 Fabian Elgner
Fabian Elgner Eberhard Hildt
Eberhard Hildt- Department of Virology, Paul-Ehrlich-Institut, Langen, Germany
Although potent direct-acting antiviral drugs for the treatment of chronic hepatitis C virus (HCV) infection are licensed, there are more than 70 million individuals suffering from chronic HCV infection. In light of the limited access to these drugs, high costs, and a lot of undiagnosed cases, it is expected that the number of HCV cases will not decrease worldwide in the next years. Therefore, and due to the paradigmatic character of HCV for deciphering the crosstalk between viral pathogens and the host cell, characterization of HCV life cycle remains a challenge. HCV belongs to the family of Flaviviridae. As an enveloped virus HCV life cycle depends in many steps on intracellular trafficking. Rab GTPases, a large family of small GTPases, play a central role in intracellular trafficking processes controlling fusion, uncoating, vesicle budding, motility by recruiting specific effector proteins. This review describes the relevance of various Rab proteins for the different steps of the HCV life cycle.
Introduction
Hepatitis C virus (HCV) belongs to the genus Hepacivirus of the family Flaviviridae. HCV's positive strand RNA genome encompasses 9.6 kb and consists of a 5′-untranslated region (5′-UTR), a long open reading frame encoding a single polyprotein of about 3,010 aa and a 3′-UTR (Simmonds, 2013). Viral and host proteases cleave the polyprotein into the three structural proteins, core that forms the capsid and the two envelope proteins E1 and E2, the viroporin p7 and the non-structural (NS) proteins (NS2, NS3, NS4A, NS4B, NS5A, NS5B). NS3, NS4A, NS4B, NS5A, NS5B form the replicase complexes that are sufficient for the genome replication that does not require the presence of further HCV proteins, enabling the establishment of subgenomic replicons that allow the analysis of HCV genome replication in the absence of infectious virion production (Lohmann et al., 1999). HCV replication occurs in the cytoplasm in the “membranous web” (MW), a characteristic structure for HCV-replicating cells that is formed by reorganization of ER-membranes and lipid droplets (LDs) (Paul et al., 2014; Meyers et al., 2016). While genome replication occurs at the replicon complexes localized at the cytoplasmic face of the membrane network, morphogenesis starts at the LDs that are in close vicinity to the replicon complexes. A characteristic of the mature HCV particle is its heavy association with lipoproteins. In light of this, HCV is frequently described as lipoviroparticle (LVP). In accordance to this, it was assumed that HCV leaves the cell via the lipoprotein pathway, but recent work provided evidence that HCV can be released in form of exosomes via multivesicular body (MVB)-dependent pathways (Elgner et al., 2016; Devhare et al., 2017).
Although there has been a tremendous progress in HCV research the last 20 years, there are still a variety of open questions regarding the molecular virology of HCV. A further more and more relevant aspect is to understand the differences and similarities of HCV to other flaviviruses i.e. Zika virus (ZIKV), Dengue virus (DENV), or West Nile virus (WNV).
Infection by HCV frequently leads to a chronic infection that can cause steatosis, fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). At present there are about 71 million individuals suffering from chronic HCV infection. There is neither a preventive nor a therapeutic vaccine available. The recent development of a new generation of HCV-specific drugs, the direct acting antivirals (DAAs), marks a significant progress to treat and to cure HCV. But the availability of these DAAs is still limited and the response of the different genotypes and the potential development of resistant mutants is not fully clear (da Silva Filipe et al., 2017). Moreover, there is still a variety of open questions with respect to the HCV life cycle.
Figure 1 summarizes the current knowledge about the involvement of Rab GTPases in the life cycle of HCV.
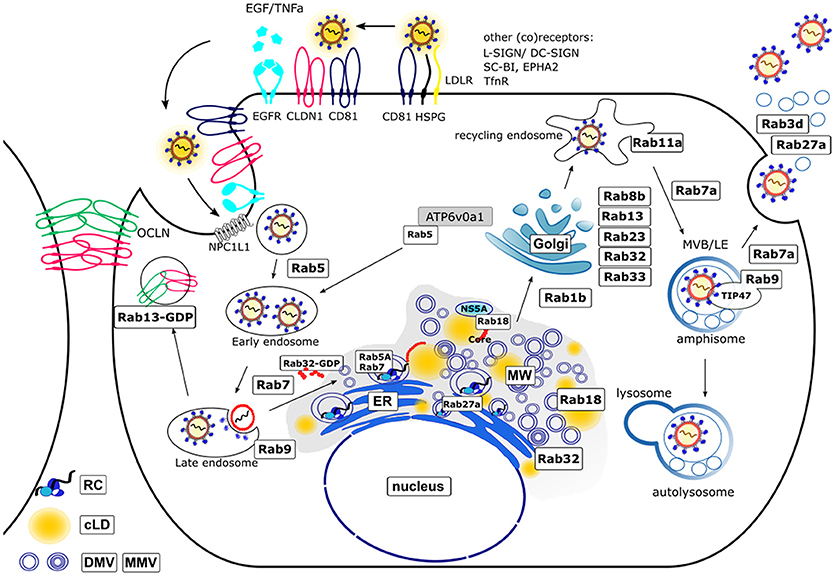
Figure 1. Schematic model of the HCV life cycle and involved Rab proteins. The entry of the HCV lipoviroparticles (LVPs) occurs in a coordinated way by binding of the LVPs to specific (co)receptors. The LVPs initially bind to heparan sulfate proteoglycans (HSPG) and the low density-lipoprotein receptor (LDLR) resulting in a local accumulation. After binding to scavenger receptor class B member 1 (SR-BI) a conformational change of E2 enables the binding to CD81 which then activates signal transduction via the epidermal growth factor receptor (EGFR). The LVP-loaded CD81 migrates by lateral diffusion to the tight junctions (TJ) where interaction with the TJ proteins occluding (OCLN) and claudin-1 (CLDN1) takes place and the LVPs are internalized via clathrin mediated endocytosis [Additional (co)receptors are: L-SIGN, DC-SIGN, ephidrin type A receptor 2 (EPHA2), Niemann-Pick C1-like receptor 1 (NPC1L1) and transferrin receptor (TnfR)]. After maturation of early endosomes to late endosomes the release of the viral nucleocapsid into the cytoplasm occurs in a pH-dependent manner. Hereby, Rab5A is required for acidification as it transports the vacuolar (H+)-ATPase (V-ATPase) ATP6v0a1 from the trans-Golgi (TGN) network toward endocytotic vesicles. The trafficking from early to late endosomes and finally lysosomes for degradation is regulated by Rab5 and Rab7. The TJ proteins OCLN and CLDN1 are recycled under control of Rab13. The uncoated positive-strand RNA is transported to the rough endoplasmic reticulum (ER) where it serves as template for the viral polyprotein synthesis. HCV infection leads to a massive rearrangement of intracellular ER derived membranes leading to the formation of a specific microenvironment, the membranous web (MW). The MW consists of double-membrane vesicles (DMVs) and multi-membrane vesicles (MMVs) embedded in a membranous matrix where HCV replication occurs in replication complexes (RCs). Rab5A and Rab7 are associated with the RCs. Another Rab protein that controls HCV replication is Rab27a. HCV assembly occurs in close proximity to the RCs. Replication and assembly sites are linked via cLDs. The Rab GTPases Rab18 and Rab32 are involved in HCV particle assembly by tethering LDs toward ER derived membranes and facilitating the trafficking of HCV Core and NS5A toward membrane structures involved in particle assembly. The exact mechanism of LVP release remains enigmatic. There is growing evidence that viral particles are released via the endosomal pathway rather than the canonical secretory route. This novel pathway includes the Rab1b-dependent transport of HCV particles through the Golgi compartment. Here, the Rab proteins Rab8b, 13, 23, 32, 33 are involved. Rab11a, a classical marker for recycling endosomes further directs the particles toward late endosomes (Rab7a-dependent) where the particles are sorted either for release by fusion of multivesicular bodies (MVBs) with the plasma membrane (Rab9a and Rab7a-dependent) in parallel to the release of exosomes (Rab3d- and Rab27a-dependent) or for autolysosomal degradation. For this, Rab9 together with TIP47 is required.
Rab (Ras-related in brain) proteins are small GTPases of the Ras-like family and regulators of the intracellular vesicle transport. In their inactive GDP (guanosine diphosphate) bound state, they are localized in the cytoplasm and get recruited to cell membranes upon activation due to binding of GTP (guanosine triphosphate). In the active state Rabs interact with distinct effector proteins to fulfill their multiple functions (Eathiraj et al., 2005). Although Rabs possess a weak intrinsic GTP-hydrolysis activity, many Rabs require the enzymatic activity of GAPs (GTPase activating proteins), which hydrolyze GTP to GDP and thereby inactivate the Rabs. GDIs (GDP dissociation inhibitors) are capable to solubilize inactive Rab proteins to provide a pool of activatable Rabs in the cytosol (Pfeffer and Aivazian, 2004). GEFs (guanine-nucleotide-exchange factors) exchange the bound GDP by GTP which leads to an activation of the Rabs. The ability to translocate into membranes is mediated by membrane bound GEFs or geranylgeranylation at the C-terminus of the Rabs. GEFs and GAPs are specific for single Rab proteins or subfamilies and thereby control the molecular switch which regulates fission, transport and fusion of membrane vesicles (Grosshans et al., 2006; Zhen and Stenmark, 2015). Rab proteins regulate amongst others the endosomal pathway, autophagy, and the interconnection of both cellular pathways (Ao et al., 2014).
HCV Entry
HCV particles are tightly associated with lipoproteins and therefore frequently designated as LVPs (Catanese et al., 2013). The sinusoidal blood stream transports the LVPs through the fenestrated endothelium into the space of Disse (the perisinusoidal space in the liver between a hepatocyte and a sinusoid). Here, an initial interaction with heparan sulfate proteoglycan and LDLR occurs leading to a local accumulation (Jiang et al., 2012; Kobayashi et al., 2012). The activity of lipopropein lipases and hepatic lipases converts very low density lipoprotein (VLDL) to low density lipoprotein (LDL) (Mahley and Ji, 1999). In the next step the LVPs bind to SR-B1 mediated by the high affinity of SR-B1 to E2 and surface exposed cholesterol esters (Zeisel et al., 2007). The SR-B1-bound LVPs accumulate ApoA and ApoC1 from HDL (Acton et al., 1996; Miao et al., 2017). The subsequent conformational change of E2 enables the release from SR-B1 and the binding to CD81, where a conformational change of E1E2 is induced. EGF-R and TGFβ-R are key factors for the control of the subsequent steps (Ströh et al., 2018; Tong et al., 2018). The LVP-loaded CD81 migrates by lateral diffusion to the tight junctions (TJ). Here, the TJs proteins Occludin (OCLN) and Claudin 1 (CLDN1) interact with the complex and thereby mediate a clustering of the tetraspanins (Evans et al., 2007; Ploss et al., 2009). In the next step the complexes that are clustered in the TJs are internalized by inward budding and clathrin-dependent endocytosis (Meertens et al., 2006; Sharma et al., 2011; Miao et al., 2017). The resulting primary endocytotic vesicles undergo a maturation process from early to late endosomes. Hereby, Rab5A is required for acidification (Meertens et al., 2006; Zhang et al., 2011). It is supposed that acidification leads to an irreversible conformational change exposing a so far not definitively identified fusogenic sequence that mediates fusion with the late endosomal membrane and release of the nucleocapsid harboring the viral genome into the cytoplasm (Tong et al., 2017, 2018; Guest and Pierce, 2018; Ströh et al., 2018). Under the reducing conditions of the cytoplasm the capsid disassembles and releases the RNA that enters the replication cycle. There is only limited evidence available describing the function of Rab proteins for the viral entry process.
Rab5A
The Rab5 and Rab7 GTPases are involved in regulation of cargo trafficking from early and late endosomes to the lysosomes for degradation. The Rab GTPases cycle between a GTP-bound active state and a GDP-bound inactive state. This process is regulated by Rab GEFs and Rab GAPs, respectively. The GTP-bound form of Rab5 and Rab7 is associated with early and late endosomes, respectively, where they are involved in recruiting effector proteins (Wandinger-Ness and Zerial, 2014).
