 Anthony K. N. Chan
Anthony K. N. Chan Chun-Wei Chen
Chun-Wei Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 15 May 2019
Sec. Epigenomics and Epigenetics
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00081
This article is part of the Research TopicEpigenetic Regulation of Stem Cell Plasticity in Tissue Regeneration and DiseaseView all 5 articles
Leukemias driven by chromosomal translocation of the mixed-lineage leukemia gene (MLL or KMT2A) are highly prevalent in pediatric oncology. The poor survival rate and lack of an effective targeted therapy for patients with MLL-rearranged (MLL-r) leukemias emphasize an urgent need for improved knowledge and novel therapeutic approaches for these malignancies. The resulting chimeric products of MLL gene rearrangements, i.e., MLL-fusion proteins (MLL-FPs), are capable of transforming hematopoietic stem/progenitor cells (HSPCs) into leukemic blasts. The ability of MLL-FPs to reprogram HSPCs toward leukemia requires the involvement of multiple chromatin effectors, including the histone 3 lysine 79 methyltransferase DOT1L, the chromatin epigenetic reader BRD4, and the super elongation complex. These epigenetic regulators constitute a complicated network that dictates maintenance of the leukemia program, and therefore represent an important cluster of therapeutic opportunities. In this review, we will discuss the role of MLL and its fusion partners in normal HSPCs and hematopoiesis, including the links between chromatin effectors, epigenetic landscapes, and leukemia development, and summarize current approaches to therapeutic targeting of MLL-r leukemias.
In the 1980s, leukemia cases were observed that showed complete phenotypic lineage switching, i.e., patients initially diagnosed as acute lymphocytic leukemia (ALL) relapsed as acute myeloid leukemia (AML) during chemotherapy (Stass et al., 1984; Mirro et al., 1985; Gagnon et al., 1989). Accordingly, the term mixed-lineage leukemia was coined (Mirro et al., 1985; Slany, 2009). Mixed-linage leukemia was reported to be associated with translocation on the long arm (q) of chromosome 11 band q23 (11q23) (Hayashi et al., 1990). Recurring 11q23 translocations are also found in both ALL (Inaba et al., 2013; Roberts and Mullighan, 2015) and AML (Döhner et al., 2015; Papaemmanuil et al., 2016). Guided by these observations, researchers located and cloned an important gene that resides on 11q23 and drives leukemogenesis when it is translocated and fused with other gene partners (Ziemin-van der Poel et al., 1991; Djabali et al., 1992; Gu et al., 1992; Tkachuk et al., 1992). In the initial discoveries, the gene was given different names: MLL (myeloid/lymphoid, or mixed-lineage leukemia) (Ziemin-van der Poel et al., 1991), ALL-1 (Gu et al., 1992), HRX (human trithorax, the human homolog of the Drosophilia trithorax, trx) (Tkachuk et al., 1992), and trithorax-like gene (Djabali et al., 1992). Subsequently, it was also re-named as KMT2A (lysine methyltransferase 2A), based on its lysine (Lys, K) methyltransferase enzymatic activity. For consistent description of the gene, we will use MLL throughout this review article.
Accordingly, leukemias that involve chromosomal rearrangement of MLL are called MLL-rearranged (MLL-r) leukemias (Rowley, 1993; Arber et al., 2016; Krivtsov et al., 2017). In adults, MLL-r leukemia accounts for approximately 5% of ALL cases (Marchesi et al., 2011) and 5–10% of AML cases (Krivtsov and Armstrong, 2007; Chowdhury and Brady, 2008; Chen and Armstrong, 2015). MLL-r leukemias are more prevalent in infant patients (<1 year of age), in which approximately 70% of infants with ALL are diagnosed as having 11q23 rearrangements (Chen et al., 1993; Heerema et al., 1999; Muntean and Hess, 2012). AML is less common than ALL in infants, in which about 50–66% of infants with AML have 11q23 translocations (Sorensen et al., 1994; Martinez-Climent et al., 1995; Hilden et al., 1997; Satake et al., 1999). Prognosis is poor for infants with MLL-r ALL, who have worse outcomes than do older children (>12 months) (Pieters et al., 2007). The five-year event-free survival rate of non-MLL-r ALL can approach 60–96% in infants (<1 year of age), but is significantly lower in MLL-r infant ALL patients—only 34–39% depending on treatment protocols (Hilden et al., 2006; Tomizawa et al., 2007). In addition, approximately 2–15% of cancer patients receiving chemotherapeutic drugs that target DNA-topoisomerase II develop treatment-related AML with 11q23 translocations (Super et al., 1993; Broeker et al., 1996; Greaves, 1997; Felix, 1998). These situations emphasize the unmet need for a deeper understanding of the underlying biology and novel therapeutic approaches for MLL-r leukemia.
Surprisingly, comprehensive genomic studies revealed that the genomes of patient-derived MLL-r leukemia cells displayed remarkable stability with only a few genetic alterations (Mullighan et al., 2007; Radtke et al., 2009). This suggests that MLL-r leukemias are largely driven by epigenetic dysregulation (Radtke et al., 2009; Bernt and Armstrong, 2011). Epigenetics is the study of heritable and reversible control of gene expression that is not dependent on the DNA sequence. In eukaryotic nuclei, DNA molecules are wrapped around histone cores consisting of eight histone proteins—H2A, H2B, H3, and H4 (two copies of each protein are present in the core). Approximately 146 bp of DNA surrounds each octameric histone core to form a nucleosome, the basic structural unit of chromatin. The nucleosomal units are packed and condensed further to form chromatin. The N-terminal peptide tails of H3 and H4 protrude from the histone core, and can be post-translationally modified in various ways (e.g., acetylation, methylation, phosphorylation, ubiquitination, sumoylation) inside eukaryotic cells (Bannister and Kouzarides, 2011). These modifications can be added by epigenetic writers, interpreted by epigenetic readers, and removed by epigenetic erasers (Arrowsmith et al., 2012; Musselman et al., 2012; Seto and Yoshida, 2014; Zhang et al., 2015). Specific modifications or epigenetic histone marks have differential effects on gene expression. For example, acetylated histone marks (e.g., H3K9ac and H3K27ac) are usually associated with gene activation (Krejčí et al., 2009; Creyghton et al., 2010; Hawkins et al., 2011; Hezroni et al., 2011). In contrast, methylated modifications are context-dependent: for instance, methylation on H3K4 or H3K79 is associated with gene activation (Schübeler et al., 2004), whereas methylation on H3K9 or H3K27 is associated with gene silencing (Musselman et al., 2012). In this review, we will introduce the mechanistic roles of MLL in normal hematopoiesis and MLL-r leukemia, describe current therapeutic targets in MLL-r leukemia, with an emphasis on chromatin epigenetic regulators, and discuss the potential of using combined epigenetic targeting strategies to treat MLL-r leukemia.
Expression of the developmentally important homeobox (HOX) cluster genes is mediated by MLL in normal hematopoietic stem/progenitor cells (HSPCs) (Kawagoe et al., 1999). Genetic knock-out of Mll in mice is embryonic lethal, with an altered Hox gene pattern, defects in yolk sac hematopoiesis, reduced proliferation and/or survival of hematopoietic progenitors, and defective HSPC activity in the aorta–gonad–mesonephros region (Yu et al., 1995; Hess et al., 1997; Yagi et al., 1998; Ernst et al., 2004). Using conditional Mll knock-out (Mll–/–) mice, McMahon et al. (2007) demonstrated that Mll was not important for the production of mature adult hematopoietic lineages, but it was required for stem cell self-renewal in fetal liver and adult bone marrow. Furthermore, Mll plays an important role in regulating transcription initiation by RNA polymerase II via H3K4 methylation (Wang et al., 2009). Although Mll was shown to influence less than 5% of promoters that carry the H3K4me3 mark in mouse embryonic fibroblasts, these Mll-regulated promoters include important developmental regulators such as the Hox genes (Wang et al., 2009). In humans, the MLL gene encodes a protein product of 3,969 amino acids (Figure 1A). This product is post-translationally cleaved by threonine aspartase 1 (taspase1) into two distinct modules (MLL-N and MLL-C), then these two modules are assembled together via the FY-rich N- and C-terminal domains (FYRN and FYRC) (García-Alai et al., 2010; Figure 1A). A recent study showed that uncleaved MLL displays higher stability than the assembled dimer (MLL-N/MLL-C) (Zhao et al., 2018). Casein kinase II (CKII) phosphorylates MLL at a location proximal to the taspase1 cleavage site, which facilitates taspase1-dependent processing of MLL into MLL-N and MLL-C (Zhao et al., 2018). This finding suggested that pharmacological targeting of MLL to enhance its stability through inhibition of CKII may present a new therapeutic opportunity in MLL-r leukemia, as uncleaved MLL can displace leukemia-causing MLL-fusion proteins (MLL-FPs) from chromatin (Zhao et al., 2018).
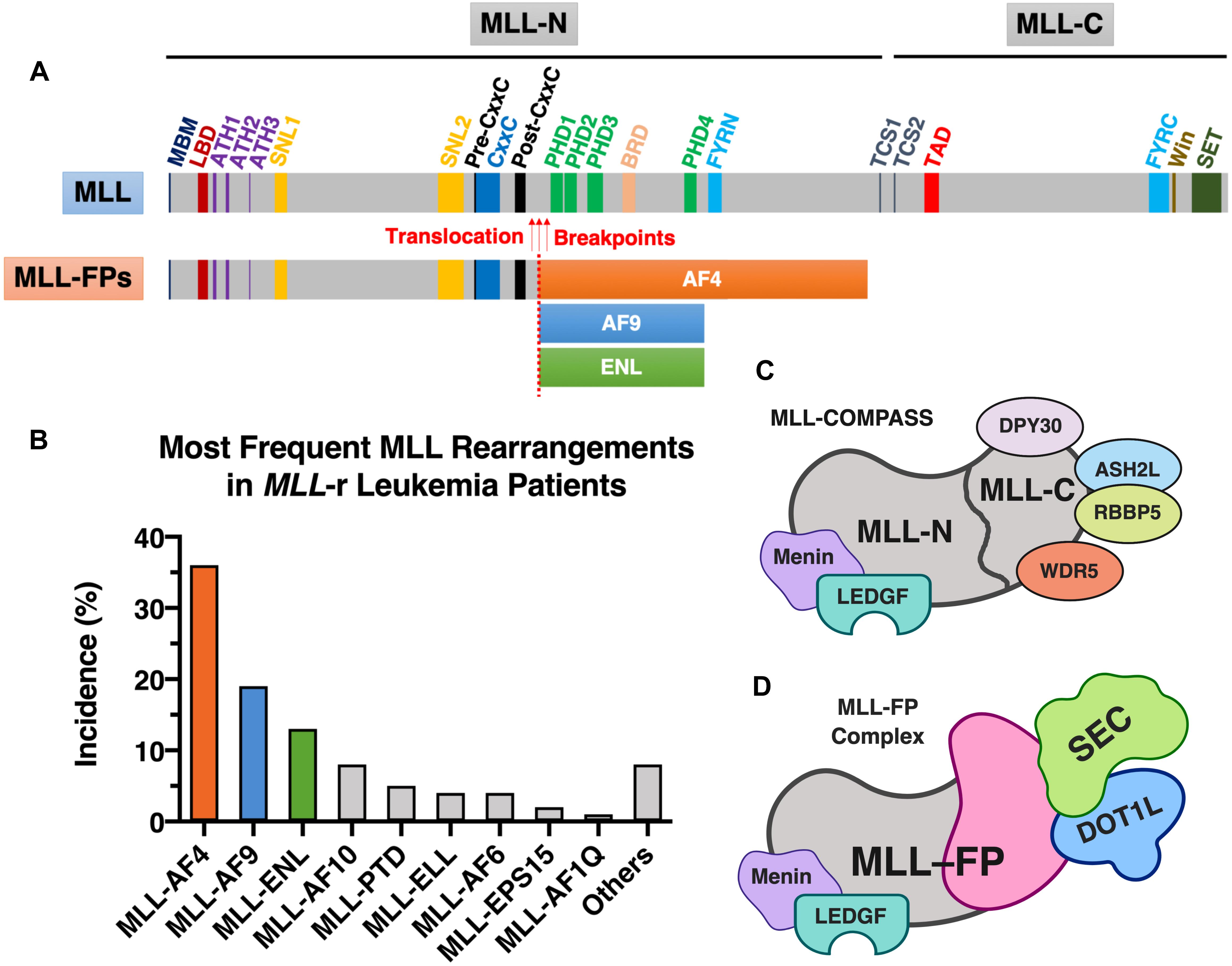
Figure 1. MLL and MLL-FPs. (A) Schematic of domain architecture of wild-type MLL and MLL-FPs. The wild-type canonical form of human MLL protein (UniProt ID: Q03164) has a total of 3,969 amino acids in length and contains several functional domains and important sites (drawn to scale): high-affinity Menin-binding motif (MBM, residue 6–10) (Yokoyama et al., 2005), LEDGF-binding domain (LBD, residue 109–153) (Yokoyama and Cleary, 2008), AT-Hook1/2/3 (ATH1, residue 169–180; ATH2, residue 217–227; ATH3, residue 301–309; UniProt annotations of Q03164); nuclear-localization signal 1/2 (SNL1, residue 400–443; SNL2, 1008–1106) (Ayton et al., 2004), pre-CxxC region (residue 1149–1154) (Muntean et al., 2010), CxxC domain (residue 1147–1242) (Bach et al., 2008), post-CxxC (residue 1298–1337) (Muntean et al., 2010), plant homology domain 1/2/3/4 (PHD1, residue 1431–1482; PHD2, residue 1479–1533, PHD3, residue 1566–1627; PHD4, residue 1931–1978; UniProt annotations of Q03164), bromodomain (BRD, residue 1703–1748; UniProt annotations of Q03164), FY-rich N-terminal domain (FYRN, residue 2018–2074; UniProt annotations of Q03164), FY-rich C-terminal domain (FYRC, residue 3666–3747; UniProt annotations of Q03164), taspase1 cleavage site 1/2 (TCS1, residue 2666–2670, D/GADD; TCS2, residue 2718–2722, D/GVDD; the exact cleavage sites are indicated by forward slashes) (Hsieh et al., 2003), transactivator domain (TAD, residue 2829–2883) (Ernst et al., 2001), WDR5 interaction motif (Win; residue 3762–3773) (Patel et al., 2008), and Su(Var)3-9, enhancer-of-zeste, trithorax domain (SET, residue 3829–2945; UniProt annotations of Q03164). The most frequently observed translocation breakpoints (indicated by red arrows) are located in the region between CxxC and PHDs. The three most common MLL-FPs (MLL-AF4, MLL-AF9, and MLL-ENL) are illustrated (the translocation breakpoints and the size of FPs are partially drawn to scale). (B) The most frequent MLL rearrangements identified in MLL-r leukemia patients. The statistics shown in this figure was obtained from a study of 2,345 MLL-r leukemia patients dated from 2003 to 2016 (Meyer et al., 2018). (C) Components of MLL-COMPASS. (D) MLL-FP (e.g., MLL-AF4, MLL-AF9, or MLL-ENL) in complex with DOT1L and SEC. Figure 1 was created with BioRender.com.
