 Javier Raboso-Gallego
Javier Raboso-Gallego Ana Casado-García
Ana Casado-García Marta Isidro-Hernández
Marta Isidro-Hernández Carolina Vicente-Dueñas
Carolina Vicente-Dueñas- 1Experimental Therapeutics and Translational Oncology Program, Instituto de Biología Molecular y Celular del Cáncer, CSIC/Universidad de Salamanca, Salamanca, Spain
- 2Institute of Biomedical Research of Salamanca (IBSAL), Salamanca, Spain
Leukemogenesis is considered to be a process by which a normal cell acquires new but aberrant identity in order to disseminate a malignant clonal population. Under this setting, the phenotype of the leukemic cells is identical to the leukemia-initiating cell in which the genetic insult is taking place. Thus, with some exceptions, B-cell and T-cell childhood leukemias are supposed to arise from B- or T-committed cells. In contrast, several recent studies have revealed that genetic alterations may act in a “hit-and-run” way in the cell-of-origin by imposing the tumor cell identity giving rise to either B-cell or T-cell leukemias. This novel mechanism of cell transformation is mediated by an epigenetic priming mechanism that is established by the initial genetic lesion. This initial hit might be unnecessary for the subsequent tumor evolution and conservation, being the epigenetic priming the engine for the tumor evolution.
Childhood Acute Lymphoblastic Leukaemia (ALL)
Childhood Acute lymphoblastic leukaemia (ALL) is the most common malignancy in children (Hunger and Mullighan, 2015). Childhood ALL is composed of two main immunophenotypes that are identified by distinctive hematopoietic lineage markers: B- and T-ALL. Approximately, 85% of ALL cases are paediatric B-ALL while T-ALL comprises the remaining 15% (Hunger and Mullighan, 2015).
Cytogenetic analyses have revealed that persistent structural and copy number genomic aberrations are hallmarks of ALL (Iacobucci and Mullighan, 2017; Pui et al., 2019b). Cytogenomic subtyping usually occurs through visual chromosome analysis by the use of fluorescence in situ hybridization (FISH), karyotyping via G-banding, and/or chromosomal microarray (Mrozek et al., 2009).
Currently, up to 90% of childhood ALL patients can be categorized into distinct genetic subtypes (Pui et al., 2019a), the prognostic characteristics and treatment response differ among them (Roberts and Mullighan, 2015; Iacobucci and Mullighan, 2017).
The genetic alterations are specific of each ALL immunophenotype being for examples hyperdiploidy (50–67 chromosomes); hypodiploidy (44 chromosomes or fewer); BCR-ABL1, ETV6-RUNX1, or TCF3-PBX1 fusions; PAX5 or ETV6 mutations, MLL rearrangements, or intrachromosomal amplification of chromosome 21 (iAMP21) specific for B-ALL, whilst alterations in LMO2, TAL1, TAL2, TLX1, TLX2, or HOXA are characteristics of T-ALL (Pui et al., 2019a).
A newly revised taxonomy of B-ALL incorporates 23 subtypes well-defined by chromosomal rearrangements, sequence mutations or heterogeneous genomic changes (Gu et al., 2019), highlighting the genetic heterogeneity of this disease. Most of these molecular changes are acquired, not inherited, as only a small number of leukaemias are associated with inherited genetic syndromes as is the case for germline mutations in PAX5 (Auer et al., 2014; Martin-Lorenzo et al., 2018), ETV6 (Moriyama et al., 2015; Noetzli et al., 2015) or IKZF1 (Churchman et al., 2018).
With few exceptions, it is thought that childhood ALL is the result of the aberrant transformation of progenitor B- or T-cells in the bone marrow into leukemic cells giving rise to B- or T-ALL. This could be in line with the fact that the genetic alterations are only present in one immunophenotype and do not share in both. However, the mechanism triggering this leukemic conversion is not well comprehended and the cell-of-origin or the leukemia-initiating cell (LIC) seems not to be necessarily a committed B- or a T-cell (Rodriguez-Hernandez et al., 2017; Garcia-Ramirez et al., 2018; Martin-Lorenzo et al., 2018). Likewise, it has been recently described for MLL-AF4+ infant B-ALL and TCF3-ZNF384+ B-ALL which both have a fetal liver hematopoietic stem cell as the cell-of-origin (Agraz-Doblas et al., 2019; Bueno et al., 2019).
Evidence from twin studies and revisions of neonatal blood spots suggest that most initiating events (“first hit”) occur during fetal development in utero (Ford et al., 1993; Gale et al., 1997; Wiemels et al., 1999; Greaves et al., 2003; Alpar et al., 2015; Bateman et al., 2015; Schafer et al., 2018; Bueno et al., 2019). But not all the preleukemic carriers will develop the disease, suggesting that a secondary event (“hit”) needs to take place in the preleukemic cell to fully transform it into a leukemic one. Under this scenario, the cancer cell-of-origin would be the initially healthy cell that will be primed by an oncogenic hit and that will give rise to a preleukemic cell. And the LIC would be the preleukemic cell that has acquired secondary mutations, and as a result, it promotes the complete transformation of the preleukemic clone. Within the context of the so-called “two-hit hypothesis,” the childhood ALL cell-of-origin and the LIC is still the focus of ongoing debate. The main problem that difficults to solve this issue is that human leukemia is diagnosed in the final stages of the transformation process when the LIC is fully converted into the leukemic one. For this reason, the identification of the cell-of-origin and the LIC, and the monitoring of the natural evolution of the disease from the first stages of the leukemia onset is almost impossible in humans (Cobaleda and Sanchez-Garcia, 2009).