Although the fusion process in the HCV entry process is not fully understood and the fusogenic sequence is still not definitively identified, it is clearly established that HCV entry occurs via receptor mediated endocytosis and depends on endosomal acidification (Tong et al., 2017). The vacuolar (H+)-ATPase (V-ATPase) ATP6v0a1 is a central factor in this process (Huotari and Helenius, 2011). The transport of ATP6v0a1 from the trans-Golgi network (TGN) to the endocytotic vesicles is mediated by Rab5A that is enriched in early endosomes. Inhibition of endosomal acidification by Bafilomycin A1 or chloroquine impairs HCV infection in a dose-dependent manner, but has no effect on HCV replication (Blanchard et al., 2006; Matsuda et al., 2014). Real-time imaging experiments using GFP-tagged Rab5A showed CD81- and CLDN1 containing vesicles moving to and co-localizing with Rab5 positive structures. Labeling with EEA1-specific antibodies confirmed the identity of this structures with early endosomes (Farquhar et al., 2012). The detailed role of Rab5A for the virus entry is currently not well-understood.
Rab13
Rab13 is involved in the regulation of the endocytotic recycling of the TJs-associated proteins CLDN1 and OCLN that are HCV entry factors (Marzesco et al., 2002; Morimoto et al., 2005; Evans et al., 2007; Ploss et al., 2009). Silencing of Rab13 expression impairs the establishment of HCV infection. However, if the expression of Rab13 is silenced after establishment of an HCV infection, there is no impact on the HCV life cycle, indicating that HCV replication is not affected by the silencing of Rab13 expression (Takeda et al., 2016). There is a crosstalk between Rab13, protein kinase A (PKA) and CLDN1 in TJs (Köhler et al., 2004; Farquhar et al., 2008). It was described that PKA promotes the CD81-CLDN1 interaction in TJs. If PKA is blocked, CLDN1 disappears from the TJs and localizes to the cytoplasm. Rab13-GTP binds to PKA and thereby impairs the phosphorylation of CLDN1. The unphosphorylated CLDN1 localizes to the cytoplasm. In contrast to this, Rab13-GDP does not bind to PKA, and PKA phosphorylates CLDN1 enabling the recycling of CLDN1 to the TJs.
The Rab GTPases involved in the entry of HCV virions are summarized in Table 1.
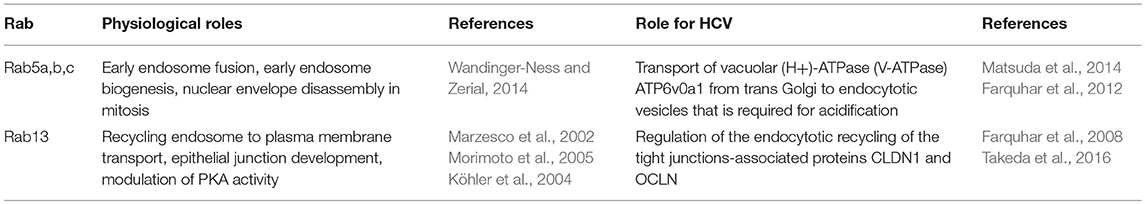
Table 1. Rab proteins involved in HCV entry.
HCV Genome Replication
The disassembly of the nucleocapsid releases the viral plus-strand RNA genome into the cytoplasm. The about 9.6 kb encompassing viral RNA is translated at the rough ER in a single polyprotein with a length of about 3,000 amino acids. It is co- and post-translationally cleaved by viral and cellular proteases in the viral structural and NS proteins (Gu and Rice, 2013; Neufeldt et al., 2018). The core protein and the two envelope glycoproteins E1 and E2 form the viral particle, whereas the p7 viroporin and non-structural protein 2 (NS2) are involved in the virus assembly that occurs on the surface of LDs. The NS proteins NS3, NS4A, NS4B, NS5A, and NS5B, the RNA-dependent RNA polymerase, form the replicase complex. These proteins are sufficient for viral RNA replication and are the base for subgenomic replicon systems (Lohmann et al., 1999). Table 2 lists the Rab proteins with involvement in the viral replication. NS5A, together with host factors, induce massive changes in the ER membrane structure leading to the formation of double membrane vesicles (DMVs). In contrast to previous reports, NS4B seems to be involved in the formation of single membrane vesicles (SMVs). The DMV formation is associated with a massive change in the ER structure leading to the formation of the MW that hosts the viral RNA replicase complexes (Paul et al., 2014; Meyers et al., 2016; Medvedev et al., 2017). The NS5B-dependent genome replication occurs via a negative-strand copy. The de novo formed plus-strand RNA copies are either packaged into capsids as viral genome that are subsequently enveloped or are translated in viral polyproteins or serve as template for the synthesis of further minus-strand intermediates (Shi and Suzuki, 2018).
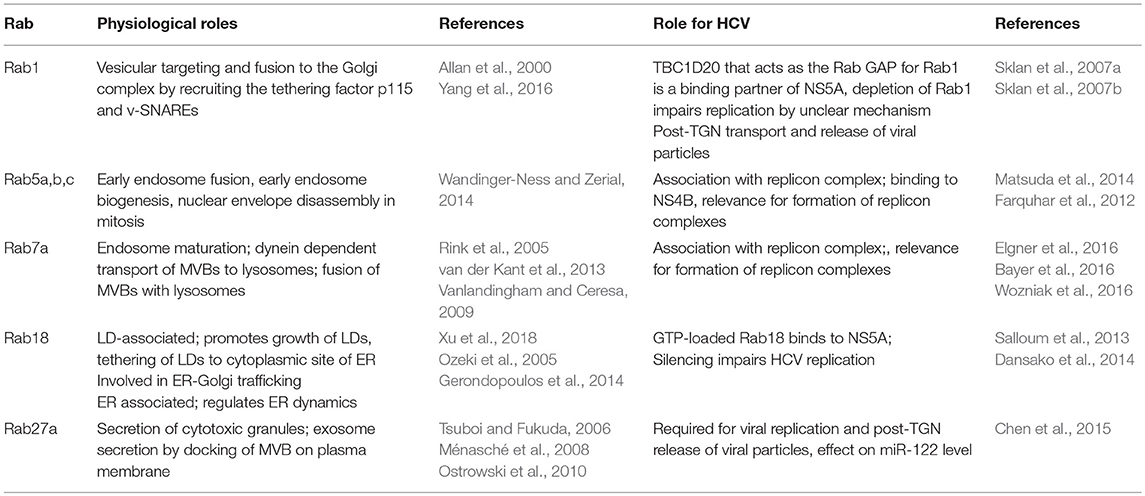
Table 2. Rab proteins involved in HCV replication.
Rab5A and Rab7
Rab5A and Rab7 were found to be associated with the replicon complex as characterized by confocal double immunofluorescence microscopy showing a co-staining with NS4B and co-purification with replicon complexes (Manna et al., 2010). Further analyses revealed that silencing of Rab5 and Rab7 expression decreases the de novo synthesis of HCV genomes. Although Rab2 was not found to be associated to the replicon complexes, silencing of Rab2 expression decreased HCV replication. In extension of these experiments it was observed that silencing of Rab1, Rab2, or Rab11 had no effect on the formation of replicon complexes. This fits to the observation that coexpression of transdominant negative (tdn) mutants of Rab11 and Rab12 does not affect the formation of replicon complexes (Manna et al., 2010).
This shows the high relevance of Rab5 and Rab7 for the functionality of replicon complexes. However, Rab5 and Rab7 are normally not associated with the ER compartment. On the first glance this seems to be a contradicting observation. In light of this it was speculated that NS4B-bound vesicles are transported to the plasma membrane (PM) and from there retrieved into the cytoplasm via endocytosis leading to the recruitment of Rab5 and Rab7. As inhibition of Rab1 or Rab 2 expression by siRNA or by co-expression of tdn mutants does not affect replicon complex formation and functionality or their association with Rab5 and Rab7, this hypothesis is not likely (Manna et al., 2010). So it is assumed that Rab5/7 are recruited to NS4B to facilitate the fusion of ER-derived vesicles in order to form replicon complexes. This is supported by the observation that Rab5 directly binds to NS4B, but so far there is no evidence for a direct interaction of NS4B with Rab7 (Stone et al., 2007). But in contrast to this model, TBC1D20, that acts as the Rab GAP for Rab1, was identified as a binding partner of NS5A. A region within the amino acids 269–403 mediates the interaction of TBC1D20 with the N-terminal part of NS5A. Based on this it was shown that TBC1D20 depletion inhibits HCV replication and prevents the accumulation of viral RNA (Sklan et al., 2007b). In accordance to this, the same group reported that integrity of Rab1 is required for HCV replication (Sklan et al., 2007a).
HCV replication leads to an induction of autophagy. A central factor for the HCV-dependent induction of autophagy is an elevated ROS level that leads to formation of phosphorylated p62. Autophagy plays a crucial role for the life cycle of HCV. Inhibition of autophagy leads to a decrease in the amount of released viral particles. Expression of tdnRab5 or silencing of Rab5 expression impairs HCV-dependent induction of autophagy and is associated with a decrease in the amount of released viral particles. As described above Rab5 was identified as a binding partner of NS4B. A mutant of NS4B (V233R and L237E) fails to bind Rab5. Rab5 is part of a macromolecular complex encompassing Rab5, Beclin-1, and Vps34 that is involved in the induction of autophagy. In accordance to the observed interaction of Rab5 and NS4B Rab5, Beclin-1, and Vps34 can be coprecipitated with NS4B using a NS4B-specific antibody (Su et al., 2011). Based on this interaction it is assumed that the NS4B/Rab5 interaction is relevant for the HCV-dependent induction of autophagy.
Rab18
In the HCV life cycle the place of genome replication (replicon complexes) and the place of nucleocapsid assembly (surface of LDs) are separated, raising the question about the efficient, and targeted transfer of the de novo synthesized RNA from the replicon complexes to the LDs. NS5A has the capacity to bind the viral RNA (Foster et al., 2010; Hwang et al., 2010). Recent reports described tail interacting protein of 47 kDA (TIP47) as a binding partner of NS5A. Due to the high affinity of TIP47 to LDs and lipoproteins, it is assumed that TIP47 is involved in targeting the RNA-complexed NS5A to the LDs (Ploen et al., 2013a,b; Vogt et al., 2013). Rab 18 was identified as a binding partner of NS5A. The exact binding domain however is not defined. The binding affinity of Rab18 to NS5A depends on the loading with GTP or GDP. The GTP-complexed Rab18 binds with higher affinity to NS5A. Rab18 is found on the surface of LDs and is assumed to promote the association of NS5A with LDs (Salloum et al., 2013). Apart from this there seems to be direct effects of Rab18 on HCV replication and release. The silencing of Rab18 expression decreases HCV replication. The overexpression of Rab18 enhances the release of HCV infectious particles, but does not affect the replication (Salloum et al., 2013; Dansako et al., 2014). From this it can be concluded that the intracellular amount of Rab18 is not limiting for HCV replication. In light of the ability of Rab18 to approximate ER membranes to LDs it is assumed that this contributes to tether NS5A-positive membranes to LDs. In accordance to this it was observed that silencing the Rab18 expression leads to a significant reduction of the proportion of LDs that are poasitive for NS5A and NS3. The relevance of Rab18 for the trafficking of core to the LDs remains unclear (Salloum et al., 2013; Dansako et al., 2014). The capacity of Rab18 to approximate ER membranes to LDs could be relevant for the formation of the MW.
Rab27a
The HCV genome is stabilized by binding of two molecules of the liver-specific miR-122 that represents a crucial host factor for HCV replication (Jopling et al., 2008; Machlin et al., 2011). Rab27a is known to modulate exosome release by docking MVBs to the PM, an aspect relevant for the release of a variety of viruses including HEV and HCV (Ostrowski et al., 2010; Nagashima et al., 2014; Elgner et al., 2016). But in addition to its effect on HCV release, Rab27a (see chapter release) affects HCV replication (Chen et al., 2015). It was observed that a decreased amount of Rab27a leads to decreased amount of miR-122 most likely by decreasing an abundance of pre-miR-122. The straight forward explanation would be that the loss of miR-122 leads to HCV RNA destabilization, but the replication of HCV genomes harboring stabilizing mutations were affected as well (Sedano and Sarnow, 2014). This indicates that the interplay between Rab27a depletion and decreased amount of intracellular HCV-RNA is more complex and does not exclusively depend on the formation of miR-122/HCV complexes. Although there is no direct experimental evidence, it is speculated that miR-122 has a Rab27a dependent function on the HCV life cycle. It is assumed that this occurs by downregulation of a cellular inhibitor that affects HCV gene expression (Chen et al., 2015).
