 Rui Benfeitas
Rui Benfeitas Mathias Uhlen1
Mathias Uhlen1 Jens Nielsen
Jens Nielsen Adil Mardinoglu
Adil Mardinoglu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 11 July 2017
Sec. Molecular and Cellular Oncology
Volume 5 - 2017 | https://doi.org/10.3389/fcell.2017.00065
This article is part of the Research Topic Precision Medicine in Oncology View all 14 articles
Reactive oxygen species (ROS) are important pathophysiological molecules involved in vital cellular processes. They are extremely harmful at high concentrations because they promote the generation of radicals and the oxidation of lipids, proteins, and nucleic acids, which can result in apoptosis. An imbalance of ROS and a disturbance of redox homeostasis are now recognized as a hallmark of complex diseases. Considering that ROS levels are significantly increased in cancer cells due to mitochondrial dysfunction, ROS metabolism has been targeted for the development of efficient treatment strategies, and antioxidants are used as potential chemotherapeutic drugs. However, initial ROS-focused clinical trials in which antioxidants were supplemented to patients provided inconsistent results, i.e., improved treatment or increased malignancy. These different outcomes may result from the highly heterogeneous redox responses of tumors in different patients. Hence, population-based treatment strategies are unsuitable and patient-tailored therapeutic approaches are required for the effective treatment of patients. Moreover, due to the crosstalk between ROS, reducing equivalents [e.g., NAD(P)H] and central metabolism, which is heterogeneous in cancer, finding the best therapeutic target requires the consideration of system-wide approaches that are capable of capturing the complex alterations observed in all of the associated pathways. Systems biology and engineering approaches may be employed to overcome these challenges, together with tools developed in personalized medicine. However, ROS- and redox-based therapies have yet to be addressed by these methodologies in the context of disease treatment. Here, we review the role of ROS and their coupled redox partners in tumorigenesis. Specifically, we highlight some of the challenges in understanding the role of hydrogen peroxide (H2O2), one of the most important ROS in pathophysiology in the progression of cancer. We also discuss its interplay with antioxidant defenses, such as the coupled peroxiredoxin/thioredoxin and glutathione/glutathione peroxidase systems, and its reducing equivalent metabolism. Finally, we highlight the need for system-level and patient-tailored approaches to clarify the roles of these systems and identify therapeutic targets through the use of the tools developed in personalized medicine.
Redox metabolism is closely intertwined with cell physiology, and reactive oxygen species (ROS) are central players in health and disease. For instance, these oxygen-derived species are involved in cancer (Reuter et al., 2010), neurodegenerative diseases (Sultana et al., 2006), aging (Höhn et al., 2013), and diabetes (Evans et al., 2002). They are produced intracellularly by several processes and dedicated enzymes, such as NADPH oxidases (Nauseef, 2008) and in multiple cellular compartments (Messner and Imlay, 2002; Chen et al., 2008; Murphy, 2009; Brown and Borutaite, 2012). Due to their high membrane permeability (Chance et al., 1979), extracellularly produced ROS (Hampton et al., 1998; Babior et al., 2002) may quickly enter cells, or they may diffuse across compartments (Bienert et al., 2007; Marchissio et al., 2012). In many diseases, imbalances in ROS metabolism lead to oxidative stress. As result, cells face toxic outcomes of protein, lipid, and nucleic acid oxidation (Garrison, 1987; Cooke et al., 2003; Smith and Murphy, 2008; Figure 1A). For instance, DNA oxidation by ROS promotes mutagenesis, cancer initiation, and progression (Shibutani et al., 1991; Cooke et al., 2003; Sabharwal and Schumacker, 2014), and at high concentrations, ROS may cause cell apoptosis (Gao et al., 2013).
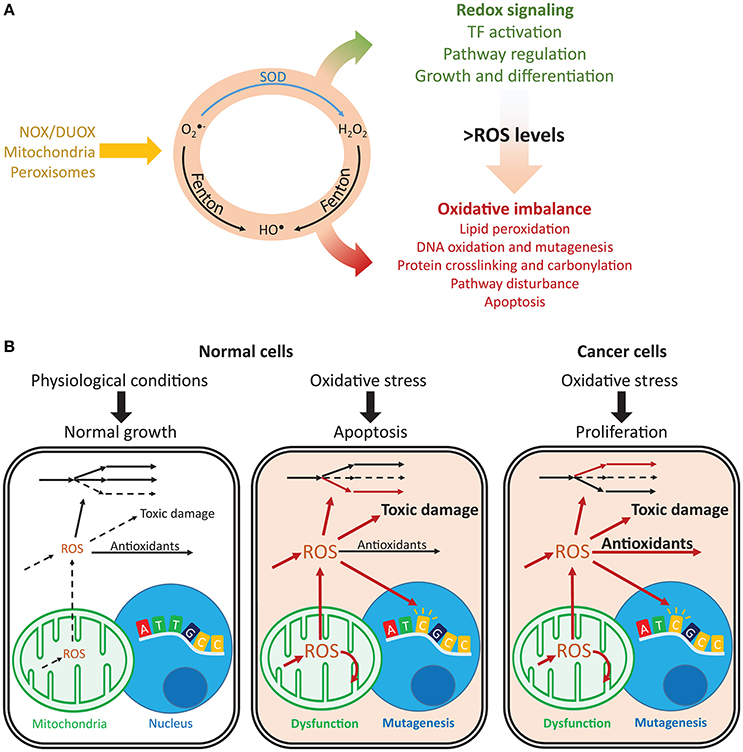
Figure 1. Imbalances in ROS and redox cycles lead to contrasting outcomes in normal and cancer cells. (A) ROS are intracellularly produced by NADPH oxidases and dual oxidases (NOX/DUOX) through mitochondrial oxidative phosphorylation and in peroxisomes. Their interconversion (orange circle) occurs through enzyme catalyzed and non-catalyzed reactions. For instance, metal-catalyzed Fenton reactions produce HO• from and H2O2. Under low ROS levels, these oxidants control important signaling reactions, activating transcription factors, regulating pathways and controlling cell growth and differentiation. Under high ROS levels, oxidation of lipids, nucleic acids, and proteins is toxic and may disturb pathways and lead to cell death. (B) Normal and cancer cells present important differences in their responses to oxidative stress. Under normal conditions, ROS production is low and antioxidant defenses are sufficient to prevent toxic damage. Under oxidative stress, the promoted production of ROS overcomes the cell's capacity for detoxification and results in increased toxic damage and pathway disruption, which may lead to mitochondrial dysfunction, mutagenesis and ultimately apoptosis. In turn, cancer cells are under constant oxidative stress, which through upregulation of antioxidant defenses, prevents apoptosis while maintaining ROS toxicity. Arrows indicate fluxes, increasing from dashed to continuous, in red. , superoxide; H2O2, hydrogen peroxide; HO•, hydroxyl radical; SOD, superoxide dismutase.
In turn, low ROS concentrations have important physiological roles (D'Autréaux and Toledano, 2007). They regulate cell-cycle progression (Havens et al., 2006), proliferation (Choe et al., 2012), growth (Arnold et al., 2001), and important signaling processes (Finkel, 2011; Rigoulet et al., 2011). For instance, hydrogen peroxide (H2O2) regulates the activity of kinases (Gotoh and Cooper, 1998; Paulsen et al., 2012), which control proliferation, differentiation, and apoptosis. NFE2L2 (also known as NRF2) responds to oxidative stress and regulates GSH biosynthesis and reduction, the expression of several proteins involved in antioxidant defense (glutathione peroxidases, transferases, peroxiredoxins, thioredoxins, and thioredoxin reductases), and NADPH production (Gorrini et al., 2013). Many of these redox-regulated processes are not directly controlled by ROS but rather by their redox partners. For instance, redox signaling transduction is often mediated by peroxiredoxins, thioredoxins, and other thiol-reacting proteins (Saitoh et al., 1998; Giannoni et al., 2005; Morinaka et al., 2011). These proteins ensure the high specificity required for efficient signaling transduction (Nagy and Winterbourn, 2010; Winterbourn, 2013; Marinho et al., 2014; Netto and Antunes, 2016). Together with other cellular antioxidants, such as catalases and dismutases, these redox systems prevent toxic ROS accumulation while permitting special-temporal selectivity and the maintenance of important redox signaling functions.
In cancer, mitochondrial dysfunction and metabolic changes promote constant oxidative stress (Szatrowski and Nathan, 1991; Hileman et al., 2004); however, this does not result in apoptosis. Cancer cells promote the expression of antioxidant defenses or reducing equivalents that enable their activity (Janssen et al., 1999; Miranda et al., 2000; Hileman et al., 2004), thus avoiding ROS-induced apoptosis and enabling proliferation, despite high mutagenesis (Toyokuni et al., 1995; Kondo et al., 1999) and metastasis (Ishikawa et al., 2008; Figure 1B). ROS closely interact with iron (Galaris et al., 2008) and central (Robbins et al., 2012; Hart et al., 2015; Miar et al., 2015) metabolism, and they are controlled by several transcription factors and tumor suppressors (Gao et al., 2007; Frohlich et al., 2008; Gupta et al., 2012; Gorrini et al., 2013; Hornsveld and Dansen, 2016). Additionally, antioxidant enzymes may display high or low expression in cancer cells (Ray et al., 2000; Oltra et al., 2001; Skrzydlewska et al., 2005; Glorieux et al., 2015), suppress tumorigenesis or promote metastasization (Zhao et al., 2001; Liu et al., 2012; Robbins et al., 2012; Miar et al., 2015), and display synergistic responses (Harris et al., 2015). As result, systematic approaches may capture these complex responses and provide insights into the mechanisms underlying the diverse phenotypic responses in cancer.
Systems biology presents promising approaches for capturing and studying complex cellular responses (Mardinoglu et al., 2013b; Ghaffari et al., 2015b). The application of such frameworks to clinical challenges is referred to as systems or network medicine (Mardinoglu and Nielsen, 2012, 2016). The complexity of biological pathways in cells and tissues may be captured through reconstruction of biological networks, including genome-scale metabolic models (GEMs), transcriptional regulatory networks, protein–protein interaction networks, and signaling networks, in an integrated approach that aims to understand entire cell processes at the systems level (Mardinoglu and Nielsen, 2015; Zhang et al., 2016). These networks may also be integrated with each other for a holistic understanding of the relationships between cellular networks, function, and disease (Bjornson et al., 2016; Lee et al., 2016; Mardinoglu and Uhlén, 2016). Generation of omics data for major human tissues enabled the generation of comprehensive biological networks (Kampf et al., 2014a; Lindskog et al., 2015; Uhlén et al., 2015, 2016; Thul et al., 2017), which have been successfully employed in revealing the underlying mechanisms involved in the occurrence of obesity (Mardinoglu et al., 2013a, 2014b, 2015a), type 2 diabetes (Väremo et al., 2015), non-alcoholic fatty liver disease (Kampf et al., 2014b; Mardinoglu et al., 2014a, in press; Hyötyläinen et al., 2016), and cancer (Agren et al., 2012, 2014; Weinstein et al., 2013; Zack et al., 2013; Leiserson et al., 2014; Yizhak et al., 2014b; Aran et al., 2015; Bjornson et al., 2015; Peng et al., 2015; Elsemman et al., 2016). Personalized models have also been used in the identification of potential therapeutic targets and biomarkers (Faratian et al., 2009; Agren et al., 2014; Bjornson et al., 2015; Mardinoglu et al., 2017; Nielsen, 2017). To date, small scale redox networks have also been analyzed (Zhang et al., 2010; Zhou et al., 2011; Zhan et al., 2012). However, despite extensive evidence highlighting the importance of ROS, antioxidants and other redox players in cancer, systems approaches have yet to systematically examine the role of redox metabolism in this disease and uncover potential personalized treatment strategies.
Here, we highlight some recent findings about important biological processes that are crucial in tumorigenesis: ROS, their redox partners, and reducing equivalents. We start by overviewing some of the main biochemical properties of ROS and their effectors. We then discuss the role of antioxidants and their reactions in tumorigenesis, focusing on thiols and reducing equivalents due to their importance in ROS and redox homeostasis. Finally, given the high heterogeneity of redox responses and the intricate crosstalk between redox and central metabolism, we highlight how system-level and patient-tailored approaches may help to identify potential cancer targets and provide mechanistic insights into redox cancer responses. These discussions do not aim to be exhaustive descriptions of all biological processes and regulators of redox homeostasis, and the interested reader may find excellent reviews on these topics elsewhere (e.g., Gao et al., 2007; Frohlich et al., 2008; Gupta et al., 2012; Gorrini et al., 2013; Hornsveld and Dansen, 2016).
Molecular oxygen freely diffuses across cell membranes and promotes the formation of intracellular ROS through electron abstraction. ROS may be classified as radicals and non-radicals. Radicals have unpaired electrons and include superoxide () and the hydroxyl (HO•) radicals. Non-radical ROS do not have unpaired electrons, and they include H2O2. Here, we focus on these three ROS due to their patho-physiological importance. ROS metabolism yields many other less reactive, abundant, or stable ROS and is highly intertwined with other important reactive species (such as Reactive Nitrogen Species, Weidinger and Kozlov, 2015).
ROS are formed in several intracellular compartments. Most notably, they are produced in peroxisomes through fatty acid oxidation (Fransen et al., 2012), in mitochondria during oxidative phosphorylation and in the cellular and intracellular membranes by NADPH oxidases (EC 1.6.3.1; Nauseef, 2008; Kowaltowski et al., 2009; Murphy, 2009; Brown and Borutaite, 2012; Fransen et al., 2012). Some crosstalk exists between these systems. For instance, mitochondrial-produced ROS promote generation by NADPH oxidases, which may have important functions during phagocytosis (Dikalov, 2011). It is currently unclear which of the compartments above contributes the most to intracellular ROS production, although mitochondria are often cited as the main cellular ROS source (Brown and Borutaite, 2012).
The reactions involving ROS and their cellular targets lead to the interconversion of various types of ROS (Figure 2). For instance, mitochondrial- and cytoplasmic-produced is dismutated to H2O2 by superoxide dismutases (SOD, EC 1.15.1.1). is fairly unreactive to most electron-rich centers due to its anionic charge, but it reacts with nitric oxide to form peroxynitrite (Huie and Padmaja, 1993) and oxidizes iron-sulfur clusters, thereby producing H2O2 and HO• (Rouault and Klausner, 1996). These clusters are found in multiple intracellular compartments (Tong et al., 2000), and their oxidation by (or H2O2) leads to iron release and the inactivation of metabolically important enzymes, such as those involved in amino acid biosynthesis (Wallace et al., 2004) or carbohydrate metabolism (Gardner et al., 1995). It is currently unclear whether most is used toward nitric oxide metabolism, if it reacts with metal clusters, or if it is dismutated to H2O2. Both dismutases and nitric oxide react with with near diffusion-limited rate constants (k > 109 M−1 s−1, Bannister et al., 1973; Huie and Padmaja, 1993), and is very reactive with some iron-sulfur cluster-bearing enzymes (k ≈ 106–107 M−1 s−1, Flint et al., 1993), but it is unreactive with amino acid residues (Bielski and Shiue, 1979). The fate of depends on the local availability of the other reactants or enzymes and likely varies between cells and under different conditions, although it is generally assumed that most is dismutated to H2O2 (Forman et al., 2010).
