 Nikolay Popgeorgiev
Nikolay Popgeorgiev Lea Jabbour
Lea Jabbour Germain Gillet
Germain Gillet
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 13 February 2018
Sec. Cell Death and Survival
Volume 6 - 2018 | https://doi.org/10.3389/fcell.2018.00013
Bcl-2 family proteins are recognized as major regulators of the mitochondrial pathway of apoptosis. They control the mitochondrial outer membrane permeabilization (MOMP) by directly localizing to this organelle. Further investigations demonstrated that Bcl-2 related proteins are also found in other intracellular compartments such as the endoplasmic reticulum, the Golgi apparatus, the nucleus and the peroxisomes. At the level of these organelles, Bcl-2 family proteins not only regulate MOMP in a remote fashion but also participate in major cellular processes including calcium homeostasis, cell cycle control and cell migration. With the advances of live cell imaging techniques and the generation of fluorescent recombinant proteins, it became clear that the distribution of Bcl-2 proteins inside the cell is a dynamic process which is profoundly affected by changes in the cellular microenvironment. Here, we describe the current knowledge related to the subcellular distribution of the Bcl-2 family of proteins and further emphasize on the emerging concept that this highly dynamic process is critical for cell fate determination.
Intracellular compartmentalization is a fundamental feature of eukaryotic cells. It allows the physical segregation and simultaneous execution of distinct biochemical processes within the same cell. In fact, proper subcellular targeting and distribution are critical for the function of intracellular proteins. B-cell lymphoma 2 (Bcl-2) family of proteins are acknowledged as major regulators of the intrinsic pathway of the programmed cell death type 1 commonly known as apoptosis. Bcl-2, the founding member of the family, was discovered more than 30 years ago in the course of a study on the t(14;18) genomic translocation. Translocations between chromosome 18 (q21) and chromosome 14 (q32) are frequently observed in human B-cell follicular lymphomas and can result in the relocation of the bcl2 open reading frame downstream of the enhancer promoter region of the igh heavy chain immunoglobulin gene (Tsujimoto et al., 1984, 1985). This translocation results in upregulation of bcl2 gene expression (Cleary et al., 1986). Previous observations on other B-cell lymphomas such as Burkitt's lymphoma or mantle cell lymphoma, in which similar translocations led to the overexpression of oncogenes such as c-myc or cyclin d1, suggested that bcl2 was another oncogene inducing uncontrolled proliferation. However, soon after this discovery, Vaux and colleagues demonstrated that Bcl-2 could sustain cell survival of lymphoid cells in absence of Interleukin-3, establishing Bcl-2 as the founding member of a new class of oncoproteins that inhibited cell death instead of promoting cell proliferation (Vaux et al., 1988). Since the discovery of Bcl-2, extensive work in mammalian cells as well as in other animal models, such as nematode and drosophila, uncovered a family of structurally related proteins involved in the control of apoptosis, reviewed in Delbridge et al. (2016). Indeed, Bcl-2 family members are globular proteins mainly composed of α-helices and characterized by conserved Bcl-2 homology (BH1-4) domains. On this basis, three Bcl-2 family subgroups have been identified: (1) the anti-apoptotic multidomain members (e.g., Bcl-2, Bcl-xL, and Mcl-1) containing all four BH domains, (2) the pro-apoptotic multidomain members (e.g., Bax, Bak, and Bok) containing three BH domain (BH1-3) and (3) the pro-apoptotic BH3-only members containing the sole BH3 domain (e.g., Bad, Bid, and Bik). In addition, numerous Bcl-2 proteins contain a hydrophobic transmembrane anchoring (TM) domain at the C-terminus end allowing them to localize to intracellular membranes. At the level of the mitochondria, proapoptotic Bax and Bak can form oligomers and induce the mitochondrial outer membrane permeabilization (MOMP), which is considered as a point of non-return in the execution of apoptosis. By direct binding to Bax and Bak, anti-apoptotic Bcl-2 proteins inhibit the MOMP. In addition, the BH3-only proteins control this process by direct activation of Bax and Bak or by repressing the anti-apoptotic Bcl-2 family members (Youle and Strasser, 2008). In fact, it rapidly became clear that Bcl-2 proteins can also localize to other organelles including the endoplasmic reticulum (ER), the Golgi apparatus, the nuclear outer membrane (NOM) or the nucleus itself. At the level of these different intracellular membranes, Bcl-2 proteins did not only control the MOMP from a distance, but also participated in a number of non-apoptotic processes. However, the implications of these “moonlighting” functions in physiological or pathological situations have been enlightened only recently.
Here, we discuss the current knowledge about the mechanisms addressing Bcl-2 family proteins to intracellular membranes and emphasize the emerging concept that this highly dynamic process is critical for cell fate determination.
Bcl-2 family members are mainly known as regulators of the mitochondrial outer membrane integrity. However, the exact distribution of Bcl-2 itself was already a matter of debate in the early 90's. Indeed, initial studies reported that Bcl-2 could be found at the level of the cytosolic leaflet of intracellular membranes. However, experiments conducted by Korsmeyer lab suggested that Bcl-2 is in fact located in the mitochondrial inner membrane (MIM) (Hockenbery et al., 1990). These observations were somehow overlooked since it was shown later on that Bcl-2 possesses a C-terminus transmembrane (TM) domain (as discussed below), addressing this protein to the MOM. However, subsequent work showed that Bcl-2 could also control the activity of MIM proteins such as Cytochrome c oxidase Va and Cyclophilin D through direct interactions. This apparent contradiction might be explained by the actual localization of Bcl-2 at the contact points between the internal and the external mitochondrial membranes (Nakai et al., 1993; Nguyen et al., 1993). Interestingly, Bcl-2 was not the only protein detected at the MIM. Indeed, seminal investigations conducted by Jonas and colleagues revealed that Bcl-xL protein localizes at the MIM in hippocampal neurons. In this compartment, Bcl-xL directly binds to the β-subunit of the F1FO ATP synthase, increasing its activity and allowing optimal synaptic transmission. Mcl-1 is another Bcl-2 homolog found to operate at the level of different mitochondrial compartments. At the level of the MOM, Mcl-1 inhibits apoptosis, whereas at the level of the mitochondrial matrix, it fosters mitochondrial bioenergetics. Of note, Mcl-1 matrix localization requires proteolytic cleavage of its N-terminal mitochondrial targeting sequence (Perciavalle et al., 2012).
During the course of these studies, it also appeared that several Bcl-2 homologs were in fact ER-residents. This includes both multidomain anti-apoptotic (Bcl-2, Bcl-xL, Mcl-1, and Bcl-2l10) (Krajewski et al., 1993; Yang et al., 1995; Aouacheria et al., 2001; White et al., 2005) and pro-apoptotic proteins (Bax, Bak and Bok) (Zong et al., 2003; Schulman et al., 2013) as well as BH3 only proteins (Bim, Bik) (Germain et al., 2002; Morishima et al., 2004; Figure 1). Thus, at the level of the ER, Bcl-2 proteins not only control MOMP in a remote fashion, but also orchestrate additional non-apoptotic pathways through direct binding with Ca2+ transporters and mediators of the Unfolded Protein Response (UPR).