HCV Morphogenesis
Infection with the HCV leads to an altered lipid metabolism and rearrangement of intracellular membranes to establish a microenvironment for efficient replication and assembly. The resulting so called “membranous web” (MW) consists of endoplasmic reticulum (ER)-derived DMVs with an average diameter of 150 nm embedded in a membranous matrix (Paul et al., 2014). DMVs are complex structures composed of two tightly associated lipid bilayers highly enriched in cholesterol. However, it is not yet known how the DMVs are formed in HCV infected cells. Due to their similarity to autophagosomes it is assumed that their biogenesis is linked to the autophagosomal pathway. The formation of the MW depends on the interplay of viral and ER-resident proteins. In this regard, the ER-associated GTPases Rab18 and Rab32 are found to be involved in membrane remodeling by tethering LDs toward ER-derived structures. HCV replication mainly occurs in DMVs that contain active viral RNA replicase complexes (Paul et al., 2013), whereas particle assembly takes place in close proximity in detergent-resistant membranes (DRMs) of the ER or mitochondrial-associated ER membranes (MAMs) that are enriched in cholesterol and sphingolipids (Horner et al., 2011, 2015; Shanmugam et al., 2015; Falcón et al., 2017). Cholesterol de novo synthesis occurs at the ER, although it shows a low cholesterol carrying capacity. To enrich ER-derived membranes in cholesterol, HCV hijacks the oxysterol binding protein (OSBP)-mediated phosphatidylinositol-4-phosphate (PI4P)-cholesterol exchange or utilizes other lipid transfer proteins, e.g., the Niemann-Pick type C1 protein (de Saint-Jean et al., 2011; Stoeck et al., 2018). Replication and assembly sites are linked via cytosolic lipid droplets (cLDs)—storage organelles for neutral lipids—that act as assembly platform. However, it is not defined whether the assembly proceeds on cLDs itself or on closely associated ER membranes. Particle assembly is a multistep process that requires the crosstalk and concerted action of viral and ER-derived proteins/factors. During the assembly process the newly synthesized viral RNA has to be transferred from the replication complex (RC) to LD-associated HCV Core proteins, followed by nucleocapsid formation and envelopment that occurs by budding into the ER lumen. In conclusion, the formation of the MW enables the virus to establish a spatio-temporal replication and assembly without being recognized by the immune system. In the following we will highlight Rab proteins that participate in HCV particle assembly.
Rab18
Using Stable Isotope Labeling by Amino acids in Cell culture (SILAC) in combination with mass spectrometry analysis Salloum et al. could identify the LD-associated small Ras-related GTPase Rab18 to interact with NS5A in HCV Jc1-infected Huh7.5.1 cells (Salloum et al., 2013). NS5A is an RNA-binding phosphoprotein that controls the switch between replication and assembly (Zayas et al., 2016; Hsu et al., 2017). It consists of a N-terminal amphipathic α-helix that mediates its membrane association (Brass et al., 2002) and three domains that are separated by low complexity sequences (Tellinghuisen et al., 2004). Domain I and II are mainly involved in RNA replication, whereas domain III participates in virion assembly. Domain III itself contains two regulatory elements: an N-terminal basic cluster that mediates the transfer of the viral RNA to the HCV Core protein and a C-terminal serine cluster essential for transport of the RC to the HCV Core protein (Zayas et al., 2016). Besides, it has been reported that hyperphosphorylation of NS5A domain III by casein kinase 1 (CK1) targets NS5A to cLDs (Tellinghuisen et al., 2008). Rab18 has been identified to be associated with LDs (Martin et al., 2005). In line with this, it is involved in the growth and maturation of LDs through enabling the exchange of lipids between specialized ER regions and tightly associated LDs in so called LD-associated membranes (LAMs) (Ozeki et al., 2005; Xu et al., 2018). Furthermore, it has been linked to ER-Golgi trafficking (Dejgaard et al., 2008). Recently TRAPPII (TRAnsport Protein Particle) has been identified as LD-associated Rab18 GEF. TRAPPII is targeted toward LDs via COPI where it activates Rab18 (Li et al., 2017; Zappa et al., 2017). In addition, the Rab3GAP complex is a Rab18 GEF involved in LD- and ER-targeting of Rab18 thereby controlling ER dynamics (Ozeki et al., 2005; Gerondopoulos et al., 2014; Xu et al., 2018). Mutations in Rab18 and the Rab3GAP complex were found in the autosomal-recessive disorder Warburg Micro syndrome that is characterized by severe developmental abnormalities in the brain and eye, developmental delay, and neurodegeneration (Bem et al., 2011). Moreover, Rab18 impairs the transport of secretory granules in neuroendocrine cells (Vazquez-Martinez et al., 2007) and plays an important role in insulin-mediated lipogenesis and ß-adrenergic-induced lipolysis (Pulido et al., 2011). In hepatocytes Rab18 triggers the association of ApoB100 with LDs after accumulation of free toxic cholesterol (Makino et al., 2016) and links retinoid and cholesterol metabolism thereby mediating hepatic stellate cell activation (O'Mahony et al., 2015). Furthermore, Rab18 enhances HBx-mediated hepatocarcinogenesis through impaired lipogenesis and proliferation (You et al., 2013).
In HCV Jc1-infected Huh7.5.1 cells the direct interaction of NS5A with GTP-Rab18 (activated) promotes the interaction of the MW with LDs thereby connecting sites of replication with sites of assembly. In adipocytes the recruitment of Rab18 toward LDs is mediated via insulin induced activation of PI3K (Pulido et al., 2011). PI3Ks are major effectors of G-protein coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs). Activation of PI3K by growth factors or cytokines generates intracellular phospholipids that in turn activate AKT and other downstream pathways (Liu et al., 2009). HCV NS5A has been reported to bind to and activate the PI3K-AKT pathway. Moreover, Liu et al. reported that HCV transiently activates the PI3K-AKT pathway early after infection to favor viral entry (Street et al., 2005; Liu et al., 2012). Based on these findings, it is tempting to specutlate that HCV might trigger translocation of Rab18 to the surface of LDs through activation of the PI3K-AKT pathway. Using immunoelectron microscopy of HCV replicating cells overexpressing Rab18, the engulfment of thin membrane cisternae around LDs could be detected (Salloum et al., 2013). Rab18 was found to co-localize with NS5A on the surface of LDs. Overexpression of Rab18 resulted in increased amounts of infectious viral particles without affecting viral replication. However, after silencing of Rab18 or expression of a dominant-negative (dn) Rab18 less NS5A as well as the RC components, NS3, and viral RNA could be detected on LDs. In addition, silencing of Rab18 results in decreased amounts of intra- and extracellular viral titers and inhibited viral replication (Salloum et al., 2013). As silencing of Rab18 affects viral replication and assembly it is hard to speculate whether Rab18 indeed affects particle assembly. In line with this, silencing of the LD-associated protein TIP47 similarly inhibits viral replication and release of viral particles. TIP47 binds NS5A and targets the de novo synthesized viral RNA to the surface of LDs allowing its interaction with the HCV Core protein, a crucial step during particle assembly (Ploen et al., 2013a). Moreover, interaction of TIP47 with Rab9 is essential for proper release of viral particles (Ploen et al., 2013b) (for more detailed information see chapters replication and release).
For efficient particle assembly the HCV Core protein itself has to be transferred to LDs. During this process the Core protein further recruits NS proteins to the LDs (Miyanari et al., 2007). The HCV Core protein is composed of three domains whereof two proline residues (aa 138, 143) and a YATG-motif in domain II are responsible for its association with LDs (Hope et al., 2002). Association of the HCV Core protein with LDs displaces the LD-associated PAT-protein PLIN2 (or ADRP—adipocyte differentiation-related protein) from LDs and induces their apposition toward the nucleus and ER-derived membranes (Boulant et al., 2008; Counihan et al., 2011). In line with this, overexpression of Rab18 in HepG2 and Balb/C 3T3 cells reduces the amount of PLIN2 on LDs and triggers the encounter of LDs and ER-derived membranes (Ozeki et al., 2005). Silencing of Rab18 in HCV JFH1-infected RSC cells resulted in decreased intra- and extracellular infectious viral particles with no effect on viral replication, which is in contrast to the observations of Salloum et al. Moreover, in immunofluorescence analyses silencing of Rab18 abrogated its co-localization with HCV Core on the LDs and less Core protein was detected around LDs corroborating the role of Rab18 for Core trafficking to LDs. In addition, overexpression of Rab18 increased the production of infectious viral particles two-fold (Dansako et al., 2014). As the observations of both groups differ, the authors explained the contradictory results due to the fact that Salloum et al. used the chimeric HCV Jc1 isolate and different Huh7 subclones. It has been reported that the HCV Jc1 Core protein does not localize to LDs (Shavinskaya et al., 2007) supporting the data of Salloum et al. that could not observe an involvement of Rab18 in HCV Core trafficking.
Taken together, these studies highlight the important role of the small GTPase Rab18 in viral morphogenesis by connecting viral replication and assembly through tethering LDs with ER-associated membranes and trafficking of the HCV Core protein and NS5A from sites of replication toward assembly sites.
Rab32
Another Rab protein involved in HCV assembly and release is the small GTPase Rab32. In murine melanocytes Rab32 is involved in post-Golgi trafficking of enzymes involved in melanogenesis (Wasmeier et al., 2006). In Xenopus melanophores Rab32 has been identified as a melanosome-specific A-kinase anchoring protein (AKAP) that modulates transport of melanosomes by tethering the cAMP dependent PKA toward melanosomes (Park et al., 2007). In non-melanogenic cells Rab32 is localized to ER-membranes and mitochondria and organizes TGN and mitochondria dynamics including mitochondria fission (Alto et al., 2002). Bui et al. reported that ER-resident Rab32 controls Ca2+-signaling and modulates MAM-specific characteristics. Moreover, Rab32 tethers PKA to ER and mitochondrial membranes and thereby indirectly regulates the process of programmed cell death via PKA-dependent phosphorylation of enzymes involved in apoptosis (Bui et al., 2010). In addition, ER-associated GTP-Rab32 is involved in phagosome maturation in HeLa and Cos7 cells (Hirota and Tanaka, 2009), intracellular lipid accumulation (Li et al., 2016), and activation of iPSCs (induced pluripotent stem cells) through elevated lipogenesis (Pei et al., 2015), brain inflammation (Liang et al., 2012), and is involved in post-Golgi sorting and transport of LRRK2 (leucine-rich repeat kinase 2) (Waschbüsch et al., 2014) and release of viral particles (Mankouri et al., 2016).
Analysis of the host transcriptome of HCVcc (cell culture grown)-infected Huh7 cells using high-throughput next-generation sequencing (NGS) Pham et al. found 30 genes to be upregulated, including Rab32. In HCV infected cells Rab32 is upregulated on the expression and transcriptional level probably through involvement of HCV NS3 (Pham et al., 2017). Overexpression of Rab32 induces the formation of autophagosomes (Hirota and Tanaka, 2009). As already mentioned above HCV infection results in activation of autophagy via elevated ROS levels and consequent phosphorylation of p62. In line with this, DMVs biogenesis is linked to the autophagosomal pathway corroborating the importance of Rab32 for HCV morphogenesis and the associated rearrangement of ER-derived membranes/MW formation. Interestingly, a shift of GTP-Rab32 to GDP-Rab32 can be observed in HCV-infected cells resulting in intracellular accumulation of GDP-Rab32 making it less susceptible toward degradation. GDP-Rab32 preferentially binds to the HCV Core protein targeting Core toward MAMs (mitochondria-associated membranes) in the perinuclear region where HCV particle assembly takes place. Moreover, silencing of Rab32 resulted in the production of less infectious particles further corroborating its role during assembly and release (see chapter release). Taken together, Rab proteins are involved in membrane dynamics through tethering proteins involved in membrane fusion/fission to their target membranes and finally control intracellular trafficking processes. They are found on various cellular organelles including ER, Golgi, mitochondria, endosomes, and intracellular vesicles (Takai et al., 2001). HCV particle assembly/morphogenesis is still not fully understood. HCV infection is accompanied by a massive rearrangement of intracellular membranes. In line with this, the Rab GTPases Rab18 and Rab32 are found to be involved in HCV particle assembly by tethering LDs toward ER-derived membranes and facilitating the trafficking of HCV Core and NS5A toward membrane structures involved in particle assembly (Table 3).
