 Ian E. Gentle
Ian E. Gentle
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 03 May 2019
Sec. Cell Death and Survival
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00073
This article is part of the Research Topic Connecting the Dots Between Inflammation and the Inner Workings of Programmed Cell Death View all 11 articles
Signaling activation is a tightly regulated process involving myriad posttranslational modifications such as phosphorylation/dephosphorylation, ubiquitylation/deubiquitylation, proteolytical cleavage events as well as translocation of proteins to new compartments within the cell. In addition to each of these events potentially regulating individual proteins, the assembly of very large supramolecular complexes has emerged as a common theme in signal transduction and is now known to regulate many signaling events. This is particularly evident in pathways regulating both inflammation and cell death/survival. Regulation of the assembly and silencing of these complexes plays important roles in immune signaling and inflammation and the fate of cells to either die or survive. Here we will give a summary of some of the better studied supramolecular complexes involved in inflammation and cell death, particularly with a focus on diseases caused by their autoactivation and the role autophagy either plays or may be playing in their regulation.
In order to assemble supramolecular signaling complexes in a tightly regulated fashion, certain protein–protein interaction domains and motifs have evolved. By using shared interaction mechanisms, these structures can assemble many subunits of differing function in a rapid and modular fashion to facilitate signal transduction. There are likely many other examples of proteins and domains that fall into this category, but in this review we have chosen to focus on the following structural elements due to their significant representation in the pathways regulating cell death and inflammation.
The death domain containing protein family is involved in numerous aspects of cell signaling and fate and contains several subfamilies including Caspase Activation and Recruitment Domain (CARD), Death Effector Domain (DED), Pyrin Domain (PYD), and the Death domain (DD) itself (Nanson et al., 2018). These domains share structural and sequence similarity but they show specificity with their interactions and tend to interact within each subfamily specifically, CARD–CARD or DD–DD interactions for example. Each member of the family mediates protein–protein interactions and seem to typically form Helical assemblies, some of which are fibrillar in nature such as ASC in inflammasomes (Lu et al., 2014; Li et al., 2018). Members of the Death Domain family interact through three distinct interaction types known as Types I–III (Figure 1A). Depending on the arrangement and combination of the different interaction interfaces between DD family proteins, different structural arrangements can be generated (Figure 1). Clustering of death domain family proteins leads to recruitment of effector proteins including caspases but also ubiquitin ligases and deubiquitinases (DUBs), kinases and other regulatory proteins involved in signal transduction. Through the ordered clustering provided by death domain structures, proteins requiring oligomerization such as caspases are locally concentrated to stimulate interaction and subsequent activation. Interruption of these Death Domain family interactions blocks function suggesting that their assembly is required for activity (Park et al., 2007).
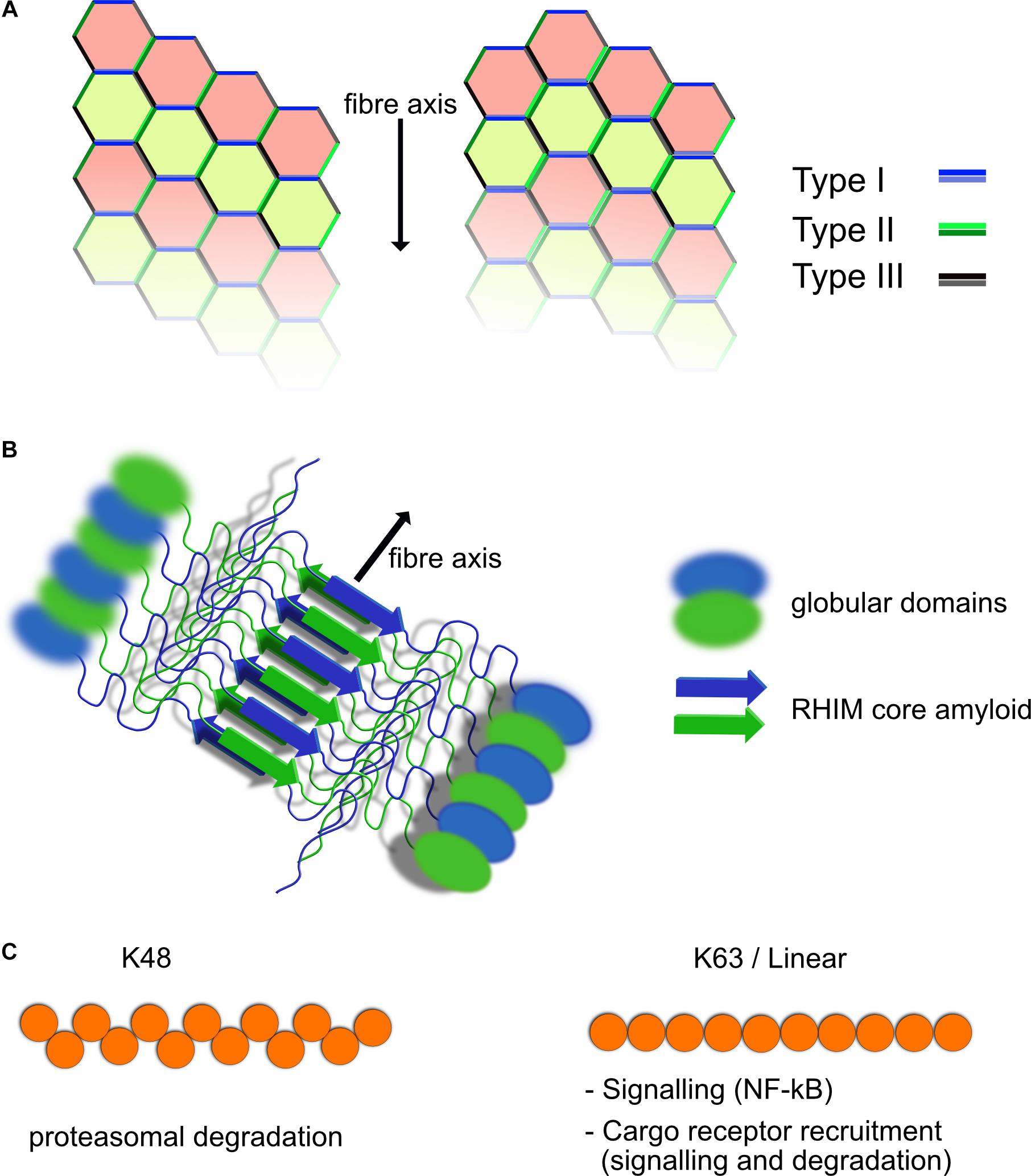
Figure 1. Structural elements of death domain family, RHIM and ubiquitin scaffolds. (A) Death domain family interaction can form fibrillar structures. Death domain family members interact with themselves through three different interaction modes known as Types I–III, depending on the surfaces of the domain that interact. Shown are two possible helical fibrillar assemblies using differing combinations of interfaces. Multiple forms of this type of interaction have been identified, allowing for modular assembly of different complexes. (B) RHIM–RHIM scaffolds form amyloid fibrils. Shown is a fibril of two proteins containing a RHIM and globular domains. The fibril is formed by two parallel beta amyloid sheets coming together. This brings the globular domains into close proximity for interaction and potential activation such as kinase domains of RIPK1/3. RHIM fibrils can be mixed or homogeneous (RIPK1–RIPK3 or RIPK3–RIPK3 fibers form example). (C) Polyubiquitin chains have different functional roles. Shown are K48 and K63/linear ubiquitin chains. The structural layout of the individual chains is different resulting in recruitment of different ubiquitin binding proteins. K63 and linear ubiquitin chains are similar in their layout, although still functionally distinct. K48 chains are predominantly used for proteasomal degradation, whereas K63 and linear ubiquitin chains are used for recruitment of NF-κB activating complexes such as TAB/TAK and IKK as well as linking to autophagic cargo receptors among other functions.
Another structural element contained in supramolecular complexes discussed in this review and which has been shown to play a crucial role in inflammatory and cell death signaling is the Receptor Homotypic Interaction Motif (RHIM) (Sun et al., 2002). RHIMs are relatively short motifs characterized by a core sequence that has the property of folding into a highly stable amyloid structure (Pham et al., 2019) (Figure 1B). Proteins that include RHIM motifs interact with each other via these RHIM–RHIM interaction and include RIPK1, RIPK3, TRIF, DAI/ZBP1. Each of these may potentially interact with the other, however, it is not clear that all combinations are seen under normal situations in the cell (Pham et al., 2018). Mutation of RHIM motifs in RIPK3 for example is enough to abolish its activity suggesting that its role in polymerizing partners together is not separable from other functions it may have (Li et al., 2012). Large structures have been demonstrated for both RIPK1 and 3 as well as TRIF, which are dependent on RHIM interactions (Li et al., 2012; Gentle et al., 2017; Samie et al., 2018). In the case of TRIF, we have shown that these structures are fibrillar complexes that contain at least RIPK1 as well and probably also RIPK3 and can activate caspase-8 and other signaling outcomes of TRIF mediated signaling (Gentle et al., 2017). RHIM–RHIM interaction also provide important scaffolds for recognition of viral infection and subsequent cell death, such as is observed in influenza A virus infections triggering a DAI/ZBP1 and RIPK3 dependent cell death (Thapa et al., 2016). RHIM containing proteins are often recruited to complexes formed through Death Domain family interactions and RIPK1 indeed, has both a RHIM and Death Domain to promote this. How the architecture of such supramolecular complexes formed through RHIM and Death Domain scaffolds looks is still an unknown question, but what is clear is that loss of either of them can drastically alter signaling outcomes.
A common component and key player in the regulation of supramolecular signaling complexes is ubiquitin. In all complexes described in this review, polyubiquitin chains are attached to one or more of the subunits. A common signal activating function of ubiquitin chains in these complexes is to recruit kinase complexes IKK and TAB/TAK to activate NF-κB. However, the same K63 chains are also involved in the eventual silencing of the signaling complexes (see later). Other linkage specific chains are also present including K48 and Met1. Met1 which is added by the Linear UBiquitin chain Assembly Complex (LUBAC) is also important for NF-κB activation through recruitment of IKK complexes in a similar fashion to and in cooperation with K63 chains (Hrdinka and Gyrd-Hansen, 2017). K48 chains are typically associated with proteasomal turnover and are also important for regulation of signals such as from TNFR1 through proteasomal degradation of RIPK1 (Grice and Nathan, 2016; Annibaldi et al., 2018). Depending on the substrate and site of attachment, these ubiquitin chains may also regulate interactions with partner proteins and thus activity, for example recruitment of the NF-kB inducing kinases. The ubiquitin network within a signaling complex is a dynamic one, with the competing actions of ligases and deubiquitinases (DUBs) modifying the overall outcome of the signaling event, regardless of the receptor complex in question. This is a very complicated system and beyond the scope of this review to cover in any depth and has been reviewed thoroughly (Swatek and Komander, 2016). We will focus instead mostly on the role ubiquitin plays regulating recruitment to the autophagy machinery and activation of NF-κB.
Assembly for supramolecular complexes such as those discussed throughout this review, presents a potential problem in terms of switching the signal off. Given the damaging outcome of overactivation of inflammatory or cell death promoting complexes, these structures need to be silenced before they can lead to cellular or tissue damage and disease. While this may in part be regulated by post translational modifications such as (de)ubiquitination, and (de)phosphorylation the likely stability and energetic requirement to break apart complexes such as RHIM or CARD mediated fibrils make this an unlikely mechanism to completely explain disassembly and inactivation of the fully assembled complexes. Degradation of subunits of these complexes by the proteasome may also play some role, however, the size of the complexes discussed in this review make it unlikely that proteasomal degradation of the assembled complexes occurs. Access to the proteasome typically requires unfolding of the substrate to allow it to fit within the catalytic barrel (Collins and Goldberg, 2017). Individually unfolding proteins assembled into supramolecular complexes, while perhaps possible, is likely an inefficient way to silence the activity of these complexes, however, is certainly a relevant regulatory mechanism for preventing their assembly through degrading members of the complexes before they can be recruited. Autophagy represents an existing mechanism capable of dealing with very large protein aggregates.