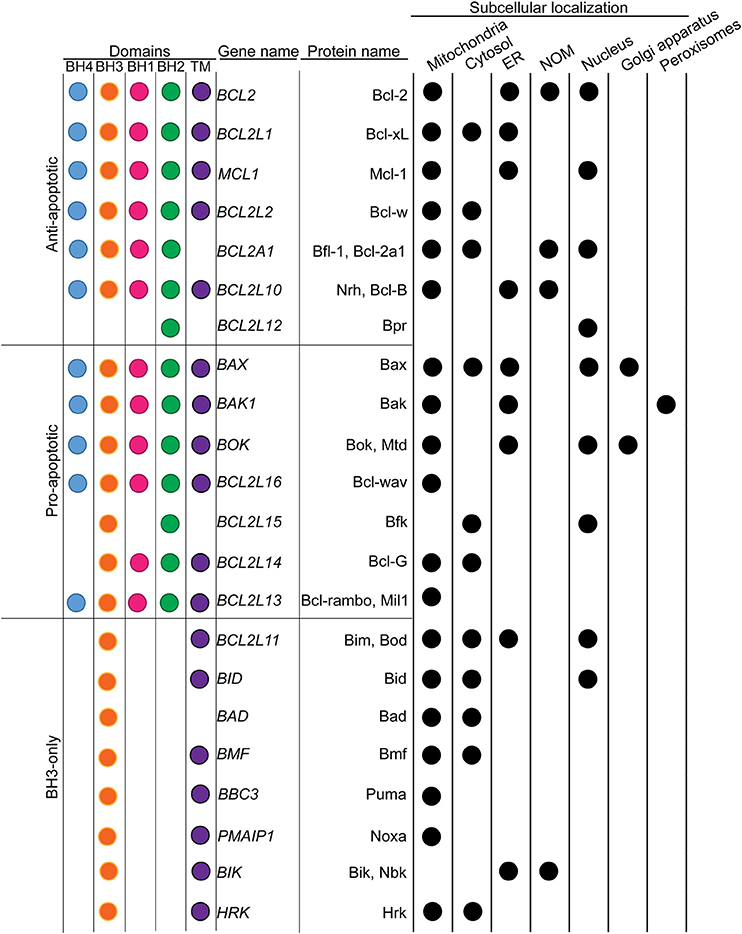
Figure 1. Classification of Bcl-2 family of proteins in respect to their subcellular localizations. Bcl-2 family members were classified based on their implication in the MOMP. The presence of conserved Bcl-2 homology (BH) and transmembrane (TM) domains were indicated with color dots. The main subcellular localizations were presented in the table on the right. Black dots indicate the presence of the protein to the corresponding subcellular localization.
Interestingly, Bcl-2 proteins were found to translocate inside the nucleus as well. In the case of Bcl-2 itself, such localization depends on the phosphorylation status of Thr56. Nuclear Bcl-2 appears to take part in a multiprotein complex comprising CDK1, PP1 and Nucleolin (Barboule et al., 2005, 2009). However, the mechanisms governing Bcl-2 nuclear translocation as well as their actual biological relevance remain poorly understood.
Finally, Bcl-2 family members may also possess some “exotic” subcellular localizations including the Golgi apparatus (Dumitru et al., 2012), the lysosomes (Guan et al., 2015) and the peroxisomes (Hosoi et al., 2017).
Their possible localization at the level of these diverse internal membranes is an emerging concept. Actually, it might illustrate the dynamics of Bcl-2 family of proteins action range and their pleiotropic functions inside the cell.
The first evidence of the membrane localization of Bcl-2 arose from the work of Cleary and colleagues. By performing subcellular fractionation and immunofluorescence experiments on lymphoid cell lines, they demonstrated that Bcl-2 was associated almost exclusively with cellular membranes, as no Bcl-2 was detected in the cytosolic fraction, whereas only a minor fraction was detected in the nucleus (Chen-Levy et al., 1989). Furthermore, the Bcl-2 protein was recovered almost solely in a detergent-rich phase, confirming its membrane anchoring properties. Sequence analyses identified a 23 amino acid-long hydrophobic sequence at the C-terminus end, referred to as transmembrane (TM) domain (Chen-Levy et al., 1989). With the exception of Bfl-1, in which the TM domain is less well defined, all mutidomain Bcl-2 homologs possess a similar TM domain. Of note, this domain seems to be conserved throughout evolution since it is found in numerous Bcl-2 homologs from distantly-related metazoan species (Quinn et al., 2003; Tan et al., 2007). The TM domain is organized as a hydrophobic α-helix, which was initially proposed to contribute to the docking of Bcl-2 into membranes. Indeed, when deleted from its C-terminal hydrophobic domain, Bcl-2 becomes cytosolic (Nguyen et al., 1993). Interestingly, this observation is not restricted to Bcl-2 itself but appears to be a genuine rule for almost all multidomain Bcl-2 homologs. However, the sole presence of a hydrophobic C-terminal region does not explain why closely related Bcl-2 homologs exhibit different subcellular distributions. For instance, Bcl-2, and Bcl-xL both are anti-apoptotic and possess a hydrophobic α-helix. Nonetheless, Bcl-xL is mainly found in the mitochondria -although being in part cytosolic and ER resident- whereas Bcl-2 is found at the level of the ER, the NOM, the nucleus and the mitochondrial membranes (Krajewski et al., 1993; Akao et al., 1994; Kaufmann et al., 2003). By analyzing the TM domains of these proteins, Borner and colleagues discovered that the apparent localization differences came from the residues flanking the hydrophobic α-helix. Using targeted mutagenesis, they demonstrated that, in the Bcl-xL sequence, the presence of at least two positively charged residues—located at each side of the hydrophobic α-helix—is critical for Bcl-xL insertion into the MOM (Kaufmann et al., 2003). Indeed, such residues are absent in the Bcl-2 TM domain, which presumably allows Bcl-2 to interact with other intracellular membranes. In this respect, TM swapping between Bcl-xL and Bcl-2 resulted in inversion of their subcellular localization. Finally, Borner and colleagues demonstrated that the TM domain of Bcl-xL represents a bona fide MOM targeting signal since it is sufficient to address recombinant GFP to the MOM, in contrast to that of Bcl-2 that addresses the GFP to both mitochondrial and ER membranes (Kaufmann et al., 2003).
It is worth to note that the TM domain of Bcl-xL also contributes to homodimerization (Jeong et al., 2004). Indeed, substantial amounts of Bcl-xL are found in the cytosol where it forms homodimers through its TM domain (Jeong et al., 2004). Moreover, the deletion of the last two C-terminal amino acids (namely R233 & K234) significantly decreased homodimer formation, whereas the deletion of the last four amino acids completely abolished this process (Jeong et al., 2004).
Bcl-xL is not the only Bcl-2 homolog residing in the cytosol. For example in healthy cells, Bax is mainly cytosolic but becomes mitochondrial upon cell death induction (Hsu et al., 1997). This is due to the fact that under non-stressful conditions, the TM domain (α9 helix) of Bax is sequestered within its hydrophobic surface groove (Wolter et al., 1997). Upon stress induction, Bax α9 helix unmasks and anchors the protein to the MOM. Interestingly, in such conditions, Bax ΔTM deletion mutant can still translocate to the mitochondria, suggesting that additional internal signals contribute to the proper subcellular localization of Bax (Er et al., 2007). It has been proposed that the N-terminus α1 helix can directly address activated Bax toward the mitochondrial surface. Indeed, Vallette and colleagues demonstrated that Bax α1 helix is capable of docking red fluorescent protein (RFP) to the mitochondria (Cartron et al., 2003). It was also shown that the α5 helix was necessary and sufficient for Bax oligomerization and MOMP induction (George et al., 2007). Additional studies further demonstrated that fusions of GFP with Bax fragments containing the α5, α6, and/or α9 helices, either alone or within the α5α6 or α5-α9 constructs, all localize to the mitochondria. Specifically, α5 and α6 helices not only bind intrinsically to the MOM, but also show high membrane destabilization properties. On this basis, mitochondria-targeted cytotoxic agents, referred to as “poropeptides,” could be developed in our lab from natural Bcl-2-like membrane-active segments such as Bax α5 and α6 helices (Valero et al., 2011).
