 Sarah R. Chadwick
Sarah R. Chadwick Patrick Lajoie
Patrick Lajoie- Department of Anatomy and Cell Biology, The University of Western Ontario, London, ON, Canada
Multiple factors lead to proteostatic perturbations, often resulting in the aberrant accumulation of toxic misfolded proteins. Cells, from yeast to humans, can respond to sudden accumulation of secretory proteins within the endoplasmic reticulum (ER) through pathways such as the Unfolded Protein Response (UPR). The ability of cells to adapt the ER folding environment to the misfolded protein burden ultimately dictates cell fate. The aging process is a particularly important modifier of the proteostasis network; as cells age, both their ability to maintain this balance in protein folding/degradation and their ability to respond to insults in these pathways can break down, a common element of age-related diseases (including neurodegenerative diseases). ER stress coping mechanisms are central to lifespan regulation under both normal and disease states. In this review, we give a brief overview of the role of ER stress response pathways in age-dependent neurodegeneration.
Introduction
Protein homeostasis (or proteostasis) is the sum of cellular processes involving protein transcription, translation, folding, and degradation (Balch et al., 2008). In order for a cell to remain functional and capable of adapting to changing biochemical and environmental signals, proteostasis must remain uncompromised (Ben-Zvi et al., 2009). Protein folding is particularly important for cellular processes, as the final conformation of a folded protein is essential to its function. Cellular membrane dynamics are a pivotal aspect of protein folding; adaptations in the ER membrane’s composition and size are required to maintain proteostasis, and proper protein folding, in turn, is required to maintain this membrane integrity (Hou and Taubert, 2014).
Under normal circumstances, proteins destined for the secretory pathway are translated directly into the ER via ribosomes embedded in the ER membrane, bound by chaperone proteins, folded, and then packaged into vesicles for secretion (Novick et al., 1981). This includes proteins destined for the plasma membrane, such as membrane-linked receptors, or secreted factors released into the extracellular environment. In some cases, however, this pathway can go awry; proteins may become misfolded or unfolded in the ER, and unable to be recovered by the protein quality control machinery. In this instance, the improperly folded protein is targeted for degradation, exported into the cytosol, and degraded by a proteasome (Werner et al., 1996). Again, however, this process is imperfect. Some environmental, cellular, or molecular factors can cause disruptions in this pathway, preventing the proper turnover of misfolded or unfolded proteins, potentially leading to their accumulation and aggregation. This generates a cellular condition known as ER stress (Friedlander et al., 2000; Walter and Ron, 2011; Karagöz et al., 2019).
Endoplasmic reticulum stress and the failure to correctly fold proteins are associated with loss of protein function and cell death (Zinszner et al., 1998; Hetz et al., 2006; Upton et al., 2012). To avoid this, the cell resolves misfolded protein stress via two major stress response pathways: the heat shock response (HSR) (Verghese et al., 2012), which handles misfolded proteins in the cytoplasm, and the unfolded protein response (UPR), which takes place in the ER (Kohno et al., 1993; Cox and Walter, 1996; Liu and Chang, 2008). These protein quality control mechanisms are essential for maintaining the function and integrity of cellular processes. When perturbed, they can lead to whole-cell dysfunction and toxicity (Ruis and Schüller, 1995; Voellmy, 2004). Under normal conditions, both lead to resolution of the cellular stress caused by the presence of misfolded proteins. In some cases, such as in several misfolded protein-associated diseases (Yoshida, 2007; Torres et al., 2015), these stress response pathways themselves can become impaired. This leads to further accumulation of misfolded proteins, which in turn causes further UPR or HSR impairment (Delépine et al., 2000; Zhang et al., 2002). Misfolded protein aggregates have also been shown to bind and sequester machinery important for degrading misfolded proteins via ER-associated degradation (ERAD), a protein quality-control mechanism which recognizes unfolded or misfolded proteins synthesized in the ER (Lippincott-Schwartz et al., 1988; McCracken and Brodsky, 1996). This ERAD impairment induces further stress in the ER and causes induction of the UPR. Proteostatic dysfunction essentially leads to a vicious cycle of increasing ER stress, protein accumulation, and stress response impairment.
The UPR is a complicated signaling pathway which works to resolve ER stress and allow protein synthesis and folding to continue and has been shown to interact with multiple cellular pathways and processes to do so, including (but not limited to) those occurring in the ER (Welihinda et al., 1999; Travers et al., 2000; Walter and Ron, 2011; Snapp, 2012). It has also been shown to be impacted by several seemingly unrelated external influences, including aging and lipid metabolism, and dysfunction in this pathway has been linked with shortened cellular lifespan and cell death (Jazwinski, 2002; Hou et al., 2014; Labunskyy et al., 2014). Because of this, the study of the molecular mechanisms behind ER stress and the UPR is essential to the understanding of how protein homeostasis impacts the entire cell and its processes, including response to stressors, aging, and cell death.
Activation of the Unfolded Protein Response
As previously mentioned, the UPR is a stress response pathway specifically activated in response to ER stress, which is a condition that can be generated by things such as small molecules, environmental factors, or the accumulation of misfolded proteins in the ER (Welihinda et al., 1999). The UPR is activated when ER stress sensors embedded in the ER membrane detect the stressors and respond. Interestingly, the ultimate function of the UPR depends on the degree of activation and the length of time before the stress is resolved (Rutkowski et al., 2006; Rutkowski and Kaufman, 2007; Vidal and Hetz, 2012). It is primarily an adaptive response, which rescues cells from ER stress, but prolonged ER stress or high amplitude of UPR signaling causes the response to become maladaptive. In these circumstances, the UPR can activate alternate signaling pathways that result in apoptosis (Hetz et al., 2006; Rutkowski et al., 2006; Lin et al., 2007; Upton et al., 2012; Lu et al., 2014; Hetz and Papa, 2018).