For chromosomal translocations, like the ones found in lymphoid tumors, the chromosome breaks are usually caused by either AID or the RAG complex (Mahowald et al., 2008; Nussenzweig and Nussenzweig, 2010; Gostissa et al., 2011). As a matter of fact, Rag-1 expression has been detected in early progenitors in both mice and humans (Boiers et al., 2013, 2018). This early immune-restricted progenitor co-expressed myeloid and lymphoid lineage programs and contributed to both myeloid and lymphoid lineages in the embryo. Rag-1, therefore, provides a mechanistic possibility for translocations to happen at very early hematopoietic developmental stages in ALL. Thus, the cell-of-origin that suffers the initial hit of transformation (eg. Chromosomal translocation) will have the potential of transformation into a myeloid or lymphoid committed cell. But as there is genotype-phenotype correlation between the genetic alteration and the tumor phenotype in ALL, it seems to be reasonable that the genetic insult is setting the tumor phenotype in the uncommitted cell-of-origin.
Besides aberrant genetic modifications, epigenetic irregularities have been shown to trigger the pathogenesis of childhood ALL (Taylor et al., 2007; Chaber et al., 2017). It has been described that aberrant methylation is associated with prognosis (Wong et al., 2000), cytogenetic subtype (Zheng et al., 2004), prediction of treatment outcome (Milani et al., 2010) and relapse (Matsushita et al., 2004; Hogan et al., 2011) in childhood acute leukemias. And also, most different cytogenetic subtypes of ALL share recurrent DNA methylation changes presumably because of those changes are involved in their pathogenesis (Wong et al., 2012). In this sense, aberrant epigenetic modifications (DNA methylation, histone modification, and microRNA alterations) have been extensively reviewed elsewhere (Burke and Bhatla, 2014; Nordlund and Syvanen, 2018), but the focus of the present review is to pay attention to the epigenetic modifications taking place at the beginning of the transformation process, being the epigenetic change the step that leads to the preleukemic stage establishment.
Epigenetic Priming as Cell Transformation Process
There is a new accepted explanation that clarifies the connection between the specific immunophenotype in childhood ALL and the specific genetic disorder provoking it. As we have mentioned before, there are B- and T-ALL specific genetic lesions that are not shared between these two biological entities, there is a strong genotype-phenotype association. It has been traditionally thought that the driving genetic event was taking place in a committed B- or T-cell and this was the reason for the immunophenotype correlation. In recent years, it has been proposed that the genetic insult has the capacity to induce genetic priming in the cell-of-origin that imposes the ultimate phenotype of the transformed cell. This last option highlights a different role for the oncogenic hit. In this new concept of transformation mechanism, the first aberrant genetic insult is imposing a specific cell-differentiation program in the cancer cell-of-origin that will define the later phenotype of the tumor cells (for instance, being a B- or a T-cell). But of note, is that the initial oncogenic alteration essential for tumor initiation will not be necessary upon the differentiation program is started. So, in the later stages of transformation, the oncogene is not required and for this reason, it is not a good target for therapies because it does not have a critical role during tumor progression (Figure 1).
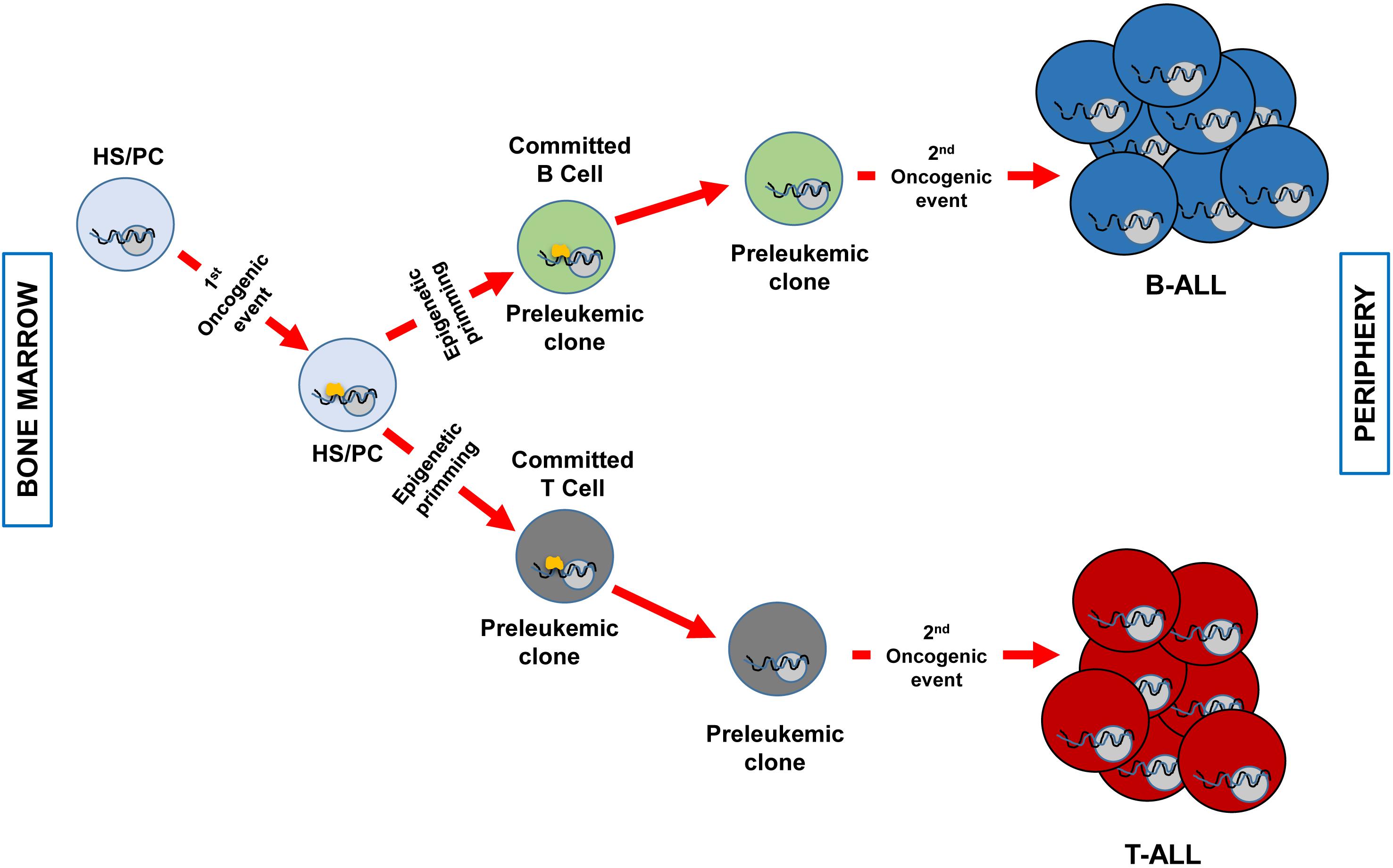
Figure 1. Clonal evolution of B and T-ALL imposed by epigenetic priming in genetically predisposed mouse models. The first genetic event that takes place in an HS/PC induces specific epigenetic priming that will determine the commitment of the preleukemic clone phenotype. For example, ETV6-RUNX1 (Rodriguez-Hernandez et al., 2017) or BCR-ABLp190 (Martin-Lorenzo et al., 2018) expression will prime a B-cell commitment, whilst the overexpression of LMO2 will command T-cell priming (Garcia-Ramirez et al., 2018). Afterward, a second hit will transform the preleukemic clone in a fully transformed leukemic cell (B o T). At this stage, the first genetic event (represented in yellow) is not required anymore and has already primed the cell-of-origin into a specific differentiation cell program. A less efficient epigenetic priming could also occur in a more differentiated cell compartment as it has been described for LMO2 in T-ALL (Garcia-Ramirez et al., 2018).