Bcl-2 α1 helix, also referred to as Bcl-2 BH4 domain, may also participate in subcellular targeting. For instance, the human FK506-binding protein 38 (FKBP38), a mitochondrial chaperone, is able to interact with and effectively target Bcl-2 to the mitochondria (Shirane and Nakayama, 2003). Importantly, a Bcl-2 mutant lacking the BH4 domain fails to interact with FKBP38 and is translocated to the nucleus, where it promotes apoptosis (Portier and Taglialatela, 2006).
Bcl-2 BH4 domain also interacts with the inositol trisphosphate receptor (IP3R) at the level of the ER (Monaco et al., 2012a). This domain interacts with the MTDII of IP3R and regulates its activity. Interestingly, subtle differences in the amino acid composition of Bcl-2 and Bcl-xL BH4 domains explain—at least partly—these distinct localizations. Bultynck and colleagues identified Lys17 in the BH4 domain of Bcl-2 as critical for IP3R binding (Monaco et al., 2012b). Interestingly enough, the Bcl-xL BH4 domain, which lacks Lys17, binds instead to the mitochondrial voltage dependent anion channel (VDAC). In fact, Bcl-xL α1 helix is able to target GFP to the mitochondria (McNally et al., 2013). Moreover, studies by Cheng and colleagues suggested that VDAC2, a minor VDAC isoform, is critical for Bak localization to the mitochondria (Cheng et al., 2003). Indeed, VDAC2 interaction with Bak was shown to prevent Bak oligomerization, compromising its pro-apoptotic activity. This model was further developed by the team of Hajnóczky who showed that VDAC2 is critical for the exclusive recruitment into the MOM of newly synthesized Bak molecules (Roy et al., 2009).
Finally, Bcl-2 proteins are major trans-regulating elements that control Bax subcellular localization. Indeed, BH3-only proteins such as Bim, Puma, or the pro-apoptotic Bcl-2 homolog tBid, have been suggested to mediate the insertion of cytosolic Bax into the OMM by directly interacting with Bax (Desagher et al., 1999; Letai et al., 2002; Kuwana et al., 2005; Kim et al., 2009). In this respect it has recently been shown that, with the notable exception of Bad, all canonical BH3-only proteins possess a functional C-terminus TM domain (Wilfling et al., 2012; Andreu-Fernández et al., 2016).
The observations that upon apoptotic stress Bcl-xL and Bax translocate from the cytosol to the mitochondria suggested that their localization is not static but rather reflects dynamic processes depending on the cellular status and microenvironment. Indeed, Youle and colleagues (Wolter et al., 1997) while exploring the localization of Bcl-2, Bcl-xL, and Bax in live cells using GFP fusion proteins, revealed that Bax translocates from the cytosol to the mitochondria upon apoptosis induction. Confocal microscopy followed by photobleaching experiments confirmed the cytosolic localization of GFP-Bax, contrary to organelle-bound GFP-Bcl-2. Apoptosis induced by Staurosporine (STS) led to significant changes of GFP-Bax distribution, as illustrated by the switch from a diffuse to a punctate mitochondrial fluorescence pattern. Importantly, they noticed that GFP-Bax redistribution took place before apoptotic cells shrinkage. Moreover, deletion of the C-terminal hydrophobic tail both prevented GFP-Bax redistribution and suppressed its death-promoting activity, confirming the importance of the TM domain. Nevertheless, the sole targeting of Bak or Bax to the MOM, does not necessarily imply that these proteins are spontaneously active once the TM regions are membrane-integrated. Activation of Bak and Bax appears to request some additional conformational changes, such as (1) α1 helix exposure, (2) transient BH3 exposure, (3) protection of the membrane-embedded Bcl-2 core, and (4) increased proximity of embedded monomers (George et al., 2007). Overall, these data raised the following major issue: “if Bax resides on the mitochondrial membranes upon apoptosis induction, could it be that it diffuses back to the cytosol, and thus how, when the microenvironment returns to normal physiological conditions?”
A few years later, Youle and colleagues directly demonstrated that the retrotranslocation of Bax from the mitochondria to the cytosol could be induced by the anti-apoptotic protein Bcl-xL (Edlich et al., 2011). The original purpose of their work was to understand how anti-apoptotic Bcl-2 proteins controlled Bax localization without directly interacting with cytosolic Bax. To this aim, they hindered conformational changes involving the α1 and α2 helices of Bax, which encompass the BH3 domain, to check their contribution to Bax activation. Indeed, such conformational changes were supposed to be required for Bax integration into the MOM. Thus, to maintain Bax inactive, residues F30 and L63 were replaced with cysteines, leading to the formation of intramolecular disulphide bonds between α1 and α2, along with other substitutions. Such intramolecular tethers were proven to maintain the protein in an inactive conformation, similar to that of wild type (WT) Bax. Caspase 3/7 apoptosis assays showed that these tethers compromise the activation of Bax by BH3-only proteins and affect its regulation by Bcl-xL, which must be added to their capacity to interfere with detergent-induced Bcl-xL binding. Furthermore, tethered Bax appeared to locate to the mitochondria, which was rather unexpected since Bax was supposed to be cytosolic, when maintained inactive. In fact, Fluorescence Loss In Photobleaching (FLIP) experiments revealed that Bcl-xL increased mitochondrial Bax off rates, without any detectable competition between Bax and Bcl-xL for MOM binding, proving that Bax effectively retrotranslocates from the mitochondria to the cytosol, when inactivated.
Bcl-xL retrotranslocation was later on analyzed by FLIP, using HCT116 cells (knocked out for bax and bak genes) expressing GFP-Bcl-xL. Apparently, in the absence of Bax, Bcl-xL localized predominantly to the mitochondria, slowly translocating back to the cytosol. However, the overexpression of Bax accelerated Bcl-xL translocation suggesting that both proteins interact at the level of the mitochondria and retro-translocate to the cytosol as a heterocomplex, which eventually dissociates (Figure 2). Of note, the overexpression of Bcl-2 and Mcl-1 accelerated Bax retrotranslocation in a similar manner to that of Bcl-xL. Thus, the retrotranslocation of Bax into the cytosol caused by apoptosis inhibitors may account for the observed cytosolic distribution of Bax in healthy cells (Edlich et al., 2011). Shortly after, Todt and colleagues showed that Bcl-xL C-terminal residues are important for Bax retrotranslocation, which is itself preceded by a conformational change unmasking Bax BH3 domain (Todt et al., 2013). Indeed, Bax retrotranslocation appears to depend on two types of interactions: (1) recognition of the Bax BH3 domain by the hydrophobic groove of Bcl-xL and (2) binding of Bcl-xL membrane anchor to Bax. The proposed model suggests that “Bax is the freight and Bcl-xL is the carrier for shuttling from the mitochondria into the cytoplasm.”
