 David M. Gonzalez
David M. Gonzalez Jill Gregory4
Jill Gregory4 Kristen J. Brennand
Kristen J. Brennand- 1Medical Scientist Training Program, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Department of Developmental and Stem Cell Biology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 3Black Family Stem Cell Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 4Instructional Technology Group, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 5Department of Neuroscience, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 6Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 7Department of Genetics and Genomics, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Current applications of human induced pluripotent stem cell (hiPSC) technologies in patient-specific models of neurodegenerative and neuropsychiatric disorders tend to focus on neuronal phenotypes. Here, we review recent efforts toward advancing hiPSCs toward non-neuronal cell types of the central nervous system (CNS) and highlight their potential use for the development of more complex in vitro models of neurodevelopment and disease. We present evidence from previous works in both rodents and humans of the importance of these cell types (oligodendrocytes, microglia, astrocytes) in neurological disease and highlight new hiPSC-based models that have sought to explore these relationships in vitro. Lastly, we summarize efforts toward conducting high-throughput screening experiments with hiPSCs and propose methods by which new screening platforms could be designed to better capture complex relationships between neural cell populations in health and disease.
Introduction
The development of successful treatments for neurological disease is hampered by a constellation of unique challenges that has resulted in a historically poor rate of success in drug development in this area (Ringel et al., 2013). Many neurological diseases are complex and heterogeneous in nature, exhibiting a breadth of genetic and epigenetic variants of small effect sizes which incompatible and/or impractical to model using traditional in vitro and in vivo models (Sullivan et al., 2012; Fass et al., 2014). Furthermore, the use of rodent models may be insufficient to capture the complexity of human disease, as large differences in neurogenesis, neuroanatomy and distribution of neural cell types within the brain exist between mice and humans. Lastly, the lack of accessibility of neural tissue (both living and post-mortem) combined with difficulties in culturing these cell types in vitro has made developing cell culture models of neurological disease exceedingly difficult and slowed understanding of the pathophysiology underlying a number of these conditions. Human induced pluripotent stem cells (hiPSCs) now offer a nearly limitless potential for disease modeling and drug screening applications. Their great self-renewal and wide differentiation capacity, coupled with the relative ease of producing patient-specific hiPSCs harboring genetic variations implicated in disease, makes possible the generation of large quantities of diverse cell types in a controlled and iterative manner, ideal for high throughput screens to discover and evaluate the efficacy and safety of novel therapeutics (Haggarty and Perlis, 2014; Mertens et al., 2016). In recent years, stem cell models of complex genetic diseases have helped to shed new light on the pathology of various neurodegenerative and neuropsychiatric disorders (Di Giorgio et al., 2008; Dimos et al., 2008; Park et al., 2008; Soldner et al., 2009; Brennand et al., 2011; Yagi et al., 2011; Yoon et al., 2014).
Until recently, studies of mechanism and pathology of neuropsychiatric disorders have tended to focus predominantly on neurons, with little recognition of the complex milieu of cell types that interact with these cells and influence their function. Fortuitously, our newfound ability to generate a variety of cell types found in the central nervous system (CNS) from hiPSCs now presents an exciting opportunity to explore how these various cell types interact with one another in a controlled manner. In this review, we briefly overview current advances toward generating the major neural cell types—neurons, astrocytes, oligodendrocytes and microglia—using stem cell technologies. Further, we highlight recent advancements in understanding non-neuron cell-autonomous effects in the pathology of three representative neuropsychiatric disorders—Amyotrophic Lateral Sclerosis (ALS), schizophrenia and Rett Syndrome—both in vitro and in vivo, with an eye toward using this information to develop hiPSC-based drug screening platforms that better capture disease pathology.
Progress in Cellular Reprogramming Strategies to Generate Major CNS Cell Types
Neurons
The lack of live patient neural tissue, combined with the limitations of post-mortem brain tissue, has spurred efforts to find new methods of models of CNS diseases. Stem cell based technologies, particularly the ability to reprogram induced pluripotent stem cells (hiPSCs), presents a powerful opportunity to create scalable, easily perturbed platforms to study how various genetic abnormalities implicated in disease manifest in altered neural cell function. There exist two general strategies for the creation of neurons using hiPSC-based technologies, directed differentiation and induction, each having advantages and disadvantages that Should be carefully considered when developing an appropriate platform for drug development.
Directed differentiation of iPSCs involves the sequential addition of growth factors and small molecules intended to recapitulate the embryonic developmental cues that drive the generation of mature tissue and different cell types in vivo. Neuronal differentiation protocols typically proceed via a neural progenitor cell (NPC) intermediate, a population of cells with the capacity to self-renew and to generate both neurons and glia. The application of a variety of mitogens can confer different positional identities and enrich for specific subtypes of neural cells. For a more in depth review of these differentiation methods and their developmental basis, an excellent review has been published elsewhere (Mertens et al., 2016). Conversely, neuronal induction proceeds via the forced overexpression of key neuronal transcription factors. When first reported, induced neurons were generated through the overexpression of three factors—BRN2 (also known as POU3F2), achaetescute homolog 1 (ASCL1) and myelin transcriptional factor1-like protein (MYT1L)—in mouse or human fibroblasts (Vierbuchen et al., 2010; Pang et al., 2011; Son et al., 2011; Vadodaria et al., 2016). Collectively known as the BAM factors, they rapidly yielded a heterogeneous population of induced neurons that formed synapses and fired action potentials. A growing understanding of the key transcription factors responsible for assigning regional specificity and neurotransmitter identity has expanded our ability to induce specific populations of excitatory (NGN2) (Zhang et al., 2013), inhibitory (ASCL1, LHX6, DHX2, miR9/124) (Sun et al., 2016), dopaminergic (MASH1, NURR1, LMX1A) (Caiazzo et al., 2011) and serotonergic (ASCL1, NGN2, NKX2.2, FEV, GATA2, LMX1B) (Vadodaria et al., 2016; Xu et al., 2016) neurons for fibroblasts, hiPSCs and/or NPCs.
The choice between directed differentiation and neuronal induction approaches generally reflects needs of the experiment in question. From a practical standpoint, differentiation of iPSCs is costly and time consuming, often requiring 4–6 months to yield functional neurons. In contrast, while induced neurons can be generated from fibroblasts within 1–3 weeks (without the need to reprogram and validate hiPSCs), the populations produced tend to include large numbers of fibroblast-like contaminants and the absolute number of neurons that can be generated is less, reflecting the more limited replicative capacity of fibroblasts. A compromise of inducing neurons directly from hiPSCs promises a larger number of rapidly maturing neurons and may grow more common as subtype-specific induction methodologies become more refined.
Several fundamental differences distinguish neurons generated by directed differentiation or induction. First, chromatin remodeling is an essential part of the cellular reprogramming process; while important epigenetic markers associated with complex genetic diseases may be lost during hiPSC reprogramming, this does not seem to occur to the same extent during neuronal induction (Huh et al., 2016; Mertens et al., 2016). It is thought that cellular division during directed neuronal differentiation requires broad transcription factor access to previously closed regions, thus changing the epigenetic landscape of the cell in question. In contrast, neuronal induction does not require proliferation; the pioneer transcription factors seem to not require extensive chromatin remodeling to complete this process (Liu et al., 2013; Iwafuchi-Doi and Zaret, 2014; Fishman et al., 2015) and so may better preserve epigenetic signatures. Second, the establishment of an embryonic-like state during the reprogramming process is now understood to result in terminally differentiated phenotypes that resemble fetal rather than adult cells (Mariani et al., 2012; Nicholas et al., 2013; Brennand et al., 2015), suggesting that hiPSC-based models may better capture the genetic elements of disease predisposition rather than the disease-state itself. Although novel strategies to accelerate aging in vitro are being uncovered (Miller et al., 2013; Studer et al., 2015), the generation of induced neurons directly from fibroblasts more faithfully maintains epigenetic markers associated with the aging state (Mertens et al., 2016). Third, although neuronal induction bypasses key neurodevelopmental processes, perhaps failing to capture critical biology relevant for disease pathology, induced neurons have now been successfully applied to query neuronal deficits in autism (Chanda et al., 2013; Pak et al., 2015; Yi et al., 2016), bipolar disorder (Bavamian et al., 2015), Alzheimer's disease (Hu et al., 2015).
Astrocytes
Once regarded as a population of cells providing little more than structural support to neuronal networks, the known roles of astrocytes in regulating neuronal function in the CNS is growing. It is now well-recognized that perturbed astrocyte function can exacerbate, and even cause, CNS diseases (Chung et al., 2015); for example, neuroinflammation and ischemia induce two different types of reactive astrocytes, termed A1 (“harmful”) and A2 (“helpful”) (Liddelow and Barres, 2017; Liddelow et al., 2017). Astrocytes are the most abundant cell type in the CNS and perform a wide variety of functions, including axonal guidance, response to inflammation, wound healing, and the formation of the blood brain barrier (Barres, 2008; Zhang and Barres, 2010; Verkhratsky et al., 2012; Freeman and Rowitch, 2013). Importantly, astrocytes are involved in recycling of glutamate and molecular regulation of ion, neurotransmitter and neurohormone concentrations, as well as synaptic pruning and maturity, underscoring their vital role in neuronal communication (Newman, 1995; Danbolt, 2001; Pfrieger, 2009). Astrocytes seem to function in an ordered manner to cover independent territory, contacting thousands of synapses through their multiple processes and branches (Bushong et al., 2002). In addition, these processes can be used to create connections with local capillaries and develop independent neurovascular units in which astrocytes mediate changes in CNS blood flow in response to neuronal activity (Schummers et al., 2008; Wolf and Kirchhoff, 2008; Koehler et al., 2009). Mirroring the diversity of their function, astrocytes display extraordinary heterogeneity in morphology, physiology, gene expression and developmental origin (Zhang and Barres, 2010). The number and complexity of astrocytes increase significantly with neuronal complexity in higher vertebrates, with important differences between rodents and humans that underscore the importance for cell-based systems to understand the contribution of this important cell type in disease pathology (Zhang et al., 2016). The astrocyte-to-neuron ratio increases with evolutionary complexity from low vertebrates to rodents and primates, with humans having a 46% higher glia-neuron ratio even when compared to other primates (Sherwood et al., 2006). Compared to their rodent counterparts, human cortical astrocytes are larger, display greater heterogeneity and diversity, and exhibit marked differences in their electrophysiological properties (Oberheim et al., 2009). When transplanted into mice, human glia (relative to transplanted mouse glia) improved learning and activity-dependent plasticity compared to controls (Han et al., 2013). These results underscore large and important differences between human and murine astrocytes, emphasizing the importance of developing clinically relevant, human-specific in vitro models that can help uncover important roles of human astrocytes in health and disease.
