 Alex Pezzotta
Alex Pezzotta Anna Pistocchi
Anna Pistocchi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 27 February 2019
Sec. Epigenomics and Epigenetics
Volume 7 - 2019 | https://doi.org/10.3389/fcell.2019.00021
This article is part of the Research TopicChromatin Traits in Human DiseasesView all 9 articles
The genes of the cohesin complex exert different functions, ranging from the adhesion of sister chromatids during the cell cycle, DNA repair, gene expression and chromatin architecture remodeling. In recent years, the improvement of DNA sequencing technologies allows the identification of cohesin mutations in different tumors such as acute myeloid leukemia (AML), acute megakaryoblastic leukemia (AMKL), and myelodysplastic syndromes (MDS). However, the role of cohesin dysfunction in cancer insurgence remains elusive. In this regard, cells harboring cohesin mutations do not show any increase in aneuploidy that might explain their oncogenic activity, nor cohesin mutations are sufficient to induce myeloid neoplasms as they have to co-occur with other causative mutations such as NPM1, FLT3-ITD, and DNMT3A. Several works, also using animal models for cohesin haploinsufficiency, correlate cohesin activity with dysregulated expression of genes involved in myeloid development and differentiation. These evidences support the involvement of cohesin mutations in myeloid neoplasms.
One of the biological meaning of a living organism is the possibility to divide by replicating DNA and generate a new organism. To accomplish this, the genome duplication should be error free and the daughter cell should properly inherit the genetic material from the mother cell. The cohesin proteins are required during this multistep process: in interphase to maintain genome stability during DNA double strand break repair (Watrin and Peters, 2009; Losada, 2014), in S-phase to enforce Sister Chromatid Cohesion (SCC) throughout DNA replication (Peters et al., 2008), and in M-phase to ensure proper chromosome distribution into dividing cells (Jeppsson et al., 2014). Since the cohesin protein complex has essential roles in the cell, members of the cohesin complex are found from bacteria to humans and are evolutionary and functionally conserved. The ability of cohesin to perform these functions resides in their property to encircle the DNA, creating topological links between chromatin fibers. To mediate sister chromatids tethering and segregation or DNA double strand break repair, the cohesin complex binds to the DNA in a trans conformation. However, cohesin might also encircle the DNA in cis, forming chromatin loops and contributing to gene regulation by modulating genome architecture or joining two distant segments of the genome. In vertebrates, the ring that embraces the DNA is formed by coiled-coil heterodimers of Structural Maintenance of Chromosomes (SMC) subunits SMC1 and SMC3, by the alpha-kleisin subunit RAD21 that brings in connection the ATPase head domains of SMC proteins and stabilizes their interactions (Nasmyth, 2011), and by the stromal antigens STAG1/STAG2 (SA1/SA2) (Canudas and Smith, 2009; Remeseiro et al., 2012). Although cohesin proteins are intrinsically able to topologically bind to the DNA (Gligoris et al., 2014), the loading of the complex is not efficient in the absence of the NIPBL/MAU2 heterodimer. As stated by their name, cohesin becomes “cohesive” only when the SMC3 head domain subunits are acetylated by the acetyl-transferase ESCO1/ESCO2. The release of the complex from the DNA is achieved by the separase-mediated proteolytic cleavage of RAD21 (Uhlmann et al., 2000), the HDAC8-mediated de-acetylation of SMC3, or the opening of the RAD21-SMC3 complex controlled by accessory proteins such as CDCA5 (soronin), PDS5 and WAPL (Ben-Shahar et al., 2008; Rowland et al., 2009; Nishiyama et al., 2010; Tedeschi et al., 2013; Beckouët et al., 2016; Figure 1A).
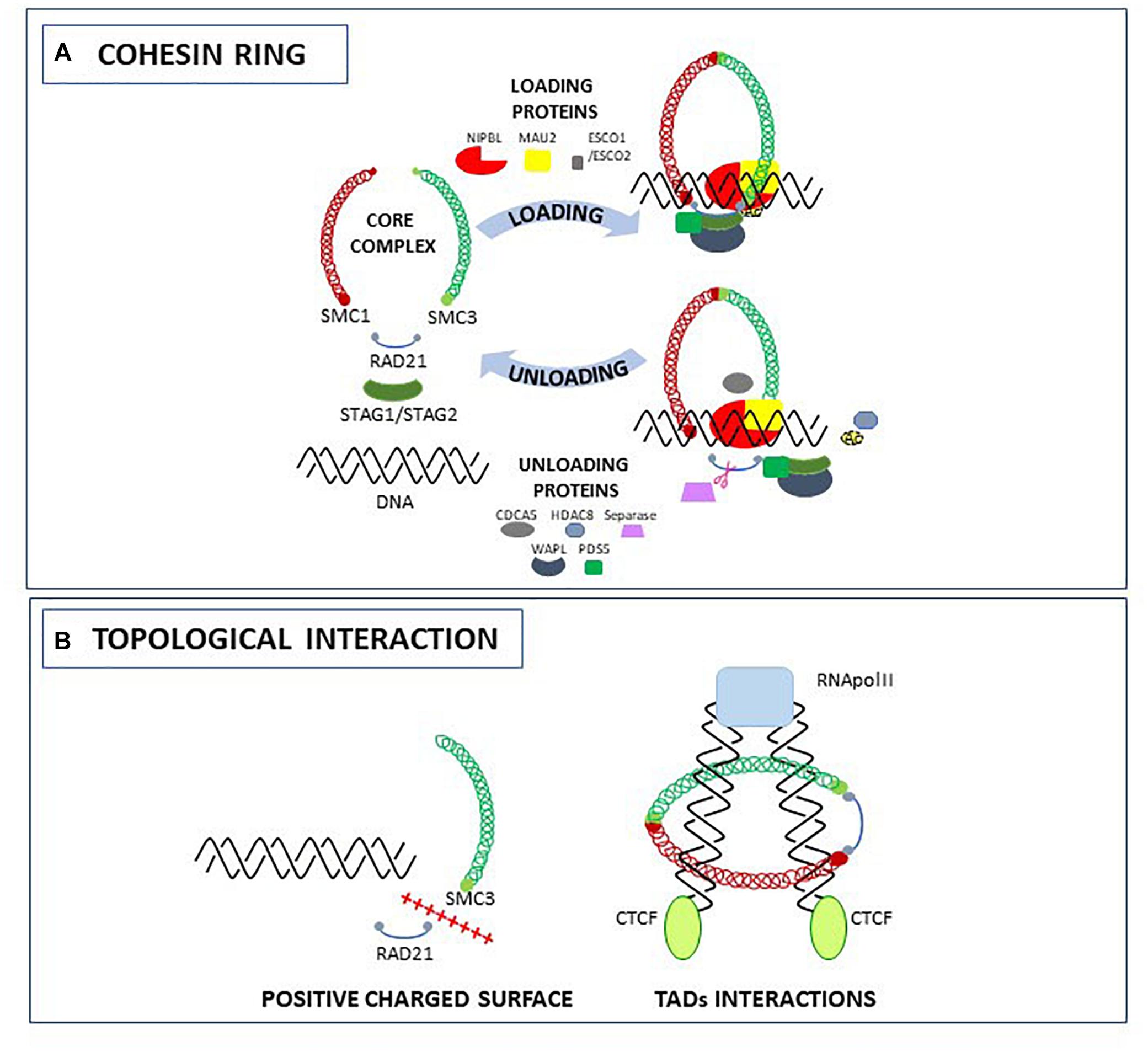
Figure 1. The cohesin complex and accessory proteins: structure (A) and topological interactions with the DNA (B).