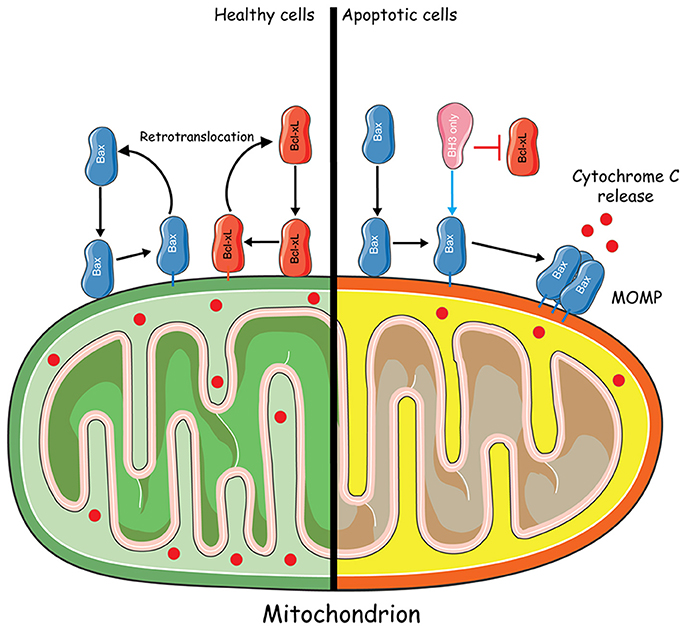
Figure 2. Mitochondria-to-cytosol Bax trafficking. In healthy cells, Bax constantly translocates from the cytosol to the mitochondria. Retrotranslocation of Bax from the mitochondria to the cytosol could be induced by the anti-apoptotic protein Bcl-xL through the formation of a heterocomplex. In apoptotic conditions, Bcl-xL is inhibited by BH3 only sensitizers. Bax translocates to the mitochondria where it becomes activated by BH3 only activators and forms a Bax pore which leads to MOMP and Cytochrome C release. Some elements of the figure were produced using Servier Medical art image bank (www.servier.com).
In 2015, the same team undertook a detailed comparison of Bak and Bax translocation mechanisms (Todt et al., 2015). They found that Bak is actually present in the cytosol and, using FLIP measurements, that Bcl-xL is responsible for Bak retrotranslocation from mitochondria to the cytosol. Overexpression of Mcl-1, but not Bcl-2, was found to accelerate this shuttling, as well as Bax. Thus, the same retrotranslocation process appears to take place for both Bax and Bak, Bax/Bak BH3 domain interaction with the hydrophobic groove of Bcl-xL being critical. Furthermore, the differential localization of Bak in the tissues might partly result from different Bak shuttling rates.
In fact, shuttling rates seem to depend more on the hydrophobicity rather than on the sequence of the TM domain. Using Bax and Bak proteins with swapped TM domains, Todt and colleagues demonstrated that increased Bax retrotranslocation to the cytosol protects the cells from Bax activation. Actually, not only Bax and Bak, but also MOM-integrated Bcl-2 proteins such as Bcl-xL, seem to shuttle similarly.
Although being predominantly mitochondrial, Bak leads to apoptosis only in the presence of apoptotic stimuli (Todt et al., 2015). On the other hand, when activated, Bax cannot be translocated anymore from the MOM to the cytosol (Edlich et al., 2011). Therefore, under non-apoptotic circumstances, Bax-dependent MOMP is blocked due to increased Bax retro-translocation to the cytosol, further reducing the time spent at the level of the MOM in order to prevent Bax activation (Edlich et al., 2011; Todt et al., 2015).
Although Bax and Bak shuttle between the cytosol and the mitochondria, it remains unclear if this process could occur between the mitochondria and the ER. Indeed both proteins were shown to be ER-resident where they control ER-mediated Ca2+ release and apoptosis induction (Scorrano et al., 2003; Zong et al., 2003). As a matter of fact, mitochondria and ER are continually moving along the cytoskeleton and undergo morphological changes via processes like membrane fission, fusion, degradation and renewal. In this regard, peroxisomes play an important role in ER-mitochondria protein exchanges. Indeed mitochondria-derived vesicles (MDVs) were shown to transport phospholipids and proteins from the mitochondria to the peroxisomes (Braschi et al., 2010) whereas peroxisomal proteins may be produced on membrane-bound polysomes of the ER and then transported to the peroxisomes (Dimitrov et al., 2013). Furthermore, it was recently shown that peroxisomes could also arise “de novo” from the fusion of organelle-derived membrane vesicles from the ER and mitochondria, highlighting the dynamic interplay between these organelles (Sugiura et al., 2017). Interestingly, some pro-apoptotic proteins, including Bak, were shown to localize to peroxisomes. This was found to occur when Bak could no longer interact with VDAC2 at the MOM (Cheng et al., 2003; Roy et al., 2009; Hosoi et al., 2017). Moreover, loss of VDAC2 diverts Bak into the peroxisomes, revealing a new activity for Bak in the control of peroxisome membrane integrity and the release of soluble peroxisomal matrix proteins (Hosoi et al., 2017).
Bcl-2 proteins may also navigate between the mitochondria and the Golgi apparatus. Indeed pro-apoptotic Bax and Bok were found to be addressed to this later compartment (Figure 3). Bok was suggested to act upstream of Bax and Bak, controlling the communication between ER-Golgi compartments and the mitochondria through apoptotic signals. In fact, Echeverry and colleagues observed that Bok, when overexpressed, contributed to the fragmentation of the Golgi and the ER before the activation of caspases. On the other hand, Bok silencing resulted in Golgi disassembly under stressful conditions, thus boosting ER stress and subsequently activating BH3-only proteins and apoptosis (Echeverry et al., 2013).
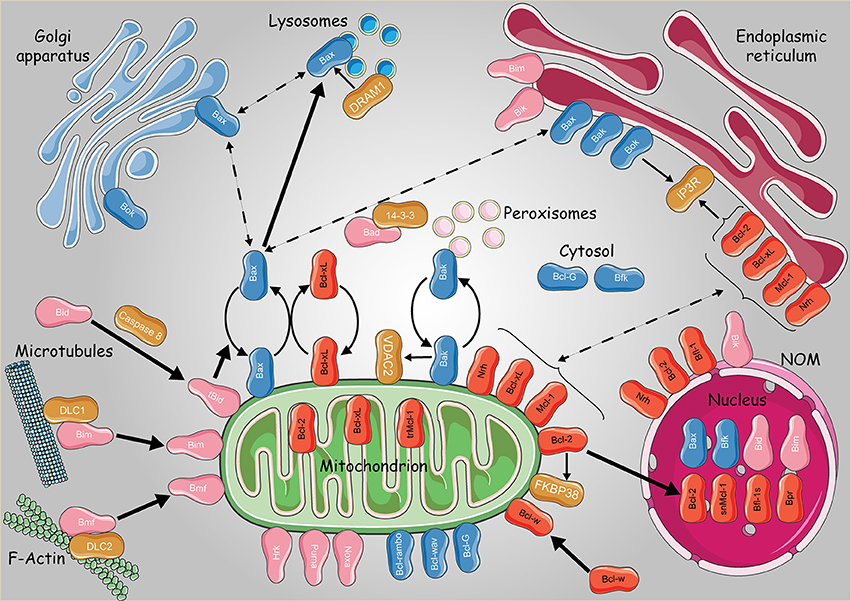
Figure 3. Subcellular dynamics of the Bcl-2 family of proteins. Schematic representation of the intracellular localizations and dynamics of anti-apoptotic (in red) pro-apoptotic multidomain (in blue) and BH3 only proteins (pink). Non Bcl-2 homologs were presented with dark orange boxes. Continuous and bold lines represent protein translocation in physiological and stress conditions, respectively. Dashed lines represent hypothetical subcellular translocation. trMcl-1, truncated Mcl-1; snMcl-1, shortened nuclear Mcl-1; Bfl-1s, Bfl-1 short isoform. Some elements of the figure were produced using Servier Medical art image bank (www.servier.com).