Autophagy is at its heart a cellular recycling mechanism to provide energy in times of nutrient stress, but has been adapted in multicellular organisms to also regulate multiple aspects of cellular biology. The canonical pathway also known as Macroautophagy creates so-called autophagosomes, membranous vesicles, which engulf random parts of the cytosol and organelles (Zhao and Zhang, 2018) (Figure 2A). These autophagosomes fuse with lysosomes and degrade the contents which are then recycled for use in energy catabolism or for synthesis of new molecules. More specific forms of autophagy have evolved to degrade particular targets including mitochondria (mitophagy), intracellular bacteria and viruses (Xenophagy) and also aggregated proteins (aggrephagy) among others. In each of these targeted autophagic pathways, the target is ubiquitylated, typically by K63 ubiquitin chains (Sharma et al., 2018). These ubiquitylated targets are detected by so-called cargo receptors through binding to ubiquitin chains. This process is now known to be regulated by phosphorylation by TBK1, which will be discussed later in the review more specifically (Figure 2A). The receptors then recruit LC3, a component of the forming autophagophores causing engulfment of the cargo receptor bound target in autophagosomes (Figure 2B). The cargo containing autophagosomes can then fuse with lysosomes and degrade the contents (Figure 2B). Thus, autophagy is ideal for switching off signals from supramolecular signaling complexes. It is of note that many of the signaling pathways using supramolecular complexes such as toll like receptors (TLR) and TNF Receptor family signals, also induce autophagy, thus stimulating the pathways involved in their silencing (Lee et al., 2018; Liu Y. et al., 2018). Examples of specific autophagy of signaling molecules are given throughout this review.
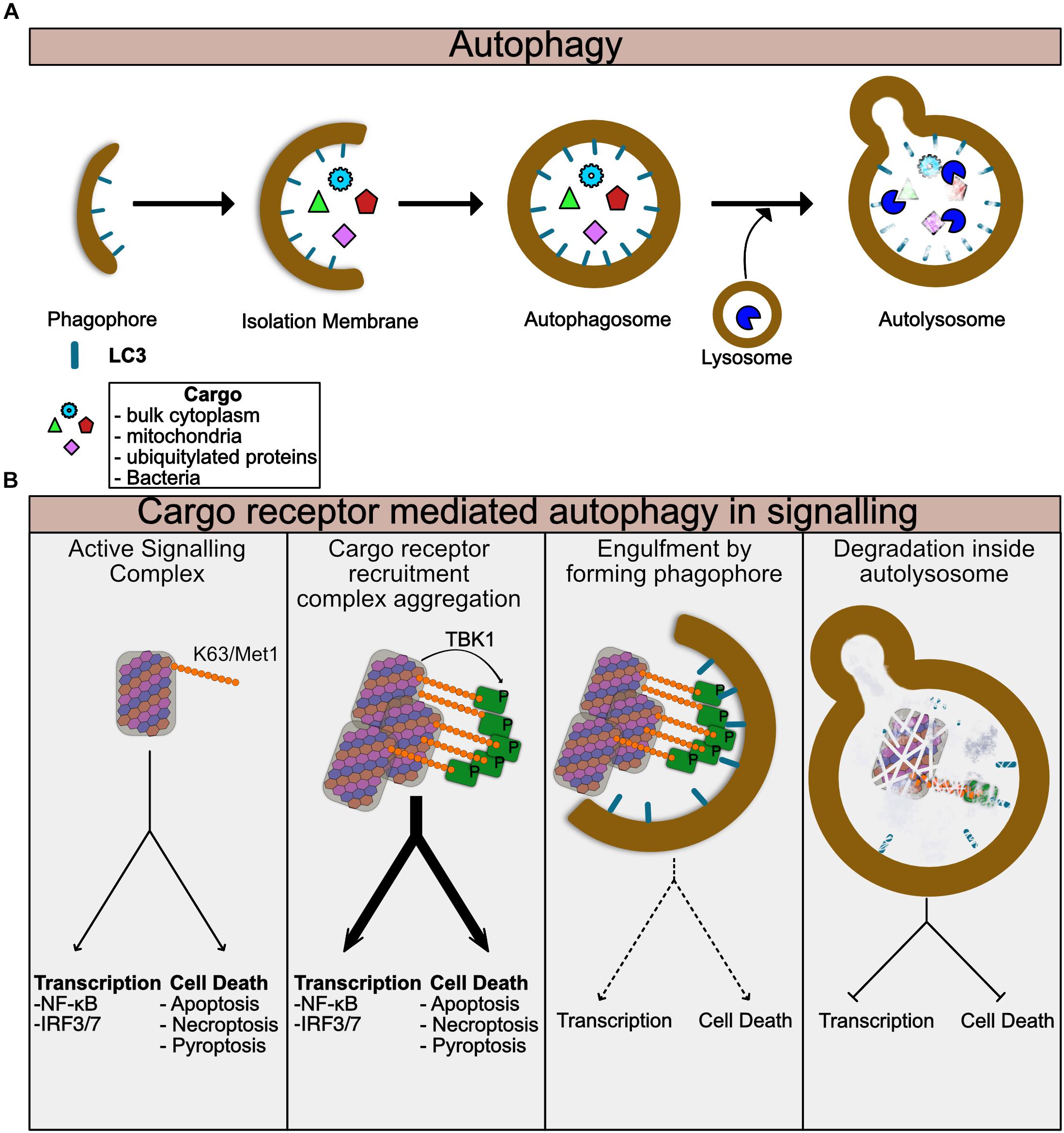
Figure 2. Autophagy regulates turnover of Supramolecular complexes. (A) Summary of general autophagic process. The autophagic machinery including LC3 is recruited to the donour membrane and forms the phagophore. This then extends to form the isolation membrane which begins to engulf cytoplasmic or ubiquitylated contents. Eventually the enclosed autophagosome is formed which then can fuse with lysosomes to form the autolysosome. Lysosomal enzymes then degrade the contents of the autolysosome. (B) Summary of specific autophagy mediated by cargo receptors. Assembled supramolecular signaling complexes begin their signaling response. Cargo receptors such as p62 are recruited via K63 ubiquitin chains on the complex. TBK1 which has been activated and recruited to the complex phosphorylates the cargo receptor to enhance its recruitment. At this stage the signal from the complex may be amplified due to clustering of multiple complexes together or perhaps further enhancement of the localized proximity of kinases, caspases, and ubiquitin ligases for example. As the cargo:cargo-receptor complexes are engulfed by the forming autophagosome, signaling will be reduced. Finally, degradation of the complex within the autolysosome completes the cycle and the complex is destroyed.
Much of the best studied signaling complexes are in the TNF Super Family (TNFSFR), including TNFR1, FAS, TRAIL among others. Each of these receptors ultimately leads to activation of caspase-8 as well as activating transcriptional programs, particularly NF-κB. TNFSFR use death domain family interactions to recruit both scaffolding proteins such as TRADD and FADD, as well as effector proteins such as Caspase-8/10, and RIPK1, although many of the effector proteins also exhibit some scaffolding function, independent of their catalytic activities. Using TNFR1 as a well-studied example, TNFR1 recruits the adapter TRADD, which in turn recruits both RIPK1 and/or FADD. Recruitment of TRAF2/5 along with cIAP1/2 triggers k63 linked ubiquitylation of RIPK1 as well as other components. These ubiquitin chains recruit IKK as well as the TAB/TAK complexes and the linear ubiquitin chain assembly complex (LUBAC) which further adds linear ubiquitin chains. The IKK and TAB/TAK complexes then both activate NF-κB as well as phosphorylate RIPK1 to prevent its activation (Figure 3). This results in upregulation of inflammatory cytokines and cell survival. Loss of RIPK1 ubiquitylation, and thus recruitment of the IKK and TAB/TAK complexes, results in cell death via apoptosis when the so-called complex-II containing RIPK1-FADD-caspase-8 separates from the receptor and activates cytosolic caspase-8 leading to apoptosis (Figure 4) (Vince et al., 2007; Dondelinger et al., 2013, 2015, 2017; Jaco et al., 2017; Menon et al., 2017; Annibaldi et al., 2018). This cytosolic amplification of complex-II formation is likely also mediated by supramolecular assembly of the complex-II through DD and RHIM interactions. Structural models have been developed for the assembly of FADD and Caspase-8 into fibrillar complexes in response to TRAIL ligand (Dickens et al., 2012) or FasL (Fu et al., 2016). While these models differ in their assembly, the principle remains that large elongated networks of Caspase-8 are assembled to trigger its activation through proximity (Figure 4). It seems likely that RIPK1 dependent Caspase-8 activation in the cytosol via Complex-II follows a similar mechanism. To date no auto-activating mutants of FADD or caspase-8 have been identified in disease, this is likely due to their propensity to trigger apoptosis, however, somatic loss of function mutants or repression of expression of FADD and caspase-8 are associated with numerous cancers, and seem to drive NF-κB (Teitz et al., 2000; Shin et al., 2002; Kim et al., 2003; Tourneur et al., 2004; Soung et al., 2005; Ando et al., 2013). This speaks to the role that supramolecular complex assembly has in regulating the complicated network of signaling molecules that are recruited and that loss of function of one, can lead to hyperactivation of another or vice versa.
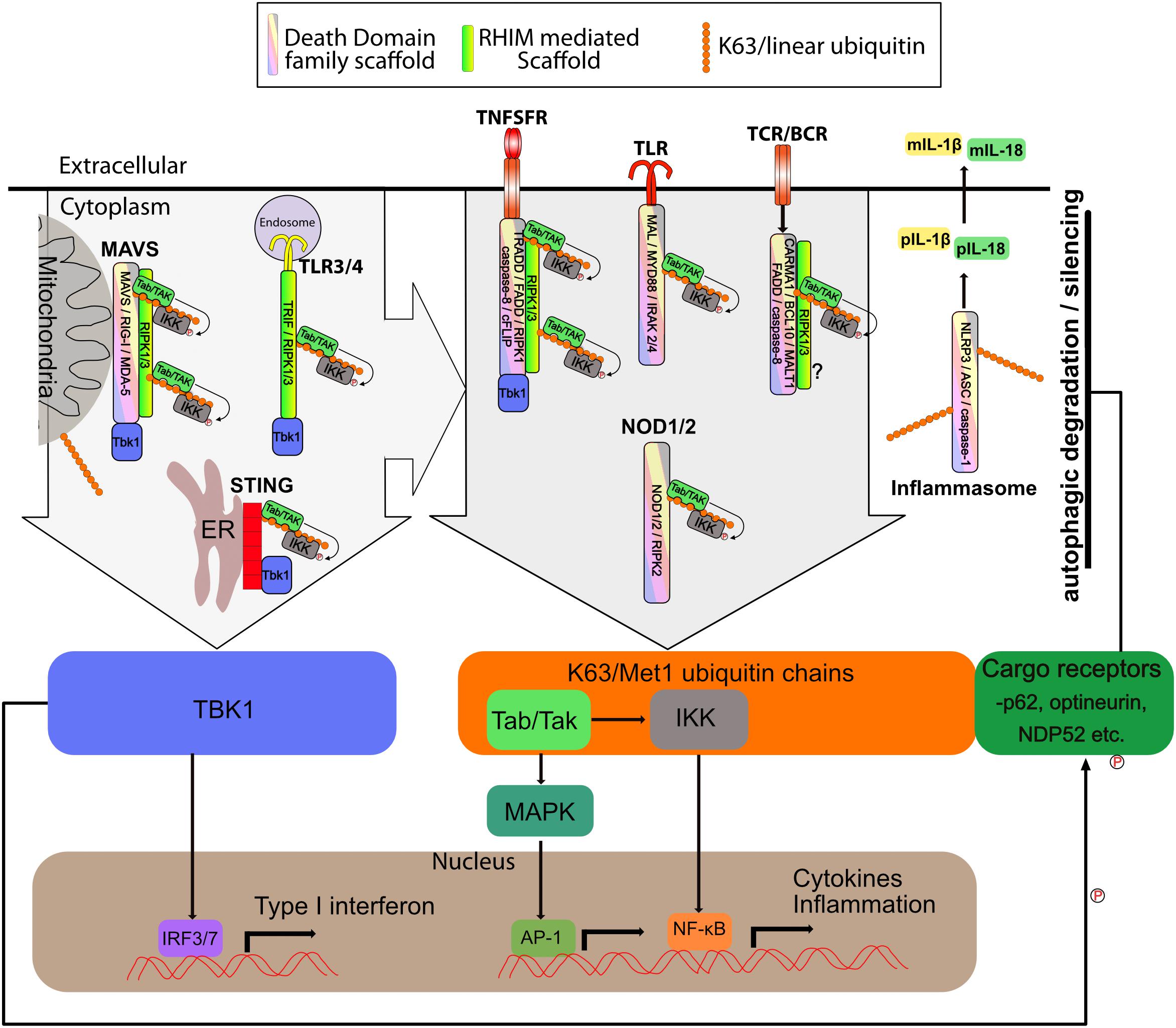
Figure 3. Supramolecular signaling complexes in inflammation share common scaffolds and signaling pathways. The supramolecular signaling complexes formed by TLR, TNFSR, NOD1/2, STING, MAVS, Carma, BCL-10, MALT1 (CBM) and inflammasome complexes are indicated. Shown are the core scaffolds formed through death domain family interactions as well as RHIM interactions. STING contains no death domain family member or RHIM. Specific proteins known to interact through death domain interactions and RHIM interaction are indicated within each complex. Recruitment of TBK1 and/or K63/linear ubiquitin is indicated. TBK1 recruitment activates IRF3 to induce interferon responses. K63/linear ubiquitin recruits the TAB/TAK and IKK complexes which result in activation of NF-kB and Map kinase transcriptional responses. Also shown is linear/K63 ubiquitination of ASC of inflammasomes which can promote their assembly and activation or degradation. Complexes and organelles such as mitochondria that are k63 ubiquitylated recruit autophagy cargo receptors such as p62. This leads ultimately to degradation and silencing of the complexes. This is enhanced by TBK1 mediated phosphorylation of the cargo receptors. Other scaffolds such as those mediated via TRAF proteins are also present, but for simplicity have been omitted from the figure. DD family and RHIM scaffold are not meant to be to scale or reflect the actual organization of the scaffold, but simply indicate that each of these scaffolds are present.