Recently, several studies were carried out to elucidate the role of the cohesin ring in topological entrapment of DNA. Biochemical and crystallographic studies in yeast led to the identification of an interlocking gate for the transportation of DNA across the cohesin ring (Murayama and Uhlmann, 2015), and of a specific HEAT-domain required for the binding of the DNA to the SMC3-RAD21 complex due to a positively charged surface (Li et al., 2018). Other studies using Chromosome Conformation Capture-derived approaches such as 5C and Hi-C, allowed the identification of a role for cohesin in the formation and stabilization of Topologically Associating Domains (TADs), genomic regions spanning form 200 kb to 1 Mb in mammals, that are thought to contribute to gene expression by remodeling chromosome architecture (Dixon et al., 2016). In a recent work using the smallest human chromosome 21, Bernardi demonstrated that the formation of TADs is related to the 3D structures of the corresponding GC-rich isochores. These “primary TADs” are bound to the cohesin complex that actively slides down, generating an extrusion loop (Bernardi, 2018). In another work it has been hypothesized that the movement of the cohesin complex along the extrusion loop is mediated by the pushing of the RNA polymerase (Björkegren and Baranello, 2018). Indeed, in Drosophila and mammals, cohesin can activate or silence genes by interacting with RNA polymerase II (RNA Pol II) (Misulovin et al., 2008; Schaaf et al., 2009; Fay et al., 2011). For example, by comparing NIPBL and RNA Pol II binding sites, it has been shown that NIPBL binds 100–200 nucleotides upstream of RNA Pol II (Zuin et al., 2014). The extrusion loop made by cohesin stops when DNA is occupied by the CCTC-binding factor (CTCF). In human and mammals, cohesin and CTCF co-localize at several loci contributing to topological organization of the genome: when CTCF is depleted, the cohesin complex is not found at CTCF sites (Wendt et al., 2008) but is still able to bind to other chromatin regions (Figure 1B).
Hence, cohesin proteins regulate both positively or negatively chromatin architecture and gene expression, by recognizing specific sites on the genome alone or in combination with different proteins, and modifying the interaction between enhancers and promoters.
Several evidences suggest that altered gene expression due to cohesin dysfunction could affect tumourigenesis. Indeed, cohesin or its regulators are frequently mutated in different types of tumors such as colorectal cancer (NIPBL) (Barber et al., 2008), glioblastoma, Ewing’s sarcoma, urothelial bladder carcinoma, melanoma (STAG2) (Solomon et al., 2011, 2013; Balbás-Martínez et al., 2013) and myeloid neoplasms (STAG1, STAG2, SMC3, and RAD21) (Dolnik et al., 2012; Kon et al., 2013). In addition, mutations in genomic binding sites of CTCF/cohesin (Katainen et al., 2015) or changes in cohesin protein levels (RAD21) have been associated to cancers (Yan et al., 2012). Since the cohesin complex is involved in chromosome segregation and DNA repair, it is not surprising that cohesin mutations or dysfunctions could enhance tumorigenesis. However, the association between cohesin mutations and genome instability in cancer is still controversial and not always reported (Balbás-Martínez et al., 2013; Taylor et al., 2014; Thol et al., 2014). To note, patients affected by the Cornelia de Lange Syndrome (CdLS) caused by germline mutations in cohesin genes (∼65% NIPBL, ∼5% SMC1A and HDAC8, 1–2% SMC3 and RAD21) (Mannini et al., 2013; Singh and Gerton, 2015), rarely develop cancers and likely not for genomic instability but as a consequence of their clinical features (e.g., gastric reflux) (Schrier et al., 2011; Deardorff et al., 2012). Since CdLS and tumors share same types of mutations in cohesin genes (for NIPBL heterozygous mutations, mainly non-sense, leading to haploinsufficiency; for SMC1A hemizygous mutations, mainly missense, probably leading to a dominant negative effect) (Mannini et al., 2013; Singh and Gerton, 2015), it has been hypothesized that they differ in their physiopathological contest. Indeed, in tumors somatic cohesin mutations occur in adult and terminally differentiated cells while in CdLS germline cohesin mutations occur in a developing and embryonic tissue. In addition, conversely to CdLS in which cohesin mutations are causative, in cancer cohesin mutations do not initiate but contribute to tumorigenesis when they co-occur with additional mutations. A study concerning AML identified 37 patients with mutation in one of the cohesin genes and, among them, the 81.1% had an additional mutation in genes causative for AML insurgence such as FLT3-ITD (21.6%), NPM1 (21.6%), RUNX1 or ASXL1 (Tsai et al., 2017).
