accommodate essential ER chaperones such as GRP78/BiP, calnexin, calreticulin, ERp44, ERp57, and the Sigma 1 receptor (Mori et al., 2013). Interestingly, along with ER chaperones, MERCs also host the UPR transducers IRE1 and PERK. In particular at MERCs, IRE1 interacts with Sig1R to mediate ER-to-MT survival (Hayashi and Su, 2007; Mori et al., 2013). Furthermore, at MERCs, PERK acts either as tethering factor or as a regulator of ROS transport and, as a consequence, transmission of the apoptotic signaling (Verfaillie et al., 2012). Remarkably, its activity is negatively controlled by the MERCs tethering factor Mfn2 (Muñoz et al., 2013).
In recent years, many compounds have been identified for their ability to reduce or induce ER stress but their effect on MERCs has not been yet examined (Remondelli and Renna, 2017). As we previously discussed, in many of neurodegenerative diseases, the PERK signaling pathway is overactive and this event is considered responsible for neuronal cell death in either AD or PD models. As a consequence, suppression of PERK signaling by using specific PERK inhibitors (i.e., GSK2606414) has neuroprotective effect (Moreno et al., 2013). Interestingly, in AD, PERK activation is correlated to enhanced memory loss and β-amyloidogenesis, which occurs at the ER–MT interface. Remarkably, in vivo models of AD, memory impairment is restored by silencing PERK expression (Ma et al., 2013). Similarly, genetic or chemical inhibition of IRE1 signaling retains a protective role against AD by reducing the expression of APP and, as a consequence, βA deposition (Duran-Aniotz et al., 2017).
More interestingly, PERK inhibition was also shown to reduce the MFN contacts at the ER–MT interface. Recently, in an early-onset PD model (PARK20) (Fasano et al., 2018), in which PERK is constitutively activated inhibition of PERK phosphorylation by GSK2606414 generates beneficial effects on the ER stress mitochondrial dysfunction (Amodio et al., 2019). Intriguingly, in pink1 and parkin mutants PD models, suppression of PERK signaling produces neuroprotective effect, most probably by reducing the MFN contacts with the ER that cause enhanced ER stress signaling (Celardo et al., 2016).
All together, these evidences convincingly reinforce the concept that strategies planned to reduce ER stress levels may impact positively on neurodegenerative diseases and strongly stimulate further investigation to identify in MERCs components the most advantageous molecular targets for therapeutic intervention.
Another age-related condition is the MetS, a clinical state affecting about 20% of the aged population in technologically advanced countries. MetS that is characterized by the concomitant presence of cardiometabolic risk factors such as obesity, insulin resistance, dyslipidemia, and hypertension (O’Neill and O’Driscoll, 2015). Several studies underlined the effect of such risk factors on the ER–MT membrane connections. Also in this regard, MERCs organization and function shows different response. As an example, in animal models of obesity, liver cells show increased number of MERCs which in turn causes mitochondrial Ca2+ overload followed by mitochondrial dysfunction (Arruda et al., 2014). Instead, in hypothalamic cells the reduction of MERCs obtained by the ablation tethering factor Mfn2 results in ER stress-induced leptin resistance, hyperphagia, reduced energy expenditure, and finally obesity (Schneeberger et al., 2013).
Insulin play essential role in aging and structural integrity of MERCs is a crucial requirement for efficient insulin pathway (Tubbs et al., 2014) as revealed by the observation that depletion of MERCs structural components impairs insulin signaling. Alternatively, overexpression of MERCs proteins increases insulin signaling, as well as pharmacologic rescue of insulin sensitivity re-establishes ER–MT connections, indicating the reciprocal control of both processes (Tubbs et al., 2014). In addition, and as we would expect, ER stress contributes to impaired insulin synthesis in pancreatic β cells and insulin resistance (Shang et al., 2014), suggesting that the pharmacological inhibition of the ER stress pathway could ameliorate insulin deficiency.
Cardiovascular diseases are often associated to MetS and, in particular to obesity, insulin resistance and type 2 diabetes (T2D). In cardiomyocytes, MERCs control of mitochondrial Ca2+ uptake is essential to guarantee insulin signaling. As a result, altered cardiomyocyte metabolism and insulin resistance are associated with cardiac hypertrophy (Kolwicz and Tian, 2011; Gutiérrez et al., 2014). Interestingly, in either patients or in animal models of pulmonary artery hypertrophy (PAH), deficiency of MERCs tethering factor Mfn-2 disrupts ER–MT membrane contacts and contributes to pulmonary artery smooth muscle cells hyperproliferation (Ryan et al., 2013).
Essential hypertension is one of the most frequent disturb in the aging. One possibility by which hypertension and other CVDs might arise is the endothelial dysfunction (ED) associated with aging through enhanced oxidative stress. Several studies describe the interconnection between ER stress, UPR, and oxidative stress (OS) in the pathogenesis of ED-derived CVDs (Amodio et al., 2018). Thus, in ED, ER stress and the UPR pathways represent a promising system to test new molecules and develop new therapeutic methodologies for the treatment of ED in aged patients.