MLL-N consists of a Menin-binding motif and a lens epithelium-derived growth factor (LEDGF)-binding domain (Yokoyama and Cleary, 2008); three DNA-binding AT-hook motifs (Zeleznik-Le et al., 1994); two nuclear-localization signals, SNL1 and SNL2 (Yano et al., 1997; Ayton et al., 2004); a pre-CxxC domain (Muntean et al., 2010); a non-methyl-CpG recognizing the CxxC domain (Birke et al., 2002; Ayton et al., 2004; Allen et al., 2006); a post-CxxC domain (Muntean et al., 2010); a bromodomain (BRD); four plant homology domains (PHD fingers) (Fair et al., 2001; Ali et al., 2014); and the homodimerization-facilitating domain FYRN (García-Alai et al., 2010; Figure 1A). The sequences flanking the CxxC domain—the pre-CxxC and post-CxxC domains—were demonstrated to be important for direct interaction of MLL with the polymerase associated factor complex (PAFc) (Muntean et al., 2010). PAFc promotes MLL and MLL-FP recruitment to target loci to activate transcription of target genes such as HOXA9 (Muntean et al., 2010). Therefore, PAFc is a crucial cofactor for both transcriptional regulation by MLL and leukemogenesis mediated by MLL-FPs (Muntean et al., 2010). The BRD of MLL recognizes acetylated lysine residues, whereas the third PHD finger of MLL specifically interacts with H3K4me2/3 (Chang P.-Y. et al., 2010). Binding of the third PHD finger of MLL to H3K4me3 is required for MLL-dependent gene transcription (Chang P.-Y. et al., 2010).
MLL-C possesses two domains capable of modifying chromatin: a transactivator domain (TAD), followed by a SET [Su(Var)3-9, enhancer-of-zeste, trithorax] domain (Figure 1A). The MLL SET domain confers methyltransferase activity that catalyzes the transfer of a methyl group from S-adenosylmethionine to H3K4 (Milne et al., 2002). MLL-C is further assembled into a larger protein complex that contains several cofactors: WD repeat protein 5 (WDR5), retinoblastoma-binding protein 5 (RBBP5), Set1/Ash2 histone methyltransferase complex subunit ASH2 (ASH2L), and protein dpy-30 homolog (DPY30) (Rao and Dou, 2015). WDR5, RBBP5, ASH2L, and DPY30 form a core entity with the MLL SET domain, and enhance the H3K4 dimethylation activity of the MLL SET domain by ∼600-fold (Dou et al., 2006; Patel et al., 2009). Although complete deletion of the Mll gene in mice results in embryonic lethality (Yu et al., 1995), mice that harbor a homozygous SET domain deletion (MllΔSET) survive into adulthood and maintain relatively normal hematopoiesis (Mishra et al., 2014). Because the profile of H3K4 methylation at the Hoxa loci remains normal in HSPCs isolated from MllΔSET mice, Mishra et al. (2014) speculated that MLL is not the dominant H3K4 methyltransferase that controls Hox gene expression. In addition to MLL, five more MLL family members of H3K4 methyltransferases (MLL2, MLL3, MLL4, SETD1A, and SETD1B) are found in mammals, and they associate with other protein factors to form larger macromolecular complexes called COMPASS (complex of proteins associated with Set1; named for the single yeast homolog) (Rao and Dou, 2015; Li et al., 2016; Slany, 2016; Meeks and Shilatifard, 2017). All of the MLL proteins physically associate with four conserved factors—WDR5, RBBP5, ASH2L, and DPY30 (Figure 1C), which stimulates the H3K4 methyltransferase activity of MLL proteins (Rao and Dou, 2015; Li et al., 2016). Among the six MLL proteins, MLL and MLL2 share two unique factors—Menin and LEDGF (Figure 1C), which mediate the recruitment of MLL/MLL2 to their gene targets (Rao and Dou, 2015). Using mouse embryonic fibroblasts as a cell model, Wang et al. (2009) showed that Menin-interacting Mll and Mll2 are key regulators of Hox genes, however, the loss of Mll3/Mll4 had little to no effect on H3K4 methylation of Hox loci and the expression of Hox genes. This suggests that individual MLL family member may play different functional roles. The MLL TAD interacts with the histone acetyltransferases CBP/p300, MOZ, and MOF, which transfer acetyl groups to H3K27, H3K9, and H4K16, respectively (Slany, 2016). These acetyltransferase activities are associated with Hox gene activation by the normal MLL protein (Ernst et al., 2001; Dou et al., 2005; Paggetti et al., 2010).
As described above, the presence of MLL rearrangements at the 11q23 chromosomal location is associated with poor clinical prognosis, and certain subgroups of MLL-r leukemia are associated with worse therapeutic outcomes (Balgobind et al., 2009; Szczepański et al., 2010). As a result of different 11q23 chromosomal translocation events, a total of 135 different MLL rearrangements (of which 84 translocations generated in-frame MLL-FPs) were identified in patients with acute leukemia (Meyer et al., 2018). According to the report, the nine most frequent fusion partners of MLL are AF4 (∼36%), AF9 (∼19%), ENL (∼13%), AF10 (∼8%), PTD (∼5%), ELL (∼4%), AF6 (∼4%), EPS15 (∼2%), and AF1Q (∼1%), which together represent more than 92% of the MLL-FPs found in MLL-r leukemia patients (Meyer et al., 2018). We will focus on MLL-AF4, MLL-AF9, and MLL-ENL, the three most common MLL-FPs discovered in MLL-r leukemia patients (Meyer et al., 2018; Figures 1A,B).
Animal models of human disease are important for studying underlying disease mechanisms and testing therapeutic agents/approaches. Following discovery of numerous MLL-FPs in human patients, biomedical researchers attempted to recapitulate the leukemogenic effects driven by various MLL-FPs in mouse models (Milne, 2017). In various reports, the genetic introduction of MLL-AF4 (Chen et al., 2006; Metzler et al., 2006; Krivtsov et al., 2008; Tamai et al., 2011; Lin et al., 2016), MLL-AF9 (Corral et al., 1996; Dobson et al., 1999; Drynan et al., 2005; Barabé et al., 2007; Krivtsov et al., 2013; Buechele et al., 2015), or MLL-ENL (Lavau et al., 1997; Forster et al., 2003; Drynan et al., 2005; Barabé et al., 2007; Buechele et al., 2015), were demonstrated to be leukemogenic in mice. Further studies revealed the ability of these MLL-FPs to efficiently transform hematopoietic cells at different developmental stages (e.g., hematopoietic stem cells, common myeloid progenitors, and granulocyte and macrophage progenitors) into leukemic cells possessing stem-cell-like properties, such as being capable of self-renewal and leukemia initiation and maintenance (Cozzio et al., 2003; Krivtsov et al., 2006, 2008; Krivtsov and Armstrong, 2007).
MLL-FPs have different chromatin-modifying activities than normal MLL proteins. The C-terminal SET domain of wild-type MLL that harbors H3K4 methyltransferase activity is lost in MLL-FPs (Figure 1). Because multiple MLL fusion partners such as AF4/AF9/ENL/ELL are also components of the super elongation complex [SEC; composed of AF4 (or AFF4), AF9 (or ENL), EAF, ELL, and P-TEFb (positive transcription elongation factor b)] (Luo et al., 2012; Cucinotta and Arndt, 2016), chimeric MLL-FPs recruit the SEC (a crucial regulator of transcriptional elongation) (Figure 1D) and result in aberrant gene expression (Collins and Hess, 2016). Furthermore, several components of the SEC interact with the histone H3K79 methyltransferase DOT1L (disruptor of telomeric silencing 1-like) (Park et al., 2010; Biswas et al., 2011; Shen et al., 2013). Increased levels of H3K79 methylation by DOT1L are found at MLL-FP targeted genomic loci such as HOXA9 and MEIS1, which are associated with leukemic transformation (Okada et al., 2005; Krivtsov et al., 2008; Bernt et al., 2011; Li and Ernst, 2014; Chen et al., 2015; Collins and Hess, 2016). Given that histone H3K79 methylation is associated with active gene transcription (Schübeler et al., 2004), this suggests that DOT1L activity and the altered H3K79 epigenetic signature observed at MLL-FP binding loci may also contribute to the expression of the oncogenic program in MLL-r leukemias (Bernt et al., 2011; Jo et al., 2011; Chen et al., 2013; Deshpande et al., 2013). Thus, current evidence suggests that MLL-FPs connect MLL to the SEC and the DOT1L-H3K79 methyltransferase complex (consisting of DOT1L, AF9, AF10, and ENL) (Bernt et al., 2011), thereby contributing to the ectopic expression of the leukemic program (Figure 1D).
In recent years, chemical inhibitors that target chromatin epigenetic regulators have undergone active development for treatment of various cancers (Filippakopoulos and Knapp, 2014; Shi and Vakoc, 2014; Cai et al., 2015; Fujisawa and Filippakopoulos, 2017; Ribich et al., 2017; Schiedel and Conway, 2018). As discussed above, MLL-FPs associate with cofactors and recruit epigenetic effectors, which eventually lead to aberrant gene expression and leukemic transformation. Therefore, pharmacological disruption of the key proteins in this MLL-FP epigenetic network represents a therapeutic opportunity for the treatments of MLL-r leukemias (Figure 2; bottom). Furthermore, MYC is a well-known proto-oncogene that is frequently over-expressed in cancer, including in ALL, AML, and MLL-r leukemias (Schreiner et al., 2001; Langenau et al., 2003; Luo et al., 2005; Delgado and León, 2010; Dang, 2012; Li L. et al., 2014; Sanchez-Martin and Ferrando, 2017). Indeed, the MYC gene is a direct transcriptional target of MLL-FPs (Dawson et al., 2011; Li and Ernst, 2014). Despite its known oncogenic role in MLL-r leukemias, direct inhibition of the MYC transcription factor (TF) is challenging, because of its difficult-to-drug three-dimensional structure. Thus, down-regulating MYC gene expression by targeting more easily druggable chromatin-binding domain structures on the regulatory proteins that affect MYC expression is an active area of research (Figure 2; top). In the following sections, we will discuss the latest approaches to therapeutic targeting of the MLL-FP epigenetic network.