In mammals, three distinct ER stress sensors exist: inositol requiring kinase 1 (IRE1) (Sidrauski and Walter, 1997; Yoshida et al., 2001; Calfon et al., 2002), double-stranded RNA-activated protein kinase like endoplasmic reticulum kinase (PERK) (Harding et al., 2000), and activating transcription factor 6 (ATF6) (Yoshida et al., 1998). When the UPR is activated, ER chaperone proteins (such as BIP) dissociate from these sensors, allowing their activation which in turn activates downstream signaling pathways (Welihinda et al., 1999; Shen et al., 2002; Ma and Hendershot, 2004; Pincus et al., 2010). Effector proteins from each of the three pathways bind to UPR response element (UPRE) sequences in gene promoters. A cell may activate over 400 UPR target genes involved in responding to ER stress, such as chaperone proteins, ribosome biogenesis genes, ERAD effectors, and genes to expand the ER lumen (Welihinda et al., 1999; Ma and Hendershot, 2004; Aragón et al., 2009). Upregulation of such genes contributes to adapt the ER folding environment to the new misfolded protein burden.
Despite its name, the UPR can be activated by stresses unrelated to misfolded or unfolded proteins. In addition to increased misfolded protein burden, ER stress can be induced by environmental factors; glucose deprivation/caloric restriction, for example, has been shown to mildly induce ER stress (Kaeberlein et al., 2005; Goldberg et al., 2009). Lipid concentration and composition in cells or in the extracellular environment have also been shown to significantly impact ER stress and UPR induction (Pineau et al., 2009; Promlek et al., 2011; Thibault et al., 2012). There is evidence to suggest that the UPR sensors IRE1 and PERK can detect perturbations of ER membrane lipid composition, independently of their luminal sensing domains, through their transmembrane domain (Promlek et al., 2011; Volmer et al., 2013; Kono et al., 2017). Other studies have also shown that the UPR is highly involved in responding to perturbation of lipid homeostasis (Thibault et al., 2012) and controls lipid synthesis and ER membrane proliferation in response to various cell stresses (Bernales et al., 2006; Schuck et al., 2009). Thus, UPR activation in the absence of unfolded proteins, via perturbation in the lipid composition of ER membrane, represents another regulatory mechanism (Promlek et al., 2011; Lajoie et al., 2012; Snapp, 2012; Volmer et al., 2013; Volmer and Ron, 2015) which may be important for UPR activation in ER stress-associated diseases.
Macroautophagy (henceforth, “autophagy”) is another protein quality control process which relies heavily upon functional membrane dynamics and proper membrane lipid composition. It is a non-specific maintenance process by which protein aggregates and damaged, defective, or aging cellular contents and organelles are transported to the lysosome for degradation (Klionsky and Emr, 2000; Mizushima, 2007; Glick et al., 2010). The membrane around the cargo destined for degradation by autophagy, the autophagosome, is derived from the ER membrane [with apparent contributions from the mitochondrial and plasma membranes (Nascimbeni et al., 2017)], wherein autophagic cargo are degraded by lysosomal hydrolases (Dunn, 1990). Autophagy occurs at a basal level in cells, but can be strongly induced by nutrient deprivation due to autophagy’s role in nutrient conservation and starvation adaptation, using bulk degradation to replenish amino acid availability for other cellular functions (Munafó and Colombo, 2001; Mizushima, 2007). ER stress has been shown to trigger autophagy, indicating parallel proteostatic responses to ER stress in the form of the UPR and autophagy (Bernales et al., 2006; Yorimitsu et al., 2006; Vidal and Hetz, 2012; Hou and Taubert, 2014). Importantly, activation of autophagy has been shown to be important for maintenance of proteostasis and lifespan regulation in multiple organisms and experimental models (Meléndez et al., 2003; Alvers et al., 2009a; Lee et al., 2012; Carroll et al., 2013).
ER Stress and Aging
Aging has been shown to modulate some of the factors leading to ER stress. It is an important modifier of the proteostasis network, meaning that aging cells may have altered capacity to properly carry out protein transcription, translation, folding, and degradation (Naidoo, 2009; Brown and Naidoo, 2012; Figure 1). Aging cells have been shown to have decreased total levels of a number of ER proteins, including protein chaperones (such as PDI, BIP, etc.) which normally supervise and ensure proper protein folding, and assist in targeting misfolded proteins for degradation (Paz Gavilán et al., 2006; Hussain and Ramaiah, 2007; Naidoo et al., 2008). This usually prevents the accumulation and aggregation of misfolded proteins and prevents them from having toxic effects on the cell. In addition, the limited chaperones that are still present in the aging ER appear to be impaired. This is possibly due to an increased rate of oxidation of these chaperones in aged cells, leading to structural changes and consequently decreased function (van der Vlies et al., 2003; Snapp et al., 2006; Naidoo et al., 2008). For example, both BIP ATPase activity and PDI enzymatic function have been shown to be significantly decreased in aged mouse livers (Nuss et al., 2008), and similar results have been seen in a number of other models as well, such as aged mouse cerebral cortex (Naidoo et al., 2008). Other components of UPR signaling have also shown to be reduced during aging. PERK mRNA, for example, has been shown to be reduced in aged rat hippocampi, indicating less efficient UPR signaling (Paz Gavilán et al., 2006).