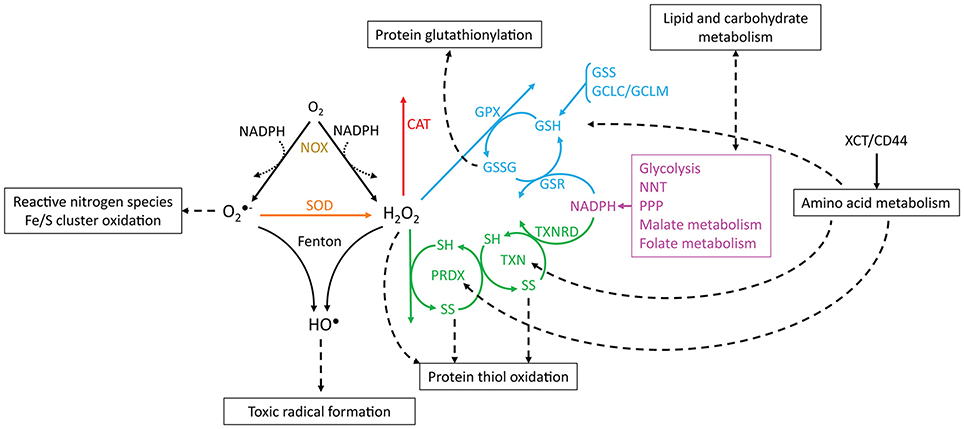
Figure 2. Reactions involving ROS, antioxidant systems and energy metabolism. and H2O2 are produced from oxygen through reactions that may oxidize NADPH (dotted arrows, e.g., catalysis by NADPH oxidases). is dismutated to H2O2 by SOD, and and H2O2 may be converted to HO• by Fenton reactions. CAT, PRDX and GPX scavenge H2O2. The catalytic cycles of PRDX and TXN are represented, where SH and SS, respectively, indicate reduced and oxidized (disulfide) thiols. Boxes and dashed arrows indicate the external processes with which the metabolites are associated. For example, XCT/CD44 mediates cysteine import, which may then be incorporated into proteins, such as TXN and PRDX, or may be metabolized to yield GSH. Colors indicate proteins or processes from the same pathway.
In turn, H2O2 reacts slowly with most biological compounds, such as free glutathione (k < 10 M−1 s−1, Winterbourn and Metodiewa, 1999) and phosphatases (k ≈ 10–200 M−1 s−1, LaButti et al., 2007; Marinho et al., 2014). However, it may display extremely high reactivities with selected protein thiols due to their neighboring chemical environment (k ≈ 105–108 M−1s−1, Peskin et al., 2007; Trujillo et al., 2007; Manta et al., 2009). It is decomposed into water and molecular oxygen as a result of dismutation by catalases (EC 1.11.1.6), or it is reduced to water by peroxidases and peroxiredoxins (EC 1.11.1.15). Often, several of these mechanisms are present in the same cells. Protection against H2O2 is accomplished through glutathione peroxidase, catalase, and peroxiredoxin 2 in human erythrocytes (Johnson et al., 2005; Low et al., 2007; Benfeitas et al., 2014). These defenses are unlikely to be redundant in their functions: while catalase is an efficient H2O2 scavenger, even under high oxidative loads, peroxiredoxin 2 has limited reduction under such conditions (Low et al., 2007), resulting in a lower contribution for H2O2 consumption. However, peroxiredoxin 2 and its coupled cycles display desirable redox signaling properties (Benfeitas et al., 2014), which are further discussed below.
and H2O2 are also involved in the production of HO• through iron-catalyzed Fenton reactions involving heme peroxidases or iron/sulfur clusters (Fenton, 1894; Chen and Schopfer, 1999; Koppenol, 2001). Iron accumulation and HO• production have been extensively associated with carcinogenesis, and iron chelators have been employed as therapeutic drugs in cancer (reviewed by Torti and Torti, 2013; Bystrom and Rivella, 2015). Due to its electrophilic nature, HO• preferably oxidizes electron-rich sites, reacting with nucleic acids, lipids, and proteins with diffusion-limited rate constants (Von Sonntag, 1987; Buxton et al., 1988; Stadtman and Levine, 2003; Sharma and Rokita, 2013). This promotes DNA strand breaks, lipid peroxidation and protein carbonylation, crosslinking, and cleavage. The products of these reactions are toxic, and they often promote radical propagation and damage to nearby molecules through subsequent chain reactions. Importantly, due to HO•'s very high and unselective reactions with biological compounds, no cellular antioxidants can feasibly scavenge this oxidant before it reacts with cellular contents. Instead, protection against the toxic outcomes of HO• comes from preventing its formation by shielding iron from ROS or by scavenging H2O2 and before they yield HO•. The role of H2O2 in HO• formation and consequential radical formation is also thought to be one of the main reasons behind H2O2's toxicity (Winterbourn, 1995).
As result of their different reactivities with biological compounds, the above ROS present varying stabilities and cellular roles. For instance, the fast and indiscriminate reactions of HO• result in very small diffusion distances (≈80 Å, Roots and Okada, 1975) that are approximately the size of a small peptide. For this reason, HO• is likely to oxidize molecules near its formation site (estimated half-life of 10−9 s, Pryor, 1986), and it is unfit to behave as a signaling molecule. is also regarded as a poor signaling effector because it does not permeate cell membranes and is quickly dismutated to H2O2, or it reacts with iron/sulfur clusters and nitric oxide. This results in low intracellular stability, hindering its diffusion across large distances. Therefore, while has some regulatory properties, these properties are possibly due to its role in nitric oxide and H2O2 metabolism (Brune, 2005; Thomas et al., 2006; D'Autréaux and Toledano, 2007; Kaewpila et al., 2008; Labunskyy and Gladyshev, 2013). In turn, H2O2 has emerged as the ROS that displays the best signaling properties. Its high stability and selective reactions with cellular compounds permit diffusion over distances of several micrometers (Winterbourn, 2008) and enable cell membrane crossing, which is also facilitated through specific channels (Bienert et al., 2007). H2O2 reacts with cellular thiols, including those contained in low molecular weight compounds, such as glutathione, and protein thiols, such as peroxiredoxins and thioredoxins (Box 1 and Figure 3). These reactions convert an oxidizing equivalent into a redox signal, which may be transduced from protein to protein via thiol disulfide exchange or between glutathione and proteins, forming mixed disulfides. Together with intracellular thiols, H2O2 regulates the redox state and activity of several target proteins and has pivotal importance in both physiological and pathological conditions (D'Autréaux and Toledano, 2007). Due to this intricate association, any discussion about the involvement of ROS in tumorigenesis also needs to consider the role of thiols and other antioxidant defenses. Although these systems have been widely studied in cancer, the role of ROS, antioxidant defenses, and redox signaling transducing partners in cancer is only now emerging beyond antioxidant activities. Further ROS-centered studies aimed at clarifying these properties and their involvement in cancer are still required.
BOX 1. Thiols as important redox signaling sensors and effectors.
Cysteine's thiol side chains (R-SH) are often very reactive with H2O2. They undergo a series of reversible or irreversible redox transitions, which are represented here through the catalytic cycles of a typical 2-Cys peroxiredoxin/thioredoxin (Figure 3). In this cycle, the reduced form is subsequently oxidized by H2O2 to sulfenic (R-SOH) and sulfinic (R-SO2H) acids. The sulfinic form may be irreversibly oxidized to sulfonic (R-SO3H) forms in vitro, but it is currently unclear whether this process occurs in vivo. Sulfinic acids may be reduced to sulfenic acids by specific proteins (e.g., sulfiredoxins, SRX) at the expense of ATP and the oxidation of TXN and GSH (Chang et al., 2004). Sulfenic acids may also conjugate to form intra- or inter-molecular disulfide bonds (R-SS-R'). Disulfides are then reduced at the expense of reducing equivalents, such as those found in NADPH (e.g., oxidized GSH or thioredoxin reduction by reductases), or by disulfide exchange with other proteins. Therefore, cysteine oxidation by H2O2 may be transduced to partner proteins or small molecular weight compounds.
Due to their chemical and kinetic properties, the systems above have potentially different signaling properties. Although they exhibit slow reactivities with H2O2 when isolated (k ≈ 2.9 M−1 s−1 for free Cys, Winterbourn and Metodiewa, 1999), some cysteine thiols display extremely high reactivities (k ≈ 105–108 M−1 s−1 for peroxiredoxins, Trujillo et al., 2007; Manta et al., 2009). GSH is very abundant, but it is relatively unreactive with H2O2 per se, so the kinetics of glutathione peroxidases should be considered when assessing glutathione's intracellular role in H2O2 detoxification and signaling. These differences in reactivity also manifest within the same pathway. Reduced and sulfenic forms quickly react with H2O2, unlike sulfinic and sulfonic acids, which are relatively unreactive with H2O2. The process of thiol oxidation to disulfide exchange may transduce oxidative equivalents to target proteins, as observed in the proteins above (Jarvis et al., 2012; Naticchia et al., 2013; Sobotta et al., 2015). For instance, thiol-disulfide exchange between peroxiredoxins and phosphatases/kinases is a mechanism for explaining H2O2-induced signaling regulation despite the low reactivity of H2O2 with phosphatases/kinases (Ray et al., 2012; Marinho et al., 2014; Sobotta et al., 2015; Latimer and Veal, 2016). Importantly, peroxiredoxin-mediated disulfide exchange controls the activity of several proteins involved in cancer (Park et al., 2007; Jarvis et al., 2012; Sobotta et al., 2015), which reinforces the role of H2O2 and redox metabolism in this disease. Similar to disulfide exchange, the oxidation of glutathione may lead to S-glutathionylation of proteins, which also regulates their activities (e.g., peroxiredoxin 2, Peskin et al., 2016). These and other properties possibly explain the involvement of PRDXs and TXNs in redox signaling, and they point toward these proteins as good redox sensors and signaling transducers (Benfeitas et al., 2014; Latimer and Veal, 2016; Netto and Antunes, 2016; Tomalin et al., 2016).
It should be noted that many of these redox systems depend on reducing equivalents to maintain their activity. Reducing equivalents are any molecules that act as electron donors in reactions, typically in reference to NADH and NADPH. These species are used by several enzymes, such as reductases, which couple their oxidation to the reduction of thioredoxin or glutathione. This provides another possible layer of selectivity in redox homeostasis: should NADPH utilization be prioritized toward one system over another, the physiological role of the former would also be prioritized over the latter. Interestingly, the link between energy metabolism and redox metabolism goes beyond NAD(P)H-enabling reductase activity. For instance, glyceraldehyde-3-phosphate dehydrogenase, an NADH-producing enzyme essential to glycolysis, is inactivated by glutathionylation and H2O2-induced disulfide formation (Little and O'brien, 1969; Mohr et al., 1999). ROS and redox-coupled processes thus not only consume reducing equivalents but also regulate energy metabolism.
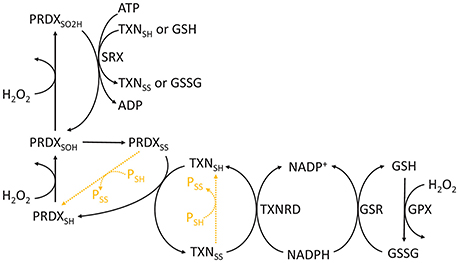
Figure 3. Chemical cycle of PRDXs, TXNs, and GSH. The redox state of PRDXs and TXN is indicated as follows: SH, reduced cysteine thiol; SOH, sulfenic acid; SO2H, sulfinic acid; SS, disulfide. Orange dashed reactions highlight disulfide exchange between peroxiredoxins and thioredoxins and other proteins.
The role of ROS in cell physiology is highly dependent on their levels. Under physiological levels, ROS regulate a number of signaling processes by reacting with proteins, genes, and transcription factors. ROS control adaptation to hypoxia, regulation of differentiation, immunity, and longevity (Sena and Chandel, 2012). However, the accumulation of ROS beyond physiological levels promotes cell proliferation, angiogenesis, and even apoptosis (D'Autréaux and Toledano, 2007; Cairns et al., 2011; Figure 4), and ROS also control cell-cycle progression (Menon and Goswami, 2007). Oncogene-induced senescence promotes AMP-activated protein kinase activation, mitochondrial dysfunction and ROS production, which trigger senescence, thereby forming a positive feedback loop. Cancer cells display high ROS production (Szatrowski and Nathan, 1991; Ray et al., 2000), which is often also associated with antioxidant imbalances (Skrzydlewska et al., 2005). This results in damage to nuclear (Shibutani et al., 1991) and mitochondrial DNA (Ishikawa et al., 2008; Weinberg et al., 2010). Mutations in nucleic acids may be particularly toxic for the cell if they occur in tumor suppressors or oncogenes. DNA mutations (Higinbotham et al., 1992; Du et al., 1994), in turn, promote ROS generation, thereby resulting in a vicious cycle of ROS production and mutagenesis concomitant with high proliferation. Mitochondrial-generated ROS are also essential for tumor aggressiveness and metastasis (Ishikawa et al., 2008; Weinberg et al., 2010; Goh et al., 2011), and increased cytoplasmic and mitochondrial ROS levels are observed in metastatic nodules and circulating tumors when compared to subcutaneous tumors (Piskounova et al., 2015).
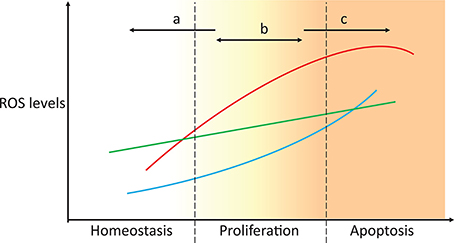
Figure 4. Targeting ROS homeostasis as a strategy for changing cell fate. In proliferative conditions, such as cancer, targeting antioxidant systems could move the redox state of the cell to either promote normal redox homeostasis or apoptosis (strategies a and c, respectively). Unsuccessful tackling of antioxidant metabolism results in the cells maintaining a proliferative state, which potentially enhances malignancy. The three lines represent the high heterogeneity between individuals, tissues, and cancer types.
ROS also stabilize the factors that drive tumor initiation and progression (Gao et al., 2007), and promote protein oxidation and the formation of toxic protein carbonyls (Stadtman and Levine, 2003). Protein carbonylation is an irreversible process present in cancer cells (Thanan et al., 2012). Carbonyls may propagate to other proteins or lipids, which may result in the formation of toxic byproducts through chain reactions. ROS-induced lipid peroxidation products, such as 4-hydroxy-2-nonenals, also have multiple physiological roles under low levels, but they become toxic upon accumulation. These species also accumulate in cancer cells (Skrzydlewska et al., 2005; Ayala et al., 2014; Zhong and Yin, 2015).
Due to the toxic effects of ROS accumulation in cancer cells and the fact that some ROS responses may be exclusive to cancer cells but not to their healthy counterparts (Hileman et al., 2004), antioxidants were envisioned as potentially important drug targets in cancer treatment. Initial ROS-focused clinical trials aimed to prevent ROS accumulation (Figure 4, strategy a). In the Linxian study (Blot et al., 1993) stomach cancer patients supplemented with selenium, vitamin E and β-carotene exhibited lower mortality. However, antioxidant-supplemented diets often failed to yield significant changes in cancer development, and in some cases, these diets even promoted tumorigenesis and metastasis (Omenn et al., 1996; Goodman et al., 2011; Klein et al., 2011; Sayin et al., 2014). This is because cancer cells often cope with increased ROS production by increasing the levels of antioxidant defenses or reducing equivalents that maintain their activity (Weinberg et al., 2010; DeNicola et al., 2011). This effect is also observed in metastases, where metastatic melanoma nodules are exposed to additional oxidative stress that is not observed in established subcutaneous tumors, and the nodules cope with this stress by promoting the expression of multiple NADPH-producing pathways (Piskounova et al., 2015). High antioxidant activities enable fast ROS-driven proliferation and metastasization, but the increased oxidative stress is insufficient to lead to apoptosis (Figure 4, strategy b). Higher antioxidant expression is also associated with the radioresistance observed in certain cancer stem cell populations (Diehn et al., 2009). In turn, recent antioxidant-targeted therapeutic strategies have shifted their focus in the opposite direction, exploiting ROS toxicity as a means leading to cancer cell apoptosis (Figure 4, strategy c). These drugs often mimic ROS-generating enzymes (e.g., NADPH oxidases and superoxide dismutases), inhibit antioxidant enzymes (e.g., catalase), deplete thiol pools, such as GSH, or shift redox buffer ratios (e.g., GSSG/2 GSH), thereby promoting a more oxidizing intracellular state and cell apoptosis. These strategies have been discussed in great detail in recent reviews (Ushio-Fukai and Nakamura, 2008; Gupta et al., 2012; Gorrini et al., 2013; Tong et al., 2015).
The targets of many of these drugs are not completely understood, nor is it known whether the drugs are targeting the best redox effectors. Are the targeted compounds selective in treating cancer, but not healthy cells? Are they the best target in that pathway? Which combined drug treatments could improve treatment? These questions stem from an insufficient understanding of redox metabolism in tumorigenesis, which is greatly caused by the high variability in metabolic and redox responses. For instance, different antioxidant defenses that target the same ROS are up- and down-regulated in cancer cells (Skrzydlewska et al., 2005), complicating the interpretation of their role and that of their target ROS in cancer progression. This high variability is even observed between individuals with the same cancer type (Figure 5). Furthermore, genes involved in the same processes are differentially expressed. For instance, a high dispersion in gene expression levels is observed within peroxiredoxins (PRDX1-6), thioredoxins (TXN and TXN2), and thioredoxin reductases (TXNRD1-3), which are highly conserved protein systems involved in H2O2 scavenging and redox signaling transduction. As result of this variability, redox-focused therapeutic strategies (Figure 4) must consider patient-specific data to determine the best approach.