Strategies for the directed differentiation of astrocytes from hiPSCs can either rely on an NPC (Haidet-Phillips et al., 2011; Krencik and Zhang, 2011; McGivern et al., 2013; Serio et al., 2013; Shaltouki et al., 2013) or oligodendrocyte progenitor cell (Jiang et al., 2013) intermediate. Transplanted hiPSC-derived astrocytes integrate and function in the mouse brain in vivo (Haidet-Phillips et al., 2011; Krencik and Zhang, 2011; Jiang et al., 2013; Chen et al., 2015). Consistent with the late emergence of astrocytes during corticogenesis (Tabata, 2015), existing methods tend to be slow (up to 6 months), thus limiting their use for in vitro modeling (Krencik and Zhang, 2011; Jiang et al., 2013; Shaltouki et al., 2013). Induction of astrocytes from mouse fibroblasts occurs more rapidly (within 16 days), via overexpression of the transcription factors NF1A, NF1B, and Sox9 (Caiazzo et al., 2015), the utility of this protocol in generating functional human astrocytes has not been demonstrated. hiPSCs have been differentiated to functional astrocytes for cell-based models of neuropsychiatric disorders in vitro (McGivern et al., 2013; Roybon et al., 2013; Serio et al., 2013; Shaltouki et al., 2013). Resulting hiPSC-derived astrocytes express canonical markers, participate in glucose homeostasis (Shaltouki et al., 2013), and can be engrafted into mouse striatum and upregulate expression of classical reactive astrocytic chemokines when treated with TNFα (Roybon et al., 2013). McGivern et al differentiated hiPSCs from patients with spinal muscular atrophy (SMA) into astrocytes, demonstrating that mutated SMA astrocytes had enlarged cell bodies with shorter processes and more pronounced GFAP expression, indicative of a reactive astrocyte phenotype (McGivern et al., 2013). In a similar fashion, Serio et al created astrocytes from hiPSCs carrying the TDP-43 mutation associated with ALS, and found that the resulting astrocytes had decreased survival, increased levels of TDP-43, and intracellular mislocalization of TDP-43 (Serio et al., 2013). Together, these studies demonstrate the utility of human and disease specific models for probing changes in astrocytic phenotype due to genetic or environmental challenges in vitro.
Oligodendrocytes
Oligodendrocytes wrap neuronal axons in a thick membrane of myelin, enabling rapid conductance of electrical signals through neural networks. Myelination occurs as multi-step process that begins with proliferation and migration of oligodendrocyte precursor cells across large distances to the appropriate axon, synthesis of the myelin sheath itself, and finally wrapping and compaction of the insulating layer around the axon (Barateiro et al., 2016). The final steps of wrapping and compaction occur in the last weeks of gestation and the first postnatal months, with the bulk of the white matter in the CNS being produced during the first year of birth (Barateiro et al., 2016). Interestingly, myelinating capacity appears to be restricted to shortly after oligodendrocyte differentiation is completed (Watkins et al., 2008), such that the appearance of white matter in specific neuroanatomic regions appears to accompany maturation of cognitive function of that area (Nagy et al., 2004; Mabbott et al., 2006; Fields, 2008). Maintenance of myelin occurs throughout life, owing to the proliferation and activation of adult oligodendrocyte precursor cells (Zhu et al., 2007; Young et al., 2013); the inability to repair damaged myelin is implicated in myelin disorders such as ALS and multiple sclerosis (MS) (Barateiro et al., 2016). Oligodendrocytes communicate in a variety of ways with other cell types in the CNS; trophic factors secreted by oligodendrocytes modulate neuron axonal size and regulate distribution of ion channels in the axon (Barres, 2008), whereas gap junctions between astrocytes and oligodendrocytes allow exchange of small molecules that affect electrical coupling, and it appears that astrocytes play a role in the initiation of myelination (Orthmann-Murphy et al., 2008).
Although human oligodendrocytes can be generated from hiPSCs (Goldman and Kuypers, 2015) and are seemingly highly active once transplanted in vivo (Windrem et al., 2014), they myelinate less than 3% of axons in vitro (Kerman et al., 2015). Oligodendrocytes can also be generated through direct conversion methods in mice, via the overexpression of Sox10, oligodendrocyte transcription factor 2 (Olig2) and either Nkx6.2 (Najm et al., 2013) or zinc-finger protein 536 (ZPF536) (Yang et al., 2013); the first method yielded oligodendrocyte restricted induced oligodendrocyte precursor cells, the second retained competence to generate astrocytes as well. It remains uncertain to what extent these same factors will be sufficient to generate induced oligodendrocyte precursor cells from human fibroblasts. Moving forward, methods to regionally pattern the fate and function of differentiated or induced oligodendrocyte precursor cells will be necessary.
Microglia
Microglia maintain homeostasis throughout the CNS through active surveillance following by phagocytic clearance of debris and elimination of synapses during development in a process known as “synaptic pruning”; they also perform vital immune functions as the first line of defense in the nervous system. Using their extensive processes, microglia sweep the CNS parenchyma in search of unhealthy or diseased astrocytes and neurons that fail to express particular receptors and/or secrete “healthy” signals (Davalos et al., 2005; Sieger et al., 2012; Aguzzi et al., 2013; Prinz and Priller, 2014). Microglia measure neuronal activity using neurotransmitter receptors and immune activity by expressing receptors for chemokines and complement factors (Hanisch and Kettenmann, 2007); they respond to stimuli by altering migration, inflammatory response, cytokine release and phagocytic activity (Prinz and Priller, 2014). Microglia are the primary antigen presenting cells of the CNS, presenting antigens to infiltrating T lymphocytes through major histocompatibility complex (MHC) class II complexes (Butovsky et al., 2005). Activation of microglia in response to injury or disease results in a morphological change that differs depending on the signals detected and microglial modulators present (Hanisch and Kettenmann, 2007). Following tissue injury or in response to pathogens, non-neural macrophages undergo polarization into either (pro-inflammatory) or M2 (anti-inflammatory) macrophages. Likewise, activated microglia become polarized into M1-like and M2-like phenotypes; co-culture with M1 microglia leads to a cytotoxic phenotype in neurons and oligodendrocytes, while co-culture with M2 microglia promotes neurite outgrowth cells (Kigerl et al., 2009; Hu et al., 2012). However, microglia have difficulty undergoing polarization to an M2-like phenotype in vitro when compared to non-neural macrophages, hampering a complete understanding of these two different phenotypes (Kim et al., 2004; Durafourt et al., 2012). Interestingly, differences between M1 and M2 macrophages and microglia become difficult to appreciate in inflammatory and neurodegenerative diseases, with microglia co-expressing markers of both subtypes (Dal Bianco et al., 2008; Vogel et al., 2013).
Until recently, no directed differentiation protocols for the creation of microglia existed, limiting our understanding of this cell type to those studies performed using mouse models or post-mortem human tissue. Because microglia are derived from the primitive macrophages in the yolk sac of myeloid lineage (Alliot et al., 1999), they cannot be generated using neural progenitor cells (NPCs) as in other lineages discussed previously. Three recent reports have now detailed the creation of microglia from hiPSCs via a hematopoietic progenitor-like intermediate cell (Muffat et al., 2016; Abud et al., 2017; Pandya et al., 2017). While the protocols differed in their reliance on embryoid body differentiation and/or FACS, all three methods yielded immature microglia-like cells expressing canonical microglial markers and demonstrating phagocytic and migratory functionality (Muffat et al., 2016; Abud et al., 2017; Pandya et al., 2017).
Summary
With a newfound ability to generate all of the major cell types of the CNS - neurons, astrocytes, oligodendrocytes and microglia—human hiPSC-based models are now primed to explore how the interactions of these various cell types contribute to risk for a variety of neuropsychiatric disorders. While post-mortem, animal models and hiPSCs have identified a number of cell autonomous deficits that underlie neurodegenerative (Marchetto et al., 2011; Bahmad et al., 2017; Poon et al., 2017) or psychiatric (Brennand and Gage, 2012; Ho et al., 2015; Wen, 2017) disorders, complex interactions between neural cells can now be explored in a fully human and patient-derived platform (Table 1). Below, we discuss evidence for non-cell autonomous effects in neurodegenerative disease (represented by ALS), psychiatric disease (represented by schizophrenia) and neurodevelopmental disease (represented by Rett Syndrome).
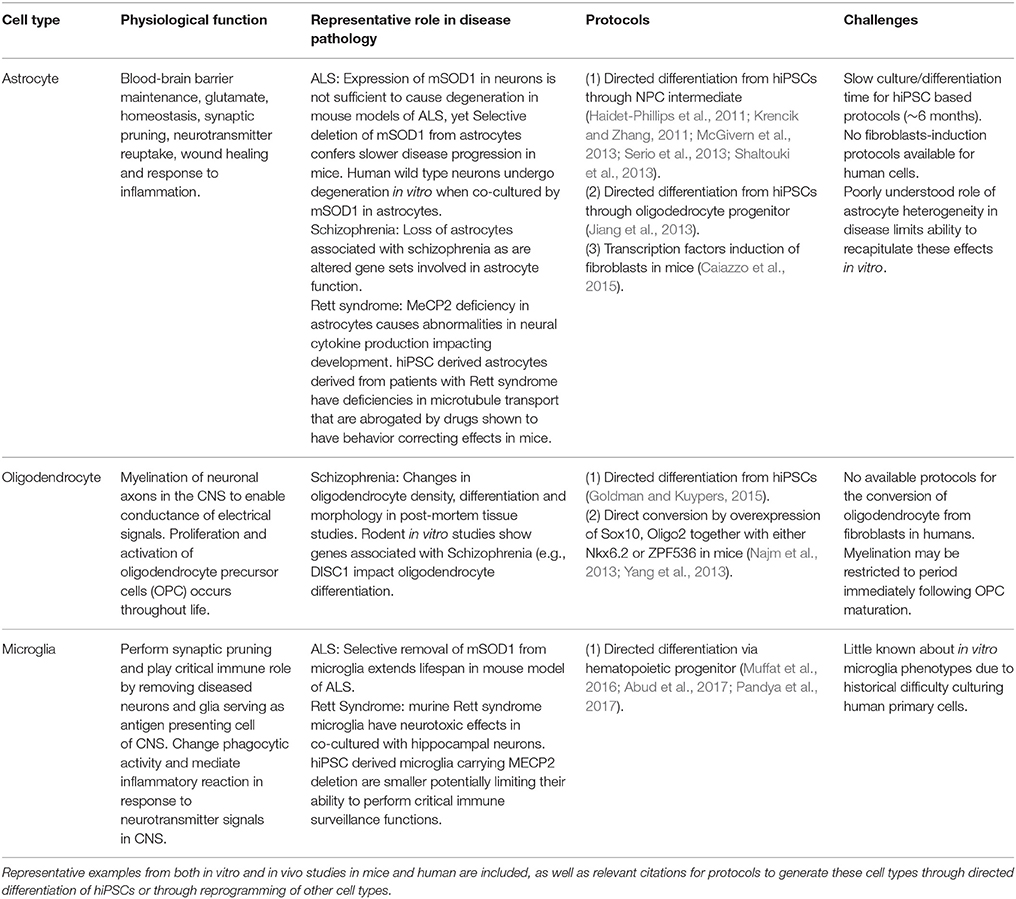
Table 1. Contribution of non-neuron cell types in neurodegenerative and neuropsychiatric diseases.