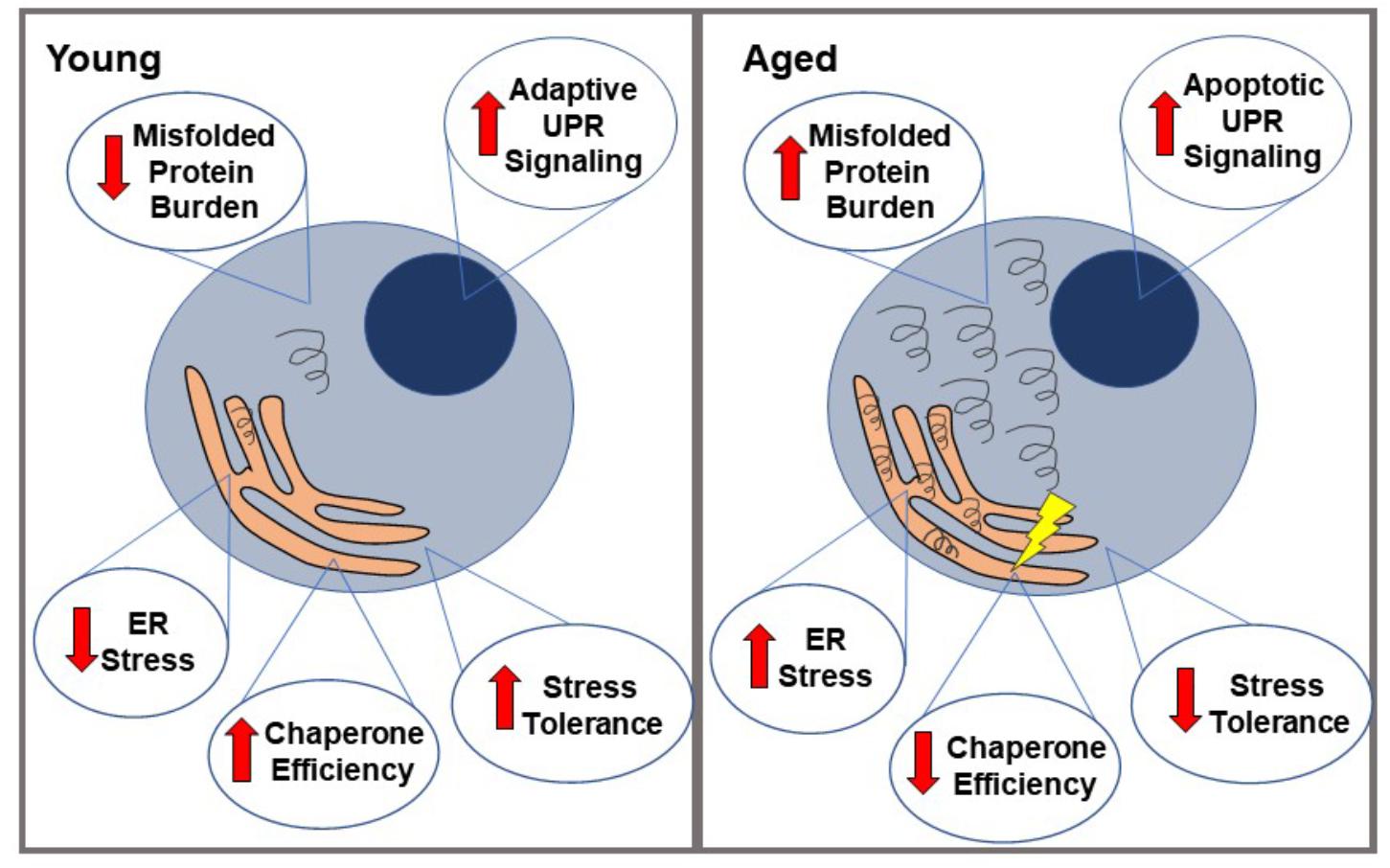
Figure 1. Proteostatic changes in aged vs. young cells. When faced with misfolded proteins, young cells demonstrate relatively low ER stress, high chaperone efficiency and stress tolerance, and primarily adaptive UPR signaling. This generally leads to a resolution of the misfolded proteins and therefore, ER stress. In contrast, aged cells are more likely to accumulate these misfolded proteins (partially due to loss of chaperone protein efficiency), leading to a state of ER stress, which they are less able to resolve. Their lower stress tolerance eventually leads to a relative increase in apoptotic UPR signaling over adaptive, ultimately causing cell death.
Aging also appears to alter the threshold at which the UPR switches from the adaptive pathway to the apoptotic pathway, which is perhaps related to the changes to proteostasis previously mentioned. When PERK signaling is decreased during aging, for example, there is evidence of an increase in GADD34 expression, which helps remove the translational block that occurs through PERK phosphorylating eIF2. This allows the expression of pro-apoptotic proteins, such as CHOP (Brown and Naidoo, 2012). CHOP has been shown to be increased with stress during aging and at baseline in aged muscular tissue in rats (Hussain and Ramaiah, 2007; Naidoo et al., 2008; Baehr et al., 2016); caspase-12 is also increased with stress in aged cells, but not during stress in younger cells (Paz Gavilán et al., 2006). The apoptotic protein JNK (which is activated by IRE1 during prolonged UPR signaling) is also upregulated during aging, as are JNK kinases that phosphorylate other apoptotic transcription factors such as ATF-2 and c-Jun (Hussain and Ramaiah, 2007; Brown and Naidoo, 2012). Calcium-mediated cell death pathways are also altered during aging. Aging has been linked to increased calcium flux between the ER and mitochondria, and consequently increased exposure to reactive oxygen species and sensitivity to cell death in the case of mitochondrial calcium overload (Fernandez-Sanz et al., 2014; Calvo-Rodríguez et al., 2016; Madreiter-Sokolowski et al., 2019). These factors, in turn, lead to a decreased threshold for the activation of calcium-mediated apoptosis. This aging-related decrease in adaptive UPR signaling and increase in apoptotic signaling may account for the apparent sensitivity of aged cells to ER stress, and the increased rate of cell death amongst stressed cells when aged (Rutkowski et al., 2006; Lu et al., 2014; Tay et al., 2014). Autophagy has also been shown to become less efficient at clearing damaged organelles and misfolded proteins during aging in yeast (as well as other models), and lifespan extension has been demonstrated when autophagy is heavily induced (Zhang and Cuervo, 2008; Alvers et al., 2009b; Caramés et al., 2010; Koga and Cuervo, 2011; Martinez-Lopez et al., 2015).
ER Stress and Age-Dependent Human Diseases
Aging is a common risk factor for a number of protein misfolding diseases, including several neurodegenerative diseases (Martínez et al., 2017), a number of which have links to UPR function as described in the previous section. While not all aging-related diseases are directly linked to breakdown of UPR signaling, this breakdown may still contribute to disease pathogeneses. For example, type 2 diabetes is known to develop more frequently due to both obesity and aging, with the two factors often coexisting in patients (Ozcan et al., 2004). As aging has also been linked to the decreases in UPR effector proteins associated with diabetes and insulin resistance, these results suggest that aging-related UPR defects may be linked to these diseases as well. Obesity and insulin resistance are also linked to heart disease and atherosclerosis, both of which increase in prevalence with age and have also been linked to ER stress and the UPR (Han et al., 2006). Huntington’s disease (HD), Parkinson’s disease, and Alzheimer’s disease have been clearly and repeatedly linked to UPR dysfunction which increases with age, thus increasing disease severity (Vidal and Hetz, 2012; Carroll et al., 2013). Though the three diseases have different causative genes, they all share misfolded protein accumulation and aggregation as part of their pathology, leading to impaired proteostasis and ER stress responses, and then cellular toxicity.