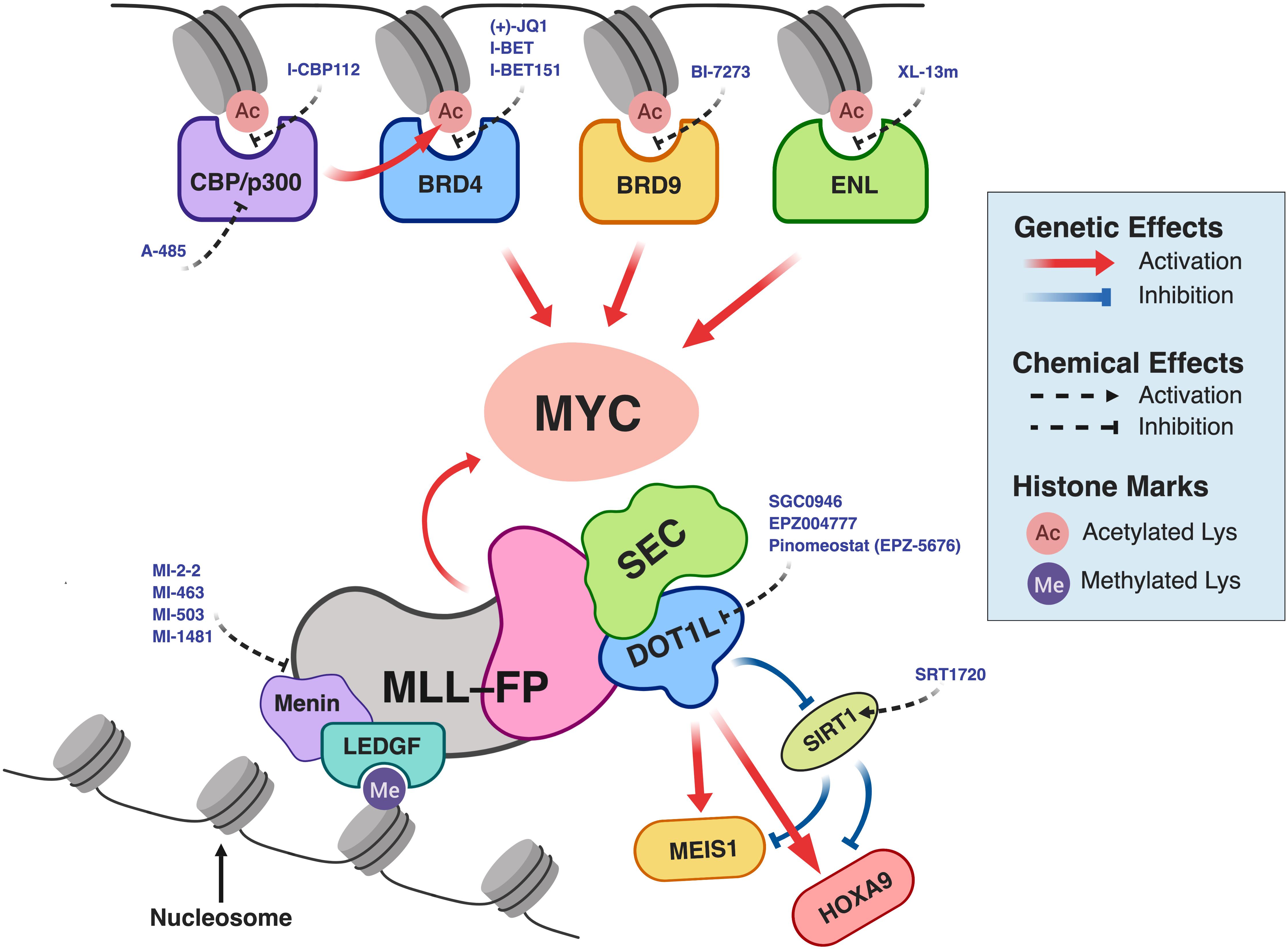
Figure 2. Pharmacological intervention in the MLL-FP epigenetic network in MLL-r leukemia. For simplicity, the components of protein complexes interacting with CBP/p300, BRD4, BRD9, or ENL are not illustrated. BRD4 and MLL-FPs (MLL-AF4 or MLL-AF9) together form a complex with SEC and PAFc (Dawson et al., 2011), which directs MYC expression. This schematic was created using BioRender.com.
Menin, encoded by the MEN1 gene, acts as a molecular adaptor to tether both MLL and LEDGF and is required for the oncogenic transformation of MLL-r leukemia mediated by MLL-FPs (Yokoyama et al., 2005; Yokoyama and Cleary, 2008). Genetic knockout of Men1 in mouse embryonic fibroblasts demonstrated that Menin is essential for H3K4 trimethylation at Hox loci and expression of nearly all Hox genes (Wang et al., 2009). Borkin et al. (2015) developed two potent small-molecule inhibitors (MI-463 and MI-503) that block the MLL-binding site on Menin, resulting in down-regulation of MLL-fusion targets (including Hoxa9 and Meis1 genes), differentiation of leukemic blasts, and prolonged survival of mouse models of MLL-r leukemia. A study reported that MLL1 does not require interaction with Menin to sustain hematopoietic stem cell hematopoiesis (Li et al., 2013). This provides a good rational for developing inhibitors of the Menin–MLL interaction to treat MLL-r leukemia without affecting normal cells. Indeed, Borkin et al. (2015) revealed that Menin inhibition by MI-463 and MI-503 resulted in no impairment of normal murine hematopoiesis after 10 days of continuous Menin inhibition. Guided by the molecular scaffolds of the two Menin inhibitors, the same group recently reported the development of a more potent Menin inhibitor, MI-1481, which showed low nanomolar inhibition (IC50 = 3.6 nM, measured by fluorescence polarization assay using fluorescein-labeled MLL4–43) against Menin–MLL interactions (Borkin et al., 2018). Compound MI-1481 showed potent activity in cells and in vivo models of MLL-r leukemias (Borkin et al., 2018).
Lens epithelium-derived growth factor is a transcriptional coactivator that specifically recognizes H3K36me2/3 (histone modifications that are associated with actively transcribed gene loci) through its PWWP domain (Pradeepa et al., 2012; Eidahl et al., 2013; Zhu et al., 2016). Through association with LEDGF, MLL is brought to chromatin to regulate transcription (Yokoyama and Cleary, 2008). MLL-ENL lacking the high-affinity Menin-binding motif (i.e., it cannot interact with Menin efficiently) failed to co-precipitate with endogenous LEDGF, suggesting that LEDGF interacts conjointly with MLL-ENL and Menin, but does not associate with either one of them separately (Yokoyama and Cleary, 2008). Recent reports suggest that LEDGF is dispensable for normal hematopoiesis but important for leukemogenesis; therefore, LEDGF is being considered as a potential drug target for MLL-r leukemias (Blokken et al., 2017; Ashkar et al., 2018).
Although wild-type MLL and MLL-FPs share the same MLL N-terminal and retain Menin/LEDGF-binding ability (Figures 1C,D), two studies suggested that MLL and MLL-FPs have different chromatin-tethering mechanisms (Wang et al., 2011; Xu et al., 2016). Wang et al. (2011) reported that MLL-FPs preferentially regulate only a small subset of wild-type MLL target genes using patient-derived leukemic cells and inducible MLL-ENL cellular system. Moreover, Xu et al. (2016) showed by ChIP-seq experiments that MLL and MLL-AF9 localized at different chromatin regions in a murine cell model of MLL-AF9, and they further demonstrated that these differences were due to the ability of MLL C-terminal domain to interact with WDR5. The C-terminal domain of MLL is lost in MLL-FPs, because of 11q23 translocations (Figure 1). These may explain why targeting either Menin or LEDGF could be a therapeutic approach in treating MLL-r leukemias, since MLL and MLL-FPs have differential dependence on Menin or LEDGF for activating gene expression.
Disruptor of telomeric silencing 1-like is a methyltransferase that catalyzes H3K79 mono-, di-, and tri-methylation (H3K79me1/2/3), which are associated with active gene expression (Barski et al., 2007; Jo et al., 2011). DOT1L binds to the nucleosomal disk surface and methylates H3K79 located in the globular histone core (Min et al., 2003). Because the aberrant methyltransferase activity of DOT1L is required for the leukemogenesis of MLL-r leukemia, DOT1L inhibition is a promising therapeutic intervention (Chang M.-J. et al., 2010; Bernt et al., 2011; Daigle et al., 2011; Nguyen et al., 2011). A number of small-molecule DOT1L inhibitors have been developed in recent years (Anglin and Song, 2013). One inhibitor, pinometostat (EPZ-5676), was identified as a highly potent and selective inhibitor of the DOT1L active site (Daigle et al., 2013). Pinometostat showed moderate to high clearance and low oral bioavailability in a non-clinical pharmacokinetic and metabolic study (Basavapathruni et al., 2014). A phase I clinical study recently revealed that administration of pinometostat was generally safe; however, its efficacy as a single-agent treatment was modest (Stein et al., 2018). Further studies are needed to investigate whether the combination of pinometostat with other pharmacological inhibitors can potentiate its therapeutic activity in patients with MLL-r leukemias.
Lysine-specific demethylase 1 (LSD1 or KDM1A), the first identified histone demethylase (Shi et al., 2004), is an epigenetic eraser of H3K4me1/2 and H3K9me1/2 histone marks (Shi et al., 2004; Forneris et al., 2005; Metzger et al., 2005; Amente et al., 2013). LSD1 is overexpressed in various solid cancers (bladder, breast, lung, and colorectal cancers) (Lim et al., 2010; Hayami et al., 2011), and in AML as well as lymphoid malignancies and myeloproliferative neoplasms (Lokken and Zeleznik-Le, 2012; Schenk et al., 2012; Niebel et al., 2014; Przespolewski and Wang, 2016). Harris et al. (2012) showed that LSD1 is required for sustaining the expression of the MLL-AF9 oncogenic program and identified LSD1 as a therapeutic target for MLL-AF9 leukemia. Genetic knock-down of Lsd1 by RNAi induced apoptosis and terminal macrophage differentiation of murine MLL-AF9 cells, and reduced AML leukemia stem cell potential in mice transplanted with murine MLL-AF9 cells (Harris et al., 2012). Moreover, Harris et al. (2012) found that pharmacological inhibition of LSD1 by OG-86 (also known as Compound B) (Maiques-Diaz and Somervaille, 2016) resulted in inhibition of colony formation and promotion of leukemic cell differentiation in murine MLL-AF9 cells and primary human MLL-AF6 and MLL-AF9 leukemic blasts (Harris et al., 2012). In 2018, ORY-1001 was identified as a highly potent and selective LSD1 covalent inhibitor with good bioavailability after intra-peritoneal and oral administration (Maes et al., 2018). ORY-1001 binds irreversibly and rapidly to the cofactor flavin adenine dinucleotide (FAD) when it is covalently bound to LSD1, but not to free FAD. ORY-1001 showed nanomolar inhibition (IC50 = 18 nM) of recombinant LSD1 and low nanomolar activity (EC50 < 1 nM in a FACS-based differentiation assay) against cellular LSD1 in THP1 cells (a human MLL-AF9 cell line) (Maes et al., 2018). ORY-1001 induced blast differentiation in leukemic cell lines (including MLL-r leukemic cell lines), primary AML samples, and two AML patients participating in a clinical trial of ORY-1001, and prolonged survival in murine models of acute leukemia (Maes et al., 2018). ORY-1001 is currently under ongoing phase I/IIa study in relapsed or refractory acute leukemia (EUDRACT no. 2013-002447-29) (Maes et al., 2018). Cusan et al. (2018) demonstrated that LSD1 inhibition using GSK-LSD1 (a LSD1 chemical inhibitor) induces an increase in chromatin accessibility on a global level. They also showed that the antileukemic effect exerted by GSK-LSD1 requires the TFs PU.1 and C/EBPα, as the reduction or depletion of PU.1 or C/EBPα expression level, respectively, rendered murine MLL-AF9 leukemia cells resistant to GSK-LSD1 inhibition (Cusan et al., 2018).