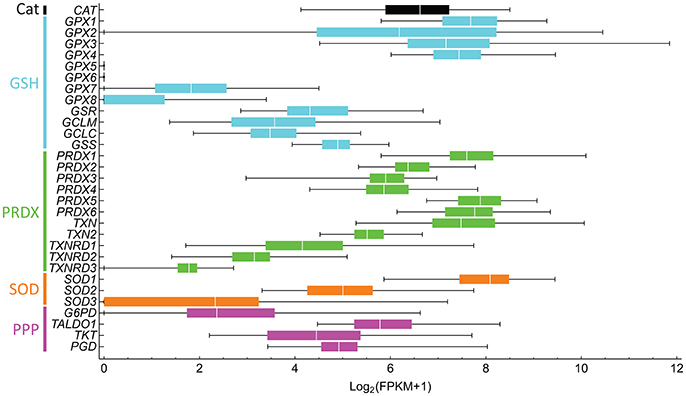
Figure 5. Antioxidant gene expression greatly varies between liver hepatocellular carcinoma in different subjects. The gene expression of 50 subjects was downloaded from NCI's Genomic Data Commons, and fragments per kilobase transcript per million (FPKM) were computed. FPKM-values lower than one were considered to be non-expressed and were assigned a value of 0. The Log2(FPKM + 1) were then computed. Bars are colored according to processes of the same pathway, as indicated on the left. Genes and respective proteins: CAT, catalase; GPX1-8, glutathione peroxidase; GSR, glutathione reductase; GCLM and GCLC, glutamate-cysteine ligase modifier and catalytic subunits, respectively; GSS, glutathione synthetase; PRDX1-6, peroxiredoxin; TXN and TXN2, thioredoxin; TXNRD1-3, thioredoxin reductase; SOD, superoxide dismutase; G6PD, glucose-6-phosphate dehydrogenase; TALDO1, transaldolase 1, TKT, transketolase, and PGD, 6-phosphogluconate dehydrogenase.
Imbalances in antioxidant defenses are one of the hallmarks of cancer (Oberley and Oberley, 1997; Huang et al., 2000; Chung-man Ho et al., 2001; Hu et al., 2005; Murawaki et al., 2008). For instance, increased expression of SOD has been observed in multiple cancers (Ray et al., 2000; Skrzydlewska et al., 2005; Holley et al., 2012; Miar et al., 2015) and metastatic tissues (Miar et al., 2015). Its overexpression promotes carcinogenesis (Lu et al., 1997) and aggressiveness (Hempel et al., 2011). In turn, SOD deficiency is associated with a higher cancer incidence and DNA damage in mice (Van Remmen et al., 2003; Elchuri et al., 2005), and it has also been observed in some cancers (Oltra et al., 2001). SOD levels fluctuate throughout the cell cycle and regulate growth factor cancer signaling (Nelson et al., 2003; Juarez et al., 2008), cell cycle progression, and the energetic changes of cells upon cancer transformation (Hempel et al., 2011; Sarsour et al., 2014). Catalases, often regarded as the main cellular defenses against H2O2 in human cells, may be up- or down-regulated in cancer cells (Ray et al., 2000; Oltra et al., 2001; Skrzydlewska et al., 2005; Glorieux et al., 2015). Catalase treatment of highly metastatic cancer cell lines decreases migration and invasion (Liu et al., 2012). Due to their antioxidant activities and correlation with decreased aggressiveness in certain tumors, catalases have been envisaged as potentially important therapeutic agents (Glorieux et al., 2011; de Oliveira et al., 2016). For instance, a recent study shows that catalase activity correlates well with the ability of pancreatic cancers to resist chemotherapeutic H2O2 treatment with ascorbate (Doskey et al., 2016). In turn, altered levels of glutathione peroxidases are amply reported in cancer (Lu et al., 1997; Skrzydlewska et al., 2005), and the reduction potential of glutathione is associated with the proliferative and apoptotic state of a cell (Buettner et al., 2013). For instance, several glutathione peroxidases are upregulated in hepatocellular cancer (Carlson et al., 2012), and GPX4-deficient mice die shortly after birth (Carlson et al., 2016). GPXs are responsible not only for reducing H2O2 but also for reducing other ROS, such as lipid peroxides (Thomas et al., 1990; Esworthy et al., 1993). Peroxiredoxins have also been associated with tumorigenesis. PRDX1-4 and 6 display significantly altered levels in some prostate cancers (Basu et al., 2011; Whitaker et al., 2013). PRDXs may act as tumor suppressors (Egler et al., 2005), and their increased gene expression is associated with metastasis and aggressiveness (Park et al., 2006; Chang et al., 2007; Stresing et al., 2012). For instance, PRDX2 is highly expressed in lung metastases, and its knockdown decreases the formation of lung metastasis (Stresing et al., 2012). PRDX2 is also highly expressed in breast carcinoma, which correlates with the formation of lung metastases. PRDXs are often upregulated in cancers, and they contribute to cancer survival and resistance to oxidative stress (Lu et al., 2014) and radiotherapy (Wang et al., 2005). Interestingly, knockouts of the PRDX genes do not always result in a favorable outcome. For instance, PRDX1 knockout mice show premature death, increased DNA oxidation and increased tumorigenesis and malignancy, and PRDX3 knockout results in increased protein carbonylation in adipose tissues (see Cao et al., 2009; Perkins et al., 2014 and references therein). These observations show the crucial role of antioxidant enzymes in cancerogenesis and survivability.
Alterations in antioxidant enzymes and ROS levels occur throughout cancer progression. Overexpression of mitochondrial SOD2 acts as a tumor suppressor in skin and breast cancers (Zhao et al., 2001; Robbins et al., 2012), suggesting that high SOD activity promotes tumor initiation. However, SOD levels also increase with tumor progression in these and other cancers (Ray et al., 2000; Chung-man Ho et al., 2001; Dhar et al., 2011; Miar et al., 2015), with similar changes occurring in other antioxidants, such as cytoplasmic SOD1 and catalase (Miar et al., 2015). Similarly, metastases show increased SOD2 protein levels when compared with matched tissues, less metastatic cell lines, or primary tumors (Liu et al., 2012; Miar et al., 2015). These changes are often accompanied by directly proportional changes in intracellular concentrations of H2O2, but they are not always followed by changes in other antioxidants. Changes in ratios of antioxidant proteins (e.g., SOD/catalase and SOD/GPX1) translate into differential intracellular concentrations of H2O2, and they vary with cancer stage and between metastatic and primary tumor cells (Miar et al., 2015). These observations show that while a comparison of the antioxidant expression levels between cancer and matched tissues is informative, such a comparison has to consider the developmental stage of the cancer.
Importantly, these antioxidant defenses are not found in all cell compartments, which should also differentially affect the responses of cancer cells to therapeutic targeting. For instance, mitochondria rely on superoxide dismutase 2, peroxiredoxin 3, thioredoxin 2, and thioredoxin reductase 2 for antioxidant defense and signaling transduction, but they do not rely on catalase (Rabilloud et al., 2001; Jones, 2006). Considering the crucial role that mitochondrial-generated ROS have in cancer initiation, progression, and apoptosis, the targeting of key antioxidant and redox signaling transduction systems in this compartment (e.g., PRDX3 Li and Yu, 2015) may have important consequences for global cell metabolism. However, it should be noted that targeting these antioxidants may lead to compensatory responses by the other antioxidants. For instance, knockdown of PRDX3 promotes the upregulation of other peroxiredoxins, including those located in the cytoplasm (PRDX1-2 and PRDX6; Li et al., 2008, 2013; Goncalves et al., 2012). This is particularly important because these proteins may catalyze similar reactions in H2O2 scavenging and disulfide exchange, albeit with different mechanisms and specificities (Perkins et al., 2014).
In addition, cancer cells cope with increased intracellular ROS by promoting the synthesis of compounds that enable their activity (e.g., glutathione for GPXs, Buettner et al., 2013 and TXNs for PRDXs, Arnér and Holmgren, 2006; Kaimul et al., 2007). GSSG/GSH ratios are higher in many cancers (Oltra et al., 2001; Skrzydlewska et al., 2005), in metastatic nodules and in circulating tumor cells (Piskounova et al., 2015), and the enzymes involved in GSH recycling or synthesis are often upregulated (Carlson et al., 2012; Harris et al., 2015; Lien et al., 2016) as response to oxidative stress. Mutations in PI(3)K/Akt, which are common in some cancer types, such as breast cancer, stabilize and activate NFE2L2, thereby promoting the upregulation of enzymes involved in synthesizing or reducing GSH (glutathione synthetase GSS, and glutathione reductase GSR; Lien et al., 2016). Upregulation of GSS and GSR is also associated with the increased resistance to oxidative stress observed in breast cancer. The inhibition of GSH biosynthesis sensitizes cancer cells to H2O2 and is potentiated by the utilization of other antioxidant inhibitors (Lien et al., 2016). High intracellular GSH concentrations also block drug-induced cytotoxicity in myeloma cells (Starheim et al., 2016). Cumulative evidence thus points toward GSH biosynthesis and homeostasis as a therapeutic target. However, in some cancers, inhibition of the GSH pathway alone does not prevent tumor progression. In addition to GSS, GSH is also synthesized by glutamate-cysteine ligase (GCL), an enzyme that consists of a heavy catalytic (GCLC) subunit and a light regulatory (modifier, GCLM) subunit. In mouse models of breast cancer, GCLM-deficiency or GLCM inhibition by Buthionine-[S,R]-sulfoximine (BSO) significantly prevented cancer initiation. However, this effect occurs only before tumor onset, and BSO treatment after onset does not alter tumor burden due to a compensatory role of TXN (Harris et al., 2015). GSH and TXN both serve as substrates for proteins with redox-important roles, including glutathione peroxidases, peroxiredoxins, glutathione and thioredoxin reductases, glutaredoxins, and sulfiredoxins (Björnstedt et al., 1994; Sun et al., 2001; Chang et al., 2004; Johansson et al., 2004; Peskin et al., 2016). Several cancers display increased expression of TXN and thioredoxin reductase 1 (TXNRD1) to compensate for GSH deficiency in GCLM−/− cells (Mandal et al., 2010; Harris et al., 2015). The inverse is also observed, where TXNRD1-deficiency promotes the expression of GSR and GCLC, but not of PRDX1, CAT, or SOD1, in liver cancer cells (Carlson et al., 2012). Given that GSH and thioredoxins are relatively unreactive with ROS per se (Chae et al., 1994; Winterbourn and Metodiewa, 1999), the promotion of GSH or TXN biosynthesis is possibly promoting the peroxidase activities of GPX and PRDX.
GLCM-deficient cells also present increased expression of the cystine transporters and stabilizers XCT and CD44 (Lu et al., 2015). This increased import is used toward promoting cysteine biosynthesis, which, in turn, is used toward TXN biosynthesis. Other observations show that chemotherapy treatment promotes XCT and GCLM expression with a concomitant increase in GSH biosynthesis in a HIF1-dependent mechanism related to therapeutic resistance (Lu et al., 2015). Targeting XCT, GCLM, and other pluripotency-involved transcription factors impaired malignant transformation. It is currently unclear whether other cysteine-based antioxidants, such as peroxiredoxins, also benefit from increased cystine import. Importantly, it was also observed that TXNs and thioredoxin reductases are upregulated and co-localize in several cancers, particularly in more aggressive cancers (Soini et al., 2001; Lincoln et al., 2003). These enzymes are associated with tumor initiation (Shen et al., 2016), and they correlated with worse prognosis (Cadenas et al., 2010). TXNRD1 knockdown significantly slowed tumor progression and metastasis in lung carcinomas (Yoo et al., 2006), but it promoted cancer incidence in liver (Carlson et al., 2012).
Due to their chemical similarities and synergistic properties, recent approaches have simultaneously targeted both TXN and GSH metabolism, and they are significantly more effective at reducing tumor volumes than when they are applied individually (Harris et al., 2015). Multiple other studies have shown this efficacy in combination with common cancer drugs or radiotherapy for multiple cancers (e.g., Lu et al., 2007; Sobhakumari et al., 2012; Rodman et al., 2016; Roh et al., 2016; Tanaka et al., 2016). Together, the intracellular redox states expressed in terms of reducing equivalents and thiol compounds (Box 1) not only influence several signaling processes but also control the reactivities of ROS and their redox partners by regulating ROS homeostasis. It is currently unclear whether the involvement of some of these antioxidant defenses in cancer is related to their detoxification role, redox signaling properties or both. For instance, PRDX2 has increased levels in some cancer cells, which correlates with lower cytoplasmic H2O2 concentrations and cellular resistance to oxidative stress (Stresing et al., 2012). PRDX2 displays peroxidase activity and is also involved in redox signaling transduction (Neumann and Fang, 2007), such as the positive regulation of JNK-dependent DNA repair (Lee et al., 2011). Since these H2O2-scavenging and redox signaling properties stem from the kinetic properties of PRDX2/TXN/TXNRD1, GPX1/GSH/GSR, and catalase systems (Benfeitas et al., 2014; Tomalin et al., 2016), further studies are required to understand whether the culprit of PRDX2's tumorigenic association is its role as a peroxidase, chaperone, or redox signaling transducer. It is unclear whether the alterations in GSH/TXN biosynthesis and PRDX/GPX levels are more important toward controlling redox signaling, detoxification, or both. For instance, some observations indicate that sulfiredoxins and peroxiredoxins promote tumor growth and metastasis by modulating phosphokinase signaling cascades (Wei et al., 2011). Do these proteins directly interact with their targets? PTEN binds to PRDX1, but not to PRDX2, and this promotes Akt-mediated proliferation (Cao et al., 2009). The PRDX1-PTEN complex dissociates upon H2O2-mediated oxidation. A localized accumulation of H2O2, such as that occurring near cell membranes or near ROS sources, would thereby alter PTEN-mediated signaling transduction and proliferation by direct PRDX1 oxidation (Woo et al., 2010) or by relaying a redox signal from another more abundant, H2O2-reactive PRDX. PRDX2 is a good candidate as a H2O2 sensor due to its high reactivity with H2O2 (k ≈ 107–108 M−1 s−1), and it was recently observed to transmit oxidative equivalents to the transcription factor STAT3 (Sobotta et al., 2015), thereby controlling tumor proliferation and survival (Yu et al., 2014). While disulfide exchange between PRDX1 and 2 remains to be shown, the above observations indicate that the direct reactions of peroxiredoxins with transcription factors are important proliferative processes controlled by H2O2. It also remains to be seen whether the promotion of TXN biosynthesis (Harris et al., 2015), which is often linked to added ROS protection, is instead enabling secondary signaling transduction reactions, and whether multiple PRDX isoforms act synergistically in this process. Studying cancer ROS metabolism should therefore consider the toxicity of these oxidants and signaling disruption.
Overall, the observations above highlight important features that should be considered in cancer studies. First, targeting one antioxidant defense may elicit compensatory behaviors by other antioxidant defenses. Second, the close relationship between antioxidant proteins (e.g., GPX and PRDX) and their redox partners (e.g., GSH and TXN) requires that the choice of suitable therapeutic targets considers possible synergisms between them. Third, the high variability in responses, even for the same tumor, requires that cancer treatment is designed in a case- and stage-specific manner, rather than a cancer-type approach. Finally, all of these considerations need to be considered to identify antioxidant pathways that are differentially regulated by cancer, but not by normal cells. The targeting of antioxidant defenses as an approach for cancer treatment should therefore require tissue- and subject-specific phenotypic characterization.