Finally, the progressive deterioration of physiological organ function occurring during aging is a primary risk factor for cancer development. Cancer cells exhibit altered expression of proteins, including oncogenes that directly affect MERCs functionality and, in particular, the ER–MT Ca2+ transport (Marchi et al., 2014; Stewart et al., 2015). This perturbation results in various characteristics of cancer cells such as resistance to apoptosis, deregulation of cell proliferation, metastatic activity, and a metabolic rewiring. Generally speaking, this happens throughout the expression of oncogenes, such as Mcl-1, Bcl-2, and Bcl-XL, which, by diverse mechanisms, prevent mitochondrial Ca2+ overload and cancer cell death (Bittremieux et al., 2016). This event happens by enhancing the IP3R-dependent Ca2+ outflow and eluding the ER-dependent mitochondrial Ca2+ overload upon stress conditions. On the other hand, expression of tumor suppressor genes, encoding for example the protein phosphatase and tensin homolog (PTEN), PML, BRCA2, favors Ca2+ transfer from the ER to MT thus having as consequence pro-apoptotic effects on cancer cells (Bononi et al., 2013; Marchi et al., 2014).
Thus, MERCs are advantageous sites where chemotherapy, hormone therapies, targeted cancer drugs, and bisphosphonates or other anti-tumoral therapies (De Marco et al., 2018; Iorio et al., 2018) might operate to interfere with the function of oncogenes or to restore ER–mitochondrial Ca2+ transfer in order to re-establish apoptosis sensitivity of cancer cells or inhibit pro-tumorigenic effects.
As a final point, MERCs participate to the regulation of many functions perturbed in the various age-related diseases. Further examinations of MERCs alterations in these disorders could certainly help to discover novel therapeutic targets to restore the correct ER–MT interplay and prevent the defects by which the diverse pathological features might develop.
Despite the fact that all the findings we argued are on their own significant, the identification of further mechanisms involved in the control of MERCs function in health and disease will supply critical information on how such a conserved function is regulated and will hopefully provide us with an even more detailed picture of the molecular environment at MERCs. This could, ultimately, pave the way for the discovery of novel therapeutic targets. Indeed, the identification of pharmacological therapies for age-associates dysfunctions remains an ambitious task in biomedical science and such a necessity will become even more serious in consideration of the increased lifespan estimated in the future for the human population.
Overall, the findings discussed in this review confirm that aging is indeed associated to mitochondrial dysfunction as a consequence of the dysregulation of mitochondrial fission, fusion, and mitophagy. Notably, MERCs, holding many of the proteins involved in these processes, may participate directly to the development or prevention of MT-mediated deterioration of cellular physiology, which is observed during aging.
In this context, the identification of novel components and additional factors involved in the MERCs dependent control of cross-talks between ER and MT could be employed to find more pharmacological approaches to be utilized to attenuate or delay the onset of age-associated diseases.
Certainly, a better understanding of the molecular events leading to pathological state for each specific age-related condition is critical in order to adopt the targeting of MERCs-dependent pathways for the cure of age-related disorders. Therefore, additional work should be addressed to identify putative MERCs targets in order to develop novel drugs for the treatment of specific clinical conditions. Finally, identifying cell type-specific regulators of the MERCs could be another approach to precisely modulate the mitochondrial fitness in the desired cellular context (i.e., tissue, organ), without affecting the homeostasis of other cells. For instance, the identification of the MERCs components selectively operating in neuronal cells would be helpful to discover more specific and potentially safe targets in the context of neurodegenerative diseases.
All authors drafted the manuscript. OM generated the illustration. GA and PR wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ATG 14/5, autophagy related 14/5; BAP31, B-cell receptor-associated protein 31; BECLIN-1, BCL-2-interacting protein; Ca2+, calcium; Cer, ceramide; Chol, cholesterol; DFCP1, double FYVE-containing protein; Drp1, dynamin-related protein; ER, endoplasmic reticulum; Ero1- α, ER oxidoreductase 1 alpha; ETC, electron transport chain; Fis1, fission 1 protein; Grp75, glucose-regulated protein 75; IM, isolation membranes; INF2, inverted formin 2; IP3R, inositol 1,4,5-trisphosphate receptor; Mfn1/2, mitofusin-1/-2; MT, mitochondria; OPA1, optic atrophy protein 1; p66Shc, 66 kDa proto-oncogene Src homologous-collagen homolog; PACS2, phosphofurin acidic cluster sorting protein 2; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI3P, phosphatidylinositol 3-phosphate; PINK1, PTEN-induced putative kinase 1; PS, phosphatidylserine; PTPIP51, protein tyrosine phosphatase interacting protein-51; ROS, reactive oxygen species; Stx17, syntaxin 17; VAPB, vesicle-associated membrane protein-associated protein B; VDAC, voltage-dependent anion-selective channel; VPS34/15, vacuolar protein sorting-associated protein 34/15.