Bax localization to the Golgi was observed in human embryonic stem cells (Dumitru et al., 2012). In fact, using the 6A7 antiBax monoclonal and the TGN46 trans-Golgi network protein, Dumitru and colleagues demonstrated that Bax locates in the trans Golgi compartment in a specific manner, being totally absent from the cis compartment. In addition, after DNA damage, Bax seemed to translocate from the Golgi to the mitochondria in a p53-dependent manner (Dumitru et al., 2012). Another study performed by Guan and colleagues showed that the lysosomes are also involved in Bax-mediated apoptosis, revealing a novel crosstalk between autophagy and apoptosis through DRAM1 (DNA damage-regulated autophagy modulator 1) (Guan et al., 2015). Actually, Bax affects the permeability of the lysosomes and the rate of lysosomal cathepsins release (Feldstein et al., 2006; Oberle et al., 2010). Furthermore, upon treatment with the mitochondrial complex II inhibitor 3-nitropropionic acid (3NP), DRAM1 and Bax expression levels were increased, contributing to lysosomal Bax relocalization in a DRAM1-dependent manner, causing the release of lysosomal cathepsin B as well as Bid cleavage (Guan et al., 2015). Accordingly, the next questions were: “How does Bax migrate in all these cellular compartments? Is there a sort of retrotranslocation similar to that of cytosol-mitochondria?”
The lysosomes are formed by the fusion of transport vesicles budded from the trans Golgi network with endosomes, which contain molecules taken up by endocytosis at the level of the plasma membrane. On this basis, the presence of Bax in the trans Golgi compartments can be clarified by suggesting that lysosomes could act as transport vesicles of Bax. However, this latter issue needs further investigations.
Thus, two hypotheses are open regarding the intracellular dynamics of Bcl-2 proteins: either (1) each protein has a specific localization signal that targets to a specific compartment, or (2) the same protein can a priori locate in different compartments, inter-organelle translocation being ensured by dedicated carriers.
Actually, both hypotheses are non-mutually exclusive. Intracellular vesicles—along with protein-protein interactions—could be responsible for the transit of Bcl-2 proteins inside the cell as suggested in the case of peroxisomal Bak. On the other hand, protein-protein interactions and stress signals can also trigger the unmasking of specific sequences addressing the protein to a specific compartment as in the case of Bcl-xL and Bax.
Bcl-2 proteins are obviously able to shuttle between different cellular compartments. However, until now, it has been generally admitted that their primary role is to control apoptosis at the level of the mitochondria. After more than 30 years of research, it is now acknowledged that Bcl-2 proteins play multiple non-canonical roles beyond apoptosis (Bonneau et al., 2013). They appear to be able do so by means of their various subcellular localizations.
Besides their mitochondrial localization, Bcl-2 proteins can also localize to the ER. At the level of this later compartment, Bcl-2 family members regulate apoptosis through the control of Ca2+ fluxes through the ER-mitochondria contact sites. Indeed, mitochondria constantly take up Ca2+ ions, stimulating biogenesis and ATP production. They also act as an intracellular Ca2+ buffer due to their rapid uptake of Ca2+ ions, when massively released from the ER. Excessive accumulation of mitochondrial Ca2+ may lead to mitochondria swelling and cell death. Bcl-2 proteins control Ca2+ trafficking through direct interactions with ER Ca2+ transporters. For example, Bcl-2 has long been studied for its interaction with IP3R. This interaction, either with the BH4 domain of Bcl-2 (Rong et al., 2008, 2009) or with its trans-membrane domain (Ivanova et al., 2016), inhibits ER Ca2+ release. On the other hand, the hydrophobic pocket of Bcl-xL (Yang et al., 2016) sensitizes IP3R to low levels of IP3, leading to the opening of the channel (White et al., 2005).
Using the zebrafish model, we demonstrated that a Bcl-2 homolog, namely Nrz (Nr-13 ortholog in zebrafish), is critical during the early stages of zebrafish development (Arnaud et al., 2006). In zebrafish Nrz protein possesses a dual subcellular localization both at the ER and the mitochondria. Nrz silencing causes premature gastrulation arrest followed by detachment of the entire blastomeres from the yolk sac (Popgeorgiev et al., 2011). By performing a series of time lapse and confocal microscopy experiments, we demonstrated that this phenotype is due to the premature formation of the actin-myosin contractile ring, a supramolecular structure, which squeezes the yolk cell at the level of the margin and cuts the embryo in half. Furthermore, we showed that the ER-resident Nrz, but not mitochondrial-resident Nrz, was critical for gastrulation progression. We showed that, at the level of the ER, Nrz interacts with IP3R1 Ca2+ channels (Popgeorgiev et al., 2011) and regulates the time course of the release of Ca2+ into the yolk sac by competing with IP3 (Bonneau et al., 2014). In addition, we established that, by doing so, Nrz controls the formation of the contractile actin-myosin ring via a Calmodulin-myosin light chain kinase (MLCK)-dependent pathway. This process seems evolutionary conserved since Nrh, the human Nrz ortholog, also prevents IP3R opening. Interestingly, Nrh anti-apoptotic activity was reported to be under the control of inositol 1,4,5-trisphosphate (IP3) receptor-binding protein (IRBIT), another partner of IP3R (Bonneau et al., 2016). In fact, Nrh may indirectly prevent apoptosis by controlling ER-stress. Indeed, there is evidence that Nrh acts upstream of the Unfolded Protein Response (UPR) by inhibiting ERCa2+ release through IP3R closure (Nougarede et al., 2018).
On the other hand, regarding cell death accelerators, the only pro-apoptotic protein reported to directly interact with IP3R is Bok. This occurs through the BH4 domain of Bok which binds the coupling domain of IP3R1 and IP3R2. However, this interaction does not affect Ca2+release, although it protects the channel from proteolytic cleavage (Schulman et al., 2013).
Numerous Bcl-2-related proteins were found to localize to the nucleus, contributing to vital processes. For example, Gross and colleagues reported that a fraction of the BH3-only protein Bid located to the nucleus, plays a role in the DNA damage response (Kamer et al., 2005; Zinkel et al., 2005). In fact, the ATM and ATR kinases are activated following DNA damage, causing cell cycle arrest and subsequent DNA repair or apoptosis. Interestingly, one of the targets of ATM is nuclear Bid, which is critical for cell cycle arrest at the S phase and apoptosis induction (Zinkel et al., 2005). It was proposed that, following DNA damage, Bid could be transported to the nucleus as part of a protein complex since it lacks an obvious nuclear localization signal. Accordingly, Bid might either help stabilize the complex or facilitate subsequent enzymatic steps. In this respect, Bid phosphorylation is critical for maintaining genomic stability at S phase check points. Overall, these data suggest that Bid acts as a mediator between apoptosis and cell cycle regulation during S phase.
Regarding Bcl-2, much evidence support existing interactions—either direct or indirect—with transcription factors. Indeed, de Moissac and colleagues reported an interaction between Bcl-2 and IκBα, the cytoplasmic inhibitor of the ubiquitously expressed transcription factor Nuclear Factor κB (NFκB). They could show that Bcl-2 facilitates IκBα degradation, activating in turn NFκB (de Moissac et al., 1998). Another study provided direct evidence that NFκB transcriptionally regulates bcl2 gene expression, directly linking the TNF-α/NFκB signaling pathway to bcl2 expression in human prostate carcinoma cells (Catz and Johnson, 2001). This regulation was later shown to control cell invasiveness in estrogen receptor alpha (ERα)-negative breast cancer cells (Wang et al., 2007). A recent study delineated the regulation of gene transcription by extra-mitochondrial Bcl-2 proteins, notably Bcl-2, Bcl-xL, and Mcl-1. In fact, these proteins engage a BH domain found in SUFU, the tumor suppressor and antagonist of GLI DNA-binding proteins. By doing so, they promote SUFU turnover, impede its interaction with GLI and allow the expression of GLI target genes (including Bcl-2, Bcl-xL and Mcl-1) thus prompting the survival and growth of cancer cells (Wu et al., 2017).