Non-cell Autonomous Effects in Neuropsychiatric Disorders
Amyotrophic Lateral Sclerosis (ALS)
ALS is a debilitating neurodegenerative condition causing muscle atrophy and loss of control of motor function, rendering patients paralyzed and eventually unable to breathe (Brooks, 1996; Appel et al., 2011). The symptomatic phase of ALS is associated with massive activation of microglia and astrocytes, which destroy motor neurons of the CNS (Kushner et al., 1991; Nagy et al., 1994; Schiffer et al., 1996). Though there is undoubtedly a genetic component to the disease, the majority of cases are idiopathic and with no family history, although a handful of highly penetrant monogenic forms of ALS follow Mendelian inheritance patterns (Brown, 1997; Cole and Siddique, 1999). The initial signs of ALS are mild, and so many patients are not identified until the damage to the tissue is quite significant. Altogether, the complex genetics, unclear environmental compounds and late diagnoses of ALS patients have made understanding the onset of disease pathology very difficult to date.
Mutations in the Super Oxide Dismutase gene (SOD1) lead to a dominant, inherited form of ALS (Rosen et al., 1993), and are well-studied with the hope that by focusing on the most penetrant and significant genetic defects associated with ALS, we may come to understand broader aspects of ALS disease onset and progression that are relevant to other ALS-associated mutations. Initial experiments in rats with mutant SOD1 gene (mSOD1) displayed degenerative symptoms and pathology consistent with ALS (Nagai et al., 2007). Moreover, introduction of the mutant gene into motor neurons in the CNS did not cause neurodegeneration when motor neurons were surrounded by healthy support cells, but even those wild type neurons proximal to mutant glia underwent degeneration (Clement et al., 2003). These data suggest that ALS disease onset and progression might be mediated by negative interactions between various cell types of the CNS, rather than an intrinsic dysfunction in neurons themselves. In fact, selective deletion of the mSOD1 gene from mouse astrocytes in transgenic mice conferred a slower disease progression (Yamanaka et al., 2008). More recently, microglia have also been identified as key mediators of ALS progression; selective removal of the mSOD1 gene from microglia significantly extended lifespan (Boillée et al., 2006). Interestingly, while the interaction between neurons and microglia appears to be protective at first, cellular stress resulting from misfolded mSOD1 ultimately activates microglia to a proinflammatory and neurotoxic phenotype (Appel et al., 2011).
Consistent findings were subsequently observed using in vitro models of ALS. Mouse astrocytes expressing the mSOD1 gene have a toxic effect on wild type neurons in vitro; this effect was stronger in motor neurons than neuronal cell types (Di Giorgio et al., 2007; Nagai et al., 2007). Expression of mSOD1 in neurons alone is insufficient for neurodegeneration. To observe significant injury, mSOD1 must be expressed in glia; moreover, the presence of wild type non-neuronal cells delays degeneration (Clement et al., 2003; Zhao et al., 2010). Sandwich co-cultures of mouse embryonic stem cell (ESC)-derived motor neurons and primary glia allows controlled interaction and subsequent separation of these two cell types, identifying striking differences in autonomous and non-autonomous changes in gene expression (Phatnani et al., 2013).
Ultimately, many of these results have now been reproduced in a human context. As in mouse ESC-derived motor neurons, human ESC-derived motor neurons are also selectively more sensitive to toxic non-cell autonomous effects than interneurons (Di Giorgio et al., 2008). This toxicity was correlated with changes in glial gene expression, which was used to identify candidate molecules associated with the toxic mutant glia-associated effects. For example, when prostaglandin D2 was added exogenously to motor neurons co-cultured with unaffected glia there was a reduction in survival of motor neurons; however, when administered in the presence of a prostaglandin D2 inhibitors, those co-cultures containing mutant astrocytes had better survival relative to control. Similar experiments in which ESC-derived motor neurons were co-cultured with primary mSOD1-expressing human glia also showed a specific decrease in motor neurons, with no adverse effects on other neural cell types (Marchetto et al., 2008). The toxicity of mSOD1-expressing astrocytes was due, at least in part, to reactive oxygen species (ROS) generated by the glia; overexpression of NOX2 increased oxygen radicals, an effect could be reversed using apocynin, a NOX2 inhibitor, preventing the loss of motor neurons when co-cultured with mSOD1-expressing glia.
Although the mSOD1 gene has now been conclusively linked to aberrant glia function and subsequent motor neuron death in ALS, it remains unclear to what effect the other familial and sporadic forms of ALS arise due to astrocyte malfunction. The ongoing generation of a library of ALS patient-specific hiPSCs should provide a powerful tool to interrogate how diverse mutations identified in ALS patients influence glial/neuron interplay within the CNS, and how this contributes to disease pathology. A growing number of recent studies have reported significant changes in mitochondrial function, synapse organization, receptor binding, and neuronal health in ALS-patient derived motor neurons (Dimos et al., 2008; Egawa et al., 2012; Chestkov et al., 2014; Alves et al., 2015). Recently, Meyer et al. reported a method to induce fibroblasts from ALS patients with two independent diseases-associated mutations into astrocytes; relative to controls, these induced astrocytes showed increased toxicity in co-culture with mouse motor neurons in vitro (Meyer et al., 2014). Yet the story of astrocyte/neuron interplay in ALS may be mutation-specific, as co-culture of hiPSC-derived astrocytes and motor neurons carrying a separate ALS causing mutation, TDP-43, revealed that mutant TDP-43 astrocytes did not cause adverse effects on neuronal survival. These results highlight the importance of modeling a variety of ALS-associated mutations, in order to understand the full complexity of processes contributing to neurodegeneration. As gene editing and hiPSC technologies improve, it will become increasingly feasible and important to conduct larger high throughput experiments, in order to understand how various ALS causing mutations affect neuronal health and function in a human disease context.
Schizophrenia
Schizophrenia is a debilitating neuropsychiatric disorder present in 1% of the world population and is associated with increased risk of homelessness, unemployment and suicide (Kooyman et al., 2007; Foster et al., 2012). Symptoms typically present in the early stages of adult life and include but are not limited to psychosis, reduced social engagement and lack of motivation that present a high personal cost and risk to the patient (Lewis and Lieberman, 2000). Post-mortem studies of neural tissue, as well as MRI studies on patients with schizophrenia reveal reduced brain volume, spine density and abnormal neural distribution and connectivity, particularly in the prefrontal cortex and hippocampus (Benes et al., 1991; McCarley et al., 1999; Lewis and Lieberman, 2000; Hulshoff Pol et al., 2002). Both the molecular mechanisms and cell types involved in schizophrenia pathology are complex, confounding the development of novel pharmacological treatments. While the high heritability of schizophrenia (80–85%) points to a strong genetic component, the disorder is highly polygenic and associated with rare highly penetrant mutations as well as common variations of smaller effect; schizophrenia is significantly associated with at least 108 different genetic loci (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) of various effect sizes. The genetic complexity of this disorder necessarily limits the development and applicability of mouse models for this psychiatric disorder, making necessary the creation of human cell-based scalable platforms capable of recapitulating critical aspects of disease initiation and progression.
The diverse neural regions implicated in its pathology suggest that symptoms may be indicative of underlying problems with neural communication involving various cell types. hiPSC-based studies of schizophrenia to date have generally focused on cell-autonomous deficiencies in neuron differentiation (Robicsek et al., 2013; Yoon et al., 2014; Brennand et al., 2015) maturation (Brennand et al., 2011) and function (Wen et al., 2014; Yu et al., 2014). Nonetheless, a growing role for aberrant astrocyte function (Matute et al., 2005), microglia activity (van Berckel et al., 2008) and decreased myelination (Bernstein et al., 2015) is now being appreciated in clinical studies, although it is unclear whether these reflect a cause or consequence of symptom onset. Our hope is that hiPSC-based technologies will provide a tractable, modular platform to establish causal links between schizophrenia genetic predisposition, cell-type interactions and disease-relevant phenotypes.
Magnetic resonance imaging (MRI) and non-volumetric diffusion tensor imaging (DTI) of post-mortem tissue have revealed abnormalities in white matter on a macro and microstructure level respectively (Walterfang et al., 2011; Whitford et al., 2011), prompting an increased interest in the role that oligodendrocytes may play in schizophrenia. Numerous studies have now associated schizophrenia with decreased oligodendrocyte differentiation (Mauney et al., 2015) and density (as much as 30% reduced in some brain regions) (Vikhreva et al., 2016), as well as changes in oligodendrocyte morphology and spatial distribution in patient brains (Uranova et al., 2001; Schmitt et al., 2009). Moreover, in vitro rodent studies demonstrated that the schizophrenia-associated gene DISC1 impacts oligodendrocyte differentiation (Hattori et al., 2014) and human genetics have identified oligodendrocyte gene sets associated with schizophrenia, most of them related to fatty acid and cholesterol metabolism (Goudriaan et al., 2014). To date, no hiPSC- or ESC-based technologies have examined the effect of schizophrenia-associated variants on oligodendrocyte differentiation and function, which could shed light on how the differences in white matter organization and myelination observed in post-mortem tissue arise on the cellular level.
Astrocytes are believed to contribute primarily to neuroinflammation and synaptic maturation/pruning in the human brain (Freeman and Rowitch, 2013), processes that are increasingly linked to schizophrenia (Jaaro-Peled et al., 2010; Fineberg and Ellman, 2013). Astrocyte loss has also been associated with schizophrenia in various cortical and subcortical regions of the brain, particularly in the white matter (Rajkowska et al., 2002; Williams et al., 2013a,b). Along with the gene sets involved in oligodendrocyte function, additional gene sets involving astrocyte function are altered in schizophrenia (Goudriaan et al., 2014). Post-mortem pathology inevitably leads to tissue shrinkage (“fixation artifacts”), particularly in regions that have abundant astrocyte cell processes (Garman, 2011); despite this technical limitation, a few post-mortem studies have observed subtle reductions in glial cell volume (Rajkowska et al., 2002) and cell number (Stark et al., 2004) in schizophrenia cortical brain tissue, although these results are not yet widely accepted. Postmortem cortical expression (Barley et al., 2009) and protein levels (Steffek et al., 2008) of major astrocyte associated genes such as glial fibrillary acidic protein (GFAP) are perturbed in schizophrenia. Critically, the relevance of these post-mortem observations is clouded by the fact that antipsychotic treatments may impact astrocyte levels and/or function in the CNS, as these cells express dopamine receptors (Hertz et al., 1984).