Acute myeloid leukemia (AML) is a heterogeneous group of hematologic aggressive neoplasms of bone marrow, characterized by irreversible expansion of precursor myeloid blasts defective in their differentiation and function (Löwenberg et al., 1999; Ley et al., 2008; Naoe and Kiyoi, 2013). The leukemic mutations are serially acquired in clones of long-lived self-renewing hematopoietic stem cells (HSCs), termed pre-leukemic HSCs (Jan and Majeti, 2013; Corces-Zimmerman and Majeti, 2014).
Among the novel recurrently mutated genes in AML patients, there are the members of the cohesin complex which occur in approximately 15% of AML cases (Thota et al., 2014). Interestingly, no correlation between mutated cohesin genes and prognosis was observed, and most of clinical features of AML patients with or without mutations in cohesin were similar (Thol et al., 2014). Thol and colleagues characterized the genomes of 389 uniformly treated AML patients in order to dissect the clinical and prognostic implications of mutated cohesin. A total of 23 patients (5.9%) had mutations in the cohesin genes and the most frequently mutated were STAG1 (1.8%), STAG2 (1.3%), and SMC3 (1.3%), while mutations in RAD21 and SMC1A were rarer events. Previously, The Cancer Genome Atlas identified mutations in cohesin in 26 out of 200 (13%) primary AML adult patients, with a higher mutations frequency in STAG2, SMC1A, SMC3, RAD21 meanwhile no mutation in STAG1 were observed in comparison to Thol and colleagues’ analysis (Ley et al., 2013; Thol et al., 2014). This discrepancy could be due to different approaches used to validate mutations (Thol et al., 2014). In myelodysplastic syndromes (MDS), a heterogeneous group of clonal hematopoietic disorders characterized by cytopenia, ineffective hematopoiesis and an increased risk of progression to AML (Haferlach et al., 2014; Shallis et al., 2018), RAD21, STAG2, and SMC1A are the most frequently mutated genes (∼15%) and are associated with poor survival (Cazzola et al., 2013; Kon et al., 2013; Haferlach et al., 2014; Malcovati et al., 2014; Thota et al., 2014). The identified cohesin mutations in AML patients are typically classified in two categories: mutations in RAD21 and STAG2 are mainly truncation or frame-shift while mutations in SMC3 and SMC1A are mainly missense (Kon et al., 2013; Thota et al., 2014). Genomic deletions have also been found for RAD21 and STAG2 (Rocquain et al., 2010). Importantly, cohesin mutations are mutually exclusive suggesting that a single altered component is sufficient to affect the tumor suppressive function of the whole cohesin complex in myeloid leukemogenesis (Welch et al., 2012; Solomon et al., 2014).
The frequency of cohesin mutations is surprisingly higher in acute megakaryoblastic leukemia (AMKL), a rare sub-type of AML (AML-M7) characterized by defective megakaryocytes proliferation and differentiation (Ley et al., 2013; Yoshida et al., 2013). AMKL represents 4–15% of pediatric AML and is predominantly found in Down Syndrome children (DS) together with somatic GATA1 mutations (Gruber and Downing, 2015). GATA1 mutated isoforms cause the hyper-proliferation of megakaryocyte progenitors during early fetal liver hematopoiesis and lead to transient abnormal myelopoiesis (TAM) (Hitzler et al., 2003; Rainis et al., 2003). Up to 10% of TAM cases typically resolve spontaneously while 30% of them develop in AMKL during childhood with the accumulation of somatic mutations in different genomic regions (Rainis et al., 2003; Yoshida et al., 2013). Sequencing analysis revealed mutations and deletions in STAG2, RAD21, SMC3, SMC1A, and NIPBL in almost 53% of DS-AMKL patients but none in the TAM clones, indicating that cohesin mutations are involved in neoplastic transformation from TAM to AMKL. Only few non-DS-AMKL patients had cohesin mutations, suggesting that mutated cohesin is a DS-AMKL feature. Different works tried to dissect the effects of cohesin haploinsufficiency and altered GATA1 binding to the chromatin (Yoshida et al., 2013; Fisher et al., 2017). GATA1 mutations were strictly linked to the context of trisomy 21 in DS patients (Shimizu et al., 2008) and this condition provided the cellular setting for the persistence and eventual transformation of GATA1 mutant cells. Moreover, GATA1 mutations involve a region that mediates GATA1-RUNX1 interaction during normal megakaryopoiesis (Elagib et al., 2003). Interestingly, the dosage effect caused by trisomy 21 hyper-activated the Wnt signaling in DS-AMKL patients. This is accomplished by the downregulation of the tumor suppressor gene APC and stabilization of the active form of beta-catenin via miR-99a/100-125 (Emmrich et al., 2014). The interaction between cohesin and Wnt/beta-catenin signaling has been described in different models of cancer (Ghiselli et al., 2003), and in our zebrafish model with cohesin haploinsufficiency (Pistocchi et al., 2013; Fazio et al., 2016; Mazzola et al., 2019), suggesting a possible effect of cohesin mutations and Wnt/beta-catenin signaling dysregulation in DS-AMKL.