Aaltonen, M. J., Friedman, J. R., Osman, C., Salin, B. J., di Rago, P., Nunnari, J., et al. (2016). Micos and phospholipid transfer by Ups2-Mdm35 organize membrane lipid synthesis in mitochondria. J. Cell Biol. 213, 525–534. doi: 10.1083/jcb.201602007
Amodio, G., Moltedo, O., Faraonio, R., and Remondelli, P. (2018). Targeting the endoplasmic reticulum unfolded protein response to counteract the oxidative stress-induced endothelial dysfunction. Oxid. Med. Cell Longev. 2018:4946289. doi: 10.1155/2018/4946289
Amodio, G., Moltedo, O., Fasano, D., Zerillo, L., Oliveti, M., Di Pietro, P., et al. (2019). PERK-mediated unfolded protein response activation and oxidative stress in PARK20 fibroblasts. Front. Neurosci. 13:673. doi: 10.3389/fnins.2019.00673
Amodio, G., Moltedo, O., Monteleone, F., D’Ambrosio, C., Scaloni, A., Remondelli, P., et al. (2011). Proteomic signatures in thapsigargin-treated hepatoma cells. Chem. Res. Toxicol. 24, 1215–1222. doi: 10.1021/tx200109y
Andersson, D. C., Betzenhauser, M. J., Reiken, S., Meli, A. C., Umanskaya, A., Xie, W., et al. (2011). Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab. 14, 196–207. doi: 10.1016/j.cmet.2011.05.014
Anelli, T., Bergamelli, L., Margittai, E., Rimessi, A., Fagioli, C., Malgaroli, A., et al. (2012). Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (Mam). Antioxid. Redox Signal. 16, 1077–1087. doi: 10.1089/ars.2011.4004
Arasaki, K., Shimizu, H., Mogari, H., Nishida, N., Hirota, N., Furuno, A., et al. (2015). A role for the ancient snare syntaxin 17 in regulating mitochondrial division. Dev. Cell 32, 304–317. doi: 10.1016/j.devcel.2014.12.011
Area-Gomez, E., de Groof A. J., Boldogh, I., Bird, T. D., Gibson, G. E., Koehler, C. M., et al. (2009). Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 175, 1810–1816. doi: 10.2353/ajpath.2009.090219
Arruda, A. P., Pers, B. M., Parlakgü, G., Güney, E., Inouye, K., and Hotamisligil, G. (2014). Chronic enrichment of hepatic endoplasmic reticulum–mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 20, 1427–1435. doi: 10.1038/nm.3735
Axe, E. L., Walker, S. A., Manifava, M., Chandra, P., Roderick, H. L., Habermann, A., et al. (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 182, 685–701. doi: 10.1083/jcb.200803137
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Barja, G. (2014). The mitochondrial free radical theory of aging. Prog. Mol. Biol. Transl. Sci. 127, 1–27. doi: 10.1016/B978-0-12-394625-6.00001-5
Bernard-Marissal, N., Chrast, R., and Schneider, B. L. (2018). Endoplasmic reticulum and mitochondria in diseases of motor and sensory neurons: a broken relationship? Cell Death Dis. 9:333. doi: 10.1038/s41419-017-0125-1
Bionda, C., Portoukalian, J., Schmitt, D., Rodriguez-Lafrasse, C., and Ardail, D. (2004). Subcellular compartmentalization of ceramide metabolism: mam (Mitochondria-Associated Membrane) and/or mitochondria? Biochem. J. 382(Pt 2), 527–533. doi: 10.1042/BJ20031819
Bittremieux, M., Parys, J. B., Pinton, P., and Bultynck, G. (2016). ER functions of oncogenes and tumor suppressors: modulators of intracellular Ca(2+) signaling. Biochim. Biophys. Acta 1863(6 Pt B), 1364–1378. doi: 10.1016/j.bbamcr.2016.01.002
Bockler, S., and Westermann, B. (2014). Er-Mitochondria contacts as sites of mitophagosome formation. Autophagy 10, 1346–1347. doi: 10.4161/auto.28981
Bononi, A., Bonora, M., Marchi, S., Missiroli, S., Poletti, F., Giorgi, C., et al. (2013). Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ. 20, 1631–1643. doi: 10.1038/cdd.2013.77
Booth, D. M., Enyedi, B., Geiszt, M., Varnai, P., and Hajnoczky, G. (2016). Redox nanodomains are induced by and control calcium signaling at the Er-Mitochondrial interface. Mol. Cell 63, 240–248. doi: 10.1016/j.molcel.2016.05.040
Bosch, M., Mari, M., Herms, A., Fernandez, A., Fajardo, A., Kassan, A., et al. (2011). Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr. Biol. 21, 681–686. doi: 10.1016/j.cub.2011.03.030
Brown, G. C., and Borutaite, V. (2012). There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 12, 1–4. doi: 10.1016/j.mito.2011.02.001
Cabiscol, E., Tamarit, J., and Ros, J. (2014). Protein carbonylation: proteomics, specificity and relevance to aging. Mass Spectrom. Rev. 33, 21–48. doi: 10.1002/mas.21375
Cali, T., Ottolini, D., Negro, A., and Brini, M. (2012). Alpha-synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 287, 17914–17929. doi: 10.1074/jbc.M111.302794
Cali, T., Ottolini, D., Negro, A., and Brini, M. (2013). Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca(2+) transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 1832, 495–508. doi: 10.1016/j.bbadis.2013.01.004
Cao, J., Liu, Y., Lockwood, J., Burn, P., and Shi, Y. (2004). A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-coa: lysocardiolipin acyltransferase (Alcat1) in mouse. J. Biol. Chem. 279, 31727–31734. doi: 10.1074/jbc.M402930200
Cao, S. S., and Kaufman, R. J. (2014). Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 21, 396–413. doi: 10.1089/ars.2014.5851