In Alzheimer’s disease, for example, tau neurofibrillary tangles and amyloid-β plaques accumulate in neurons and lead to neurodegeneration (Lee et al., 2010). Studies have shown that cells with these protein aggregates have high UPR induction identified through high levels of phosphorylated eIF2α, PERK, and IRE1 (Hoozemans et al., 2005, 2009; Gerakis and Hetz, 2018a,b). This has been identified in early stages of protein accumulation and linked to later stage neurodegeneration, suggesting an early beneficial role for the UPR that may later become maladaptive (Hoozemans et al., 2009). Indeed, as cells age, the UPR’s capacity to cope with the misfolded protein load decreases in an fly model of Alzheimer’s disease; decreased signaling through the IRE1 branch of the UPR has been identified in this model, leading to decreased misfolded protein clearance (Marcora et al., 2017). Similar findings have also been reported in spinal cord tissue from patients with sporadic Amyotrophic Lateral Sclerosis (ALS) (Atkin et al., 2008). Enhancing UPR signaling and/or reducing ER stress through genetic and pharmacological modulation of UPR effectors such as eIF2α, PERK, XBP1, ATF4, and heat shock proteins have all been shown to have positive effects on various models of ALS (Hetz et al., 2009; Saxena et al., 2009; Castillo et al., 2013; Matus et al., 2013; Saxena et al., 2013; Jiang et al., 2014; Wang et al., 2014a,b; Das et al., 2015; Vieira et al., 2015; Nagy et al., 2016). Huntington’s disease is another neurodegenerative disease which is characterized by the accumulation of misfolded huntingtin protein, which undergoes abnormal expansion of a segment of polyQ repeats (Penney et al., 1997). Longer polyQ tracts are associated with earlier onset and more severe symptoms (Macdonald, 1993), and are also more prone to aggregation and are associated with a higher degree of UPR induction but a lower degree of HSR induction (Martindale et al., 1998; Chafekar and Duennwald, 2012). These aggregates have also been shown to cause ER stress and impaired ERAD due to sequestration of ERAD machinery, leading to UPR hyperactivation (Duennwald and Lindquist, 2008; Lajoie and Snapp, 2011; Leitman et al., 2013; Jiang et al., 2016). In agreement with dysregulated UPR in HD, restoration of “normal” XBP1 and PERK activity has been shown to improve disease phenotypes in both cell and animal models (Vidal et al., 2011, 2012; Leitman et al., 2014; Rivas et al., 2015). Importantly, activation of ER stress pathways has been detected in post-mortem patient samples (Carnemolla et al., 2009). Similarly, UPR has been associated with the onset of Parkinson’s disease (Mercado et al., 2016). Accumulation of α-synuclein has been shown to block ER to Golgi trafficking and consequently activate the UPR in both yeast and humans (Cooper et al., 2006; Heman-Ackah et al., 2017). PERK inhibition showed positive effects in a mouse model of Parkinson’s disease (Celardo et al., 2016; Mercado et al., 2018). In Parkinson’s disease, misfolded proteins accumulate in the substantia nigra region of the brain, leading to loss of dopaminergic neurons in this region. Similar to the studies performed on Alzheimer’s disease, it has been shown that the UPR is highly activated in these areas and that this UPR activation may be causally linked to the neurodegeneration seen in this disease (Hoozemans et al., 2007). ER stress is therefore a common determinant of multiple neurodegenerative diseases. Thus, targeting the UPR has emerged as an attractive therapeutic approach these disorders (Rivas et al., 2015; Valenzuela et al., 2016).
Conclusion
As more connections are drawn between ER proteostasis and aging, it becomes clear that these interactions reach much further than previously thought – from neurodegeneration to temperature adaptation, and from simple model organisms like yeast up to higher mammals. Future research on these fields (individually and as a whole) will hopefully address some yet-unanswered questions on how and why these connections exist. For example, could age-related changes in membrane composition and fluidity explain age-related increases in UPR signaling? What advantage would be conferred to the cell by UPR stress-sensing proteins responding to changes in membrane lipids as well as ER stress? What other cellular functions and pathways intersect with these processes, in both baseline and stressed/aged states? In all likelihood, there is not one single cause for the breakdown of ER homeostasis during aging, but instead a combination of factors contributes to overall increased sensitivity to ER stress. Increased misfolded protein accumulation, decreased effectiveness of the adaptive UPR, and an altered threshold for apoptotic UPR signaling likely all play a role (Naidoo et al., 2008; Brown and Naidoo, 2012; Madreiter-Sokolowski et al., 2019). Finally, ER stress sensitivity is not only dictated by the amplitude of the UPR response but also by upregulation of a specific set of target genes required to adapt the ER folding environment for a given stress-causing situation (Thibault et al., 2011). Therefore, the question of what categories of UPR target genes define the aging UPR is a crucial one that needs to be addressed. As technology and research methods advance and our understanding of these areas improves, this future research will likely have important implications for basic science and therapeutic approaches to human diseases.
Author Contributions
PL and SC wrote the manuscript. SC generated the figure.