Cohesin mutations are associated to myeloid neoplasms, but are not causative of tumor onset indicating that other pathways might be relevant. Since cohesin mutations are often described as early, founder mutations in pre-leukemic HSCs (Corces-Zimmerman and Majeti, 2014), several groups investigated the role of cohesin complex in differentiation and self-renewal of HSCs and of their progenitor cells (HSPCs), and leukemogenic transformation using in-vitro and in-vivo models (Figures 2A–C).
Mullenders and colleagues described the role of cohesin in HSCs homeostasis (Mullenders et al., 2015). Firstly, they transduced murine bone marrow c-Kit+ HSPCs with shRNA against Rad21, Smc3, and Smc1a demonstrating a rapid increase of their replating capacity and defects in myeloid differentiation. Then, they generated a mouse model with cohesin down-regulation showing clinically features of myeloid neoplasia but not genomic alteration due to cohesin dysregulation. Moreover, they demonstrated that HSCs and myeloid precursors were subjected to changes in gene regulation and chromatin accessibility as a consequence of cohesin down-regulation (Mullenders et al., 2015). The correlation between the increase of hematopoietic progenitors and cohesin downregulation has been also described in a mouse model generated by Viny and colleagues. Cells derived from this mouse with combined effects of Smc3 haploinsufficiency and FLT3-ITD mutation, showed increased HSPCs proliferation and survival rates and a simultaneous decrease in myeloid progenitors (Viny et al., 2015). The role of the cohesin complex as a major regulator of HSCs has also been described by Galeev and colleagues with genome wide RNAi analyses on primary human cord blood derived CD34+ cells with cohesin knock-down (Galeev et al., 2016). They demonstrated that in sorted CD34+ cells, the transfection of shRNA against RAD21, STAG1-2 and SMC3 negatively affected differentiation and enhanced HSCs expansion in vitro and in vivo when these cells were transplanted in immunodeficient mice (Figure 2B). Transcriptome analyses on cohesin-deficient CD34+ cells demonstrated an increased expression of genes responsible for the stem cell phenotype. Mazumdar and colleagues identified genomic regions with altered chromatin accessibility following RAD21 or SMC1A mutations in primary human HSPCs. Using the transposase-accessible chromatin and sequencing (ATAC-seq) technique, they found increased accessibility of GATA2, RUNX1 and ERG DNA binding motifs when RAD21 and SMC1A were mutated in comparison to controls. They also demonstrated that the block in the HSPCs differentiation was specifically due to the increased activity of these transcription factors as their shRNA-mediated silencing rescued the differentiation defects of cohesin-mutated-HSPCs (Mazumdar and Majeti, 2017) (Figure 2A). In a murine model, the loss of the Asxl1 gene that is frequently mutated in different myeloid malignancies with poor prognosis, reduced the genome binding of RAD21 and SMC1A and altered the expression of their target genes in cells enriched for myeloid progenitors Lin-cKit+ (LK) (Li et al., 2017).
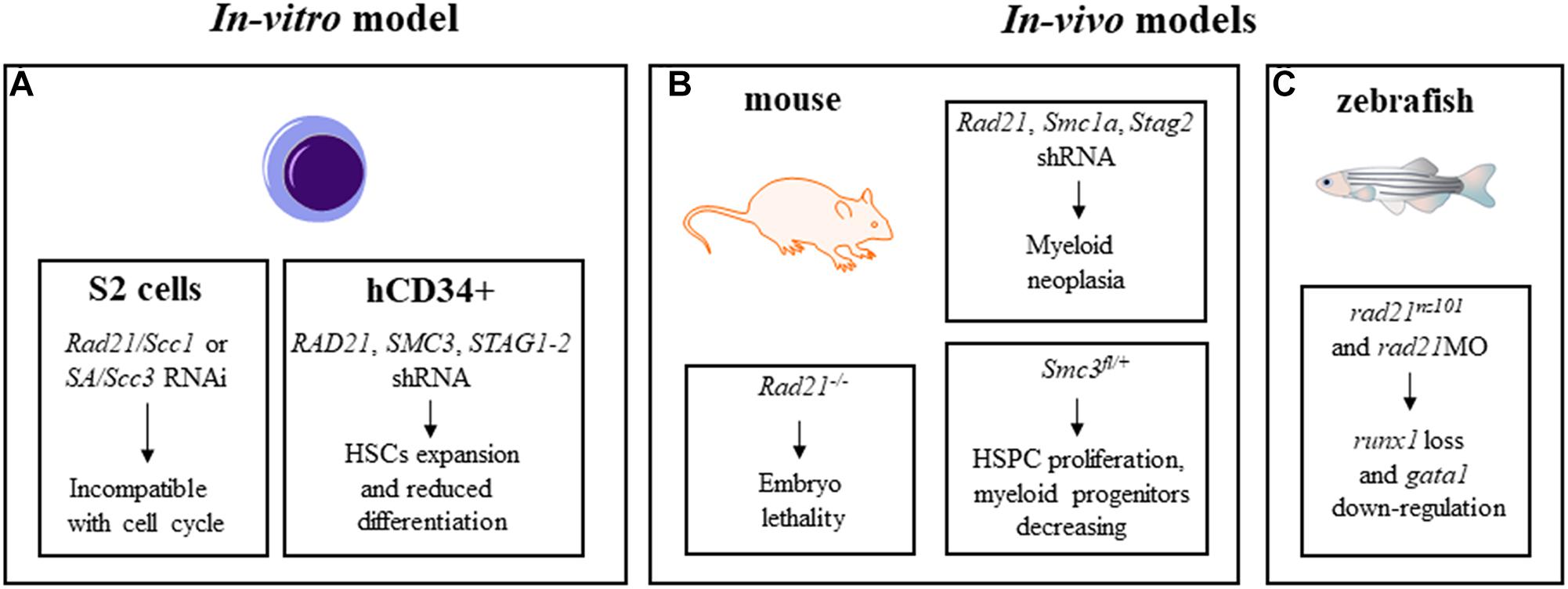
Figure 2. In-vitro (A) and in-vivo (B,C) models for the study of cohesin roles in hematopoiesis.