Celardo, I., Costa, A. C., Lehmann, S., Jones, C., Wood, N., Mencacci, N. E., et al. (2016). Mitofusin-mediated. ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 7:e2271. doi: 10.1038/cddis.2016.173
Chakkarapani, S. K., Zhang, P., and Kang, S. H. (2018). 3D super-localization of intracellular organelle contacts at live single cell by dual-wavelength synchronized fluorescence-free imaging. Anal. Bioanal. Chem. 410, 1551–1560. doi: 10.1007/s00216-017-0805-9
Chakrabarti, R., Ji, W. K., Stan, R. V., de Juan Sanz, J., Ryan, T. A., and Higgs, H. N. (2018). Inf2-mediated actin polymerization at the Er Stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J. Cell Biol. 217, 251–268. doi: 10.1083/jcb.201709111
Chan, D. C. (2006). Dissecting mitochondrial fusion. Dev. Cell 11, 592–594. doi: 10.1016/j.devcel.2006.10.009
Chen, Y., and Dorn, G. W. II (2013). Pink1-Phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science 340, 471–475. doi: 10.1126/science.1231031
Claypool, S. M., and Koehler, C. M. (2012). The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41. doi: 10.1016/j.tibs.2011.09.003
Copeland, D. E., and Dalton, A. J. (1959). An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J. Biophys. Biochem. Cytol. 5, 393–396. doi: 10.1083/jcb.5.3.393
Cosson, P., Marchetti, A., Ravazzola, M., and Orci, L. (2012). Mitofusin-2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS One 7:e46293. doi: 10.1371/journal.pone.0046293
Csordas, G., Renken, C., Varnai, P., Walter, L., Weaver, D., Buttle, K. F., et al. (2006). Structural and functional features and significance of the physical linkage between Er and mitochondria. J. Cell Biol. 174, 915–921. doi: 10.1083/jcb.200604016
Danese, A., Patergnani, S., Bonora, M., Wieckowski, M. R., Previati, M., Giorgi, C., et al. (2017). Calcium regulates cell death in cancer: roles of the mitochondria and mitochondria-associated membranes (Mams). Biochim. Biophys. Acta Bioenerg. 1858, 615–627. doi: 10.1016/j.bbabio.2017.01.003
de Brito, O. M., and Scorrano, L. (2008). Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610. doi: 10.1016/j.celrep.2017.10.060
De la Fuente, S., and Sheu, S. S. (2019). Sr-Mitochondria communication in adult cardiomyocytes: a close relationship where the Ca(2+) has a lot to say. Arch. Biochem. Biophys. 663, 259–268. doi: 10.1016/j.abb.2019.01.026
De Marco, M., Basile, A., Iorio, V., Festa, M., FalcoA, B., Ranieri, B., et al. (2018). Role of Bag3 in cancer progression: a therapeutic opportunity. Semin. Cell Dev. Biol. 78, 85–92. doi: 10.1016/j.semcdb.2017.08.049
De Vos, K. J., Mórotz, G. M., Stoica, R., Tudor, E. L., Lau, K. F., Ackerley, S., et al. (2012). VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 21, 1299–1311. doi: 10.1093/hmg/ddr559
Debattisti, V., Gerencser, A. A., Saotome, M., Das, S., and Hajnoczky, G. (2017). Ros control mitochondrial motility through P38 and the motor adaptor miro/trak. Cell Rep. 21, 1667–1680. doi: 10.1016/j.celrep.2017.10.060
Duran-Aniotz, C., Cornejo, V. H., Espinoza, S., Ardiles, Á. O., Medinas, D. B., Salazar, C., et al. (2017). IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 34, 489–506. doi: 10.1007/s00401-017-1694-x
Elgass, K. D., Smith, E. A., LeGros, M. A., Larabell, C. A., and Ryan, M. T. (2015). Analysis of Er-mitochondria contacts using correlative fluorescence microscopy and Soft X-Ray tomography of mammalian cells. J. Cell Sci. 128, 2795–2804. doi: 10.1242/jcs.169136
Fasano, D., Parisi, S., Pierantoni, G. M., De Rosa, A., Picillo, M., Amodio, G., et al. (2018). Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by synj1 mutations. Cell Death Dis. 9:385. doi: 10.1038/s41419-018-0410-7
Filadi, R., Greotti, E., Turacchio, G., Luini, A., Pozzan, T., and Pizzo, P. (2015). Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. U.S.A. 112, E2174–E2181. doi: 10.1073/pnas.1504880112
Filadi, R., Pendin, D., and Pizzo, P. (2018). Mitofusin 2: from functions to disease. Cell Death Dis. 9:330. doi: 10.1038/s41419-017-0023-6
Flis, V. V., and Daum, G. (2013). Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 5:a013235. doi: 10.1101/cshperspect.a013235
Frank, S., Gaume, B., Bergmann-Leitner, E. S., Leitner, W. W., Robert, E. G., Catez, F., et al. (2001). The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell 1, 515–525. doi: 10.1016/S1534-5807(01)00055-7
Friedman, J. R., Lackner, L. L., West, M., Di Benedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). Er Tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Fujimoto, M., Hayashi, T., and Su, T. P. (2012). The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 417, 635–639. doi: 10.1016/j.bbrc.2011.12.022
Garofalo, T., Matarrese, P., Manganelli, V., Marconi, M., Tinari, A., Gambardella, L., et al. (2016). Evidence for the involvement of lipid rafts localized at the er-mitochondria associated membranes in autophagosome formation. Autophagy 12, 917–935. doi: 10.1080/15548627.2016.1160971
Giacomello, M., and Pellegrini, L. (2016). The coming of age of the mitochondria-er contact: a matter of thickness. Cell Death Differ. 23, 1417–1427. doi: 10.1038/cdd.2016.52
