 Ashley Ramsawhook1
Ashley Ramsawhook1 Beth Coyle
Beth Coyle
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 22 March 2018
Sec. Epigenomics and Epigenetics
Volume 6 - 2018 | https://doi.org/10.3389/fcell.2018.00026
This article is part of the Research TopicBeyond CpG Methylation: New Modifications in Eukaryotic DNAView all 8 articles
The patterns of 5-methylcytosine (5mC) and its oxidized derivatives, 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine (5caC) are reportedly altered in a range of cancers. Likewise, Wilms' Tumor protein 1 (WT1), a transcription factor essential for urogenital, epicardium, and kidney development exhibits aberrant expression in multiple tumors. Interestingly, WT1 directly interacts with TET proteins that catalyze the enzymatic oxidation of 5mC and exhibits high affinity for 5caC-containing DNA substrates in vitro. Here we review recent developments in the fields of Tet-dependent 5mC oxidation and WT1 biology and explore potential perspectives for studying the interplay between TETs and WT1 in brain tumors.
Wilms' Tumor protein 1 (WT1) is a Kruppel-like transcription factor important for development of the mammalian embryonic kidney, urogenital system and epicardium (Roberts, 2005) that exhibits developmental and tissue specific expression patterns in mammals (Dressler, 2009; Hastie, 2017). Inactivating mutations occurring in WT1 during embryonic kidney development result in the formation of the pediatric renal neoplasm Wilms' tumor (WT) (Pritchard-Jones et al., 1990).
Onset of WT1 expression in murine embryos commences at 9 days post coitum (dpc) where the protein is detectable in the parietal epithelium lining the coelomic cavity (Armstrong et al., 1993). At this stage WT1 mRNA transcripts (Pritchard-Jones et al., 1990) localize to the intermediate mesenchyme/mesoderm (Pelletier et al., 1991b; Vize et al., 1997). At 11 dpc, WT1 expression is detected in the metanephric mesenchyme, presumptive spinal cord motor neurons, gonads, and ureter (Armstrong et al., 1993). Significant increase in WT1 expression is observed in the metanephric mesenchyme and nephrogenic condensates at 12.5 dpc, whereas gonadal WT1 expression remains level at this stage (Armstrong et al., 1993). Between 13.5 and 15 dpc, developing epicardium, podocytes of the glomerulus, endothelial, and stromal cells of the ovaries and uterus, Sertoli cells of the testis, retinal ganglia of the eye and ependymal cells of the fourth ventricle in the brain all possess positive immunostaining for WT1 (Sharma et al., 1992). At 20 dpc, WT1 is strongly expressed in kidney glomeruli and is weakly present in the eye and tongue (Mundlos et al., 1993). These periods of murine embryonic development correlate to 28–70 dpc in human embryos (Reidy and Rosenblum, 2009). In murine and human adult tissues, WT1 is present in multiple genitourinary structures, haematopoietic stem cells, kidney glomeruli, and podocytes, ependymal cells of the spinal cord and the area postrema of the medulla (Huang et al., 1990; Mundlos et al., 1993; Ramani and Cowell, 1996; Clark, 2006; Nakatsuka et al., 2006).
Homozygous deletion of WT1 (wt1−/−) in mice of C57BL/6 genetic background is embryonically lethal mainly due to genitourinary, kidney, and cardiac complications occurring between 12 and 15 dpc (Herzer et al., 1999). Specifically, wt1−/- mice exhibit lack of seminiferous tubules, complete lack of spleen and uretic bud formation in kidneys and failure of epicardium to undergo epithelial to mesenchymal transition (EMT) (Herzer et al., 1999). In a developmental context, during embryonic kidney formation, WT1 functions as a proto-oncogene, inducing proliferation of metanephric mesenchyme pluripotent progenitor cells required to respond to inductive WNT9b signals from the invading uretic bud to undergo mesenchymal to epithelial transitioning (MET) into glomerular podocytes (Hohenstein and Hastie, 2006). Metanephric mesenchyme cells originate from intermediate mesoderm arising at the gradient boundary of inductive bone morphogenetic protein (BMP) signals from the Splanchnic/lateral plate mesoderm and repressive retinoic acid (RA) signaling from the sixth somite of the paraxial mesoderm (Dressler, 2009).
WT1 exhibits a biphasic (at 10 and 12 dpc) pattern of expression in the developing kidney, specifically in (1) intermediate mesenchymal stem cells and mesonephric progenitors prior to their epithelial differentiation, and (2) during differentiation of epithelia in cap mesenchyme to glomerular podocytes, S-shaped bodies, comma shaped bodies, and renal vesicles at 12 dpc (Armstrong et al., 1993; Wilm et al., 2005). At 10 dpc, WT1 is required for the differentiation of mesonephric mesenchyme progenitors into transiently existing caudal tubular structures which function as a primitive temporary kidney (Wilm et al., 2005). In concordance with studies revealing that mesonephric progenitor induction at 10 dpc is regulated by the WT1 signaling-related genes including Pax2 and Six2 (Dressler et al., 1990; Dressler and Douglass, 1992), WT1 ablation in these progenitors results in significant reduction of caudal mesonephric tubules (Kreidberg et al., 1993). At 12 dpc, wt1−/− murine embryos fail to induce uretic bud formation and hence patterning of the metanephric mesenchyme and subsequent glomerular structures cannot develop, culminating in embryonic lethality (Kreidberg et al., 1993). In both instances of WT1-contingent development (i.e., mesonephric and metanephric mesenchyme differentiation), WT1 is required to regulate the mesenchymal to epithelial transition of mesodermally derived blastemal progenitors toward nephrogenic capillary epithelium (Sainio et al., 1997). In the developing human fetus circa 11–12 weeks of gestation, WT1 protein expression has been observed in kidney glomerular podocytes, Bowman's Capsule parietal epithelium, mesothelial cells covering surfaces of ovaries and testis, somatic skeletal muscle, the tongue, and radial glia of the spinal cord (Parenti et al., 2013; Magro et al., 2015). WT1 expression in glomerular podocytes is maintained throughout adult life which may implicate a role in metanephric mesenchyme maintenance of stemness and organ repair (Guo et al., 2002).
Approximately 5–20% of Wilms' Tumors cases exhibit somatic WT1 gene mutations which tend to be sporadic bi-allelic aberrations occurring in blastemal progenitors (Kaneko et al., 2015). The inherited nature of mono-allelic germline WT1 mutations or deletions occurring within chromosomal regions 11p13 predisposes individuals to Wilms' Tumor formation and account for ~5% of WT cases (Ruteshouser and Huff, 2004; Charlton et al., 2017). WT1 null mutants in mesonephros pluripotent progenitors of the intermediate mesoderm exhibit morphology and genotypic characteristics of paraxial mesoderm-derived mesenchymal stem cells i.e., they possess adipogenic, chondrogenic, and osteogenic lineage differentiation potential (Royer-Pokora et al., 2010). Histologically, Wilms' Tumors exhibit a highly disorganized heterogeneous cell population with blastemal, stromal, undifferentiated mesenchymal, and epithelial cells represented (Grosfeld, 1999; Scott et al., 2006). Failure of nephrogenic mesenchyme progenitors to differentiate into pretubular aggregates, renal vesicles, and eventually glomerula podocytes is attributable to WT1 mutations (Morizane et al., 2015). However, mutation of early nephrogenic progenitor specific genes such as Cited1 and Six2 results in stabilization and accumulation of β-catenin, thus inducing oncogenic targets of WNT signaling (Beckwith et al., 1990; Rivera and Haber, 2005; Huang et al., 2016). Mutations in WT1, Cited1, and Six2 result in Wilms' Tumors when they occur in pluripotent nephrogenic progenitors but not in stromal progenitors (Charlton et al., 2017). Moreover, Wilms' tumors exhibit elevated expression of genes pertaining to early kidney development such as those involved in uretic bud induction and nephrogenic mesenchyme patterning (e.g., Pax2, Pax8, Gata3, GDNF, and Wnts 4 and 9b) and down-regulation of late development genes (Reidy and Rosenblum, 2009; Morizane et al., 2015).