RUNX1, an essential regulator of hematopoiesis frequently involved in leukemia insurgence, is also related to cohesin, as demonstrated by Horsfield and colleagues using a zebrafish model. The zebrafish rad21nz171 mutant completely lacked early runx1 expression in hematopoietic compartments, while monoallelic loss of rad21 reduced the transcription of runx1, suggesting a dose-dependent modulation controlled by Rad21. Early hematopoiesis in rad21nz171 mutant was not dramatically affected except for the gata1 downregulated expression, while later differentiation was severely reduced, indicating that the altered expression of markers of late hematopoiesis depends on the loss of runx1 and reduction of gata1 (Horsfield et al., 2007; Figure 2C).
The hypothesis that cohesin dysfunction in HSPCs might alter the expression of hematopoietic genes was further investigated going inside the mechanism through which cohesin accomplish this. Fisher and colleagues demonstrated that cohesin complex binds to and regulates the Polycomb Repressive Complex 2 (PRC2) to silence Hoxa7 and Hoxa9 genes involved in HSPCs proliferation. Mutations in cohesin (RAD21) enhanced HSPCs proliferation but the phenotype could be rescued when Hoxa7 or Hoxa9 genes were simultaneously knocked-down (Fisher et al., 2017). The authors argued that the cohesin-mediated regulation of PRC2 interaction with Hoxa locus was accomplished through remodeling of chromatin architecture. Indeed, cohesin proteins interact with CTCF in DNA binding, specifically in the establishment of TADs (Merkenschlager and Nora, 2016; Bernardi, 2018). The binding of CTCF, together with the epigenetic chromatin-remodeling factor Smarca5 and cohesin was also found at upstream regulatory element (URE) of SPI1 gene, a master transcription factor of myeloid cell differentiation. This recruitment was disrupted in AML blasts suggesting its involvement in tumor insurgence (Dluhosova et al., 2014). The specific action of the cohesin complex on myeloid cells was also observed in one case of gene fusion. Murine hematopoietic cells transfected with the fusion gene NIPBL-HOXB9 exhibited increased in vitro colony replating capacity with hallmarks of myeloid progenitors (Dang et al., 2017). In our recent work, we used zebrafish to confirm that other mutations might dysregulate cohesin expression. In this regard, we observed that AML patients carrying NPM1 mutations showed a specific NIPBL downregulation and a zebrafish model with NIPBL haploinsufficiency presented defects in myeloid cell differentiation, demonstrating that animal models could enhance the comprehension of the action of multiple mutations/dysregulations (Mazzola et al., 2019).
In myeloid neoplasms cohesin mutations occur with low frequency in comparison to other more frequently mutated genes and are not sufficient, alone, to drive to tumourigenesis. However, cohesin mutations occur early in the clonal hierarchy and cohesin dysfunction enhances HSC and HSPCs proliferation and controls the expression of genes involved in myeloid differentiation. Therefore, cohesin might be considered as promising pharmacological target for myeloid malignancies. Some drugs already used in AML clinical trials such as Dot1I methyltransferase inhibitors (Bernt et al., 2011) or azacitidine, have been proved to be efficient in the rescue of the phenotype caused by cohesin mutations (Fisher et al., 2017; Tothova et al., 2017). Thus, the dissection of molecular pathways altered by cohesin dysfunction might allow the discovery of new therapeutic targets downstream of cohesin. In this regard, the screening for cohesin mutations of larger cohorts of patients or the development of animal models with cohesin haploinsufficiency are required to address this intriguing possibility. Moreover, the co-occurrence of mutations in cohesin and causative genes of myeloid neoplasms (e.g., FLT3-ITD, DNMT3A, NPM1, and TET2), leads to the hypothesis that the efforts to develop therapies for AML might be improved by combining those targeting specific genes and those directed on shared targets, as well as by combining multiple therapies to treat diverse sub-clones.
AP, MM, and MS contributed to writing and figures. AM contributed to figures and supervision. AP conceived, wrote, and supervised the manuscript.
This work was supported by My First AIRC Grant (MFAG#18714).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Balbás-Martínez, C., Sagrera, A., Carrillo-De-Santa-Pau, E., Earl, J., Márquez, M., Vazquez, M., et al. (2013). Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat. Genet. 45, 1464–1469. doi: 10.1038/ng.2799
Barber, T. D., McManus, K., Yuen, K. W. Y., Reis, M., Parmigiani, G., Shen, D., et al. (2008). Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 105, 3443–3448. doi: 10.1073/pnas.0712384105
Beckouët, F., Srinivasan, M., Roig, M. B., Chan, K. L., Scheinost, J. C., Batty, P., et al. (2016). Releasing activity disengages Cohesin’s Smc3/Scc1 interface in a process blocked by acetylation. Mol. Cell 61, 563–574. doi: 10.1016/j.molcel.2016.01.026