Gilady, S. Y., Bui, M., Lynes, E. M., Benson, M. D., Watts, R., Vance, J. E., et al. (2010). Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (Mam). Cell Stress Chaperones 15, 619–629. doi: 10.1007/s12192-010-0174-1
Giorgio, M., Migliaccio, E., Orsini, F., Paolucci, D., Moroni, M., Contursi, C., et al. (2005). Electron transfer between cytochrome C and P66shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221–233. doi: 10.1016/j.cell.2005.05.011
Gkogkas, C., Middleton, S., Kremer, A. M., Wardrope, C., Hannah, M., Gillingwate, T. H., et al. (2008). VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 17, 1517–1526. doi: 10.1093/hmg/ddn040
Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D. P., and Miller, C. C. J. (2017). The Er-Mitochondria tethering complex Vapb-Ptpip51 regulates autophagy. Curr. Biol. 27, 371–385. doi: 10.1016/j.cub.2016.12.038
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Gutiérrez, T., Parra, V., Troncoso, R., Pennanen, C., Contreras-Ferrat, A., Vasquez-Trincado, C., et al. (2014). Alteration in mitochondrial Ca2+ uptake disrupts insulin signaling in hypertrophic cardiomyocytes. Cell Commun. Signal. 12:68. doi: 10.1186/s12964-014-0068-4
Hamasaki, M., Furuta, N., Matsuda, A., Nezu, A., Yamamoto, A., Fujita, N., et al. (2013). Autophagosomes form at er-mitochondria contact sites. Nature 495, 389–393. doi: 10.1038/nature11910
Hayashi, T., and Fujimoto, M. (2010). Detergent-Resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol. Pharmacol. 77, 517–528. doi: 10.1124/mol.109.062539
Hayashi, T., and Su, T. P. (2007). Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610. doi: 10.1016/j.cell.2007.08.036
He, Q., and Han, X. (2014). Cardiolipin remodeling in diabetic heart. Chem. Phys. Lipids 179, 75–81. doi: 10.1016/j.chemphyslip.2013.10.007
Hedskog, L., Pinho, C. M., Filadi, R., Rönnbäck, A., Hertwig, L., Wiehager, B., et al. (2013). Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. U.S.A. 110, 7916–7921. doi: 10.1073/pnas.1300677110
Holmström, K. M., and Finkel, T. (2014). Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 15, 411–421. doi: 10.1038/nrm3801
Hsu, P., and Shi, Y. (2017). Regulation of autophagy by mitochondrial phospholipids in health and diseases. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 114–129. doi: 10.1016/j.bbalip.2016.08.003
Hung, V., Lam, S. S., Udeshi, N. D., Svinkina, T., Guzman, G., Mootha, V. K., et al. (2017). Proteomic mapping of cytosol-facing outer mitochondrial and Er Membranes in living human cells by proximity biotinylation. eLife 6:e24463. doi: 10.7554/eLife.24463
Iorio, V., Rosati, A., D’Auria, R., De Marco, M., Marzullo, L., Basile, A., et al. (2018). Combined effect of Anti-Bag3 and Anti-Pd-1 treatment on macrophage infiltrate, Cd8(+) T cell number and tumour growth in pancreatic cancer. Gut 67, 780–782. doi: 10.1136/gutjnl-2017-314225
Issop, L., Fan, J., Lee, S., Rone, M. B., Basu, K., Mui, J., et al. (2015). Mitochondria-associated membrane formation in hormone-stimulated leydig cell steroidogenesis: role of Atad3. Endocrinology 156, 334–345. doi: 10.1210/en.2014-1503
Iwasawa, R., Mahul-Mellier, L. A., Datler, C., Pazarentzos, E., and Grimm, S. (2011). Fis1 and Bap31 bridge the mitochondria-Er interface to establish a platform for apoptosis induction. EMBO J. 30, 556–568. doi: 10.1038/emboj.2010.346
Janikiewicz, J., Hanzelka, K., Dziewulska, A., Kozinski, K., Dobrzyn, P., Bernas, T., et al. (2015). Inhibition of Scd1 impairs palmitate-derived autophagy at the step of autophagosome-lysosome fusion in pancreatic beta-cells. J. Lipid Res. 56, 1901–1911. doi: 10.1194/jlr.M059980
Joseph, A. M., Adhihetty, P. J., Wawrzyniak, N. R., Wohlgemuth, S. E., Picca, A., Kujoth, G. C., et al. (2013). Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS One 8:e69327. doi: 10.1371/journal.pone.0069327
Kanekura, K., Nishimoto, I., Aiso, S., and Matsuoka, M. (2006). Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J. Biol. Chem. 28, 30223–30232. doi: 10.1074/jbc.M605049200
Katajisto, P., Dohla, J., Chaffer, C. L., Pentinmikko, N., Marjanovic, N., Iqbal, S., et al. (2015). Stem cells. asymmetric apportioning of aged mitochondriabetween daughter cells is required for stemness. Science 348, 340–343. doi: 10.1126/science.1260384
Kojima, R., Endo, T., and Tamura, Y. (2016). A phospholipid transfer function of er-mitochondria encounter structure revealed in vitro. Sci. Rep. 6:30777. doi: 10.1038/srep30777
Kolwicz, S. C. Jr., and Tian, R. (2011). Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 90, 194–201. doi: 10.1093/cvr/cvr071
Korobova, F., Ramabhadran, V., and Higgs, H. N. (2013). An actin-dependent step in mitochondrial fission mediated by the er-associated formin inf2. Science 339, 464–467. doi: 10.1126/science.1228360
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi: 10.1126/science.1099793
Krols, M., van Isterdael, G., Asselbergh, B., Kremer, A., Lippens, S., Timmerman, V., et al. (2016). Mitochondria-Associated membranes as hubs for neurodegeneration. Acta Neuropathol. 131, 505–523. doi: 10.1007/s00401-015-1528-7