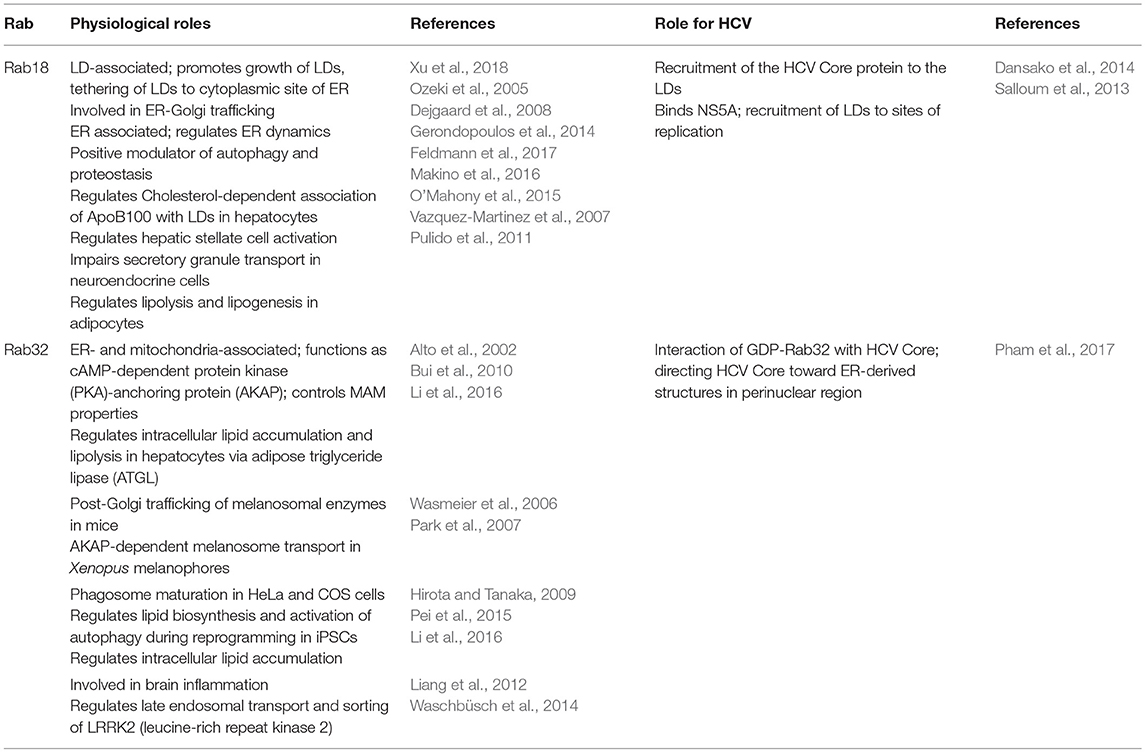
Table 3. Rab proteins involved in HCV morphogenesis.
Release
The egress of infectious HCV particles remains enigmatic. Because of their high similarity to VLDLs regarding morphological and physicochemical properties and the incorporation of lipids and lipoproteins as ApoE and ApoB100, HCV virions are proposed to be LVPs (Catanese et al., 2013). This close functional relationship has led to the assumption that HCV virions may use the canonical secretory pathway to leave the host cells just as VLDLs (Moradpour et al., 2007). However, this hypothesis has never been validated. Quite contrary, recent studies describe differences in the post-ER transport and release between HCV virions and lipoproteins, even though lipoproteins are essential for the proper assembly of the LVPs (Chang et al., 2007; Cun et al., 2010; Bayer et al., 2016; Takacs et al., 2017). Moreover, several key components of the endosomal transport system have been described to play essential roles in the egress of HCV particles, including SNAREs, ESCRT components, and several Rab proteins (Corless et al., 2010; Ariumi et al., 2011; Tamai et al., 2012; Ren et al., 2016, 2017). The later will be discussed here in more detail.
Rab3d and Rab11a
Using an siRNA-screen Coller et al. identified multiple components of the secretory pathway to be involved in the secretion of HCV virions including proteins required for Golgi trafficking, Golgi sorting, and TGN budding (Coller et al., 2012). Remarkably, knockdown of the exocytotic GTPases Rab3d and Rab11a, which regulate post-TGN to PM transport, strongly decreased the amount of released viral particles. Rab3d is not well-characterized but it has been described to regulate the exocytosis of vesicle cargoes in non-neuronal cells (Millar et al., 2002). It remains unclear, whether this exocytosis represents the release of exosomes by fusion of MVBs with the PM which came into focus just in the past years. Since Rab11a is a classical marker for recycling endosomes (REs), the authors state that trafficking of HCV particles include these sorting organelles (Coller et al., 2012). This is further corroborated by live cell imaging analyses visualizing the HCV Core protein co-trafficking with Rab11a. Knockdown of Rab11a resulted in the accumulation of the Core protein in the Golgi compartment, explaining on the one hand the decreased virion egress and on the other hand validating the involvement of post-Golgi REs in the release route. These results led to the hypothesis, that HCV particles leave the host cell via the secretory pathway, starting at the ER followed by the Golgi, budding from the TGN, and finally the transport via REs to the PM (Coller et al., 2012). Interestingly, the role of Rab11a in the regulation of MVB transport and exosome secretion in human K562 cells and Drosophila melanogaster has been described by several groups (Savina et al., 2002, 2005; Fader et al., 2008; Koles et al., 2012). However, whether this function of Rab11a in addition contributes to the proper release of HCV particles has not been discussed by the authors.
Rab9
With the generation of an E1-mCherry tagged HCV genome, Bayer et al. were able to observe a co-trafficking of the envelope protein E1 with Rab9 (Bayer et al., 2016). Using Fluorescence Recovery After Photobleaching (FRAP) experiments it was confirmed that E1 is present inside membrane enclosed Rab9-positive vesicles, since E1-mCherry fluorescence was not able to recover after bleaching in Rab9-positive loci. The presence of a structural protein does not directly prove the presence of infectious viral particles. However, since there is no regulatory role of E1 known, it can be assumed, that the majority of trafficking E1 resembles assembled HCV virions. A role of Rab9 in the release of infectious particles was further corroborated by recent work from our group. We mutated or deleted the Rab9-binding domains of the intracellular sorting factor TIP47 to inhibit the interaction of these proteins while keeping functional Rab9 available. Besides, TIP47 is a LD-associated protein located on the surface of LDs. In accordance with its role as a sorting factor we could identify that TIP47 recruits the viral RNA bound to NS5A to the surface of LDs contributing to HCV particle assembly, thereby getting incorporated into the viral particle itself. Impairment of the TIP47-Rab9 interaction by expression of the above mentioned mutants did not affect the viral replication but strongly decreased the egress of infectious particles. Moreover, the HCV Core protein accumulates in lysosomal structures (Ploen et al., 2013b). Taken together, these findings highlight a role of Rab9 in concert with TIP47 in the sorting of infectious HCV particles between their release and lysosomal degradation.
Rab7
Several studies documented the sorting of HCV particles for either lysosomal degradation or release from the host cell (Elgner et al., 2016; Ren et al., 2016). Wozniak et al. found that the amount of the Rab7-dynein adapter Rab-interacting lysosomal protein (RILP) is heavily decreased in HCV infected cells (Wozniak et al., 2016). This comes along with a dramatically shorter half-life of RILP (7 h in infected cells, whereas 62 h in the control cells) and the appearance of a cleavage product of RILP (cRILP) which is missing the dynein-binding motif. The exact mechanism of RILP cleavage remains unclear, but it seems not to be a consequence of apoptosis, ER stress or innate immunity. Interestingly, inhibition of c-Jun N-terminal kinase (JNK) blocks the formation of cRILP also in control cells leading to the hypothesis that RILP cleavage is an intrinsic cellular process. After electroporation of the HCV genome into Huh7.5 cells the virus mainly replicates and assembles, but the release is at a low level for 3 days. At day 5 post electroporation (dpe) the cells convert into an efficient virion secretion state which directly correlates with the appearance of cRILP. The expression of a non-cleavable RILP (ncRILP) 5 dpe reduced the amount of released virions by 60% compared to control cells, whereas cRILP had no effect. In contrast to this, the expression of cRILP strongly increased the amount of released viral particles 3 dpe, but ncRILP had no effect at this time point. This suggests that the cleavage of RILP renders the host cell to be an efficient virion secreting cell. Overexpression of cRILP leads to a delayed vesicular trafficking to lysosomes compared to ncRILP, which is localized perinuclear in vesicular structures. Inhibition of kinesins abolished the cRILP effect and rescued intracellular virion levels back to control values. This indicates that cRILP renders Rab7-containing vesicles to bind to kinesins which traffic anterograde to the cell periphery to execute virion release. In contrast, full length RILP promotes binding of Rab7-containing vesicles to dyneins moving retrograde to fuse with lysosomes ending in the degradation of the cargo (Wozniak et al., 2016). This study is an example of how viruses exploit also Rab effector proteins to modify Rab protein functions for their benefit.
Rab7 is involved in the maturation of early to late endosomes and regulates the fusion of the later with autophagosomes and lysosomes (Rink et al., 2005; Vanlandingham and Ceresa, 2009). Rab7-positive vesicles were also observed to co-traffick with the above mentioned E1-mCherry HCV virus in live-cell imaging studies corroborating the role of Rab7 in the release of infectious particles (Bayer et al., 2016). In addition, our group observed in a recent study that treatment of HCV positive cells with the substance U18666A leads to the formation of large multilaminar bodies (MLBs) positive for Rab7, the lysosomal protein LAMP2 and the tetraspanin CD63 which is a common exosome marker (Elgner et al., 2016). U18666A is described as MVB/cholesterol-transport inhibitor and fulfills this function by direct inhibition of the cholesterol transporter NPC1 which is resident in the MVB membrane (Appelqvist et al., 2011). Inhibition of NPC1 by U18666A results in an accumulation of cholesterol in these organelles, sensed by ORP1 resulting in a dynein-dependent transport of MVBs to lysosomes (Rocha et al., 2009). The fusion of lysosomes with cholesterol overloaded MVBs triggers the formation of lysosomal storage compartments, MLBs, which are not degradation competent. Interestingly, MLBs were also found to include E1, ApoE, and neutral lipids highlighting the role of Rab7-positive MVBs in the egress of HCV VLPs (Elgner et al., 2016).
Rab27a
Regarding the impact of Rab7a and Rab11a, there is raising evidence, that HCV particles might use an exosome-like release pathway. Indeed, knockdown of the exosome controlling Rab27a by Shrivastava et al. reduced the amount of released exosomes and HCV virions. As knockdown of Rab27a also diminished HCV replication, no direct influence of Rab27a knockdown on the release of HCV particles should be stated here (Shrivastava et al., 2016).
Rab1b
In a lentiviral large-scale screening including 62 dominant negative (dn) Rab mutants Takac et al. observed that dn versions of Rab35, Rab8a, Rab13, and Rab43 reduced the secretion of ApoE, ApoB100 and albumin whilst dn mutants Rab8b, Rab11a, Rab11b, and Rab12 augment the secretion of these proteins (Takacs et al., 2017). Most interestingly dnRab1b reduced the secretion of ApoB100 and stimulated that of ApoE whilst dnRab23 inhibited secretion of albumin and ApoB100 and stimulated ApoE release. These differential results suggest that the investigated cargoes use distinct routes to leave the cell. The authors focused on the characterization of Rab1b and generated two mutants of the protein. Rab1bS22N is locked in its inactive GDP-bound form and RabN121I is unable to bind nucleotides. Therefore, both mutants are potent ER-to-Golgi transport inhibitors. RabN121I impaired the secretion of all three tested cargoes, confirming that their secretion involves Rab1b function. The mutant Rab1S22N inhibits the secretion of albumin and ApoB100, but not that of ApoE and the same phenotype is seen by Rab1b inhibition by the bacterial protein DrrA. This difference can be explained by the fact that Rab1b functions at several stages of ER-to-Golgi transport: ApoB100 and albumin may exit the ER in a Rab1b-dependent process whereas ApoE does not require Rab1b to leave the ER (Plutner, 1991; Schwaninger et al., 1991). In a later step, during ER-to-Golgi transport, all three cargos may depend on Rab1b function to reach the Golgi. To investigate the role of the Rab1b mutants in the release of HCV particles, a TetON system was used, avoiding proposed Rab1 effects on the viral replication. The expression of the Rab1b inhibitors was induced 2 dpe, when the viral replication machinery has been established. The expression of DrrA, Rab1bS22N, and Rab1bN122I resulted in ER-retention and thus an impaired release of infectious viral particles. This reflects the effect of the Rab1b inhibitors on the secretion of ApoB100 but not that on ApoE, depicting that HCV particles rely on the early function of Rab1b facilitating ER exit. Therefore, despite the correct assembly and infectivity of HCV virions depends on ApoE, they use distinct routes that are differentially regulated release pathways (Takacs et al., 2017).