This also fits with the fact that specific epigenomic profiles can distinguish between cell lineage differentiation stages in the hematopoietic system and also in leukemic cells (Hu and Shilatifard, 2016; Carrillo-de-Santa-Pau et al., 2017). This epigenetic reprogramming has been proved in several animal cancer models that resemble human disease quite well. Conducting genome-wide DNA methylation profiling in these models [MafB in multiple myeloma (MM) (Vicente-Duenas et al., 2012), Bcl6 in Diffuse Large B cell Lymphoma (DLBCL) (Green et al., 2014), BCR-ABLp210 in chronic myeloid leukemia (CML) (Perez-Caro et al., 2009)] it has been identified a molecular reprogramming profile in the tumor-initiating cells (the hematopoietic stem/progenitor cells (HS/PC)), where the oncogene was expressed, that was still present in the tumor cells where the oncogene was switched off. In these models, the oncogene function in a “hit-and-run” way that works at an initial step of cell development to reprogram HS/PCs for malignancy. This epigenetic memory would maintain gene expression profiles throughout several cell generations, but not including modifications in the DNA sequence of the cells and in the absence of the initiating drivers of cell transformation, the expression of the oncogene. Interestingly, this tumoral epigenetic rewiring in progenitors cells is not limited to the hematopoietic system, but it is also a shared process that likewise has a role in non-hematopoietic tumors and has been reviewed elsewhere (Vicente-Duenas et al., 2018).
Epigenetic Priming in Childhood B-ALL
B-ALL is a clonal malignant disease that initiates in a single cell and is characterized by an enlargement of precursor B-cells that are phenotypically similar to healthy precursors B-cells. This is why it has been argued that a committed B cell should be the B-ALL cell-of-origin (le Viseur et al., 2008), but its origin is still the subject of continuing discussion.
The ETV6-RUNX1 chromosomal alteration is the most common aberrant genetic finding in childhood B-ALL (Pui et al., 2019a). One of the most extreme examples of genotype-phenotype relationship is the ETV6-RUNX1 genetic alterations, as its detection is only associated with B-ALL and not to T-ALL or other types of cancer. The ETV6-RUNX1 oncogenic lesion is found in neonatal cord blood in up to 5% of new-borns (Lausten-Thomsen et al., 2011; Schafer et al., 2018), those carrying the oncogenic lesion are the preleukemic clones. But only a few ETV6-RUNX1 carriers will develop the disease, as the incidence of the disease in human beings is much lower than the detection of the preleukemic clone in healthy children. These findings suggest that the preleukemic clone (the ETV6-RUNX1 positive cell) is susceptible to malignant transformation through the acquisition of supplementary secondary genetic events that will be acting as drivers of leukemogenesis. Consequently, ETV6-RUNX1 does not give the impression to be a traditional dominant oncogene as it acts as a driver at the initial stage of the preleukemic cell appearance, in the hematopoietic stem cell subset. But later on, further drivers taking place in the already predisposed cell, the preleukemic clone, are necessary to fully transform the cell in a leukemic one and, at that time, ETV6-RUNX1 fusion gene will not be necessary for leukemic propagation. But its role might be prompting an epigenetic priming in an uncommitted cell subset of the hematopoietic system that induces an aberrant B cell differentiation program that is later on susceptible of transformation. This has been proved in an ETV6-RUNX1 mouse model where the fusion gene was restricted to hematopoietic stem/progenitor cells (HS/PCs) with the purpose of reveal this new possible way of action of ETV6-RUNX1 (the Sca1-ETV6-RUNX1 mice) (Rodriguez-Hernandez et al., 2017). This mouse model mimicked human disease quite well and settled solely pB-ALL with a low incidence, similar to human leukemias, but only after being exposed to common infections (Rodriguez-Hernandez et al., 2017). Hence, this model resembles human disease not only in incidence, phenotype and genetics features but also linking the exposure to environmental factors, infections, to the development of the disease as it has been hypothesized in humans after epidemiological studies (Greaves, 2018). Leukemic cells arising in these mice not only resemble the human phenotype but also share the genetic defects identity in ETV6-RUNX1 human leukemia (Rodriguez-Hernandez et al., 2017). As the genetic alteration is not expressed in the bulk of the B committed tumoral cells, ETV6-RUNX1 is acting as a “hit and run” oncogene that primes the cell-of-origin (HS/PC) to a B cell program preleukemic clone that due to a second hit (exposure to infection) is fully transformed into a leukemic cell (Figure 1).