Cancer cells display increased glycolytic activity and lower mitochondrial oxidative phosphorylation. Glucose uptake by breast, liver, colorectal, lung, and pancreatic cancers may reach 8–15 times the fluxes observed by surrounding normal tissues (see Boros et al., 1998 and references therein). This metabolic shift, characterized by increased ATP production from glycolytic pathways rather than respiratory pathways, even under aerobiosis, is one of the most well-known metabolic hallmarks of cancer cells, and it is generally referred to as the Warburg effect (Warburg, 1956). This process is crucial to maintaining the high energetic demand of fast proliferative cells. However, the energetic changes extend beyond the Warburg effect and are intimately related to the redox responses of cancer cells. This is the case of the metabolic changes that alter NADPH production. For instance, the MYC-controlled expression of pyruvate kinase type M2 (PKM2) is higher in cancer cells and promotes the diversion of carbohydrate metabolism from glycolytic pathways to other pathways (Vander Heiden et al., 2009), including the pentose phosphate pathway (PPP). Carbohydrates are thus diverted from ATP production to generate reducing equivalents and building blocks, such as phosphopentoses and ribonucleotides, supporting the fast proliferation of cancer cells (Boros et al., 1997; Raïs et al., 1999). Because ROS also regulate carbohydrate metabolism (Robbins et al., 2012; Hart et al., 2015; Miar et al., 2015) and some enzymes couple redox metabolism and ATP phosphorylation (Chang et al., 2004), the crosstalk between energetic and redox metabolism extends beyond enabling NADPH-driven peroxidase antioxidant activities.
Many of the glycolytic and PPP enzymes that are involved in NADPH production are elevated in cancer cells. Glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD), which are both enzymes of the oxidative branch of PPP, catalyze the production of NADPH from hexoses entering the PPP from either glycolysis or the non-oxidative PPP. G6PD's activity is promoted by multiple oncogenic pathways upregulated in cancer (Stanton et al., 1991; Tian et al., 1994; Au et al., 2000; Wang et al., 2012; Zhang et al., 2014), and multiple studies have proposed that G6PD has pro-oncogenic activities (Wang et al., 2012; Patra and Hay, 2014; Zhang et al., 2014). G6PD overexpression leads to higher levels of intracellular NADPH, GSH, and nucleotide precursors, increased health span, and lower nucleotide oxidation (Nóbrega-pereira et al., 2016). These observations provide a clear link between PPP-mediated NADPH production and oxidative stress. In turn, G6PD deficiency severely limits cell resistance to oxidative stress (Pandolfi et al., 1995) and promotes oxidative damage to DNA (Jeng et al., 2013). These observations raise the hypothesis that a targeted inhibition of G6PD may be conducive to oxidative imbalance and ROS-mediated cell death. Considering that G6PD catalyzes the first and rate-limiting step of PPP and that it has a role in controlling the intracellular redox environment, this enzyme has been envisaged as one of the potentially most important therapeutic cancer redox targets (Wang et al., 2012; Patra and Hay, 2014; Zhang et al., 2014; Nóbrega-pereira et al., 2016), and it has been included in pre-clinical trials (Budihardjo et al., 1998; De Preter et al., 2015). 6PGD is also upregulated in many cancers, including thyroid (Giusti et al., 2008), lung (Sukhatme and Chan, 2012), and cervical (Jonas et al., 1992) tumors. This enzyme is important for proliferation and tumor growth (Sukhatme and Chan, 2012; Shan et al., 2014), and its inhibition promotes senescence in lung cancer (Sukhatme and Chan, 2012). This phenotype results from altered glucose levels, but not altered NADPH levels (Sukhatme and Chan, 2012; Lin et al., 2015), suggesting that NADPH metabolism, and ultimately glucose metabolism, may adapt in such a way that compensates for the selective targeting of the PPP's enzymes. Interestingly, 6PGD suppression limits lipid biosynthesis and elevates intracellular ROS levels, and this effect translates into decreased tumor growth (Lin et al., 2015). However, conflicting observations regarding its importance in cancerogenesis (Sukhatme and Chan, 2012; Lin et al., 2015) also suggest that its role may vary depending on tissue and oncogenic background (Lin et al., 2015).
Other enzymes also promote downstream NADPH production. For instance, transketolase (TKT) and transaldolase (TALDO) are both enzymes of the non-oxidative PPP. While neither of them catalyze NADPH production, they are both important in directing the phosphorylated pentoses generated in the PPP back to glycolysis. Both enzymes are upregulated in cancer (Heinrich et al., 1976; Liu et al., 2010). TKT is required for cancer growth and controls resistance to oxidative stress by modulating NADPH levels. Its inhibition leads to higher intracellular ROS and decreased NADPH/NADP+ ratios (Xu et al., 2016), and it sensitizes cells to drug treatment. Importantly, TKT knockdown increases oxidative PPP fluxes, but it also leads to lower NADPH levels, which is a striking observation considering that NADPH is produced through the oxidative PPP; this point remains to be clarified. TALDO's expression is linked to metastasis in hepatocellular carcinoma (Wang et al., 2011). TALDO deficiency also elicits hepatocellular carcinoma and promotes the formation of malignant tumors (Hanczko et al., 2009). These outcomes are associated with redox imbalances (lower NADPH and GSH levels) due to the insufficient recycling of PPP metabolites to support NADPH production, and they are reverted with dietary supplementation of antioxidants. This insufficient recycling exposes the liver to added oxidative stress and decreases lifespan. Other NADPH-producing enzymes are also upregulated in cancer cells, and some of the isoforms are exclusive to proliferating cells (Mazurek et al., 2005), suggesting that these enzymes may be selective therapeutic targets. Altered glycolytic and PPP metabolism has been proposed for potential therapeutic targeting in cancer (Wang et al., 2012; Patra and Hay, 2014; Zhang et al., 2014; Wen et al., 2015; Hay, 2016). Importantly, the crucial role of the PPP in cancer development seems to be associated with its redox homeostasis properties rather than its production of ribonucleotide precursors, as observed in hepatocellular carcinoma (Xu et al., 2016). Further studies are required to understand whether similar observations are present in other cancers.
While the oxidative PPP represents the main source of cytoplasmic NADPH in proliferating cells (Fan et al., 2014), other sources contribute significantly. Serine (Mehrmohamadi et al., 2014), folate (Tedeschi et al., 2013; Fan et al., 2014; Piskounova et al., 2015), and malate (Jiang et al., 2013) pathways also produce and regenerate NADPH and have crucial roles in maintaining the redox status and buffering oxidative stress in cancer cells. A system-wide comparison the contributions of these pathways to NADPH production and ROS metabolism in cancer is beginning to emerge (e.g., Tedeschi et al., 2013; Mehrmohamadi et al., 2014). In the context of antioxidant defense, most of these studies have focused on GSH-mediated ROS protection due to the close relationship between the PPP, serine/glycine metabolism, and de novo GSH biosynthesis. However, an analysis addressing the role of these pathways in supporting the activities of other important antioxidant defenses and redox signaling processes is currently missing. This becomes an important issue because defenses have different reducing equivalent requirements for activity, which also reflect their different antioxidant capacities and redox signaling roles. For instance, while antioxidant systems like PRDX/TXN/TXNRD and GPX/GSH/GSR stoichiometrically couple ROS detoxification to NADPH consumption, others, such as catalase, scavenge ROS while oxidizing virtually no NADPH. In normal cells, where energetic metabolism is limited, NADPH must be utilized toward lipid and cholesterol biosynthesis, and ROS decomposition by NADPH-consuming systems is thus an energetically expensive process. In cancer cells, where NADPH-producing fluxes are promoted, NADPH may be sufficiently abundant for cells to afford utilizing PRDX- or GPX-mediated detoxification. Importantly, the differences in the energetic requirements and kinetics of the PRDX/TXN/TXNRD, GPX/GSH/GSR, and catalase systems become particularly important if cancer treatments are targeting ROS metabolism by inhibiting NADPH production. ROS defenses may then be maintained by catalase or other energetically inexpensive processes, which is similar to what is observed in non-cancerous cells (Johnson et al., 2005; Benfeitas et al., 2014). Nevertheless, the lower cancer progression and increased ROS levels induced by the inhibition of NADPH-producing pathways indicate that this is a viable cancer therapy. A further understanding of the energetic requirements of antioxidant defense (e.g., PRDX/TXN vs. GSH/GPX pathways) may permit an efficient combination of anti-oxidant- and energetic-focused drug utilization for effective cancer treatment. Importantly, the heterogeneous gene expression and synergistic responses that may occur between alternative metabolic pathways at different cancer stages require an assessment of possible targets that considers specific oncogenic backgrounds.
The observations above highlight the extensive crosstalk within and between ROS detoxification, redox signaling transduction, energy metabolism, and central metabolism. As such, the therapeutic targeting of cancer is more effectively strategized by addressing multi-pathway dysregulation (Pawson and Linding, 2008). Thus, while targeting the activity of specific enzymes may yield promising results in vitro and to a certain extent, in vivo, methods that encompass global metabolism are required to devise viable, cancer-specific treatment targets. Furthermore, these redox responses are highly heterogeneous, as has been observed by the different redox responses displayed by different cancer types, between individuals with the same cancer type, and between different cancer stages. Finding the best targets (Figure 4) and elucidating the mechanisms behind cancer phenotypes hence requires integrative analysis of a large number of biological networks, together with tissue- and patient-tailored data.
Systems biology aims at analyzing assorted biological data (e.g., genomics, proteomics, metabolomics, fluxomics), and it has consistently assisted in understanding the complex underlying mechanisms in health and disease (Mardinoglu and Nielsen, 2012; Agren et al., 2014; Benfeitas et al., 2014; Ghaffari et al., 2015b; Mardinoglu et al., 2017). Using systems biology approaches, others successfully clarified the role of small molecular decision circuits (e.g., Faratian et al., 2009; Gaglio et al., 2011; Tyson et al., 2011) and found commonalities across different cancers through pan-cancer analyses (e.g., Weinstein et al., 2013; Zack et al., 2013; Leiserson et al., 2014; Aran et al., 2015; Peng et al., 2015). However, few studies have addressed ROS metabolism using systems approaches (Zhang et al., 2010; Zhou et al., 2011; Zhan et al., 2012). For instance, modeling of the NFE2L2 pathway suggests that the high NFE2L2 expression that typically occurs in cancer cells promotes chemoresistance (Zhan et al., 2012) and suggests apparently opposite roles in antioxidant and ROS-mediated cancer signaling (Zhang et al., 2010). However, certain antioxidants, such as peroxiredoxins, display both scavenging and redox sensor and signaling transducer properties (Box 1). Mathematical modeling of H2O2 metabolism indicates that cancer-related transcription factors are unlikely to be activated by direct reaction with H2O2, and it points toward protein thiols as the likely signaling sensors and transducers. Others have observed that the glutathione and NADPH synthesis pathways are simultaneously up- or down-regulated in breast, ovary, colon and lung cancers, establishing important interactions with de novo nucleotide synthesis (Mehrmohamadi et al., 2014), which suggests that cancers utilize redox homeostasis and biosynthesis pathways in parallel. ROS and redox responses of cancer cells have yet to be more extensively examined.
With the advent of big data, recent approaches aimed at understanding cell metabolism now incorporate large reaction networks derived from omics technologies (genomics, transcriptomics, proteomics, and others). By encompassing whole-cell reaction networks, GEMs have helped identify important redox alterations in metabolic diseases and physiological processes. For instance, disturbed H2O2 metabolism is observed (Mardinoglu et al., 2017) in non-alcoholic fatty liver disease, specifically due to deficient GSH biosynthesis (by GCLC/GCLM, GSR) and NADPH production (NNT). A lower abundance of plasma glycine, a substrate for de novo GSH biosynthesis, is also found in subjects with high hepatic steatosis. Observations in mice (Mardinoglu et al., 2015b) indicate that commensal gut microbes decrease glycine availability in the gastro-intestinal tract of the host, which results in decreased de novo GSH synthesis and promotes NNT and GSR expression, possibly to compensate for the decreased GSH pool. In the context of cancer, publicly deposited genomic data permit the stratification of cancer patients based on network-specific mutations (Hofree et al., 2013), and these data have been used to find biomarkers and potential cancer therapeutic targets (Jerby and Ruppin, 2012; Agren et al., 2014). Others have combined experimental and interactome data with stochastic modeling to find that ROS and DNA damage are necessary and sufficient for senescent growth arrest (Passos et al., 2010). Chronic, non-toxic ROS supplementation reverses drug resistance in carcinoma cells (Maiti, 2010), which, through pathway analysis, identified several genes (e.g., TP53, Rac/Cdc42 guanine nucleotide exchange factor 6 ARHGEF6, and a DNA-activated protein kinase PRKDC) that mediate ROS-related apoptosis. Reconstruction of a generic human whole-cell GEMs encompassing >9,000 reactions and >3,000 metabolites and genes (Mardinoglu et al., 2014a) enabled context-specific integration and interpretation of cancer omics data. Comparisons between hepatocellular carcinoma and healthy liver samples using these models indicate that tumors display an increased abundance of NADPH-producing enzymes (e.g., ME1, G6PD, TALDO1, and TKT) and that H2O2 may be used as reporter metabolite in some of the patients (Bjornson et al., 2015). Personalized medicine approaches would greatly benefit from clarifying redox cancer responses. GEMs were previously used to identify novel anticancer drugs by reconstructing patient- and tissue-specific GEMs (Agren et al., 2014) and to identify anti-growth factors in human cancer cell lines (Ghaffari et al., 2015a). Cell-specific GEMs have also been used to identify malonyl-CoA decarboxylase, an important enzyme in fatty acid metabolism, as a selective and effective cancer therapeutic target (Yizhak et al., 2014a). Its inhibition drains reducing equivalents, decreases GSH/GSSG ratios, and promotes oxidative stress, which may help in chemotherapeutic approaches. Patient-specific breast and lung cancer GEMs were also able to predict patient survivability, and they predicted that knockout of GSR significantly affects cancer growth.
Interestingly, despite an enzyme- or process-specific focus in redox cancer metabolism in recent years, system-wide studies of ROS and redox metabolism and their interactions with central metabolism in multiple cancers are lacking. Antioxidant profiling of tumor cells and their surrounding cells may be used in conjunction with patient-specific GEMs to identify the best therapeutic targets in this disease (Figure 6). Studies using reaction network information with tissue- and patient-specific models will be useful in (1) Redox profiling of patient-specific cancer tissues; (2) Understanding mechanistic properties of redox responses; (3) Devising effective, selective and patient-specific therapeutic strategies to regulate redox responses; (4) Establishing redox-based therapies to synergize with existent drugs (Kasiappan and Safe, 2016) and identifying and averting drug-resistance mechanisms; and (5) Shortening the gap between pre-clinical and clinical trials, potentially overcoming issues faced by previous trials, such as those that assessed the impact of dietary antioxidants on cancerogenesis (Omenn et al., 1996; Goodman et al., 2011; Klein et al., 2011; Sayin et al., 2014). The adopted strategies (Figure 6) will benefit from patient-specific tumor profiling to identify single or multiple targetable processes within the same pathway or to identify processes that serve as metabolic central hubs. The combination of these approaches with drugs that target other metabolic processes may promote desirable synergisms. Redox systems medicine is thus an interesting emerging field with potentially important implications for disease treatment.
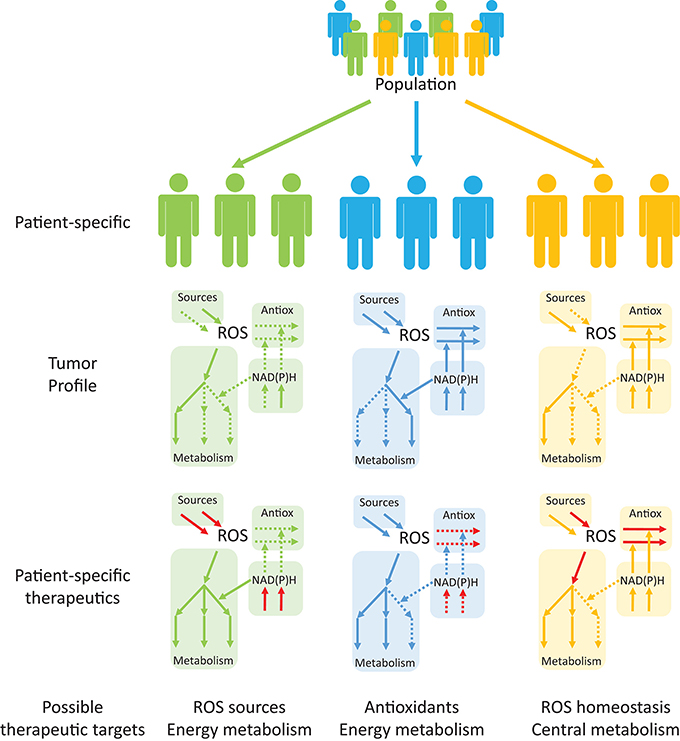
Figure 6. Personalized systems medicine approaches are emerging as useful tools in devising patient-specific, rather than population-based, therapeutic targets in cancer. Tumor profiling of patients may help in identifying up- and down-regulated pathways (continuous and dashed arrows, respectively) that are suitable for therapeutic targeting. Drug targeting of specific processes (red arrows), either to promote or inhibit the processes, will permit alterations in the consequences of redox processes in cancer and other diseases.