Exocytotic Rabs
Another screen with dn mutants of several Rab proteins identified the secretory/exocytotic Rab proteins Rab8b, 13, 23, 27, 32, and 33 to be required for HCV particle release, whereas the endocytotic/recycling GTPases Rab7, 9, 11, and 35 did not seem to play a role in virion egress (Mankouri et al., 2016). This is in contrast to previous studies, as discussed above. The authors explain the different observations due to the fact that they use the original JFH-1 isolate which poorly replicates and releases infectious HCV particles compared to the chimeric virus Jc1 used in the other studies. However, according to the post-TGN-transport functions of the above mentioned Rab proteins (Rab8b, 13, 23, 27, 32, and 33), the authors claimed that assembled HCV particles transit the Golgi compartment on the way to the defined secretory endosomal pathway. Remarkably, none of the dnRab proteins impaired the release of ApoB and ApoE further contributing to the theory that HCV virions and lipoproteins leave the cell by distinct routes, suggesting a post-release association of VLDLs and HCV particles, rather than the formation and secretion of a hybrid LVP.
Taken together, since several years there is growing evidence that HCV is using an endosomal pathway for the release of infectious particles rather than the canonical secretion pathway. In addition, this HCV-release pathway might share some commonalities with the secretion pathway of VLDLs, including the lipoproteins ApoE and ApoB. HCV exploits several Rab proteins and Rab effector proteins to establish or modulate this pathway for its own benefit as summarized in Table 4. Currently based on the above described data, a novel model pathway could be established including the transport of HCV virions from the ER to the Golgi compartment (Rab1b) followed by transport of the virions through the Golgi compartment and TGN (Rab8b, 13, 23, 32, 33) into REs (Rab11a) and finally to late endosomes (Rab7a). In this sorting platform the virions may be sorted for either lysosomal degradation or release by fusion of the MVB membrane with the PM (RILP, Rab7a, Rab9a) which occurs in parallel to the release of exosomes (Rab3d, Rab27a). The Golgi compartment may represent a crossing point for the HCV-release pathway and the canonical pathway where the HCV virions and the VLDLs might get in contact to form the proposed LVPs. In addition, an HCV egress pathway completely independent from the Golgi compartment has been proposed (Bayer et al., 2016). Whether this Golgi-bypass is an additional- or the main viral release pathway needs further investigation.
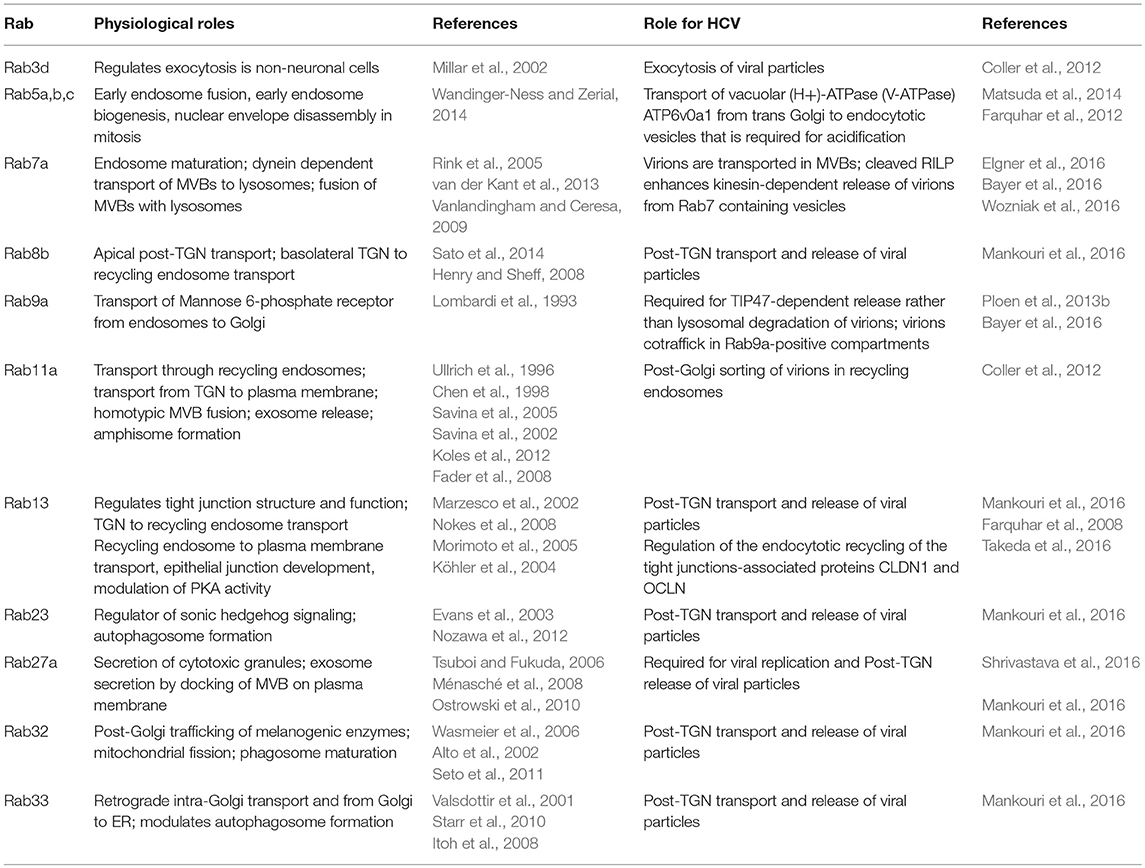
Table 4. Rab proteins involved in HCV release.
Conclusions and Future Perspectives
Although there has been a tremendous increase in our knowledge about HCV, mainly based on the establishment of subgenomic (Lohmann et al., 1999) and full length genomic (Blight et al., 2002; Wakita et al., 2005; Zhong et al., 2005) replication systems, there are still a variety of open questions regarding the HCV life cycle. This concerns the understanding of the entry process and characterization of the fusion process or the release of HCV with respect to the relevance of MVB-dependent processes and the release in form of exosomal vesicles. It should be considered that the biggest part of our knowledge is based on the use of immortalized hepatoma cell lines and a very limited number of isolates. In light of this, the comparative analysis of various genotypes with respect to their dependency on Rab-mediated processes and the use of primary hepatocytes will be challenging. Our present view on the molecular virology of HCV is in many aspects a static view i.e., based on the analysis of fixed cells. Just with respect to the central role of RabGTPases in controlling trafficking processes, live cell imaging to investigate the spatiotemporal regulation of the HCV life cycle will contribute to a deeper understanding of the pathogen host interaction.
In light of the central role of RabGTPases for a variety of cellular processes targeting of Rab GTPase to control HCV replication seems not to be a very promising approach since a variety of side effects can be expected. Strategies directly affecting the interaction between viral proteins and Rab GTPases e.g., by the development of small molecules or competitive peptides that are pathogen specific will be more straightforward. However, different strategies to target GTPase activity have been under investigation. One approach is to address post-translational modifications by inhibition of lipid transferases (farnesyl- and geranylgeranyltransferases) that tether GTPases toward their target membranes. Albeit, clinical trials reported about toxicities of these compounds making them less attractive for therapeutical applications (Konstantinopoulos et al., 2007). Moreover, inhibition of the mevalonate pathway interferes with the biosynthesis of isoprenoids [farnesylpyrophosphate (FPP) and geranylgeranylpyrophate (GGPP)], which are substrates for prenylation, and cholesterol synthesis. In context of HCV, statins, inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase in the mevalonate pathway, inhibit HCV replication, and were reported with a lower risk of HCC (El-Serag et al., 2009) and lower portal pressure in patients with cirrhosis (Abraldes et al., 2009). Treatment with statins inhibits the geranylgeranylation of host proteins that play a crucial role during the HCV life-cycle via inhibition of GGPP and not cholesterol (Ikeda et al., 2006, 2011). Another possibility to modulate GTPase activity is to regulate their function by addressing GAPs, GEFs, and GDIs. Recently, the small molecule pan-GTPase inhibitor CID1067700 has been identified to inhibit multiple GTPases (Agola et al., 2012; Hong et al., 2015). However, the specific application of inhibitors and their progress in clinical trials is limited. In addition, the small globular structure of GTPases and their high affinity toward ligands depicts GTPases as difficult target for inhibtors (for more detailed information see Konstantinopoulos et al., 2007).
HCV belongs to the family of flaviviridae that encompasses a variety of emerging viruses like ZIKV or WNV. Thus it will be interesting to see whether insights obtained for HCV can be transferred to other flaviviruses and allow the development of antivirals with a “pan-flavivirus” spectrum.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. The authors contributed equally to all sections of this work. EH focused on HCV entry and replication, DB on morphogenesis, and FE on release. DB illustrated the figure.