Ben-Shahar, T. R., Heeger, S., Lehane, C., East, P., Flynn, H., Skehel, M., et al. (2008). Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321, 563–566. doi: 10.1126/science.1157774
Bernardi, G. (2018). The formation of chromatin domains involves a primary step based on the 3-D structure of DNA. Sci. Rep. 8:17821. doi: 10.1038/s41598-018-35851-0
Bernt, K. M., Zhu, N., Sinha, A. U., Vempati, S., Faber, J., Krivtsov, A. V., et al. (2011). MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78. doi: 10.1016/j.ccr.2011.06.010
Björkegren, C., and Baranello, L. (2018). DNA supercoiling, topoisomerases, and cohesin: partners in regulating chromatin architecture? Int. J. Mol. Sci. 19:E884. doi: 10.3390/ijms19030884
Canudas, S., and Smith, S. (2009). Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J. Cell Biol. 187, 165–173. doi: 10.1083/jcb.200903096
Cazzola, M., Della Porta, M. G., and Malcovati, L. (2013). The genetic basis of myelodysplasia and its clinical relevance. Blood 122, 4021–4034. doi: 10.1182/blood-2013-09-381665
Corces-Zimmerman, M. R., and Majeti, R. (2014). Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia 28, 2276–2282. doi: 10.1038/leu.2014.211
Dang, J., Nance, S., Ma, J., Cheng, J., Walsh, M. P., Vogel, P., et al. (2017). AMKL chimeric transcription factors are potent inducers of leukemia. Leukemia 31, 2228–2234. doi: 10.1038/leu.2017.51
Deardorff, M. A., Bando, M., Nakato, R., Watrin, E., Itoh, T., Minamino, M., et al. (2012). HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317. doi: 10.1038/nature11316
Dixon, J. R., Gorkin, D. U., and Ren, B. (2016). Chromatin domains: the unit of chromosome organization. Mol. Cell 62, 668–680. doi: 10.1016/j.molcel.2016.05.018
Dluhosova, M., Curik, N., Vargova, J., Jonasova, A., Zikmund, T., and Stopka, T. (2014). Epigenetic control of SPI1 gene by CTCF and ISWI ATPase SMARCA5. PLoS One 9:e87448. doi: 10.1371/journal.pone.0087448
Dolnik, A., Engelmann, J. C., Scharfenberger-Schmeer, M., Mauch, J., Kelkenberg-Schade, S., Haldemann, B., et al. (2012). Commonly altered genomic regions in acute myeloid leukemia are enriched for somatic mutations involved in chromatin remodeling and splicing. Blood 120, e83–e92. doi: 10.1182/blood-2011-12-401471
Elagib, K. E., Racke, F. K., Mogass, M., Khetawat, R., Delehanty, L. L., and Goldfarb, A. N. (2003). RUNX1 and GATA-1 coexpression and cooperation in megakaryocytic differentiation. Blood 101, 4333–4341. doi: 10.1016/j.vetmic.2017.07.023
Emmrich, S., Rasche, M., Schoning, J., Reimer, C., Keihani, S., Maroz, A., et al. (2014). miR-99a/100∼125b tricistrons regulate hematopoietic stem and progenitor cell homeostasis by shifting the balance between TGFβ and Wnt signaling. Genes Dev. 28, 858–874. doi: 10.1101/gad.233791.113
Fay, A., Misulovin, Z., Li, J., Schaaf, C. A., Gause, M., Gilmour, D. S., et al. (2011). Cohesin selectively binds and regulates genes with paused RNA polymerase. Curr. Biol. 21, 1624–1634. doi: 10.1016/j.cub.2011.08.036
Fazio, G., Gaston-Massuet, C., Bettini, L. R., Graziola, F., Scagliotti, V., Cereda, A., et al. (2016). CyclinD1 down-regulation and increased apoptosis are common features of cohesinopathies. J. Cell. Physiol. 231, 613–622. doi: 10.1002/jcp.25106
Fisher, J. B., Peterson, J., Reimer, M., Stelloh, C., Pulakanti, K., Gerbec, Z. J., et al. (2017). The cohesin subunit Rad21 is a negative regulator of hematopoietic self-renewal through epigenetic repression of Hoxa7 and Hoxa9. Leukemia 31, 712–719. doi: 10.1038/leu.2016.240
Galeev, R., Baudet, A., Kumar, P., Rundberg Nilsson, A., Nilsson, B., Soneji, S., et al. (2016). Genome-wide RNAi screen identifies cohesin genes as modifiers of renewal and differentiation in human HSCs. Cell Rep. 14, 2988–3000. doi: 10.1016/j.celrep.2016.02.082
Ghiselli, G., Coffee, N., Munnery, C. E., Koratkar, R., and Siracusa, L. D. (2003). The cohesin SMC3 is a target the for β-catenin/TCF4 transactivation pathway. J. Biol. Chem. 278, 20259–20267. doi: 10.1074/jbc.M209511200
Gligoris, T. G., Scheinost, J. C., Bürmann, F., Petela, N., Chan, K. L., Uluocak, P., et al. (2014). Closing the cohesin ring: structure and function of its Smc3-kleisin interface. Science 346, 963–967. doi: 10.1126/science.1256917
Gruber, T. A., and Downing, J. R. (2015). The biology of pediatric acute megakaryoblastic leukemia. Blood 126, 943–949. doi: 10.1182/blood-2015-05-567859
Haferlach, T., Nagata, Y., Grossmann, V., Okuno, Y., Bacher, U., Nagae, G., et al. (2014). Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 28, 241–247. doi: 10.1038/leu.2013.336
Hitzler, J. K., Cheung, J., Li, Y., Scherer, S. W., and Zipursky, A. (2003). GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood 101, 4301–4304. doi: 10.1182/blood-2003-01-0013
Horsfield, J. A., Anagnostou, S. H., Hu, J. K.-H., Cho, K. H. Y., Geisler, R., Lieschke, G., et al. (2007). Cohesin-dependent regulation of Runx genes. Development 134, 2639–2649. doi: 10.1242/dev.002485
Jan, M., and Majeti, R. (2013). Clonal evolution of acute leukemia genomes. Oncogene 32, 135–140. doi: 10.1038/onc.2012.48