Lambert, A. J., Portero-Otin, M., Pamplona, R., and Merry, B. J. (2004). Effect of ageing and caloric restriction on specific markers of protein oxidative damage and membrane peroxidizability in rat liver mitochondria. Mech. Ageing Dev. 125, 529–538. doi: 10.1016/j.mad.2004.06.002
Lebiedzinska, M., Duszynski, J., Rizzuto, R., Pinton, P., and Wieckowski, M. R. (2009). Age-Related changes in levels of P66shc and serine 36-Phosphorylated P66shc in organs and mouse tissues. Arch. Biochem. Biophys. 486, 73–80. doi: 10.1016/j.abb.2009.03.007
Lee, H. J., Mayette, J., Rapoport, S. I., and Bazinet, R. P. (2006). Selective remodeling of cardiolipin fatty acids in the aged rat heart. Lipids Health Dis. 5:2. doi: 10.1186/1476-511X-5-2
Lewis, S. C., Uchiyama, L. F., and Nunnari, J. (2016). Er-Mitochondria contacts couple mtdna synthesis with mitochondrial division in human cells. Science 353:aaf5549. doi: 10.1126/science.aaf5549
Li, G., Mongillo, M., Chin, K. T., Harding Ron, H. D., Marks, A. R., and Tabas, I. (2009). Role of Ero1-Alpha-Mediated stimulation of inositol 1,4,5-Triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186, 783–792. doi: 10.1083/jcb.200904060
Liu, C. C., Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Lopez-Lluch, G., and Navas, P. (2016). Calorie restriction as an intervention in ageing. J. Physiol. 594, 2043–2060. doi: 10.1113/JP270543
Ma, T., Trinh, M. A., Wexler, A. J., Bourbon, C., Gatti, E., Pierre, P., et al. (2013). Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. doi: 10.1038/nn.3486
Man, W. C., Miyazaki, M., Chu, K., and Ntambi, J. (2006). Colocalization of Scd1 and Dgat2: implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J. Lipid Res. 47, 1928–1939. doi: 10.1194/jlr.M600172-JLR200
Marchi, S., Giorgi, C., Oparka, M., Duszynski, J., Wieckowski, M. R., and Pinton, P. (2014). Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol. Cell Oncol. 1:e956469. doi: 10.4161/23723548.2014.956469
Mendes, C. C., Gomes, D. A., Thompson, M., Souto, N. C., Goes, T. S., Goes, A. M., et al. (2005). The Type III Inositol 1,4,5-Trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 280, 40892–40900. doi: 10.1074/jbc.M506623200
Migliaccio, E., Giorgio, M., Mele, S., Pelicci, G., Reboldi, P., Pandolfi, P. P., et al. (1999). The P66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 402, 309–313. doi: 10.1038/46311
Mitchell, W. D., and Thompson, T. L. II (1990). Psychiatric distress in systemic lupus erythematosus outpatients. Psychosomatics 31, 293–300. doi: 10.1016/s0033-3182(90)72167-6
Mogk, A., and Bukau, B. (2014). Mitochondria tether protein trash to rejuvenate cellular environments. Cell 159, 471–472. doi: 10.1016/j.cell.2014.10.007
Morciano, G., Marchi, S., Morganti, C., Sbano, L., Bittremieux, M., Kerkhofs, M., et al. (2018). Role of mitochondria-associated Er Membranes in calcium regulation in cancer-specific settings. Neoplasia 20, 510–523. doi: 10.1016/j.neo.2018.03.005
Moreno, J. A., Halliday, M., Molloy, C., Radford, R., Verity, N., Axten, J. M., et al. (2013). Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 5:206ra138. doi: 10.1126/scitranslmed.3006767
Mori, T., Hayashi, T., Hayashi, E., and Su, T. P. (2013). Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 8:e76941. doi: 10.1371/journal.pone.0076941
Muñoz, J. P., Ivanova, S., Sánchez-Wandelmer, J., Martínez-Cristóbal, P., Noguera, E., Sancho, A., et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 32, 2348–2361. doi: 10.1038/emboj.2013.168
Murley, A., Lackner, L. L., Osman, C., West, M., Voeltz, G. K., Walter, P., et al. (2013). Er-Associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in Yeast. eLife 2:e00422. doi: 10.7554/eLife.00422
Naon, D., Zaninello, M., Giacomello, M., Varanita, T., Grespi, F., Lakshminaranayan, S., et al. (2016). Critical reappraisal confirms that mitofusin 2 is an endoplasmic reticulum-mitochondria tether. Proc. Natl. Acad. Sci. U.S.A. 113, 11249–11254. doi: 10.1073/pnas.1606786113
Napoli, C., Martin-Padura, I., de Nigris, F., Giorgio, M., Mansueto, G., Somma, P., et al. (2003). Deletion of the P66shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high-fat diet. Proc. Natl. Acad. Sci. U.S.A. 100, 2112–2116. doi: 10.1073/pnas.0336359100
Nuss, J. E., Choksi, K. B., DeFord, J. H., and Papaconstantinou, J. (2008). Decreased enzyme activities of chaperones Pdi and Bip in aged mouse livers. Biochem. Biophys. Res. Commun. 365, 355–361. doi: 10.1016/j.bbrc.2007.10.194
Ogasawara, Y., Itakura, E., Kono, N., Mizushima, N., Arai, H., Nara, A., et al. (2014). Stearoyl-Coa desaturase 1 activity is required for autophagosome formation. J. Biol. Chem. 289, 23938–23950. doi: 10.1074/jbc.M114.591065
Okada, S., Kao, A. W., Ceresa, B. P., Blaikie, P., Margolis, B., and Pessin, J. E. (1997). The 66-Kda Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. J. Biol. Chem. 272, 28042–28049. doi: 10.1074/jbc.272.44.28042
O’Neill, S., and O’Driscoll, L. (2015). Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes. Rev. 16, 1–12. doi: 10.1111/obr.12229