Bcl-2 family proteins are arguably one of the main regulators of the mitochondrial outer membrane permeability. Since their discovery it became abundantly clear that the vast majority of these proteins can localize in multiple subcellular compartments where they not only remotely control MOMP but also participate in additional non apoptotic functions (Bonneau et al., 2013). Although structurally related, Bcl-2 family proteins present important differences in their subcellular localizations, which were attributed to subtle intramolecular variations of their TM amino acid composition. These observations led to a rather predeterministic view of Bcl-2 family intracellular distribution in which once synthetized, these proteins were directly targeted to precise subcellular compartments (Kaufmann et al., 2003). However, more recent data proposed that at least part of the Bcl-2 family can translocate and change their subcellular localizations depending on the cellular status and apoptotic stimuli (Edlich et al., 2011; Todt et al., 2013, 2015). How exactly Bcl-2 proteins navigate through the intracellular membrane network remains currently unexplained. Two recent reports demonstrated that at least the pro-apoptotic Bax and Bak can localize to lysosomes and peroxisomes respectively (Oberle et al., 2010; Guan et al., 2015; Fujiki et al., 2017). Peroxisomes are critical in ER-mitochondria protein exchanges and lysosomes are derived from ER and Golgi membranes. Thus, it is tempting to speculate that Bcl-2 proteins trafficking may occur through vesicular transport inside the cell. However, further studies are expected to reveal the exact mechanisms of Bcl-2 family subcellular dynamics.
Is there a difference between subcellular distribution of Bcl-2 proteins between normal and cancer cells and if so, does this difference contribute to cancer progression or chemoresistance represents another open question. Early studies suggested that Bcl-2 is similarly distributed in normal peripheral blood lymphocytes and CLL cells (de Jong et al., 1994). However, more recently, differences in the intracellular distribution have been observed for individual Bcl-2 homologs. For example, nuclear localization of Bcl-2 and Bcl-xL was observed in various cancer cells including breast cancer, endometrial carcinoma, squamous cell carcinoma, and astrocytoma (Chan et al., 1995; Mosnier et al., 1996; Choi et al., 2016). Interestingly, nuclear Bcl-xL localization was found to correlate with epithelial–mesenchymal transition and increased metastasis formation. Thus, it would be of interest to further analyse the molecular mechanisms behind Bcl-2 intracellular distribution in tumor cells (Choi et al., 2016).
Globally, and to conclude, this review provides new insights into the multiple subcellular localizations of Bcl-2 proteins and the way-too-long masked importance of their extra-mitochondrial distribution.
NP, LJ, and GG wrote the manuscript and approved it for publication.
This work is supported by AFM telethon, Ligue contre le cancer, Fondation ARC. LJ is recipient of a Fellowship from the French Ministry for Higher Education and Research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Akao, Y., Otsuki, Y., Kataoka, S., Ito, Y., and Tsujimoto, Y. (1994). Multiple subcellular localization of bcl-2: detection in nuclear outer membrane, endoplasmic reticulum membrane, and mitochondrial membranes. Cancer Res. 54, 2468–2471.
Andreu-Fernández, V., García-Murria, M. J., Bañó-Polo, M., Martin, J., Monticelli, L., Orzáez, M., et al. (2016). The C-terminal domains of apoptotic BH3-only proteins mediate their insertion into distinct biological membranes. J. Biol. Chem. 291, 25207–25216. doi: 10.1074/jbc.M116.733634
Aouacheria, A., Arnaud, E., Venet, S., Lalle, P., Gouy, M., Rigal, D., et al. (2001). Nrh, a human homologue of Nr-13 associates with Bcl-Xs and is an inhibitor of apoptosis. Oncogene 20, 5846–5855. doi: 10.1038/sj.onc.1204740
Arnaud, E., Ferri, K. F., Thibaut, J., Haftek-Terreau, Z., Aouacheria, A., Le Guellec, D., et al. (2006). The zebrafish bcl-2 homologue Nrz controls development during somitogenesis and gastrulation via apoptosis-dependent and -independent mechanisms. Cell Death Differ. 13, 1128–1137. doi: 10.1038/sj.cdd.4401797
Barboule, N., Demeter, K., Benmeradi, N., and Larminat, F. (2009). Bcl-2 is an integral component of mitotic chromosomes. Cell Biol. Int. 33, 572–577. doi: 10.1016/j.cellbi.2009.02.012
Barboule, N., Truchet, I., and Valette, A. (2005). Localization of phosphorylated forms of Bcl-2 in mitosis: co-localization with Ki-67 and nucleolin in nuclear structures and on mitotic chromosomes. Cell Cycle 4, 590–596. doi: 10.4161/cc.4.4.1587
Bonneau, B., Ando, H., Kawaai, K., Hirose, M., Takahashi-Iwanaga, H., and Mikoshiba, K. (2016). IRBIT controls apoptosis by interacting with the Bcl-2 homolog, Bcl2l10, and by promoting ER-mitochondria contact. eLife 5:e19896. doi: 10.7554/eLife.19896
Bonneau, B., Nougarède, A., Prudent, J., Popgeorgiev, N., Peyriéras, N., Rimokh, R., et al. (2014). The Bcl-2 homolog Nrz inhibits binding of IP3 to its receptor to control calcium signaling during zebrafish epiboly. Sci. Signal. 7:ra14. doi: 10.1126/scisignal.2004480
Bonneau, B., Prudent, J., Popgeorgiev, N., and Gillet, G. (2013). Non-apoptotic roles of Bcl-2 family: the calcium connection. Biochim. Biophys. Acta 1833, 1755–1765. doi: 10.1016/j.bbamcr.2013.01.021
Braschi, E., Goyon, V., Zunino, R., Mohanty, A., Xu, L., and McBride, H. M. (2010). Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 20, 1310–1315. doi: 10.1016/j.cub.2010.05.066
Cartron, P. F., Priault, M., Oliver, L., Meflah, K., Manon, S., and Vallette, F. M. (2003). The N-terminal end of Bax contains a mitochondrial-targeting signal. J. Biol. Chem. 278, 11633–11641. doi: 10.1074/jbc.M208955200
Catz, S. D., and Johnson, J. L. (2001). Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 20, 7342–7351. doi: 10.1038/sj.onc.1204926
Chan, W. K., Mole, M. M., Levison, D. A., Ball, R. Y., Lu, Q. L., Patel, K., et al. (1995). Nuclear and cytoplasmic bcl-2 expression in endometrial hyperplasia and adenocarcinoma. J. Pathol. 177, 241–246. doi: 10.1002/path.1711770305
Cheng, E. H., Sheiko, T. V., Fisher, J. K., Craigen, W. J., and Korsmeyer, S. J. (2003). VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517. doi: 10.1126/science.1083995
Chen-Levy, Z., Nourse, J., and Cleary, M. L. (1989). The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14;18) translocation. Mol. Cell. Biol. 9, 701–710. doi: 10.1128/MCB.9.2.701
Choi, S., Chen, Z., Tang, L. H., Fang, Y., Shin, S. J., Panarelli, N. C., et al. (2016). Bcl-xL promotes metastasis independent of its anti-apoptotic activity. Nat. Commun. 7:10384. doi: 10.1038/ncomms10384
Cleary, M. L., Smith, S. D., and Sklar, J. (1986). Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell 47, 19–28. doi: 10.1016/0092-8674(86)90362-4
de Jong, D., Prins, F. A., Mason, D. Y., Reed, J. C., van Ommen, G. B., and Kluin, P. M. (1994). Subcellular localization of the bcl-2 protein in malignant and normal lymphoid cells. Cancer Res. 54, 256–260.