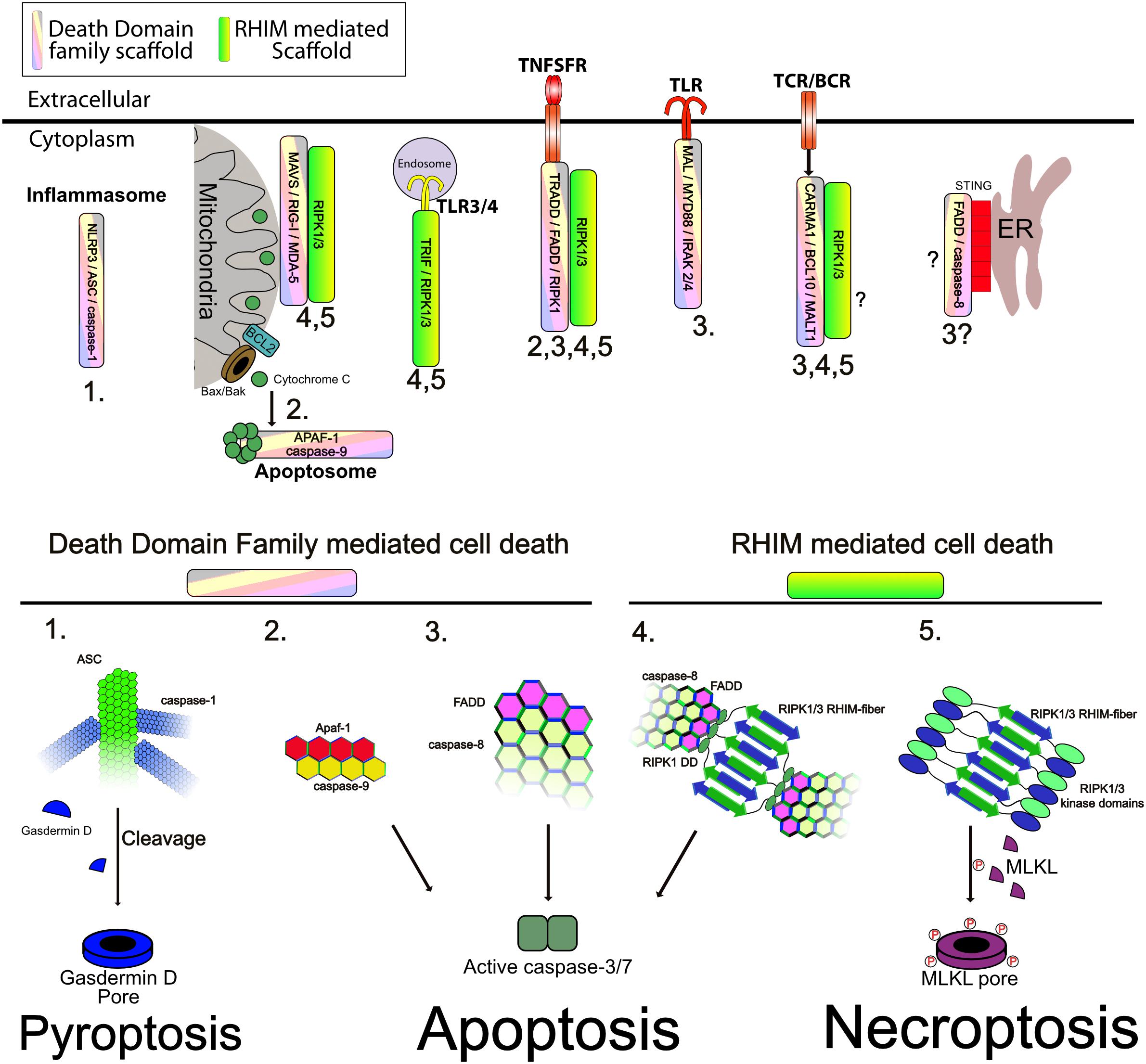
Figure 4. Supramolecular signaling complexes can activate cell death. (Upper) The indicated complexes and the respective scaffolds recruited are indicated. Numbers correspond to the modes of cell death shown in the lower panel ad indicate the modes shown to be triggered by the respective complexes. (Lower) Different modes of cell death and the role of the respective scaffold in activating caspases, or RIP kinases is shown. (1) Inflammasomes assemble a core filament of ASC which in turn recruits filaments of caspase-1, to cluster it and trigger its activation. Active caspase-1 then cleaves Gasdermin D to allow the cleaved form to assemble into multimeric pores in the plasma membrane, causing pyroptosis. (2) Release of cytochrome C through Bax/Bak pores on the mitochondrial outer membrane triggers formation of the apoptosome. The apoptosome consists of a core of APAF1 and caspase-9 interacting through CARD–CARD interactions as indicated. This allows activation of caspase-9. Caspase-9 then cleaves and activates caspase-3/7 to trigger apoptosis. (3) Caspase-8 clustering onto FADD causes its autoactivation. Clustering is mediated through DED–DED interactions between FADD and caspase-8 and caspase-8 itself (not shown is cFLIP which can commingle with caspase-8 to regulate its activation). Active caspase-8 can cleave caspase-3/7 and trigger apoptosis. This scaffold can be activated by numerous receptors as indicated. (4) Recruitment and activation of RIPK1/3 through RHIM–RHIM mediated amyloid fibers can trigger FADD recruitment and clustering through the DD of RIPK1. Clustered FADD recruits caspase-8, probably causing similar aggregation as shown in mode 3. Active caspase-8 cleaves caspase-3/7 to trigger apoptosis. (5) The kinase domains of RIPK1 and RIPK3 are also brought into close proximity by RHIM fibers. Normally, the capase-8 indicated in mode 4 will inactivate these through cleavage of their kinase domains, however, when no caspase-8 activity is present, as is shown, RIPK3 is activated and phosphorylates MLKL. Phosphorylated MLKL assembles into pore complexes in the plasma membrane, causing necroptosis.
In the case of TNF family receptors, one important aspect of their activity is their endocytosis. Endocytosis is required for apoptosis from TNFR1 for example (Micheau and Tschopp, 2003; Schütze et al., 2008). Endocytosed receptors are thought to be able to either traffic back to the plasma membrane or are packaged into multivesicular bodies (MVB) that ultimately fuse with lysosomes and are degraded. In this regard, endocytosis and trafficking via vesicles acts as an in-built silencing mechanism for TNFSFR proteins. However, targeted autophagy was recently identified to regulate the levels of fn14 suggesting that autophagic degradation may also occur in other TNFSFR too (Winer et al., 2018). Supporting this, optineurin (a homolog of NEMO and a functioning autophagic cargo receptor) blocks TNF induced NF-κB by binding ubiquitylated RIPK1 (Zhu et al., 2007). While not shown in this study, this likely also results in degradation of RIPK1/3 containing complexes. More recently, optineurin, mutations of which are associated with amyotrophic lateral sclerosis (ALS), was shown to trigger degradation of RIPK1 and its loss lead to axonal degeneration (Ito et al., 2016). Optineurin loss also sensitized L929 cells to necroptosis induction supporting its role in targeting RIPK1 and RIPK3 for autophagic degradation to prevent their accumulation and activation (Ito et al., 2016). Optineurin loss has also been shown to sensitize to TNF induced caspase-8 activation (Nakazawa et al., 2016), supporting it as having a negative regulation of TNFR1 signaling.
Toll Like receptors, like TNFSFR, also signal through large complexes which may promote both inflammation and cell death. TLR’s, use specific adapter proteins to recruit signaling complexes to the receptor. More specifically, TLRs that signal through the adaptor TRIF/TICAM1 are able to recruit RIPK1 and through RIPK1 activate caspase-8 and or RIPK3 to trigger apoptosis or necroptosis (Kaiser and Offermann, 2005; Weber et al., 2010; Kaiser et al., 2013) (Figures 3, 4) TRIF is recruited to both TLR3, where it is the sole adapter, and TLR4, which also uses MYD88 like all other TLR’s (Hoebe et al., 2003; Oshiumi et al., 2003). Normally, TRIF mediated signaling results in assembly of K63 and linear ubiquitin chains that recruit the TAB/TAK and IKK complexes to trigger NF-κB (Figure 3) (Shim et al., 2005; Zinngrebe et al., 2016). TRIF also activates IRF3/7 mediated interferon responses via TBK1 activation (Fitzgerald et al., 2003; Hemmi et al., 2004) (Figure 3). In either case TRIF signaling complexes are restricted to endosomes (Figures 3, 4). Myd88 dependent signaling occurs through formation of the myddosome (Motshwene et al., 2009). The myddosome is a helical complex consisting of Myd88, IRAK4, and IRAK1/2 stacked together through Death Domain interactions in a manner similar to that shown in Figure 1A (Lin et al., 2010). This platform recruits TRAF6, leading to IKK activation and NF-κB (Häcker et al., 2006) (Figure 3). While some evidence exists for apoptosis induced through MYD88 from TLR2 through recruitment of FADD and caspase-8, this seems to be the exception rather than the rule (Figure 4) (Aliprantis et al., 2000).
In either case, both MYDD88 and TRIF adapters have been shown to form supramolecular complexes which are required for several aspects of their signaling activity (Funami et al., 2007; Tatematsu et al., 2010; Guven-Maiorov et al., 2015; Gentle et al., 2017; Samie et al., 2018). Indeed Mutations in Myd88 that are associated with Lymphoma trigger auto-assembly of the TIR domain and the myddosome without receptor ligation (Avbelj et al., 2014). To date no auto-activating mutations of TRIF have been identified, possibly due to its capacity to trigger cell death when overactive. Mutations in FADD that are associated with cancers have also been shown to interfere with the association of FADD with MYD88, which may in turn promote myddosome assembly (Guven-Maiorov et al., 2015).
A number of studies have now associated regulation of TLR signaling via autophagy. Specifically Myd88 was reported to be recruited to p62 and Histone deacetylase 6 (HDAC6) positive structures upon TLR4 stimulation (Into et al., 2010; Fujita et al., 2011). p62 acts as a cargo receptor for selective autophagy and HDAC6 is thought to help traffic the ubiquitylated complexes to bring them together and promote fusion of the autophagosomes with lysosomes (Kawaguchi et al., 2003; Lee et al., 2010). Loss of p62 or HDAC6 resulted in enhanced JNK, p38, and ERK signaling in response to TLR ligands, but appeared to have little effect on NF-κB. While it was not specifically demonstrated in these studies, the enhanced signaling, albeit through specific pathways, is suggestive of the myddosome being targeted by autophagy, loss of which prevents the signals being dampened.
TRIF can form large fibrillar complexes that have been variably reported to interact with the autophagy cargo receptors p62, Tax1BP1, and NDP52 to target them for degradation (Inomata et al., 2012; Gentle et al., 2017; Yang et al., 2017; Samie et al., 2018). Our own study showed that if autophagy is inhibited, not only did the transcriptional activity of TRIF signaling become enhanced resulting in increased cytokine production, but also the cell death inducing activity, with enhanced caspase-8 activation being detected upon TLR3 stimulation of melanoma cells (Gentle et al., 2017). Similar results were shown in the other studies indicating enhanced IFN production in response to polyI:C or LPS when autophagy was inhibited (Samie et al., 2018). TRIF can also interact with the ubiquitin like protein ubiquitilin1 driving association with autophagosomes and degradation of TRIF (Biswas et al., 2011). Loss of ubiquitilin1 leads to excessive type I interferon responses from TLR4 and TLR3 (Biswas et al., 2011). Additionally TRIF can also induce autophagy as is the case with most of the complexes discussed here (Gentle et al., 2017).