Bromodomains are specific epigenetic reader modules that recognize ε-N-acetylation of lysine motifs, the key epigenetic marks for maintaining open chromatin structure, which is associated with transcriptional activation. In the human genome, there are 46 known BRD-containing proteins. Some of these proteins possess more than one BRD, so there are a total of 61 different BRDs found in humans (Filippakopoulos et al., 2012). Large-scale structural and binding studies demonstrated that BRDs do not specifically read a particular acetylated lysine (Kac) sequence, but instead recognize a combination of different Kac motifs (Filippakopoulos et al., 2012). Although BRDs differ in the primary amino acid sequence, they all possess similar structural folds: a left-handed bundle of four α helices (designated αZ, αA, αB, αC) linked by loop regions of various lengths (ZA and BC loops) that contribute to specificity in Kac substrate recognition (Filippakopoulos et al., 2012; Filippakopoulos and Knapp, 2014; Fujisawa and Filippakopoulos, 2017). Large-scale co-crystallization of various Kac peptides and BRDs showed that Kac peptides bind in the central hydrophobic pocket and form hydrogen bonding with an asparagine residue, present in most BRDs (Filippakopoulos et al., 2012; Filippakopoulos and Knapp, 2014; Fujisawa and Filippakopoulos, 2017). Bromodomain-containing protein 4 (BRD4), a member of the bromodomain and extra-terminal domain (BET) family, is a chromatin reader that recognizes Kac residues through two BRDs. It plays key roles in the maintenance of epigenetic memory, cell cycle control, and transcriptional regulation (Dey et al., 2000, 2009; Yang et al., 2005; Devaiah et al., 2012). In 2010, two independent groups discovered the first BRD4 inhibitors, (+)-JQ1 (Filippakopoulos et al., 2010) and I-BET (Nicodeme et al., 2010). These two inhibitors also represent the first examples of small molecules that target epigenetic readers; before the discovery of (+)-JQ1 and I-BET, only epigenetic writers and erasers had been targeted in the field of epigenetics. This discovery opened up a new possibility of treating cancers by targeting epigenetic reader modules.
In Zuber et al. (2011) used an RNA interference (RNAi) genetic screen to identify BRD4 as a therapeutic target in AMLs, including MLL-AF9-induced AML. They demonstrated that the use of either short-hairpin RNAs (shRNAs) or (+)-JQ1 to induce knock-down or inhibition of Brd4, respectively, led to anti-leukemic effects in cells and in vivo. In a murine MLL-AF9 AML cell model, shRNA or (+)-JQ1 treatments induced cell differentiation and led to depletion of leukemia stem cells and a global reduction in the expression of Myc target genes. Moreover, Zuber’s group used ChIP-qPCR to show that Brd4 occupancy approximately 2 kb upstream of the Myc promoter was reduced after exposure to (+)-JQ1. These data suggest that the effects of Brd4 inhibition are linked at least in part to suppression of the Myc-dependent leukemia sustainment program. Indeed, ectopic over-expression of Myc cDNA rescued cell cycle arrest and terminal differentiation induced by Brd4-targeting shRNAs and (+)-JQ1 treatment. However, over-expression of Myc could not rescue cell death induced by (+)-JQ1, suggesting a Myc-independent function of Brd4 in regulating cell survival (Zuber et al., 2011). Consistent with this, another research group employed a large-scale global proteomic strategy to reveal that MLL-FPs that form part of the PAFc and SEC are associated with the epigenetic reader BRD4 (Dawson et al., 2011). Dawson et al. (2011) suggested that BRD4 may function to recruit leukemogenic MLL-FPs (e.g., MLL-AF4 or MLL-AF9) to chromatin for activating expression of oncogenic genes, and therefore they hypothesized that the displacement of BRD4 using chemical inhibitors may have anti-leukemic effect in MLL-r leukemias. The authors showed that I-BET151 displaced BRD4 from Kac substrates on the chromatin and resulted in down-regulation of the antiapoptotic gene BCL2, cell cycle regulator CDK6, and MYC (Dawson et al., 2011). Furthermore, I-BET151 showed anti-leukemic activity in cells and in vivo, and possessed better pharmacokinetic properties in mice than (+)-JQ1 (Dawson et al., 2011).
Mechanistically, the pronounced inhibition of MYC expression by (+)-JQ1 is thought to result from dissociation of BRD4 from MYC super-enhancers (Lovén et al., 2013). Super-enhancers are large clusters of transcriptional enhancers packed with TFs, cofactors, transcription apparatus, and chromatin regulators such as BRD4 (Hnisz et al., 2013). Sustainment of oncogenic MYC expression is associated with MYC super-enhancers in multiple myeloma (MM1.S) and MLL-r leukemia (MLL-AF9/NrasG12D) cells (Lovén et al., 2013; Shi et al., 2013; Shi and Vakoc, 2014). BRD4 molecules densely bind to these active super-enhancers and promote transcriptional elongation through physical association with the Mediator coactivator complex and P-TEFb (Lovén et al., 2013; Shi et al., 2013; Xu and Vakoc, 2017). BRD4 recruits and activate P-TEFb, a multi-protein kinase complex that functions to promote transcriptional elongation (Slany, 2016; Xu and Vakoc, 2017). Chemical inhibition of BRD4 by (+)-JQ1 preferentially displaced BRD4 molecules, as well as Mediator and P-TEFb, from the super-enhancers, thereby down-regulating MYC and MYC-target gene expression (Lovén et al., 2013).
Roe et al. (2015) provided evidence that lineage-specific TFs recruit the lysine acetyltransferases CBP or p300 to acetylate the TFs and histone lysine residues at lineage-specific promoters and enhancers in mouse MLL-AF9/NrasG12D AML cells. BRD4 was shown to bind to hyperacetylated histone residues and acetylated TFs through its BRDs, then promote transcriptional activation via its association with Mediator and P-TEFb. This chromatin-based signaling cascade provides an additional mechanistic explanation for the rapid down-regulation of gene expression by (+)-JQ1, which functions to displace densely localized BRD4 molecules from transcriptionally activated cancer-promoting genes including BCL2, CDK6, and MYC (Roe et al., 2015; Xu and Vakoc, 2017). Given that CBP/p300 play important roles in maintaining MLL-r leukemia, this suggests that chemical inhibition of CBP or p300 may present a therapeutic strategy for treating MLL-r AMLs (Roe et al., 2015). Small molecules targeting the BRDs of CBP/p300 have been developed (Borah et al., 2011; Hay et al., 2014; Rooney et al., 2014; Brand et al., 2015; Picaud et al., 2015; Schiedel and Conway, 2018). For example, I-CBP112, a potent and selective CBP/p300 inhibitor with modest pharmacokinetic properties, was reported in 2015 (Picaud et al., 2015). I-CBP112 showed inhibitory effects on cancer cell growth in both human and mouse MLL-AF9 AML cell lines and prolonged the survival of mice injected with MLL-AF9 AML cells. Picaud et al. (2015) also reported that I-CBP112 sensitized MOLM-13 MLL-r leukemia cells to (+)-JQ1. The combined use of both epigenetic inhibitors could achieve greater antileukemic effects than employing a single agent alone (Picaud et al., 2015). In Lasko et al. (2017), a major breakthrough was achieved in the development of a highly potent, selective, and cell-permeable CBP/p300 inhibitor. Instead of targeting BRDs, the new compound, A-485, binds and inhibits the catalytic core of CBP/p300 and shows inhibitory effects in various cancer cell lines, including MOLM-13 MLL-r leukemia cells. Importantly, A-485 was also found to robustly reduce MYC expression and show tumor-inhibitory effects in a prostate cancer mouse model.
Bromodomain-containing protein 9 (BRD9) is a component of the SWI-SNF chromatin remodeling complex, also known as the BRG1-associated factor (BAF) chromatin remodeling complex in mammals. BRD9 is important for sustaining MYC expression via its BRD in diverse MLL-r leukemia cell lines, including MV4-11, MOLM-13, ML-2, EoL-1, and NOMO-1 (Hohmann et al., 2016). Hohmann et al. (2016) showed that application of RNAi or the small molecule BI-7273 (Martin et al., 2016) to achieve genetic knock-down or chemical inhibition of BRD9, respectively, induced cell growth inhibition and differentiation of MLL-r leukemia cell lines through down-regulation of MYC. The same team also devised a domain-swapping strategy to assess the on-target specificity of the chemical probe (BI-7273) on BRD9. In this method, they swapped the BRD of BRD9 with the first BRD of BRD4, to generate a new chimeric protein, BRD9-BET. The full biological function of BRD9 was preserved in BRD9-BET after the domain-swap; however, BI-7273 could no longer inhibit BRD9-BET. This validated the on-target specificity of BI-7273 for the BRD of BRD9.
ENL (eleven-nineteen-leukemia protein or MTTL1) is the third most frequent MLL translocation partner identified in MLL-r leukemia patients (Meyer et al., 2018; Figure 1B). ENL is also a component of the SEC, which is observed in the MLL-FP complexes (Zeisig et al., 2005; Yokoyama et al., 2010; Figure 1D). The N-terminal of ENL possesses a YEATS domain. In additional to BRDs recognizing Kac (Dhalluin et al., 1999; Filippakopoulos et al., 2012), in 2014, YEATS domains were discovered to be epigenetic readers of Kac (Li Y. et al., 2014). YEATS domains were named after the five founding members of the YEATS-containing protein family—Yaf9, ENL, AF9, Taf14, and Sas5 (Li Y. et al., 2014). Structurally, the YEATS domains of ENL and AF9 adopt an eight-stranded β-sandwich fold (Li Y. et al., 2014; Wan et al., 2017), which is different from the four-α-helical fold of BRDs (Dhalluin et al., 1999; Filippakopoulos et al., 2012). ENL was found to be important in maintaining AMLs (including MLL-r leukemias) by two independent groups in 2017 (Erb et al., 2017; Wan et al., 2017). Depletion of ENL results in down-regulation of key leukemic drivers such as MYC, cell growth inhibition, reduced expression of the leukemia stem cell signature, and terminal differentiation of MLL-r leukemia cell models (Erb et al., 2017; Wan et al., 2017). Cas9-mediated depletion of ENL prolonged survival in leukemic mouse models, which were xenotransplanted with either MV4-11 or MOLM-13 cells transduced with single-guide RNA targeting the ENL gene (Erb et al., 2017; Wan et al., 2017). The two groups also demonstrated that the YEATS reader domain of ENL plays an important role in oncogenic expression and leukemia maintenance through YEATS-Kac interactions. This suggests that pharmacological inhibition of the ENL YEATS domain is a potential therapeutic target for AMLs such as MLL-r leukemias (Erb et al., 2017; Wan et al., 2017). Subsequently, another two research groups separately described small molecules capable of inhibiting the YEATS domains of AF9/ENL (Christott et al., 2018; Li et al., 2018). Christott et al. (2018) screened a library of 24,000 compounds using a peptide displacement assay and discovered a small molecule (XS018661) that binds to AF9 and ENL with equilibrium dissociation constant (Kd) values of 523 ± 53 and 745 ± 45 nM, respectively, as determined by isothermal calorimetry (ITC). Li et al. (2018), on the other hand, used structure-guided development to produce selective peptide-based AF9 and ENL inhibitors, termed XL-13a and XL-13m, respectively. They also showed that XL-13m down-regulated key leukemic driver genes including MYC, MYB (a transcriptional activator), HOXA9, and MEIS1 in MOLM-13 cells (Li et al., 2018).
Although the DOT1L methyltransferase inhibitor, pinometostat, has cell-inhibitory effect in cells and in vivo models of MLL-r leukemia (Daigle et al., 2013), recent results from a phase I clinical trial indicated that the small molecule has only modest efficacy in treating MLL-r leukemias (Stein et al., 2018). This suggests that combining DOT1L inhibitors with other pharmacological agents may be necessary to boost anti-leukemic efficacy. A genome-wide RNAi screen showed that SIRT1, an NAD+-dependent deacetylase, is required to establish a chromatin-repressive state after inhibition of DOT1L (Chen et al., 2015). Consistent with this, a potent activator of SIRT1, SRT1720 (Milne et al., 2007; Mitchell et al., 2014), was demonstrated to synergize with EPZ004777, a DOT1L inhibitor, and enhance anti-proliferative activity against MLL-r leukemia cells (Chen et al., 2015). Another study by Gilan et al. (2016) showed that although DOT1L and BRD4 occupy distinct molecular complexes, they functionally cooperate with each other, and that such cooperation is particularly important for highly transcribed genes with proximity to super-enhancers. They found that combined targeting of both DOT1L and BRD4 using SGC0946 (a small-molecule inhibitor of DOT1L) and I-BET, respectively, resulted in growth inhibitory synergy against MLL-r cell lines, primary human leukemia cells, and mouse leukemia models (Gilan et al., 2016). In a more recent study, Dafflon et al. (2017) employed an epigenome-focused shRNA library to identify epigenetic regulators that sensitize MLL-r leukemia cells treated with EPZ004777. They reported that combination of EPZ004777 and MI-2-2 (an inhibitor of MLL–Menin interaction) enhanced the down-regulation of important leukemia genes (i.e., MYC, HOXA9, and MEIS1) and anti-proliferative effects against MLL-r leukemia cells, compared to either single-agent treatment (Dafflon et al., 2017). Maes et al. (2018) recently showed that the LSD1 demethylase inhibitor ORY-1001 could synergize with a retinoid derivative (ATRA), a nucleoside analog [cytosine arabinoside (ARA-C)], a FLT3 inhibitor (quizartinib), DOT1L inhibitors (EPZ-5676; SGC0946), DNMT1 inhibitors (decitabine; azacitidine), an HDAC inhibitor (SAHA), and a BCL2 inhibitor (ABT-737) to inhibit the proliferation of MV4-11 and MOLM-13 MLL-r leukemic cell lines.