Redox, energetic and central metabolism are closely intertwined, and the view that ROS are simple secondary products of cell metabolism is long gone. Together with their redox partners, ROS and antioxidant defenses are now regarded as crucial processes in tumorigenic initiation, progression and aggressiveness. However, the redox cancer responses are highly heterogeneous, manifesting not only between different cancer types but also between patients who suffer from the same cancer, and they are altered throughout cancer progression. The influence of ROS on different biochemical levels makes it necessary to seek an integrative analysis of these systems at the genomic, proteomic, and metabolomic levels. Approaches that are able to encompass these levels and integrate the crosstalk between antioxidant, redox, energetic, and central metabolism are able to capture and understand these complex responses. Systems biology approaches may be used to analyze omics data and understand the roles of each redox system in cancer. These approaches may be tissue- and patient-tailored, which enables the identification of the best therapeutic targets, while taking in account patient-specific oncogenic backgrounds. This is the aim of personalized systems medicine (Mardinoglu and Nielsen, 2015; Schork, 2015), an emerging field that presents a high potential to overcome some of the problems in therapeutic treatments, including the low (< 25%) drug efficacies caused by population—rather than patient-wise data (Schork, 2015). Together with existing drugs, novel or existing redox-targeting drugs may be identified to produce synergistic responses for the treatment or prevention of cancer. Personalized medicine may thus enable an understanding of the role of redox systems in cancer and other diseases and may assist in drug discovery.
RB has written the manuscript. All authors actively contributed in writing and editing of the manuscript.
This work was financially supported by the Knut and Alice Wallenberg Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Agren, R., Bordel, S., Mardinoglu, A., Pornputtapong, N., Nookaew, I., and Nielsen, J. (2012). Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput. Biol. 8:e1002518. doi: 10.1371/journal.pcbi.1002518
Agren, R., Mardinoglu, A., Asplund, A., Kampf, C., Uhlen, M., and Nielsen, J. (2014). Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol. Syst. Biol. 10, 1–13. doi: 10.1002/msb.145122
Aran, D., Sirota, M., and Butte, A. J. (2015). Systematic pan-cancer analysis of tumour purity. Nat. Commun. 6:8971. doi: 10.1038/ncomms9971
Arnér, E. S. J., and Holmgren, A. (2006). The thioredoxin system in cancer. Semin. Cancer Biol. 16, 420–426. doi: 10.1016/j.semcancer.2006.10.009
Arnold, R. S., Shi, J., Murad, E., Whalen, A. M., Sun, C. Q., Polavarapu, R., et al. (2001). Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc. Natl. Acad. Sci. U.S.A. 98, 5550–5555. doi: 10.1073/pnas.101505898
Au, S. W. N., Gover, S., Lam, V. M. S., and Adams, M. J. (2000). Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP(+) molecule and provides insights into enzyme deficiency. Struct. Fold. Des. 8, 293–303. doi: 10.1016/S0969-2126(00)00104-0
Ayala, A., Muñoz, M. F., and Argüelles, S. (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014:360438. doi: 10.1155/2014/360438
Babior, B. M., Lambeth, J. D., and Nauseef, W. (2002). The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 397, 342–344. doi: 10.1006/abbi.2001.2642
Bannister, J. V., Bannister, W. H., Bray, R. C., Fielden, E. M., Roberts, P. B., and Rotilio, G. (1973). The superoxide dismutase activity of human erythrocuprein. FEBS Lett. 32, 303–306. doi: 10.1016/0014-5793(73)80859-2
Basu, A., Banerjee, H., Rojas, H., Martinez, S. R., Roy, S., Jia, Z., et al. (2011). Differential expression of peroxiredoxins in prostate cancer: consistent upregulation of PRDX3 and PRDX4. Prostate 71, 755–765. doi: 10.1002/pros.21292
Benfeitas, R., Selvaggio, G., Antunes, F., Coelho, P. M., and Salvador, A. (2014). Hydrogen peroxide metabolism and sensing in human erythrocytes: a validated kinetic model and reappraisal of the role of peroxiredoxin II. Free Radic. Biol. Med. 74, 35–49. doi: 10.1016/j.freeradbiomed.2014.06.007
Bielski, B. H., and Shiue, G. G. (1979). “Reaction rates of superoxide radicals with the essential aminoacids,” in Oxygen Free Radicals and Tissue Damage, ed I. Fridovich (Amsterdam: Excerpta Medica), 43–56.
Bienert, G. P., Møller, A. L. B., Kristiansen, K. A., Schulz, A., Møller, I. M., Schjoerring, J. K., et al. (2007). Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 282, 1183–1192. doi: 10.1074/jbc.M603761200
Bjornson, E., Boren, J., and Mardinoglu, A. (2016). Personalized cardiovascular disease prediction and treatment—a review of existing strategies and novel systems medicine tools. Front. Physiol. 7:2. doi: 10.3389/fphys.2016.00002
Bjornson, E., Mukhopadhyay, B., Asplund, A., Pristovsek, N., Cinar, R., Romeo, S., et al. (2015). Stratification of hepatocellular carcinoma patients based on acetate utilization. Cell Rep. 13, 2014–2026. doi: 10.1016/j.celrep.2015.10.045
Björnstedt, M., Xue, J., Huang, W., Åkesson, B., and Holmgren, A. (1994). The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J. Biol. Chem. 269, 29382–29384.
Blot, W. J., Li, J.-Y. Y., Taylor, P. R., Guo, W., Dawsey, S., Wang, G.-Q., et al. (1993). Nutrition intervention trials in linxian, china: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J. Natl. Cancer Inst. 85, 1483–1492. doi: 10.1093/jnci/85.18.1483
Boros, L. G., Lee, P. W., Brandes, J. L., Cascante, M., Muscarella, P., Schirmer, W. J., et al. (1998). Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: is cancer a disease of cellular glucose metabolism? Med. Hypotheses 50, 55–59. doi: 10.1016/S0306-9877(98)90178-5
Boros, L. G., Puigjaner, J., Cascante, M., Lee, W. N. P., Brandes, J. L., Bassilian, S., et al. (1997). Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 2, 4242–4248.
Brown, G. C., and Borutaite, V. (2012). There is no evidence that mitochondria are the main source of reactive oxygen species in mammalian cells. Mitochondrion 12, 1–4. doi: 10.1016/j.mito.2011.02.001
Brune, B. (2005). The intimate relation between nitric oxide and superoxide in apoptosis and cell survival. Antioxid. Redox Signal. 7, 497–507. doi: 10.1089/ars.2005.7.497
Budihardjo, I. I., Walker, D. L., Svingen, P. A., Buckwalter, C. A., Desnoyers, S., Eckdahl, S., et al. (1998). 6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin. Clin. Cancer Res. 4, 117–130.
Buettner, G. R., Wagner, B. A., and Rodgers, V. G. J. (2013). Quantitative redox biology: an approach to understand the role of reactive species in defining the cellular redox environment. Cell Biochem. Biophys. 67, 477–483. doi: 10.1007/s12013-011-9320-3
Buxton, G. V., Greenstock, C. L., Helman, W. P., and Ross, A. B. (1988). Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (· OH/· O−) in aqueous solution. J. Phys. Chem. Ref. Data 17, 513–886. doi: 10.1063/1.555805
Bystrom, L. M., and Rivella, S. (2015). Cancer cells with irons in the fire. Free Radic. Biol. Med. 79, 337–342. doi: 10.1016/j.freeradbiomed.2014.04.035
Cadenas, C., Franckenstein, D., Schmidt, M., Gehrmann, M., Hermes, M., Geppert, B., et al. (2010). Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 12, R44. doi: 10.1186/bcr2599
Cairns, R. A., Harris, I. S., and Mak, T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95. doi: 10.1038/nrc2981
Cao, J., Schulte, J., Knight, A., Leslie, N. R., Zagozdzon, A., Bronson, R., et al. (2009). Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 28, 1505–1517. doi: 10.1038/emboj.2009.101
Carlson, B. A., Tobe, R., Yefremova, E., Tsuji, P. A., Hoffmann, V. J., Schweizer, U., et al. (2016). Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 9, 22–31. doi: 10.1016/j.redox.2016.05.003
Carlson, B. A., Yoo, M. H., Tobe, R., Mueller, C., Naranjo-Suarez, S., Hoffmann, V. J., et al. (2012). Thioredoxin reductase 1 protects against chemically induced hepatocarcinogenesis via control of cellular redox homeostasis. Carcinogenesis 33, 1806–1813. doi: 10.1093/carcin/bgs230
Chae, H. Z., Chung, S. J., and Rhee, S. G. (1994). Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 269, 27670–27678.
Chance, B., Sies, H., and Boveris, A. (1979). Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 59, 527–605.
Chang, T.-S., Jeong, W., Hyun, A. W., Sun, M. L., Park, S., Sue, G. R., et al. (2004). Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 279, 50994–51001. doi: 10.1074/jbc.M409482200
Chang, X. Z., Li, D. Q., Hou, Y. F., Wu, J., Lu, J. S., Di, G. H., et al. (2007). Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 9, R76. doi: 10.1186/bcr1789
Chen, K., Kirber, M. T., Xiao, H., Yang, Y., and Keaney, J. F. Jr. (2008). Regulation of ROS signal transduction by NADPH oxidase 4 localization. J. Cell Biol. 181, 1129–1139. doi: 10.1083/jcb.200709049
Chen, S. X., and Schopfer, P. (1999). Hydroxyl-radical production in physiological reactions. A novel function of peroxidase. Eur. J. Biochem. 260, 726–735. doi: 10.1046/j.1432-1327.1999.00199.x
Choe, Y., Yu, J., Son, Y., Park, S., Kim, J., Shi, X., et al. (2012). Continuously generated H2O2 stimulates the proliferation and osteoblastic differentiation of human periodontal ligament fibroblasts. J. Cell. Biochem. 113, 1426–1436. doi: 10.1002/jcb.24017
Chung-man Ho, J., Zheng, S., Comhair, S. A., Farver, C., and Erzurum, S. C. (2001). Differential expression of manganese superoxide dismutase and catalase in lung cancer. Cancer Res. 61, 8578–8585.
Cooke, M. S., Evans, M. D., Dizdaroglu, M., and Lunec, J. (2003). Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17, 1195–1214. doi: 10.1096/fj.02-0752rev
D'Autréaux, B., and Toledano, M. B. (2007). ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 8, 813–824. doi: 10.1038/nrm2256
DeNicola, G. M., Karreth, F. A., Humpton, T. J., Gopinathan, A., Wei, C., Frese, K., et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. doi: 10.1038/nature10189
de Oliveira, V. A., da Motta, L. L., de Bastiani, M. A., Lopes, F. M., Müller, C. B., Gabiatti, B. P., et al. (2016). In vitro evaluation of antitumoral efficacy of catalase in combination with traditional chemotherapeutic drugs against human lung adenocarcinoma cells. Tumor Biol. 37, 10775–10784. doi: 10.1007/s13277-016-4973-6
De Preter, G., Neveu, M.-A., Danhier, P., Brisson, L., Payen, V. L., Porporato, P. E., et al. (2015). Inhibition of the pentose phosphate pathway by dichloroacetate unravels a missing link between aerobic glycolysis and cancer cell proliferation. Oncotarget 7, 2910–2920. doi: 10.18632/oncotarget.6272
Dhar, S. K., Tangpong, J., Chaiswing, L., Oberley, T. D., and Clair, D. K. S. (2011). Manganese superoxide dismutase is a p53-regulated gene that switches cancers between early and advanced stages. Cancer Res. 71, 6684–6695. doi: 10.1158/0008-5472.CAN-11-1233
Diehn, M., Cho, R. W., Lobo, N. A., Kalisky, T., Dorie, M. J., Kulp, A. N., et al. (2009). Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780–783. doi: 10.1038/nature07733
Dikalov, S. (2011). Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 51, 1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033
Doskey, C. M., Buranasudja, V., Wagner, B. A., Wilkes, J. G., Du, J., Cullen, J. J., et al. (2016). Tumor cells have decreased ability to metabolize H2O2: implications for pharmacological ascorbate in cancer therapy. Redox Biol. 10, 274–284. doi: 10.1016/j.redox.2016.10.010
Du, M. Q., Carmichael, P. L., and Phillips, D. H. (1994). Induction of activating mutations in the human c-Ha-ras-1 proto-oncogene by oxygen free radicals. Mol. Carcinog. 11, 170–175. doi: 10.1002/mc.2940110308
Egler, R. A., Fernandes, E., Rothermund, K., Sereika, S., de Souza-Pinto, N., Jaruga, P., et al. (2005). Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene 24, 8038–8050. doi: 10.1038/sj.onc.1208821
Elchuri, S., Oberley, T. D., Qi, W. B., Eisenstein, R. S., Roberts, L. J., Van Remmen, H., et al. (2005). CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24, 367–380. doi: 10.1038/sj.onc.1208207
Elsemman, I. E., Mardinoglu, A., Shoaie, S., Soliman, T. H., and Nielsen, J. (2016). Systems biology analysis of hepatitis C virus infection reveals the role of copy number increases in regions of chromosome 1q in hepatocellular carcinoma metabolism. Mol. Biosyst. 12, 1496–1506. doi: 10.1039/C5MB00827A
Esworthy, R. S., Chu, F. F., Geiger, P., Girotti, A. W., and Doroshow, J. H. (1993). Reactivity of plasma glutathione peroxidase with hydroperoxide substrates and glutathione. Arch. Biochem. Biophys. 307, 29–34. doi: 10.1006/abbi.1993.1555
Evans, J. L., Goldfine, I. D., Maddux, B. A., and Grodsky, G. M. (2002). Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr. Rev. 23, 599–622. doi: 10.1210/er.2001-0039
Fan, J., Ye, J., Kamphorst, J. J., Shlomi, T., Thompson, C. B., and Rabinowitz, J. D. (2014). Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302. doi: 10.1038/nature13236
Faratian, D., Goltsov, A., Lebedeva, G., Sorokin, A., Moodie, S., Mullen, P., et al. (2009). Systems biology reveals new strategies for personalizing cancer medicine and confirms the role of PTEN in resistance to trastuzumab. Cancer Res. 69, 6713–6720. doi: 10.1158/0008-5472.CAN-09-0777
Fenton, H. J. H. (1894). LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 65, 899–910. doi: 10.1039/CT8946500899
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
Flint, D. H., Tuminello, J. F., and Emptage, M. H. (1993). The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 268, 22369–22376.
Forman, H. J., Maiorino, M., and Ursini, F. (2010). Signaling Functions of Reactive Oxygen Species. Biochemistry 49, 835–842. doi: 10.1021/bi9020378
Fransen, M., Nordgren, M., Wang, B., and Apanasets, O. (2012). Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim. Biophys. Acta Mol. Basis Dis. 1822, 1363–1373. doi: 10.1016/j.bbadis.2011.12.001
Frohlich, D. A., McCabe, M. T., Arnold, R. S., and Day, M. L. (2008). The role of Nrf2 in increased reactive oxygen species and DNA damage in prostate tumorigenesis. Oncogene 27, 4353–4362. doi: 10.1038/onc.2008.79
Gaglio, D., Metallo, C. M., Gameiro, P. A., Hiller, K., Danna, L. S., Balestrieri, C., et al. (2011). Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 7, 523. doi: 10.1038/msb.2011.56
Galaris, D., Skiada, V., and Barbouti, A. (2008). Redox signaling and cancer: the role of “labile” iron. Cancer Lett. 266, 21–29. doi: 10.1016/j.canlet.2008.02.038
Gao, P., Zhang, H., Dinavahi, R., Li, F., Xiang, Y., Raman, V., et al. (2007). HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell 12, 230–238. doi: 10.1016/j.ccr.2007.08.004
Gao, X., Liu, Y., Deeb, D., Liu, P., Liu, A., Arbab, A. S., et al. (2013). ROS mediate proapoptotic and antisurvival activity of oleanane triterpenoid CDDO-Me in ovarian cancer cells. Anticancer Res. 33, 215–221.