Nephrogenic rests (precursors to malignant Wilms' tumors) formed as a consequence of mono-allelic inactivation transformed into the eponymous renal malignancy upon secondary WT1 allele inactivation, a classic example of Knudson's two hit hypothesis (Ruteshouser et al., 2008; Royer-Pokora et al., 2010; Kaneko et al., 2015). The nephrogenic rests themselves occur more frequently as a consequence of somatic mono-allelic WT1 mutation in sporadic Wilms' Tumors (90–95%) compared to familial Wilms' tumor germline WT1 mutations (1–2%; Cardoso et al., 2013). Nephrogenic rests consist of primitive undifferentiated embryonic blastemal cells which are observed predominately in intralobar nephrogenic rests (ILNR) whereas undifferentiated mesenchymal stromal progenitors populate perilobar nephrogenic rests (PLNR) (Royer-Pokora et al., 2010; Charlton et al., 2017). WT1 mutations are associated with ILNR which persist in early kidney development (10 dpc) and is commensurate with induction of WT1 expression in pluripotent mesonephros during this time (Wilm and Muñoz-Chapuli, 2016). The absence of WT1 mutations in PLNR may be due to a WT1-independent and restricted lineage differentiation potential of metanephric stromal progenitors (Wilm and Muñoz-Chapuli, 2016). Collectively, this is suggestive of a developmentally early WT1-dependent paradigm of patterning and differentiation of the intermediate mesoderm and metanephric mesenchyme (Pelletier et al., 1991a; Royer-Pokora et al., 2010). However, the increased cellular proliferation rate resulting in the formation of neoplasms with WT1 mutant genotype evident in 5–20% of Wilms' Tumor cases implicates a tumor suppressor role for WT1, indicating both proto-oncogene and tumor suppressor functions for this transcription factor (Algar et al., 1996; Yamagami et al., 1996; Inoue et al., 1998; Menke and Van Der Eb, 1998; Tsuboi et al., 1999; Loeb and Sukumar, 2002; Li et al., 2003; Tatsumi et al., 2008).
WT1 wildtype mRNA has been detected in a range of malignancies including oesophageal, gastric, colorectal, pancreatic, biliary, lung, prostate, renal, breast, cervical, ovarian, endometrial, soft tissue, and brain cancers, indicative of a possible oncogenic contribution of ectopic WT1 expression in tumorigenesis (Nakatsuka et al., 2006). Studies performed on mouse lung cancer cells revealed a WT1-dependent mechanism of oncogenic KRAS induced proliferation (Vicent et al., 2010). Human and mouse lung cancer cells with ablated WT1 reduced proliferation and triggered senescence, highlighting anti-apoptotic consequences of WT1 ectopic overexpression (Vicent et al., 2010). These results have been corroborated by immunohistochemistry by both N- and C-terminus-specific WT1 antibodies (Nakatsuka et al., 2006). Interestingly, although WT1 is not expressed in astrocytes and is found only in brain endothelium in healthy adult humans (Bourne et al., 2010), its elevated expression is common for brain tumors such as pilocytic astrocytoma (grade I), anaplastic astrocytoma (grade III), and glioblastoma multiforme (grade IV) (Nakahara et al., 2004; Izumoto et al., 2008; Chiba et al., 2010; Rauscher et al., 2014). Increasing WT1 levels are commensurate with increasing tumor grade and associated with poor patient prognosis (Nakahara et al., 2004; Izumoto et al., 2008; Chiba et al., 2010; Rauscher et al., 2014). Increased incidence of high WT1 expression levels have been observed to correlate with severity of tumor grade of pediatric ependymoma with highest levels present in grade III anaplastic ependymoma (Yeung et al., 2013). WT1 overexpression was observed in 98% of glioblastoma primary cell samples and 83% of anaplastic astrocytomas compared to 53% of grade II oligodendroglioma and pilocytic astrocytomas (Schittenhelm et al., 2008). Short hairpin RNA (shRNA) molecules targeting WT1 mRNA transiently silence WT1 gene expression and reduce glioblastoma cell proliferation, viability, and invasion ability suggesting an oncogenic role for WT1 in these malignancies as opposed to the tumor suppressor role this protein plays in Wilms' tumor (Schittenhelm et al., 2008; Clark et al., 2010; Kijima et al., 2016).
Closer inspection of the WT1 structure provides clues to its functional role in tumorigenesis. WT1 is encoded by the corresponding gene located on chromosome 11p13 that is composed of 10 exons with exons 5 and 9 undergoing alternate splicing to form functionally different isoforms (Call et al., 1990). The WT1 gene harbors two ATG start codons, with one at the conventional +1 site and the second residing between exons 1 and 2, translation from which, synthesizes a curtailed protein variant at the N terminal (Dallosso et al., 2004). Full length WT1 protein consists of an N terminal RNA recognition motif (RRM), dimerization domain, activation domain, alternatively spliced 17 amino acid auxiliary interaction domain encoded by exon 5 and 4 and C-terminal Cysteine-Histidine (C2-H2) zinc finger domains (Bickmore et al., 1992). Zinc fingers 1–4 are encoded by exons 7, 8, 9, and 10, respectively (Ladomery and Dellaire, 2002). Alternate splicing between zinc fingers 3 and 4 generates two isoforms either possessing or absent for lysine-threonine-serine (KTS+/−−) amino acid triplet sequence located between zinc fingers 3 and 4 (Clark et al., 2007). WT1 Zinc fingers 1 & 2 can interact with p53 facilitating its sequestration and stabilization for subsequent ubiquitination and proteolysis (Maheswaran et al., 1995). Stabilization of p53 by WT1 prevents induction of pro-apoptotic pathways (Maheswaran et al., 1995). This occurs in a temporally and spatially regulated fashion during embryonic kidney development as nephrogenic progenitor cells (NPCs) within the metanephric mesenchyme are permitted to undergo differentiation to pretubular aggregates, renal vesicles and eventually glomerula podocytes at E8.0 (Kreidberg et al., 1993; Dressler, 2009; Brown et al., 2011; Short et al., 2014; Kann et al., 2015). Full length WT1 DNA binding domain is required for its physical interaction with p53 and as such, only +KTS and not –KTS isoforms can bind to wildtype p53 to stabilize the protein (Maheswaran et al., 1995). Surprisingly, studies in osteosarcoma models indicated that the pro-apoptotic effect of p53 expression can be inhibited by WT1 +KTS isoforms but not –KTS isoforms (Maheswaran et al., 1995; Mayo et al., 1999; Loeb, 2006). One explanation for this observation may include the WT1 facilitated inhibition of E6/E6AP mediated p53 ubiquitination and consequent proteolysis. Upon complexing with p53, WT1, may sterically prevent proteolytic degradation of p53 by occluding its ubiquitination sites from targeting by E6 ubiquitin ligase (Yamanouchi et al., 2014). In a murine model of ovarian cancer, transient overexpression of WT1 −17aa/-KTS isoform was linked to significant increases in cellular proliferation, migration, and angiogenesis leading to a significant reduction of mouse survival time (Yamanouchi et al., 2014). These results were in stark contrast to over-expression of +KTS isoforms which did not significantly affect these parameters and the +17aa/–KTS splice variant which induced apoptosis via suppression of EGFR transcription (Yamanouchi et al., 2014).
DNA methylation, denoted by 5-methylcytosine (5mC) occurring on the 5′ carbon of cytosine within a CpG dinucleotide has been well characterized as a transcriptionally repressive mark (Bird, 2002). Owing to its mutagenic potential to deaminate to thymine, 5mC is present on 70% of CpG cytosines, accounting for ~1% of mammalian genomes (Youssoufian et al., 1986). Methylation of CpG cytosines is catalyzed by maintenance and de novo DNA methyltransferase (DNMT) enzymes DNMT1 and DNMT3A/B, respectively (Kim et al., 2002). Genome wide methylation studies have compared 5mC distribution among DNA sequences of varying CpG density including low CpG density, intermediate CpG density, high CpG density, differentially methylated regions (DMRs), and long terminal repeats (LTRs) (Weber et al., 2007). Reduced representation bisulphite sequencing (RRBS) analysis of these regions revealed an inversely proportional relationship between CpG density and methylation level in sequenced fragments of 40–220 base pairs (bp) across 21 million reads (Weber et al., 2007). In contrast, methylation levels were significantly higher among low density CpG regions which correlated with the presence of transcriptionally permissive or active histone mark histone 3, lysine 4 trimethylation (H3K4me3), and dimethylated lysine 4 tails on histone 3 (H3K4me2) (Weber et al., 2007). Amongst the minute fraction of methylated CpGs (0.3%) in the vastly unmethylated high density CpG island sequences, occurrence of CpG methylation correlated with transcriptionally repressive histone mark H3K27me3 (Weber et al., 2007).