The ETV6-RUNX1 model is not the only case of B-ALL epigenetic priming (Table 1). The BCR-ABLp190 can also confer genetic susceptibility to B-ALL in a similar way to ETV6-RUNX1 (Martin-Lorenzo et al., 2018). In this case, the restriction of BCR-ABLp190 to HS/PC also primed the cells to a preleukemic stage and afterward this genetic insult is not required anymore. In this model, the secondary genetic event is not induced by the exposure to infection, and forcing the presence of the second hit (Pax5 deletion) from the beginning of the preleukemic clone appearance, induces cell transformation in 100% of the cases (Martin-Lorenzo et al., 2018). This suggests that the first hit (BCR-ABLp190) primes the cells but the second hit (Pax5 deletion) is acting as the real driver of the pB-ALL. It looks as if the first genetic lesion (ETV6-RUNX1 or BCR-ABLp190) primes the cell-of-origin in a precise cell program that makes the emergence of oncogene-specific- preleukemic clones. The specific priming of the preleukemic clone will make it be susceptible to fully transformation only by exact secondary hits. Consequently, the second insult will depend on the type of priming imposed by the oncogene to be able to fully transform the preleukemic clone into the LIC. In this sense, the ETV6-RUNX1-preleukemic clones are susceptible to transformation upon exposure to infections whilst BCR-ABLp190- preleukemic clones are not.
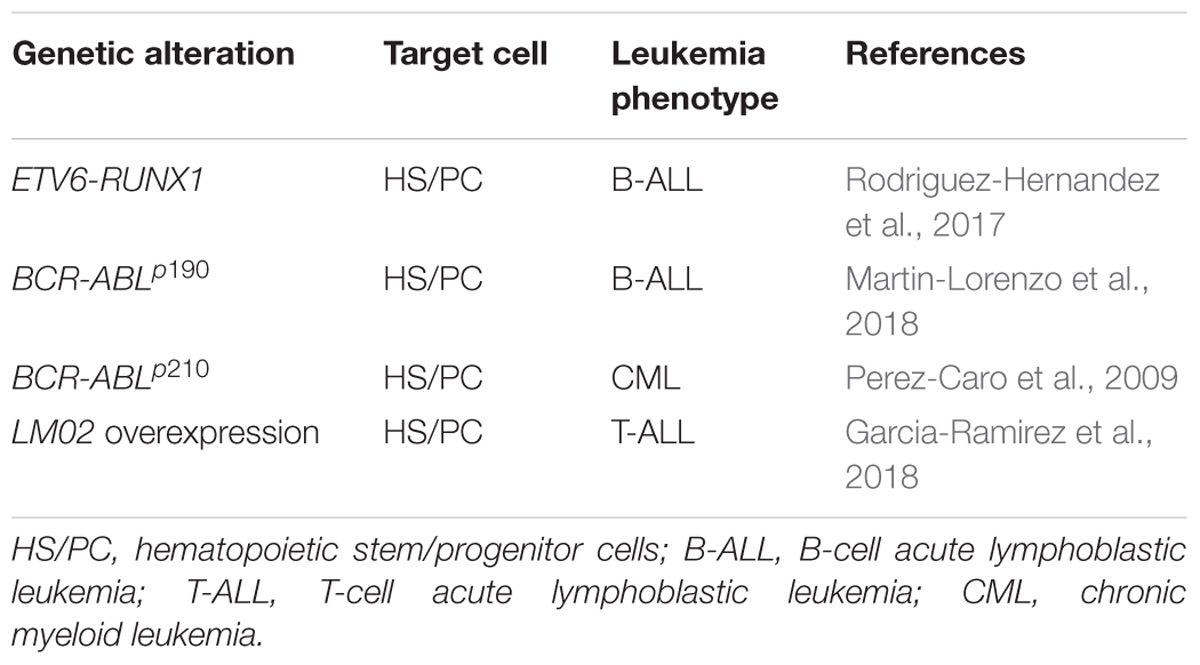
Table 1. Driving of leukaemia by a malignant epigenetic stem cell priming.
It has been shown that cytogenetic subtypes of ALL showed a clear correlation with methylation profile (Milani et al., 2010; Figueroa et al., 2013; Nordlund et al., 2013; Gabriel et al., 2015; Chaber et al., 2017), such signatures have been used to design DNA methylation classifiers (Nordlund et al., 2015). These observations are in line with the fact that the initial genetic insult is imposing a specific leukemic phenotype mediated by the priming of distinctive epigenetic profiles suggesting a genetic basis for the tumor specific epigenetic changes. Those epigenetic programs are also decisive for normal B cell differentiation (Martin-Subero and Oakes, 2018).
Another recent finding connecting the epigenetic priming in childhood B-ALL comes from the study of concordant BCP-ALL twins with TCF3-ZNF384 and PTPN11 mutations (Bueno et al., 2019). In this study, the cell-of-origin was identified as a stem cell or a nascent fetal progenitor. The preleukemic clone harboring the first hit, the identical TCF3-ZNF384 rearrangement shared by both twins, evolved to the leukemic stage in parallel in both patients (Bueno et al., 2019). The described acquisition of a shared hypomethylation profile in the tumor cells of this pair of monozygotic twins fits with the idea that the first hit is priming the cell-of-origin in a specific epigenetic route that depends on the initial event of transformation. In addition, the initial translocation models the cell-of-origin in such a way that favors the acquisition of secondary mutations that do not have to be identical, although they do affect the same target (D61Y and T507K activating mutations of PTPN11). Later on, leukemia evolves independently of the initial hit and secondary twin-specific mutations accumulate during tumor progression (Bueno et al., 2019). The conserved hypomethylation profile described in Bueno et al. (2019) in the leukemic cells also fits with previous findings in mouse models of epigenetic reprogramming (Vicente-Duenas et al., 2012, 2019; Green et al., 2014). As an example, restricted expression of BCR-ABLp210 to hematopoietic stem-progenitor cells (HS/PC) leads to CML in vivo by inducing a global hypomethylation in the stem cells (the cell-of-origin) that is maintained in the mature myeloid tumor cells (Vicente-Duenas et al., 2019).