Gardner, P. R., Raineri, I., Epstein, L. B., and White, C. W. (1995). Superoxide radical and iron modulate aconitase activity in mammalian cells. J. Biol. Chem. 270, 13399–13405. doi: 10.1074/jbc.270.22.13399
Garrison, W. (1987). Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins. Chem. Rev. 87, 381–398. doi: 10.1021/cr00078a006
Ghaffari, P., Mardinoglu, A., Asplund, A., Shoaie, S., Kampf, C., Uhlen, M., et al. (2015a). Identifying anti-growth factors for human cancer cell lines through genome-scale metabolic modeling. Sci. Rep. 5:8183. doi: 10.1038/srep08183
Ghaffari, P., Mardinoglu, A., and Nielsen, J. (2015b). Cancer metabolism: a modeling perspective. Front. Physiol. 6:382. doi: 10.3389/fphys.2015.00382
Giannoni, E., Buricchi, F., Raugei, G., Ramponi, G., and Chiarugi, P. (2005). Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 25, 6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005
Giusti, L., Iacconi, P., Ciregia, F., Giannaccini, G., Donatini, G. L., Basolo, F., et al. (2008). Fine-needle aspiration of thyroid nodules: proteomic analysis to identify cancer biomarkers. J. Proteome Res. 7, 4079–4088. doi: 10.1021/pr8000404
Glorieux, C., Dejeans, N., Sid, B., Beck, R., Calderon, P. B., and Verrax, J. (2011). Catalase overexpression in mammary cancer cells leads to a less aggressive phenotype and an altered response to chemotherapy. Biochem. Pharmacol. 82, 1384–1390. doi: 10.1016/j.bcp.2011.06.007
Glorieux, C., Zamocky, M., Sandoval, J. M., Verrax, J., and Calderon, P. B. (2015). Regulation of catalase expression in healthy and cancerous cells. Free Radic. Biol. Med. 87, 84–97. doi: 10.1016/j.freeradbiomed.2015.06.017
Goh, J., Enns, L., Fatemie, S., Hopkins, H., Morton, J., Pettan-Brewer, C., et al. (2011). Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 11:191. doi: 10.1186/1471-2407-11-191
Goncalves, K., Sullivan, K., and Phelan, S. (2012). Differential expression and function of peroxiredoxin 1 and peroxiredoxin 6 in cancerous MCF-7 and noncancerous MCF-10A breast epithelial cells. Cancer Invest. 30, 38–47. doi: 10.3109/07357907.2011.629382
Goodman, M., Bostick, R. M., Kucuk, O., and Jones, D. P. (2011). Clinical trials of antioxidants as cancer prevention agents: past, present, and future. Free Radic. Biol. Med. 51, 1068–1084. doi: 10.1016/j.freeradbiomed.2011.05.018
Gorrini, C., Harris, I. S., and Mak, T. W. (2013). Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. doi: 10.1038/nrd4002
Gotoh, Y., and Cooper, J. A. (1998). Reactive oxygen species-and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-α signal transduction. J. Biol. Chem. 273, 17477–17482. doi: 10.1074/jbc.273.28.17477
Gupta, S. C., Hevia, D., Patchva, S., Park, B., Koh, W., and Aggarwal, B. B. (2012). Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 16, 1295–1322. doi: 10.1089/ars.2011.4414
Hampton, M. B., Kettle, A. J., and Winterbourn, C. C. (1998). Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92, 3007–3017.
Hanczko, R., Fernandez, D. R., Doherty, E., Qian, Y., Vas, G., Niland, B., et al. (2009). Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J. Clin. Invest. 119, 1546–1557. doi: 10.1172/JCI35722
Harris, I. S., Treloar, A. E., Inoue, S., Sasaki, M., Gorrini, C., Lee, K., et al. (2015). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222. doi: 10.1016/j.ccell.2014.11.019
Hart, P. C., Mao, M., de Abreu, A. L. P., Ansenberger-Fricano, K., Ekoue, D. N., Ganini, D., et al. (2015). MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 6:6053. doi: 10.1038/ncomms7053
Havens, C. G., Ho, A., Yoshioka, N., and Dowdy, S. F. (2006). Regulation of late G1/S phase transition and APCCdh1 by reactive oxygen species. Mol. Cell. Biol. 26, 4701–4711. doi: 10.1128/MCB.00303-06
Hay, N. (2016). Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat. Rev. Cancer 16, 635–649. doi: 10.1038/nrc.2016.77
Heinrich, P. C., Morris, H. P., and Weber, G. (1976). Behavior of transaldolase (EC 2.2.1.2) and transketolase (EC 2.2.1.1) activities in normal, neoplastic, differentiating, and regenerating liver. Cancer Res. 36, 3189–3197.
Hempel, N., Carrico, P. M., and Melendez, J. A. (2011). Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer. Agents Med. Chem. 11, 191–201. doi: 10.2174/187152011795255911
Higinbotham, K. G., Rice, J. M., Kasprzak, K. S., Reed, C. D., and Perantoni, A. O. (1992). GGT to GTT transversions in codon 12 of the K-ras oncogene in rat renal sarcomas induced with nickel subsulfide or nickel subsulfide/iron are consistent with oxidative damage to DNA. Cancer Res. 52, 4747–4751.
Hileman, E. O., Liu, J., Albitar, M., Keating, M. J., and Huang, P. (2004). Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother. Pharmacol. 53, 209–219. doi: 10.1007/s00280-003-0726-5
Hofree, M., Shen, J. P., Carter, H., Gross, A., and Ideker, T. (2013). Network-based stratification of tumor mutations. Nat. Methods 10, 1108–1115. doi: 10.1038/nmeth.2651
Höhn, A., König, J., and Grune, T. (2013). Protein oxidation in aging and the removal of oxidized proteins. J. Proteomics 92, 132–159. doi: 10.1016/j.jprot.2013.01.004
Holley, A. K., Dhar, S. K., Xu, Y., and Clair, D. K. S. (2012). Manganese superoxide dismutase: beyond life and death. Amino Acids 42, 139–158. doi: 10.1007/s00726-010-0600-9
Hornsveld, M., and Dansen, T. B. (2016). The hallmarks of cancer from a redox perspective. Antioxid. Redox Signal. 25, 300–325. doi: 10.1089/ars.2015.6580
Hu, Y., Rosen, D. G., Zhou, Y., Feng, L., Yang, G., Liu, J., et al. (2005). Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J. Biol. Chem. 280, 39485–39492. doi: 10.1074/jbc.M503296200
Huang, P., Feng, L., Oldham, E. A., Keating, M. J., and Plunkett, W. (2000). Superoxide dismutase as a target for the selective killing of cancer cells. Nature 407, 390–395. doi: 10.1038/35030140
Huie, R. E., and Padmaja, S. (1993). The reaction of NO with superoxide. Free Radic. Res. 18, 195–199. doi: 10.3109/10715769309145868
Hyötyläinen, T., Jerby, L., Petäjä, E. M., Mattila, I., Jäntti, S., Auvinen, P., et al. (2016). Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat. Commun. 7:8994. doi: 10.1038/ncomms9994
Ishikawa, K., Takenaga, K., Akimoto, M., Koshikawa, N., Yamaguchi, A., Imanishi, H., et al. (2008). ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320, 661–664. doi: 10.1126/science.1156906
Janssen, A. M. L., Bosman, C. B., Kruidenier, L., Griffioen, G., Lamers, C., Van Krieken, J., et al. (1999). Superoxide dismutases in the human colorectal cancer sequence. J. Cancer Res. Clin. Oncol. 125, 327–335. doi: 10.1007/s004320050282
Jarvis, R. M., Hughes, S. M., and Ledgerwood, E. C. (2012). Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 53, 1522–1530. doi: 10.1016/j.freeradbiomed.2012.08.001
Jeng, W., Loniewska, M. M., and Wells, P. G. (2013). Brain glucose-6-phosphate dehydrogenase protects against endogenous oxidative DNA damage and neurodegeneration in aged mice. ACS Chem. Neurosci. 4, 1123–1132. doi: 10.1021/cn400079y
Jerby, L., and Ruppin, E. (2012). Predicting drug targets and biomarkers of cancer via genome-scale metabolic modeling. Clin. Cancer Res. 18, 5572–5584. doi: 10.1158/1078-0432.CCR-12-1856
Jiang, P., Du, W., Mancuso, A., Wellen, K. E., and Yang, X. (2013). Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493, 689–693. doi: 10.1038/nature11776
Johansson, C., Lillig, C. H., and Holmgren, A. (2004). Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 279, 7537–7543. doi: 10.1074/jbc.M312719200
Johnson, R. M., Goyette, G., Ravindranath, Y., and Ho, Y.-S. S. (2005). Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radic. Biol. Med. 39, 1407–1417. doi: 10.1016/j.freeradbiomed.2005.07.002
Jonas, S. K., Benedetto, C., Flatman, A., Hammond, R. H., Micheletti, L., Riley, C., et al. (1992). Increased activity of 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase in purified cell suspensions and single cells from the uterine cervix in cervical intraepithelial neoplasia. Br. J. Cancer 66, 185–191. doi: 10.1038/bjc.1992.240
Jones, D. P. (2006). Disruption of mitochondrial redox circuitry in oxidative stress. Chem. Biol. Interact. 163, 38–53. doi: 10.1016/j.cbi.2006.07.008
Juarez, J. C., Manuia, M., Burnett, M., Betancourt, O., Boivin, B., Shaw, D. E., et al. (2008). Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 7147–7152. doi: 10.1073/pnas.0709451105
Kaewpila, S., Venkataraman, S., Buettner, G. R., and Oberley, L. W. (2008). Manganese superoxide dismutase modulates hypoxia-inducible factor-1 alpha induction via superoxide. Cancer Res. 68, 2781–2788. doi: 10.1158/0008-5472.CAN-07-2635
Kaimul, A. M., Nakamura, H., Masutani, H., and Yodoi, J. (2007). Thioredoxin and thioredoxin-binding protein-2 in cancer and metabolic syndrome. Free Radic. Biol. Med. 43, 861–868. doi: 10.1016/j.freeradbiomed.2007.05.032
Kampf, C., Mardinoglu, A., Fagerberg, L., Hallström, B. M., Danielsson, A., Nielsen, J., et al. (2014a). Defining the human gallbladder proteome by transcriptomics and affinity proteomics. Proteomics 14, 2498–2507. doi: 10.1002/pmic.201400201
Kampf, C., Mardinoglu, A., Fagerberg, L., Hallström, B. M., Edlund, K., Lundberg, E., et al. (2014b). The human liver-specific proteome defined by transcriptomics and antibody-based profiling. FASEB J. 28, 1–14. doi: 10.1096/fj.14-250555
Kasiappan, R., and Safe, S. (2016). ROS-inducing agents for cancer chemotherapy. React. Oxyg. Species 1, 22–37. doi: 10.20455/ros.2016.805
Klein, E. A., Thompson, I. M. Jr., Tangen, C. M., John, J., Lucia, M. S., Goodman, P. J., et al. (2011). Vitamin E and the risk of prostate cancer: updated results of the selenium and vitamin E cancer prevention trial (SELECT). J. Am. Med. Assoc. 306, 1549–1556. doi: 10.1001/jama.2011.1437
Kondo, S., Toyokuni, S., Iwasa, Y., Tanaka, T., Onodera, H., Hiai, H., et al. (1999). Persistent oxidative stress in human colorectal carcinoma, but not in adenoma. Free Radic. Biol. Med. 27, 401–410. doi: 10.1016/S0891-5849(99)00087-8
Koppenol, W. H. (2001). The Haber-Weiss cycle – 70 years later. Redox Rep. 6, 229–234. doi: 10.1179/135100001101536373
Kowaltowski, A. J., de Souza-Pinto, N. C., Castilho, R. F., and Vercesi, A. E. (2009). Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 47, 333–343. doi: 10.1016/j.freeradbiomed.2009.05.004
Labunskyy, V. M., and Gladyshev, V. N. (2013). Role of reactive oxygen species-mediated signaling in aging. Antioxid. Redox Signal. 19, 1362–1372. doi: 10.1089/ars.2012.4891
LaButti, J. N., Chowdhury, G., Reilly, T. J., and Gates, K. S. (2007). Redox regulation of protein tyrosine phosphatase 1B by peroxymonophosphate (O3POOH). J. Am. Chem. Soc. 129, 5320–5321. doi: 10.1021/ja070194j
Latimer, H. R., and Veal, E. A. (2016). Peroxiredoxins in regulation of MAPK signalling pathways; sensors and barriers to signal transduction. Mol. Cells 39, 40–45. doi: 10.14348/molcells.2016.2327
Lee, K. W., Lee, D. J., Lee, J. Y., Kang, D. H., Kwon, J., and Kang, S. W. (2011). Peroxiredoxin II restrains DNA damage-induced death in cancer cells by positively regulating JNK-dependent DNA repair. J. Biol. Chem. 286, 8394–8404. doi: 10.1074/jbc.M110.179416
Lee, S., Zhang, C., Kilicarslan, M., Piening, B. D. D., Bjornson, E., Hallström, B. M. M., et al. (2016). integrated network analysis reveals an association between plasma mannose levels and insulin resistance. Cell Metab. 24, 1–13. doi: 10.1016/j.cmet.2016.05.026
Leiserson, M. D. M., Vandin, F., Wu, H.-T., Dobson, J. R., Eldridge, J. V., Thomas, J. L., et al. (2014). Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 47, 106–114. doi: 10.1038/ng.3168
Li, L., Shoji, W., Oshima, H., Obinata, M., Fukumoto, M., and Kanno, N. (2008). Crucial role of peroxiredoxin III in placental antioxidant defense of mice. FEBS Lett. 582, 2431–2434. doi: 10.1016/j.febslet.2008.05.050
Li, L., and Yu, A. Q. (2015). The functional role of peroxiredoxin 3 in reactive oxygen species, apoptosis, and chemoresistance of cancer cells. J. Cancer Res. Clin. Oncol. 141, 2071–2077. doi: 10.1007/s00432-015-1916-3
Li, L., Zhang, Y. G., and Chen, C. L. (2013). Anti-apoptotic role of peroxiredoxin III in cervical cancer cells. FEBS Open Bio. 3, 51–54. doi: 10.1016/j.fob.2012.12.002
Lien, E. C., Lyssiotis, C. A., Juvekar, A., Hu, H., Asara, J. M., Cantley, L. C., et al. (2016). Glutathione biosynthesis is a metabolic vulnerability in PI(3)K/Akt-driven breast cancer. Nat. Cell Biol. 18, 572–578. doi: 10.1038/ncb3341
Lin, R., Elf, S., Shan, C., Kang, H.-B., Ji, Q., Zhou, L., et al. (2015). 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1-AMPK signalling. Nat. Cell Biol. 17, 1484–1496. doi: 10.1038/ncb3255
Lincoln, D. T., Ali Emadi, E. M., Tonissen, K. F., and Clarke, F. M. (2003). The thioredoxin-thioredoxin reductase system: over-expression in human cancer. Anticancer Res. 23, 2425–2433.
Lindskog, C., Linné, J., Fagerberg, L., Hallström, B. M., Sundberg, C. J., Lindholm, M., et al. (2015). The human cardiac and skeletal muscle proteomes defined by transcriptomics and antibody-based profiling. BMC Genomics 16:475. doi: 10.1186/s12864-015-1686-y
Little, C., and O'brien, P. J. (1969). Mechanism of peroxide-inactivation of the sulphydryl enzyme glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 10, 533–538.