Funding
DB and EH obtained funding by the LOEWE Center DRUID (Novel Drug Targets against Poverty-Related and Neglected Tropical Infectious Diseases).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraldes, J. G., Albillos, A., Bañares, R., Turnes, J., González, R., García-Pagán, J. C., et al. (2009). Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension. A randomized controlled trial. Gastroenterology 136, 1651–1658. doi: 10.1053/j.gastro.2009.01.043
Acton, S., Rigotti, A., Landschulz, K. T., Xu, S., Hobbs, H. H., and Krieger, M. (1996). Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520. doi: 10.1126/science.271.5248.518
Agola, J. O., Hong, L., Surviladze, Z., Ursu, O., Waller, A., Strouse, J. J., et al. (2012). A competitive nucleotide binding inhibitor. in vitro characterization of Rab7 GTPase inhibition. ACS Chem. Biol. 7, 1095–1108. doi: 10.1021/cb3001099
Allan, B. B., Moyer, B. D., and Balch, W. E. (2000). Rab1 recruitment of p115 into a cis-SNARE complex. Programming budding COPII vesicles for fusion. Science 289, 444–448. doi: 10.1126/science.289.5478.444
Alto, N. M., Soderling, J., and Scott, J. D. (2002). Rab32 is an A-kinase anchoring protein and participates in mitochondrial dynamics. J. Cell Biol. 158, 659–668. doi: 10.1083/jcb.200204081
Ao, X., Zou, L., and Wu, Y. (2014). Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 21, 348–358. doi: 10.1038/cdd.2013.187
Appelqvist, H., Nilsson, C., Garner, B., Brown, A. J., Kågedal, K., and Ollinger, K. (2011). Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Amer. J. Pathol. 178, 629–639. doi: 10.1016/j.ajpath.2010.10.030
Ariumi, Y., Kuroki, M., Maki, M., Ikeda, M., Dansako, H., Wakita, T., and Kato, N. (2011). The ESCRT system is required for hepatitis C virus production. PLoS ONE 6:e14517. doi: 10.1371/journal.pone.0014517
Bayer, K., Banning, C., Bruss, V., Wiltzer-Bach, L., and Schindler, M. (2016). Hepatitis C virus is released via a non-canonical secretory route. J. Virol. 90, 10558–10573. doi: 10.1128/JVI.01615-16
Bem, D., Yoshimura, S., Nunes-Bastos, R., Bond, F. C., Bond, F. F., Kurian, M. A., et al. (2011). Loss-of-function mutations in RAB18 cause Warburg micro syndrome. Am. J. Hum. Genet. 88, 499–507. doi: 10.1016/j.ajhg.2011.03.012
Blanchard, E., Belouzard, S., Goueslain, L., Wakita, T., Dubuisson, J., Wychowski, C., et al. (2006). Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 80, 6964–6972. doi: 10.1128/JVI.00024-06
Blight, K. J., McKeating, J. A., and Rice, C. M. (2002). Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76, 13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002
Boulant, S., Douglas, M. W., Moody, L., Budkowska, A., Targett-Adams, P., and McLauchlan, J. (2008). Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 9, 1268–1282. doi: 10.1111/j.1600-0854.2008.00767.x
Brass, V., Bieck, E., Montserret, R., Wölk, B., Hellings, J. A., Blum, H. E., et al. (2002). An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277, 8130–8139. doi: 10.1074/jbc.M111289200
Bui, M., Gilady, S. Y., Fitzsimmons, R. E. B., Benson, M. D., Lynes, E. M., Gesson, K., et al. (2010). Rab32 modulates apoptosis onset and mitochondria-associated membrane (MAM) properties. J. Biol. Chem. 285, 31590–31602. doi: 10.1074/jbc.M110.101584
Catanese, M. T., Uryu, K., Kopp, M., Edwards, T. J., Andrus, L., Rice, W. J., et al. (2013). Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. U.S.A. 110, 9505–9510. doi: 10.1073/pnas.1307527110
Chang, K.-S., Jiang, J., Cai, Z., and Luo, G. (2007). Human apolipoprotein e is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81, 13783–13793. doi: 10.1128/JVI.01091-07
Chen, T.-C., Hsieh, C.-H., and Sarnow, P. (2015). Supporting role for GTPase Rab27a in hepatitis C virus RNA replication through a novel miR-122-mediated effect. PLoS Pathog. 11:e1005116. doi: 10.1371/journal.ppat.1005116
Chen, W., Feng, Y., Chen, D., and Wandinger-Ness, A. (1998). Rab11 is required for trans-golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol. Biol. Cell 9, 3241–3257. doi: 10.1091/mbc.9.11.3241
Coller, K. E., Heaton, N. S., Berger, K. L., Cooper, J. D., Saunders, J. L., and Randall, G. (2012). Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 8:e1002466. doi: 10.1371/journal.ppat.1002466
Corless, L., Crump, C. M., Griffin, S. D. C., and Harris, M. (2010). Vps4 and the ESCRT-III complex are required for the release of infectious hepatitis C virus particles. J. Gen. Virol. 91(Pt 2), 362–372. doi: 10.1099/vir.0.017285-0
Counihan, N. A., Rawlinson, S. M., and Lindenbach, B. D. (2011). Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 7:e1002302. doi: 10.1371/journal.ppat.1002302
Cun, W., Jiang, J., and Luo, G. (2010). The C-terminal alpha-helix domain of apolipoprotein E is required for interaction with nonstructural protein 5A and assembly of hepatitis C virus. J. Virol. 84, 11532–11541. doi: 10.1128/JVI.01021-10
da Silva Filipe, A., Sreenu, V., Hughes, J., Aranday-Cortes, E., Irving, W. L., Foster, G. R., et al. (2017). Response to DAA therapy in the NHS England early access programme for rare HCV subtypes from low and middle income countries. J. Hepatol. 67, 1348–1350. doi: 10.1016/j.jhep.2017.06.035
Dansako, H., Hiramoto, H., Ikeda, M., Wakita, T., and Kato, N. (2014). Rab18 is required for viral assembly of hepatitis C virus through trafficking of the core protein to lipid droplets. Virology 462–463, 166–174. doi: 10.1016/j.virol.2014.05.017
de Saint-Jean, M., Delfosse, V., Douguet, D., Chicanne, G., Payrastre, B., Bourguet, W., et al. (2011). Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J. Cell Biol. 195, 965–978. doi: 10.1083/jcb.201104062
Dejgaard, S. Y., Murshid, A., Erman, A., Kizilay, O., Verbich, D., Lodge, R., et al. (2008). Rab18 and Rab43 have key roles in ER-Golgi trafficking. J. Cell Sci. 121 (Pt 16), 2768–2781. doi: 10.1242/jcs.021808
Devhare, P. B., Sasaki, R., Shrivastava, S., Di Bisceglie, A. M., Ray, R., and Ray, R. B. (2017). Exosome mediated intercellular communication between hepatitis C virus infected hepatocytes and hepatic stellate cells. J. Virol. 91:e02225-16. doi: 10.1128/JVI.02225-16
Eathiraj, S., Pan, X., Ritacco, C., and Lambright, D. G. (2005). Structural basis of family-wide Rab GTPase recognition by rabenosyn-5. Nature 436, 415–419. doi: 10.1038/nature03798
Elgner, F., Ren, H., Medvedev, R., Ploen, D., Himmelsbach, K., Boller, K., et al. (2016). The intra-cellular cholesterol transport inhibitor U18666A inhibits the exosome-dependent release of mature hepatitis C virus. J. Virol. 90, 11181–11196. doi: 10.1128/JVI.01053-16
El-Serag, H. B., Johnson, M. L., Hachem, C., and Morgana, R. O. (2009). Statins are associated with a reduced risk of hepatocellular carcinoma in a large cohort of patients with diabetes. Gastroenterology 136, 1601–1608. doi: 10.1053/j.gastro.2009.01.053
Evans, M. J., von Hahn, T., Tscherne, D. M., Syder, A. J., Panis, M., Wölk, B., et al. (2007). Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805. doi: 10.1038/nature05654
Evans, T. M., Ferguson, C., Wainwright, B. J., Parton, R. G., and Wicking, C. (2003). Rab23, a negative regulator of hedgehog signaling, localizes to the plasma membrane and the endocytic pathway. Traffic 4, 869–884. doi: 10.1046/j.1600-0854.2003.00141.x
Fader, C. M., Sanchez, D., Furlan, M., and Colombo, M. I. (2008). Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 9, 230–250. doi: 10.1111/j.1600-0854.2007.00677.x
Falcón, V., Acosta-Rivero, N., González, S., Dueñas-Carrera, S., Martinez-Donato, G., Menéndez, I., et al. (2017). Ultrastructural and biochemical basis for hepatitis C virus morphogenesis. Virus Genes 53, 151–164. doi: 10.1007/s11262-017-1426-2
Farquhar, M. J., Harris, H. J., Diskar, M., Jones, S., Mee, C. J., Nielsen, S. U., et al. (2008). Protein kinase A-dependent step(s) in hepatitis C virus entry and infectivity. J. Virol. 82, 8797–8811. doi: 10.1128/JVI.00592-08
Farquhar, M. J., Hu, K., Harris, H. J., Davis, C., Brimacombe, C. L., Fletcher, S. J., et al. (2012). Hepatitis C virus induces CD81 and claudin-1 endocytosis. J. Virol. 86, 4305–4316. doi: 10.1128/JVI.06996-11
Feldmann, A., Bekbulat, F., Huesmann, H., Ulbrich, S., Tatzelt, J., Behl, C., and Kern, A. (2017). The RAB GTPase RAB18 modulates macroautophagy and proteostasis. Biochem. Biophys. Res. Commun. 486, 738–743. doi: 10.1016/j.bbrc.2017.03.112
Foster, T. L., Belyaeva, T., Stonehouse, N. J., Pearson, A. R., and Harris, M. (2010). All three domains of the hepatitis C virus nonstructural NS5A protein contribute to RNA binding. J. Virol. 84, 9267–9277. doi: 10.1128/JVI.00616-10
Gerondopoulos, A., Bastos, R. N., Yoshimura, S., Anderson, R., Carpanini, S., Aligianis, I., et al. (2014). Rab18 and a Rab18 GEF complex are required for normal ER structure. J. Cell Biol. 205, 707–720. doi: 10.1083/jcb.201403026
Grosshans, B. L., Ortiz, D., and Novick, P. (2006). Rabs and their effectors: achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. U.S.A. 103, 11821–11827. doi: 10.1073/pnas.0601617103
Gu, M., and Rice, C. M. (2013). Structures of hepatitis C virus nonstructural proteins required for replicase assembly and function. Curr. Opin. Virol. 3, 129–136. doi: 10.1016/j.coviro.2013.03.013
Guest, J. D., and Pierce, B. G. (2018). Computational modeling of hepatitis c virus envelope glycoprotein structure and recognition. Front. Immunol. 9:1117. doi: 10.3389/fimmu.2018.01117
Henry, L., and Sheff, D. R. (2008). Rab8 regulates basolateral secretory, but not recycling, traffic at the recycling endosome. Mol. Biol. Cell 19, 2059–2068. doi: 10.1091/mbc.e07-09-0902
Hirota, Y., and Tanaka, Y. (2009). A small GTPase, human Rab32, is required for the formation of autophagic vacuoles under basal conditions. Cell. Mol. Life Sci. 66, 2913–2932. doi: 10.1007/s00018-009-0080-9
Hong, L., Guo, Y., BasuRay, S., Agola, J. O., Romero, E., Simpson, D. S., et al. (2015). A Pan-GTPase inhibitor as a molecular probe. PLoS ONE 10:e0134317. doi: 10.1371/journal.pone.0134317
Hope, R. G., Murphy, D. J., and McLauchlan, J. (2002). The domains required to direct core proteins of hepatitis C virus and GB virus-B to lipid droplets share common features with plant oleosin proteins. J. Biol. Chem. 277, 4261–4270. doi: 10.1074/jbc.M108798200
Horner, S. M., Liu, H. M., Park, H. S., Briley, J., and Gale, M. (2011). Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 108, 14590–14595. doi: 10.1073/pnas.1110133108
Horner, S. M., Wilkins, C., Badil, S., Iskarpatyoti, J., and Gale, M. (2015). Proteomic analysis of mitochondrial-associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS ONE 10:e0117963. doi: 10.1371/journal.pone.0117963
Hsu, S.-C., Lo, C.-W., Pan, T.-C., Lee, K.-Y., and Yu, M.-J. (2017). Serine 235 is the primary NS5A hyperphosphorylation site responsible for hepatitis C virus replication. J. Virol. 91:e00194-17. doi: 10.1128/jvi.00194-17
Huotari, J., and Helenius, A. (2011). Endosome maturation. EMBO J. 30, 3481–3500. doi: 10.1038/emboj.2011.286
Hwang, J., Huang, L., Cordek, D. G., Vaughan, R., Reynolds, S. L., Kihara, G., et al. (2010). Hepatitis C virus nonstructural protein 5A. Biochemical characterization of a novel structural class of RNA-binding proteins. J. Virol. 84, 12480–12491. doi: 10.1128/JVI.01319-10
Ikeda, M., Abe, K., Yamada, M., Dansako, H., Naka, K., and Kato, N. (2006). Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology 44, 117–125. doi: 10.1002/hep.21232
Ikeda, M., Kawai, Y., Mori, K., Yano, M., Abe, K., Nishimura, G., et al. (2011). Anti-ulcer agent teprenone inhibits hepatitis C virus replication. Potential treatment for hepatitis C. Liver Int. 31, 871–880. doi: 10.1111/j.1478-3231.2011.02499.x
Itoh, T., Fujita, N., Kanno, E., Yamamoto, A., Yoshimori, T., and Fukuda, M. (2008). Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol. Biol. Cell 19, 2916–2925. doi: 10.1091/mbc.e07-12-1231
Jiang, J., Cun, W., Wu, X., Shi, Q., Tang, H., and Luo, G. (2012). Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J. Virol. 86, 7256–7267. doi: 10.1128/JVI.07222-11
Jopling, C. L., Schütz, S., and Sarnow, P. (2008). Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe 4, 77–85. doi: 10.1016/j.chom.2008.05.013
Kobayashi, F., Yamada, S., Taguwa, S., Kataoka, C., Naito, S., Hama, Y., et al. (2012). Specific interaction of the envelope glycoproteins E1 and E2 with liver heparan sulfate involved in the tissue tropismatic infection by hepatitis C virus. Glycoconj. J. 29, 211–220. doi: 10.1007/s10719-012-9388-z
Köhler, K., Louvard, D., and Zahraoui, A. (2004). Rab13 regulates PKA signaling during tight junction assembly. J. Cell Biol. 165, 175–180. doi: 10.1083/jcb.200312118
Koles, K., Nunnari, J., Korkut, C., Barria, R., Brewer, C., Li, Y., et al. (2012). Mechanism of evenness interrupted (Evi)-exosome release at synaptic boutons. J. Biol. Chem. 287, 16820–16834. doi: 10.1074/jbc.M112.342667
Konstantinopoulos, P. A., Karamouzis, M. V., and Papavassiliou, A. G. (2007). Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 6, 541–555. doi: 10.1038/nrd2221
Li, C., Luo, X., Zhao, S., Siu, G. K., Liang, Y., Chan, H. C., et al. (2017). COPI-TRAPPII activates Rab18 and regulates its lipid droplet association. EMBO J. 36, 441–457. doi: 10.15252/embj.201694866
Li, Q., Wang, J., Wan, Y., and Chen, D. (2016). Depletion of Rab32 decreases intracellular lipid accumulation and induces lipolysis through enhancing ATGL expression in hepatocytes. Biochem. Biophys. Res. Commun. 471, 492–496. doi: 10.1016/j.bbrc.2016.02.047
Liang, Y., Lin, S., Zou, L., Zhou, H., Zhang, J., Su, B., and Wan, Y. (2012). Expression profiling of Rab GTPases reveals the involvement of Rab20 and Rab32 in acute brain inflammation in mice. Neurosci. Lett. 527, 110–114. doi: 10.1016/j.neulet.2012.08.039
Liu, P., Cheng, H., Roberts, T. M., and Zhao, J. J. (2009). Targeting the phosphoinositide 3-kinase pathway in cancer. Na. Rev. Drug Discov. 8, 627–644. doi: 10.1038/nrd2926
Liu, Z., Tian, Y., Machida, K., Lai, M. M. C., Luo, G., Foung, S. K. H., and Ou, J. J. (2012). Transient activation of the PI3K-AKT pathway by hepatitis C virus to enhance viral entry. J. Biol. Chem. 287, 41922–41930. doi: 10.1074/jbc.M112.414789
Lohmann, K. F., Koch, J. O., Herian, U., Theilmann, L., and Batenschlager, R. (1999). Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999. doi: 10.1126/science.285.5424.110
Lombardi, D., Soldati, T., Riederer, M. A., Goda, Y., Zerial, M., and Pfeffer, S. R. (1993). Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 12, 677–682. doi: 10.1002/j.1460-2075.1993.tb05701.x
Machlin, E. S., Sarnow, P., and Sagan, S. M. (2011). Masking the 5' terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. U.S.A. 108, 3193–3198. doi: 10.1073/pnas.1012464108
Mahley, R. W., and Ji, Z. S. (1999). Remnant lipoprotein metabolism. Key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 40, 1–16.