Epigenetic Priming in Childhood T-ALL
The oncogene Lim Domain Only 2 (LMO2) is one of the most frequent drivers of childhood T-cell acute lymphoblastic leukemia (T-ALL) and accounts for about 15 to 25% of T-ALLs in children and adults (Van Vlierberghe et al., 2006; Qian et al., 2019). The intimate relationship between the aberrant expression of LMO2 and human T-ALL development can be supported by two alternative explanations. The first one is the classical interpretation that considers that the initiating genetic alteration, the aberrant expression of LMO2, arises in a differentiated T-cell and has been supported by several studies (McCormack et al., 2010; Cleveland et al., 2013; Smith et al., 2014; Ruggero et al., 2016). Under these conditions, the oncogene (LMO2) is fundamental and necessary for the transformation and maintenance of the cell where the tumor originates (the committed-T cell). Although during the evolution of the tumor, the cells will acquire secondary alterations that will define the clinical characteristics of the disease and further decontrol its behavior. But the expression of the oncogene (LMO2) is necessary for all the steps of the disease. Because cell transformation arises in a differentiated cell (a T cell), this is the reason why tumor cells have such a phenotype.
Nevertheless, if we consider the epigenetic priming mechanism, we can explain the relationship between LMO2 and T-ALL in a different way. Under this hypothesis, LMO2 oncogene is directly capable of priming the phenotypic characteristics of the tumor in a non-committed T cell that could be an HS/PC (Figure 1). And the T-cell phenotype of the leukemic cell is the result of the priming of the cell-of-origin imposed by the aberrant expression of LMO2. In this sense, T-ALL is also a scenario for epigenetic priming. This second possibility has been proved in mouse models that ectopically express LMO2 in non-T target cells (Garcia-Ramirez et al., 2018). These new T-ALL models driven by LMO2, reveal that the tumor T-cell personality is imposed by the oncogene instead of by the cell-of-origin phenotype (Garcia-Ramirez et al., 2018). In this model, LMO2 operates as a “hit-and-run” oncogene. In this sense, is able to prime the hematopoietic stem/progenitor cells (HS/PCs) into a T-cell developmental program. This priming to T-ALL is affected by the vulnerability of the cancer cell-of-origin. This means that the capability of reprogramming is decreased when the oncogene is expressed in more differentiated progenitors in the hematopoietic system (Garcia-Ramirez et al., 2018). The LMO2 genetic lesion that initiates the T-ALL process is replaceable for the subsequent progression and maintenance of the tumor. While evolved tumor cells are dependent on secondary oncogenic procedures, like Notch1 mutations. Despite the absence of Lmo2 protein within the leukemic T-cells, these mice progress to a clonal and aggressive T-ALL (Garcia-Ramirez et al., 2018).
The reprogramming capacity of LMO2 has been also proved as is one of the six transcription factors whose transient expression is mandatory for the reprogramming of terminally differentiated blood cells in the mouse into induced hematopoietic stem cells (Batta et al., 2014; Riddell et al., 2014). In the same direction, LMO2 expression due to retroviral insertion and transactivation in CD34+ HSCs of X-SCID patients caused T-ALL but no other hematopoietic tumors (Hacein-Bey-Abina et al., 2008; Howe et al., 2008). Also, supporting this theory is the fact that epigenomic regulators are recurrently mutated in T-ALL (68% of cases). And if we focus on T-ALL driven by LMO2, recurrent mutations in USP7, PHF6, and EZH2 have been found (Liu et al., 2017; Qian et al., 2019).
A similar reprogramming process of transformation is conducted by the Human T Cell Lymphoma Leukemia Virus (HTLV-1) responsible for the Adult T cell Leukemia (ATL). In this regard, the virus-oncoprotein Tax is essential for the initial transformation process with pleiotropic actions in the infected cell, but upon initial infection and transformation of the cell, the oncoprotein is no longer expressed by the retrovirus. It has been described genetic and epigenetic abnormalities that replace the function of Tax and are responsible for the maintenance of the leukemic cells after the initial malignancy process imposed by Tax (Yamagishi and Watanabe, 2012). On the other hand, it has always been thought that the HTLV-1 virus preferentially infects CD4+CCR4+ T cells, and this is why ATL leukemias are developed. But, interestingly, it has been shown that HTLV-1 is able to infect hematopoietic stem cells (HSCs) and converts the precursor infected cells into specialized T cells (Furuta et al., 2017). Taken together these results, it seems that the epigenetic priming is also behind the ATL development.
Leukemia-Epigenetic Priming Implications
The leukemia-epigenetic priming mechanism opens new possibilities to treat ALL. As epigenetic modifications can be manipulating, and somehow reinitiated, maybe in the near future it could be possible to identify them in the preleukemic clones and modify in order to interrupt the malignant priming. In this line, B-ALL cells have been reprogrammed using a DNA methyl-transferase inhibitor to an alternative lineage cell fate and making the disappearance of its malignant behavior (Bhatla et al., 2012). Also, epigenetic reprogramming has been used as a therapy to kill leukemia stem cells (Scott et al., 2016).
In any case, a better understanding of the epigenetic priming in leukemia is a prerequisite to offer and develop novel potential therapeutic approaches. To this aim, the development of preclinical models as the ones summarized in this review are unique tools to this purpose.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
Research in CV-D’s group was partially supported by FEDER, “Miguel Servet” Grant (CP14/00082 – AES 2013–2016) from the Instituto de Salud Carlos III (Ministerio de Economía y Competitividad), the “Fondo de Investigaciones Sanitarias/Instituto de Salud Carlos III” (PI17/00167), and by the German Federal Office for Radiation Protection (BfS)-Germany (FKZ: 3618S32274). JR-G was supported by Instituto de Salud Carlos III (Ministerio de Economía y Competitividad-FEDER) (PI17/00167) (ERDF-European Regional Development Fund). AC-G was supported by FSE-Consejería de Educación de la Junta de Castilla y León 2019 (ESF-European Social Fund) fellowship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all members of our groups for useful suggestions and for their critical reading of the manuscript.