The use of hiPSC-based models for schizophrenia continues to be increasingly useful; we and others have demonstrated increases in oxidative stress, (Paulsen et al., 2012; Robicsek et al., 2013; Brennand et al., 2014) deficits in adherens junctions and polarity (Yoon et al., 2014), as well as marked differences in migration and responses to environmental stressors in NPCs derived from schizophrenic patients (Brennand et al., 2014; Hashimoto-Torii et al., 2014). Similarly, schizophrenia hiPSC neurons exhibit decreased synaptic maturation and neurite number (Brennand et al., 2011; Robicsek et al., 2013; Wen et al., 2014; Yu et al., 2014), as well as a reduction in synaptic activity (Wen et al., 2014; Yu et al., 2014). Given the success of these previous hiPSC models and the strong evidence presented above on the role of glial cell-types in schizophrenia disease pathology, it may be very informative to extend these in vitro systems to query glial function using patient-specific hiPSC lines to develop better, more human-specific models of schizophrenia.
Rett Syndrome
Rett Syndrome is a rare X-linked neurodevelopmental disorder affecting the gray matter of the brain, primarily in female patients, and characterized by an initial period of normal development during early infancy followed by a sudden attenuation of developmental growth and the loss a number of motor and language skills (Hagberg et al., 2002). This regression of physical development is associated with differences in cognitive function; many patients exhibit autistic-like behaviors, sleep disorders and increased anxiety (Hagberg et al., 2002; Bienvenu and Chelly, 2006). Rett Syndrome is frequently caused by de novo mutations in the methyl CpG binding protein 2 gene (MECP2), though other genetic variations account for a minority of cases. MECP2 encodes the regulatory protein MeCP2, which binds to methylated DNA to regulate transcription of a number of genes (Amir et al., 1999; Bienvenu and Chelly, 2006). How such broad changes in transcriptional regulation leads to the neurodevelopmental and neurobehavioral phenotypes of Rett syndrome is not fully clear, but the unigenic nature of the disorder makes it particularly tractable to study using hiPSC-based technologies.
The role of MECP2 mutations in neurons has been well-examined in a variety of studies (Marchetto et al., 2010; Kim et al., 2011; Farra et al., 2012), but mutant MECP2 also exerts its effect through non-cell autonomous events involving astrocytes. MeCP2 deficiency in astrocytes causes significant abnormalities in cytokine production and neuronal dendritic induction that could impact neurodevelopment in mice (Maezawa et al., 2009). Mutant astrocytes have an adverse effect when co-cultured with either wild type or mutant hippocampal neurons, an effect that can be recapitulated using conditioned media alone (Ballas et al., 2009), suggesting that astrocyte targets of MeCP2 regulation are involved in glial maintenance of neuronal function and that astrocytes are a key player in Rett syndrome pathology. Importantly re-expression of MECP2 preferentially in astrocytes restores normal neuronal dendritic morphology, improves locomotion and anxiety, and rescues respiratory symptoms in a mouse model of Rett syndrome (Lioy et al., 2011).
While the role of astrocytes in non-cell autonomous effects on neuronal function in Rett syndrome is well-established in mice, it was unclear whether the same effects held true in a human context. Two recent studies using Rett syndrome patient-derived hiPSCs have now not only confirmed that astrocyte role in Rett Syndrome, but also identified molecular targets involved in astrocyte-mediated neuronal deficits. Astrocytes derived from patient-specific hiPSCs recapitulate the negative effects on neuronal morphology observed in mouse studies, an effect that can again be reproduced using only conditioned media (Williams et al., 2014). Using combinations of control and Rett Syndrome-derived astrocytes and interneurons, the authors show that the glial effect on neuronal function is independent of intrinsic deficits in neurons themselves and demonstrated that insulin-like growth factor (IGF-1) and GPE (a short peptide containing the first 3 amino acids of IGF-1) are capable of partially rescuing the astrocyte-mediated neuronal phenotypes. The power of hiPSC-based systems to probe non-cell autonomous systems in Rett syndrome is underscored by recent studies demonstrating that MECP2-mutant astrocytes from patient-derived hiPSCs have deficiencies in microtubule-dependent vesicle transport, and that administration of Epothilone D, a microtubule stabilizing agent capable of crossing the blood brain barrier is sufficient to restore microtubule dynamics in these cells (Delépine et al., 2016). Critically, weekly doses of Epothilone D was capable of reversing the reduced exploratory behavior in a mouse model of Rett Syndrome (Delépine et al., 2016), demonstrating the power of hiPSC-based platforms to identify new targets that are capable of improving animal behavior when administered in vivo.
Microglia have also been implicated in Rett syndrome pathology through a variety of in vitro and in vivo mouse models. Selective correction of Rett syndrome microglia with wild type microglia using a Cre-Lox based system in mice produced an improvement of symptoms, one that could be reversed using annexin IV, suggesting that microglia-mediated phagocytosis is a key mechanism of Rett syndrome pathology (Derecki et al., 2012). In addition, murine Rett syndrome microglia appear to have a direct neurotoxic effect on hippocampal neurons in co-culture models, releasing increased glutamate that causes stunted dendritic morphology and disruption of microtubule organization that ultimately disrupts synaptic function (Maezawa and Jin, 2010). While these findings have not yet been tested in a human context, it was recently shown that hiPSC-derived microglia carrying the MECP2 deletion are significantly smaller than their wild type counterparts (Muffat et al., 2016), which may impart a decreased ability to “patrol” the parenchyma and clear apoptotic tissue. Further studies using this newly developed pathology may offer important insight into Rett syndrome pathology and provide a powerful platform to identify molecular targets for drug development.
Altogether, while Rett syndrome is arguably the first autism spectrum disorder to be convincingly linked to aberrant astrocyte and microglial function, there is no reason to suspect that other forms of autism will not soon be associated with neuron non-cell autonomous effects as well. Complex genetic neuropsychiatric disorders, from ALS to schizophrenia to Rett Syndrome, may in fact represent a convergence of clinical phenotypes arising through a diverse range of genetic and cellular mechanism, suggesting that a “personalized” or genotype-dependent understanding of disease mechanisms may be critical for properly matching patients with appropriate therapeutics.
The Future of hiPSC-Based Drug Screening
The development of new pharmaceutical treatments for neuropsychiatric diseases has been severely hampered by the poor availability of preclinical models that adequately capture the complex pathophysiology of these disorders. It has been difficult to translate the success of the high throughput drug screens used to identify novel targets and lead structures for therapeutics in other fields to neuroscience. Moving forward, we hope that this will be addressed by this new ability to create disease specific, patient-derived hiPSC lines, which serve as genetically relevant models that are scalable and easily perturbed through genetic and chemical approaches. Importantly, the ability to create and bank large stores of hiPSC-derived cells will permit repeated experiments across genetically isogenic human cell types, hopefully improving reproducibility. As advancements in high content imaging technologies develop alongside hiPSC technology, this integrated approach may help identify new therapeutic targets for drug development and advance our understanding of genotype-phenotype correlations in neuropsychiatric disease in ways that were not previously possible. This approach is of particular importance for complex genetic diseases such as schizophrenia and autism, where simplistic knock in models fail to accurately capture each patient's complex and potentially unique genetic background.
Pluripotent stem cell based screens have shown notable recent successes in the context of neuropsychiatric disease, reviewed elsewhere (Haggarty and Perlis, 2014; Haggarty et al., 2016). In the context of mitochondrial DNA (mtDNA) disorders, NPCs derived from patients carrying a single base pair mutation in the MT-ATP6 gene have defective ATP production, elevated mitochondrial membrane potential, and dysregulation of calcium handling (Lorenz et al., 2017); a large high-throughput screen using FDA approved drugs identified avanafil as a potential therapeutic owing to its ability to restore normal calcium homeostasis. The introduction of a TCF/LEF-responsive luciferase reporter into hiPSC-derived NPCs permitting screening of a small pilot library of 1500 compounds, identifying a number of compounds that potentiate Wnt or lithium signaling (Zhao et al., 2012), with potential relevance to a number of psychiatric disorders (including schizophrenia and Fragile X) and many common antidepressants and antipsychotics. A similar approach, applying an ATP bioluminescence end-point assay to screen a 1,000 compound library, identified five novel compounds that enhance proliferation and viability of hiPSC-derived NPCs (McLaren et al., 2013); these compounds could be used to further expand populations of NPCs and facilitate larger screens moving forward.
While the screens outlined above are unique in their methods and targets of interest, they are unified in their focus on a single cell type, limiting their relevance to the physiological context. With advancements in hiPSC differentiation protocols now encompassing many different neural cell types, we hope that future screening experiments will combine several distinct pre-differentiated cell types. Such a proof-of concept screen was conducted using Nestin-positive progenitor cells that were differentiated into neurons and/or astrocytes, cultured both in isolation and together (Efthymiou et al., 2014); a high-throughput MTT assay measured viability, while live-cell imaging tracked organelles within neurons and examined neurite length as a proxy for neuronal maturation. Likewise, a large 2.4 million compound screen using mouse ESC derived NPCs differentiated into a mix of neuron and glial subtypes used a fluorimetric imaging plate reader to measure calcium influx and screen for potentiators of AMPA receptor signaling (McNeish et al., 2010); however, the analysis did not stratify hits based on cell type, potentially diluting more modest effects that may have had large effects within key cell types. Overall, given our growing understanding of non-cell autonomous effects on neuronal health, next generation screening platforms must address the challenge of stratifying by cell type and considering interactions between cell types within a single population of cells. Toward this, an elegant platform that allows for the targeting of safe harbor loci in hiPSCs to introduce multiplexed lineage-specific reporter systems on the same isogenic background (Pei et al., 2015) could be combined with high content imaging technologies, providing a valuable context to resolve which cell populations are affected by candidate molecules. The importance of this analysis is demonstrated in a follow-up paper by the same group, which showed that of an initial panel of 80 compounds, 50 were observed to have a cytotoxic effect in at least one cell type but only four of those showed cytotoxicity in four different neural subtypes (Pei et al., 2016). Given the disparate and cell-specific phenotypes discussed above in a variety of neuropsychiatric disorders, qualifying successful “hits” on high throughput screens by matching the drug target with the cell type will be of critical importance for pharmaceutical development.
A number of coculture platforms have been developed for the study of neurological disease, shedding light on the interactions that occur between neural cell types (excellently reviewed by Meyer and Kaspar, 2017). These vary in their design and complexity, from focusing on paracrine signaling through transwell interactions to culturing disparate cell populations in pre-determined orientations to modeling physiological structures such as the blood brain barrier. Recently, a number of groups have reported protocols for the generation of endothelial cells, pericytes, neurons and astrocytes involved in the blood-brain barrier (BBB) from hiPSCs and other sources, using transwells to study efflux of a variety of drugs across the newly formed barrier (Lippmann et al., 2012; Boyer-Di Ponio et al., 2014; Appelt-Menzel et al., 2017; Yamamizu et al., 2017). When combined with microfluidic systems similar to those reported in Wang et al, these coculture systems may provide an efficient and cost-effective model to screen large pharmaceutical libraries (Wang et al., 2017). Newer, more sophisticated screening platforms should take inspiration from these co-culture models, adapting platforms to a high-throughput format that can be combined with fluorescent reporters and high content imaging software, allowing exploration of how the interactions between cell types change under the influence of pharmaceutical compounds (Figure 1). With ever improving cellular differentiation protocols allowing us to generate larger numbers of defined neural cell types with increasing efficiency, future screens should improve our understanding of neurological disease pathology and identify new treatment modalities for neurodegenerative and neuropsychiatric disease.