Jeppsson, K., Kanno, T., Shirahige, K., and Sjögren, C. (2014). The maintenance of chromosome structure: positioning and functioning of SMC complexes. Nat. Rev. Mol. Cell Biol. 15, 601–614. doi: 10.1038/nrm3857
Katainen, R., Dave, K., Pitkänen, E., Palin, K., Kivioja, T., Välimäki, N., et al. (2015). CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 47, 818–821. doi: 10.1038/ng.3335
Kon, A., Shih, L. Y., Minamino, M., Sanada, M., Shiraishi, Y., Nagata, Y., et al. (2013). Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 45, 1232–1237. doi: 10.1038/ng.2731
Ley, T. J., Mardis, E. R., Ding, L., Fulton, B., McLellan, M. D., Chen, K., et al. (2008). DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456, 66–72. doi: 10.1038/nature07485
Ley, T. J., Miller, C., Ding, L., Raphael, B. J., Mungall, A. J., Robertson, A. G., et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074. doi: 10.1056/NEJMoa1301689
Li, Y., Muir, K. W., Bowler, M. W., Metz, J., Haering, C. H., and Panne, D. (2018). Structural basis for scc3-dependent cohesin recruitment to chromatin. eLife 7:e38356. doi: 10.7554/eLife.38356
Li, Z., Zhang, P., Yan, A., Guo, Z., Ban, Y., Li, J., et al. (2017). ASXL1 interacts with the cohesin complex to maintain chromatid separation and gene expression for normal hematopoiesis. Sci. Adv. 3:e1601602. doi: 10.1126/sciadv.1601602
Losada, A. (2014). Cohesin in cancer: chromosome segregation and beyond. Nat. Rev. Cancer 14, 389–393. doi: 10.1038/nrc3743
Löwenberg, B., Downing, J. R., and Burnett, A. (1999). Acute myeloid leukemia. N. Engl. J. Med. 341, 1051–1062. doi: 10.1056/NEJM199909303411407
Malcovati, L., Papaemmanuil, E., Ambaglio, I., Elena, C., Gallì, A., Della Porta, M. G., et al. (2014). Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood 124, 1513–1521. doi: 10.1182/blood-2014-03-560227
Mannini, L., Cucco, F., Quarantotti, V., Krantz, I. D., and Musio, A. (2013). Mutation spectrum and genotype-phenotype correlation in cornelia de lange syndrome. Hum. Mutat. 34, 1589–1596. doi: 10.1002/humu.22430
Mazumdar, C., and Majeti, R. (2017). The role of mutations in the cohesin complex in acute myeloid leukemia. Int. J. Hematol. 105, 31–36. doi: 10.1007/s12185-016-2119-7
Mazzola, M., Deflorian, G., Pezzotta, A., Ferrari, L., Fazio, G., Bresciani, E., et al. (2019). NIPBL: a new player in myeloid cells differentiation. Haematologica doi: 10.3324/haematol.2018.200899 [Epub ahead print].
Merkenschlager, M., and Nora, E. P. (2016). CTCF and cohesin in genome folding and transcriptional gene regulation. Annu. Rev. Genomics Hum. Genet. 17, 17–43. doi: 10.1146/annurev-genom-083115-022339
Misulovin, Z., Schwartz, Y. B., Li, X. Y., Kahn, T. G., Gause, M., MacArthur, S., et al. (2008). Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 117, 89–102. doi: 10.1007/s00412-007-0129-1
Mullenders, J., Aranda-Orgilles, B., Lhoumaud, P., Keller, M., Pae, J., Wang, K., et al. (2015). Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 212, 1833–1850. doi: 10.1084/jem.20151323
Murayama, Y., and Uhlmann, F. (2015). DNA entry into and exit out of the cohesin ring by an interlocking gate mechanism. Cell 163, 1628–1640. doi: 10.1016/j.cell.2015.11.030
Naoe, T., and Kiyoi, H. (2013). Gene mutations of acute myeloid leukemia in the genome era. Int. J. Hematol. 97, 165–174. doi: 10.1007/s12185-013-1257-4
Nasmyth, K. (2011). Cohesin: a catenase with separate entry and exit gates? Nat. Cell Biol. 13, 1170–1177. doi: 10.1038/ncb2349
Nishiyama, T., Ladurner, R., Schmitz, J., Kreidl, E., Schleiffer, A., Bhaskara, V., et al. (2010). Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell 143, 737–749. doi: 10.1016/j.cell.2010.10.031
Peters, J. M., Tedeschi, A., and Schmitz, J. (2008). The cohesin complex and its roles in chromosome biology. Genes Dev. 22, 3089–3114. doi: 10.1101/gad.1724308
Pistocchi, A., Fazio, G., Cereda, A., Ferrari, L., Bettini, L. R., Messina, G., et al. (2013). Cornelia de lange syndrome: NIPBL haploinsufficiency downregulates canonical Wnt pathway in zebrafish embryos and patients fibroblasts. Cell Death Dis. 4:e866. doi: 10.1038/cddis.2013.371
Rainis, L., Bercovich, D., Strehl, S., Teigler-Schlegel, A., Stark, B., Trka, J., et al. (2003). Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood 102, 981–986. doi: 10.1182/blood-2002-11-3599
Remeseiro, S., Cuadrado, A., Carretero, M., Martínez, P., Drosopoulos, W. C., Cañamero, M., et al. (2012). Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 31, 2076–2089. doi: 10.1038/emboj.2012.11
Rocquain, J., Gelsi-Boyer, V., Adélaïde, J., Murati, A., Carbuccia, N., Vey, N., et al. (2010). Alteration of cohesin genes in myeloid diseases. Am. J. Hematol. 85, 717–719. doi: 10.1002/ajh.21798
Rowland, B. D., Roig, M. B., Nishino, T., Kurze, A., Uluocak, P., Mishra, A., et al. (2009). Building sister chromatid cohesion: Smc3 acetylation counteracts an antiestablishment activity. Mol. Cell 33, 763–774. doi: 10.1016/j.molcel.2009.02.028