References
Agraz-Doblas, A., Bueno, C., Bashford-Rogers, R., Roy, A., Schneider, P., Bardini, M., et al. (2019). Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica 104, 1176–1188. doi: 10.3324/haematol.2018.206375
Alpar, D., Wren, D., Ermini, L., Mansur, M. B., van Delft, F. W., Bateman, C. M., et al. (2015). Clonal origins of ETV6-RUNX1(+) acute lymphoblastic leukemia: studies in monozygotic twins. Leukemia 29, 839–846. doi: 10.1038/leu.2014.322
Auer, F., Ruschendorf, F., Gombert, M., Husemann, P., Ginzel, S., Izraeli, S., et al. (2014). Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547GA. Leukemia 28, 1136–1138. doi: 10.1038/leu.2013.363
Bateman, C. M., Alpar, D., Ford, A. M., Colman, S. M., Wren, D., Morgan, M., et al. (2015). Evolutionary trajectories of hyperdiploid ALL in monozygotic twins. Leukemia 29, 58–65. doi: 10.1038/leu.2014.177
Batta, K., Florkowska, M., Kouskoff, V., and Lacaud, G. (2014). Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep. 9, 1871–1884. doi: 10.1016/j.celrep.2014.11.002
Bhatla, T., Wang, J., Morrison, D. J., Raetz, E. A., Burke, M. J., Brown, P., et al. (2012). Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood 119, 5201–5210. doi: 10.1182/blood-2012-01-401687
Boiers, C., Carrelha, J., Lutteropp, M., Luc, S., Green, J. C., Azzoni, E., et al. (2013). Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell 13, 535–548. doi: 10.1016/j.stem.2013.08.012
Boiers, C., Richardson, S. E., Laycock, E., Zriwil, A., Turati, V. A., Brown, J., et al. (2018). A human IPS model implicates embryonic b-myeloid fate restriction as developmental susceptibility to b acute lymphoblastic leukemia-associated ETV6-RUNX1. Dev. Cell 44, 362.e7–377.e7. doi: 10.1016/j.devcel.2017.12.005
Bueno, C., Tejedor, J. R., Bashford-Rogers, R., Gonzalez-Silva, L., Valdes-Mas, R., Agraz-Doblas, A., et al. (2019). Natural history and cell-of-origin of TCF3-ZNF384 and PTPN11 mutations in monozygotic twins with concordant BCP-ALL. Blood. doi: 10.1182/blood.2019000893 [Epub ahead of print].
Burke, M. J., and Bhatla, T. (2014). Epigenetic modifications in pediatric acute lymphoblastic leukemia. Front. Pediatr. 2:42. doi: 10.3389/fped.2014.00042
Carrillo-de-Santa-Pau, E., Juan, D., Pancaldi, V., Were, F., Martin-Subero, I., Rico, D., et al. (2017). Automatic identification of informative regions with epigenomic changes associated to hematopoiesis. Nucleic Acids Res. 45, 9244–9259. doi: 10.1093/nar/gkx618
Chaber, R., Gurgul, A., Wrobel, G., Haus, O., Tomon, A., Kowalczyk, J., et al. (2017). Whole-genome DNA methylation characteristics in pediatric precursor B cell acute lymphoblastic leukemia (BCP ALL). PLoS One 12:e0187422. doi: 10.1371/journal.pone.0187422
Churchman, M. L., Qian, M., Te Kronnie, G., Zhang, R., Yang, W., Zhang, H., et al. (2018). Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell 33, 937.e8–948.e8.
Cleveland, S. M., Smith, S., Tripathi, R., Mathias, E. M., Goodings, C., Elliott, N., et al. (2013). Lmo2 induces hematopoietic stem cell-like features in T-cell progenitor cells prior to leukemia. Stem Cells 31, 882–894. doi: 10.1002/stem.1345
Cobaleda, C., and Sanchez-Garcia, I. (2009). B-cell acute lymphoblastic leukaemia: towards understanding its cellular origin. Bioessays 31, 600–609. doi: 10.1002/bies.200800234
Figueroa, M. E., Chen, S. C., Andersson, A. K., Phillips, L. A., Li, Y., Sotzen, J., et al. (2013). Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J. Clin. Invest. 123, 3099–3111. doi: 10.1172/JCI66203
Ford, A. M., Ridge, S. A., Cabrera, M. E., Mahmoud, H., Steel, C. M., Chan, L. C., et al. (1993). In utero rearrangements in the trithorax-related oncogene in infant leukaemias. Nature 363, 358–360. doi: 10.1038/363358a0
Furuta, R., Yasunaga, J. I., Miura, M., Sugata, K., Saito, A., Akari, H., et al. (2017). Human T-cell leukemia virus type 1 infects multiple lineage hematopoietic cells in vivo. PLoS Pathog. 13:e1006722. doi: 10.1371/journal.ppat.1006722
Gabriel, A. S., Lafta, F. M., Schwalbe, E. C., Nakjang, S., Cockell, S. J., Iliasova, A., et al. (2015). Epigenetic landscape correlates with genetic subtype but does not predict outcome in childhood acute lymphoblastic leukemia. Epigenetics 10, 717–726. doi: 10.1080/15592294.2015.1061174
Gale, K. B., Ford, A. M., Repp, R., Borkhardt, A., Keller, C., Eden, O. B., et al. (1997). Backtracking leukemia to birth: identification of clonotypic gene fusion sequences in neonatal blood spots. Proc. Natl. Acad. Sci. U.S.A. 94, 13950–13954. doi: 10.1073/pnas.94.25.13950
Garcia-Ramirez, I., Bhatia, S., Rodriguez-Hernandez, G., Gonzalez-Herrero, I., Walter, C., Gonzalez de Tena-Davila, S., et al. (2018). Lmo2 expression defines tumor cell identity during T-cell leukemogenesis. EMBO J. 37:e98783. doi: 10.15252/embj.201798783
Gostissa, M., Alt, F. W., and Chiarle, R. (2011). Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu. Rev. Immunol. 29, 319–350. doi: 10.1146/annurev-immunol-031210-101329
Greaves, M. (2018). A causal mechanism for childhood acute lymphoblastic leukaemia. Nat. Rev. Cancer 18, 471–484. doi: 10.1038/s41568-018-0015-6
Greaves, M. F., Maia, A. T., Wiemels, J. L., and Ford, A. M. (2003). Leukemia in twins: lessons in natural history. Blood 102, 2321–2333. doi: 10.1182/blood-2002-12-3817
Green, M. R., Vicente-Duenas, C., Romero-Camarero, I., Long Liu, C., Dai, B., Gonzalez-Herrero, I., et al. (2014). Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat. Commun. 5:3904. doi: 10.1038/ncomms4904
Gu, Z., Churchman, M. L., Roberts, K. G., Moore, I., Zhou, X., Nakitandwe, J., et al. (2019). PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat. Genet. 51, 296–307.