High-performance liquid chromatography (HPLC) analysis of 5mC distribution between different tissues detected highest levels of this modification in the brain (Kriaucionis and Heintz, 2009). Isotope labeled liquid chromatography-coupled mass spectrometry studies investigating 5mC levels in the adult human brain depicted significant differences in the distribution of this epigenetic mark between different brain regions (Kraus et al., 2012). Frontal and occipital lobes of the cerebral cortex scored highest for 5mC levels whilst frontal and occipital white matter tracts exhibited significantly lower 5mC presence (Kraus et al., 2012). Clues as to the nature of this observed disparity in 5mC yield between different organs and even distinct regions of the same tissue may be attributable to the relatively recent discovery of oxidized forms of 5mC and the mechanism governing their generation (Kriaucionis and Heintz, 2009; Khare et al., 2012).
The Ten-Eleven Translocase (TET) proteins, homologous to J-Base binding proteins (JBP) discovered in Trypanosome bruceii, can recognize and oxidize 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) (Tahiliani et al., 2009; Ito et al., 2011). According to a growing body of experimental evidence, these oxidized forms of 5mC (referred together as oxi-mCs) may play specific roles in epigenetic regulation of gene expression (Wu and Zhang, 2010; Seisenberger et al., 2012; Hackett et al., 2013; Smith and Meissner, 2013; Guo et al., 2014; Hu et al., 2014; Lewis et al., 2017). Importantly, both HPLC and the restriction endonuclease enzyme-facilitated oligonucleotide probe array hybridization mapping at single base resolution data demonstrate the relative enrichment of 5hmC within healthy human brains relative to other organs such as liver, kidney, pancreas, and heart (Khare et al., 2012).
In a recent study, Brain tumors representing all World Health Organization (WHO) classifications (grades I-IV) ranging from grade I temporal lobe pilocytic astrocytoma and grade II cervical spine ependymoma to grade III cerebellum anaplastic astrocytoma and grade IV parietal lobe glioblastoma multiforme were interrogated for the presence and magnitude of 5mC and 5hmC marks (Kraus et al., 2012). The WHO tumor grades increase with disease severity as characterized by tumor cell proliferative index, nuclear abnormality, necrosis, micro-vascularization, invasiveness, and anti-correlate with patient prognosis and survival rate (Louis et al., 2007). In these experiments examining the quantity and distribution of 5mC and 5hmC between healthy brain and brain tumor specimens, mass spectrometry-validated immunohistochemical analysis revealed no significant difference in 5mC levels between healthy brains and tissue matched tumors of varying grades, isolated from multiple locations (Kraus et al., 2012). Contrasting with this, the same study revealed heterogeneous 5hmC signatures with greater proportion of positive staining in lower (grade I) tumors (16.7%) relative to higher (grade IV) ones (1.43%) in the analyzed brain tumors. Highest 5hmC signal intensity (5hmC/dG ratio) captured via mass spectrometry was observed also in grade I tumors (0.22%) whereas grade IV tumors exhibited the lowest 5hmC signal intensity (0.078%). Frontal and occipital cortex regions and even their lower white matter tracts scored considerably higher 5hmC levels relative to all brain tumors analyzed (Kraus et al., 2012).
Bisulphite and oxidative bisulphite sequencing employing sodium bisulphite and potassium perruthenate chemistries, respectively, facilitate the discrimination of 5mC from unmodified cytosine and 5fC from 5hmC (Johnson et al., 2016). Sequencing of methylated and demethylation intermediate attached bases is possible at single base resolution and has consequently revealed the overall depletion of 5hmC from glioblastoma cells (Johnson et al., 2016). Examination of 30 glioblastoma cell lines and primary tissues exposed distinct 5hmC occupancy at CpG oceans and shores which are comprised of low density CpG frequency relative to CpG islands and shores and, thus, possess intermediate levels of 5mC enrichment. This highlights a DNA demethylation intermediate role for 5hmC, as 5mC erasure from sequences augments transcriptional status of genes under regulation (Johnson et al., 2016). The CpG shelves which displayed highest 5hmC enrichment (75%) localized within a window of ~5 kilobases (Kb) upstream from transcriptional start sites coinciding with enhancer and super-enhancer territory (Johnson et al., 2016). These genomic features characterized by association with active chromatin elements such as H3K27 acetylation (H3K27Ac), positively correlated with transcription of pathogenicity related genes in the aggressive glioblastoma cell line U87 (Johnson et al., 2016). In concurrence with previously mentioned studies highlighting the correlation of depletion of 5hmC levels with the tumor grade increase (Orr et al., 2012), Recursive Partition Mixture Model (RPMM) clustering analysis of 5hmC occupancy in glioblastoma cells linked the cytosine modification to prolonged patient survival, an index associated with pro-neural subtypes of glioblastoma (Verhaak et al., 2010; Johnson et al., 2016). Despite 5hmC depletion within aggressive brain tumor grades, its presence within genomic features appears critical to glioblastoma pathogenicity with the enrichment of 5hmC identified in gene regulatory regions upstream of core glioblastoma signaling genes such as epidermal growth factor receptor vIII (EGFRvIII) and cyclin-dependent kinase 6 (CDK6) (Johnson et al., 2016).
Whilst WT1 over-expression and null mutations have both been documented in cancer, implicating its dual properties as a proto-oncogene and tumor suppressor (Algar et al., 1996; Yamagami et al., 1996; Inoue et al., 1998; Menke and Van Der Eb, 1998; Tsuboi et al., 1999; Loeb and Sukumar, 2002; Li et al., 2003; Tatsumi et al., 2008), recent evidence has begun to demystify potential epigenetic regulatory functions of this protein in malignant neoplasms (Akpa et al., 2015). Thus, according to several studies, WT1 is able to prime metanephric mesenchyme progenitors for differentiation triggered by inductive WNT9b signals from the uretic bud via the repression of Enhancer of Zeste Homologue 2 (EZH2) transcription (Akpa et al., 2015). The consequent reduction of trimethylated lysine 27 on histone 3 (H3K27me3) enables transcription of beta-catenin (CTNNB1) in these progenitors and their subsequent nephrogenic differentiation (Akpa et al., 2015). In Wt1+/− mutant cells, progenitors cannot respond to WNT9b induction and thus proliferate into nephrogenic rests that transform into malignant tumors upon wt1 ablation (Dressler, 2009; Akpa et al., 2015).