Liu, H., Huang, D., McArthur, D. L., Boros, L. G., Nissen, N., and Heaney, A. P. (2010). Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 70, 6368–6376. doi: 10.1158/0008-5472.CAN-09-4615
Liu, Z., Li, S., Cai, Y., Wang, A., He, Q., Zheng, C., et al. (2012). Manganese superoxide dismutase induces migration and invasion of tongue squamous cell carcinoma via H2O2-dependent Snail signaling. Free Radic. Biol. Med. 53, 44–50. doi: 10.1016/j.freeradbiomed.2012.04.031
Low, F. M., Hampton, M. B., Peskin, A. V., and Winterbourn, C. C. (2007). Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 109, 2611–2617. doi: 10.1182/blood-2006-09-048728
Lu, H., Samanta, D., Xiang, L., Zhang, H., Hu, H., Chen, I., et al. (2015). Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. U.S.A. 112, E4600–E4609. doi: 10.1073/pnas.1513433112
Lu, P., Vogel, C., Wang, R., Yao, X., and Marcotte, E. M. (2007). Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25, 117–124. doi: 10.1038/nbt1270
Lu, W., Fu, Z., Wang, H., Feng, J., Wei, J., and Guo, J. (2014). Peroxiredoxin 2 is upregulated in colorectal cancer and contributes to colorectal cancer cells' survival by protecting cells from oxidative stress. Mol. Cell. Biochem. 387, 261–270. doi: 10.1007/s11010-013-1891-4
Lu, Y. P., Lou, Y. R., Yen, P., Newmark, H. L., Mirochnitchenko, O. I., Inouye, M., et al. (1997). Enhanced skin carcinogenesis in transgenic mice with high expression of glutathione peroxidase or both glutathione peroxidase and superoxide dismutase. Cancer Res. 57, 1468–1474.
Maiti, A. K. (2010). Gene network analysis of oxidative stress-mediated drug sensitivity in resistant ovarian carcinoma cells. Pharmacogenomics J. 10, 94–104. doi: 10.1038/tpj.2009.49
Mandal, P. K., Seiler, A., Perisic, T., Kölle, P., Canak, A. B., Förster, H., et al. (2010). System xc- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 285, 22244–22253. doi: 10.1074/jbc.M110.121327
Manta, B., Hugo, M., Ortiz, C., Ferrer-Sueta, G., Trujillo, M., and Denicola, A. (2009). The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Arch. Biochem. Biophys. 484, 146–154. doi: 10.1016/j.abb.2008.11.017
Marchissio, M. J., Frances, D. E., Carnovale, C. E., and Marinelli, R. A. (2012). Mitochondrial aquaporin-8 knockdown in human hepatoma HepG2 cells causes ROS-induced mitochondrial depolarization and loss of viability. Toxicol. Appl. Pharmacol. 264, 246–254. doi: 10.1016/j.taap.2012.08.005
Mardinoglu, A., Agren, R., Kampf, C., Asplund, A., Nookaew, I., Jacobson, P., et al. (2013a). Integration of clinical data with a genome-scale metabolic model of the human adipocyte. Mol. Syst. Biol. 9:649. doi: 10.1038/msb.2013.5
Mardinoglu, A., Agren, R., Kampf, C., Asplund, A., Uhlen, M., and Nielsen, J. (2014a). Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 5:3083. doi: 10.1038/ncomms4083
Mardinoglu, A., Bjornson, E., Zhang, C., Klevstig, M., Söderlund, S., Ståhlman, M., et al. (2017). Personal model-assisted identification of NAD+ and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 13, 1–17. doi: 10.15252/msb.20167422
Mardinoglu, A., Boren, J., Ulf, S., Uhlén, M., and Nielsen, J. (in press). The employment of systems biology in gastroenterology hepatology. Nat. Rev. Gastroenterol. Hepatol.
Mardinoglu, A., Gatto, F., and Nielsen, J. (2013b). Genome-scale modeling of human metabolism - a systems biology approach. Biotechnol. J. 8, 985–996. doi: 10.1002/biot.201200275
Mardinoglu, A., Heiker, J. T., Gärtner, D., Björnson, E., Schön, M. R., Flehmig, G., et al. (2015a). Extensive weight loss reveals distinct gene expression changes in human subcutaneous and visceral adipose tissue. Sci. Rep. 5:14841. doi: 10.1038/srep14841
Mardinoglu, A., Kampf, C., Asplund, A., Fagerberg, L., Hallström, B. M., Edlund, K., et al. (2014b). Defining the human adipose tissue proteome to reveal metabolic alterations in obesity. J. Proteome Res. 13, 5106–5119. doi: 10.1021/pr500586e
Mardinoglu, A., and Nielsen, J. (2012). Systems medicine and metabolic modelling. J. Intern. Med. 271, 142–154. doi: 10.1111/j.1365-2796.2011.02493.x
Mardinoglu, A., and Nielsen, J. (2015). New paradigms for metabolic modeling of human cells. Curr. Opin. Biotechnol. 34, 91–97. doi: 10.1016/j.copbio.2014.12.013
Mardinoglu, A., and Nielsen, J. (2016). Editorial: the impact of systems medicine on human health and disease. Front. Physiol. 7:552. doi: 10.3389/fphys.2016.00552
Mardinoglu, A., Shoaie, S., Bergentall, M., Ghaffari, P., Zhang, C., Larsson, E., et al. (2015b). The gut microbiota modulates host amino acid and glutathione metabolism in mice. Mol. Syst. Biol. 11:834. doi: 10.15252/msb.20156487
Mardinoglu, A., and Uhlén, M. (2016). Liver: phenotypic and genetic variance: a systems approach to the liver. Nat. Rev. Gastroenterol. Hepatol. 13, 439–440. doi: 10.1038/nrgastro.2016.93
Marinho, H. S., Real, C., Cyrne, L., Soares, H., and Antunes, F. (2014). Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2, 535–562. doi: 10.1016/j.redox.2014.02.006
Mazurek, S., Boschek, C. B., Hugo, F., and Eigenbrodt, E. (2005). Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 15, 300–308. doi: 10.1016/j.semcancer.2005.04.009
Mehrmohamadi, M., Liu, X., Shestov, A. A., and Locasale, J. W. (2014). Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 9, 1507–1519. doi: 10.1016/j.celrep.2014.10.026
Menon, S. G., and Goswami, P. C. (2007). A redox cycle within the cell cycle: ring in the old with the new. Oncogene 26, 1101–1109. doi: 10.1038/sj.onc.1209895
Messner, K. R., and Imlay, J. A. (2002). Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 277, 42563–42571. doi: 10.1074/jbc.M204958200
Miar, A., Hevia, D., Muñoz-Cimadevilla, H., Astudillo, A., Velasco, J., Sainz, R. M., et al. (2015). Manganese superoxide dismutase (SOD2/MnSOD)/catalase and SOD2/GPx1 ratios as biomarkers for tumor progression and metastasis in prostate, colon, and lung cancer. Free Radic. Biol. Med. 85, 45–55. doi: 10.1016/j.freeradbiomed.2015.04.001
Miranda, A., Janssen, L., Bosman, C. B., van Duijn, W., Oostendorp-van de Ruit, M. M., Kubben, F. J., et al. (2000). Superoxide dismutases in gastric and esophageal cancer and the prognostic impact in gastric cancer. Clin. Cancer Res. 6, 3183–3192.
Mohr, S., Hallak, H., de Boitte, A., Lapetina, E. G., and Brune, B. (1999). Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 274, 9427–9430. doi: 10.1074/jbc.274.14.9427
Morinaka, A., Funato, Y., Uesugi, K., and Miki, H. (2011). Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene 30, 4208–4218. doi: 10.1038/onc.2011.139
Murawaki, Y. Y., Tsuchiya, H., Kanbe, T., Harada, K., Yashima, K., Nozaka, K., et al. (2008). Aberrant expression of selenoproteins in the progression of colorectal cancer. Cancer Lett. 259, 218–230. doi: 10.1016/j.canlet.2007.10.019
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Nagy, P., and Winterbourn, C. C. (2010). Redox chemistry of biological thiols. Adv. Mol. Toxicol. 4, 183–222. doi: 10.1016/S1872-0854(10)04006-3
Naticchia, M. R., Brown, H. A., Garcia, F. J., Lamade, A. M., Justice, S. L., Herrin, R. P., et al. (2013). Bifunctional electrophiles cross-link thioredoxins with redox relay partners in cells. Chem. Res. Toxicol. 26, 490–497. doi: 10.1021/tx4000123
Nauseef, W. M. (2008). Biological roles for the NOX family NADPH oxidases. J. Biol. Chem. 283, 16961–16965. doi: 10.1074/jbc.R700045200
Nelson, K. K., Ranganathan, A. C., Mansouri, J., Rodriguez, A. M., Providence, K. M., Rutter, J. L., et al. (2003). Elevated Sod2 activity augments matrix metalloproteinase expression: evidence for the involvement of endogenous hydrogen peroxide in regulating metastasis. Clin. Cancer Res. 9, 424–432.
Netto, L. E. S., and Antunes, F. (2016). The roles of peroxiredoxin and thioredoxin in hydrogen peroxide sensing and in signal transduction. Mol. Cells 39, 65–71. doi: 10.14348/molcells.2016.2349
Neumann, C. A., and Fang, Q. (2007). Are peroxiredoxins tumor suppressors? Curr. Opin. Pharmacol. 7, 375–380. doi: 10.1016/j.coph.2007.04.007
Nielsen, J. (2017). Systems biology of metabolism: a driver for developing personalized and precision medicine. Cell Metab. 25, 572–579. doi: 10.1016/j.cmet.2017.02.002
Nóbrega-pereira, S., Fernandez-marcos, P. J., Brioche, T., Gomez-cabrera, M. C., Salvador, A., Flores, J. M., et al. (2016). G6PD protects from oxidative damage and improves healthspan in mice. Nat. Commun. 7, 1–9. doi: 10.1038/ncomms10894
Oberley, T. D., and Oberley, L. W. (1997). Antioxidant enzyme levels in cancer. Histol. Histopathol. 12, 525–535.
Oltra, A. M., Carbonell, F., Tormos, C., Iradi, A., and Sáez, G. T. (2001). Antioxidant enzyme activities and the production of MDA and 8-oxo-dG in chronic lymphocytic leukemia. Free Radic. Biol. Med. 30, 1286–1292. doi: 10.1016/S0891-5849(01)00521-4
Omenn, G., Goodman, G., Thornquist, M., Balmes, J., Cullen, M., and Glass, A. (1996). Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. New Engl. J. Med. 334, 1150–1155. doi: 10.1056/NEJM199605023341802
Pandolfi, P. P., Sonati, F., Rivi, R., Mason, P., Grosveld, F., and Luzzatto, L. (1995). Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 14, 5209–5215.
Park, J. H., Kim, Y. S., Lee, H. L., Shim, J. Y., Lee, K. S., Oh, Y. J., et al. (2006). Expression of peroxiredoxin and thioredoxin in human lung cancer and paired normal lung. Respirology 11, 269–275. doi: 10.1111/j.1440-1843.2006.00849.x
Park, S. Y., Yu, X., Ip, C., Mohler, J. L., Bogner, P. N., and Park, Y. M. (2007). Peroxiredoxin 1 interacts with androgen receptor and enhances its transactivation. Cancer Res. 67, 9294–9303. doi: 10.1158/0008-5472.CAN-07-0651
Passos, J. F., Nelson, G., Wang, C., Richter, T., Simillion, C., Proctor, C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6:347. doi: 10.1038/msb.2010.5
Patra, K. C., and Hay, N. (2014). The pentose phosphate pathway and cancer. Trends Biochem. Sci. 39, 347–354. doi: 10.1016/j.tibs.2014.06.005
Paulsen, C. E., Truong, T. H., Garcia, F. J., Homann, A., Gupta, V., Leonard, S. E., et al. (2012). Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 8, 57–64. doi: 10.1038/nchembio.736
Pawson, T., and Linding, R. (2008). Network medicine. FEBS Lett. 582, 1266–1270. doi: 10.1016/j.febslet.2008.02.011
Peng, L., Bian, X. W., Li, D. K., Xu, C., Wang, G. M., Xia, Q. Y., et al. (2015). Large-scale RNA-Seq transcriptome analysis of 4043 cancers and 548 normal tissue controls across 12 TCGA cancer types. Sci. Rep. 5:13413. doi: 10.1038/srep13413
Perkins, A., Poole, L. B., and Karplus, P. A. (2014). Tuning of peroxiredoxin catalysis for various physiological roles. Biochemistry 53, 7693–7705. doi: 10.1021/bi5013222
Peskin, A. V., Low, F. M., Paton, L. N., Maghzal, G. J., Hampton, M. B., and Winterbourn, C. C. (2007). The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 282, 11885–11892. doi: 10.1074/jbc.M700339200
Peskin, A. V., Pace, P. E., Behring, J. B., Paton, L. N., Soethoudt, M., Bachschmid, M. M., et al. (2016). Glutathionylation of the active site cysteines of peroxiredoxin 2 and recycling by glutaredoxin. J. Biol. Chem. 291, 3053–3062. doi: 10.1074/jbc.M115.692798
Piskounova, E., Agathocleous, M., Murphy, M. M., Hu, Z., Huddlestun, S. E., Zhao, Z., et al. (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. doi: 10.1038/nature15726
Pryor, W. A. (1986). Oxy-radicals and related species: their formation, lifetimes, and reactions. Annu. Rev. Physiol. 48, 657–667. doi: 10.1146/annurev.ph.48.030186.003301
Rabilloud, T., Heller, M., Rigobello, M. P., Bindoli, A., Aebersold, R., and Lunardi, J. (2001). The mitochondrial antioxidant defence system and its response to oxidative stress. Proteomics 1, 1105–1110. doi: 10.1002/1615-9861(200109)1:9<1105::AID-PROT1105>3.0.CO;2-M
Raïs, B., Comin, B., Puigjaner, J., Brandes, J. L., Creppy, E., Saboureau, D., et al. (1999). Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich's tumor cells through inhibition of the pentose cycle. FEBS Lett. 456, 113–118. doi: 10.1016/S0014-5793(99)00924-2
Ray, G., Batra, S., Shukla, N. K., Deo, S., Raina, V., Ashok, S., et al. (2000). Lipid peroxidation, free radical production and antioxidant status in breast cancer. Breast Cancer Res. Treat. 59, 163–170. doi: 10.1023/A:1006357330486
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 24, 981–990. doi: 10.1016/j.cellsig.2012.01.008
Reuter, S., Gupta, S. C., Chaturvedi, M. M., and Aggarwal, B. B. (2010). Oxidative stress, inflammation, and cancer: how are they linked? Free Radic. Biol. Med. 49, 1603–1616. doi: 10.1016/j.freeradbiomed.2010.09.006
Rigoulet, M., Yoboue, E. D., and Devin, A. (2011). Mitochondrial ROS generation and its regulation: mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 14, 459–468. doi: 10.1089/ars.2010.3363
Robbins, D., Wittwer, J. A., Codarin, S., Circu, M. L., Aw, T. Y., Huang, T. T., et al. (2012). Isocitrate dehydrogenase 1 is downregulated during early skin tumorigenesis which can be inhibited by overexpression of manganese superoxide dismutase. Cancer Sci. 103, 1429–1433. doi: 10.1111/j.1349-7006.2012.02317.x
Rodman, S. N., Spence, J. M., Ronnfeldt, T. J., Zhu, Y., Solst, S. R., O'Neill, R. A., et al. (2016). Enhancement of radiation response in breast cancer stem cells by inhibition of thioredoxin-and glutathione-dependent metabolism. Radiat. Res. 186, 385–395. doi: 10.1667/RR14463.1
Roh, J.-L., Jang, H. J., Kim, E. H., and Shin, D. (2016). Targeting of the glutathione, thioredoxin, and Nrf2 antioxidant systems in head and neck cancer. Antioxid. Redox Signal. 27, 106–114. doi: 10.1089/ars.2016.6841
Roots, R., and Okada, S. (1975). Estimation of life times and diffusion distances of radicals involved in x-ray-induced DNA strand breaks of killing of mammalian cells. Radiat. Res. 64, 306–320. doi: 10.2307/3574267
Rouault, T. A., and Klausner, R. D. (1996). Iron-sulfur clusters as biosensors of oxidants and iron. Trends Biochem. Sci. 21, 174–177. doi: 10.1016/S0968-0004(96)10024-4
Sabharwal, S. S., and Schumacker, P. T. (2014). Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat. Rev. Cancer 14, 709–721. doi: 10.1038/nrc3803
Saitoh, M., Nishitoh, H., Fujii, M., Takeda, K., Tobiume, K., Sawada, Y., et al. (1998). Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606. doi: 10.1093/emboj/17.9.2596
Sarsour, E. H., Kalen, A. L., and Goswami, P. C. (2014). Manganese superoxide dismutase regulates a redox cycle within the cell cycle. Antioxid. Redox Signal. 20, 1618–1627. doi: 10.1089/ars.2013.5303
Sayin, V. I., Ibrahim, M. X., Larsson, E., Nilsson, J. A., Lindahl, P., and Bergo, M. O. (2014). Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 6:221ra15. doi: 10.1126/scitranslmed.3007653
Schork, N. J. (2015). Personalized medicine: time for one-person trials. Nature 520, 609–611. doi: 10.1038/520609a
Sena, L. A., and Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–166. doi: 10.1016/j.molcel.2012.09.025
Shan, C., Elf, S., Ji, Q., Kang, H. B., Zhou, L., Hitosugi, T., et al. (2014). Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth. Mol. Cell 55, 552–565. doi: 10.1016/j.molcel.2014.06.020
Sharma, V. K., and Rokita, S. E. (2013). Oxidation of Amino Acids, Peptides, and Proteins: Kinetics and Mechanism. Hoboken, NJ: John Wiley and Sons, Inc.