In advance of identifying the first-in-class YEATS domain inhibitors, scientists used genetic and chemical biology tools to determine if ENL YEATS inhibition synergized with known inhibitors of epigenetic regulators (Erb et al., 2017; Wan et al., 2017). Wan et al. (2017) proved that Cas9-mediated depletion of ENL sensitized MOLM-13 cells to (+)-JQ1 treatment. In a proof-of-concept experiment, Li et al. (2018) used small-molecule chemical probes to demonstrate that the ENL YEATS domain inhibitor XL-13m synergized with either the DOT1L inhibitor pinometostat or the BRD4 inhibitor (+)-JQ1 in reducing the expression of MYC in MOLM-13 cells. Erb et al. (2017) applied a novel approach to induce proteasome-mediated acute degradation of ENL using degradation tag (dTAG) technology (Winter et al., 2015; Nabet et al., 2018) in leukemic cells. The dTAG system is a new technology that uses an FKBP12F36V-fused target protein-of-interest and a heterobifunctional ligand degrader—a dTAG molecule (e.g., dTAG-13)—that binds FKBP12F36V and cereblon (CRBN) E3 ligase (Winter et al., 2015; Nabet et al., 2018; Mayor-Ruiz and Winter, 2019). This offers a generalized strategy to fuse any target protein to an engineered variant of the immunophilin FKBP12, then tag FKBP12F36V-fused target proteins for acute proteasome-mediated degradation. The F36V mutation in FKBP12F36V affords a “bump-hole” strategy that allows specific tagging of the FKBP12F36V-fusion proteins, without affecting endogenous FKBP12 proteins. The FKBP12F36V-target fusion gene can be genetically introduced via transgene expression or CRISPR-mediated locus-specific knock-in Nabet et al. (2018). The dTAG-13 molecule induces dimerization of the FKBP12F36V-fused target protein with CRBN E3 ligase, which leads to polyubiquitination of the target protein and proteasomal degradation. After degradation, the dTAG-13 molecules are recycled (i.e., without being degraded) for subsequent rounds of proteasome-mediated degradation of additional FKBP12F36V-fused target proteins. Erb et al. (2017) demonstrated that combining dTAG-mediated acute degradation of ENL with pinometostat had additive inhibitory effects on the expression of Myc and Hoxa9 in MLL-r cells (MV4-11, Cas9+, ENL-FKBP12F36V–HA+, ENL–/–).
Chimeric MLL-FPs form complexes with different epigenetic regulators that are capable of rewiring the epigenetic networks and driving the leukemic programs in MLL-r leukemias. For example, MLL-FPs recruit DOT1L (an epigenetic writer), and cause aberrant expression of HOXA9 and MEIS1, which leads to leukemic transformation. Moreover, MLL-FPs are able to form a molecular complex with BRD4 (an epigenetic reader), PAFc and SEC to sustain the oncogenic expression of BCL2, CDK6, and MYC in MLL-r leukemias (Dawson et al., 2011). Pharmacological intervention with small molecules disrupting the epigenetic networks rewired by the MLL-FPs (Figure 2), holds promises in treating MLL-r leukemias. However, in some circumstances, cancer monotherapies using single chemical agents may have sub-optimal efficacy in eliminating cancer cells and are likely limited by the emergence of resistant cell populations (Fong et al., 2015; Rathert et al., 2015; Doroshow et al., 2017). Recent studies have shown that combining epigenetic drug treatments offers improved therapeutic responses over monotherapies in treating various cancers including MLL-r leukemias (Doroshow et al., 2017). It is of crucial importance to understand the mechanistic actions of drugs, in order to build a comprehensive rationale for using the drugs in a combinatory setting. This is especially important in targeting context-dependent epigenetic regulatory proteins. The advent of massive parallel sequencing technologies and genetic screening technologies such as RNAi and CRISPR-Cas9 has enabled biomedical scientists to uncover cancer vulnerabilities and discover new therapeutic targets for cancer treatments in a high-throughput manner (Shalem et al., 2015; McDonald et al., 2017; Meyers et al., 2017; Tsherniak et al., 2017). With the emergence of dTAG technology, cancer researchers are equipped with an acute proteasome-mediated protein knock-out method for interrogating biological function and early validation of therapeutic targets, before a valid binding ligand is identified (Winter et al., 2015; Nabet et al., 2018; Mayor-Ruiz and Winter, 2019; Schiedel et al., 2019). With such genetic and chemical discovery tools in hand, we foresee that researchers are empowered to discover and validate important therapeutic targets to treat diseases driven by epigenetic abnormalities such as MLL-r leukemias.
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the American Society of Hematology, and National Institutes of Health Grants CA197498, CA233691, and CA236626 to C-WC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We wish to thank Dr. Sarah T. Wilkinson for editing and critical comments on our manuscript.
Ali, M., Hom, R. A., Blakeslee, W., Ikenouye, L., and Kutateladze, T. G. (2014). Diverse functions of PHD fingers of the MLL/KMT2 subfamily. Biochim. Biophys. Acta 1843, 366–371. doi: 10.1016/j.bbamcr.2013.11.016
Allen, M. D., Grummitt, C. G., Hilcenko, C., Min, S., Tonkin, L. M., Johnson, C. M., et al. (2006). Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J. 25, 4503–4512. doi: 10.1038/sj.emboj.7601340
Amente, S., Lania, L., and Majello, B. (2013). The histone LSD1 demethylase in stemness and cancer transcription programs. Biochim. Biophys. Acta 1829, 981–986. doi: 10.1016/j.bbagrm.2013.05.002
Anglin, J. L., and Song, Y. (2013). A medicinal chemistry perspective for targeting histone H3 lysine-79 methyltransferase DOT1L. J. Med. Chem. 56, 8972–8983. doi: 10.1021/jm4007752
Arber, D. A., Orazi, A., Hasserjian, R., Thiele, J., Borowitz, M. J., Beau, M. M., et al. (2016). The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405. doi: 10.1182/blood-2016-03-643544
Arrowsmith, C. H., Bountra, C., Fish, P. V., Lee, K., and Schapira, M. (2012). Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 11, 384–400. doi: 10.1038/nrd3674
Ashkar, S., Schwaller, J., Pieters, T., Goossens, S., Demeulemeester, J., Christ, F., et al. (2018). LEDGF/p75 is dispensable for hematopoiesis but essential for MLL-rearranged leukemogenesis. Blood 131, 95–107. doi: 10.1182/blood-2017-05-786962
Ayton, P. M., Chen, E. H., and Cleary, M. L. (2004). Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol. Cell. Biol. 24, 10470–10478. doi: 10.1128/mcb.24.23.10470-10478.2004
Bach, C., Mueller, D., Buhl, S., Garcia-Cuellar, M. P., and Slany, R. K. (2008). Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene 28, 815–823. doi: 10.1038/onc.2008.443
Balgobind, B. V., Raimondi, S. C., Harbott, J., Zimmermann, M., Alonzo, T. A., Auvrignon, A., et al. (2009). Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood 114, 2489–2496. doi: 10.1182/blood-2009-04-215152
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Barabé, F., Kennedy, J. A., Hope, K. J., and Dick, J. E. (2007). Modeling the initiation and progression of human acute leukemia in mice. Science 316, 600–604. doi: 10.1126/science.1139851
Barski, A., Cuddapah, S., Cui, K., Roh, T.-Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi: 10.1016/j.cell.2007.05.009
Basavapathruni, A., Olhava, E. J., Daigle, S. R., Therkelsen, C. A., Jin, L., Boriack-Sjodin, A. P., et al. (2014). Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm. Drug Dispos. 35, 237–252. doi: 10.1002/bdd.1889
Bernt, K. M., and Armstrong, S. A. (2011). Targeting epigenetic programs in MLL-rearranged leukemias. Hematol. Am. Soc. Hematol. Educ. Program 2011, 354–360. doi: 10.1182/asheducation-2011.1.354
Bernt, K. M., Zhu, N., Sinha, A. U., Vempati, S., Faber, J., Krivtsov, A. V., et al. (2011). MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78. doi: 10.1016/j.ccr.2011.06.010
Birke, M., Schreiner, S., García-Cuéllar, M.-P., Mahr, K., Titgemeyer, F., and Slany, R. K. (2002). The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 30, 958–965. doi: 10.1093/nar/30.4.958
Biswas, D., Milne, T. A., Basrur, V., Kim, J., Elenitoba-Johnson, K. S., Allis, D. C., et al. (2011). Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc. Natl. Acad. Sci. U.S.A. 108, 15751–15756. doi: 10.1073/pnas.1111498108
Blokken, J., Rijck, J., Christ, F., and Debyser, Z. (2017). Protein–protein and protein–chromatin interactions of LEDGF/p75 as novel drug targets. Drug Discov. Today Technol. 24, 25–31. doi: 10.1016/j.ddtec.2017.11.002
Borah, J. C., Mujtaba, S., Karakikes, I., Zeng, L., Muller, M., Patel, J., et al. (2011). A small molecule binding to the coactivator CREB-binding protein blocks apoptosis in cardiomyocytes. Chem. Biol. 18, 531–541. doi: 10.1016/j.chembiol.2010.12.021
Borkin, D., He, S., Miao, H., Kempinska, K., Pollock, J., Chase, J., et al. (2015). Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 27, 589–602. doi: 10.1016/j.ccell.2015.02.016
Borkin, D., Klossowski, S., Pollock, J., Miao, H., Linhares, B., Kempinska, K., et al. (2018). Complexity of blocking bivalent protein-protein interactions: development of a highly potent inhibitor of the menin-mixed-lineage leukemia interaction. J. Med. Chem. 61, 4832–4850. doi: 10.1021/acs.jmedchem.8b00071
Brand, M., Measures, A. R., Measures, A. M., Wilson, B. G., Cortopassi, W. A., Alexander, R., et al. (2015). Small molecule inhibitors of bromodomain–acetyl-lysine interactions. ACS Chem. Biol. 10, 22–39. doi: 10.1021/cb500996u
Broeker, P., Super, H., Thirman, M., Pomykala, H., Yonebayashi, Y., Tanabe, S., et al. (1996). Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood 87, 1912–1922.
Buechele, C., Breese, E. H., Schneidawind, D., Lin, C.-H., Jeong, J., Duque-Afonso, J., et al. (2015). MLL leukemia induction by genome editing of human CD34+ hematopoietic cells. Blood 126, 1683–1694. doi: 10.1182/blood-2015-05-646398
Cai, S. F., Chen, C.-W., and Armstrong, S. A. (2015). Drugging chromatin in cancer: recent advances and novel approaches. Mol. Cell 60, 561–570. doi: 10.1016/j.molcel.2015.10.042
Chang, M.-J., Wu, H., Achille, N. J., Reisenauer, M., Chou, C.-W., Zeleznik-Le, N. J., et al. (2010). Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 70, 10234–10242. doi: 10.1158/0008-5472.can-10-3294
Chang, P.-Y., Hom, R. A., Musselman, C. A., Zhu, L., Kuo, A., Gozani, O., et al. (2010). Binding of the MLL PHD3 finger to histone H3K4me3 is required for MLL-dependent gene transcription. J. Mol. Biol. 400, 137–144. doi: 10.1016/j.jmb.2010.05.005
Chen, C., Sorensen, P., Domer, P., Reaman, G., Korsmeyer, S., Heerema, N., et al. (1993). Molecular rearrangements on chromosome 11q23 predominate in infant acute lymphoblastic leukemia and are associated with specific biologic variables and poor outcome. Blood 81, 2386–2393.