As with TNFRSF receptors that become endocytosed, TLRs, in part at least, are associated with endosomes, an aspect which is essential for their activity (Petes et al., 2017). In addition to the autophagic regulation discussed above, a similar fate probably awaits activated TLRs that are present on endosomes such as TLR3 and TLR4 containing TRIF complexes (Wang et al., 2007). What ultimately happens to plasma membrane bound TLRs after assembly of myddosomes is unclear. They may also be trafficked via general endocytosis or they may, as is the case with TNFR1 complex II dissociate from the receptors themselves and then become targeted by autophagy separately.
Inflammasomes are another example of a death domain based supramolecular complex that has potent inflammatory signaling activity but can also trigger death of cells. There are a number of inflammasomes, however, the majority share a core structure, with a receptor subunit such as NLRP3 activating and recruiting Apoptosis-associated speck-like protein containing a CARD (ASC), which is able to then polymerize into a helical fibrillar structure that recruits Caspase-1 (Lu et al., 2014; Li et al., 2018). This brings caspase-1 into close proximity with other caspase-1 molecules, triggering auto-processing and activation of the zymogen into its functional form. Caspase-1 then cleaves the cytokines IL-1β and IL-18 to trigger further inflammation, but can also cleave substrates including Gasdermin D which then forms pores in the plasma membrane leading to a type of cell death called Pyroptosis (Martinon et al., 2002; He et al., 2015; Shi et al., 2015; Liu et al., 2016) (Figures 3, 4). Pyroptotic death is additionally thought to release danger associated molecular patterns (DAMPS) that trigger inflammation in and of themselves, thus promoting an inflammatory environment (Frank and Vince, 2018). Auto-activating mutations in inflammasome components are known and cause diseases such as Familial Mediterranean Fever (FMF) and cryopyrin-associated periodic syndrome (CAPS) (Cordero et al., 2018). These mutations typically lead to auto-assembly of the inflammasome and lead to severe inflammatory pathologies due to the increased secretion of IL-1β and II-18 and pyroptosis (Cordero et al., 2018). Efficiently regulating the signal strength and length of signaling through inflammasomes is therefore essential. Autophagy plays a number of roles in regulating inflammasomes. Both the AIM2 and NLRP3 inflammasomes can be recruited to p62 and engulfed by autophagosomes and later associate with lysosomes for degradation (Shi et al., 2012). Blocking autophagy enhances inflammasome activity and stimulating autophagy reduces it (Shi et al., 2012). Loss of autophagy is also associated with enhanced NLRP3 activation and IL-1β secretion (Saitoh et al., 2008), although the mechanism behind this activation is not yet clear. A number of studies have suggested that mitochondrial defects, possibly through insufficient mitophagy, may promote inflammasome activation through excessive ROS production or possibly release of ligands such as mtDNA (Ip et al., 2017). Stimuli that induce inflammasome activation also induce autophagy (Shi et al., 2012), supporting its role as a negative feedback system for these complexes.
STING is activated by cyclic dinucleotides (cGAMP) produced by cGAS upon detection of cytoplasmic DNA (Ablasser et al., 2013). cGAMP binding causes STING to assemble into a large ER associated complex that leads to activation of NF-κB through IKK activation upon recruitment to K63 and linear polyubiquitin chains, but also to IRF3 through recruitment and activation of TBK1 (Abe and Barber, 2014) (Figure 3).
STING activation can also induce apoptosis in T cells and B-cell lymphomas although the mechanism is still unclear (Tang et al., 2016; Gulen et al., 2017; Larkin et al., 2017). Auto activating mutations of STING cause STING-associated vasculopathy with onset in infancy (SAVI), which among other problems results in T-cell cytopenia. This is independent of IRF-3 and could be due to cell death of developing or mature T-cells based on mouse knock-in models (Warner et al., 2017; Wu and Yan, 2018). Autoactivating mutations of STING trigger dimerization in the absence of ligand, allowing the signaling complex to form (Konno et al., 2018). FADD deficiency can block IFN activation in STING activated cells, suggesting that STING requires FADD and providing a possible mechanism for how STING may induce cell death through apoptosis (Ishikawa and Barber, 2008) (Figure 4). Additionally, STING can also cause necroptotic cell death indirectly through induction of TNF and interferon in macrophages and dendritic cells (Brault et al., 2018).
STING has also recently been identified as an autophagic substrate. Upon activation it binds to p62 and is targeted for degradation through autophagy (Liu D. et al., 2018; Prabakaran et al., 2018). STING itself can also act as an autophagy cargo receptor through an LIR motif to directly recruit LC3 and form autophagosomes around it after activation (Liu D. et al., 2018), although why it should need its own LIR when p62 is also recruited remains to be seen. Loss of p62 causes strongly enhanced IFN stimulated gene expression in response to cytosolic DNA indicating that STING activity is much higher. STING also induces autophagy in order to stimulate its own degradation (Liu D. et al., 2018) and the autophagy inducing kinase ULK1 also phosphorylates activated STING to negatively regulate its function (Konno et al., 2013). STING directly promotes the lipidation of LC3 at the ER to promote autophagosome production (Gui et al., 2019). This function of STING appears to have evolved before interferon activation as demonstrated by a lack of TBK1 and IRF activation by both Xenopus and sea anemone STING homologs, and is important for clearance of HSV-1 (Gui et al., 2019). Together, these studies support the picture of autophagy regulating signaling platforms and vice versa.
Mitochondrial antiviral-signaling protein (MAVS) is a signaling scaffold for detection of viral RNA products. The receptors RIG-I and MDA5 bind to viral dsRNA molecules and then oligomerize (Berke et al., 2012; Jiang et al., 2012). Exposure of their CARD domains allows them to bind to the card domains of MAVS on the mitochondrial outer membrane whereby MAVS polymerizes into supramolecular fibrillar complexes to act as a scaffold for recruitment of IKK and TBK1 complexes for the activation of NF-κB and IRF3 (Figure 3) (Seth et al., 2005; Hou et al., 2011; Xu et al., 2014). Additionally FADD, RIPK1, RIPK3, and caspase-8 can be recruited to these complexes (Kawai et al., 2005; Downey et al., 2017). Infection with Semliki Forest virus induces apoptotic cell death through caspase-8 activation via the MAVS pathway highlighting that there can be a direct death inducing signal triggered by MAVS (Figure 4) (El Maadidi et al., 2014). Again, the MAVS complexes assemble through CARD–CARD interactions, and while no auto-activating mutations are known in humans for MAVS itself, gain of function mutations are found in both mda5 and RIG-I and are associated with type I interferonopathies such as Aicardi-Goutières syndrome (AGS) and Atypical Singleton-Merten syndrome (SMS) (Rice et al., 2014; Jang et al., 2015; Rutsch et al., 2015). These mutations can lead to enhanced fibril formation of the MAVS complex through an unknown mechanism that may be due to enhanced activation of the receptors themselves leading to excessive MAVS polymerization (Funabiki et al., 2014; Rice et al., 2014).
While MAVS appears to be an autophagy substrate it is perhaps a somewhat special case in this regard due to its location on the mitochondria. MAVS is reported to contain an LIR motif for direct recruitment of LC3 and MAVS activation and assembly into its large fibrillar state can stimulate mitophagy (Sun et al., 2016), thus removing MAVS along with the mitochondrial it is associated with. Additionally, MAVS can be degraded by autophagy through an NDP52 dependent mechanism regulated by the interferon response gene tetherin and loss of NDP52 gives an enhanced interferon response, albeit minor (Jin et al., 2017). The relatively minor effect that loss of NDP52 has on MAVS signaling, may be due to MAVS acting as its own cargo receptor and inducing autophagy/mitophagy. Clearly, however, MAVS adheres to the pattern shown for the other complexes so far discussed in using autophagy as a silencing mechanism.
NOD2 is another CARD containing PRR that recognizes bacterial muramyl dipeptide (MDP) (Girardin et al., 2003). Through its CARD domain NOD2 recruits RIPK2, which is then ubiquitylated by XIAP resulting in the recruitment of the TAB/TAK and IKK complexes and NF-κB activation through addition of linear ubiquitin chains via LUBAC (Figure 3) (Damgaard et al., 2012, 2013). Recently, RIPK2 was shown to also form filaments through CARD–CARD interactions and filament formation and scaffolding function is required for NOD2 activity (Hrdinka et al., 2018; Pellegrini et al., 2018). Interestingly, crosstalk between the NOD2 and MAVS pathway has been demonstrated during viral infection. ssRNA is recognized by NOD2 and it then interacts with MAVS triggering IRF3 activation through TBK1, although surprisingly, not through CARD–CARD interactions (Sabbah et al., 2009). Mutations in NOD2 are associated predominantly with Crohn’s Disease and result in a failure to induce NF-κB in response to ligand (Hampe et al., 2001; Hugot et al., 2001; Ogura et al., 2001; Bonen et al., 2003). However, there is some question as to whether this is the causative defect in Crohn’s Disease (Eckmann and Karin, 2005). Another disease thought to be caused by autoactivating mutations of NOD2 is Blau syndrome. Patients with Blau syndrome exhibit granulomatous dermatitis, arthritis, and uveitis (Kanazawa et al., 2005; Parkhouse et al., 2014) Although, again the autoactivation of NOD2 driving disease has also been questioned (Dugan et al., 2015). Given the fibril forming nature of NOD2-RIPK2 complexes, it seems likely that hyperactivation could result from aberrant assembly of RIPK2 fibrils, however, this has not been demonstrated yet.
NOD2 has a complex interaction with the autophagy machinery. As is the case with most of the complexes discussed here, NOD2 can induce autophagy. It is thought that this occurs through a RIPK2 kinase dependent mechanism (Homer et al., 2012). Additionally, NOD2 is also involved in the recruitment of autophagic machinery to sites of invading bacteria at the plasma membrane through interaction with Atg16. This targets the bacteria for degradation via xenophagy (Cooney et al., 2010; Homer et al., 2010, 2012; Travassos et al., 2010; Anand et al., 2011; Negroni et al., 2016). To date no degradation of NOD2 or its complexes have been shown via autophagy. NOD2 is thought to be a proteasomal substrate under conditions where HSP90 is blocked or its access to NOD2 is restricted (Normand et al., 2018). RIPK2, however, has been identified as interacting with p62, although autophagic degradation was not shown in this study (Park et al., 2013). The NOD2-RIPK2 fibril may therefore also be a target of autophagic degradation given this association and may play a role in dampening NOD driven inflammation.
In a similar manner to inflammasomes, the CARMA-BCL10-MALT1 (CBM) signaling complex consists of a core of BCL10 that recruits the paracaspase MALT-1. Different CARD containing adapters can recruit BCL10 to trigger its assembly including CARD9, -10, -11, and CARD14 (Gehring et al., 2018; Juilland and Thome, 2018). CARD11/CARMA1 is one of the best studied to date and is activated by TCR and BCR signaling. CARMA1 recruits BCL10 through CARD–CARD interactions, whereupon BCL10 forms filamentous helical structures in a manner similar to what has been described here for other CARD mediated structures such as inflammasomes (Qiao et al., 2013; David et al., 2018; Schlauderer et al., 2018). These filaments assemble in a star like formation radiating out from CARMA1 nucleation points (David et al., 2018). MALT1 is a paracaspase which requires recruitment to BCL10 for activation. TRAF6 is also recruited via interaction with MALT1 and NF-κB is subsequently activated through recruitment of IKK complexes to ubiquitin chains (Figure 3) (Sun et al., 2004). Recently it was shown that LUBAC is also required for NF-κB activation, although this may be due to a scaffolding role as catalytically inactive HOIP could also restore NF-κB signals in HOIP deficient cells (Dubois et al., 2014). FADD and caspase-8 are also recruited to the CBM complex and caspase-8 catalytic activity is required for effective activation of NF-κB by the CBM (Su et al., 2005). As loss of caspase-8 activity is a known trigger for necroptosis in stimulated T cells, these data also suggest that recruitment of FADD and caspase-8 are accompanied by RIPK1 and that this could be the route for activation of necroptosis in the absence of caspase-8 in stimulated lymphocytes (Figure 4).
Interestingly, mutations in CARD11 (Carma1) are causative of B cell expansion with NF-κB and T cell anergy (BENTA), an immunodeficiency caused by overactivation of the CBM complex. These mutations cause CARD11 to aggregate and recruit BCL10 and MALT1 into large complexes (Snow et al., 2012; Brohl et al., 2015). They are also often associated with diffuse large B cell lymphoma and other lymphomas (Lenz et al., 2008; Chan et al., 2013). CARMA3 has also been identified as a negative regulator of MAVS oligomerization and IRF3 activation (Jiang et al., 2016). This highlights again, that supramolecular signaling complexes can be prone to autoactivating mutations due to their ability to rapidly assemble into ordered signaling hubs, and the complexity with which they share common interactions and regulation.