Hacein-Bey-Abina, S., Garrigue, A., Wang, G. P., Soulier, J., Lim, A., Morillon, E., et al. (2008). Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 118, 3132–3142. doi: 10.1172/JCI35700
Hogan, L. E., Meyer, J. A., Yang, J., Wang, J., Wong, N., Yang, W., et al. (2011). Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 118, 5218–5226. doi: 10.1182/blood-2011-04-345595
Howe, S. J., Mansour, M. R., Schwarzwaelder, K., Bartholomae, C., Hubank, M., Kempski, H., et al. (2008). Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 118, 3143–3150. doi: 10.1172/JCI35798
Hu, D., and Shilatifard, A. (2016). Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 30, 2021–2041. doi: 10.1101/gad.284109.116
Hunger, S. P., and Mullighan, C. G. (2015). Acute lymphoblastic leukemia in children. N. Engl. J. Med. 373, 1541–1552.
Iacobucci, I., and Mullighan, C. G. (2017). Genetic basis of acute lymphoblastic leukemia. J. Clin. Oncol. 35, 975–983. doi: 10.1200/JCO.2016.70.7836
Lausten-Thomsen, U., Madsen, H. O., Vestergaard, T. R., Hjalgrim, H., Nersting, J., and Schmiegelow, K. (2011). Prevalence of t(12;21)[ETV6-RUNX1]-positive cells in healthy neonates. Blood 117, 186–189. doi: 10.1182/blood-2010-05-282764
le Viseur, C., Hotfilder, M., Bomken, S., Wilson, K., Rottgers, S., Schrauder, A., et al. (2008). In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell 14, 47–58. doi: 10.1016/j.ccr.2008.05.015
Liu, Y., Easton, J., Shao, Y., Maciaszek, J., Wang, Z., Wilkinson, M. R., et al. (2017). The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 49, 1211–1218. doi: 10.1038/ng.3909
Mahowald, G. K., Baron, J. M., and Sleckman, B. P. (2008). Collateral damage from antigen receptor gene diversification. Cell 135, 1009–1012. doi: 10.1016/j.cell.2008.11.024
Martin-Lorenzo, A., Auer, F., Chan, L. N., Garcia-Ramirez, I., Gonzalez-Herrero, I., Rodriguez-Hernandez, G., et al. (2018). Loss of Pax5 exploits Sca1-BCR-ABL(p190) susceptibility to confer the metabolic shift essential for pB-ALL. Cancer Res. 78, 2669–2679. doi: 10.1158/0008-5472.CAN-17-3262
Martin-Subero, J. I., and Oakes, C. C. (2018). Charting the dynamic epigenome during B-cell development. Semin. Cancer Biol. 51, 139–148. doi: 10.1016/j.semcancer.2017.08.008
Matsushita, C., Yang, Y., Takeuchi, S., Matsushita, M., Van Dongen, J. J., Szczepanski, T., et al. (2004). Aberrant methylation in promoter-associated CpG islands of multiple genes in relapsed childhood acute lymphoblastic leukemia. Oncol. Rep. 12, 97–99.
McCormack, M. P., Young, L. F., Vasudevan, S., de Graaf, C. A., Codrington, R., Rabbitts, T. H., et al. (2010). The Lmo2 oncogene initiates leukemia in mice by inducing thymocyte self-renewal. Science 327, 879–883. doi: 10.1126/science.1182378
Milani, L., Lundmark, A., Kiialainen, A., Nordlund, J., Flaegstad, T., Forestier, E., et al. (2010). DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood 115, 1214–1225. doi: 10.1182/blood-2009-04-214668
Moriyama, T., Metzger, M. L., Wu, G., Nishii, R., Qian, M., Devidas, M., et al. (2015). Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 16, 1659–1666. doi: 10.1016/S1470-2045(15)00369-1
Mrozek, K., Harper, D. P., and Aplan, P. D. (2009). Cytogenetics and molecular genetics of acute lymphoblastic leukemia. Hematol. Oncol. Clin. North. Am. 23, 991–1010.
Noetzli, L., Lo, R. W., Lee-Sherick, A. B., Callaghan, M., Noris, P., Savoia, A., et al. (2015). Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat. Genet. 47, 535–538. doi: 10.1038/ng.3253
Nordlund, J., Backlin, C. L., Wahlberg, P., Busche, S., Berglund, E. C., Eloranta, M. L., et al. (2013). Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 14, r105. doi: 10.1186/gb-2013-14-9-r105
Nordlund, J., Backlin, C. L., Zachariadis, V., Cavelier, L., Dahlberg, J., Ofverholm, I., et al. (2015). DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin. Epigenet. 7:11. doi: 10.1186/s13148-014-0039-z
Nordlund, J., and Syvanen, A. C. (2018). Epigenetics in pediatric acute lymphoblastic leukemia. Semin. Cancer Biol. 51, 129–138. doi: 10.1016/j.semcancer.2017.09.001
Nussenzweig, A., and Nussenzweig, M. C. (2010). Origin of chromosomal translocations in lymphoid cancer. Cell 141, 27–38. doi: 10.1016/j.cell.2010.03.016
Perez-Caro, M., Cobaleda, C., Gonzalez-Herrero, I., Vicente-Duenas, C., Bermejo-Rodriguez, C., Sanchez-Beato, M., et al. (2009). Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 28, 8–20. doi: 10.1038/emboj.2008.253
Pui, C. H., Nichols, K. E., and Yang, J. J. (2019a). Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 16, 227–240. doi: 10.1038/s41571-018-0136-6
Pui, C. H., Rebora, P., Schrappe, M., Attarbaschi, A., Baruchel, A., Basso, G., et al. (2019b). Outcome of children with hypodiploid acute lymphoblastic leukemia: a retrospective multinational study. J. Clin. Oncol. 37, 770–779. doi: 10.1200/JCO.18.00822
Qian, M., Zhao, X., Devidas, M., Yang, W., Gocho, Y., Smith, C., et al. (2019). Genome-wide association study of susceptibility loci for T-cell acute lymphoblastic leukemia in children. J. Natl. Cancer Inst. doi: 10.1093/jnci/djz043 [Epub ahead of print].