Aberrant DNA methylation has been well documented in cancers where both hypermethylation of tumor suppressor genes and hypomethylation of retrotransposons, IAPs and proto-oncogenes are considered bona fide tumorigenic conditions (Ehrlich, 2002; Baylin, 2005; Baylin and Ohm, 2006; Hinoue et al., 2012). In addition, as oxi-mCs may serve as intermediates in the DNA demethylation process (Jones and Liang, 2009; Tahiliani et al., 2009; He et al., 2011; Shen et al., 2013; Dawlaty et al., 2014) these modifications may also be functionally involved in tumorigenesis (Dawson and Kouzarides, 2012; Ehrlich and Lacey, 2013; Eleftheriou et al., 2015; Tian et al., 2016; Ramsawhook et al., 2017). Correspondingly, TET proteins may display tumor suppressor activity and their mutations, generating defective or non-functional proteins may result in tumorigenesis (Ko et al., 2010). Thus, TET2 protein has been implicated in the development of hematological malignancies such as acute myeloid leukemia (AML), chronic myelomonocytic leukemia (CMML), and Myelodysplastic syndrome (MDS) (Tefferi et al., 2009). Interestingly, WT1 has been demonstrated to directly recruit DNMT3A to unmodified CpG cytosines (Figure 1) within gene regulatory sequences (Szemes et al., 2012). Moreover, WT1 has been demonstrated to bind directly to TET2 and TET3 recruiting them to their potential target sequences in acute myeloid leukemia models where mutation of WT1 resulting in its inactivation is accompanied with significant locus specific diminishing of 5hmC levels, a phenotype mimicked by mutations in the isocitrate dehydrogenase genes (IDH1/2) (Verhaak et al., 2010; Rampal et al., 2014; Kelly et al., 2017). The IDH1 mutations have been linked to aberrant DNA methylation signatures in AML (Rampal et al., 2014) and secondary pro-neural glioblastoma (Verhaak et al., 2010; Turcan et al., 2012; Rampal et al., 2014; Kelly et al., 2017). In AML and pro-neural glioblastoma, a CpG island methylator phenotype (G-CIMP) is common (Noushmehr et al., 2010; Turcan et al., 2012) and hypermethylation of non-housekeeping genes has been observed (Sturm et al., 2012; Hughes et al., 2013; Weisenberger, 2014). Substitution mutations such as arginine 132 replaced by histidine (R132H) mutate IDH1, erroneously converting isocitrate to 2-hydroxyglutarate instead of alpha-ketoglutarate, the canonical co-factor required by TET enzymes for oxidation of 5mC (Rampal et al., 2014). The consequent inhibition of TET enzyme activity correlates with significant 5mC hypermethylation and reduction in 5hmC observed in AML cells (Rampal et al., 2014). Similar effects of hypomethylation and 5hmC reduction are characteristic of Tet2 missense mutations that either result in its attenuated or abrogated catalytic ability (Konstandin et al., 2011). Interestingly, the aberrant methylation signatures observed in AML can be recapitulated in cells with wildtype IDH1&2 and Tet2 but mutant WT1 (Rampal et al., 2014). Correspondingly, a negative correlation of IDH1&2 and Tet2 mutations with mutations of WT1 was reported for a cohort of 398 AML patients (Rampal et al., 2014). Moreover, a significant reduction in 5hmC levels was demonstrated for these WT1 mutants compared to IDH1&2, TET2, and WT1 wildtype AML background controls (Rampal et al., 2014). Furthermore, next generation sequencing-based analysis revealed the existence of differentially methylated and hydroxymethylated regions between the wildtype AML controls and WT1 mutants, with the majority of 5hmC peaks localizing to enhancers and distal regulatory regions whereas differential 5mC peaks between wildtype and mutants clustered around transcriptional start sites (Rampal et al., 2014). The 5hmC levels at differentially hydroxymethylated distal enhancers were lower in WT1 AML mutants compared to controls and differentially methylated regions at transcriptional start sites were more greatly enriched for 5mC in WT1 mutants (Rampal et al., 2014). This may suggest a transcriptional priming role for 5hmC in haematopoiesis which when perturbed by WT1 mutations, may contribute to tumorigenesis (Ko et al., 2010; Yang et al., 2013). In glioblastoma multiforme, WT1 over-expression may facilitate competition or co-operative binding with other transcription factors capable of recognizing 5hmC occupancy at promoter and enhancer sequences (Takai et al., 2014). Intriguingly, DNA immunoprecipitation and subsequent sequencing (DIPSeq) demonstrate possible interactions between cytosine modifications and protein complexes capable of “reading” them and altering transcriptional status of associated genes (Takai et al., 2014). Core GBM signaling pathway genes e.g., BRAF, AKT, EGFR, & CDK6 possessed 5hmC enrichment at their promoters and intergenic regions which positively correlated with chromatin target of PRMT1 (CHTOP) promoter presence and binding and consequent transcriptional activation of these genes (Takai et al., 2014). Excitingly, WT1 and CHTOP share binding affinity for the same promoter sequence (Figure 1), implicating WT1 as a strong contender for influencing sustained pathogenic transcriptional signaling in glioblastoma (Hashimoto et al., 2015).
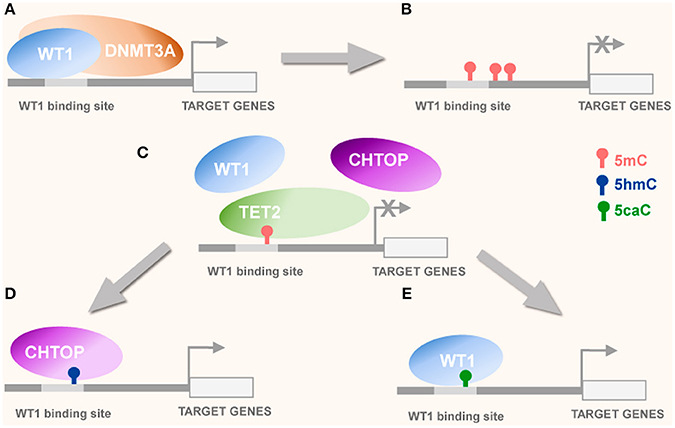
Figure 1. Potential interplay of WT1 with DNMT3A and CHTOP. (A) WT1 recruits DNMT3A to unmethylated CpGs or nearby WT1 binding sequences (Szemes et al., 2012) leading to de novo methylation of unmethylated cytosines by DNMT3A and consequently, transcriptional repression of WT1 target genes (B). (C) Tet protein mediated oxidation of 5 mC to oxi-mC derivatives attracts oxi-mC reader proteins WT1 and CHTOP competing for the same DNA binding sequence. (D) 5mC oxidation to 5hmC attracts CHTOP to WT1 binding site leading to histone demethylation and transcriptional activation. (E) 5mC oxidation to 5caC, attracting WT1, which possesses high 5caC binding affinity may also result in transcriptional activation of its target genes.
In addition to its interaction with TET proteins, recent studies suggest that WT1 can also specifically bind certain oxi-mCs (Hashimoto et al., 2014, 2016). WT1, a member of the Early Growth Response family (EGR) of transcription factors shares DNA binding consensus sequence 5′-GCG(T/G)GGGCG-3′ (EGR-1 consensus) with its fellow family members EGR1 and Zif268 (Stoll et al., 2007). WT1 possesses oxi-mC vs. mC discrimination capabilities in contrast to 5mC vs. unmodified cytosine recognition as demonstrated by EGR1 and Zif268 (Hashimoto et al., 2014). Amongst oxi-mC derivatives, WT1 displays preferential binding affinity for 5caC over 5hmC and 5fC. Additionally, preferential binding affinity for 5caC is enhanced by presence of asymmetric methylation on the same transcription factor “recognition strand” and by occupancy of 5mC at CpG sites on the complementary strand (Hashimoto et al., 2016). This is indicative of specific epigenetic configurations on DNA sequences which can be “read” by transcription factors such as WT1 (Hashimoto et al., 2016). Moreover, the markedly reduced binding affinity for 5caC and 5mC displayed by WT1 +KTS isoform may demonstrate a splice variant-specific requirement of certain gene promoters to undergo transcriptional activation (Hashimoto et al., 2016). Ergo, this may suggest that oxi-mC presence or absence on promoters and enhancers may perturb or enhance WT1-contingent gene expression, thus contributing WT1-implicated pathologies (Hashimoto et al., 2016). In this context, it is striking that our recent studies have highlighted an enrichment of 5caC not only in certain types of pediatric brain tumors such as medulloblastomas and ependymomas (Ramsawhook et al., 2017) but also in a number of samples of glioblastoma multiforme (Eleftheriou et al., 2015), a cancer previously reported to possess elevated levels of WT1 (Schittenhelm et al., 2008). Considering the observations across the board that 5hmC levels are significantly reduced in malignancies compared to their healthy matched tissue (Figueroa et al., 2010; Ko et al., 2010; Jin et al., 2011), the presence of detectable 5caC in brain tumors may seem rather unexpected and may depend on certain specific features of their methylation/demethylation machinery influenced by overexpression of WT1 and or TET2/IDH1 & 2 mutations in these cancers.
Collectively, these studies imply two potential modes of WT1 interplay with TET-dependent 5mC oxidation that may influence tumorigenesis. Thus, WT1 can either (1) serve as a binding partner of TET proteins participating in their recruitment to target sequences (Wang et al., 2015) or (2) act as a specific “reader”- of certain oxi-mCs (most notably 5caC) (Hashimoto et al., 2014, 2016; Figure 2). Presently available experimental evidence suggests that both these scenarios may take place during the pathogenesis of brain tumors, particularly glioblastoma multiforme (Schittenhelm et al., 2008; Hashimoto et al., 2009; Eleftheriou et al., 2015; Ramsawhook et al., 2017).
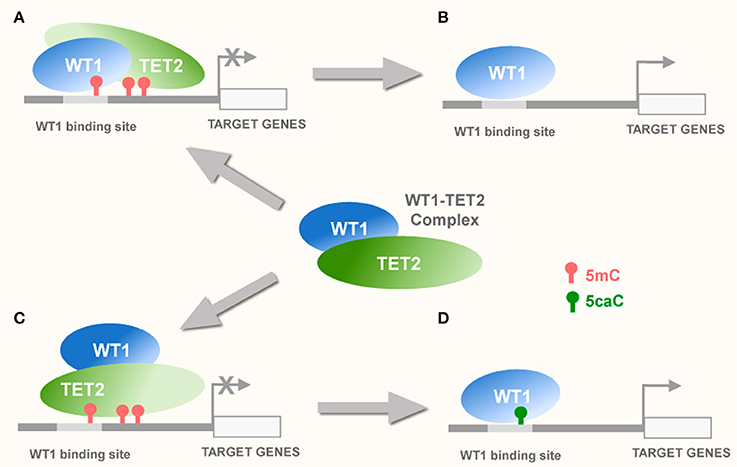
Figure 2. Potential interplay of WT1 with TET proteins. (A) WT1 can recruit TET2 to the methylated CpGs within or nearby WT1 binding sites (Rampal et al., 2014) causing demethylation of these CpGs via TET-dependent 5mC oxidation and activation of transcription of the target genes (B). Alternatively, recruitment of TET2 to WT1 binding sequences by WT1 within transcriptionally inactive promoters may lead to oxidation of 5mC to 5caC (C). This may enable WT1 that possesses high affinity for 5caC (Hashimoto et al., 2014), to stabilize this modification and hence potentially facilitate transcriptional activation of its target genes (D).