As with the other supramolecular complexes discussed so far, a link with autophagy has been shown with CBM signalosomes too. Degradation of BCL10 in response to TCR stimulation occurs through a proteasome independent lysosomal dependent mechanism (Scharschmidt et al., 2004), suggesting autophagy as a likely route. As discussed later, the CBM complex also associates with p62 during TCR signaling in order to regulate the intensity of the signal (Paul et al., 2012).
Mitochondria act not only as the metabolic engine of the cell, but also as a key signaling hub filtering signals for cellular growth and energetics, innate immune and inflammatory signals, and also decisions to survive or die. The classical intrinsic mitochondrial apoptotic machinery comprised of Bax and Bak mediated pores regulated by BCL2 family pro and anti-apoptotic proteins is well characterized, if not completely understood (Edlich, 2018). As with the death domain family, the BCL2 family of proteins share common domains that they use for their interaction, namely the BCL2 Homology (BH) domain. Mechanistically, Bax and Bak form pores in the outer membrane of mitochondria upon an apoptotic stimulus. Contents of the mitochondrial intermembrane space then leak out, including cytochrome C and SMAC/DIABLO. Cytochrome C then binds APAF1 triggering the formation of the apoptosome thus activating caspase-9. Caspase-9 in turn activates effector caspases such as caspase-3 which carry out the end points of apoptosis (Figure 4). The BCL2 family proteins also form large complexes which are quite dynamic in their composition and function depending on the state of the cell, and may be antiapoptotic or proapoptotic [reviewed in Westphal et al. (2014), Cosentino and García-Sáez (2017), and Kale et al. (2018)]. As such they can be considered a kind of dynamic supramolecular complexes themselves that also use common domains (BH) to interact. In another link between mitochondria and inflammatory signaling, release of mitochondrial DNA (mtDNA) after Bak/Bak mediated permeabilization can also activate STING to trigger inflammation (McArthur et al., 2018; Riley et al., 2018).
The central role mitochondria play in regulating so many aspects of cellular homeostasis and immunity mean that they can in a sense be considered as a supramolecular signaling hub themselves. Given their importance it is not surprising then, that they are also heavily regulated and prone to quality control. Mitophagy is a specialized form of autophagy that causes the degradation of damaged mitochondria (Narendra et al., 2014). Mitophagy is analogous to aggrephagy in that targets on the mitochondria are ubiquitylated and cargo receptors such as p62 are recruited to the mitochondria followed by engulfment in autophagosomes and subsequent degradation in lysosomes (Figures 2, 3) (Geisler et al., 2010). Recent work by a number of labs has shown that mitophagy plays a role in regulating aspects of apoptosis and also associated inflammation. A recent study has shown the activation of Bax and Bak on mitochondria is associated with induction of autophagy, and that subsequently, apoptotic mitochondria are engulfed and degraded through autophagy (Lindqvist et al., 2017). Blockade of autophagy lead to enhanced production of interferon β (Lindqvist et al., 2017). Release of other mitochondrial contents in to the cytosol after outer membrane permeabilization can also be a stimulatory event, inducing cytokine production and DNA damage (Ichim et al., 2015; McArthur et al., 2018; Riley et al., 2018). BCL2 family of proteins integration into the outer membranes of mitochondria likely makes them a difficult target for direct action by specific degradation through autophagy, however, evidence exists for a role of Parkin (E3 ubiquitin ligase responsible for ubiquitylating damaged mitochondria during mitophagy) in regulating BCL2 complexes as well as mitophagy. Parkin can ubiquitylate MCL-1 leading to its degradation and cell death in response to mitochondrial damaging agents such as valinomycin (Zhang et al., 2014). Less damaging treatments such as cccp are reported to induce mitophagy instead (Zhang et al., 2014). Loss of mitophagy may contribute to inflammation in a number of ways including accumulation of ROS, release of mtDNA from damaged mitochondria as well as persistence of signaling platforms such as MAVS that are localized on the mitochondrial membrane.
An interesting connection between the activation of these receptor signaling complexes and their degradation by autophagy is the activation of the Tank Binding Kinase 1 (TBK1). TBK1 is required for IRF activation from many of the receptors complexes described in this review including TLR3/4 via TRIF, STING and MAVS. TBK1 is also a well characterized activator of autophagic cargo receptors including p62, optineurin and NDP52 (Pilli et al., 2012; Korac et al., 2013; Matsumoto et al., 2015; Yang et al., 2016; Oakes et al., 2017; Cho et al., 2018). TBK1 phosphorylates these receptors, enhancing their recruitment of the autophagic machinery and therefore promoting degradation of the target complexes (Figure 2). This was shown directly for STING, but also for mitochondria and bacteria. Recently TBK1 was also identified as being recruited to TNFR1 where it does not activate IRF3, but instead played an essential role in regulating survival (Figure 3). Loss of TBK1 expression leads to RIPK1 dependent apoptosis and reduced levels can replicate ALS in mice (Lafont et al., 2018; Xu et al., 2018). This was shown to be dependent on TBK1 mediated phosphorylation of RIPK1. Given the recent observations about TNFR1 and TBK1 mediated survival, it is possible that TBK1 activation may also promote the degradation of RIPK1 complexes in reducing the amount of activated complex-II in response to TNF, however, this remains to be shown. Of note is that TBK1 is also a critical component of enhanced autophagy and NF-κB activation in K-Ras-dependent non-small cell lung carcinoma (NSCLC) (Newman et al., 2012). The role of TBK1 in activating autophagy receptors and its recruitment to and activation by supramolecular complexes described here is indicative of the importance of this pathway in dampening inflammatory and cell death signals.
In another twist to the role of autophagy in regulating supramolecular signaling complexes, some cargo receptors such as p62 can promote signaling and assembly of the complexes prior to their degradation (Figure 2B). This has been reported for caspase-8 activation upon TRAIL stimulation (Jin et al., 2009), whereby p62 promoted the aggregation of caspase-8 in a cullin-3 dependent fashion, thus suggesting that aggregation by p62 enhanced the signal triggering apoptosis, but at the same time lead to the ultimate degradation of the complex and its silencing.
Recently a role for HDAC6 has been shown in clearance of Listeria monocytogenes (Moreno-Gonzalo et al., 2017). In this context loss of HDAC6 in DCs resulted in enhanced bacterial load, due to defects in autophagy, and also strongly reduced activation of NF-κB and MAPK pathways. It was proposed that by interacting with MYD88 that HDAC6 promoted its aggregation and activation of downstream signaling pathways. While no specific mechanism for this is given, HDAC6 may promote association of MYD88 with autophagy receptors such as p62 to enhance its activity before being degraded. Additionally loss of p62 was shown to reduce cytokine production, NF-κB and ERK activation in response to TLR2 and TLR6 activation in keratinocytes (Lee et al., 2011). In a similar fashion to the above examples, p62 promoted NF-κB activation prior to degradation of BCL10 in TCR signaling (Paul et al., 2012, 2014), however, BCl10 degradation ultimately silences NF-κB activation (Scharschmidt et al., 2004). NOD2 also shows reduced NF-κB activation in response to ligand in the absence of p62 (Park et al., 2013). Of note is that is required for TRAF6 dependent ubiquitylation of NEMO/IKKγ, and loss of p62 blocks IL-1β induced NF-κB substantially (Zotti et al., 2014). Additionally p62 is required for RAS induced NF-κB in cancer through TRAF6 ubiquitylation and IKK activation (Durán et al., 2008). Together these data support the idea that these large signaling complexes that become ubiquitylated also use this aggregation phase to enhance signaling prior to silencing. While p62 is by far the most studied of the autophagy cargo receptors, it is likely that there is some redundancy and that the other cargo receptors also exhibit signal amplifying activities prior to their degradation. Thinking of these adapters as cargo receptors may actually be too simplistic for their role in signal regulation, and instead perhaps they should be thought of more as generalized modulators or scaffolds for tuning signal strength and duration.
A number of diseases are associated with deficiencies in autophagy, many of which are inflammatory in nature and in a number of cases show direct links to proteins from supramolecular signaling complexes involved in cell death and inflammatory signaling. Gaucher’s Disease is a lipid storage disease caused by mutations in glucocerebrosidase that results in accumulation of the sphingolipid glucocerebroside in lysosomes, effectively blocking their function. Thus, as a byproduct, the autophagy pathway is also backed-up and blocked by a failure to degrade targets in the lysosome (Settembre et al., 2008). Gaucher’s disease is associated with a strong hyperinflammation and splenomegaly and interestingly, in mouse models, it was shown that it could be largely blocked by loss of RIPK3, suggesting a potential role for RIPK3 mediated cell death as a possible driver of the disease (Vitner et al., 2014). While it has yet to be shown, it is intriguing to speculate that active RIPK3, assembled into fibrillar complexes through the RHIM domain do not get degraded, and then promote either cell death or inflammation directly. Indeed increased RIPK3 levels are seen in Gaucher’s patients (Vitner et al., 2014).
Niemann Pick disease is another lysosomal disease that is associated with inflammatory pathology, particularly Crohn’s disease like symptoms. Niemann Pick diseases are caused by failure to metabolize Sphingomyelin for various reasons, leading to lysosomal disfunction (Guo et al., 2016). While it has not directly been shown that Niemann Pick is regulated by RIPK3 in a similar fashion to Gaucher’s disease, the possibility remains. As mentioned, a particular pathology associated with Niemann-Pick is the development of Crohn’s Disease like pathology. This was reported to be associated with decreased xenophagy in a manner similar to loss of function of two other well-known Crohn’s Disease associated proteins, Nod2 and XIAP (Schwerd et al., 2017), both of which also positively regulate autophagy (Homer et al., 2010; Gradzka et al., 2018). Mutations in NOD2 lead to loss of NF-κB activation, as do many of the mutations in XIAP that are associated with disease, suggesting that failure to activate this pathway is the initial cause of the pathology, however, recent studies have shown that NOD2 and XIAP both also have roles in targeting invasive bacteria for xenophagy (Homer et al., 2010; Gradzka et al., 2018). How this may be coordinated is not clear, but the association of other autophagy related mutations such as the Atg16L1, NDP52, Optineurin, and others CD suggest that specific defects in the autophagy process may be a key trigger as well. At least part of the inflammation seen may be associated with a concomitant failure to silence assembled signaling complexes within the usual time frame. Additionally, there are a number of neuronal diseases associated with defects in autophagy that may also have an inflammatory element. These include Alzheimer’s disease and Parkinson’s disease to give just two prominent examples. Again, a role for degradation of signaling complexes may also play a significant role in the inflammation and cell death seen in these diseases.
Formation of large supramolecular complexes as signaling hubs is emerging as a unifying theme in signaling and it is likely that many more receptor complexes will demonstrate this capacity. The fact that so many receptors all share common structural domains and signaling targets supports this and highlights the crosstalk that many of these receptors have in regulating the strength, and severity of signals that are produced. The propensity of these complexes to assemble is also highlighted by the numerous, although rare, genetic immune diseases associated with auto-activation of components of the complexes discussed here. The common theme of induction of autophagy, recruitment to cargo receptors to amplify signals prior to their degradation provides a tightly regulatable circuit for negative regulation of these important signaling complexes and suggest that autophagy induction may provide a useful target in helping to prevent some of these diseases at their source.