Riddell, J., Gazit, R., Garrison, B. S., Guo, G., Saadatpour, A., Mandal, P. K., et al. (2014). Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell 157, 549–564. doi: 10.1016/j.cell.2014.04.006
Roberts, K. G., and Mullighan, C. G. (2015). Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat. Rev. Clin. Oncol. 12, 344–357. doi: 10.1038/nrclinonc.2015.38
Rodriguez-Hernandez, G., Hauer, J., Martin-Lorenzo, A., Schafer, D., Bartenhagen, C., Garcia-Ramirez, I., et al. (2017). Infection exposure promotes ETV6-RUNX1 precursor B-cell leukemia via impaired H3K4 demethylases. Cancer Res. 77, 4365–4377. doi: 10.1158/0008-5472.CAN-17-0701
Ruggero, K., Al-Assar, O., Chambers, J. S., Codrington, R., Brend, T., and Rabbitts, T. H. (2016). LMO2 and IL2RG synergize in thymocytes to mimic the evolution of SCID-X1 gene therapy-associated T-cell leukaemia. Leukemia 30, 1959–1962. doi: 10.1038/leu.2016.116
Schafer, D., Olsen, M., Lahnemann, D., Stanulla, M., Slany, R., Schmiegelow, K., et al. (2018). Five percent of healthy newborns have an ETV6-RUNX1 fusion as revealed by DNA-based GIPFEL screening. Blood 131, 821–826. doi: 10.1182/blood-2017-09-808402
Scott, M. T., Korfi, K., Saffrey, P., Hopcroft, L. E., Kinstrie, R., Pellicano, F., et al. (2016). Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. 6, 1248–1257. doi10.1158/2159-8290.cd-16-0263
Smith, S., Tripathi, R., Goodings, C., Cleveland, S., Mathias, E., Hardaway, J. A., et al. (2014). LIM domain only-2 (LMO2) induces T-cell leukemia by two distinct pathways. PLoS One 9:e85883. doi: 10.1371/journal.pone.0085883
Taylor, K. H., Pena-Hernandez, K. E., Davis, J. W., Arthur, G. L., Duff, D. J., Shi, H., et al. (2007). Large-scale CpG methylation analysis identifies novel candidate genes and reveals methylation hotspots in acute lymphoblastic leukemia. Cancer Res. 67, 2617–2625. doi: 10.1158/0008-5472.can-06-3993
Van Vlierberghe, P., van Grotel, M., Beverloo, H. B., Lee, C., Helgason, T., Buijs-Gladdines, J., et al. (2006). The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood 108, 3520–3529. doi: 10.1182/blood-2006-04-019927
Vicente-Duenas, C., Gonzalez-Herrero, I., Sehgal, L., Garcia-Ramirez, I., Rodriguez-Hernandez, G., Pintado, B., et al. (2019). Dnmt1 links BCR-ABLp210 to epigenetic tumor stem cell priming in myeloid leukemia. Leukemia 33, 249–278. doi: 10.1038/s41375-018-0192-z
Vicente-Duenas, C., Hauer, J., Cobaleda, C., Borkhardt, A., and Sanchez-Garcia, I. (2018). Epigenetic priming in cancer initiation. Trends Cancer 4, 408–417. doi: 10.1016/j.trecan.2018.04.007
Vicente-Duenas, C., Romero-Camarero, I., Gonzalez-Herrero, I., Alonso-Escudero, E., Abollo-Jimenez, F., Jiang, X., et al. (2012). A novel molecular mechanism involved in multiple myeloma development revealed by targeting MafB to haematopoietic progenitors. EMBO J. 31, 3704–3717. doi: 10.1038/emboj.2012.227
Wiemels, J. L., Cazzaniga, G., Daniotti, M., Eden, O. B., Addison, G. M., Masera, G., et al. (1999). Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 354, 1499–1503.
Wong, I. H., Ng, M. H., Huang, D. P., and Lee, J. C. (2000). Aberrant p15 promoter methylation in adult and childhood acute leukemias of nearly all morphologic subtypes: potential prognostic implications. Blood 95, 1942–1949.
Wong, N. C., Ashley, D., Chatterton, Z., Parkinson-Bates, M., Ng, H. K., Halemba, M. S., et al. (2012). A distinct DNA methylation signature defines pediatric pre-B cell acute lymphoblastic leukemia. Epigenetics 7, 535–541. doi: 10.4161/epi.20193
Yamagishi, M., and Watanabe, T. (2012). Molecular hallmarks of adult T cell leukemia. Front. Microbiol. 3:334. doi: 10.3389/fmicb.2012.00334
Keywords: epigenetic priming, reprogramming, childhood leukemia, B-ALL, T-ALL, stem cells
Citation: Raboso-Gallego J, Casado-García A, Isidro-Hernández M and Vicente-Dueñas C (2019) Epigenetic Priming in Childhood Acute Lymphoblastic Leukemia. Front. Cell Dev. Biol. 7:137. doi: 10.3389/fcell.2019.00137
Received: 22 May 2019; Accepted: 05 July 2019;
Published: 17 July 2019.
Edited by:
Takaomi Sanda, National University of Singapore, SingaporeReviewed by:
Hirotaka Matsui, Kumamoto University, JapanPablo Menendez, Andalusian Health Service, Spain
Copyright © 2019 Raboso-Gallego, Casado-García, Isidro-Hernández and Vicente-Dueñas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carolina Vicente-Dueñas, Y3ZkQHVzYWwuZXM=