Considering the high mortality rate of this tumor and an immediate need for an efficient strategy for its therapy (Smoll et al., 2013; Thakkar et al., 2014; Ostrom et al., 2015), elucidating potential roles of WT1 and its potential interaction with TETs/oxi-mCs in glioblastomas should represent an important direction for future research. Commensurate with this perspective, two groups in Japan have developed an anti-WT1 peptide vaccine aimed at reducing WT1 protein activity in glioblastoma multiforme patients that is currently undergoing phase II clinical trial (Izumoto et al., 2008; Oji et al., 2016).
Considering the crucial spatiotemporal expression patterns of WT1 as a developmental master-regulator, tumor suppressor and proto-oncogene as well as its potential involvement in the TET/oxi-mCs-related demethylation and transcriptional repression, it is highly likely that ectopic and anachronistic involvement of WT1 in biological processes may perturb normal functioning of cellular machinery, resulting in methylome reprogramming and ultimately, tumorigenesis. Therefore, targeted therapies against WT1 in tumors exiting its over-expression may help curtail disease malignancy and improve patient survival.
AsR: researched and wrote the initial draft; BC and AR: conceived and edited the final review manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Lara Lewis for aiding in proof reading and editing the manuscript. The work was supported by Biotechnology and Biological Sciences Research Council [grant number BB/N005759/1] to AR.
Akpa, M. M., Iglesias, D. M., Chu, L. L., Cybulsky, M., Bravi, C., and Goodyer, P. R. (2015). Wilms tumor suppressor, WT1, suppresses epigenetic silencing of the β-catenin gene. J. Biol. Chem. 290, 2279–2288. doi: 10.1074/jbc.M114.573576
Algar, E. M., Khromykh, T., Smith, S., Blackburn, D. M., Bryson, G. J., and Smith, P. J. (1996). A WT1 antisense oligonucleotide inhibits proliferation and induces apoptosis in myeloid leukaemia cell lines. Oncogene 12, 1005–1014.
Armstrong, J. F., Pritchard-Jones, K., Bickmore, W. A., Hastie, N. D., and Bard, J. B. (1993). The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech. Dev. 40, 85-97. doi: 10.1016/0925-4773(93)90090-K
Baylin, S. B. (2005). DNA methylation and gene silencing in cancer. Nat. Rev. Clin. Oncol. 2, S4–S11. doi: 10.1038/ncponc0354
Baylin, S. B., and Ohm, J. E. (2006). Epigenetic gene silencing in cancer-a mechanism for early oncogenic pathway addiction? Nat. Rev. Cancer 6, 107-116. doi: 10.1038/nrc1799
Beckwith, J. B., Kiviat, N. B., and Bonadio, J. F. (1990). Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms' tumor. Pediatr. Pathol. 10, 1–36. doi: 10.3109/15513819009067094
Bickmore, W., Oghene, K., Little, M., Seawright, A., Van Heyningen, V., and Hastie, N. (1992). Modulation of DNA binding specificity by alternative splicing of the Wilms tumor wt1 gene transcript. Science 257, 235–237. doi: 10.1126/science.1321494
Bird, A. (2002). DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21. doi: 10.1101/gad.947102
Bourne, T. D., Elias, W. J., Lopes, M. B., and Mandell, J. W. (2010). WT1 is not a reliable marker to distinguish reactive from neoplastic astrocyte populations in the central nervous system. Brain Pathol. 20, 1090–1095. doi: 10.1111/j.1750-3639.2010.00415.x
Brown, A. C., Adams, D., de Caestecker, M., Yang, X., Friesel, R., and Oxburgh, L. (2011). FGF/EGF signaling regulates the renewal of early nephron progenitors during embryonic development. Development 138, 5099–5112. doi: 10.1242/dev.065995
Call, K. M., Glaser, T., Ito, C. Y., Buckler, A. J., Pelletier, J., Haber, D. A., et al. (1990). Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell 60, 509–520. doi: 10.1016/0092-8674(90)90601-A
Cardoso, L. C., De Souza, K. R., Reis, D. O., Helena, A., Andrade, R. C., Britto, A. C., et al. (2013). WT1, WTX and CTNNB1 mutation analysis in 43 patients with sporadic Wilms' tumor. Oncol. Rep. 29, 315–320. doi: 10.3892/or.2012.2096
Charlton, J., Irtan, S., Bergeron, C., and Pritchard-Jones, K. (2017). Bilateral Wilms tumour: a review of clinical and molecular features. Expert Rev. Mol. Med. 19:e8. doi: 10.1017/erm.2017.8
Chiba, Y., Hashimoto, N., Tsuboi, A., Rabo, C., Oka, Y., Kinoshita, M., et al. (2010). Prognostic value of WT1 protein expression level and MIB-1 staining index as predictor of response to WT1 immunotherapy in glioblastoma patients. Brain Tumor Pathol. 27, 29–34. doi: 10.1007/s10014-010-0265-9
Clark, A. J. (2006). The Expression and Function of Wilms' Tumor 1 in Malignant Glioma. Virginia Commonwealth University.
Clark, A. J., Dos Santos, W. G., McCready, J., Chen, M. Y., Van Meter, T. E., Ware, J. L., et al. (2007). Wilms tumor 1 expression in malignant gliomas and correlation of + KTS isoforms with p53 status. J. Neurosurg. 107, 586–592. doi: 10.3171/JNS-07/09/0586
Clark, A. J., Ware, J. L., Chen, M. Y., Graf, M. R., Van Meter, T. E., Dos Santos, W. G., et al. (2010). Effect of WT1 gene silencing on the tumorigenicity of human glioblastoma multiforme cells. J. Neurosurg. 112, 18–25. doi: 10.3171/2008.11.JNS08368
Dallosso, A., Hancock, A., Brown, K., Williams, A., Jackson, S., and Malik, K. (2004). Genomic imprinting at the WT1 gene involves a novel coding transcript (AWT1) that shows deregulation in Wilms' tumours. Hum. Mol. Genet. 13, 405–415. doi: 10.1093/hmg/ddh038
Dawlaty, M. M., Breiling, A., Le, T., Barrasa, M. I., Raddatz, G., Gao, Q., et al. (2014). Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 29, 102–111. doi: 10.1016/j.devcel.2014.03.003
Dawson, M. A., and Kouzarides, T. (2012). Cancer epigenetics: from mechanism to therapy. Cell 150, 12–27. doi: 10.1016/j.cell.2012.06.013
Dressler, G. R. (2009). Advances in early kidney specification, development and patterning. Development 136, 3863–3874. doi: 10.1242/dev.034876
Dressler, G. R., Deutsch, U., Chowdhury, K., Nornes, H. O., and Gruss, P. (1990). Pax2, a new murine paired-box-containing gene and its expression in the developing excretory system. Development 109, 787–795.
Dressler, G. R., and Douglass, E. C. (1992). Pax-2 is a DNA-binding protein expressed in embryonic kidney and Wilms tumor. Proc. Natl. Acad. Sci. U.S.A. 89, 1179–1183. doi: 10.1073/pnas.89.4.1179
Ehrlich, M. (2002). DNA methylation in cancer: too much, but also too little. Oncogene 21:5400. doi: 10.1038/sj.onc.1205651
Ehrlich, M., and Lacey, M. (2013). DNA hypomethylation and hemimethylation in cancer. Adv. Exp. Med. Biol. 754, 31–56. doi: 10.1007/978-1-4419-9967-2_2
Eleftheriou, M., Pascual, A. J., Wheldon, L. M., Perry, C., Abakir, A., Arora, A., et al. (2015). 5-Carboxylcytosine levels are elevated in human breast cancers and gliomas. Clin. Epigenetics 7:88. doi: 10.1186/s13148-015-0117-x
Figueroa, M. E., Abdel-Wahab, O., Lu, C., Ward, P. S., Patel, J., Shih, A., et al. (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567. doi: 10.1016/j.ccr.2010.11.015
Grosfeld, J. L. (1999). Risk-based management: current concepts of treating malignant solid tumors of childhood. J. Am. Coll. Surg. 189, 407–425. doi: 10.1016/S1072-7515(99)00167-2
Guo, F., Li, X., Liang, D., Li, T., Zhu, P., Guo, H., et al. (2014). Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 15, 447–458. doi: 10.1016/j.stem.2014.08.003
Guo, J. K., Hammes, A., Chaboissier, M. C., Vidal, V., Xing, Y., Wong, F., et al. (2002). Early gonadal development: exploring Wtl and Sox9 function. Novartis Found. Symp. 44, 23–31.