Chen, C.-W., and Armstrong, S. A. (2015). Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp. Hematol. 43, 673–684. doi: 10.1016/j.exphem.2015.05.012
Chen, C.-W., Koche, R. P., Sinha, A. U., Deshpande, A. J., Zhu, N., Eng, R., et al. (2015). DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 21, 335–343. doi: 10.1038/nm.3832
Chen, L., Deshpande, A., Banka, D., Bernt, K., Dias, S., Buske, C., et al. (2013). Abrogation of MLL–AF10 and CALM–AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 27:813. doi: 10.1038/leu.2012.327
Chen, W., Li, Q., Hudson, W. A., Kumar, A., Kirchhof, N., and Kersey, J. H. (2006). A murine Mll-AF4 knock-in model results in lymphoid and myeloid deregulation and hematologic malignancy. Blood 108, 669–677. doi: 10.1182/blood-2005-08-3498
Chowdhury, T., and Brady, H. (2008). Insights from clinical studies into the role of the MLL gene in infant and childhood leukemia. Blood Cells Mol. Dis. 40, 192–199. doi: 10.1016/j.bcmd.2007.07.005
Christott, T., Bennett, J., Coxon, C., Monteiro, O., Giroud, C., Beke, V., et al. (2018). Discovery of a selective inhibitor for the YEATS domains of ENL/AF9. SLAS Discov 24, 133–141. doi: 10.1177/2472555218809904
Collins, C. T., and Hess, J. L. (2016). Deregulation of the HOXA9/MEIS1 axis in acute leukemia. Curr. Opin. Hematol. 23, 354–361. doi: 10.1097/moh.0000000000000245
Corral, J., Lavenir, I., Impey, H., Warren, A. J., Forster, A., Larson, T. A., et al. (1996). An Mll–AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: a method to create fusion oncogenes. Cell 85, 853–861. doi: 10.1016/s0092-8674(00)81269-6
Cozzio, A., Passegué, E., Ayton, P. M., Karsunky, H., Cleary, M. L., and Weissman, I. L. (2003). Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 17, 3029–3035. doi: 10.1101/gad.1143403
Creyghton, M. P., Cheng, A. W., Welstead, G. G., Kooistra, T., Carey, B. W., Steine, E. J., et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 107, 21931–21936. doi: 10.1073/pnas.1016071107
Cucinotta, C. E., and Arndt, K. M. (2016). SnapShot: transcription elongation. Cell 166:1058.e1. doi: 10.1016/j.cell.2016.07.039
Cusan, M., Cai, S. F., Mohammad, H. P., Krivtsov, A., Chramiec, A., Loizou, E., et al. (2018). LSD1 inhibition exerts its antileukemic effect by recommissioning PU.1- and C/EBPα-dependent enhancers in AML. Blood 131, 1730–1742. doi: 10.1182/blood-2017-09-807024
Dafflon, C., Craig, V., Méreau, H., Gräsel, J., Engstler, S. B., Hoffman, G., et al. (2017). Complementary activities of DOT1L and Menin inhibitors in MLL-rearranged leukemia. Leukemia 31, 1269–1277. doi: 10.1038/leu.2016.327
Daigle, S. R., Olhava, E. J., Therkelsen, C. A., Basavapathruni, A., Jin, L., Boriack-Sjodin, A. P., et al. (2013). Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025. doi: 10.1182/blood-2013-04-497644
Daigle, S. R., Olhava, E. J., Therkelsen, C. A., Majer, C. R., Sneeringer, C. J., Song, J., et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. doi: 10.1016/j.ccr.2011.06.009
Dawson, M. A., Prinjha, R. K., Dittmann, A., Giotopoulos, G., Bantscheff, M., Chan, W.-I., et al. (2011). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478:529. doi: 10.1038/nature10509
Delgado, D. M., and León, J. (2010). Myc roles in hematopoiesis and leukemia. Genes Cancer 1, 605–616. doi: 10.1177/1947601910377495
Deshpande, A. J., Chen, L., Fazio, M., Sinha, A. U., Bernt, K. M., Banka, D., et al. (2013). Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 121, 2533–2541. doi: 10.1182/blood-2012-11-465120
Devaiah, B. N., Lewis, B. A., Cherman, N., Hewitt, M. C., Albrecht, B. K., Robey, P. G., et al. (2012). BRD4 is an atypical kinase that phosphorylates Serine2 of the RNA Polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. U.S.A. 109, 6927–6932. doi: 10.1073/pnas.1120422109
Dey, A., Ellenberg, J., Farina, A., Coleman, A. E., Maruyama, T., Sciortino, S., et al. (2000). A bromodomain protein, MCAP, associates with mitotic chromosomes and fffects G2-to-M transition. Mol. Cell. Biol. 20, 6537–6549. doi: 10.1128/mcb.20.17.6537-6549.2000
Dey, A., Nishiyama, A., Karpova, T., McNally, J., and Ozato, K. (2009). Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell. 20, 4899–4909. doi: 10.1091/mbc.e09-05-0380
Dhalluin, C., Carlson, J. E., Zeng, L., He, C., Aggarwal, A. K., and Zhou, M.-M. (1999). Structure and ligand of a histone acetyltransferase bromodomain. Nature 399:491. doi: 10.1038/20974
Djabali, M., Selleri, L., Parry, P., Bower, M., Young, B. D., and Evans, G. A. (1992). A trithorax–like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 2, 113–118. doi: 10.1038/ng1092-113
Dobson, C. L., Warren, A. J., Pannell, R., Forster, A., Lavenir, I., Corral, J., et al. (1999). The Mll–AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 18, 3564–3574. doi: 10.1093/emboj/18.13.3564
Döhner, H., Weisdorf, D. J., and Bloomfield, C. D. (2015). Acute myeloid leukemia. N. Engl. J. Med. 373, 1136–1152. doi: 10.1056/nejmra1406184
Doroshow, D., Eder, J., and LoRusso, P. (2017). BET inhibitors: a novel epigenetic approach. Ann. Oncol. 28, 1776–1787. doi: 10.1093/annonc/mdx157
Dou, Y., Milne, T. A., Ruthenburg, A. J., Lee, S., Lee, J., Verdine, G. L., et al. (2006). Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 13:nsmb1128. doi: 10.1038/nsmb1128
Dou, Y., Milne, T. A., Tackett, A. J., Smith, E. R., Fukuda, A., Wysocka, J., et al. (2005). Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121, 873–885. doi: 10.1016/j.cell.2005.04.031
Drynan, L. F., Pannell, R., Forster, A., Chan, N. M., Cano, F., Daser, A., et al. (2005). Mll fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reassignment in tumorigenesis. EMBO J. 24, 3136–3146. doi: 10.1038/sj.emboj.7600760
Eidahl, J. O., Crowe, B. L., North, J. A., McKee, C. J., Shkriabai, N., Feng, L., et al. (2013). Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 41, 3924–3936. doi: 10.1093/nar/gkt074
Erb, M. A., Scott, T. G., Li, B. E., Xie, H., Paulk, J., Seo, H.-S., et al. (2017). Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543:270. doi: 10.1038/nature21688
Ernst, P., Fisher, J. K., Avery, W., Wade, S., Foy, D., and Korsmeyer, S. J. (2004). Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev. Cell 6, 437–443. doi: 10.1016/s1534-5807(04)00061-9
Ernst, P., Wang, J., Huang, M., Goodman, R. H., and Korsmeyer, S. J. (2001). MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell. Biol. 21, 2249–2258. doi: 10.1128/mcb.21.7.2249-2258.2001
Fair, K., Anderson, M., Bulanova, E., Mi, H., Tropschug, M., and Diaz, M. O. (2001). Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol. Cell. Biol. 21, 3589–3597. doi: 10.1128/mcb.21.10.3589-3597.2001
Felix, C. A. (1998). Secondary leukemias induced by topoisomerase-targeted drugs. Biochim. Biophys. Acta 1400, 233–255. doi: 10.1016/s0167-4781(98)00139-0
Filippakopoulos, P., and Knapp, S. (2014). Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 13, 337–356. doi: 10.1038/nrd4286
Filippakopoulos, P., Picaud, S., Mangos, M., Keates, T., Lambert, J.-P., Barsyte-Lovejoy, D., et al. (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231. doi: 10.1016/j.cell.2012.02.013
Filippakopoulos, P., Qi, J., Picaud, S., Shen, Y., Smith, W. B., Fedorov, O., et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073. doi: 10.1038/nature09504
Fong, C., Gilan, O., Lam, E. Y., Rubin, A. F., Ftouni, S., Tyler, D., et al. (2015). BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542. doi: 10.1038/nature14888
Forneris, F., Binda, C., Vanoni, M., Battaglioli, E., and Mattevi, A. (2005). Human histone demethylase LSD1 reads the histone code. J. Biol. Chem. 280, 41360–41365. doi: 10.1074/jbc.m509549200
Forster, A., Pannell, R., Drynan, L. F., McCormack, M., Collins, E. C., Daser, A., et al. (2003). Engineering de novo reciprocal chromosomal translocations associated with Mll to replicate primary events of human cancer. Cancer Cell 3, 449–458. doi: 10.1016/s1535-6108(03)00106-5
Fujisawa, T., and Filippakopoulos, P. (2017). Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 18, 246–262. doi: 10.1038/nrm.2016.143
Gagnon, G., Childs, C., LeMaistre, A., Keating, M., Cork, A., Trujillo, J., et al. (1989). Molecular heterogeneity in acute leukemia lineage switch. Blood 74, 2088–2095.
García-Alai, M. M., Allen, M. D., Joerger, A. C., and Bycroft, M. (2010). The structure of the FYR domain of transforming growth factor beta regulator 1. Protein Sci. 19, 1432–1438. doi: 10.1002/pro.404
Gilan, O., Lam, E. Y., Becher, I., Lugo, D., Cannizzaro, E., Joberty, G., et al. (2016). Functional interdependence of BRD4 and DOT1L in MLL leukemia. Nat. Struct. Mol. Biol. 23, 673–681. doi: 10.1038/nsmb.3249
Greaves, M. (1997). Aetiology of acute leukaemia. Lancet 349, 344–349. doi: 10.1016/s0140-6736(96)09412-3
Gu, Y., Nakamura, T., Alder, H., Prasad, R., Canaani, O., Cimino, G., et al. (1992). The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell 71, 701–708. doi: 10.1016/0092-8674(92)90603-a
Harris, W. J., Huang, X., Lynch, J. T., Spencer, G. J., Hitchin, J. R., Li, Y., et al. (2012). The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487. doi: 10.1016/j.ccr.2012.03.014
Hawkins, D. R., Hon, G. C., Yang, C., Antosiewicz-Bourget, J. E., Lee, L. K., Ngo, Q.-M., et al. (2011). Dynamic chromatin states in human ES cells reveal potential regulatory sequences and genes involved in pluripotency. Cell Res. 21, 1393–1409. doi: 10.1038/cr.2011.146
Hay, D. A., Fedorov, O., Martin, S., Singleton, D. C., Tallant, C., Wells, C., et al. (2014). Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 136, 9308–9319. doi: 10.1021/ja412434f
Hayami, S., Kelly, J. D., Cho, H., Yoshimatsu, M., Unoki, M., Tsunoda, T., et al. (2011). Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 128, 574–586. doi: 10.1002/ijc.25349
Hayashi, Y., Sugita, K., Nakazawa, S., Abe, T., Kojima, S., Inaba, T., et al. (1990). Karyotypic patterns in acute mixed lineage leukemia. Leukemia 4, 121–126.
Heerema, N., Sather, H., Ge, J., Arthur, D., Hilden, J., Trigg, M., et al. (1999). Cytogenetic studies of infant acute lymphoblastic leukemia: poor prognosis of infants with t(4;11) – a report of the children’s cancer group. Leukemia 13, 679–686. doi: 10.1038/sj.leu.2401413
Hess, J., Yu, B., Li, B., Hanson, R., and Korsmeyer, S. (1997). Defects in yolk sac hematopoiesis in Mll-null embryos. Blood 90, 1799–1806.
Hezroni, H., Sailaja, B., and Meshorer, E. (2011). Pluripotency-related, valproic acid (VPA)-induced genome-wide histone H3 lysine 9 (H3K9) acetylation patterns in embryonic stem cells. J. Biol. Chem. 286, 35977–35988. doi: 10.1074/jbc.m111.266254
Hilden, J., Smith, F., Frestedt, J., McGlennen, R., Howells, W., Sorensen, P., et al. (1997). MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood 89, 3801–3805.