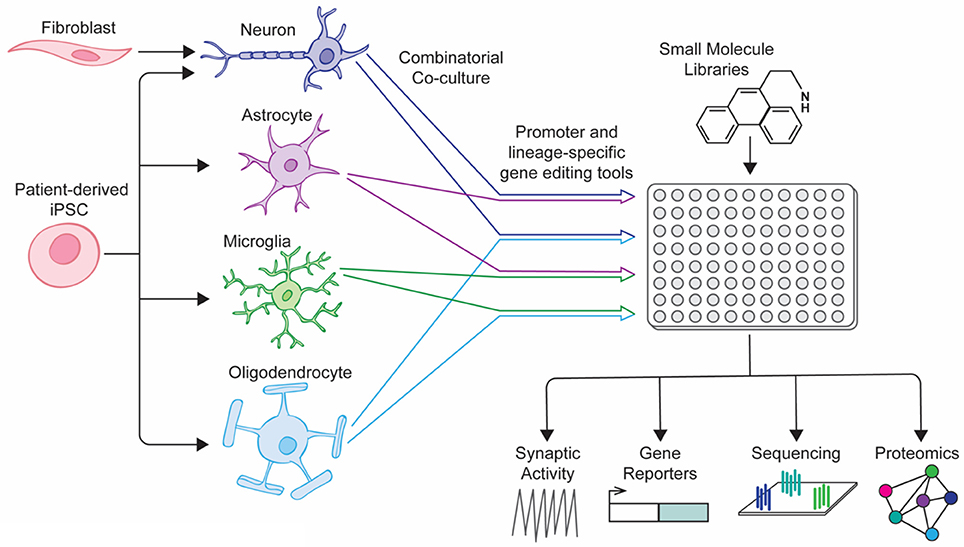
Figure 1. Next-generation drug screening platform for neuropsychiatric disorders. Directed differentiation and reprogramming of patient-specific cell types into various neural cell types allows for combination culture of cell types of interest. When combined with lineage-specific reporter systems, mixed neural cultures could be screened using small-molecule libraries and the results of downstream assays correlated with the cell-type of interest.
Summary
New advances in the field of stem cell biology and reprogramming are allowing for the creation of once inaccessible neural cell types, improving our ability to model complex neuropsychiatric and neurodegenerative diseases in ways not previously possible. The development of these models has begun to shed light on important interactions between neural cell-types in the context of human disease pathology. As the importance of these non-cell autonomous effects becomes increasingly clear, drug discovery platforms that pair new stem cell technologies with high throughput assays and high content imaging software capable of analyzing individual cell populations within a heterogenous pool may be an invaluable resource for the development of new therapies for these complex disorders.
Author Contributions
DG and KB: wrote the manuscript; JG: prepared Figure 1.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LJ and handling Editor declared their shared affiliation.
Acknowledgments
KB is a New York Stem Cell Foundation—Robertson Investigator. This work was partially supported by National Institute of Health (NIH) grants R01 MH101454 (KB), as well as the New York Stem Cell Foundation and the Brain Behavior Research Foundation.
References
Abud, E. M., Ramirez, R. N., Martinez, E. S., Healy, L. M., Nguyen, C. H. H., Newman, S. A., et al. (2017). iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278–293.e9. doi: 10.1016/j.neuron.2017.03.042
Aguzzi, A., Barres, B. A., and Bennett, M. L. (2013). Microglia: scapegoat, saboteur, or something else? Science 339, 156–161. doi: 10.1126/science.1227901
Alliot, F., Godin, I., and Pessac, B. (1999). Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res. Dev. Brain Res. 117, 145–152. doi: 10.1016/S0165-3806(99)00113-3
Alves, C. J., Dariolli, R., Jorge, F. M., Monteiro, M. R., Maximino, J. R., Martins, R. S., et al. (2015). Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 9:289. doi: 10.3389/fncel.2015.00289
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Appel, S. H., Zhao, W., Beers, D. R., and Henkel, J. S. (2011). The microglial-motoneuron dialogue in ALS. Acta Myol. 30, 4–8.
Appelt-Menzel, A., Cubukova, A., Günther, K., Edenhofer, F., Piontek, J., Krause, G., et al. (2017). Establishment of a human blood-brain barrier co-culture model mimicking the neurovascular unit using induced pluri- and multipotent stem cells. Stem Cell Rep. 8, 894–906. doi: 10.1016/j.stemcr.2017.02.021
Bahmad, H., Hadadeh, O., Chamaa, F., Cheaito, K., Darwish, B., Makkawi, A.-K., et al. (2017). Modeling human neurological and neurodegenerative diseases: from induced pluripotent stem cells to neuronal differentiation and its applications in neurotrauma. Front. Mol. Neurosci. 10:50. doi: 10.3389/fnmol.2017.00050
Ballas, N., Lioy, D. T., Grunseich, C., and Mandel, G. (2009). Non–cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12, 311–317. doi: 10.1038/nn.2275
Barateiro, A., Brites, D., and Fernandes, A. (2016). Oligodendrocyte development and myelination in neurodevelopment: molecular mechanisms in health and disease. Curr. Pharm. Des. 22, 656–679. doi: 10.2174/1381612822666151204000636
Barley, K., Dracheva, S., and Byne, W. (2009). Subcortical oligodendrocyte- and astrocyte-associated gene expression in subjects with schizophrenia, major depression and bipolar disorder. Schizophr. Res. 112, 54–64. doi: 10.1016/j.schres.2009.04.019
Barres, B. A. (2008). The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60, 430–440. doi: 10.1016/j.neuron.2008.10.013
Bavamian, S., Mellios, N., Lalonde, J., Fass, D. M., Wang, J., Sheridan, S. D., et al. (2015). Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol. Psychiatry 20, 573–584. doi: 10.1038/mp.2014.176
Benes, F. M., McSparren, J., Bird, E. D., SanGiovanni, J. P., and Vincent, S. L. (1991). Deficits in small interneurons in prefrontal and cingulate cortices of schizophrenic and schizoaffective patients. Arch. Gen. Psychiatry 48, 996–1001. doi: 10.1001/archpsyc.1991.01810350036005
Bernstein, H. G., Steiner, J., Guest, P. C., Dobrowolny, H., and Bogerts, B. (2015). Glial cells as key players in schizophrenia pathology: recent insights and concepts of therapy. Schizophr. Res. 161, 4–18. doi: 10.1016/j.schres.2014.03.035
Bienvenu, T., and Chelly, J. (2006). Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 7, 415–426. doi: 10.1038/nrg1878
Boillée, S., Yamanaka, K., Lobsiger, C. S., Copeland, N. G., Jenkins, N. A., Kassiotis, G., et al. (2006). Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. doi: 10.1126/science.1123511
Boyer-Di Ponio, J., El-Ayoubi, F., Glacial, F., Ganeshamoorthy, K., Driancourt, C., Godet, M., et al. (2014). Instruction of circulating endothelial progenitors in vitro towards specialized blood-brain barrier and arterial phenotypes. PLoS ONE 9:e84179. doi: 10.1371/journal.pone.0084179
Brennand, K. J., and Gage, F. H. (2012). Modeling psychiatric disorders through reprogramming. Dis. Model. Mech. 5, 26–32. doi: 10.1242/dmm.008268
Brennand, K. J., Landek-Salgado, M. A., and Sawa, A. (2014). Modeling heterogeneous patients with a clinical diagnosis of schizophrenia with induced pluripotent stem cells. Biol. Psychiatry 75, 936–944. doi: 10.1016/j.biopsych.2013.10.025
Brennand, K. J., Simone, A., Jou, J., Gelboin-Burkhart, C., Tran, N., Sangar, S., et al. (2011). Modelling schizophrenia using human induced pluripotent stem cells. Nature 473, 221–225. doi: 10.1038/nature09915
Brennand, K., Savas, J. N., Kim, Y., Tran, N., Simone, A., Hashimoto-Torii, K., et al. (2015). Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 20, 361–368. doi: 10.1038/mp.2014.22
Brooks, B. R. (1996). Natural history of ALS symptoms, strength, pulmonary function, and disability. Neurology 47, 71S–82S. doi: 10.1212/WNL.47.4_Suppl_2.71S
Brown, R. H. (1997). Amyotrophic lateral sclerosis. Insights from genetics. Arch. Neurol. 54, 1246–1250. doi: 10.1001/archneur.1997.00550220050013
Bushong, E. A., Martone, M. E., Jones, Y. Z., and Ellisman, M. H. (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192.
Butovsky, O., Talpalar, A. E., Ben-Yaakov, K., and Schwartz, M. (2005). Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol. Cell. Neurosci. 29, 381–393. doi: 10.1016/j.mcn.2005.03.005
Caiazzo, M., Dell'Anno, M. T., Dvoretskova, E., Lazarevic, D., Taverna, S., Leo, D., et al. (2011). Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227. doi: 10.1038/nature10284
Caiazzo, M., Giannelli, S., Valente, P., Lignani, G., Carissimo, A., Sessa, A., et al. (2015). Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Rep. 4, 25–36. doi: 10.1016/j.stemcr.2014.12.002
Chanda, S., Marro, S., Wernig, M., and Südhof, T. C. (2013). Neurons generated by direct conversion of fibroblasts reproduce synaptic phenotype caused by autism-associated neuroligin-3 mutation. Proc. Natl. Acad. Sci. U.S.A. 110, 16622–16627. doi: 10.1073/pnas.1316240110
Chen, H., Qian, K., Chen, W., Hu, B., Blackbourn, L. W., Du, Z., et al. (2015). Human-derived neural progenitors functionally replace astrocytes in adult mice. J. Clin. Invest. 125, 1033–1042. doi: 10.1172/JCI69097
Chestkov, I. V., Vasilieva, E. A., Illarioshkin, S. N., Lagarkova, M. A., and Kiselev, S. L. (2014). Patient-specific induced pluripotent stem cells for SOD1-associated amyotrophic lateral sclerosis pathogenesis studies. Acta Nat. 6, 54–60.