Funding
This work was supported by a grant from the Canadian Institutes for Health Research (MOP-137041). SC is the recipient of an Ontario Graduate Scholarship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alvers, A. L., Fishwick, L. K., Wood, M. S., Hu, D., Chung, H. S., Dunn, W. A., et al. (2009a). Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell 8, 353–369. doi: 10.1111/j.1474-9726.2009.00469.x
Alvers, A. L., Wood, M. S., Hu, D., Kaywell, A. C., Dunn, W. A., and Aris, J. P. (2009b). Autophagy is required for extension of yeast chronological life span by rapamycin. Autophagy 5, 847–849. doi: 10.4161/auto.8824
Aragón, T., van Anken, E., Pincus, D., Serafimova, I. M., Korennykh, A. V., Rubio, C. A., et al. (2009). Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature 457, 736–740. doi: 10.1038/nature07641
Atkin, J. D., Farg, M. A., Walker, A. K., McLean, C., Tomas, D., and Horne, M. K. (2008). Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 30, 400–407. doi: 10.1016/j.nbd.2008.02.009
Baehr, L. M., West, D. W. D., Marcotte, G., Marshall, A. G., De Sousa, L. G., Baar, K., et al. (2016). Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging 8, 127–146. doi: 10.18632/aging.100879
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Ben-Zvi, A., Miller, E. A., and Morimoto, R. I. (2009). Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc. Natl. Acad. Sci. U.S.A. 106, 14914–14919. doi: 10.1073/pnas.0902882106
Bernales, S., McDonald, K. L., and Walter, P. (2006). Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4:e423. doi: 10.1371/journal.pbio.0040423
Brown, M. K., and Naidoo, N. (2012). The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 3:263. doi: 10.3389/fphys.2012.00263
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Calvo-Rodríguez, M., García-Durillo, M., Villalobos, C., and Núñez, L. (2016). In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 1863, 2637–2649. doi: 10.1016/j.bbamcr.2016.08.001
Caramés, B., Taniguchi, N., Otsuki, S., Blanco, F. J., and Lotz, M. (2010). Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 62, 791–801. doi: 10.1002/art.27305
Carnemolla, A., Fossale, E., Agostoni, E., Michelazzi, S., Calligaris, R., De Maso, L., et al. (2009). Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J. Biol. Chem. 284, 18167–18173. doi: 10.1074/jbc.M109.018325
Carroll, B., Hewitt, G., and Korolchuk, V. I. (2013). Autophagy and ageing: implications for age-related neurodegenerative diseases. Essays Biochem. 55, 119–131. doi: 10.1042/bse0550119
Castillo, K., Nassif, M., Valenzuela, V., Rojas, F., Matus, S., Mercado, G., et al. (2013). Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 9, 1308–1320. doi: 10.4161/auto.25188
Celardo, I., Costa, A. C., Lehmann, S., Jones, C., Wood, N., Mencacci, N. E., et al. (2016). Mitofusin-mediated ER stress triggers neurodegeneration in PINK1/Parkin models of Parkinson’s disease. Cell Death Dis. 7:e2271. doi: 10.1038/cddis.2016.173
Chafekar, S. M., and Duennwald, M. L. (2012). Impaired heat shock response in cells expressing full-length polyglutamine-expanded huntingtin. PLoS One 7:e37929. doi: 10.1371/journal.pone.0037929
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328. doi: 10.1126/science.1129462
Cox, J. S., and Walter, P. (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404. doi: 10.1016/s0092-8674(00)81360-4
Das, I., Krzyzosiak, A., Schneider, K., Wrabetz, L., Antonio, M. D., Barry, N., et al. (2015). Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 348, 239–242. doi: 10.1126/science.aaa4484
Delépine, M., Nicolino, M., Barrett, T., Golamaully, M., Lathrop, G. M., and Julier, C. (2000). EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat. Genet. 25, 406–409. doi: 10.1038/78085
Duennwald, M. L., and Lindquist, S. (2008). Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 22, 3308–3319. doi: 10.1101/gad.1673408
Dunn, W. A. (1990). Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J. Cell Biol. 110, 1935–1945. doi: 10.1083/jcb.110.6.1935
Fernandez-Sanz, C., Ruiz-Meana, M., Miro-Casas, E., Nuñez, E., Castellano, J., Loureiro, M., et al. (2014). Defective sarcoplasmic reticulum-mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 5:e1573. doi: 10.1038/cddis.2014.526
Friedlander, R., Jarosch, E., Urban, J., Volkwein, C., and Sommer, T. (2000). A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat. Cell Biol. 2, 379–384. doi: 10.1038/35017001
Gerakis, Y., and Hetz, C. (2018a). A decay of the adaptive capacity of the unfolded protein response exacerbates Alzheimer’s disease. Neurobiol. Aging 63, 162–164. doi: 10.1016/j.neurobiolaging.2017.09.012
Gerakis, Y., and Hetz, C. (2018b). Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 285, 995–1011. doi: 10.1111/febs.14332
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Goldberg, A. A., Bourque, S. D., Kyryakov, P., Gregg, C., Boukh-Viner, T., Beach, A., et al. (2009). Effect of calorie restriction on the metabolic history of chronologically aging yeast. Exp. Gerontol. 44, 555–571. doi: 10.1016/j.exger.2009.06.001
Han, S., Liang, C.-P., DeVries-Seimon, T., Ranalletta, M., Welch, C. L., Collins-Fletcher, K., et al. (2006). Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 3, 257–266. doi: 10.1016/j.cmet.2006.02.008
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi: 10.1016/s1097-2765(00)00108-8
Heman-Ackah, S. M., Manzano, R., Hoozemans, J. J. M., Scheper, W., Flynn, R., Haerty, W., et al. (2017). Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 26, 4441–4450. doi: 10.1093/hmg/ddx331
Hetz, C., Bernasconi, P., Fisher, J., Lee, A.-H., Bassik, M. C., Antonsson, B., et al. (2006). Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 312, 572–576. doi: 10.1126/science.1123480
Hetz, C., and Papa, F. R. (2018). The unfolded protein response and cell fate control. Mol. Cell 69, 169–181. doi: 10.1016/j.molcel.2017.06.017
Hetz, C., Thielen, P., Matus, S., Nassif, M., Court, F., Kiffin, R., et al. (2009). XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 23, 2294–2306. doi: 10.1101/gad.1830709
Hoozemans, J. J. M., van Haastert, E. S., Eikelenboom, P., de Vos, R. A. I., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 354, 707–711. doi: 10.1016/j.bbrc.2007.01.043
Hoozemans, J. J. M., van Haastert, E. S., Nijholt, D. A. T., Rozemuller, A. J. M., Eikelenboom, P., and Scheper, W. (2009). The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am. J. Pathol. 174, 1241–1251. doi: 10.2353/ajpath.2009.080814
Hoozemans, J. J. M., Veerhuis, R., Van Haastert, E. S., Rozemuller, J. M., Baas, F., Eikelenboom, P., et al. (2005). The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 110, 165–172.