IG conceived of and wrote the manuscript.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abe, T., and Barber, G. N. (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 88, 5328–5341. doi: 10.1128/JVI.00037-14
Ablasser, A., Goldeck, M., Cavlar, T., Deimling, T., Witte, G., Röhl, I., et al. (2013). cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. doi: 10.1038/nature12306
Aliprantis, A. O., Yang, R. B., Weiss, D. S., Godowski, P., and Zychlinsky, A. (2000). The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 19, 3325–3336. doi: 10.1093/emboj/19.13.3325
Anand, P. K., Tait, S. W. G., Lamkanfi, M., Amer, A. O., Nunez, G., Pages, G., et al. (2011). TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation. J. Biol. Chem. 286, 42981–42991. doi: 10.1074/jbc.M111.310599
Ando, M., Kawazu, M., Ueno, T., Fukumura, K., Yamato, A., Soda, M., et al. (2013). Cancer-associated missense mutations of caspase-8 activate nuclear factor-κB signaling. Cancer Sci. 104, 1002–1008. doi: 10.1111/cas.12191
Annibaldi, A., Wicky John, S., Vanden Berghe, T., Swatek, K. N., Ruan, J., Liccardi, G., et al. (2018). Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol. Cell 69, 566–580.e5. doi: 10.1016/j.molcel.2018.01.027
Avbelj, M., Wolz, O.-O., Fekonja, O., Benčina, M., Repič, M., Mavri, J., et al. (2014). Activation of lymphoma-associated MyD88 mutations via allostery-induced TIR-domain oligomerization. Blood 124, 3896–3904. doi: 10.1182/blood-2014-05-573188
Berke, I. C., Yu, X., Modis, Y., and Egelman, E. H. (2012). MDA5 assembles into a polar helical filament on dsRNA. Proc. Natl. Acad. Sci. U.S.A. 109, 18437–18441. doi: 10.1073/pnas.1212186109
Biswas, N., Liu, S., Ronni, T., Aussenberg, S. E., Liu, W., Fujita, T., et al. (2011). The ubiquitin-like protein PLIC-1 or ubiquilin 1 inhibits TLR3-trif signaling. PLoS One 6:e21153. doi: 10.1371/journal.pone.0021153
Bonen, D. K., Ogura, Y., Nicolae, D. L., Inohara, N., Saab, L., Tanabe, T., et al. (2003). Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 124, 140–146. doi: 10.1053/gast.2003.50019
Brault, M., Olsen, T. M., Martinez, J., Stetson, D. B., and Oberst, A. (2018). Intracellular nucleic acid sensing triggers necroptosis through synergistic type I IFN and TNF signaling. J. Immunol. 200, 2748–2756. doi: 10.4049/jimmunol.1701492
Brohl, A. S., Stinson, J. R., Su, H. C., Badgett, T., Jennings, C. D., Sukumar, G., et al. (2015). Germline CARD11 mutation in a patient with severe congenital B cell lymphocytosis. J. Clin. Immunol. 35, 32–46. doi: 10.1007/s10875-014-0106-4
Chan, W., Schaffer, T. B., and Pomerantz, J. L. (2013). A quantitative signaling screen identifies CARD11 mutations in the CARD and LATCH domains that induce Bcl10 ubiquitination and human lymphoma cell survival. Mol. Cell. Biol. 33, 429–443. doi: 10.1128/MCB.00850-12
Cho, C. S., Park, H. W., Ho, A., Semple, I. A., Kim, B., Jang, I., et al. (2018). Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1–mediated p62/sequestosome 1 phosphorylation. Hepatology 68, 1331–1346. doi: 10.1002/hep.29742
Collins, G. A., and Goldberg, A. L. (2017). The logic of the 26S proteasome. Cell 169, 792–806. doi: 10.1016/j.cell.2017.04.023
Cooney, R., Baker, J., Brain, O., Danis, B., Pichulik, T., Allan, P., et al. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97. doi: 10.1038/nm.2069
Cordero, M. D., Alcocer-Gómez, E., and Ryffel, B. (2018). Gain of function mutation and inflammasome driven diseases in human and mouse models. J. Autoimmun. 91, 13–22. doi: 10.1016/j.jaut.2018.03.002
Cosentino, K., and García-Sáez, A. J. (2017). Bax and Bak Pores: are we closing the circle? Trends Cell Biol. 27, 266–275. doi: 10.1016/j.tcb.2016.11.004
Damgaard, R. B., Fiil, B. K., Speckmann, C., Yabal, M., Stadt, U. Z., Bekker-Jensen, S., et al. (2013). Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol. Med. 5, 1278–1295. doi: 10.1002/emmm.201303090
Damgaard, R. B., Nachbur, U., Yabal, M., Wong, W. W.-L., Fiil, B. K., Kastirr, M., et al. (2012). The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 46, 746–758. doi: 10.1016/j.molcel.2012.04.014
David, L., Li, Y., Ma, J., Garner, E., Zhang, X., and Wu, H. (2018). Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc. Natl. Acad. Sci. U.S.A. 115, 1499–1504. doi: 10.1073/pnas.1721967115
Dickens, L. S., Boyd, R. S., Jukes-Jones, R., Hughes, M. A., Robinson, G. L., Fairall, L., et al. (2012). A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 47, 291–305. doi: 10.1016/j.molcel.2012.05.004
Dondelinger, Y., Aguileta, M. A., Goossens, V., Dubuisson, C., Grootjans, S., Dejardin, E., et al. (2013). RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20, 1381–1392. doi: 10.1038/cdd.2013.94
Dondelinger, Y., Delanghe, T., Rojas-Rivera, D., Priem, D., Delvaeye, T., Bruggeman, I., et al. (2017). MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247. doi: 10.1038/ncb3608
Dondelinger, Y., Jouan-Lanhouet, S., Divert, T., Theatre, E., Bertin, J., Gough, P. J., et al. (2015). NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76. doi: 10.1016/j.molcel.2015.07.032
Downey, J., Pernet, E., Coulombe, F., Allard, B., Meunier, I., Jaworska, J., et al. (2017). RIPK3 interacts with MAVS to regulate type I IFN-mediated immunity to Influenza A virus infection. PLoS Pathog. 13:e1006326. doi: 10.1371/journal.ppat.1006326
Dubois, S. M., Alexia, C., Wu, Y., Leclair, H. M., Leveau, C., Schol, E., et al. (2014). A catalytic-independent role for the LUBAC in NF-κB activation upon antigen receptor engagement and in lymphoma cells. Blood 123, 2199–2203. doi: 10.1182/blood-2013-05-504019
Dugan, J., Griffiths, E., Snow, P., Rosenzweig, H., Lee, E., Brown, B., et al. (2015). Blau syndrome–associated NOD2 mutation alters expression of full-length NOD2 and limits responses to muramyl dipeptide in knock-in mice. J. Immunol. 194, 349–357. doi: 10.4049/jimmunol.1402330
Durán, A., Linares, J. F., Galvez, A. S., Wikenheiser, K., Flores, J. M., Diaz-Meco, M.-T., et al. (2008). The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 13, 343–354. doi: 10.1016/j.ccr.2008.02.001
Eckmann, L., and Karin, M. (2005). NOD2 and Crohn’s Disease: loss or gain of function? Immunity 22, 661–667. doi: 10.1016/j.immuni.2005.06.004
Edlich, F. (2018). BCL-2 proteins and apoptosis: recent insights and unknowns. Biochem. Biophys. Res. Commun. 500, 26–34. doi: 10.1016/j.bbrc.2017.06.190
El Maadidi, S., Faletti, L., Berg, B., Wenzl, C., Wieland, K., Chen, Z. J., et al. (2014). A novel mitochondrial MAVS/Caspase-8 platform links RNA virus-induced innate antiviral signaling to Bax/Bak-independent apoptosis. J. Immunol. 192, 1171–1183. doi: 10.4049/jimmunol.1300842
Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E., Golenbock, D. T., et al. (2003). IKK𝜀 and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496. doi: 10.1038/ni921
Frank, D., and Vince, J. E. (2018). Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 26, 99–114. doi: 10.1038/s41418-018-0212-6
Fu, T.-M., Li, Y., Lu, A., Li, Z., Vajjhala, P. R., Cruz, A. C., et al. (2016). Cryo-EM structure of caspase-8 tandem DED filament reveals assembly and regulation mechanisms of the death-inducing signaling complex. Mol. Cell 64, 236–250. doi: 10.1016/j.molcel.2016.09.009
Fujita, K.-I., Maeda, D., Xiao, Q., and Srinivasula, S. M. (2011). Nrf2-mediated induction of p62 controls Toll-like receptor-4–driven aggresome-like induced structure formation and autophagic degradation. Proc. Natl. Acad. Sci. U.S.A. 108, 1427–1432. doi: 10.1073/pnas.1014156108
Funabiki, M., Kato, H., Miyachi, Y., Toki, H., Motegi, H., Inoue, M., et al. (2014). Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40, 199–212. doi: 10.1016/j.immuni.2013.12.014
Funami, K., Sasai, M., Ohba, Y., Oshiumi, H., Seya, T., and Matsumoto, M. (2007). Spatiotemporal mobilization of Toll/IL-1 receptor domain-containing adaptor molecule-1 in response to dsRNA. J. Immunol. 179, 6867–6872. doi: 10.4049/jimmunol.179.10.6867
Gehring, T., Seeholzer, T., and Krappmann, D. (2018). BCL10 – bridging CARDs to immune activation. Front. Immunol. 9:1539. doi: 10.3389/fimmu.2018.01539
Geisler, S., Holmström, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–131. doi: 10.1038/ncb2012
Gentle, I. E., McHenry, K. T., Weber, A., Metz, A., Kretz, O., Porter, D., et al. (2017). TIR-domain-containing adapter-inducing interferon-β (TRIF) forms filamentous structures, whose pro-apoptotic signalling is terminated by autophagy. FEBS J. 284, 1987–2003.
Girardin, S. E., Boneca, I. G., Viala, J., Chamaillard, M., Labigne, A., Thomas, G., et al. (2003). NOD2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872. doi: 10.1074/jbc.C200651200
Gradzka, S., Thomas, O. S., Kretz, O., Haimovici, A., Vasilikos, L., Wong, W. W.-L., et al. (2018). Inhibitor of apoptosis proteins are required for effective fusion of autophagosomes with lysosomes. Cell Death Dis. 9:529. doi: 10.1038/s41419-018-0508-y
Grice, G. L., and Nathan, J. A. (2016). The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 73, 3497–3506. doi: 10.1007/s00018-016-2255-5
Gui, X., Yang, H., Li, T., Tan, X., Shi, P., Li, M., et al. (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. doi: 10.1038/s41586-019-1006-9
Gulen, M. F., Koch, U., Haag, S. M., Schuler, F., Apetoh, L., Villunger, A., et al. (2017). Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8:427. doi: 10.1038/s41467-017-00573-w
Guo, H., Zhao, M., Qiu, X., Deis, J. A., Huang, H., Tang, Q.-Q., et al. (2016). Niemann-Pick type C2 deficiency impairs autophagy-lysosomal activity, mitochondrial function, and TLR signaling in adipocytes. J. Lipid Res. 57, 1644–1658. doi: 10.1194/jlr.M066522
Guven-Maiorov, E., Keskin, O., Gursoy, A., VanWaes, C., Chen, Z., Tsai, C.-J., et al. (2015). The architecture of the TIR domain signalosome in the toll-like receptor-4 signaling pathway. Sci. Rep. 5:13128. doi: 10.1038/srep13128
Häcker, H., Redecke, V., Blagoev, B., Kratchmarova, I., Hsu, L.-C., Wang, G. G., et al. (2006). Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439, 204–207. doi: 10.1038/nature04369
Hampe, J., Cuthbert, A., Croucher, P. J., Mirza, M. M., Mascheretti, S., Fisher, S., et al. (2001). Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 357, 1925–1928. doi: 10.1016/S0140-6736(00)05063-7
He, W.-T., Wan, H., Hu, L., Chen, P., Wang, X., Huang, Z., et al. (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25, 1285–1298. doi: 10.1038/cr.2015.139
Hemmi, H., Takeuchi, O., Sato, S., Yamamoto, M., Kaisho, T., Sanjo, H., et al. (2004). The roles of two iκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650. doi: 10.1084/jem.20040520
Hoebe, K., Du, X., Georgel, P., Janssen, E., Tabeta, K., Kim, S. O., et al. (2003). Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748. doi: 10.1038/nature01889
Homer, C. R., Kabi, A., Marina-Garcia, N., Sreekumar, A., Nesvizhskii, A. I., Nickerson, K. P., et al. (2012). A dual role for receptor-interacting protein kinase 2 (RIP2) kinase activity in nucleotide-binding oligomerization domain 2 (n.d.)-dependent autophagy. J. Biol. Chem. 287, 25565–25576. doi: 10.1074/jbc.M111.326835
Homer, C. R., Richmond, A. L., Rebert, N. A., Achkar, J.-P., and McDonald, C. (2010). ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139, 1630–1641.e2. doi: 10.1053/j.gastro.2010.07.006