Chung, W. S., Welsh, C. A., Barres, B. A., and Stevens, B. (2015). Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci. 18, 1539–1545. doi: 10.1038/nn.4142
Clement, A. M., Nguyen, M. D., Roberts, E. A., Garcia, M. L., Boillée, S., Rule, M., et al. (2003). Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117. doi: 10.1126/science.1086071
Cole, N., and Siddique, T. (1999). Genetic disorders of motor neurons. Semin. Neurol. 19, 407–418. doi: 10.1055/s-2008-1040855
Dal Bianco, A., Bradl, M., Frischer, J., Kutzelnigg, A., Jellinger, K., and Lassmann, H. (2008). Multiple sclerosis and Alzheimer's disease. Ann. Neurol. 63, 174–183. doi: 10.1002/ana.21240
Danbolt, N. C. (2001). Glutamate uptake. Prog. Neurobiol. 65, 1–105. doi: 10.1016/S0301-0082(00)00067-8
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Delépine, C., Meziane, H., Nectoux, J., Opitz, M., Smith, A. B., Ballatore, C., et al. (2016). Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Hum. Mol. Genet. 25, 146–157. doi: 10.1093/hmg/ddv464
Derecki, N. C., Cronk, J. C., Lu, Z., Xu, E., Abbott, S. B. G., Guyenet, P. G., et al. (2012). Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105–109. doi: 10.1038/nature10907
Di Giorgio, F. P., Boulting, G. L., Bobrowicz, S., and Eggan, K. C. (2008). Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 3, 637–648. doi: 10.1016/j.stem.2008.09.017
Di Giorgio, F. P., Carrasco, M. A., Siao, M. C., Maniatis, T., and Eggan, K. (2007). Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat. Neurosci. 10, 608–614. doi: 10.1038/nn1885
Dimos, J. T., Rodolfa, K. T., Niakan, K. K., Weisenthal, L. M., Mitsumoto, H., Chung, W., et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218–1221. doi: 10.1126/science.1158799
Durafourt, B. A., Moore, C. S., Zammit, D. A., Johnson, T. A., Zaguia, F., Guiot, M.-C., et al. (2012). Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia 60, 717–727. doi: 10.1002/glia.22298
Efthymiou, A., Shaltouki, A., Steiner, J. P., Jha, B., Heman-Ackah, S. M., Swistowski, A., et al. (2014). Functional screening assays with neurons generated from pluripotent stem cell-derived neural stem cells. J. Biomol. Screen. 19, 32–43. doi: 10.1177/1087057113501869
Egawa, N., Kitaoka, S., Tsukita, K., Naitoh, M., Takahashi, K., Yamamoto, T., et al. (2012). Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci. Transl. Med. 4:145ra104. doi: 10.1126/scitranslmed.3004052
Farra, N., Zhang, W. B., Pasceri, P., Eubanks, J. H., Salter, M. W., and Ellis, J. (2012). Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol. Psychiatry 17, 1261–1271. doi: 10.1038/mp.2011.180
Fass, D. M., Schroeder, F. A., Perlis, R. H., and Haggarty, S. J. (2014). Epigenetic mechanisms in mood disorders: targeting neuroplasticity. Neuroscience 264, 112–130. doi: 10.1016/j.neuroscience.2013.01.041
Fields, R. D. (2008). White matter in learning, cognition and psychiatric disorders. Trends Neurosci. 31, 361–370. doi: 10.1016/j.tins.2008.04.001
Fineberg, A. M., and Ellman, L. M. (2013). Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol. Psychiatry 73, 951–966. doi: 10.1016/j.biopsych.2013.01.001
Fishman, V. S., Shnayder, T. A., Orishchenko, K. E., Bader, M., Alenina, N., and Serov, O. L. (2015). Cell divisions are not essential for the direct conversion of fibroblasts into neuronal cells. Cell Cycle 14, 1188–1196. doi: 10.1080/15384101.2015.1012875
Foster, A., Gable, J., and Buckley, J. (2012). Homelessness in schizophrenia. Psychiatr. Clin. North Am. 35, 717–734. doi: 10.1016/j.psc.2012.06.010
Freeman, M. R., and Rowitch, D. H. (2013). Evolving concepts of gliogenesis: a look way back and ahead to the next 25 years. Neuron 80, 613–623. doi: 10.1016/j.neuron.2013.10.034
Garman, R. H. (2011). Histology of the central nervous system. Toxicol. Pathol. 39, 22–35. doi: 10.1177/0192623310389621
Goldman, S. A., and Kuypers, N. J. (2015). How to make an oligodendrocyte. Development 142, 3983–3995. doi: 10.1242/dev.126409
Goudriaan, A., de Leeuw, C., Ripke, S., Hultman, C. M., Sklar, P., Sullivan, P. F., et al. (2014). Specific glial functions contribute to schizophrenia susceptibility. Schizophr. Bull. 40, 925–935. doi: 10.1093/schbul/sbt109
Hagberg, B., Hanefeld, F., Percy, A., and Skjeldal, O. (2002). An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome clinical criteria consensus panel satellite to European paediatric neurology society meeting, Baden Baden, Germany, 11 September 2001. Eur. J. Paediatr. Neurol. 6, 293–297. doi: 10.1053/ejpn.2002.0612
Haggarty, S. J., and Perlis, R. H. (2014). Translation: screening for novel therapeutics with disease-relevant cell types derived from human stem cell models. Biol. Psychiatry 75, 952–960. doi: 10.1016/j.biopsych.2013.05.028
Haggarty, S. J., Silva, M. C., Cross, A., Brandon, N. J., and Perlis, R. H. (2016). Advancing drug discovery for neuropsychiatric disorders using patient-specific stem cell models. Mol. Cell. Neurosci. 73, 104–115. doi: 10.1016/j.mcn.2016.01.011
Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., et al. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 29, 824–828. doi: 10.1038/nbt.1957
Han, X., Chen, M., Wang, F., Windrem, M., Wang, S., Shanz, S., et al. (2013). Forebrain engraftment by human glial progenitor cells enhances synaptic plasticity and learning in adult mice. Cell Stem Cell 12, 342–353. doi: 10.1016/j.stem.2012.12.015
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Hashimoto-Torii, K., Torii, M., Fujimoto, M., Nakai, A., El Fatimy, R., Mezger, V., et al. (2014). Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron 82, 560–572. doi: 10.1016/j.neuron.2014.03.002
Hattori, T., Shimizu, S., Koyama, Y., Emoto, H., Matsumoto, Y., Kumamoto, N., et al. (2014). DISC1 (disrupted-in-schizophrenia-1) regulates differentiation of oligodendrocytes. PLoS ONE 9:e88506. doi: 10.1371/journal.pone.0088506
Hertz, L., Schousboe, I., Hertz, L., and Schousboe, A. (1984). Receptor expression in primary cultures of neurons or astrocytes. Prog. Neuropsychopharmacol. Biol. Psychiatry 8, 521–527. doi: 10.1016/0278-5846(84)90010-1
Ho, S. M., Topol, A., and Brennand, K. J. (2015). From “directed differentiation” to “neuronal induction”: modeling neuropsychiatric disease. Biomark Insights 10, 31–41. doi: 10.4137/BMI.S20066
Hu, W., Qiu, B., Guan, W., Wang, Q., Wang, M., Li, W., et al. (2015). Direct conversion of normal and Alzheimer's disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell 17, 204–212. doi: 10.1016/j.stem.2015.07.006
Hu, X., Li, P., Guo, Y., Wang, H., Leak, R. K., Chen, S., et al. (2012). Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 43, 3063–3070. doi: 10.1161/STROKEAHA.112.659656
Huh, C. J., Zhang, B., Victor, M. B., Dahiya, S., Batista, L. F., Horvath, S., et al. (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. ELife 5:e18648. doi: 10.7554/eLife.18648
Hulshoff Pol, H. E., Schnack, H. G., Bertens, M. G. B. C., van Haren, N. E. M., van der Tweel, I., Staal, W. G., et al. (2002). Volume changes in gray matter in patients with schizophrenia. Am. J. Psychiatry 159, 244–250. doi: 10.1176/appi.ajp.159.2.244
Iwafuchi-Doi, M., and Zaret, K. S. (2014). Pioneer transcription factors in cell reprogramming. Genes Dev. 28, 2679–2692. doi: 10.1101/gad.253443.114
Jaaro-Peled, H., Ayhan, Y., Pletnikov, M. V., and Sawa, A. (2010). Review of pathological hallmarks of schizophrenia: comparison of genetic models with patients and nongenetic models. Schizophr. Bull. 36, 301–313. doi: 10.1093/schbul/sbp133
Jiang, P., Chen, C., Wang, R., Chechneva, O. V., Chung, S.-H., Rao, M. S., et al. (2013). hESC-derived Olig2+ progenitors generate a subtype of astroglia with protective effects against ischaemic brain injury. Nat. Commun. 4:2196. doi: 10.1038/ncomms3196
Kerman, B. E., Kim, H. J., Padmanabhan, K., Mei, A., Georges, S., Joens, M. S., et al. (2015). In vitro myelin formation using embryonic stem cells. Development 142, 2213–2225. doi: 10.1242/dev.116517
Kigerl, K. A., Gensel, J. C., Ankeny, D. P., Alexander, J. K., Donnelly, D. J., and Popovich, P. G. (2009). Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009
Kim, H. J., Ifergan, I., Antel, J. P., Seguin, R., Duddy, M., Lapierre, Y., et al. (2004). Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J. Immunol. Baltim. 172, 7144–7153. doi: 10.4049/jimmunol.172.11.7144
Kim, K. Y., Hysolli, E., and Park, I. H. (2011). Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 14169–14174. doi: 10.1073/pnas.1018979108
Koehler, R. C., Roman, R. J., and Harder, D. R. (2009). Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 32, 160–169. doi: 10.1016/j.tins.2008.11.005
Kooyman, I., Dean, K., Harvey, S., and Walsh, E. (2007). Outcomes of public concern in schizophrenia. Br. J. Psychiatry Suppl. 50, s29–s36. doi: 10.1192/bjp.191.50.s29
Krencik, R., and Zhang, S. C. (2011). Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat. Protoc. 6, 1710–1717. doi: 10.1038/nprot.2011.405
Kushner, P. D., Stephenson, D. T., and Wright, S. (1991). Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J. Neuropathol. Exp. Neurol. 50, 263–277. doi: 10.1097/00005072-199105000-00008
Lewis, D. A., and Lieberman, J. A. (2000). Catching up on schizophrenia: natural history and neurobiology. Neuron 28, 325–334. doi: 10.1016/S0896-6273(00)00111-2