Hilden, J. M., Dinndorf, P. A., Meerbaum, S. O., Sather, H., Villaluna, D., Heerema, N. A., et al. (2006). Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the children’s oncology group. Blood 108, 441–451. doi: 10.1182/blood-2005-07-3011
Hnisz, D., Abraham, B. J., Lee, T., Lau, A., Saint-André, V., Sigova, A. A., et al. (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. doi: 10.1016/j.cell.2013.09.053
Hohmann, A. F., Martin, L. J., Minder, J. L., Roe, J.-S., Shi, J., Steurer, S., et al. (2016). Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 12, 672–679. doi: 10.1038/nchembio.2115
Hsieh, J., Cheng, E., and Korsmeyer, S. J. (2003). Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell 115, 293–303. doi: 10.1016/s0092-8674(03)00816-x
Inaba, H., Greaves, M., and Mullighan, C. G. (2013). Acute lymphoblastic leukaemia. Lancet 381, 1943–1955. doi: 10.1016/s0140-6736(12)62187-4
Jo, S. Y., Granowicz, E. M., Maillard, I., Thomas, D., and Hess, J. L. (2011). Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood 117, 4759–4768. doi: 10.1182/blood-2010-12-327668
Kawagoe, H., Humphries, R., Blair, A., Sutherland, H., and Hogge, D. (1999). Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 13, 687–698. doi: 10.1038/sj.leu.2401410
Krejčí, J., Uhlířová, R., Galiová, G., Kozubek, S., Šmigová, J., and Bártová, E. (2009). Genome-wide reduction in H3K9 acetylation during human embryonic stem cell differentiation. J. Cell Physiol. 219, 677–687. doi: 10.1002/jcp.21714
Krivtsov, A. V., and Armstrong, S. A. (2007). MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 7, nrc2253. doi: 10.1038/nrc2253
Krivtsov, A. V., Feng, Z., Lemieux, M. E., Faber, J., Vempati, S., Sinha, A. U., et al. (2008). H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14, 355–368. doi: 10.1016/j.ccr.2008.10.001
Krivtsov, A. V., Figueroa, M., Sinha, A., Stubbs, M., Feng, Z., Valk, P., et al. (2013). Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia 27, 852–860. doi: 10.1038/leu.2012.363
Krivtsov, A. V., Hoshii, T., and Armstrong, S. A. (2017). Mixed-lineage leukemia fusions and chromatin in leukemia. Cold Spring Harb. Perspect. Med. 7, a026658. doi: 10.1101/cshperspect.a026658
Krivtsov, A. V., Twomey, D., Feng, Z., Stubbs, M. C., Wang, Y., Faber, J., et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 442, 818–822. doi: 10.1038/nature04980
Langenau, D. M., Traver, D., Ferrando, A. A., Kutok, J. L., Aster, J. C., Kanki, J. P., et al. (2003). Myc-induced T cell leukemia in transgenic zebrafish. Science 299, 887–890. doi: 10.1126/science.1080280
Lasko, L. M., Jakob, C. G., Edalji, R. P., Qiu, W., Montgomery, D., Digiammarino, E. L., et al. (2017). Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132. doi: 10.1038/nature24028
Lavau, C., Szilvassy, S. J., Slany, R., and Cleary, M. L. (1997). Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX–ENL. EMBO J. 16, 4226–4237. doi: 10.1093/emboj/16.14.4226
Li, B. E., and Ernst, P. (2014). Two decades of leukemia oncoprotein epistasis: the MLL1 paradigm for epigenetic deregulation in leukemia. Exp. Hematol. 42, 995–1012. doi: 10.1016/j.exphem.2014.09.006
Li, B. E., Gan, T., Meyerson, M., Rabbitts, T. H., and Ernst, P. (2013). Distinct pathways regulated by menin and by MLL1 in hematopoietic stem cells and developing B cells. Blood 122, 2039–2046. doi: 10.1182/blood-2013-03-486647
Li, L., Osdal, T., Ho, Y., Chun, S., Nald, T., Agarwal, P., et al. (2014). SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell 15, 431–446. doi: 10.1016/j.stem.2014.08.001
Li, Y., Wen, H., Xi, Y., Tanaka, K., Wang, H., Peng, D., et al. (2014). AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 159, 558–571. doi: 10.1016/j.cell.2014.09.049
Li, X., Li, X.-M., Jiang, Y., Liu, Z., Cui, Y., Fung, K., et al. (2018). Structure-guided development of YEATS domain inhibitors by targeting π-π-π stacking. Nat. Chem. Biol. 14, 1140–1149. doi: 10.1038/s41589-018-0144-y
Li, Y., Han, J., Zhang, Y., Cao, F., Liu, Z., Li, S., et al. (2016). Structural basis for activity regulation of MLL family methyltransferases. Nature 530, 447–452. doi: 10.1038/nature16952
Lim, S., Janzer, A., Becker, A., Zimmer, A., Schüle, R., Buettner, R., et al. (2010). Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 31, 512–520. doi: 10.1093/carcin/bgp324
Lin, S., Luo, R. T., Ptasinska, A., Kerry, J., Assi, S. A., Wunderlich, M., et al. (2016). Instructive role of MLL-fusion proteins revealed by a model of t(4;11) pro-B acute lymphoblastic leukemia. Cancer Cell 30, 737–749. doi: 10.1016/j.ccell.2016.10.008
Lokken, A. A., and Zeleznik-Le, N. J. (2012). Breaking the LSD1/KDM1A addiction: therapeutic targeting of the epigenetic modifier in AML. Cancer Cell 21, 451–453. doi: 10.1016/j.ccr.2012.03.027
Lovén, J., Hoke, H. A., Lin, C. Y., Lau, A., Orlando, D. A., Vakoc, C. R., et al. (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. doi: 10.1016/j.cell.2013.03.036
Luo, H., Li, Q., O’Neal, J., Kreisel, F., Beau, M. M., and Tomasson, M. H. (2005). c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood 106, 2452–2461. doi: 10.1182/blood-2005-02-0734
Luo, Z., Lin, C., and Shilatifard, A. (2012). The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol 13, 543–547. doi: 10.1038/nrm3417
Maes, T., Mascaró, C., Tirapu, I., Estiarte, A., Ciceri, F., Lunardi, S., et al. (2018). ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell 33, 495–511.e12. doi: 10.1016/j.ccell.2018.02.002
Maiques-Diaz, A., and Somervaille, T. C. (2016). LSD1: biologic roles and therapeutic targeting. Epigenomics 8, 1103–1116. doi: 10.2217/epi-2016-0009
Marchesi, F., Girardi, K., and Avvisati, G. (2011). Pathogenetic, clinical, and prognostic features of adult t(4;11)(q21;q23)/MLL-AF4 positive B-cell acute lymphoblastic leukemia. Adv. Hematol. 2011:621627. doi: 10.1155/2011/621627
Martin, L. J., Koegl, M., Bader, G., Cockcroft, X.-L., Fedorov, O., Fiegen, D., et al. (2016). Structure-based design of an in vivo active selective BRD9 inhibitor. J. Med. Chem. 59, 4462–4475. doi: 10.1021/acs.jmedchem.5b01865
Martinez-Climent, J., Thirman, M., Espinosa, R., Beau, L. M., and Rowley, J. (1995). Detection of 11q23/MLL rearrangements in infant leukemias with fluorescence in situ hybridization and molecular analysis. Leukemia 9, 1299–1304.
Mayor-Ruiz, C., and Winter, G. E. (2019). Identification and characterization of cancer vulnerabilities via targeted protein degradation. Drug Discov. Today Technol. doi: 10.1016/j.ddtec.2018.12.003
McDonald, R. E., de Weck, A., Schlabach, M. R., Billy, E., Mavrakis, K. J., Hoffman, G. R., et al. (2017). Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170, 577–592.e10. doi: 10.1016/j.cell.2017.07.005
McMahon, K. A., Hiew, S., Hadjur, S., Veiga-Fernandes, H., Menzel, U., Price, A. J., et al. (2007). Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 1, 338–345. doi: 10.1016/j.stem.2007.07.002
Meeks, J. J., and Shilatifard, A. (2017). Multiple roles for the MLL/COMPASS family in the epigenetic regulation of gene expression and in cancer. Annu. Rev. Cancer Biol. 1, 425–446. doi: 10.1146/annurev-cancerbio-050216-034333
Metzger, E., Wissmann, M., Yin, N., Müller, J. M., Schneider, R., Peters, A. H., et al. (2005). LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437, 436–439. doi: 10.1038/nature04020
Metzler, M., Forster, A., Pannell, R., Arends, M., Daser, A., Lobato, M., et al. (2006). A conditional model of MLL-AF4 B-cell tumourigenesis using invertor technology. Oncogene 25, 3093–3103. doi: 10.1038/sj.onc.1209636
Meyer, C., Burmeister, T., Gröger, D., Tsaur, G., Fechina, L., Renneville, A., et al. (2018). The MLL recombinome of acute leukemias in 2017. Leukemia 32, 273–284. doi: 10.1038/leu.2017.213
Meyers, R. M., Bryan, J. G., McFarland, J. M., Weir, B. A., Sizemore, A. E., Xu, H., et al. (2017). Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779–1784. doi: 10.1038/ng.3984
Milne, J. C., Lambert, P. D., Schenk, S., Carney, D. P., Smith, J. J., Gagne, D. J., et al. (2007). Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450, 712–716. doi: 10.1038/nature06261
Milne, T. A. (2017). Mouse models of MLL leukemia: recapitulating the human disease. Blood 129, 2217–2223. doi: 10.1182/blood-2016-10-691428
Milne, T. A., Briggs, S. D., Brock, H. W., Martin, M., Gibbs, D., Allis, C. D., et al. (2002). MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10, 1107–1117. doi: 10.1016/s1097-2765(02)00741-4
Min, J., Feng, Q., Li, Z., Zhang, Y., and Xu, R.-M. (2003). Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723. doi: 10.1016/s0092-8674(03)00114-4
Mirro, J., Zipf, T., Pui, C., Kitchingman, G., Williams, D., Melvin, S., et al. (1985). Acute mixed lineage leukemia: clinicopathologic correlations and prognostic significance. Blood 66, 1115–1123.
Mishra, B. P., Zaffuto, K. M., Artinger, E. L., Org, T., Mikkola, H., Cheng, C., et al. (2014). The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 7, 1239–1247. doi: 10.1016/j.celrep.2014.04.015
Mitchell, S. J., Martin-Montalvo, A., Mercken, E. M., Palacios, H. H., Ward, T. M., Abulwerdi, G., et al. (2014). The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep. 6, 836–843. doi: 10.1016/j.celrep.2014.01.031
Mullighan, C. G., Goorha, S., Radtke, I., Miller, C. B., Coustan-Smith, E., Dalton, J. D., et al. (2007). Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446, 758–764. doi: 10.1038/nature05690
Muntean, A. G., and Hess, J. L. (2012). The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. Mech. Dis. 7, 283–301. doi: 10.1146/annurev-pathol-011811-132434
Muntean, A. G., Tan, J., Sitwala, K., Huang, Y., Bronstein, J., Connelly, J. A., et al. (2010). The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell 17, 609–621. doi: 10.1016/j.ccr.2010.04.012
Musselman, C. A., Lalonde, M.-E., Côté, J., and Kutateladze, T. G. (2012). Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 19, 1218–1227. doi: 10.1038/nsmb.2436
Nabet, B., Roberts, J. M., Buckley, D. L., Paulk, J., Dastjerdi, S., Yang, A., et al. (2018). The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441. doi: 10.1038/s41589-018-0021-8
Nguyen, A., Taranova, O., He, J., and Zhang, Y. (2011). DOT1L, the H3K79 methyltransferase, is required for MLL-AF9–mediated leukemogenesis. Blood 117, 6912–6922. doi: 10.1182/blood-2011-02-334359
Nicodeme, E., Jeffrey, K. L., Schaefer, U., Beinke, S., Dewell, S., Chung, C., et al. (2010). Suppression of inflammation by a synthetic histone mimic. Nature 468, 1119–1123. doi: 10.1038/nature09589
Niebel, D., Kirfel, J., Janzen, V., Höller, T., Majores, M., and Gütgemann, I. (2014). Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood 124, 151–152. doi: 10.1182/blood-2014-04-569525
Okada, Y., Feng, Q., Lin, Y., Jiang, Q., Li, Y., Coffield, V. M., et al. (2005). hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178. doi: 10.1016/j.cell.2005.02.020
Paggetti, J., Largeot, A., Aucagne, R., Jacquel, A., Lagrange, B., Yang, X.-J., et al. (2010). Crosstalk between leukemia-associated proteins MOZ and MLL regulates HOX gene expression in human cord blood CD34+ cells. Oncogene 29, 5019–5031. doi: 10.1038/onc.2010.254
Papaemmanuil, E., Gerstung, M., Bullinger, L., Gaidzik, V. I., Paschka, P., Roberts, N. D., et al. (2016). Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221. doi: 10.1056/nejmoa1516192
Park, G., Gong, Z., Chen, J., and Kim, J.-E. (2010). Characterization of the DOT1L network: implications of diverse roles for DOT1L. Protein J. 29, 213–223. doi: 10.1007/s10930-010-9242-8
Patel, A., Dharmarajan, V., and Cosgrove, M. S. (2008). Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem. 283, 32158–32161. doi: 10.1074/jbc.c800164200
Patel, A., Dharmarajan, V., Vought, V. E., and Cosgrove, M. S. (2009). On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 284, 24242–24256. doi: 10.1074/jbc.m109.014498
Picaud, S., Fedorov, O., Thanasopoulou, A., Leonards, K., Jones, K., Meier, J., et al. (2015). Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 75, 5106–5119. doi: 10.1158/0008-5472.can-15-0236
Pieters, R., Schrappe, M., Lorenzo, P., Hann, I., Rossi, G., Felice, M., et al. (2007). A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet 370, 240–250. doi: 10.1016/s0140-6736(07)61126-x
Pradeepa, M. M., Sutherland, H. G., Ule, J., Grimes, G. R., and Bickmore, W. A. (2012). Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. Plos. Genet. 8:e1002717. doi: 10.1371/journal.pgen.1002717
Przespolewski, A., and Wang, E. S. (2016). Inhibitors of LSD1 as a potential therapy for acute myeloid leukemia. Expert Opin. Investig. Drug 25, 771–780. doi: 10.1080/13543784.2016.1175432
Radtke, I., Mullighan, C. G., Ishii, M., Su, X., Cheng, J., Ma, J., et al. (2009). Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc. Natl. Acad. Sci. U.S.A. 106, 12944–12949. doi: 10.1073/pnas.0903142106
Rao, R. C., and Dou, Y. (2015). Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 15, 334–346. doi: 10.1038/nrc3929
Rathert, P., Roth, M., Neumann, T., Muerdter, F., Roe, J.-S., Muhar, M., et al. (2015). Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547. doi: 10.1038/nature14898
Ribich, S., Harvey, D., and Copeland, R. A. (2017). Drug discovery and chemical biology of cancer epigenetics. Cell Chem. Biol. 24, 1120–1147. doi: 10.1016/j.chembiol.2017.08.020
Roberts, K. G., and Mullighan, C. G. (2015). Genomics in acute lymphoblastic leukaemia: insights and treatment implications. Nat. Rev. Clin. Oncol. 12, 344–357. doi: 10.1038/nrclinonc.2015.38
Roe, J.-S., Mercan, F., Rivera, K., Pappin, D. J., and Vakoc, C. R. (2015). BET bromodomain inhibition suppresses the function of hematopoietic transcription factors in acute myeloid leukemia. Mol. Cell 58, 1028–1039. doi: 10.1016/j.molcel.2015.04.011
Rooney, T. P., Filippakopoulos, P., Fedorov, O., Picaud, S., Cortopassi, W. A., Hay, D. A., et al. (2014). A series of potent CREBBP bromodomain ligands reveals an induced-fit pocket stabilized by a cation–π interaction. Angew. Chem. Int. Ed. Engl. 53, 6126–6130. doi: 10.1002/anie.201402750
Rowley, J. (1993). Rearrangements involving chromosome band 11Q23 in acute leukaemia. Semin. Cancer Biol. 4, 377–385.