Delbridge, A. R., Grabow, S., Strasser, A., and Vaux, D. L. (2016). Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 16, 99–109. doi: 10.1038/nrc.2015.17
de Moissac, D., Mustapha, S., Greenberg, A. H., and Kirshenbaum, L. A. (1998). Bcl-2 activates the transcription factor NFκB through the degradation of the cytoplasmic inhibitor IκBα. J. Biol. Chem. 273, 23946–23951. doi: 10.1074/jbc.273.37.23946
Desagher, S., Osen-Sand, A., Nichols, A., Eskes, R., Montessuit, S., Lauper, S., et al. (1999). Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144, 891–901. doi: 10.1083/jcb.144.5.891
Dimitrov, L., Lam, S. K., and Schekman, R. (2013). The role of the endoplasmic reticulum in peroxisome biogenesis. Cold Spring Harb. Perspect. Biol. 5:a013243. doi: 10.1101/cshperspect.a013243
Dumitru, R., Gama, V., Fagan, B. M., Bower, J. J., Swahari, V., Pevny, L. H., et al. (2012). Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol. Cell 46, 573–583. doi: 10.1016/j.molcel.2012.04.002
Echeverry, N., Bachmann, D., Ke, F., Strasser, A., Simon, H. U., and Kaufmann, T. (2013). Intracellular localization of the BCL-2 family member BOK and functional implications. Cell Death Differ. 20, 785–799. doi: 10.1038/cdd.2013.10
Edlich, F., Banerjee, S., Suzuki, M., Cleland, M. M., Arnoult, D., Wang, C., et al. (2011). Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 145, 104–116. doi: 10.1016/j.cell.2011.02.034
Er, E., Lalier, L., Cartron, P. F., Oliver, L., and Vallette, F. M. (2007). Control of Bax homodimerization by its carboxyl terminus. J. Biol. Chem. 282, 24938–24947. doi: 10.1074/jbc.M703823200
Feldstein, A. E., Werneburg, N. W., Li, Z., Bronk, S. F., and Gores, G. J. (2006). Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1339–G1346. doi: 10.1152/ajpgi.00509.2005
Fujiki, Y., Miyata, N., Mukai, S., Okumoto, K., and Cheng, E. H. (2017). BAK regulates catalase release from peroxisomes. Mol. Cell. Oncol. 4:e1306610. doi: 10.1080/23723556.2017.1306610
George, N. M., Evans, J. J., and Luo, X. (2007). A three-helix homo-oligomerization domain containing BH3 and BH1 is responsible for the apoptotic activity of Bax. Genes Dev. 21, 1937–1948. doi: 10.1101/gad.1553607
Germain, M., Mathai, J. P., and Shore, G. C. (2002). BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem. 277, 18053–18060. doi: 10.1074/jbc.M201235200
Guan, J. J., Zhang, X. D., Sun, W., Qi, L., Wu, J. C., and Qin, Z. H. (2015). DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis. 6:e1624. doi: 10.1038/cddis.2014.546
Hockenbery, D., Nuñez, G., Milliman, C., Schreiber, R. D., and Korsmeyer, S. J. (1990). Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348, 334–336. doi: 10.1038/348334a0
Hosoi, K. I., Miyata, N., Mukai, S., Furuki, S., Okumoto, K., Cheng, E. H., et al. (2017). The VDAC2-BAK axis regulates peroxisomal membrane permeability. J. Cell Biol. 216, 709–722. doi: 10.1083/jcb.201605002
Hsu, Y. T., Wolter, K. G., and Youle, R. J. (1997). Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. U.S.A. 94, 3668–3672. doi: 10.1073/pnas.94.8.3668
Ivanova, H., Ritaine, A., Wagner, L., Luyten, T., Shapovalov, G., Welkenhuyzen, K., et al. (2016). The trans-membrane domain of Bcl-2α, but not its hydrophobic cleft, is a critical determinant for efficient IP3 receptor inhibition. Oncotarget 7, 55704–55720. doi: 10.18632/oncotarget.11005
Jeong, S. Y., Gaume, B., Lee, Y. J., Hsu, Y. T., Ryu, S. W., Yoon, S. H., et al. (2004). Bcl-x(L) sequesters its C-terminal membrane anchor in soluble, cytosolic homodimers. EMBO J. 23, 2146–2155. doi: 10.1038/sj.emboj.7600225
Kamer, I., Sarig, R., Zaltsman, Y., Niv, H., Oberkovitz, G., Regev, L., et al. (2005). Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 122, 593–603. doi: 10.1016/j.cell.2005.06.014
Kaufmann, T., Schlipf, S., Sanz, J., Neubert, K., Stein, R., and Borner, C. (2003). Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J. Cell Biol. 160, 53–64. doi: 10.1083/jcb.200210084
Kim, H., Tu, H. C., Ren, D., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., et al. (2009). Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499. doi: 10.1016/j.molcel.2009.09.030
Krajewski, S., Tanaka, S., Takayama, S., Schibler, M. J., Fenton, W., and Reed, J. C. (1993). Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 53, 4701–4714.
Kuwana, T., Bouchier-Hayes, L., Chipuk, J. E., Bonzon, C., Sullivan, B. A., Green, D. R., et al. (2005). BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17, 525–535. doi: 10.1016/j.molcel.2005.02.003
Letai, A., Bassik, M. C., Walensky, L. D., Sorcinelli, M. D., Weiler, S., and Korsmeyer, S. J. (2002). Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192. doi: 10.1016/S1535-6108(02)00127-7
McNally, M. A., Soane, L., Roelofs, B. A., Hartman, A. L., and Hardwick, J. M. (2013). The N-terminal helix of Bcl-xL targets mitochondria. Mitochondrion 13, 119–124. doi: 10.1016/j.mito.2013.01.004
Monaco, G., Beckers, M., Ivanova, H., Missiaen, L., Parys, J. B., De Smedt, H., et al. (2012a). Profiling of the Bcl-2/Bcl-X(L)-binding sites on type 1 IP(3) receptor. Biochem. Biophys. Res. Commun. 428, 31–35. doi: 10.1016/j.bbrc.2012.10.002
Monaco, G., Decrock, E., Akl, H., Ponsaerts, R., Vervliet, T., Luyten, T., et al. (2012b). Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 19, 295–309. doi: 10.1038/cdd.2011.97
Morishima, N., Nakanishi, K., Tsuchiya, K., Shibata, T., and Seiwa, E. (2004). Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J. Biol. Chem. 279, 50375–50381. doi: 10.1074/jbc.M408493200
Mosnier, J. F., Perret, A. G., Brunon, J., and Boucheron, S. (1996). Expression of the bcl-2 oncoprotein in meningiomas. Am. J. Clin. Pathol. 106, 652–659. doi: 10.1093/ajcp/106.5.652
Nakai, M., Takeda, A., Cleary, M. L., and Endo, T. (1993). The bcl-2 protein is inserted into the outer membrane but not into the inner membrane of rat liver mitochondria in vitro. Biochem. Biophys. Res. Commun. 196, 233–239. doi: 10.1006/bbrc.1993.2239
Nguyen, M., Millar, D. G., Yong, V. W., Korsmeyer, S. J., and Shore, G. C. (1993). Targeting of Bcl-2 to the mitochondrial outer membrane by a COOH-terminal signal anchor sequence. J. Biol. Chem. 268, 25265–25268.