Hou, N. S., Gutschmidt, A., Choi, D. Y., Pather, K., Shi, X., Watts, J. L., et al. (2014). Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. U.S.A. 111, E2271–E2280. doi: 10.1073/pnas.1318262111
Hou, N. S., and Taubert, S. (2014). Membrane lipids and the endoplasmic reticulum unfolded protein response: an interesting relationship. Worm 3:e962405. doi: 10.4161/21624046.2014.962405
Hussain, S. G., and Ramaiah, K. V. A. (2007). Reduced eIF2alpha phosphorylation and increased proapoptotic proteins in aging. Biochem. Biophys. Res. Commun. 355, 365–370. doi: 10.1016/j.bbrc.2007.01.156
Jazwinski, S. M. (2002). Growing old: metabolic control and yeast aging. Annu. Rev. Microbiol. 56, 769–792. doi: 10.1146/annurev.micro.56.012302.160830
Jiang, H. Q., Ren, M., Jiang, H. Z., Wang, J., Zhang, J., Yin, X., et al. (2014). Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience 277, 132–138. doi: 10.1016/j.neuroscience.2014.03.047
Jiang, Y., Chadwick, S. R., and Lajoie, P. (2016). Endoplasmic reticulum stress: the cause and solution to Huntington’s disease? Brain Res. 1648, 650–657. doi: 10.1016/j.brainres.2016.03.034
Kaeberlein, M., Hu, D., Kerr, E. O., Tsuchiya, M., Westman, E. A., Dang, N., et al. (2005). Increased life span due to calorie restriction in respiratory-deficient yeast. PLoS Genet. 1:e69. doi: 10.1371/journal.pgen.0010069
Karagöz, G. E., Acosta-Alvear, D., and Walter, P. (2019). The unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. [Epub ahead of print].
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721. doi: 10.1126/science.290.5497.1717
Koga, H., and Cuervo, A. M. (2011). Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol. Dis. 43, 29–37. doi: 10.1016/j.nbd.2010.07.006
Kohno, K., Normington, K., Sambrook, J., Gething, M. J., and Mori, K. (1993). The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol. Cell. Biol. 13, 877–890. doi: 10.1128/MCB.13.2.877
Kono, N., Amin-Wetzel, N., and Ron, D. (2017). Generic membrane-spanning features endow IRE1α with responsiveness to membrane aberrancy. Mol. Biol. Cell 28, 2318–2332. doi: 10.1091/mbc.e17-03-0144
Labunskyy, V. M., Gerashchenko, M. V., Delaney, J. R., Kaya, A., Kennedy, B. K., Kaeberlein, M., et al. (2014). Lifespan extension conferred by endoplasmic reticulum secretory pathway deficiency requires induction of the unfolded protein response. PLoS Genet. 10:e1004019. doi: 10.1371/journal.pgen.1004019
Lajoie, P., Moir, R. D., Willis, I. M., and Snapp, E. L. (2012). Kar2p availability defines distinct forms of endoplasmic reticulum stress in living cells. Mol. Biol. Cell 23, 955–964. doi: 10.1091/mbc.E11-12-0995
Lajoie, P., and Snapp, E. L. (2011). Changes in BiP availability reveal hypersensitivity to acute endoplasmic reticulum stress in cells expressing mutant huntingtin. J. Cell Sci. 124, 3332–3343. doi: 10.1242/jcs.087510
Lee, H., Noh, J.-Y., Oh, Y., Kim, Y., Chang, J.-W., Chung, C.-W., et al. (2012). IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 21, 101–114. doi: 10.1093/hmg/ddr445
Lee, J. H., Won, S. M., Suh, J., Son, S. J., Moon, G. J., Park, U. J., et al. (2010). Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 42, 386–394. doi: 10.3858/emm.2010.42.5.040
Leitman, J., Barak, B., Benyair, R., Shenkman, M., Ashery, U., Hartl, F. U., et al. (2014). ER stress-induced eIF2-alpha phosphorylation underlies sensitivity of striatal neurons to pathogenic huntingtin. PLoS One 9:e90803. doi: 10.1371/journal.pone.0090803
Leitman, J., Ulrich Hartl, F., and Lederkremer, G. Z. (2013). Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun. 4:2753. doi: 10.1038/ncomms3753
Lin, J. H., Li, H., Yasumura, D., Cohen, H. R., Zhang, C., Panning, B., et al. (2007). IRE1 signaling affects cell fate during the unfolded protein response. Science 318, 944–949. doi: 10.1126/science.1146361
Lippincott-Schwartz, J., Bonifacino, J. S., Yuan, L. C., and Klausner, R. D. (1988). Degradation from the endoplasmic reticulum: disposing of newly synthesized proteins. Cell 54, 209–220. doi: 10.1016/0092-8674(88)90553-3
Liu, Y., and Chang, A. (2008). Heat shock response relieves ER stress. EMBO J. 27, 1049–1059. doi: 10.1038/emboj.2008.42
Lu, M., Lawrence, D. A., Marsters, S., Acosta-Alvear, D., Kimmig, P., Mendez, A. S., et al. (2014). Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345, 98–101. doi: 10.1126/science.1254312
Ma, Y., and Hendershot, L. M. (2004). ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 28, 51–65. doi: 10.1016/j.jchemneu.2003.08.007
Macdonald, M. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983. doi: 10.1016/0092-8674(93)90585-E
Madreiter-Sokolowski, C. T., Waldeck-Weiermair, M., Bourguignon, M.-P., Villeneuve, N., Gottschalk, B., Klec, C., et al. (2019). Enhanced inter-compartmental Ca2+ flux modulates mitochondrial metabolism and apoptotic threshold during aging. Redox Biol. 20, 458–466. doi: 10.1016/j.redox.2018.11.003
Marcora, M. S., Belfiori-Carrasco, L. F., Bocai, N. I., Morelli, L., and Castaño, E. M. (2017). Amyloid-β42 clearance and neuroprotection mediated by X-box binding protein 1 signaling decline with aging in the Drosophila brain. Neurobiol. Aging 60, 57–70. doi: 10.1016/j.neurobiolaging.2017.08.012
Martindale, D., Hackam, A., Wieczorek, A., Ellerby, L., Wellington, C., McCutcheon, K., et al. (1998). Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat. Genet. 18, 150–154. doi: 10.1038/ng0298-150