Sanchez-Martin, M., and Ferrando, A. (2017). The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 129, 1124–1133. doi: 10.1182/blood-2016-09-692582
Satake, N., Maseki, N., Nishiyama, M., Kobayashi, H., Sakurai, M., Inaba, H., et al. (1999). Chromosome abnormalities and MLL rearrangements in acute myeloid leukemia of infants. Leukemia 13, 1013–1017. doi: 10.1038/sj.leu.2401439
Schenk, T., Chen, W., Göllner, S., Howell, L., Jin, L., Hebestreit, K., et al. (2012). Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611. doi: 10.1038/nm.2661
Schiedel, M., and Conway, S. J. (2018). Small molecules as tools to study the chemical epigenetics of lysine acetylation. Curr. Opin. Chem. Biol. 45, 166–178. doi: 10.1016/j.cbpa.2018.06.015
Schiedel, M., Moroglu, M., Ascough, D. M., Chamberlain, A. E., Kamps, J. J., Sekirnik, A. R., et al. (2019). Chemical epigenetics: the impact of chemical- and chemical biology techniques on bromodomain target validation. Angew. Chem. Int. Ed. Engl. [Epub ahead of print].Google Scholar
Schreiner, S., Birke, M., García-Cuéllar, M., Zilles, O., Greil, J., and Slany, R. (2001). MLL-ENL causes a reversible and myc-dependent block of myelomonocytic cell differentiation. Cancer Res. 61, 6480–6486.
Schübeler, D., MacAlpine, D. M., Scalzo, D., Wirbelauer, C., Kooperberg, C., van Leeuwen, F., et al. (2004). The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Gene Dev. 18, 1263–1271. doi: 10.1101/gad.1198204
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6:a018713. doi: 10.1101/cshperspect.a018713
Shalem, O., Sanjana, N. E., and Zhang, F. (2015). High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet. 16, 299–311. doi: 10.1038/nrg3899
Shen, C., Jo, S. Y., Liao, C., Hess, J. L., and Nikolovska-Coleska, Z. (2013). Targeting recruitment of disruptor of telomeric silencing 1-like (DOT1L): characterizing the interactions between DOT1L and mixed lineage leukemia (MLL) fusion proteins. J. Biol. Chem. 288, 30585–30596. doi: 10.1074/jbc.m113.457135
Shi, J., and Vakoc, C. R. (2014). The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 54, 728–736. doi: 10.1016/j.molcel.2014.05.016
Shi, J., Whyte, W. A., Zepeda-Mendoza, C. J., Milazzo, J. P., Shen, C., Roe, J.-S., et al. (2013). Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Gene Dev. 27, 2648–2662. doi: 10.1101/gad.232710.113
Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J. R., Cole, P. A., et al. (2004). Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953. doi: 10.1016/j.cell.2004.12.012
Slany, R. K. (2009). The molecular biology of mixed lineage leukemia. Haematologica 94, 984–993. doi: 10.3324/haematol.2008.002436
Slany, R. K. (2016). The molecular mechanics of mixed lineage leukemia. Oncogene 35, 5215–5223. doi: 10.1038/onc.2016.30
Sorensen, P., Chen, C., Smith, F., Arthur, D., Domer, P., Bernstein, I., et al. (1994). Molecular rearrangements of the MLL gene are present in most cases of infant acute myeloid leukemia and are strongly correlated with monocytic or myelomonocytic phenotypes. J. Clin. Invest. 93, 429–437. doi: 10.1172/jci116978
Stass, S., Mirro, J., Melvin, S., Pui, C., Murphy, S., and Williams, D. (1984). Lineage switch in acute leukemia. Blood 64, 701–706.
Stein, E., Garcia-Manero, G., Rizzieri, D. A., Tibes, R., Berdeja, J. G., Savona, M. R., et al. (2018). The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131, 2661–2669. doi: 10.1182/blood-2017-12-818948
Super, H., McCabe, N., Thirman, M., Larson, R., Beau, L. M., Pedersen-Bjergaard, J., et al. (1993). Rearrangements of the MLL gene in therapy-related acute myeloid leukemia in patients previously treated with agents targeting DNA-topoisomerase II. Blood 82, 3705–3711.
Szczepański, T., Harrison, C. J., and van Dongen, J. J. (2010). Genetic aberrations in paediatric acute leukaemias and implications for management of patients. Lancet Oncol. 11, 880–889. doi: 10.1016/s1470-2045(09)70369-9
Tamai, H., Miyake, K., Takatori, M., Miyake, N., Yamaguchi, H., Dan, K., et al. (2011). Activated K-Ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia 25, 888–891. doi: 10.1038/leu.2011.15
Tkachuk, D. C., Kohler, S., and Cleary, M. L. (1992). Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 71, 691–700. doi: 10.1016/0092-8674(92)90602-9
Tomizawa, D., Koh, K., Sato, T., Kinukawa, N., Morimoto, A., Isoyama, K., et al. (2007). Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia 21, 2258–2263. doi: 10.1038/sj.leu.2404903
Tsherniak, A., Vazquez, F., Montgomery, P. G., Weir, B. A., Kryukov, G., Cowley, G. S., et al. (2017). Defining a cancer dependency map. Cell 170, 564–576.e16. doi: 10.1016/j.cell.2017.06.010
Wan, L., Wen, H., Li, Y., Lyu, J., Xi, Y., Hoshii, T., et al. (2017). ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. doi: 10.1038/nature21687
Wang, P., Lin, C., Smith, E. R., Guo, H., Sanderson, B. W., Wu, M., et al. (2009). Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by RNA polymerase II. Mol. Cell. Biol. 29, 6074–6085. doi: 10.1128/mcb.00924-09
Wang, Q., Wu, G., Mi, S., He, F., Wu, J., Dong, J., et al. (2011). MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood 117, 6895–6905. doi: 10.1182/blood-2010-12-324699
Winter, G. E., Buckley, D. L., Paulk, J., Roberts, J. M., Souza, A., Dhe-Paganon, S., et al. (2015). Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381. doi: 10.1126/science.aab1433
Xu, J., Li, L., Xiong, J., denDekker, A., Ye, A., Karatas, H., et al. (2016). MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov. 2:16008. doi: 10.1038/celldisc.2016.8
Xu, Y., and Vakoc, C. R. (2017). Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb. Perspect. Med. 7:a026674. doi: 10.1101/cshperspect.a026674
Yagi, H., Deguchi, K., Aono, A., Tani, Y., Kishimoto, T., and Komori, T. (1998). Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood 92, 108–117.
Yang, Z., Yik, J., Chen, R., He, N., Jang, M., Ozato, K., et al. (2005). Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell. 19, 535–545. doi: 10.1016/j.molcel.2005.06.029
Yano, T., Nakamura, T., Blechman, J., Sorio, C., Dang, C. V., Geiger, B., et al. (1997). Nuclear punctate distribution of ALL-1 is conferred by distinct elements at the N terminus of the protein. Proc. Natl. Acad. Sci. U.S.A. 94, 7286–7291. doi: 10.1073/pnas.94.14.7286
Yokoyama, A., and Cleary, M. L. (2008). Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 14, 36–46. doi: 10.1016/j.ccr.2008.05.003
Yokoyama, A., Lin, M., Naresh, A., Kitabayashi, I., and Cleary, M. L. (2010). A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 17, 198–212. doi: 10.1016/j.ccr.2009.12.040
Yokoyama, A., Somervaille, T., Smith, K. S., Rozenblatt-Rosen, O., Meyerson, M., and Cleary, M. L. (2005). The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123, 207–218. doi: 10.1016/j.cell.2005.09.025
Yu, B. D., Hess, J. L., Horning, S. E., Brown, G. A., and Korsmeyer, S. J. (1995). Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378, 505–508. doi: 10.1038/378505a0
Zeisig, D. T., Bittner, C. B., Zeisig, B. B., García-Cuéllar, M.-P., Hess, J. L., and Slany, R. K. (2005). The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene 24, 5525–5532. doi: 10.1038/sj.onc.1208699
Zeleznik-Le, N., Harden, A., and Rowley, J. (1994). 11q23 translocations split the “AT-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc. Natl. Acad. Sci. U.S.A. 91, 10610–10614. doi: 10.1073/pnas.91.22.10610
Zhang, T., Cooper, S., and Brockdorff, N. (2015). The interplay of histone modifications – writers that read. EMBO Rep. 16, 1467–1481. doi: 10.15252/embr.201540945
Zhao, Z., Wang, L., Volk, A. G., Birch, N. W., Stoltz, K. L., Bartom, E. T., et al. (2018). Regulation of MLL/COMPASS stability through its proteolytic cleavage by taspase1 as a possible approach for clinical therapy of leukemia. Gene Dev. 33, 61–74. doi: 10.1101/gad.319830.118
Zhu, L., Li, Q., Wong, S., Huang, M., Klein, B. J., Shen, J., et al. (2016). ASH1L links histone H3 lysine 36 dimethylation to MLL Leukemia. Cancer Discov. 6, 770–783. doi: 10.1158/2159-8290.cd-16-0058
Ziemin-van der Poel, S., McCabe, N., Gill, H., Espinosa, R., Patel, Y., Harden, A., et al. (1991). Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc. Natl. Acad. Sci. U.S.A. 88, 10735–10739. doi: 10.1073/pnas.88.23.10735
Keywords: MLL-rearranged, leukemias, epigenetics, MLL, chromatin
Citation: Chan AKN and Chen C-W (2019) Rewiring the Epigenetic Networks in MLL-Rearranged Leukemias: Epigenetic Dysregulation and Pharmacological Interventions. Front. Cell Dev. Biol. 7:81. doi: 10.3389/fcell.2019.00081
Received: 23 February 2019; Accepted: 30 April 2019;
Published: 15 May 2019.
Edited by:
Lluis Morey, Sylvester Comprehensive Cancer Center, United StatesReviewed by:
Pedro Vizan, Centre for Genomic Regulation (CRG), SpainCopyright © 2019 Chan and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun-Wei Chen, Y3dlaWNoZW5AY29oLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.