Hou, F., Sun, L., Zheng, H., Skaug, B., Jiang, Q.-X., and Chen, Z. J. (2011). MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146, 448–461. doi: 10.1016/j.cell.2011.06.041
Hrdinka, M., and Gyrd-Hansen, M. (2017). The Met1-linked ubiquitin machinery: emerging themes of (De)regulation. Mol. Cell 68, 265–280. doi: 10.1016/j.molcel.2017.09.001
Hrdinka, M., Schlicher, L., Dai, B., Pinkas, D. M., Bufton, J. C., Picaud, S., et al. (2018). Small molecule inhibitors reveal an indispensable scaffolding role of RIPK2 in NOD2 signaling. EMBO J. 37:e99372. doi: 10.15252/embj.201899372
Hugot, J.-P., Chamaillard, M., Zouali, H., Lesage, S., Cézard, J.-P., Belaiche, J., et al. (2001). Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603. doi: 10.1038/35079107
Ichim, G., Lopez, J., Ahmed, S. U., Muthalagu, N., Giampazolias, E., Delgado, M. E., et al. (2015). Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872. doi: 10.1016/j.molcel.2015.01.018
Inomata, M., Niida, S., Shibata, K.-I., and Into, T. (2012). Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell. Mol. Life Sci. 69, 963–979. doi: 10.1007/s00018-011-0819-y
Into, T., Inomata, M., Niida, S., Murakami, Y., and Shibata, K. I. (2010). Regulation of MyD88 aggregation and the MyD88-dependent signaling pathway by sequestosome 1 and histone deacetylase 6. J. Biol. Chem. 285, 35759–35769. doi: 10.1074/jbc.M110.126904
Ip, W. K. E., Hoshi, N., Shouval, D. S., Snapper, S., and Medzhitov, R. (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. doi: 10.1126/science.aal3535
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. doi: 10.1038/nature07317
Ito, Y., Ofengeim, D., Najafov, A., Das, S., Saberi, S., Li, Y., et al. (2016). RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608. doi: 10.1126/science.aaf6803
Jaco, I., Annibaldi, A., Lalaoui, N., Wilson, R., Tenev, T., Laurien, L., et al. (2017). MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol. Cell 66, 698–710.e5. doi: 10.1016/j.molcel.2017.05.003
Jang, M.-A., Kim, E. K., Now, H., Nguyen, N. T. H., Kim, W.-J., Yoo, J.-Y., et al. (2015). Mutations in DDX58, which encodes RIG-I, cause atypical singleton-merten syndrome. Am. J. Hum. Genet. 96, 266–274. doi: 10.1016/j.ajhg.2014.11.019
Jiang, C., Zhou, Z., Quan, Y., Zhang, S., Wang, T., Zhao, X., et al. (2016). CARMA3 is a host factor regulating the balance of inflammatory and antiviral responses against viral infection. Cell Rep. 14, 2389–2401. doi: 10.1016/j.celrep.2016.02.031
Jiang, X., Kinch, L. N., Brautigam, C. A., Chen, X., Du, F., Grishin, N. V., et al. (2012). Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36, 959–973. doi: 10.1016/j.immuni.2012.03.022
Jin, S., Tian, S., Luo, M., Xie, W., Liu, T., Duan, T., et al. (2017). Tetherin suppresses type I interferon signaling by targeting MAVS for NDP52-mediated selective autophagic degradation in human cells. Mol. Cell 68, 308–322.e4. doi: 10.1016/j.molcel.2017.09.005
Jin, Z., Li, Y., Pitti, R., Lawrence, D., Pham, V. C., Lill, J. R., et al. (2009). Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 137, 721–735. doi: 10.1016/j.cell.2009.03.015
Juilland, M., and Thome, M. (2018). Holding all the CARDs: how MALT1 controls CARMA/CARD-dependent signaling. Front. Immunol. 9:1927. doi: 10.3389/fimmu.2018.01927
Kaiser, W. J., and Offermann, M. K. (2005). Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952. doi: 10.4049/jimmunol.174.8.4942
Kaiser, W. J., Sridharan, H., Huang, C., Mandal, P., Upton, J. W., Gough, P. J., et al. (2013). Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 288, 31268–31279. doi: 10.1074/jbc.M113.462341
Kale, J., Osterlund, E. J., and Andrews, D. W. (2018). BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 25, 65–80. doi: 10.1038/cdd.2017.186
Kanazawa, N., Okafuji, I., Kambe, N., Nishikomori, R., Nakata-Hizume, M., Nagai, S., et al. (2005). Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood 105, 1195–1197. doi: 10.1182/blood-2004-07-2972
Kawaguchi, Y., Kovacs, J. J., McLaurin, A., Vance, J. M., Ito, A., and Yao, T.-P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738. doi: 10.1016/s0092-8674(03)00939-5
Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., et al. (2005). IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988. doi: 10.1038/ni1243
Kim, H. S., Lee, J. W., Soung, Y. H., Park, W. S., Kim, S. Y., Lee, J. H., et al. (2003). Inactivating mutations of caspase-8 gene in colorectal carcinomas. Gastroenterology 125, 708–715. doi: 10.1016/S0016-5085(03)01059-X
Konno, H., Chinn, I. K., Hong, D., Orange, J. S., Lupski, J. R., Mendoza, A., et al. (2018). Pro-inflammation associated with a gain-of-function mutation (R284S) in the innate immune sensor STING. Cell Rep. 23, 1112–1123. doi: 10.1016/j.celrep.2018.03.115
Konno, H., Konno, K., and Barber, G. N. (2013). Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698. doi: 10.1016/j.cell.2013.09.049
Korac, J., Schaeffer, V., Kovacevic, I., Clement, A. M., Jungblut, B., Behl, C., et al. (2013). Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 126, 580–592. doi: 10.1242/jcs.114926
Lafont, E., Draber, P., Rieser, E., Reichert, M., Kupka, S., de Miguel, D., et al. (2018). TBK1 and IKK𝜀 prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399. doi: 10.1038/s41556-018-0229-6
Larkin, B., Ilyukha, V., Sorokin, M., Buzdin, A., Vannier, E., and Poltorak, A. (2017). Cutting edge: activation of STING in T cells induces type I IFN responses and cell death. J. Immunol. 199, 397–402. doi: 10.4049/jimmunol.1601999
Lee, H. M., Shin, D. M., Yuk, J. M., Shi, G., Choi, D. K., Lee, S. H., et al. (2011). Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J. Immunol. 186, 1248–1258. doi: 10.4049/jimmunol.1001954
Lee, J.-Y., Koga, H., Kawaguchi, Y., Tang, W., Wong, E., Gao, Y.-S., et al. (2010). HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980. doi: 10.1038/emboj.2009.405
Lee, N.-R., Ban, J., Lee, N.-J., Yi, C.-M., Choi, J.-Y., Kim, H., et al. (2018). Activation of RIG-I-mediated antiviral signaling triggers autophagy through the MAVS-TRAF6-Beclin-1 signaling axis. Front. Immunol. 9:2096. doi: 10.3389/fimmu.2018.02096
Lenz, G., Davis, R. E., Ngo, V. N., Lam, L., George, T. C., Wright, G. W., et al. (2008). Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679. doi: 10.1126/science.1153629
Li, J., Mcquade, T., Siemer, A. B., Napetschnig, J., Moriwaki, K., Hsiao, Y.-S., et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350. doi: 10.1016/j.cell.2012.06.019
Li, Y., Fu, T.-M., Lu, A., Witt, K., Ruan, J., Shen, C., et al. (2018). Cryo-EM structures of ASC and NLRC4 CARD filaments reveal a unified mechanism of nucleation and activation of caspase-1. Proc. Natl. Acad. Sci. U.S.A. 115, 10845–10852. doi: 10.1073/pnas.1810524115
Lin, S.-C., Lo, Y.-C., and Wu, H. (2010). Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890. doi: 10.1038/nature09121
Lindqvist, L. M., Frank, D., McArthur, K., Dite, T. A., Lazarou, M., Oakhill, J. S., et al. (2017). Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ. 25, 782–794. doi: 10.1038/s41418-017-0017-z
Liu, D., Wu, H., Wang, C., Li, Y., Tian, H., Siraj, S., et al. (2018). STING directly activates autophagy to tune the innate immune response. Cell Death Differ. doi: 10.1016/j.nlm.2018.06.013 [Epub ahead of print].
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. doi: 10.1038/ni.1960
Liu, Y., Gordesky-Gold, B., Leney-Greene, M., Weinbren, N. L., Tudor, M., and Cherry, S. (2018). Inflammation-induced, STING-dependent autophagy restricts zika virus infection in the drosophila brain. Cell Host Microbe 24, 57–68.e3. doi: 10.1016/j.chom.2018.05.022
Lu, A., Magupalli, V. G., Ruan, J., Yin, Q., Atianand, M. K., Vos, M. R., et al. (2014). Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156, 1193–1206. doi: 10.1016/j.cell.2014.02.008
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426.
Matsumoto, G., Shimogori, T., Hattori, N., and Nukina, N. (2015). TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum. Mol. Genet. 24, 4429–4442. doi: 10.1093/hmg/ddv179
McArthur, K., Whitehead, L. W., Heddleston, J. M., Li, L., Padman, B. S., Oorschot, V., et al. (2018). BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359:eaao6047. doi: 10.1126/science.aao6047
Menon, M. B., Gropengießer, J., Fischer, J., Novikova, L., Deuretzbacher, A., Lafera, J., et al. (2017). p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259. doi: 10.1038/ncb3614
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. doi: 10.1016/s0092-8674(03)00521-x
Moreno-Gonzalo, O., Ramírez-Huesca, M., Blas-Rus, N., Cibrián, D., Saiz, M. L., Jorge, I., et al. (2017). HDAC6 controls innate immune and autophagy responses to TLR-mediated signalling by the intracellular bacteria Listeria monocytogenes. PLoS Pathog. 13:e1006799. doi: 10.1371/journal.ppat.1006799
Motshwene, P. G., Moncrieffe, M. C., Grossmann, J. G., Kao, C., Ayaluru, M., Sandercock, A. M., et al. (2009). An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 284, 25404–25411. doi: 10.1074/jbc.M109.022392
Nakazawa, S., Oikawa, D., Ishii, R., Ayaki, T., Takahashi, H., Takeda, H., et al. (2016). Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat. Commun. 7:12547. doi: 10.1038/ncomms12547
Nanson, J. D., Kobe, B., and Ve, T. (2018). Death, TIR, and RHIM: self-assembling domains involved in innate immunity and cell-death signaling. J. Leukoc. Biol. 105, 363–375. doi: 10.1002/JLB.MR0318-123R
Narendra, D., Tanaka, A., Suen, D.-F., and Youle, R. J. (2014). Parkin-induced mitophagy in the pathogenesis of Parkinson disease. Autophagy 5, 706–708. doi: 10.4161/auto.5.5.8505
Negroni, A., Colantoni, E., Vitali, R., Palone, F., Pierdomenico, M., Costanzo, M., et al. (2016). NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm. Res. 65, 803–813. doi: 10.1016/j.cell.2010.05.009
Newman, A. C., Scholefield, C. L., Kemp, A. J., Newman, M., McIver, E. G., Kamal, A., et al. (2012). TBK1 kinase addiction in lung cancer cells is mediated via autophagy of Tax1bp1/Ndp52 and non-canonical NF-κB signalling. PLoS One 7:e50672. doi: 10.1371/journal.pone.0050672
Normand, S., Waldschmitt, N., Neerincx, A., Martinez-Torres, R. J., Chauvin, C., Couturier-Maillard, A., et al. (2018). Proteasomal degradation of NOD2 by NLRP12 in monocytes promotes bacterial tolerance and colonization by enteropathogens. Nat. Commun. 9:5338. doi: 10.1038/s41467-018-07750-5
Oakes, J. A., Davies, M. C., and Collins, M. O. (2017). TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10:5. doi: 10.1186/s13041-017-0287-x
Ogura, Y., Bonen, D. K., Inohara, N., Nicolae, D. L., Chen, F. F., Ramos, R., et al. (2001). A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606. doi: 10.1038/35079114
Oshiumi, H., Matsumoto, M., Funami, K., Akazawa, T., and Seya, T. (2003). TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 4, 161–167. doi: 10.1038/ni886
Park, H. H., Lo, Y.-C., Lin, S.-C., Wang, L., Yang, J. K., and Wu, H. (2007). The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 25, 561–586. doi: 10.1146/annurev.immunol.25.022106.141656
Park, S., Ha, S.-D., Coleman, M. D., Meshkibaf, S., and Kim, S. O. (2013). p62/SQSTM1 enhances NOD2-mediated signaling and cytokine production through stabilizing NOD2 oligomerization. PLoS One 8:e57138. doi: 10.1371/journal.pone.0057138
Parkhouse, R., Boyle, J. P., and Monie, T. P. (2014). Blau syndrome polymorphisms in NOD2 identify nucleotide hydrolysis and helical domain 1 as signalling regulators. FEBS Lett. 588, 3382–3389. doi: 10.1016/j.febslet.2014.07.029