Martínez, G., Duran-Aniotz, C., Cabral-Miranda, F., Vivar, J. P., and Hetz, C. (2017). Endoplasmic reticulum proteostasis impairment in aging. Aging Cell 16, 615–623. doi: 10.1111/acel.12599
Martinez-Lopez, N., Athonvarangkul, D., and Singh, R. (2015). Autophagy and aging. Adv. Exp. Med. Biol. 847, 73–87. doi: 10.1007/978-1-4939-2404-2_3
Matus, S., Lopez, E., Valenzuela, V., Nassif, M., and Hetz, C. (2013). Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS One 8:e66672. doi: 10.1371/journal.pone.0066672
McCracken, A. A., and Brodsky, J. L. (1996). Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol. 132, 291–298. doi: 10.1083/jcb.132.3.291
Meléndez, A., Tallóczy, Z., Seaman, M., Eskelinen, E.-L., Hall, D. H., and Levine, B. (2003). Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391. doi: 10.1126/science.1087782
Mercado, G., Castillo, V., Soto, P., López, N., Axten, J. M., Sardi, S. P., et al. (2018). Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol. Dis. 112, 136–148. doi: 10.1016/j.nbd.2018.01.004
Mercado, G., Castillo, V., Soto, P., and Sidhu, A. (2016). ER stress and Parkinson’s disease: pathological inputs that converge into the secretory pathway. Brain Res. 1648, 626–632. doi: 10.1016/j.brainres.2016.04.042
Mizushima, N. (2007). Autophagy: process and function. Genes Dev. 21, 2861–2873. doi: 10.1101/gad.1599207
Munafó, D. B., and Colombo, M. I. (2001). A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J. Cell Sci. 114, 3619–3629.
Nagy, M., Fenton, W. A., Li, D., Furtak, K., and Horwich, A. L. (2016). Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc. Natl. Acad. Sci. U.S.A. 113, 5424–5428. doi: 10.1073/pnas.1604885113
Naidoo, N. (2009). ER and aging-Protein folding and the ER stress response. Ageing Res. Rev. 8, 150–159. doi: 10.1016/j.arr.2009.03.001
Naidoo, N., Ferber, M., Master, M., Zhu, Y., and Pack, A. I. (2008). Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 28, 6539–6548. doi: 10.1523/JNEUROSCI.5685-07.2008
Nascimbeni, A. C., Giordano, F., Dupont, N., Grasso, D., Vaccaro, M. I., Codogno, P., et al. (2017). ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J. 36, 2018–2033. doi: 10.15252/embj.201797006
Novick, P., Ferro, S., and Schekman, R. (1981). Order of events in the yeast secretory pathway. Cell 25, 461–469. doi: 10.1016/0092-8674(81)90064-7
Nuss, J. E., Choksi, K. B., DeFord, J. H., and Papaconstantinou, J. (2008). Decreased enzyme activities of chaperones PDI and BiP in aged mouse livers. Biochem. Biophys. Res. Commun. 365, 355–361. doi: 10.1016/j.bbrc.2007.10.194
Ozcan, U., Cao, Q., Yilmaz, E., Lee, A.-H., Iwakoshi, N. N., Ozdelen, E., et al. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. doi: 10.1126/science.1103160
Paz Gavilán, M., Vela, J., Castaño, A., Ramos, B., del Río, J. C., Vitorica, J., et al. (2006). Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol. Aging 27, 973–982. doi: 10.1016/j.neurobiolaging.2005.05.010
Penney, J. B., Vonsattel, J. P., MacDonald, M. E., Gusella, J. F., and Myers, R. H. (1997). CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann. Neurol. 41, 689–692. doi: 10.1002/ana.410410521
Pincus, D., Chevalier, M. W., Aragón, T., van Anken, E., Vidal, S. E., El-Samad, H., et al. (2010). BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 8:e1000415. doi: 10.1371/journal.pbio.1000415
Pineau, L., Colas, J., Dupont, S., Beney, L., Fleurat-Lessard, P., Berjeaud, J.-M., et al. (2009). Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic 10, 673–690. doi: 10.1111/j.1600-0854.2009.00903.x
Promlek, T., Ishiwata-Kimata, Y., Shido, M., Sakuramoto, M., Kohno, K., and Kimata, Y. (2011). Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 22, 3520–3532. doi: 10.1091/mbc.E11-04-0295
Rivas, A., Vidal, R. L., and Hetz, C. (2015). Targeting the unfolded protein response for disease intervention. Expert Opin. Ther. Targets 19, 1203–1218. doi: 10.1517/14728222.2015.1053869
Ruis, H., and Schüller, C. (1995). Stress signaling in yeast. Bioessays 17, 959–965. doi: 10.1002/bies.950171109
Rutkowski, D. T., Arnold, S. M., Miller, C. N., Wu, J., Li, J., Gunnison, K. M., et al. (2006). Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4:e374. doi: 10.1371/journal.pbio.0040374
Rutkowski, D. T., and Kaufman, R. J. (2007). That which does not kill me makes me stronger: adapting to chronic ER stress. Trends Biochem. Sci. 32, 469–476. doi: 10.1016/j.tibs.2007.09.003
Saxena, S., Cabuy, E., and Caroni, P. (2009). A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636. doi: 10.1038/nn.2297
Saxena, S., Roselli, F., Singh, K., Leptien, K., Julien, J.-P., Gros-Louis, F., et al. (2013). Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80, 80–96. doi: 10.1016/j.neuron.2013.07.027
Schuck, S., Prinz, W. A., Thorn, K. S., Voss, C., and Walter, P. (2009). Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 187, 525–536. doi: 10.1083/jcb.200907074
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111. doi: 10.1016/s1534-5807(02)00203-4
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039. doi: 10.1016/s0092-8674(00)80369-4
Snapp, E. L. (2012). Unfolded protein responses with or without unfolded proteins? Cells 1, 926–950. doi: 10.3390/cells1040926
Snapp, E. L., Sharma, A., Lippincott-Schwartz, J., and Hegde, R. S. (2006). Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. U.S.A. 103, 6536–6541. doi: 10.1073/pnas.0510657103