Hackett, J. A., Sengupta, R., Zylicz, J. J., Murakami, K., Lee, C., Down, T. A., et al. (2013). Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339, 448–452. doi: 10.1126/science.1229277
Hashimoto, H., Olanrewaju, Y. O., Zheng, Y., Wilson, G. G., Zhang, X., and Cheng, X. (2014). Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. 28, 2304–2313. doi: 10.1101/gad.250746.114
Hashimoto, H., Zhang, X., Vertino, P. M., and Cheng, X. (2015). The mechanisms of generation, recognition, and erasure of DNA 5-methylcytosine and thymine oxidations. J. Biol. Chem. 290, 20723–20733. doi: 10.1074/jbc.R115.656884
Hashimoto, H., Zhang, X., Zheng, Y., Wilson, G. G., and Cheng, X. (2016). Denys-drash syndrome associated WT1 glutamine 369 mutants have altered sequence-preferences and altered responses to epigenetic modifications. Nucleic Acids Res. 44, 10165–10176. doi: 10.1093/nar/gkw766
Hashimoto, N., Tsuboi, A., Chiba, Y., Izumoto, S., Oka, Y., Yoshimine, T., et al. (2009). Immunotherapy targeting the Wilms' tumor 1 gene product for patients with malignant brain tumors. Brain Nerve 61, 805–814. doi: 10.11477/mf.1416100522
Hastie, N. D. (2017). Wilms' tumour 1 (WT1) in development, homeostasis and disease. Development 144, 2862–2872. doi: 10.1242/dev.153163
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307. doi: 10.1126/science.1210944
Herzer, U., Crocoll, A., Barton, D., Howells, N., and Englert, C. (1999). The Wilms tumor suppressor gene wt1 is required for development of the spleen. Curr. Biol. 9, 837–840. doi: 10.1016/S0960-9822(99)80369-8
Hinoue, T., Weisenberger, D. J., Lange, C. P., Shen, H., Byun, H. M., Van Den Berg, D., et al. (2012). Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 22, 271–282. doi: 10.1101/gr.117523.110
Hohenstein, P., and Hastie, N. D. (2006). The many facets of the Wilms' tumour gene, WT1. Hum. Mol. Genet. 15, R196–R201. doi: 10.1093/hmg/ddl196
Hu, X., Zhang, L., Mao, S.-Q., Li, Z., Chen, J., Zhang, R.-R., et al. (2014). Tet and TDG mediate DNA demethylation essential for mesenchymal-to-epithelial transition in somatic cell reprogramming. Cell Stem. Cell 14, 512–522. doi: 10.1016/j.stem.2014.01.001
Huang, A., Campbell, C. E., Bonetta, L., McAndrews-Hill, M. S., Chilton-Macneil, S., Coppes, M. J., et al. (1990). Tissue, developmental, and tumor-specific expression of divergent transcripts in Wilms tumor. Science 250, 991–994. doi: 10.1126/science.2173145
Huang, L., Mokkapati, S., Hu, Q., Ruteshouser, E. C., Hicks, M. J., and Huff, V. (2016). Nephron progenitor but not stromal progenitor cells give rise to wilms tumors in mouse models with β-catenin activation or Wt1 ablation and Igf2 upregulation. Neoplasia 18, 71–81. doi: 10.1016/j.neo.2015.12.001
Hughes, L. A., Melotte, V., De Schrijver, J., De Maat, M., Smit, V. T., Bovée, J. V., et al. (2013). The CpG island methylator phenotype: what's in a name? Cancer Res. 73, 5858–5868. doi: 10.1158/0008-5472.CAN-12-4306
Inoue, K., Tamaki, H., Ogawa, H., Oka, Y., Soma, T., Tatekawa, T., et al. (1998). Wilms9 tumor gene (wt1) competes with differentiation-inducing signal in hematopoietic progenitor cells. Blood 91, 2969–2976.
Ito, S., Shen, L., Dai, Q., Wu, S. C., Collins, L. B., Swenberg, J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303. doi: 10.1126/science.1210597
Izumoto, S., Tsuboi, A., Oka, Y., Suzuki, T., Hashiba, T., Kagawa, N., et al. (2008). Phase II clinical trial of Wilms tumor 1 peptide vaccination for patients with recurrent glioblastoma multiforme. J. Neurosurg. 108, 963–971. doi: 10.3171/JNS/2008/108/5/0963
Jin, S.-G., Jiang, Y., Qiu, R., Rauch, T. A., Wang, Y., Schackert, G., et al. (2011). 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 71, 7360–7365. doi: 10.1158/0008-5472.CAN-11-2023
Johnson, K. C., Houseman, E. A., King, J. E., von Herrmann, K. M., Fadul, C. E., and Christensen, B. C. (2016). 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat. Commun. 7:13177. doi: 10.1038/ncomms13177
Jones, P. A., and Liang, G. (2009). Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 10, 805–811. doi: 10.1038/nrg2651
Kaneko, Y., Okita, H., Haruta, M., Arai, Y., Oue, T., Tanaka, Y., et al. (2015). A high incidence of WT1 abnormality in bilateral Wilms tumours in Japan, and the penetrance rates in children with WT1 germline mutation. Br. J. Cancer 112, 1121-1133. doi: 10.1038/bjc.2015.13
Kann, M., Bae, E., Lenz, M. O., Li, L., Trannguyen, B., Schumacher, V. A., et al. (2015). WT1 targets Gas1 to maintain nephron progenitor cells by modulating FGF signals. Development 142, 1254–1266. doi: 10.1242/dev.119735
Kelly, A. D., Kroeger, H., Yamazaki, J., Taby, R., Neumann, F., Yu, S., et al. (2017). A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia 31, 2011–2019. doi: 10.1038/leu.2017.12
Khare, T., Pai, S., Koncevicius, K., Pal, M., Kriukiene, E., Liutkeviciute, Z., et al. (2012). 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol. 19, 1037-1043. doi: 10.1038/nsmb.2372
Kijima, N., Hashimoto, N., Chiba, Y., Fujimoto, Y., Sugiyama, H., and Yoshimine, T. (2016). “Functional roles of Wilms' tumor 1 (WT1) in malignant brain tumors,” in van den Heuvel-Eibrink MM, ed T. Wilms (Brisbane, QLD: Codon Publications), 261–273.