Paul, S., Kashyap, A. K., Jia, W., He, Y.-W., and Schaefer, B. C. (2012). Selective autophagy of the adaptor protein Bcl10 modulates T cell receptor activation of NF-kB. Immunity 36, 947–958. doi: 10.1016/j.immuni.2012.04.008
Paul, S., Traver, M. K., Kashyap, A. K., Washington, M. A., Latoche, J. R., and Schaefer, B. C. (2014). T cell receptor signals to NF-κB are transmitted by a cytosolic p62-Bcl10-Malt1-IKK signalosome. Sci. Signal. 7:ra45. doi: 10.1126/scisignal.2004882
Pellegrini, E., Desfosses, A., Wallmann, A., Schulze, W. M., Rehbein, K., Mas, P., et al. (2018). RIP2 filament formation is required for NOD2 dependent NF-κB signalling. Nat. Commun. 9:4043. doi: 10.1038/s41467-018-06451-3
Petes, C., Odoardi, N., and Gee, K. (2017). The toll for trafficking: toll-like receptor 7 delivery to the endosome. Front. Immunol. 8:1075. doi: 10.3389/fimmu.2017.01075
Pham, C. L., Shanmugam, N., Strange, M., O’Carroll, A., Brown, J. W., Sierecki, E., et al. (2018). Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep. 20:e46518. doi: 10.15252/embr.201846518
Pham, C. L., Shanmugam, N., Strange, M., O’Carroll, A., Brown, J. W., Sierecki, E., et al. (2019). Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep. 20:e46518. doi: 10.15252/embr.201846518
Pilli, M., Arko-Mensah, J., Ponpuak, M., Roberts, E., Master, S., Mandell, M. A., et al. (2012). TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234. doi: 10.1016/j.immuni.2012.04.015
Prabakaran, T., Bodda, C., Krapp, C., Zhang, B. C., Christensen, M. H., Sun, C., et al. (2018). Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J. 37:e97858. doi: 10.15252/embj.201797858
Qiao, Q., Yang, C., Zheng, C., Fontán, L., David, L., Yu, X., et al. (2013). Structural architecture of the CARMA1/Bcl10/MALT1 signalosome: nucleation-induced filamentous assembly. Mol. Cell 51, 766–779. doi: 10.1016/j.molcel.2013.08.032
Rice, G. I., del Toro Duany, Y., Jenkinson, E. M., Forte, G. M. A., Anderson, B. H., Ariaudo, G., et al. (2014). Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 46, 503–509. doi: 10.1038/ng.2933
Riley, J. S., Quarato, G., Cloix, C., Lopez, J., O’Prey, J., Pearson, M., et al. (2018). Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37:e99238. doi: 10.15252/embj.201899238
Rutsch, F., MacDougall, M., Lu, C., Buers, I., Mamaeva, O., Nitschke, Y., et al. (2015). A specific IFIH1 gain-of-function mutation causes singleton-merten syndrome. Am. J. Hum. Genet. 96, 275–282. doi: 10.1016/j.ajhg.2014.12.014
Sabbah, A., Chang, T. H., Harnack, R., Frohlich, V., Tominaga, K., Dube, P. H., et al. (2009). Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 10, 1073–1080. doi: 10.1038/ni.1782
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B.-G., Satoh, T., et al. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268. doi: 10.1038/nature07383
Samie, M., Lim, J., Verschueren, E., Baughman, J. M., Peng, I., Wong, A., et al. (2018). Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat. Immunol. 19, 246–254. doi: 10.1038/s41590-017-0042-6
Scharschmidt, E., Wegener, E., Heissmeyer, V., Rao, A., and Krappmann, D. (2004). Degradation of Bcl10 induced by T-cell activation negatively regulates NF- B signaling. Mol. Cell. Biol. 24, 3860–3873. doi: 10.1128/MCB.24.9.3860-3873.2004
Schlauderer, F., Seeholzer, T., Desfosses, A., Gehring, T., Strauss, M., Hopfner, K.-P., et al. (2018). Molecular architecture and regulation of BCL10-MALT1 filaments. Nat. Commun. 9:4041. doi: 10.1038/s41467-018-06573-8
Schütze, S., Tchikov, V., and Schneider-Brachert, W. (2008). Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 655–662. doi: 10.1038/nrm2430
Schwerd, T., Pandey, S., Yang, H.-T., Bagola, K., Jameson, E., Jung, J., et al. (2017). Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut 66, 1060–1073. doi: 10.1136/gutjnl-2015-310382
Seth, R. B., Sun, L., Ea, C.-K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Settembre, C., Fraldi, A., Jahreiss, L., Spampanato, C., Venturi, C., Medina, D., et al. (2008). A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 17, 119–129. doi: 10.1093/hmg/ddm289
Sharma, V., Verma, S., Seranova, E., Sarkar, S., and Kumar, D. (2018). Selective autophagy and xenophagy in infection and disease. Front. Cell. Dev. Biol. 6:147. doi: 10.3389/fcell.2018.00147
Shi, C.-S., Shenderov, K., Huang, N.-N., Kabat, J., Abu-Asab, M., Fitzgerald, K. A., et al. (2012). Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263. doi: 10.1038/ni.2215
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shim, J.-H., Xiao, C., Paschal, A. E., Bailey, S. T., Rao, P., Hayden, M. S., et al. (2005). TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681. doi: 10.1101/gad.1360605
Shin, M. S., Kim, H. S., Lee, S. H., Lee, J. W., Song, Y. H., Kim, Y. S., et al. (2002). Alterations of Fas-pathway genes associated with nodal metastasis innon-small cell lung cancer. Oncogene 21, 4129–4136. doi: 10.1038/sj.onc.1205527
Snow, A. L., Xiao, W., Stinson, J. R., Lu, W., Chaigne-Delalande, B., Zheng, L., et al. (2012). Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J. Exp. Med. 209, 2247–2261. doi: 10.1084/jem.20120831
Soung, Y. H., Lee, J. W., Kim, S. Y., Jang, J., Park, Y. G., Park, W. S., et al. (2005). CASPASE-8 gene is inactivated by somatic mutations in gastric carcinomas. Cancer Res. 65, 815–821. doi: 10.1016/S0092-8674(00)81874-7
Su, H., Bidere, N., Zheng, L., Cubre, A., Sakai, K., Dale, J., et al. (2005). Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science 307, 1465–1468. doi: 10.1126/science.1104765
Sun, L., Deng, L., Ea, C.-K., Xia, Z.-P., and Chen, Z. J. (2004). The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301. doi: 10.1038/nri1379
Sun, X., Sun, L., Zhao, Y., Li, Y., Lin, W., Chen, D., et al. (2016). MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov. 2:16024. doi: 10.1038/celldisc.2016.24
Sun, X., Yin, J., Starovasnik, M. A., Fairbrother, W. J., and Dixit, V. M. (2002). Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 277, 9505–9511. doi: 10.1074/jbc.M109488200
Swatek, K. N., and Komander, D. (2016). Ubiquitin modifications. Cell Res. 26, 399–422. doi: 10.1038/cr.2016.39
Tang, C.-H. A., Zundell, J. A., Ranatunga, S., Lin, C., Nefedova, Y., Del Valle, J. R., et al. (2016). Agonist-mediated activation of STING induces apoptosis in malignant B cells. Cancer Res. 76, 2137–2152. doi: 10.1158/0008-5472.CAN-15-1885
Tatematsu, M., Ishii, A., Oshiumi, H., Horiuchi, M., Inagaki, F., Seya, T., et al. (2010). A molecular mechanism for toll-IL-1 receptor domain-containing adaptor molecule-1-mediated IRF-3 activation. J. Biol. Chem. 285, 20128–20136. doi: 10.1074/jbc.M109.099101
Teitz, T., Wei, T., Valentine, M. B., Vanin, E. F., Grenet, J., Valentine, V. A., et al. (2000). Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 6, 529–535. doi: 10.1038/75007
Thapa, R. J., Ingram, J. P., Ragan, K. B., Nogusa, S., Boyd, D. F., Benitez, A. A., et al. (2016). DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681. doi: 10.1016/j.chom.2016.09.014
Tourneur, L., Delluc, S., Lévy, V., Valensi, F., Radford-Weiss, I., Legrand, O., et al. (2004). Absence or low expression of Fas-associated protein with death domain in acute myeloid leukemia cells predicts resistance to chemotherapy and poor outcome. Cancer Res. 64, 8101–8108. doi: 10.1158/0008-5472.CAN-04-2361
Travassos, L. H., Carneiro, L. A. M., Ramjeet, M., Hussey, S., Kim, Y.-G., Magalhães, J. G., et al. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62. doi: 10.1038/ni.1823
Vince, J. E., Wong, W. W.-L., Khan, N., Feltham, R., Chau, D., Ahmed, A. U., et al. (2007). IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131, 682–693. doi: 10.1016/j.cell.2007.10.037
Vitner, E. B., Salomon, R., Farfel-Becker, T., Meshcheriakova, A., Ali, M., Klein, A. D., et al. (2014). RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 20, 204–208. doi: 10.1038/nm.3449
Wang, Y., Chen, T., Han, C., He, D., Liu, H., An, H., et al. (2007). Lysosome-associated small Rab GTPase Rab7b negatively regulates TLR4 signaling in macrophages by promoting lysosomal degradation of TLR4. Blood 110, 962–971. doi: 10.1182/blood-2007-01-066027
Warner, J. D., Irizarry-Caro, R. A., Bennion, B. G., Ai, T. L., Smith, A. M., Miner, C. A., et al. (2017). STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med. 214, 3279–3292. doi: 10.1084/jem.20171351
Weber, A., Kirejczyk, Z., Besch, R., Potthoff, S., Leverkus, M., and Häcker, G. (2010). Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ. 17, 942–951. doi: 10.1038/cdd.2009.190
Westphal, D., Kluck, R. M., and Dewson, G. (2014). Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 21, 196–205. doi: 10.1038/cdd.2013.139
Winer, H., Fraiberg, M., Abada, A., Dadosh, T., Tamim-Yecheskel, B.-C., and Elazar, Z. (2018). Autophagy differentially regulates TNF receptor Fn14 by distinct mammalian Atg8 proteins. Nat. Commun. 9:3744. doi: 10.1038/s41467-018-06275-1
Wu, J., and Yan, N. (2018). Gain-of-function STING mutations induce T cell death in the SAVI mouse. J. Immunol. 200:166.29.
Xu, D., Jin, T., Zhu, H., Chen, H., Ofengeim, D., Zou, C., et al. (2018). TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19. doi: 10.1016/j.cell.2018.07.041
Xu, H., He, X., Zheng, H., Huang, L. J., Hou, F., Yu, Z., et al. (2014). Structural basis for the prion-like MAVS filaments in antiviral innate immunity. eLife 3:e01489. doi: 10.7554/eLife.01489
Yang, Q., Liu, T.-T., Lin, H., Zhang, M., Wei, J., Luo, W.-W., et al. (2017). TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 13:e1006600. doi: 10.1371/journal.ppat.1006600
Yang, S., Imamura, Y., Jenkins, R. W., Cañadas, I., Kitajima, S., Aref, A., et al. (2016). Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol. Res. 4, 520–530. doi: 10.1158/2326-6066.CIR-15-0235
Zhang, C., Lee, S., Peng, Y., Bunker, E., Giaime, E., Shen, J., et al. (2014). PINK1 triggers autocatalytic activation of Parkin to specify cell fate decisions. Curr. Biol. 24, 1854–1865. doi: 10.1016/j.cub.2014.07.014
Zhao, Y. G., and Zhang, H. (2018). Autophagosome maturation: an epic journey from the ER to lysosomes. J. Cell Biol. 38:jcb.201810099. doi: 10.1083/jcb.201810099
Zhu, G., Wu, C.-J., Zhao, Y., and Ashwell, J. D. (2007). Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr. Biol. 17, 1438–1443. doi: 10.1016/j.cub.2007.07.041
Zinngrebe, J., Rieser, E., Taraborrelli, L., Peltzer, N., Hartwig, T., Ren, H., et al. (2016). –LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammation. J. Exp. Med. 213, 2671–2689. doi: 10.1084/jem.20160041
Keywords: inflammation, cell death, death domain, RHIM, supramolecular complexes, innate immunity, autophagy, cargo receptors
Citation: Gentle IE (2019) Supramolecular Complexes in Cell Death and Inflammation and Their Regulation by Autophagy. Front. Cell Dev. Biol. 7:73. doi: 10.3389/fcell.2019.00073
Received: 15 January 2019; Accepted: 10 April 2019;
Published: 03 May 2019.
Edited by:
Thomas Kaufmann, University of Bern, SwitzerlandReviewed by:
Dwayne G. Stupack, University of California, San Diego, United StatesCopyright © 2019 Gentle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian E. Gentle, aWFuLmdlbnRsZUB1bmlrbGluaWstZnJlaWJ1cmcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.