Tay, K. H., Luan, Q., Croft, A., Jiang, C. C., Jin, L., Zhang, X. D., et al. (2014). Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell. Signal. 26, 287–294. doi: 10.1016/j.cellsig.2013.11.008
Thibault, G., Ismail, N., and Ng, D. T. W. (2011). The unfolded protein response supports cellular robustness as a broad-spectrum compensatory pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20597–20602. doi: 10.1073/pnas.1117184109
Thibault, G., Shui, G., Kim, W., McAlister, G. C., Ismail, N., Gygi, S. P., et al. (2012). The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol. Cell 48, 16–27. doi: 10.1016/j.molcel.2012.08.016
Torres, M., Matamala, J. M., Duran-Aniotz, C., Cornejo, V. H., Foley, A., and Hetz, C. (2015). ER stress signaling and neurodegeneration: at the intersection between Alzheimer’s disease and Prion-related disorders. Virus Res. 207, 69–75. doi: 10.1016/j.virusres.2014.12.018
Travers, K. J., Patil, C. K., Wodicka, L., Lockhart, D. J., Weissman, J. S., and Walter, P. (2000). Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258. doi: 10.1016/s0092-8674(00)80835-1
Upton, J.-P., Wang, L., Han, D., Wang, E. S., Huskey, N. E., Lim, L., et al. (2012). IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 338, 818–822. doi: 10.1126/science.1226191
Valenzuela, V., Martínez, G., Duran-Aniotz, C., and Hetz, C. (2016). Gene therapy to target ER stress in brain diseases. Brain Res. 1648, 561–570. doi: 10.1016/j.brainres.2016.04.064
van der Vlies, D., Woudenberg, J., and Post, J. A. (2003). Protein oxidation in aging: endoplasmic reticulum as a target. Amino Acids 25, 397–407. doi: 10.1007/s00726-003-0025-9
Verghese, J., Abrams, J., Wang, Y., and Morano, K. A. (2012). Biology of the heat shock response and protein chaperones: budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol. Mol. Biol. Rev. 76, 115–158. doi: 10.1128/mmbr.05018-11
Vidal, R., Caballero, B., Couve, A., and Hetz, C. (2011). Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Curr. Mol. Med. 11, 1–12. doi: 10.2174/156652411794474419
Vidal, R. L., Figueroa, A., Court, F. A., Thielen, P., Molina, C., Wirth, C., et al. (2012). Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum. Mol. Genet. 21, 2245–2262. doi: 10.1093/hmg/dds040
Vidal, R. L., and Hetz, C. (2012). Crosstalk between the UPR and autophagy pathway contributes to handling cellular stress in neurodegenerative disease. Autophagy 8, 970–972. doi: 10.4161/auto.20139
Vieira, F. G., Ping, Q., Moreno, A. J., Kidd, J. D., Thompson, K., Jiang, B., et al. (2015). Guanabenz treatment accelerates disease in a mutant SOD1 mouse model of ALS. PLoS One 10:e0135570. doi: 10.1371/journal.pone.0135570
Voellmy, R. (2004). Transcriptional Regulation of the Metazoan Stress Protein Response. Amsterdam: Elsevier, 143–185.
Volmer, R., and Ron, D. (2015). Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 33, 67–73. doi: 10.1016/j.ceb.2014.12.002
Volmer, R., van der Ploeg, K., and Ron, D. (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A. 110, 4628–4633. doi: 10.1073/pnas.1217611110
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wang, L., Popko, B., and Roos, R. P. (2014a). An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 23, 2629–2638. doi: 10.1093/hmg/ddt658
Wang, L., Popko, B., Tixier, E., and Roos, R. P. (2014b). Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol. Dis. 71, 317–324. doi: 10.1016/j.nbd.2014.08.010
Welihinda, A. A., Tirasophon, W., and Kaufman, R. J. (1999). The cellular response to protein misfolding in the endoplasmic reticulum. Gene Expr 7, 293–300.
Werner, E. D., Brodsky, J. L., and McCracken, A. A. (1996). Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc. Natl. Acad. Sci. U.S.A. 93, 13797–13801. doi: 10.1073/pnas.93.24.13797
Yorimitsu, T., Nair, U., Yang, Z., and Klionsky, D. J. (2006). Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30299–30304. doi: 10.1074/jbc.M607007200
Yoshida, H. (2007). ER stress and diseases. FEBS J. 274, 630–658. doi: 10.1111/j.1742-4658.2007.05639.x
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and Mori, K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749. doi: 10.1074/jbc.273.50.33741
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/s0092-8674(01)00611-0
Zhang, C., and Cuervo, A. M. (2008). Restoration of chaperone-mediated autophagy in aging liver improves cellular maintenance and hepatic function. Nat. Med. 14, 959–965. doi: 10.1038/nm.1851
Zhang, P., McGrath, B., Li, S., Frank, A., Zambito, F., Reinert, J., et al. (2002). The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell Biol. 22, 3864–3874. doi: 10.1128/mcb.22.11.3864-3874.2002
Keywords: endoplasm reticulum stress, aging, neurodegeneration, unfolded protein response, proteostasis
Citation: Chadwick SR and Lajoie P (2019) Endoplasmic Reticulum Stress Coping Mechanisms and Lifespan Regulation in Health and Diseases. Front. Cell Dev. Biol. 7:84. doi: 10.3389/fcell.2019.00084
Received: 05 March 2019; Accepted: 03 May 2019;
Published: 21 May 2019.
Edited by:
Guillaume Thibault, Nanyang Technological University, SingaporeReviewed by:
Jesse C. Hay, University of Montana, United StatesMarcelo Ehrlich, Tel Aviv University, Israel
Copyright © 2019 Chadwick and Lajoie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Lajoie, cGxham9pZTNAdXdvLmNh