Nougarede, A., Popgeorgiev, N., Kassem, L., Omarjee, S., Borel, S., Mikaelian, I., et al. (2018). Breast cancer targeting through inhibition of the endoplasmic reticulum-based apoptosis regulator Nrh/BCL2L10. Cancer Res. doi: 10.1158/0008-5472.CAN-17-0846. [Epub ahead of print].
Oberle, C., Huai, J., Reinheckel, T., Tacke, M., Rassner, M., Ekert, P. G., et al. (2010). Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ. 17, 1167–1178. doi: 10.1038/cdd.2009.214
Perciavalle, R. M., Stewart, D. P., Koss, B., Lynch, J., Milasta, S., Bathina, M., et al. (2012). Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 14, 575–583. doi: 10.1038/ncb2488
Popgeorgiev, N., Bonneau, B., Ferri, K. F., Prudent, J., Thibaut, J., and Gillet, G. (2011). The apoptotic regulator Nrz controls cytoskeletal dynamics via the regulation of Ca2+ trafficking in the zebrafish blastula. Dev. Cell 20, 663–676. doi: 10.1016/j.devcel.2011.03.016
Portier, B. P., and Taglialatela, G. (2006). Bcl-2 localized at the nuclear compartment induces apoptosis after transient overexpression. J. Biol. Chem. 281, 40493–40502. doi: 10.1074/jbc.M606181200
Quinn, L., Coombe, M., Mills, K., Daish, T., Colussi, P., Kumar, S., et al. (2003). Buffy, a Drosophila Bcl-2 protein, has anti-apoptotic and cell cycle inhibitory functions. EMBO J. 22, 3568–3579. doi: 10.1093/emboj/cdg355
Rong, Y. P., Aromolaran, A. S., Bultynck, G., Zhong, F., Li, X., McColl, K., et al. (2008). Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol. Cell 31, 255–265. doi: 10.1016/j.molcel.2008.06.014
Rong, Y. P., Barr, P., Yee, V. C., and Distelhorst, C. W. (2009). Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim. Biophys. Acta 1793, 971–978. doi: 10.1016/j.bbamcr.2008.10.015
Roy, S. S., Ehrlich, A. M., Craigen, W. J., and Hajnóczky, G. (2009). VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria. EMBO Rep. 10, 1341–1347. doi: 10.1038/embor.2009.219
Schulman, J. J., Wright, F. A., Kaufmann, T., and Wojcikiewicz, R. J. (2013). The Bcl-2 protein family member Bok binds to the coupling domain of inositol 1,4,5-trisphosphate receptors and protects them from proteolytic cleavage. J. Biol. Chem. 288, 25340–25349. doi: 10.1074/jbc.M113.496570
Scorrano, L., Oakes, S. A., Opferman, J. T., Cheng, E. H., Sorcinelli, M. D., Pozzan, T., et al. (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135–139. doi: 10.1126/science.1081208
Shirane, M., and Nakayama, K. I. (2003). Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat. Cell Biol. 5, 28–37. doi: 10.1038/ncb894
Sugiura, A., Mattie, S., Prudent, J., and McBride, H. M. (2017). Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature 542, 251–254. doi: 10.1038/nature21375
Tan, F. J., Fire, A. Z., and Hill, R. B. (2007). Regulation of apoptosis by C. elegans CED-9 in the absence of the C-terminal transmembrane domain. Cell Death Differ. 14, 1925–1935. doi: 10.1038/sj.cdd.4402215
Todt, F., Cakir, Z., Reichenbach, F., Emschermann, F., Lauterwasser, J., Kaiser, A., et al. (2015). Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 34, 67–80. doi: 10.15252/embj.201488806
Todt, F., Cakir, Z., Reichenbach, F., Youle, R. J., and Edlich, F. (2013). The C-terminal helix of Bcl-x(L) mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 20, 333–342. doi: 10.1038/cdd.2012.131
Tsujimoto, Y., Finger, L. R., Yunis, J., Nowell, P. C., and Croce, C. M. (1984). Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 226, 1097–1099. doi: 10.1126/science.6093263
Tsujimoto, Y., Jaffe, E., Cossman, J., Gorham, J., Nowell, P. C., and Croce, C. M. (1985). Clustering of breakpoints on chromosome 11 in human B-cell neoplasms with the t(11;14) chromosome translocation. Nature 315, 340–343. doi: 10.1038/315340a0
Valero, J. G., Sancey, L., Kucharczak, J., Guillemin, Y., Gimenez, D., Prudent, J., et al. (2011). Bax-derived membrane-active peptides act as potent and direct inducers of apoptosis in cancer cells. J. Cell Sci. 124, 556–564. doi: 10.1242/jcs.076745
Vaux, D. L., Cory, S., and Adams, J. M. (1988). Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335, 440–442. doi: 10.1038/335440a0
Wang, X., Belguise, K., Kersual, N., Kirsch, K. H., Mineva, N. D., Galtier, F., et al. (2007). Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 9, 470–478. doi: 10.1038/ncb1559
White, C., Li, C., Yang, J., Petrenko, N. B., Madesh, M., Thompson, C. B., et al. (2005). The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat. Cell Biol. 7, 1021–1028. doi: 10.1038/ncb1302
Wilfling, F., Weber, A., Potthoff, S., Vögtle, F. N., Meisinger, C., Paschen, S. A., et al. (2012). BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ. 19, 1328–1336. doi: 10.1038/cdd.2012.9
Wolter, K. G., Hsu, Y. T., Smith, C. L., Nechushtan, A., Xi, X. G., and Youle, R. J. (1997). Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139, 1281–1292. doi: 10.1083/jcb.139.5.1281
Wu, X., Zhang, L. S., Toombs, J., Kuo, Y. C., Piazza, J. T., Tuladhar, R., et al. (2017). Extra-mitochondrial prosurvival BCL-2 proteins regulate gene transcription by inhibiting the SUFU tumour suppressor. Nat. Cell Biol. 19, 1226–1236. doi: 10.1038/ncb3616
Yang, J., Vais, H., Gu, W., and Foskett, J. K. (2016). Biphasic regulation of InsP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc. Nat. Acad. Sci. U.S.A. 113, E1953–E1962. doi: 10.1073/pnas.1517935113
Yang, T., Kozopas, K. M., and Craig, R. W. (1995). The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J. Cell Biol. 128, 1173–1184. doi: 10.1083/jcb.128.6.1173
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59. doi: 10.1038/nrm2308
Zinkel, S. S., Hurov, K. E., Ong, C., Abtahi, F. M., Gross, A., and Korsmeyer, S. J. (2005). A role for proapoptotic BID in the DNA-damage response. Cell 122, 579–591. doi: 10.1016/j.cell.2005.06.022
Keywords: Bcl-2 family, subcellular localization, mitochondria, endoplasmic reticulum, nucleus
Citation: Popgeorgiev N, Jabbour L and Gillet G (2018) Subcellular Localization and Dynamics of the Bcl-2 Family of Proteins. Front. Cell Dev. Biol. 6:13. doi: 10.3389/fcell.2018.00013
Received: 14 November 2017; Accepted: 30 January 2018;
Published: 13 February 2018.
Edited by:
You-Wen He, Duke University, United StatesReviewed by:
Gustavo P. Amarante-Mendes, University of São Paulo, BrazilCopyright © 2018 Popgeorgiev, Jabbour and Gillet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolay Popgeorgiev, bmlrb2xheS5wb3BnZW9yZ2lldkB1bml2LWx5b24xLmZy
Germain Gillet, Z2VybWFpbi5naWxsZXRAdW5pdi1seW9uMS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.