Schaaf, C. A., Misulovin, Z., Sahota, G., Siddiqui, A. M., Schwartz, Y. B., Kahn, T. G., et al. (2009). Regulation of the drosophila enhancer of split and invected-engrailed gene complexes by sister chromatid cohesion proteins. PLoS One 4:e6202. doi: 10.1371/journal.pone.0006202
Schrier, S. A., Sherer, I., Deardorff, M. A., Clark, D., Audette, L., Gillis, L., et al. (2011). Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am. J. Med. Genet. Part A 155, 3007–3024. doi: 10.1002/ajmg.a.34329
Shallis, R. M., Ahmad, R., and Zeidan, A. M. (2018). The genetic and molecular pathogenesis of myelodysplastic syndromes. Eur. J. Haematol. 101, 260–271. doi: 10.1111/ejh.13092
Shimizu, R., Engel, J. D., and Yamamoto, M. (2008). GATA1-related leukaemias. Nat. Rev. Cancer 8, 279–287. doi: 10.1038/nrc2348
Singh, V. P., and Gerton, J. L. (2015). Cohesin and human disease: lessons from mouse models. Curr. Opin. Cell Biol. 37, 9–17. doi: 10.1016/j.ceb.2015.08.003
Solomon, D. A., Kim, J. S., Bondaruk, J., Shariat, S. F., Wang, Z. F., Elkahloun, A. G., et al. (2013). Frequent truncating mutations of STAG2 in bladder cancer. Nat. Genet. 45, 1428–1430. doi: 10.1038/ng.2800
Solomon, D. A., Kim, J. S., and Waldman, T. (2014). Cohesin gene mutations in tumorigenesis: from discovery to clinical significance. BMB Rep. 47, 299–310. doi: 10.5483/BMBRep.2014.47.6.092
Solomon, D. A., Kim, T., Diaz-Martinez, L. A., Fair, J., Elkahloun, A. G., Harris, B. T., et al. (2011). Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333, 1039–1043. doi: 10.1126/science.1203619
Taylor, C. F., Platt, F. M., Hurst, C. D., Thygesen, H. H., and Knowles, M. A. (2014). Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumour grade and stage and inversely related to chromosomal copy number changes. Hum. Mol. Genet. 23, 1964–1974. doi: 10.1093/hmg/ddt589
Tedeschi, A., Wutz, G., Huet, S., Jaritz, M., Wuensche, A., Schirghuber, E., et al. (2013). Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature 501, 564–568. doi: 10.1038/nature12471
Thol, F., Bollin, R., Gehlhaar, M., Walter, C., Dugas, M., Suchanek, K. J., et al. (2014). Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood 123, 914–920. doi: 10.1182/blood-2013-07-518746
Thota, S., Viny, A. D., Makishima, H., Spitzer, B., Radivoyevitch, T., Przychodzen, B., et al. (2014). Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood 124, 1790–1798. doi: 10.1182/blood-2014-04-567057
Tothova, Z., Krill-Burger, J. M., Popova, K. D., Landers, C. C., Sievers, Q. L., Yudovich, D., et al. (2017). Multiplex CRISPR/Cas9-based genome editing in human hematopoietic stem cells models clonal hematopoiesis and myeloid neoplasia. Cell Stem Cell 21, 547–555.e8. doi: 10.1016/j.stem.2017.07.015
Tsai, C. H., Hou, H. A., Tang, J. L., Kuo, Y. Y., Chiu, Y. C., Lin, C. C., et al. (2017). Prognostic impacts and dynamic changes of cohesin complex gene mutations in de novo acute myeloid leukemia. Blood Cancer J. 7:663. doi: 10.1038/s41408-017-0022-y
Uhlmann, F., Wernic, D., Poupart, M. A., Koonin, E. V., and Nasmyth, K. (2000). Cleavage of cohesin by the CD clan protease separin triggers anaphase in yeast. Cell 103, 375–386. doi: 10.1016/S0092-8674(00)00130-6
Viny, A. D., Ott, C. J., Spitzer, B., Rivas, M., Meydan, C., Papalexi, E., et al. (2015). Dose-dependent role of the cohesin complex in normal and malignant hematopoiesis. J. Exp. Med. 212, 1819–1832. doi: 10.1084/jem.20151317
Watrin, E., and Peters, J. M. (2009). The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 28, 2625–2635. doi: 10.1038/emboj.2009.202
Welch, J. S., Ley, T. J., Link, D. C., Miller, C. A., Larson, D. E., Koboldt, D. C., et al. (2012). The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278. doi: 10.1016/j.cell.2012.06.023
Wendt, K. S., Yoshida, K., Itoh, T., Bando, M., Koch, B., Schirghuber, E., et al. (2008). Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451, 796–801. doi: 10.1038/nature06634
Yan, M., Xu, H., Waddell, N., Shield-Artin, K., Haviv, I., McKay, M. J., et al. (2012). Enhanced RAD21 cohesin expression confers poor prognosis in BRCA2 and BRCAX, but not BRCA1 familial breast cancers. Breast Cancer Res. 14:R69. doi: 10.1186/bcr3176
Yoshida, K., Toki, T., Okuno, Y., Kanezaki, R., Shiraishi, Y., Sato-Otsubo, A., et al. (2013). The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat. Genet. 45, 1293–1301. doi: 10.1038/ng.2759
Keywords: cohesin, AML, AMKL, MDS, animal models, gene expression
Citation: Pezzotta A, Mazzola M, Spreafico M, Marozzi A and Pistocchi A (2019) Enigmatic Ladies of the Rings: How Cohesin Dysfunction Affects Myeloid Neoplasms Insurgence. Front. Cell Dev. Biol. 7:21. doi: 10.3389/fcell.2019.00021
Received: 08 November 2018; Accepted: 05 February 2019;
Published: 27 February 2019.
Edited by:
Chiara Lanzuolo, Institute of Cell Biology and Neurobiology, CNR, ItalyReviewed by:
Daniel Vaiman, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2019 Pezzotta, Mazzola, Spreafico, Marozzi and Pistocchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Marozzi, YW5uYS5tYXJvenppQHVuaW1pLml0 Anna Pistocchi, YW5uYS5waXN0b2NjaGlAdW5pbWkuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.