Makino, A., Hullin-Matsuda, F., Murate, M., Abe, M., Tomishige, N., Fukuda, M., et al. (2016). Acute accumulation of free cholesterol induces the degradation of perilipin 2 and Rab18-dependent fusion of ER and lipid droplets in cultured human hepatocytes. Mol. Biol. Cell 27, 3293–3304. doi: 10.1091/mbc.e15-10-0730
Mankouri, J., Walter, C., Stewart, H., Bentham, M., Park, W. S., Heo, W. D., et al. (2016). Release of infectious hepatitis C virus from Huh7 cells occurs via a trans-Golgi network to endosome pathway independent of VLDL. J. Virol. 90:JVI.00826-16. doi: 10.1128/JVI.00826-16
Manna, D., Aligo, J., Xu, C., Park, W. S., Koc, H., Heo, W. D., and Konan, K. V. (2010). Endocytic Rab proteins are required for hepatitis C virus replication complex formation. Virology 398, 21–37. doi: 10.1016/j.virol.2009.11.034
Martin, S., Driessen, K., Nixon, S. J., Zerial, M., and Parton, R. G. (2005). Regulated localization of Rab18 to lipid droplets. Effects of lipolytic stimulation and inhibition of lipid droplet catabolism. J. Biol. Chem. 280, 42325–42335. doi: 10.1074/jbc.M506651200
Marzesco, A.-M., Dunia, I., Pandjaitan, R., Recouvreur, M., Dauzonne, D., Benedetti, E. L., et al. (2002). The small GTPase Rab13 regulates assembly of functional tight junctions in epithelial cells. Mol. Biol. Cell 13, 1819–1831. doi: 10.1091/mbc.02-02-0029
Matsuda, M., Suzuki, R., Kataoka, C., Watashi, K., Aizaki, H., Kato, N., et al. (2014). Alternative endocytosis pathway for productive entry of hepatitis C virus. J. Gen. Virol. 95(Pt 12), 2658–2667. doi: 10.1099/vir.0.068528-0
Medvedev, R., Hildt, E., and Ploen, D. (2017). Look who's talking-the crosstalk between oxidative stress and autophagy supports exosomal-dependent release of HCV particles. Cell Biol. Toxicol. 33, 211–231. doi: 10.1007/s10565-016-9376-3
Meertens, L., Bertaux, C., and Dragic, T. (2006). Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 80, 11571–11578. doi: 10.1128/JVI.01717-06
Ménasché, G., Ménager, M. M., Lefebvre, J. M., Deutsch, E., Athman, R., Lambert, N., et al. (2008). A newly identified isoform of Slp2a associates with Rab27a in cytotoxic T cells and participates to cytotoxic granule secretion. Blood 112, 5052–5062. doi: 10.1182/blood-2008-02-141069
Meyers, N. L., Fontaine, K. A., Kumar, G. R., and Ott, M. (2016). Entangled in a membranous web. ER and lipid droplet reorganization during hepatitis C virus infection. Curr. Opin. Cell Biol. 41, 117–124. doi: 10.1016/j.ceb.2016.05.003
Miao, Z., Xie, Z., Miao, J., Ran, J., Feng, Y., and Xia, X. (2017). Regulated entry of hepatitis C virus into hepatocytes. Viruses 9:E100. doi: 10.3390/v9050100
Millar, A. L., Pavios, N. J., Xu, J., and Zheng, M. H. (2002). Rab3D. A regulator of exocytosis in non-neuronal cells. Histol. Histopathol. 17, 929–936. doi: 10.14670/HH-17.929
Miyanari, Y., Atsuzawa, K., Usuda, N., Watashi, K., Hishiki, T., Zayas, M., et al. (2007). The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9, 1089–1097. doi: 10.1038/ncb1631
Moradpour, D., Penin, F., and Rice, C. M. (2007). Replication of hepatitis C virus. Nat. Rev. Microbiol. 5, 453–463. doi: 10.1038/nrmicro1645
Morimoto, S., Nishimura, N., Terai, T., Manabe, S., Yamamoto, Y., Shinahara, W., et al. (2005). Rab13 mediates the continuous endocytic recycling of occludin to the cell surface. J. Biol. Chem. 280, 2220–2228. doi: 10.1074/jbc.M406906200
Nagashima, S., Jirintai, S., Takahashi, M., Kobayashi, T., Tanggis, Nishizawa, T., et al. (2014). Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J. Gen. Virol. 95(Pt 10), 2166–2175. doi: 10.1099/vir.0.066910-0
Neufeldt, C. J., Cortese, M., Acosta, E. G., and Bartenschlager, R. (2018). Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 16, 125–142. doi: 10.1038/nrmicro.2017.170
Nokes, R. L., Fields, I. C., Collins, R. N., and Fölsch, H. (2008). Rab13 regulates membrane trafficking between TGN and recycling endosomes in polarized epithelial cells. J. Cell Biol. 182, 845–853. doi: 10.1083/jcb.200802176
Nozawa, T., Aikawa, C., Goda, A., Maruyama, F., Hamada, S., and Nakagawa, I. (2012). The small GTPases Rab9A and Rab23 function at distinct steps in autophagy during Group A Streptococcus infection. Cell. Microbiol. 14, 1149–1165. doi: 10.1111/j.1462-5822.2012.01792.x
O'Mahony, F., Wroblewski, K., O'Byrne, S. M., Jiang, H., Clerkin, K., Benhammou, J., et al. (2015). Liver X receptors balance lipid stores in hepatic stellate cells through Rab18, a retinoid responsive lipid droplet protein. Hepatology 62, 615–626. doi: 10.1002/hep.27645
Ostrowski, M., Carmo, N. B., Krumeich, S., Fanget, I., Raposo, G., Savina, A., et al. (2010). Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 12, 19–30; sup pp 1–13. doi: 10.1038/ncb2000
Ozeki, S., Cheng, J., Tauchi-Sato, K., Hatano, N., Taniguchi, H., and Fujimoto, T. (2005). Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane. J. Cell Sci. 118 (Pt 12), 2601–2611. doi: 10.1242/jcs.02401
Park, M., Serpinskaya, A. S., Papalopulu, N., and Gelfand, V. I. (2007). Rab32 regulates melanosome transport in Xenopus melanophores by protein kinase a recruitment. Curr. Biol. 17, 2030–2034. doi: 10.1016/j.cub.2007.10.051
Paul, D., Hoppe, S., Saher, G., Krijnse-Locker, J., and Bartenschlager, R. (2013). Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 87, 10612–10627. doi: 10.1128/JVI.01370-13
Paul, D., Madan, V., and Bartenschlager, R. (2014). Hepatitis C virus RNA replication and assembly: living on the fat of the land. Cell Host Microbe 16, 569–579. doi: 10.1016/j.chom.2014.10.008
Pei, Y., Yue, L., Zhang, W., Wang, Y., Wen, B., Zhong, L., et al. (2015). Improvement in mouse iPSC induction by Rab32 reveals the importance of lipid metabolism during reprogramming. Sci. Rep. 5:16539. doi: 10.1038/srep16539
Pfeffer, S., and Aivazian, D. (2004). Targeting Rab GTPases to distinct membrane compartments. Nat. Rev. Mol. Cell Biol. 5, 886–896. doi: 10.1038/nrm1500
Pham, T. M., Tran, S. C., Lim, Y.-S., and Hwang, S. B. (2017). Hepatitis C virus-induced Rab32 aggregation and its implications for virion assembly. J. Virol. 91:e01662-16. doi: 10.1128/JVI.01662-16
Ploen, D., Hafirassou, M. L., Himmelsbach, K., Sauter, D., Biniossek, M. L., Weiss, T. S., et al. (2013a). TIP47 plays a crucial role in the life cycle of hepatitis C virus. J. Hepatol. 58, 1081–1088. doi: 10.1016/j.jhep.2013.01.022
Ploen, D., Hafirassou, M. L., Himmelsbach, K., Schille, S. A., Biniossek, M. L., Baumert, T. F., et al. (2013b). TIP47 is associated with the hepatitis C virus and its interaction with Rab9 is required for release of viral particles. Eur. J. Cell Biol. 92, 374–382. doi: 10.1016/j.ejcb.2013.12.003
Ploss, A., Evans, M. J., Gaysinskaya, V. A., Panis, M., You, H., de Jong, Y. P., and Rice, C. M. (2009). Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886. doi: 10.1038/nature07684
Plutner, H. (1991). Rab1b regulates vesicular transport between the endoplasmic reticulum and successive Golgi compartments. J. Cell Biol. 115, 31–43. doi: 10.1083/jcb.115.1.31
Pulido, M. R., Diaz-Ruiz, A., Jiménez-Gómez, Y., Garcia-Navarro, S., Gracia-Navarro, F., Tinahones, F., et al. (2011). Rab18 dynamics in adipocytes in relation to lipogenesis, lipolysis and obesity. PLoS ONE 6:e22931. doi: 10.1371/journal.pone.0022931
Ren, H., Elgner, F., Himmelsbach, K., Akhras, S., Jiang, B., Medvedev, R., et al. (2017). Identification of syntaxin 4 as an essential factor for the hepatitis C virus life cycle. Eur. J. Cell Biol. 96, 542–552. doi: 10.1016/j.ejcb.2017.06.002
Ren, H., Elgner, F., Jiang, B., Himmelsbach, K., Medvedev, R., Ploen, D., and Hildt, E. (2016). The autophagosomal SNARE protein syntaxin 17 is an essential factor for the hepatitis C virus life cycle. J. Virol. 90, 5989–6000. doi: 10.1128/JVI.00551-16
Rink, J., Ghigo, E., Kalaidzidis, Y., and Zerial, M. (2005). Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749. doi: 10.1016/j.cell.2005.06.043
Rocha, N., Kuijl, C., van der Kant, R., Janssen, L., Houben, D., Janssen, H., et al. (2009). Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J. Cell Biol. 185, 1209–1225. doi: 10.1083/jcb.200811005
Salloum, S., Wang, H., Ferguson, C., Parton, R. G., and Tai, A. W. (2013). Rab18 binds to hepatitis C virus NS5A and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 9:e1003513. doi: 10.1371/journal.ppat.1003513
Sato, T., Iwano, T., Kunii, M., Matsuda, S., Mizuguchi, R., Jung, Y., et al. (2014). Rab8a and Rab8b are essential for several apical transport pathways but insufficient for ciliogenesis. J. Cell Sci. 127(Pt 2), 422–431. doi: 10.1242/jcs.136903
Savina, A., Fader, C. M., Damiani, M. T., and Colombo, M. I. (2005). Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic 6, 131–143. doi: 10.1111/j.1600-0854.2004.00257.x
Savina, A., Vidal, M., and Colombo, M. I. (2002). The exosome pathway in K562 cells is regulated by Rab11. J. Cell. Sci. 115 (Pt 12), 2505–2515.