Kim, G. D., Ni, J., Kelesoglu, N., Roberts, R. J., and Pradhan, S. (2002). Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 21, 4183–4195. doi: 10.1093/emboj/cdf401
Ko, M., Huang, Y., Jankowska, A. M., Pape, U. J., Tahiliani, M., Bandukwala, H. S., et al. (2010). Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 468, 839-843. doi: 10.1038/nature09586
Konstandin, N., Bultmann, S., Szwagierczak, A., Dufour, A., Ksienzyk, B., Schneider, F., et al. (2011). Genomic 5-hydroxymethyicytosine levels correlate with TET2 mutations and a distinct global gene expression pattern in secondary acute myeloid leukemia. Leukemia 25, 1649–1653. doi: 10.1038/leu.2011.134
Kraus, T. F., Globisch, D., Wagner, M., Eigenbrod, S., Widmann, D., Münzel, M., et al. (2012). Low values of 5-hydroxymethylcytosine (5hmC), the “sixth base,” are associated with anaplasia in human brain tumors. Int. J. Cancer 131, 1577–1590. doi: 10.1002/ijc.27429
Kreidberg, J. A., Sariola, H., Loring, J. M., Maeda, M., Pelletier, J., Housman, D., et al. (1993). WT-1 is required for early kidney development. Cell 74, 679–691. doi: 10.1016/0092-8674(93)90515-R
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. doi: 10.1126/science.1169786
Ladomery, M., and Dellaire, G. (2002). Multifunctional zinc finger proteins in development and disease. Ann. Hum. Genet. 66, 331–342. doi: 10.1017/S0003480002001215
Lewis, L. C., Lo, P. C., Foster, J. M., Dai, N., Corrêa, I. R., Durczak, P. M., et al. (2017). Dynamics of 5-carboxylcytosine during hepatic differentiation: potential general role for active demethylation by DNA repair in lineage specification. Epigenetics 12, 277–286. doi: 10.1080/15592294.2017.1292189
Li, X., Oghi, K. A., Zhang, J., and Krones, A. (2003). Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 426, 247-254. doi: 10.1038/nature02083
Loeb, D. M. (2006). WT1 influences apoptosis through transcriptional regulation of Bcl-2 family members. Cell Cycle 5, 1249–1253. doi: 10.4161/cc.5.12.2807
Loeb, D. M., and Sukumar, S. (2002). The role of WT1 in oncogenesis: tumor suppressor or oncogene? Int. J. Hematol. 76, 117-126. doi: 10.1007/BF02982573
Louis, D. N., Ohgaki, H., Wiestler, O. D., Cavenee, W. K., Burger, P. C., Jouvet, A., et al. (2007). The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109. doi: 10.1007/s00401-007-0243-4
Magro, G., Salvatorelli, L., Puzzo, L., Musumeci, G., Bisceglia, M., and Parenti, R. (2015). Oncofetal expression of Wilms' tumor 1 (WT1) protein in human fetal, adult and neoplastic skeletal muscle tissues. Acta Histochem. 117, 492–504. doi: 10.1016/j.acthis.2015.02.012
Maheswaran, S., Englert, C., Bennett, P., Heinrich, G., and Haber, D. A. (1995). The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev. 9, 2143–2156. doi: 10.1101/gad.9.17.2143
Mayo, M. W., Wang, C. Y., Drouin, S. S., Madrid, L. V., Marshall, A. F., Reed, J. C., et al. (1999). WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 proto-oncogene. EMBO J. 18, 3990–4003. doi: 10.1093/emboj/18.14.3990
Menke, A. L., Van Der Eb, A., and Jochemsen, A. (1998). The Wilms' tumor 1 gene: oncogene or tumor suppressor gene? Int. Rev. Cytol. 181, 151–212.
Morizane, R., Lam, A. Q., Freedman, B. S., Kishi, S., Valerius, M. T., and Bonventre, J. V. (2015). Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 33, 1193-1200. doi: 10.1038/nbt.3392
Mundlos, S., Pelletier, J., Darveau, A., Bachmann, M., Winterpacht, A., and Zabel, B. (1993). Nuclear localization of the protein encoded by the Wilms' tumor gene WT1 in embryonic and adult tissues. Development 119, 1329–1341.
Nakahara, Y., Okamoto, H., Mineta, T., and Tabuchi, K. (2004). Expression of the Wilms' tumor gene product WT1 in glioblastomas and medulloblastomas. Brain Tumor Pathol. 21, 113–116. doi: 10.1007/BF02482185
Nakatsuka, S., Oji, Y., Horiuchi, T., Kanda, T., Kitagawa, M., Takeuchi, T., et al. (2006). Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod. Pathol. 19, 804-814. doi: 10.1038/modpathol.3800588
Noushmehr, H., Weisenberger, D. J., Diefes, K., Phillips, H. S., Pujara, K., Berman, B. P., et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17, 510–522. doi: 10.1016/j.ccr.2010.03.017
Oji, Y., Hashimoto, N., Tsuboi, A., Murakami, Y., Iwai, M., Kagawa, N., et al. (2016). Association of WT1 IgG antibody against WT1 peptide with prolonged survival in glioblastoma multiforme patients vaccinated with WT1 peptide. Int. J. Cancer 139, 1391–1401. doi: 10.1002/ijc.30182
Orr, B. A., Haffner, M. C., Nelson, W. G., Yegnasubramanian, S., and Eberhart, C. G. (2012). Decreased 5-hydroxymethylcytosine is associated with neural progenitor phenotype in normal brain and shorter survival in malignant glioma. PLoS ONE 7:e41036. doi: 10.1371/journal.pone.0041036
Ostrom, Q. T., Gittleman, H., Fulop, J., Liu, M., Blanda, R., Kromer, C., et al. (2015). CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro Oncol. 17, iv1-iv62. doi: 10.1093/neuonc/nov189
Parenti, R., Perris, R., Vecchio, G. M., Salvatorelli, L., Torrisi, A., Gravina, L., et al. (2013). Immunohistochemical expression of Wilms' tumor protein (WT1) in developing human epithelial and mesenchymal tissues. Acta Histochem. 115, 70–75. doi: 10.1016/j.acthis.2012.04.006
Pelletier, J., Bruening, W., Kashtan, C. E., Mauer, S. M., Manivel, J. C., Striegel, J. E., et al. (1991a). Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell 67, 437–447. doi: 10.1016/0092-8674(91)90194-4
Pelletier, J., Schalling, M., Buckler, A., Rogers, A., Haber, D., and Housman, D. (1991b). Expression of the Wilms' tumor gene WT1 in the murine urogenital system. Genes Dev. 5, 1345–1356. doi: 10.1101/gad.5.8.1345
Pritchard-Jones, K., Fleming, S., Davidson, D., Bickmore, W., Porteous, D., Gosden, C., et al. (1990). The candidate Wilms' tumour gene is involved in genitourinary development. Nature 346, 194–197. doi: 10.1038/346194a0
Ramani, P., and Cowell, J. K. (1996). The expression pattern of Wilms' tumour gene (WT1) product in normal tissues and paediatric renal tumours. J. Pathol. 179, 162–168. doi: 10.1002/(SICI)1096-9896(199606)179:2<162::AID-PATH545>3.0.CO;2-0
Rampal, R., Alkalin, A., Madzo, J., Vasanthakumar, A., Pronier, E., Patel, J., et al. (2014). DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 9, 1841–1855. doi: 10.1016/j.celrep.2014.11.004
Ramsawhook, A., Lewis, L., Coyle, B., and Ruzov, A. (2017). Medulloblastoma and ependymoma cells display increased levels of 5-carboxylcytosine and elevated TET1 expression. Clin. Epigenet. 9:18. doi: 10.1186/s13148-016-0306-2
Rauscher, J., Beschorner, R., Gierke, M., Bisdas, S., Braun, C., Ebner, F. H., et al. (2014). WT1 expression increases with malignancy and indicates unfavourable outcome in astrocytoma. J. Clin. Pathol. 67, 556–561. doi: 10.1136/jclinpath-2013-202114
Reidy, K. J., and Rosenblum, N. D. (2009). Cell and molecular biology of kidney development. Semin. Nephrol. 29, 321–337. doi: 10.1016/j.semnephrol.2009.03.009
Rivera, M. N., and Haber, D. A. (2005). Wilms' tumour: connecting tumorigenesis and organ development in the kidney. Nat. Rev. Cancer 5, 699–712. doi: 10.1038/nrc1696
Roberts, S. G. (2005). Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 15, 542–547. doi: 10.1016/j.gde.2005.08.004
Royer-Pokora, B., Busch, M., Beier, M., Duhme, C., De Torres, C., Mora, J., et al. (2010). Wilms tumor cells with WT1 mutations have characteristic features of mesenchymal stem cells and express molecular markers of paraxial mesoderm. Hum. Mol. Genet. 19, 1651–1668. doi: 10.1093/hmg/ddq042
Ruteshouser, E. C., and Huff, V. (2004). Familial Wilms tumor. Am. J. Med. Genet. C Semin. Med. Genet. 129C, 29–34. doi: 10.1002/ajmg.c.30025
Ruteshouser, E. C., Robinson, S. M., and Huff, V. (2008). Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer 47, 461–470. doi: 10.1002/gcc.20553
Sainio, K., Hellstedt, P., Kreidberg, J. A., Saxén, L., and Sariola, H. (1997). Differential regulation of two sets of mesonephric tubules by WT-1. Development 124, 1293–1299.