Ottolini, D., Cali, T., Negro, A., and Brini, M. (2013). The Parkinson disease related protein DJ-1 counteracts mitochondrial impairment induced by the tumor suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 22, 2152–2168. doi: 10.1093/hmg/ddt068
Pamplona, R., Barja, G., and Portero-Otin, M. (2002). Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: a homeoviscous-longevity adaptation? Ann. N. Y. Acad. Sci. 959, 475–490. doi: 10.1111/j.1749-6632.2002.tb02118.x
Pandolfi, S., Bonafe, M., Di Tella, L., Tiberi, L., Salvioli, S., Monti, D., et al. (2005). P66(Shc) is highly expressed in fibroblasts from centenarians. Mech. Ageing Dev. 126, 839–844. doi: 10.1016/j.mad.2005.03.004
Petrosillo, G., Matera, M., Casanova, G., Ruggiero, F. M., and Paradies, G. (2008). Mitochondrial dysfunction in rat brain with aging involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 53, 126–131. doi: 10.1016/j.neuint.2008.07.001
Pinton, P., Rimessi, A., Marchi, S., Orsini, F., Migliaccio, E., Giorgio, M., et al. (2007). Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant P66shc. Science 315, 659–663. doi: 10.1126/science.1135380
Poston, C. N., Krishnan, S. C., and Bazemore-Walker, C. R. (2013). In-Depth proteomic analysis of mammalian mitochondria-associated membranes (Mam). J. Proteomics 79, 219–230. doi: 10.1016/j.jprot.2012.12.018
Puca, A. A., Andrew, P., Novelli, V., Anselmi, C. V., Somalvico, F., Cirillo, N. A., et al. (2008). Fatty acid profile of erythrocyte membranes as possible biomarker of longevity. Rejuvenation Res. 11, 63–72. doi: 10.1089/rej.2007.0566
Rabek, J. P., Boylston, W. H. III, and Papaconstantinou, J. (2003). Carbonylation of Er chaperone proteins in aged mouse liver. Biochem. Biophys. Res. Commun. 305, 566–572. doi: 10.1016/S0006-291X(03)00826-X
Ranieri, R., Ciaglia, E., Amodio, G., Picardi, P., Proto, M. C., Gazzerro, P., et al. (2018). N6-Isopentenyladenosine dual targeting of Ampk and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 25, 353–367. doi: 10.1038/cdd.2017.165
Remondelli, P., and Renna, M. (2017). The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front. Mol. Neurosci. 10:187. doi: 10.3389/fnmol.2017.00187
Rizzuto, R., Pinton, P., Carrington, W., Fay, F. S., Fogarty, K. E., Lifshitz, L. M., et al. (1998). Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766. doi: 10.1126/science.280.5370.1763
Rockenfeller, P., Koska, M., Pietrocola, F., Minois, N., Knittelfelder, O., Sica, V., et al. (2015). Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ. 22, 499–508. doi: 10.1038/cdd.2014.219
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Rubinsztein, D. C., Marino, G., and Kroemer, G. (2011). Autophagy and Aging. Cell 146, 682–695. doi: 10.1016/j.cell.2011.07.030
Ryan, J. J., Marsboom, G., Fang, Y. H., Toth, P. T., Morrow, E., Luo, N., et al. (2013). PGC1α-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 187, 865–878. doi: 10.1164/rccm.201209-1687OC
Sala-Vila, A., Navarro-Lerida, I., Sanchez-Alvarez, M., Bosch, M., Calvo, C., Lopez, J. A., et al. (2016). Interplay between hepatic mitochondria-associated membranes, lipid metabolism and caveolin-1 in mice. Sci. Rep. 6:27351. doi: 10.1038/srep27351
Schlame, M., Ren, M., Xu, Y., Greenberg, M. L., and Haller, I. (2005). Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 138, 38–49. doi: 10.1016/j.chemphyslip.2005.08.002
Schneeberger, M., Dietrich, M. O., Sebastiaìn, D., Imbernoìn, M., CastanÞo, C., Garcia, A., et al. (2013). Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155, 172–187. doi: 10.1016/j.cell.2013.09.003
Schon, E. A., and Area-Gomez, E. (2013). Mitochondria-Associated Er Membranes in alzheimer disease. Mol. Cell Neurosci. 55, 26–36. doi: 10.1016/j.mcn.2012.07.011
Schreiner, B., Hedskog, L., Wiehager, B., and Ankarcrona, M. (2015). Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 43, 369–374. doi: 10.3233/JAD-132543
Sebastian, D., Sorianello, E., Segales, J., Irazoki, A., Ruiz-Bonilla, V., Sala, D., et al. (2016). Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 35, 1677–1693. doi: 10.15252/embj.201593084
Sezgin, E. (2017). Super-resolution optical microscopy for studying membrane structure and dynamics. J. Phys. Condens. Matter 29:273001. doi: 10.1088/1361-648X/aa7185
Shang, L., Hua, H., Foo, K., Martinez, H., Watanabe, K., Zimmer, M., et al. (2014). β-Cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 63, 923–933. doi: 10.2337/db13-0717
Simmen, T., Aslan, J. E., Blagoveshchenskaya, A. D., Thomas, L., Wan, L., Xiang, Y., et al. (2005). Pacs-2 controls endoplasmic reticulum-mitochondria communication and bid-mediated apoptosis. EMBO J. 24, 717–729. doi: 10.1038/sj.emboj.7600559
Sood, A., Jeyaraju, D. V., Prudent, J., Caron, A., Lemieux, P., McBride, H. M., et al. (2014). A Mitofusin-2-dependent inactivating cleavage of opa1 links changes in mitochondria cristae and Er Contacts in the postprandial liver. Proc. Natl. Acad. Sci. U.S.A. 111, 16017–16022. doi: 10.1073/pnas.1408061111