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lioy, D. T., Garg, S. K., Monaghan, C. E., Raber, J., Foust, K. D., Kaspar, B. K., et al. (2011). A role for glia in the progression of Rett's syndrome. Nature 475, 497–500. doi: 10.1038/nature10214
Lippmann, E. S., Azarin, S. M., Kay, J. E., Nessler, R. A., Wilson, H. K., Al-Ahmad, A., et al. (2012). Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 30, 783–791. doi: 10.1038/nbt.2247
Liu, M. L., Zang, T., Zou, Y., Chang, J. C., Gibson, J. R., Huber, K. M., et al. (2013). Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 4:2183. doi: 10.1038/ncomms3183
Lorenz, C., Lesimple, P., Bukowiecki, R., Zink, A., Inak, G., Mlody, B., et al. (2017). Human iPSC-derived neural progenitors are an effective drug discovery model for neurological mtDNA disorders. Cell Stem Cell 20, 659–674.e9. doi: 10.1016/j.stem.2016.12.013
Mabbott, D. J., Noseworthy, M., Bouffet, E., Laughlin, S., and Rockel, C. (2006). White matter growth as a mechanism of cognitive development in children. Neuroimage 33, 936–946. doi: 10.1016/j.neuroimage.2006.07.024
Maezawa, I., and Jin, L. W. (2010). Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J. Neurosci. 30, 5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010
Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., and Jin, L.-W. (2009). Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J. Neurosci. 29, 5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009
Marchetto, M. C. N., Carromeu, C., Acab, A., Yu, D., Yeo, G. W., Mu, Y., et al. (2010). A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539. doi: 10.1016/j.cell.2010.10.016
Marchetto, M. C., Muotri, A. R., Mu, Y., Smith, A. M., Cezar, G. G., and Gage, F. H. (2008). Non-cell-autonomous effect of human SOD1 G37R astrocytes on motor neurons derived from human embryonic stem cells. Cell Stem Cell 3, 649–657. doi: 10.1016/j.stem.2008.10.001
Marchetto, M. C., Brennand, K. J., Boyer, L. F., and Gage, F. H. (2011). Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum. Mol. Genet. 20, R109–R115. doi: 10.1093/hmg/ddr336
Mariani, J., Simonini, M. V., Palejev, D., Tomasini, L., Coppola, G., Szekely, A. M., et al. (2012). Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. U.S.A. 109, 12770–12775. doi: 10.1073/pnas.1202944109
Matute, C., Melone, M., Vallejo-Illarramendi, A., and Conti, F. (2005). Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia 49, 451–455. doi: 10.1002/glia.20119
Mauney, S. A., Pietersen, C. Y., Sonntag, K.-C., and Woo, T.-U. W. (2015). Differentiation of oligodendrocyte precursors is impaired in the prefrontal cortex in schizophrenia. Schizophr. Res. 169, 374–380. doi: 10.1016/j.schres.2015.10.042
McCarley, R. W., Wible, C. G., Frumin, M., Hirayasu, Y., Levitt, J. J., Fischer, I. A., et al. (1999). MRI anatomy of schizophrenia. Biol. Psychiatry 45, 1099–1119. doi: 10.1016/S0006-3223(99)00018-9
McGivern, J. V., Patitucci, T. N., Nord, J. A., Barabas, M.-E. A., Stucky, C. L., and Ebert, A. D. (2013). Spinal muscular atrophy astrocytes exhibit abnormal calcium regulation and reduced growth factor production. Glia 61, 1418–1428. doi: 10.1002/glia.22522
McLaren, D., Gorba, T., Marguerie de Rotrou, A., Pillai, G., Chappell, C., Stacey, A., et al. (2013). Automated large-scale culture and medium-throughput chemical screen for modulators of proliferation and viability of human induced pluripotent stem cell-derived neuroepithelial-like stem cells. J. Biomol. Screen. 18, 258–268. doi: 10.1177/1087057112461446
McNeish, J., Roach, M., Hambor, J., Mather, R. J., Weibley, L., Lazzaro, J., et al. (2010). High-throughput screening in embryonic stem cell-derived neurons identifies potentiators of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate-type glutamate receptors. J. Biol. Chem. 285, 17209–17217. doi: 10.1074/jbc.M109.098814
Mertens, J., Marchetto, M. C., Bardy, C., and Gage, F. H. (2016). Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 17, 424–437. doi: 10.1038/nrn.2016.46
Meyer, K., and Kaspar, B. K. (2017). Glia-neuron interactions in neurological diseases: testing non-cell autonomy in a dish. Brain Res. 1656, 27–39. doi: 10.1016/j.brainres.2015.12.051
Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., McElroy, S., Renusch, S., et al. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc. Natl. Acad. Sci. U.S.A. 111, 829–832. doi: 10.1073/pnas.1314085111
Miller, J. D., Ganat, Y. M., Kishinevsky, S., Bowman, R. L., Liu, B., Tu, E. Y., et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. doi: 10.1016/j.stem.2013.11.006
Muffat, J., Li, Y., Yuan, B., Mitalipova, M., Omer, A., Corcoran, S., et al. (2016). Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 22, 1358–1367. doi: 10.1038/nm.4189
Nagai, M., Re, D. B., Nagata, T., Chalazonitis, A., Jessell, T. M., Wichterle, H., et al. (2007). Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. Neurosci. 10, 615–622. doi: 10.1038/nn1876
Nagy, D., Kato, T., and Kushner, P. D. (1994). Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 38, 336–347. doi: 10.1002/jnr.490380312
Nagy, Z., Westerberg, H., and Klingberg, T. (2004). Maturation of white matter is associated with the development of cognitive functions during childhood. J. Cogn. Neurosci. 16, 1227–1233. doi: 10.1162/0898929041920441
Najm, F. J., Lager, A. M., Zaremba, A., Wyatt, K., Caprariello, A. V., Factor, D. C., et al. (2013). Transcription factor-mediated reprogramming of fibroblasts to expandable, myelinogenic oligodendrocyte progenitor cells. Nat. Biotechnol. 31, 426–433. doi: 10.1038/nbt.2561
Newman, E. A. (1995). “Glial cell regulation of extracellular potassium,” in Neuroglia, eds H. Kettenmann and B. R. Ransom (New York, NY: Oxford University Press), 717–731.
Nicholas, C. R., Chen, J., Tang, Y., Southwell, D. G., Chalmers, N., Vogt, D., et al. (2013). Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12, 573–586. doi: 10.1016/j.stem.2013.04.005
Oberheim, N. A., Takano, T., Han, X., He, W., Lin, J. H. C., Wang, F., et al. (2009). Uniquely hominid features of adult human astrocytes. J. Neurosci. 29, 3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009
Orthmann-Murphy, J. L., Abrams, C. K., and Scherer, S. S. (2008). Gap junctions couple astrocytes and oligodendrocytes. J. Mol. Neurosci. 35, 101–116. doi: 10.1007/s12031-007-9027-5
Pak, C., Danko, T., Zhang, Y., Aoto, J., Anderson, G., Maxeiner, S., et al. (2015). Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell 17, 316–328. doi: 10.1016/j.stem.2015.07.017
Pandya, H., Shen, M. J., Ichikawa, D. M., Sedlock, A. B., Choi, Y., Johnson, K. R., et al. (2017). Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 20, 753–759. doi: 10.1038/nn.4534
Pang, Z. P., Yang, N., Vierbuchen, T., Ostermeier, A., Fuentes, D. R., Yang, T. Q., et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. doi: 10.1038/nature10202
Park, I. H., Arora, N., Huo, H., Maherali, N., Ahfeldt, T., Shimamura, A., et al. (2008). Disease-specific induced pluripotent stem cells. Cell 134, 877–886. doi: 10.1016/j.cell.2008.07.041
Paulsen Bda, S., de Moraes Maciel, R., Galina, A., Souza da Silveira, M., dos Santos Souza, C., Drummond, H., et al. (2012). Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transplant. 21, 1547–1559. doi: 10.3727/096368911X600957
Pei, Y., Peng, J., Behl, M., Sipes, N. S., Shockley, K. R., Rao, M. S., et al. (2016). Comparative neurotoxicity screening in human iPSC-derived neural stem cells, neurons and astrocytes. Brain Res. 1638, 57–73. doi: 10.1016/j.brainres.2015.07.048
Pei, Y., Sierra, G., Sivapatham, R., Swistowski, A., Rao, M. S., and Zeng, X. (2015). A platform for rapid generation of single and multiplexed reporters in human iPSC lines. Sci. Rep. 5:srep09205. doi: 10.1038/srep09205
Pfrieger, F. W. (2009). Roles of glial cells in synapse development. Cell. Mol. Life Sci. 66, 2037–2047. doi: 10.1007/s00018-009-0005-7
Phatnani, H. P., Guarnieri, P., Friedman, B. A., Carrasco, M. A., Muratet, M., O'Keeffe, S., et al. (2013). Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. U. S. A. 110, E756–E765. doi: 10.1073/pnas.1222361110
Poon, A., Zhang, Y., Chandrasekaran, A., Phanthong, P., Schmid, B., Nielsen, T. T., et al. (2017). Modeling neurodegenerative diseases with patient-derived induced pluripotent cells: possibilities and challenges. N. Biotechnol. 39(Pt B), 190–198. doi: 10.1016/j.nbt.2017.05.009
Prinz, M., and Priller, J. (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312. doi: 10.1038/nrn3722
Rajkowska, G., Miguel-Hidalgo, J. J., Makkos, Z., Meltzer, H., Overholser, J., and Stockmeier, C. (2002). Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr. Res. 57, 127–138. doi: 10.1016/S0920-9964(02)00339-0
Ringel, M., Tollman, P., Hersch, G., and Schulze, U. (2013). Does size matter in R&D productivity? If not, what does? Nat. Rev. Drug Discov. 12, 901–902. doi: 10.1038/nrd4164
Robicsek, O., Karry, R., Petit, I., Salman-Kesner, N., Müller, F.-J., Klein, E., et al. (2013). Abnormal neuronal differentiation and mitochondrial dysfunction in hair follicle-derived induced pluripotent stem cells of schizophrenia patients. Mol. Psychiatry 18, 1067–1076. doi: 10.1038/mp.2013.67
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62.