Schwaninger, R., Beckers, C. J., and Balch, W. E. (1991). Sequential transport of protein between the endoplasmic reticulum and successive Golgi compartments in semi-intact cells. J. Biol. Chem. 266, 13055–13063.
Sedano, C. D., and Sarnow, P. (2014). Hepatitis C virus subverts liver-specific miR-122 to protect the viral genome from exoribonuclease Xrn2. Cell Host Microbe 16, 257–264. doi: 10.1016/j.chom.2014.07.006
Seto, S., Tsujimura, K., and Koide, Y. (2011). Rab GTPases regulating phagosome maturation are differentially recruited to mycobacterial phagosomes. Traffic 12, 407–420. doi: 10.1111/j.1600-0854.2011.01165.x
Shanmugam, S., Saravanabalaji, D., and Yi, M. K. (2015). Detergent-resistant membrane association of NS2 and E2 during hepatitis C virus replication. J. Virol. 89, 4562–4574. doi: 10.1128/JVI.00123-15
Sharma, N. R., Mateu, G., Dreux, M., Grakoui, A., Cosset, F.-L., and Melikyan, G. B. (2011). Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 286, 30361–30376. doi: 10.1074/jbc.M111.263350
Shavinskaya, A., Boulant, S., Penin, F., McLauchlan, J., and Bartenschlager, R. (2007). The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282, 37158–37169. doi: 10.1074/jbc.M707329200
Shi, G., and Suzuki, T. (2018). Molecular basis of encapsidation of hepatitis C virus genome. Front. Microbiol. 9:396. doi: 10.3389/fmicb.2018.00396
Shrivastava, S., Devhare, P., Sujijantarat, N., Steele, R., Kwon, Y.-C., Ray, R., and Ray, R. B. (2016). Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J. Virol. 90, 1387–1396. doi: 10.1128/JVI.02383-15
Simmonds, P. (2013). The origin of hepatitis C virus. Curr. Top. Microbiol. Immunol. 369, 1–15. doi: 10.1007/978-3-642-27340-7_1
Sklan, E. H., Serrano, R. L., Einav, S., Pfeffer, S. R., Lambright, D. G., and Glenn, J. S. (2007a). TBC1D20 is a Rab1 GTPase-activating protein that mediates hepatitis C virus replication. J. Biol. Chem. 282, 36354–36361. doi: 10.1074/jbc.M705221200
Sklan, E. H., Staschke, K., Oakes, T. M., Elazar, M., Winters, M., Aroeti, B., et al. (2007b). A Rab-GAP TBC domain protein binds hepatitis C virus NS5A and mediates viral replication. J. Virol. 81, 11096–11105. doi: 10.1128/JVI.01249-07
Starr, T., Sun, Y., Wilkins, N., and Storrie, B. (2010). Rab33b and Rab6 are functionally overlapping regulators of Golgi homeostasis and trafficking. Traffic 11, 626–636. doi: 10.1111/j.1600-0854.2010.01051.x
Stoeck, I. K., Lee, J.-Y., Tabata, K., Romero-Brey, I., Paul, D., Schult, P., et al. (2018). Hepatitis C virus replication depends on endosomal cholesterol homeostasis. J. Virol. 92:JVI.01196-17. doi: 10.1128/JVI.01196-17
Stone, M., Jia, S., Heo, W. D., Meyer, T., and Konan, K. V. (2007). Participation of rab5, an early endosome protein, in hepatitis C virus RNA replication machinery. J. Virol. 81, 4551–4563. doi: 10.1128/JVI.01366-06
Street, A., Macdonald, A., McCormick, C., and Harris, M. (2005). Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol. 79, 5006–5016. doi: 10.1128/JVI.79.8.5006-5016.2005
Ströh, L. J., Nagarathinam, K., and Krey, T. (2018). Conformational flexibility in the CD81-binding site of the hepatitis C virus glycoprotein E2. Front. Immunol. 9:1396. doi: 10.3389/fimmu.2018.01396
Su, W.-C., Chao, T.-C., Huang, Y.-L., Weng, S.-C., Jeng, K.-S., and Lai, M. M. C. (2011). Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 85, 10561–10571. doi: 10.1128/JVI.00173-11
Takacs, C. N., Andreo, U., Dao Thi, V. L., Wu, X., Gleason, C. E., Itano, M. S., et al. (2017). Differential regulation of lipoprotein and hepatitis C virus secretion by Rab1b. Cell Rep. 21, 431–441. doi: 10.1016/j.celrep.2017.09.053
Takai, Y., Sasaki, T., and Matozaki, T. (2001). Small GTP-binding proteins. Physiol. Rev. 81, 153–208. doi: 10.1152/physrev.2001.81.1.153
Takeda, M., Ikeda, M., Satoh, S., Dansako, H., Wakita, T., and Kato, N. (2016). Rab13 is involved in the entry step of hepatitis C virus infection. Acta Med. Okayama. 70, 111–118. doi: 10.18926/AMO/54190
Tamai, K., Shiina, M., Tanaka, N., Nakano, T., Yamamoto, A., Kondo, Y., et al. (2012). Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology 422, 377–385. doi: 10.1016/j.virol.2011.11.009
Tellinghuisen, T. L., Foss, K. L., and Treadaway, J. (2008). Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4:e1000032. doi: 10.1371/journal.ppat.1000032
Tellinghuisen, T. L., Marcotrigiano, J., Gorbalenya, A. E., and Rice, C. M. (2004). The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 279, 48576–48587. doi: 10.1074/jbc.M407787200
Tong, Y., Chi, X., Yang, W., and Zhong, J. (2017). Functional analysis of Hepatitis C Virus (HCV) envelope protein E1 using a trans-complementation system reveals a dual role of a putative fusion peptide of E1 in both HCV entry and morphogenesis. J. Virol. 91:e02468-16. doi: 10.1128/JVI.02468-16
Tong, Y., Lavillette, D., Li, Q., and Zhong, J. (2018). Role of hepatitis C Virus envelope glycoprotein e1 in virus entry and assembly. Front. Immunol. 9:1411. doi: 10.3389/fimmu.2018.01411
Tsuboi, T., and Fukuda, M. (2006). Rab3A and Rab27A cooperatively regulate the docking step of dense-core vesicle exocytosis in PC12 cells. J. Cell. Sci. 119 (Pt 11), 2196–2203. doi: 10.1242/jcs.02962
Ullrich, O., Reinsch, S., Urb,é, S., Zerial, M., and Parton, R. G. (1996). Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135, 913–924. doi: 10.1083/jcb.135.4.913
Valsdottir, R., Hashimoto, H., Ashman, K., Koda, T., Storrie, B., and Nilsson, T. (2001). Identification of rabaptin-5, rabex-5, and GM130 as putative effectors of rab33b, a regulator of retrograde traffic between the Golgi apparatus and ER. FEBS Lett. 508, 201–209. doi: 10.1016/S0014-5793(01)02993-3
van der Kant, R., Fish, A., Janssen, L., Janssen, H., Krom, S., Ho, N., et al. (2013). Late endosomal transport and tethering are coupled processes controlled by RILP and the cholesterol sensor ORP1L. J. Cell Sci. 126(Pt 15), 3462–3474. doi: 10.1242/jcs.129270
Vanlandingham, P. A., and Ceresa, B. P. (2009). Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 284, 12110–12124. doi: 10.1074/jbc.M809277200
Vazquez-Martinez, R., Cruz-Garcia, D., Duran-Prado, M., Peinado, J. R., Castaño, J. P., and Malagon, M. M. (2007). Rab18 inhibits secretory activity in neuroendocrine cells by interacting with secretory granules. Traffic 8, 867–882. doi: 10.1111/j.1600-0854.2007.00570.x
Vogt, D. A., Camus, G., Herker, E., Webster, B. R., Tsou, C.-L., Greene, W. C., et al. (2013). Lipid droplet-binding protein TIP47 regulates hepatitis C Virus RNA replication through interaction with the viral NS5A protein. PLoS Pathog. 9:e1003302. doi: 10.1371/journal.ppat.1003302
Wakita, T., Pietschmann, T., Kato, T., Date, T., Miyamoto, M., Zhao, Z., et al. (2005). Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796. doi: 10.1038/nm1268
Wandinger-Ness, A., and Zerial, M. (2014). Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 6:a022616. doi: 10.1101/cshperspect.a022616
Waschbüsch, D., Michels, H., Strassheim, S., Ossendorf, E., Kessler, D., Gloeckner, C. J., and Barnekow, A. (2014). LRRK2 transport is regulated by its novel interacting partner Rab32. PLoS ONE 9:e111632. doi: 10.1371/journal.pone.0111632
Wasmeier, C., Romao, M., Plowright, L., Bennett, D. C., Raposo, G., and Seabra, M. C. (2006). Rab38 and Rab32 control post-Golgi trafficking of melanogenic enzymes. J. Cell Biol. 175, 271–281. doi: 10.1083/jcb.200606050
Wozniak, A. L., Long, A., Jones-Jamtgaard, K. N., and Weinman, S. A. (2016). Hepatitis C virus promotes virion secretion through cleavage of the Rab7 adaptor protein RILP. Proc. Natl. Acad. Sci. U.S.A. 113, 12484–12489. doi: 10.1073/pnas.1607277113
Xu, D., Li, Y., Wu, L., Li, Y., Zhao, D., Yu, J., et al. (2018). Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions. J. Cell Biol. 217, 975–995. doi: 10.1083/jcb.201704184
Yang, X. Z., Li, X. X., Zhang, Y. J., Rodriguez-Rodriguez, L., Xiang, M. Q., Wang, H. Y., et al. (2016). Rab1 in cell signaling, cancer and other diseases. Oncogene 35, 5699–5704. doi: 10.1038/onc.2016.81
You, X., Liu, F., Zhang, T., Li, Y., Ye, L., and Zhang, X. (2013). Hepatitis B virus X protein upregulates oncogene Rab18 to result in the dysregulation of lipogenesis and proliferation of hepatoma cells. Carcinogenesis 34, 1644–1652. doi: 10.1093/carcin/bgt089
Zappa, F., Venditti, R., and de Matteis, M. A. (2017). TRAPPing Rab18 in lipid droplets. EMBO J. 36, 394–396. doi: 10.15252/embj.201696287
Zayas, M., Long, G., Madan, V., and Bartenschlager, R. (2016). Coordination of hepatitis C virus assembly by distinct regulatory regions in nonstructural protein 5A. PLoS Pathog. 12:e1005376. doi: 10.1371/journal.ppat.1005376
Zeisel, M. B., Koutsoudakis, G., Schnober, E. K., Haberstroh, A., Blum, H. E., Cosset, F.-L., et al. (2007). Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 46, 1722–1731. doi: 10.1002/hep.21994
Zhang, C., Li, A., Zhang, X., and Xiao, H. (2011). A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J. Biol. Chem. 286, 9373–9381. doi: 10.1074/jbc.M110.207720
Zhen, Y., and Stenmark, H. (2015). Cellular functions of Rab GTPases at a glance. J. Cell Sci. 128, 3171–3176. doi: 10.1242/jcs.166074
Keywords: hepatitis C virus, rab GTPases, replication, morphogenesis, release
Citation: Elgner F, Hildt E and Bender D (2018) Relevance of Rab Proteins for the Life Cycle of Hepatitis C Virus. Front. Cell Dev. Biol. 6:166. doi: 10.3389/fcell.2018.00166
Received: 10 September 2018; Accepted: 20 November 2018;
Published: 04 December 2018.
Edited by:
Mary-Pat Stein, California State University, Northridge, United StatesReviewed by:
Christopher Stroupe, University of Virginia, United StatesGuangpu Li, University of Oklahoma Health Sciences Center, United States
Copyright © 2018 Elgner, Hildt and Bender. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eberhard Hildt, ZWJlcmhhcmQuaGlsZHRAcGVpLmRl