Schittenhelm, J., Mittelbronn, M., Nguyen, T. D., Meyermann, R., and Beschorner, R. (2008). WT1 expression distinguishes astrocytic tumor cells from normal and reactive astrocytes. Brain Pathol. 18, 344–353. doi: 10.1111/j.1750-3639.2008.00127.x
Scott, R. H., Stiller, C. A., Walker, L., and Rahman, N. (2006). Syndromes and constitutional chromosomal abnormalities associated with Wilms tumour. J. Med. Genet. 43, 705–715. doi: 10.1136/jmg.2006.041723
Seisenberger, S., Andrews, S., Krueger, F., Arand, J., Walter, J., Santos, F., et al. (2012). The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell 48, 849–862. doi: 10.1016/j.molcel.2012.11.001
Sharma, P. M., Yang, X., Bowman, M., Roberts, V., and Sukumar, S. (1992). Molecular cloning of rat Wilms' tumor complementary DNA and a study of messenger RNA expression in the urogenital system and the brain. Cancer Res. 52, 6407–6412.
Shen, L., Wu, H., Diep, D., Yamaguchi, S., D'alessio, A. C., Fung, H. L., et al. (2013). Genome-wide analysis reveals TET-and TDG-dependent 5-methylcytosine oxidation dynaics. Cell 153, 692–706. doi: 10.1016/j.cell.2013.04.002
Short, K. M., Combes, A. N., Lefevre, J., Ju, A. L., Georgas, K. M., Lamberton, T., et al. (2014). Global quantification of tissue dynamics in the developing mouse kidney. Dev. Cell 29, 188–202. doi: 10.1016/j.devcel.2014.02.017
Smith, Z. D., and Meissner, A. (2013). DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220. doi: 10.1038/nrg3354
Smoll, N. R., Schaller, K., and Gautschi, O. P. (2013). Long-term survival of patients with glioblastoma multiforme (GBM). J. Clin. Neurosci. 20, 670–675. doi: 10.1016/j.jocn.2012.05.040
Stoll, R., Lee, B. M., Debler, E. W., Laity, J. H., Wilson, I. A., Dyson, H. J., et al. (2007). Structure of the Wilms tumor suppressor protein zinc finger domain bound to DNA. J. Mol. Biol. 372, 1227–1245. doi: 10.1016/j.jmb.2007.07.017
Sturm, D., Witt, H., Hovestadt, V., Khuong-Quang, D.-A., Jones, D. T., Konermann, C., et al. (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437. doi: 10.1016/j.ccr.2012.08.024
Szemes, M., Dallosso, A. R., Melegh, Z., Curry, T., Li, Y., Rivers, C., et al. (2012). Control of epigenetic states by WT1 via regulation of de novo DNA methyltransferase 3A. Hum. Mol. Genet. 22, 74–83. doi: 10.1093/hmg/dds.403
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., and Brudno, Y. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935. doi: 10.1126/science.1170116
Takai, H., Masuda, K., Sato, T., Sakaguchi, Y., Suzuki, T., Suzuki, T., et al. (2014). 5-Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP-methylosome complex. Cell Rep. 9, 48–60. doi: 10.1016/j.celrep.2014.08.071
Tatsumi, N., Oji, Y., Tsuji, N., Tsuda, A., Higashio, M., Aoyagi, S., et al. (2008). Wilms' tumor gene WT1-shRNA as a potent apoptosis-inducing agent for solid tumors. Int. J. Oncol. 32, 701–711. doi: 10.3892/ijo.32.3.701
Tefferi, A., Lim, K., Abdel-Wahab, O., Lasho, T., Patel, J., Patnaik, M., et al. (2009). Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 23, 1343–1345. doi: 10.1038/leu.2009.59
Thakkar, J. P., Dolecek, T. A., Horbinski, C., Ostrom, Q. T., Lightner, D. D., Barnholtz-Sloan, J. S., et al. (2014). Epidemiol. and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomarkers Prev. 23, 1985–1996. doi: 10.1158/1055-9965.EPI-14-0275
Tian, Y.-P., Zhu, Y.-M., Sun, X.-H., and Lai, M.-D. (2016). Multiple functions of ten-eleven translocation 1 during tumorigenesis. Chin. Med. J. 129, 1744-1751. doi: 10.4103/0366-6999.185873
Tsuboi, A., Oka, Y., Ogawa, H., Elisseeva, O. A., Tamaki, H., Oji, Y., et al. (1999). Constitutive expression of the Wilms' tumor gene WT1 inhibits the differentiation of myeloid progenitor cells but promotes their proliferation in response to granulocyte-colony stimulating factor (G-CSF). Leuk. Res. 23, 499–505.
Turcan, S., Rohle, D., Goenka, A., Walsh, L. A., Fang, F., Yilmaz, E., et al. (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479-483. doi: 10.1038/nature10866
Verhaak, R. G., Hoadley, K. A., Purdom, E., Wang, V., Qi, Y., Wilkerson, M. D., et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110. doi: 10.1016/j.ccr.2009.12.020
Vicent, S., Chen, R., Sayles, L. C., Lin, C., Walker, R. G., Gillespie, A. K., et al. (2010). Wilms tumor 1 (WT1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J. Clin. Invest. 120, 3940-3952. doi: 10.1172/JCI44165
Vize, P. D., Seufert, D. W., Carroll, T. J., and Wallingford, J. B. (1997). Model systems for the study of kidney development: use of the pronephros in the analysis of organ induction and patterning. Dev. Biol. 188, 189–204. doi: 10.1006/dbio.1997.8629
Wang, Y., Xiao, M., Chen, X., Chen, L., Xu, Y., Lv, L., et al. (2015). WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol. Cell 57, 662–673. doi: 10.1016/j.molcel.2014.12.023
Weber, M., Hellmann, I., Stadler, M. B., Ramos, L., Pääbo, S., Rebhan, M., et al. (2007). Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457-466. doi: 10.1038/ng1990
Weisenberger, D. J. (2014). Characterizing DNA methylation alterations from the cancer genome atlas. J. Clin. Invest. 124, 17-23. doi: 10.1172/JCI69740
Wilm, B., Ipenberg, A., Hastie, N. D., Burch, J. B., and Bader, D. M. (2005). The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 132, 5317–5328. doi: 10.1242/dev.02141
Wilm, B., and Muñoz-Chapuli, R. (2016). “The role of WT1 in embryonic development and normal organ homeostasis,” in The Wilms' Tumor (WT1) Gene. Methods in Molecular Biology, Vol. 1467, ed H. Hastie (New York, NY: Humana Press), 23–39.
Wu, S. C., and Zhang, Y. (2010). Active DNA demethylation: many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 11, 607-620. doi: 10.1038/nrm2950
Yamagami, T., Sugiyama, H., Inoue, K., Ogawa, H., Tatekawa, T., Hirata, M., et al. (1996). Growth inhibition of human leukemic cells by WT1 (Wilms tumor gene) antisense oligodeoxynucleotides: implications for the involvement of WT1 in leukemogenesis. Blood 87, 2878–2884.
Yamanouchi, K., Ohta, T., Liu, Z., Oji, Y., Sugiyama, H., Shridhar, V., et al. (2014). The Wilms' tumor gene WT1– 17AA/– KTS splice variant increases tumorigenic activity through up-regulation of vascular endothelial growth factor in an in vivo ovarian cancer model. Transl. Oncol. 7, 580–589. doi: 10.1016/j.tranon.2014.07.008
Yang, H., Liu, Y., Bai, F., Zhang, J., Ma, S., Liu, J., et al. (2013). Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene 32, 663–669. doi: 10.1038/onc.2012.67
Yeung, J. T., Hamilton, R. L., Okada, H., Jakacki, R. I., and Pollack, I. F. (2013). Increased expression of tumor-associated antigens in pediatric and adult ependymomas: implication for vaccine therapy. J. Neurooncol. 111, 103–111. doi: 10.1007/s11060-012-0998-x
Keywords: Wilms' Tumor 1, DNA demethylation, epigenetics, brain tumors, glioblastoma, ependymoma, medullobastoma, TET proteins
Citation: Ramsawhook A, Ruzov A and Coyle B (2018) Wilms' Tumor Protein 1 and Enzymatic Oxidation of 5-Methylcytosine in Brain Tumors: Potential Perspectives. Front. Cell Dev. Biol. 6:26. doi: 10.3389/fcell.2018.00026
Received: 06 November 2017; Accepted: 05 March 2018;
Published: 22 March 2018.
Edited by:
Kyoko Yokomori, University of California, Irvine, United StatesReviewed by:
Abhijit Shukla, Memorial Sloan Kettering Cancer Center, United StatesCopyright © 2018 Ramsawhook, Ruzov and Coyle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexey Ruzov, bXN6YXJAZXhtYWlsLm5vdHRpbmdoYW0uYWMudWs=
Beth Coyle, bWd6ZWNAZXhtYWlsLm5vdHRpbmdoYW0uYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.