Shen, X., Burguillos, M. A., Osman, A. M., Frijhoff, J., Carrillo-Jiménez, A., Kanatani, S., et al. (2016). Glioma-induced inhibition of caspase-3 in microglia promotes a tumor-supportive phenotype. Nat. Immunol. 17, 1282–1290. doi: 10.1038/ni.3545
Shibutani, S., Takeshita, M., and Grollman, A. P. (1991). Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349, 431–434. doi: 10.1038/349431a0
Skrzydlewska, E., Sulkowski, S., Koda, M., Zalewski, B., Kanczuga-Koda, L., and Sulkowska, M. (2005). Lipid peroxidation and antioxidant status in colorectal cancer. World J. Gastroenterol. 11, 403–406. doi: 10.3748/wjg.v11.i3.403
Smith, W. L., and Murphy, R. C. (2008). Oxidized lipids formed non-enzymatically by reactive oxygen species. J. Biol. Chem. 283, 15513–15514. doi: 10.1074/jbc.R800006200
Sobhakumari, A., Love-Homan, L., Fletcher, E. V. M., Martin, S. M., Parsons, A. D., Spitz, D. R., et al. (2012). Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE 7:e48175. doi: 10.1371/journal.pone.0048175
Sobotta, M. C., Liou, W., Stöcker, S., Talwar, D., Oehler, M., Ruppert, T., et al. (2015). Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70. doi: 10.1038/nchembio.1695
Soini, Y., Kahlos, K., Näpänkangas, U., Kaarteenaho-Wiik, R., Säily, M., Koistinen, P., et al. (2001). Widespread expression of thioredoxin and thioredoxin reductase in non-small cell lung carcinoma. Clin. Cancer Res. 7, 1750–1757.
Stadtman, E. R., and Levine, R. L. (2003). Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25, 207–218. doi: 10.1007/s00726-003-0011-2
Stanton, R. C., Seifter, J. L., Boxer, D. C., Zimmerman, E., and Cantley, L. C. (1991). Rapid release of bound glucose-6-phosphate dehydrogenase by growth factors correlation with increased enzymatic activity. J. Biol. Chem. 266, 12442–12448.
Starheim, K. K., Holien, T., Misund, K., Johansson, I., Baranowska, K. A., Sponaas, A. M., et al. (2016). Intracellular glutathione determines bortezomib cytotoxicity in multiple myeloma cells. Blood Cancer J. 6, e446. doi: 10.1038/bcj.2016.56
Stresing, V., Baltziskueta, E., Rubio, N., Blanco, J., Arriba, M., Valls, J., et al. (2012). Peroxiredoxin 2 specifically regulates the oxidative and metabolic stress response of human metastatic breast cancer cells in lungs. Oncogene 32, 724–735. doi: 10.1038/onc.2012.93
Sukhatme, V. P., and Chan, B. (2012). Glycolytic cancer cells lacking 6-phosphogluconate dehydrogenase metabolize glucose to induce senescence. FEBS Lett. 586, 2389–2395. doi: 10.1016/j.febslet.2012.05.052
Sultana, R., Perluigi, M., and Butterfield, D. A. (2006). Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer's disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid. Redox Signal. 8, 2021–2037. doi: 10.1089/ars.2006.8.2021
Sun, Q. A., Kirnarsky, L., Sherman, S., and Gladyshev, V. N. (2001). Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc. Natl. Acad. Sci. U.S.A. 98, 3673–3678. doi: 10.1073/pnas.051454398
Szatrowski, T. P., and Nathan, C. F. (1991). Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 51, 794–799.
Tanaka, G., Inoue, K.-I., Shimizu, T., Akimoto, K., and Kubota, K. (2016). Dual pharmacological inhibition of glutathione and thioredoxin systems synergizes to kill colorectal carcinoma stem cells. Cancer Med. 5, 2544–2557. doi: 10.1002/cam4.844
Tedeschi, P. M., Markert, E. K., Gounder, M., Lin, H., Dvorzhinski, D., Dolfi, S. C., et al. (2013). Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 4:e877. doi: 10.1038/cddis.2013.393
Thanan, R., Oikawa, S., Yongvanit, P., Hiraku, Y., Ma, N., Pinlaor, S., et al. (2012). Inflammation-induced protein carbonylation contributes to poor prognosis for cholangiocarcinoma. Free Radic. Biol. Med. 52, 1465–1472. doi: 10.1016/j.freeradbiomed.2012.01.018
Thomas, D. D., Ridnour, L. A., Espey, M. G., Donzelli, S., Ambs, S., Hussain, S. P., et al. (2006). Superoxide fluxes limit nitric oxide-induced signaling. J. Biol. Chem. 281, 25984–25993. doi: 10.1074/jbc.M602242200
Thomas, J. P., Maiorino, M., Ursini, F., and Girotti, A. W. (1990). Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation: in situ reduction of phospholipid and cholesterol hydroperoxides. J. Biol. Chem. 265, 454–461.
Thul, P. J., Åkesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal, H., et al. (2017). A subcellular map of the human proteome. Science 356:eaal3321. doi: 10.1126/science.aal3321
Tian, W. N., Pignatare, J. N., and Stanton, R. C. (1994). Signal transduction proteins that associate with the platelet-derived growth factor (PDGF) receptor mediate the PDGF-induced release of glucose-6-phosphate dehydrogenase from permeabilized cells. J. Biol. Chem. 269, 14798–14805.
Tomalin, L. E., Day, A. M., Underwood, Z. E., Smith, G. R., Dalle Pezze, P., Rallis, C., et al. (2016). Increasing extracellular H2O2 produces a bi-phasic response in intracellular H2O2, with peroxiredoxin hyperoxidation only triggered once the cellular H2O2-buffering capacity is overwhelmed. Free Radic. Biol. Med. 95, 333–348. doi: 10.1016/j.freeradbiomed.2016.02.035
Tong, L., Chuang, C.-C., Wu, S., and Zuo, L. (2015). Reactive oxygen species in redox cancer therapy. Cancer Lett. 367, 18–25. doi: 10.1016/j.canlet.2015.07.008
Tong, W.-H., Rouault, T., Agar, J., Zheng, L., Cash, V., Dean, D., et al. (2000). Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 19, 5692–5700. doi: 10.1093/emboj/19.21.5692
Torti, S. V., and Torti, F. M. (2013). Iron and cancer: more ore to be mined. Nat. Rev. Cancer 13, 342–355. doi: 10.1038/nrc3495
Toyokuni, S., Okamoto, K., Yodoi, J., and Hiai, H. (1995). Persistent oxidative stress in cancer. FEBS Lett. 358, 1–3. doi: 10.1016/0014-5793(94)01368-B
Trujillo, M., Clippe, A., Manta, B., Ferrer-Sueta, G., Smeets, A., Declercq, J. P., et al. (2007). Pre-steady state kinetic characterization of human peroxiredoxin 5: taking advantage of Trp84 fluorescence increase upon oxidation. Arch. Biochem. Biophys. 467, 95–106. doi: 10.1016/j.abb.2007.08.008
Tyson, J. J., Baumann, W. T., Chen, C., Verdugo, A., Tavassoly, I., Wang, Y., et al. (2011). Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat. Rev. Cancer 11, 523–532. doi: 10.1038/nrc3081
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Tissue-based map of the human proteome. Science 347, 1260419–1260419. doi: 10.1126/science.1260419
Uhlén, M., Hallstrom, B. M., Lindskog, C., Mardinoglu, A., Ponten, F., and Nielsen, J. (2016). Transcriptomics resources of human tissues and organs. Mol. Syst. Biol. 12, 862–862. doi: 10.15252/msb.20155865
Ushio-Fukai, M., and Nakamura, Y. (2008). Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 266, 37–52. doi: 10.1016/j.canlet.2008.02.044
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1034. doi: 10.1126/science.1160809
Van Remmen, H., Ikeno, Y., Hamilton, M., Pahlavani, M., Wolf, N., Thorpe, S. R., et al. (2003). Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genomics 16, 29–37. doi: 10.1152/physiolgenomics.00122.2003
Väremo, L., Scheele, C., Broholm, C., Mardinoglu, A., Kampf, C., Asplund, A., et al. (2015). Proteome- and transcriptome-driven reconstruction of the human myocyte metabolic network and its use for identification of markers for diabetes. Cell Rep. 11, 921–933. doi: 10.1016/j.celrep.2015.04.010
Wallace, M. A., Liou, L. L., Martins, J., Clement, M. H. S., Bailey, S., Longo, V. D., et al. (2004). Superoxide inhibits 4Fe-4S cluster enzymes involved in amino acid biosynthesis. Cross-compartment protection by CuZn-superoxide dismutase. J. Biol. Chem. 279, 32055–32062. doi: 10.1074/jbc.M403590200
Wang, C., Guo, K., Gao, D., Kang, X., Jiang, K., Li, Y., et al. (2011). Identification of transaldolase as a novel serum biomarker for hepatocellular carcinoma metastasis using xenografted mouse model and clinic samples. Cancer Lett. 313, 154–166. doi: 10.1016/j.canlet.2011.08.031
Wang, J., Yuan, W., Chen, Z., Wu, S., Chen, J., Ge, J., et al. (2012). Overexpression of G6PD is associated with poor clinical outcome in gastric cancer. Tumour Biol. 33, 95–101. doi: 10.1007/s13277-011-0251-9
Wang, T., Tamae, D., LeBon, T., Shively, J. E., Yen, Y., and Li, J. J. (2005). The role of peroxiredoxin II in radiation-resistant MCF-7 breast cancer cells. Cancer Res. 65, 10338–10346. doi: 10.1158/0008-5472.CAN-04-4614
Warburg, O. (1956). On the origin of cancer cells. Sci. New Ser. 123, 309–314. doi: 10.1126/science.123.3191.309
Wei, Q., Jiang, H., Xiao, Z., Baker, A., Young, M. R., Veenstra, T. D., et al. (2011). Sulfiredoxin-Peroxiredoxin IV axis promotes human lung cancer progression through modulation of specific phosphokinase signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 7004–7009. doi: 10.1073/pnas.1013012108
Weidinger, A., and Kozlov, A. V. (2015). Biological Activities of reactive oxygen and nitrogen species: oxidative stress versus signal transduction. Biomolecules 5, 472–484. doi: 10.3390/biom5020472
Weinberg, F., Hamanaka, R., Wheaton, W. W., Weinberg, S., Joseph, J., Lopez, M., et al. (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793. doi: 10.1073/pnas.1003428107
Weinstein, J. N., Collisson, E. A., Mills, G. B., Shaw, K. R. M., Ozenberger, B. A., Ellrott, K., et al. (2013). The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120. doi: 10.1038/ng.2764
Wen, D., Liu, D., Tang, J., Dong, L., Liu, Y., Tao, Z., et al. (2015). Malic enzyme 1 induces epithelial-mesenchymal transition and indicates poor prognosis in hepatocellular carcinoma. Tumour Biol. 36, 6211–6221. doi: 10.1007/s13277-015-3306-5
Whitaker, H. C., Patel, D., Howat, W. J., Warren, A. Y., Kay, J. D., Sangan, T., et al. (2013). Peroxiredoxin-3 is overexpressed in prostate cancer and promotes cancer cell survival by protecting cells from oxidative stress. Br. J. Cancer 109, 983–993. doi: 10.1038/bjc.2013.396
Winterbourn, C. C. (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 82–83, 969–974. doi: 10.1016/0378-4274(95)03532-X
Winterbourn, C. C. (2008). Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286. doi: 10.1038/nchembio.85
Winterbourn, C. C. (2013). The biological chemistry of hydrogen peroxide. Meth. Enzymol. 528, 3–25. doi: 10.1016/B978-0-12-405881-1.00001-X
Winterbourn, C. C., and Metodiewa, D. (1999). Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322–328. doi: 10.1016/S0891-5849(99)00051-9
Woo, H. A., Yim, S. H., Shin, D. H., Kang, D., Yu, D.-Y., and Rhee, S. G. (2010). Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 140, 517–528. doi: 10.1016/j.cell.2010.01.009
Xu, I. M.-J., Lai, R. K.-H., Lin, S.-H., Tse, A. P.-W., Chiu, D. K.-C., Koh, H.-Y., et al. (2016). Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. USA. 113, E725–E734. doi: 10.1073/pnas.1508779113
Yizhak, K., Gaude, E., Le Dévédec, S., Waldman, Y. Y., Stein, G. Y., van de Water, B., et al. (2014a). Phenotype-based cell-specific metabolic modeling reveals metabolic liabilities of cancer. Elife 3:e03641. doi: 10.7554/eLife.03641
Yizhak, K., Le Dévédec, S. E., Rogkoti, V. M., Baenke, F., de Boer, V. C., Frezza, C., et al. (2014b). A computational study of the Warburg effect identifies metabolic targets inhibiting cancer migration. Mol. Syst. Biol. 10:744. doi: 10.15252/msb.20134993
Yoo, M. H., Xu, X. M., Carlson, B. A., Gladyshev, V. N., and Hatfield, D. L. (2006). Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 281, 13005–13008. doi: 10.1074/jbc.C600012200
Yu, H., Lee, H., Herrmann, A., Buettner, R., and Jove, R. (2014). Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746. doi: 10.1038/nrc3818
Zack, T. I., Schumacher, S. E., Carter, S. L., Cherniack, A. D., Saksena, G., Tabak, B., et al. (2013). Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140. doi: 10.1038/ng.2760
Zhan, L., Zhang, H., Zhang, Q., Woods, C. G., Chen, Y., Xue, P., et al. (2012). Regulatory role of KEAP1 and NRF2 in PPARγ expression and chemoresistance in human non-small-cell lung carcinoma cells. Free Radic. Biol. Med. 53, 758–768. doi: 10.1016/j.freeradbiomed.2012.05.041
Zhang, C., Lee, S., Mardinoglu, A., and Hua, Q. (2016). Investigating the combinatory effects of biological networks on gene co-expression. Front. Physiol. 7:160. doi: 10.3389/fphys.2016.00160
Zhang, C., Zhang, Z., Zhu, Y., and Qin, S. (2014). Glucose-6-phosphate dehydrogenase: a biomarker and potential therapeutic target for cancer. Anticancer. Agents Med. Chem. 14, 280–289. doi: 10.2174/18715206113136660337
Zhang, Q., Pi, J., Woods, C. G., and Andersen, M. E. (2010). A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol. Appl. Pharmacol. 244, 84–97. doi: 10.1016/j.taap.2009.08.018
Zhao, Y., Xue, Y., Oberley, T. D., Hines, J., and Clair, D. S. (2001). Overexpression of manganese superoxide dismutase suppresses tumor formation by modulation of activator protein-1 signaling in a multistage skin carcinogenesis model. Cancer Res. 61, 6082–6088.
Zhong, H., and Yin, H. (2015). Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol. 4, 193–199. doi: 10.1016/j.redox.2014.12.011
Keywords: cancer heterogeneity, redox biology, reactive oxygen species, systems biology, personalized medicine
Citation: Benfeitas R, Uhlen M, Nielsen J and Mardinoglu A (2017) New Challenges to Study Heterogeneity in Cancer Redox Metabolism. Front. Cell Dev. Biol. 5:65. doi: 10.3389/fcell.2017.00065
Received: 25 April 2017; Accepted: 26 June 2017;
Published: 11 July 2017.
Edited by:
Angela Re, University of Trento, ItalyReviewed by:
Frederique Gaits-Iacovoni, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2017 Benfeitas, Uhlen, Nielsen and Mardinoglu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adil Mardinoglu, YWRpbG1Ac2NpbGlmZWxhYi5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.