Roybon, L., Lamas, N. J., Garcia-Diaz, A., Yang, E. J., Sattler, R., Jackson-Lewis, V., et al. (2013). Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Rep. 4, 1035–1048. doi: 10.1016/j.celrep.2013.06.021
Schiffer, D., Cordera, S., Cavalla, P., and Migheli, A. (1996). Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 139(Suppl.), 27–33. doi: 10.1016/0022-510X(96)00073-1
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schmitt, A., Steyskal, C., Bernstein, H.-G., Schneider-Axmann, T., Parlapani, E., Schaeffer, E. L., et al. (2009). Stereologic investigation of the posterior part of the hippocampus in schizophrenia. Acta Neuropathol. 117, 395–407. doi: 10.1007/s00401-008-0430-y
Schummers, J., Yu, H., and Sur, M. (2008). Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science 320, 1638–1643. doi: 10.1126/science.1156120
Serio, A., Bilican, B., Barmada, S. J., Ando, D. M., Zhao, C., Siller, R., et al. (2013). Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U.S.A. 110, 4697–4702. doi: 10.1073/pnas.1300398110
Shaltouki, A., Peng, J., Liu, Q., Rao, M. S., and Zeng, X. (2013). Efficient generation of astrocytes from human pluripotent stem cells in defined conditions. Stem Cells 31, 941–952. doi: 10.1002/stem.1334
Sherwood, C. C., Stimpson, C. D., Raghanti, M. A., Wildman, D. E., Uddin, M., Grossman, L. I., et al. (2006). Evolution of increased glia-neuron ratios in the human frontal cortex. Proc. Natl. Acad. Sci. U.S.A. 103, 13606–13611. doi: 10.1073/pnas.0605843103
Sieger, D., Moritz, C., Ziegenhals, T., Prykhozhij, S., and Peri, F. (2012). Long-range Ca2+ waves transmit brain-damage signals to microglia. Dev. Cell 22, 1138–1148. doi: 10.1016/j.devcel.2012.04.012
Soldner, F., Hockemeyer, D., Beard, C., Gao, Q., Bell, G. W., Cook, E. G., et al. (2009). Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977. doi: 10.1016/j.cell.2009.02.013
Son, E. Y., Ichida, J. K., Wainger, B. J., Toma, J. S., Rafuse, V. F., Woolf, C. J., et al. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205–218. doi: 10.1016/j.stem.2011.07.014
Stark, A. K., Uylings, H. B. M., Sanz-Arigita, E., and Pakkenberg, B. (2004). Glial cell loss in the anterior cingulate cortex, a subregion of the prefrontal cortex, in subjects with schizophrenia. Am. J. Psychiatry 161, 882–888. doi: 10.1176/appi.ajp.161.5.882
Steffek, A. E., McCullumsmith, R. E., Haroutunian, V., and Meador-Woodruff, J. H. (2008). Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophr. Res. 103, 71–82. doi: 10.1016/j.schres.2008.04.032
Studer, L., Vera, E., and Cornacchia, D. (2015). Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell 16, 591–600. doi: 10.1016/j.stem.2015.05.004
Sullivan, P. F., Daly, M. J., and O'Donovan, M. (2012). Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet. 13, 537–551. doi: 10.1038/nrg3240
Sun, A. X., Yuan, Q., Tan, S., Xiao, Y., Wang, D., Khoo, A. T. T., et al. (2016). Direct induction and functional maturation of forebrain GABAergic neurons from human pluripotent stem cells. Cell Rep. 16, 1942–1953. doi: 10.1016/j.celrep.2016.07.035
Tabata, H. (2015). Diverse subtypes of astrocytes and their development during corticogenesis. Front. Neurosci. 9:114. doi: 10.3389/fnins.2015.00114
Uranova, N. A., Orlovskaia, D. D., Vikhreva, O. V., Zimina, I. S., and Rakhmanova, V. I. (2001). Morphometric study of ultrastructural changes in oligodendroglial cells in the postmortem brain in endogenous psychoses. Vestn. Ross. Akad. Med. Nauk 7, 42–48.
Vadodaria, K. C., Mertens, J., Paquola, A., Bardy, C., Li, X., Jappelli, R., et al. (2016). Generation of functional human serotonergic neurons from fibroblasts. Mol. Psychiatry 21, 49–61. doi: 10.1038/mp.2015.161
van Berckel, B. N., Bossong, M. G., Boellaard, R., Kloet, R., Schuitemaker, A., Caspers, E., et al. (2008). Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol. Psychiatry 64, 820–822. doi: 10.1016/j.biopsych.2008.04.025
Verkhratsky, A., Sofroniew, M. V., Messing, A., deLanerolle, N. C., Rempe, D., Rodríguez, J. J., et al. (2012). Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro 4:e00082. doi: 10.1042/AN20120010
Vierbuchen, T., Ostermeier, A., Pang, Z. P., Kokubu, Y., Südhof, T. C., and Wernig, M. (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041. doi: 10.1038/nature08797
Vikhreva, O. V., Rakhmanova, V. I., Orlovskaya, D. D., and Uranova, N. A. (2016). Ultrastructural alterations of oligodendrocytes in prefrontal white matter in schizophrenia: a post-mortem morphometric study. Schizophr. Res. 177, 28–36. doi: 10.1016/j.schres.2016.04.023
Vogel, D. Y. S., Vereyken, E. J. F., Glim, J. E., Heijnen, P. D. A. M., Moeton, M., van der Valk, P., et al. (2013). Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflamm. 10:35. doi: 10.1186/1742-2094-10-35
Walterfang, M., Velakoulis, D., Whitford, T. J., and Pantelis, C. (2011). Understanding aberrant white matter development in schizophrenia: an avenue for therapy? Expert Rev. Neurother. 11, 971–987. doi: 10.1586/ern.11.76
Wang, Y. I., Abaci, H. E., and Shuler, M. L. (2017). Microfluidic blood-brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng. 114, 184–194. doi: 10.1002/bit.26045
Watkins, T. A., Emery, B., Mulinyawe, S., and Barres, B. A. (2008). Distinct stages of myelination regulated by γ-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron 60, 555–569. doi: 10.1016/j.neuron.2008.09.011
Wen, Z. (2017). Modeling neurodevelopmental and psychiatric diseases with human iPSCs. J. Neurosci. Res. 95, 1097–1109. doi: 10.1002/jnr.24031
Wen, Z., Nguyen, H. N., Guo, Z., Lalli, M. A., Wang, X., Su, Y., et al. (2014). Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515, 414–418. doi: 10.1038/nature13716
Whitford, T. J., Kubicki, M., and Shenton, M. E. (2011). Diffusion tensor imaging, structural connectivity, and schizophrenia. Schizophr. Res. Treat 2011:709523. doi: 10.1155/2011/709523
Williams, E. C., Zhong, X., Mohamed, A., Li, R., Liu, Y., Dong, Q., et al. (2014). Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum. Mol. Genet. 23, 2968–2980. doi: 10.1093/hmg/ddu008
Williams, M. R., Hampton, T., Pearce, R. K., Hirsch, S. R., Ansorge, O., Thom, M., et al. (2013a). Astrocyte decrease in the subgenual cingulate and callosal genu in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 263, 41–52. doi: 10.1007/s00406-012-0328-5
Williams, M. R., Marsh, R., Macdonald, C. D., Jain, J., Pearce, R. K. B., Hirsch, S. R., et al. (2013b). Neuropathological changes in the nucleus basalis in schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 263, 485–495. doi: 10.1007/s00406-012-0387-7
Windrem, M. S., Schanz, S. J., Morrow, C., Munir, J., Chandler-Militello, D., Wang, S., et al. (2014). A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J. Neurosci. 34, 16153–16161. doi: 10.1523/JNEUROSCI.1510-14.2014
Wolf, F., and Kirchhoff, F. (2008). Neuroscience. Imaging astrocyte activity. Science 320, 1597–1599. doi: 10.1126/science.1160122
Xu, Z., Jiang, H., Zhong, P., Yan, Z., Chen, S., and Feng, J. (2016). Direct conversion of human fibroblasts to induced serotonergic neurons. Mol. Psychiatry 21, 62–70. doi: 10.1038/mp.2015.101
Yagi, T., Ito, D., Okada, Y., Akamatsu, W., Nihei, Y., Yoshizaki, T., et al. (2011). Modeling familial Alzheimer's disease with induced pluripotent stem cells. Hum. Mol. Genet. 20, 4530–4539. doi: 10.1093/hmg/ddr394
Yamamizu, K., Iwasaki, M., Takakubo, H., Sakamoto, T., Ikuno, T., Miyoshi, M., et al. (2017). In Vitro modeling of blood-brain barrier with human iPSC-derived endothelial cells, pericytes, neurons, and astrocytes via Notch signaling. Stem Cell Rep. 8, 634–647. doi: 10.1016/j.stemcr.2017.01.023
Yamanaka, K., Chun, S. J., Boillee, S., Fujimori-Tonou, N., Yamashita, H., Gutmann, D. H., et al. (2008). Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 11, 251–253. doi: 10.1038/nn2047
Yang, N., Zuchero, J. B., Ahlenius, H., Marro, S., Ng, Y. H., Vierbuchen, T., et al. (2013). Generation of oligodendroglial cells by direct lineage conversion. Nat. Biotechnol. 31, 434–439. doi: 10.1038/nbt.2564
Yi, F., Danko, T., Botelho, S. C., Patzke, C., Pak, C., Wernig, M., et al. (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352:aaf2669. doi: 10.1126/science.aaf2669
Yoon, K. J., Nguyen, H. N., Ursini, G., Zhang, F., Kim, N.-S., Wen, Z., et al. (2014). Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 15, 79–91. doi: 10.1016/j.stem.2014.05.003
Young, K. M., Psachoulia, K., Tripathi, R. B., Dunn, S.-J., Cossell, L., Attwell, D., et al. (2013). Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77, 873–885. doi: 10.1016/j.neuron.2013.01.006
Yu, D. X., Di Giorgio, F. P., Yao, J., Marchetto, M. C., Brennand, K., Wright, R., et al. (2014). Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Rep. 2, 295–310. doi: 10.1016/j.stemcr.2014.01.009
Zhang, Y., and Barres, B. A. (2010). Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 20, 588–594. doi: 10.1016/j.conb.2010.06.005
Zhang, Y., Pak, C., Han, Y., Ahlenius, H., Zhang, Z., Chanda, S., et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. doi: 10.1016/j.neuron.2013.05.029
Zhang, Y., Sloan, S. A., Clarke, L. E., Caneda, C., Plaza, C. A., Blumenthal, P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. doi: 10.1016/j.neuron.2015.11.013
Zhao, W., Beers, D. R., Henkel, J. S., Zhang, W., Urushitani, M., Julien, J.-P., et al. (2010). Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 58, 231–243. doi: 10.1002/glia.20919
Zhao, W.-N., Cheng, C., Theriault, K. M., Sheridan, S. D., Tsai, L.-H., and Haggarty, S. J. (2012). A high-throughput screen for Wnt/β-catenin signaling pathway modulators in human iPSC-derived neural progenitors. J. Biomol. Screen. 17, 1252–1263. doi: 10.1177/1087057112456876
Keywords: human induced pluripotent stem cells, drug screening, glia, schizophrenia, ALS, Rett syndrome
Citation: Gonzalez DM, Gregory J and Brennand KJ (2017) The Importance of Non-neuronal Cell Types in hiPSC-Based Disease Modeling and Drug Screening. Front. Cell Dev. Biol. 5:117. doi: 10.3389/fcell.2017.00117
Received: 23 October 2017; Accepted: 08 December 2017;
Published: 19 December 2017.
Edited by:
William L. Stanford, Ottawa Hospital Research Institute (OHRI), CanadaReviewed by:
Tetsuya S. Tanaka, Elixirgen LLC., United StatesFederica Sangiuolo, Università degli Studi di Roma Tor Vergata, Italy
Lisa Marie Julian, Ottawa Hospital, Canada
Copyright © 2017 Gonzalez, Gregory and Brennand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristen J. Brennand, a3Jpc3Rlbi5icmVubmFuZEBtc3NtLmVkdQ==