Stab, B. R. II, Martinez, L., Grismaldo, A., Lerma, A., Gutierrez, M. L., Barrera, L. A., et al. (2016). Mitochondrial functional changes characterization in young and senescent human adipose derived Mscs. Front. Aging Neurosci. 8:299. doi: 10.3389/fnagi.2016.00299
Steffen, J., and Koehler, C. M. (2018). Er-Mitochondria contacts: actin dynamics at the Er Control mitochondrial fission via calcium release. J. Cell Biol. 217, 15–17. doi: 10.1083/jcb.201711075
Stewart, T. A., Yapa, K. T., and Monteith, G. R. (2015). Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 1848, 2502–2511. doi: 10.1016/j.bbamem.2014.08.016
Stiban, J., Caputo, L., and Colombini, M. (2008). Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 49, 625–634. doi: 10.1194/jlr.M700480-JLR200
Stoica, R., De Vos, K. J., Paillusson, S., Mueller, S., Sancho, R. M., Lau, K. F., et al. (2014). Er-Mitochondria associations are regulated by the Vapb-Ptpip51 interaction and are disrupted by Als/Ftd-Associated Tdp-43. Nat. Commun. 5:3996. doi: 10.1038/ncomms4996
Sun, N., Youle, R. J., and Finkel, T. (2016). The mitochondrial basis of aging. Mol. Cell 61, 654–666. doi: 10.1016/j.molcel.2016.01.028
Szabadkai, G., Bianchi, K., Varnai, P., De Stefani, D., Wieckowski, M. R., Cavagna, D., et al. (2006). Chaperone-Mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 175, 901–911. doi: 10.1083/jcb.200608073
Tambini, M. D., Pera, M., Kanter, E., Yang, H., Guardia-Laguarta, C., Holtzman, D., et al. (2016). ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 17, 27–36. doi: 10.15252/embr.201540614
Theurey, P., and Rieusset, J. (2017). Mitochondria-Associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol. Metab. 28, 32–45. doi: 10.1016/j.tem.2016.09.002
Trinei, M., Giorgio, M., Cicalese, A., Barozzi, S., Ventura, A., Migliaccio, E., et al. (2002). A P53-P66shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 21, 3872–3878. doi: 10.1038/sj.onc.1205513
Tubbs, E., Theurey, P., Vial, G., Bendridi, N., Bravard, A., Chauvin, M. A., et al. (2014). Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63, 3279–3294. doi: 10.2337/db13-1751
Twig, G., Hyde, B., and Shirihai, O. S. (2008). Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta 1777, 1092–1097. doi: 10.1016/j.bbabio.2008.05.001
Umanskaya, A., Santulli, G., Xie, W., Andersson, D. C., Reiken, S. R., and Marks, A. R. (2014). Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc. Natl. Acad. Sci. U.S.A. 111, 15250–15255. doi: 10.1073/pnas.1412754111
Vance, J. E. (1990). Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265, 7248–7256.
Vance, J. E. (2014). Mam (Mitochondria-Associated Membranes) in Mammalian cells: lipids and beyond. Biochim. Biophys. Acta 1841, 595–609. doi: 10.1016/j.bbalip.2013.11.014
Verfaillie, T., Rubio, N., Garg, A. D., Bultynck, G., Rizzuto, R., Decuypere, J. P., et al. (2012). PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 19, 1880–1891. doi: 10.1038/cdd.2012.74
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, M., and Kaufman, R. J. (2016). Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335. doi: 10.1038/nature1704
Wang, W., Fernandez-Sanz, C., and Sheu, S. S. (2018). Regulation of mitochondrial bioenergetics by the non-canonical roles of mitochondrial dynamics proteins in the heart. Biochim. Biophys. Acta Mol. Basis Dis. 1864(Pt B), 1991–2001. doi: 10.1016/j.bbadis.2017.09.004
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884. doi: 10.1038/nrm3013
Westermann, B. (2011). Organelle dynamics: Er Embraces mitochondria for fission. Curr. Biol. 21, R922–R924. doi: 10.1016/j.cub.2011.10.010
Zaccagnini, G., Martelli, F., Fasanaro, P., Magenta, A., Gaetano, C., Di Carlo, A., et al. (2004). P66shca modulates tissue response to hindlimb ischemia. Circulation 109, 2917–2923. doi: 10.1161/01.CIR.0000129309.58874.0F
Zampese, E., Fasolato, C., Kipanyula, M. J., Bortolozzi, M., Pozzan, T., and Pizzo, P. (2011). Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. U.S.A. 108, 2777–2782. doi: 10.1073/pnas.1100735108
Zhou, C., Slaughter, B. D., Unruh, J. R., Guo, F., Yu, Z., Mickey, K., et al. (2014). Organelle-Based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell 159, 530–542. doi: 10.1016/j.cell.2014.09.026
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A role for mitochondria in Nlrp3 inflammasome activation. Nature 469, 221–225. doi: 10.1038/nature09663
Keywords: aging, age-associated diseases, oxidative stress, senescence, endoplasmic reticulum, mitochondria
Citation: Moltedo O, Remondelli P and Amodio G (2019) The Mitochondria–Endoplasmic Reticulum Contacts and Their Critical Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 7:172. doi: 10.3389/fcell.2019.00172
Received: 30 May 2019; Accepted: 07 August 2019;
Published: 21 August 2019.
Edited by:
Adelaide Fernandes, University of Lisbon, PortugalReviewed by:
Paola Rizzo, University of Ferrara, ItalyCopyright © 2019 Moltedo, Remondelli and Amodio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Remondelli, cHJlbW9uZGVsbGlAdW5pc2EuaXQ=; Giuseppina Amodio, Z2Ftb2Rpb0B1bmlzYS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.