 Jonathan R. Whitfield
Jonathan R. Whitfield Marie-Eve Beaulieu
Marie-Eve Beaulieu Laura Soucek
Laura Soucek
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol., 23 February 2017
Sec. Molecular and Cellular Oncology
Volume 5 - 2017 | https://doi.org/10.3389/fcell.2017.00010
This article is part of the Research TopicMolecular Mechanisms of MYC in Physiology and Cancer: New Insights, Current and Future TherapiesView all 5 articles
Myc is an oncogene deregulated in most—perhaps all—human cancers. Each Myc family member, c-, L-, and N-Myc, has been connected to tumor progression and maintenance. Myc is recognized as a “most wanted” target for cancer therapy, but has for many years been considered undruggable, mainly due to its nuclear localization, lack of a defined ligand binding site, and physiological function essential to the maintenance of normal tissues. The challenge of identifying a pharmacophore capable of overcoming these hurdles is reflected in the current absence of a clinically-viable Myc inhibitor. The first attempts to inhibit Myc used antisense technology some three decades ago, followed by small molecule inhibitors discovered through “classical” compound library screens. Notable breakthroughs proving the feasibility of systemic Myc inhibition were made with the Myc dominant negative mutant Omomyc, showing both the great promise in targeting this infamous oncogene for cancer treatment as well as allaying fears about the deleterious side effects that Myc inhibition might have on normal proliferating tissues. During this time many other strategies have appeared in an attempt to drug the undruggable, including direct and indirect targeting, knockdown, protein/protein and DNA interaction inhibitors, and translation and expression regulation. The inhibitors range from traditional small molecules to natural chemicals, to RNA and antisense, to peptides and miniproteins. Here, we briefly describe the many approaches taken so far, with a particular focus on their potential clinical applicability.
The Myc oncoproteins are a family of pleiotropic transcription factors that control several cellular functions related to efficient proliferation, growth and metabolism, as well as programs of tissue remodeling and regeneration (Dang, 2013). In a normal physiological context, the level of Myc proteins (comprising c-, L-, and N-Myc) is tightly regulated (Meyer and Penn, 2008; Conacci-Sorrell et al., 2014). Indeed, myc gene expression normally depends on growth factor signaling and both myc mRNA and Myc protein have very short half-lives (of 30 and 20 min respectively) (Dang, 2012). In tumor cells however, the cellular levels of Myc become independent from such signaling and regulation, and the resulting exacerbated Myc function drives intracellular and extracellular transcription programs that allow tumors to grow and thrive (Soucek and Evan, 2002; Dang, 2012; Whitfield and Soucek, 2012; Conacci-Sorrell et al., 2014; Fletcher and Prochownik, 2015). In this pathological condition, Myc deregulation can occur at any given stage of its expression (Meyer and Penn, 2008; Conacci-Sorrell et al., 2014). First, the myc gene itself is often subject to amplification, viral insertional events, or chromosomal translocations that provoke its exaggerated expression. Second, myc mRNA can become stabilized through both direct and indirect regulatory events. Third, the Myc protein turnover rate, which is normally dependent on Myc's phosphorylation status and on signaling from FBW7 to engage the ubiquitin-proteasome system, is also found altered in cancer. Finally, even when Myc is not itself mutated, its aberrant expression can occur as a consequence of upstream oncogenic signals (i.e., Ras, PI3K, Wnt, etc.) that converge on this central downstream node inside the nucleus (Meyer and Penn, 2008).
Myc functions within a network of similar proteins, called bHLH-Zip proteins, that all share a DNA-binding basic region and a bHLH-Zip dimerization domain. In this network, Myc forms heterodimers with its natural partner Max, recognizing DNA binding sites called E-boxes and thereby modulating the transcription of specific target genes (Meyer and Penn, 2008; Fletcher and Prochownik, 2015).
Given its crucial role in cancer progression and maintenance (Meyer and Penn, 2008; Dang, 2012; Hartl, 2016), Myc constitutes an ideal cancer target. However, no Myc inhibitor has reached the clinic yet, due in part to the general dogma that dominated the field for a long time claiming that Myc inhibition would cause catastrophic side effects in normal tissues, as well as to various technical issues. These include targeting a nuclear transcription factor displaying a predominantly intrinsically disordered structure, and notably, lacking a binding pocket that has been the typical target for traditional drug discovery approaches using small molecule inhibitor libraries. These issues have been addressed in recent years (Soucek et al., 2008; Prochownik and Vogt, 2010; McKeown and Bradner, 2014; Fletcher and Prochownik, 2015) and we are now witnessing a renewed interest in making Myc inhibition soon a reality for cancer patients. The technical difficulties with targeting Myc help explain the diversity of strategies that have been developed.
Recent reviews have focused on particular aspects of Myc inhibition or specific diseases (Fletcher and Prochownik, 2015; Li et al., 2015; Abedin et al., 2016; Koh et al., 2016; Posternak and Cole, 2016; Shalaby and Grotzer, 2016). Here we have given a concise overview of the strategies employed to inhibit Myc to date, with a particular focus on their applicability in clinical practice.
Direct Myc inhibition can be achieved either by interference with its production or function. In the first case, one could, for example, target its transcription—either interfering with promoter accessibility and/or recruitment of transcription factors—or translation (Figures 1, 2). In the second case, efforts would likely be directed instead to preventing Myc interaction with its “partner in crime” Max or its DNA recognition binding site (Figure 3). The following section describes direct inhibitors of Myc production, while indirect inhibitors of its expression are discussed later. Table 1 provides a summary of the strategies and molecules discussed in this review.
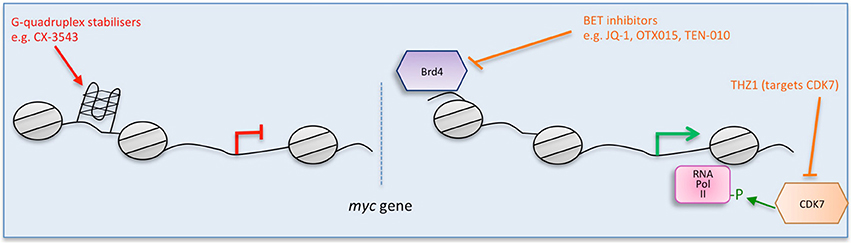
Figure 1. Multiple strategies to target Myc: impairing myc transcription. Direct (red) and indirect (orange) inhibitors are shown related to how they interfere with myc. Some examples of each inhibitor are listed. Figure adapted from Koh et al. (2016).
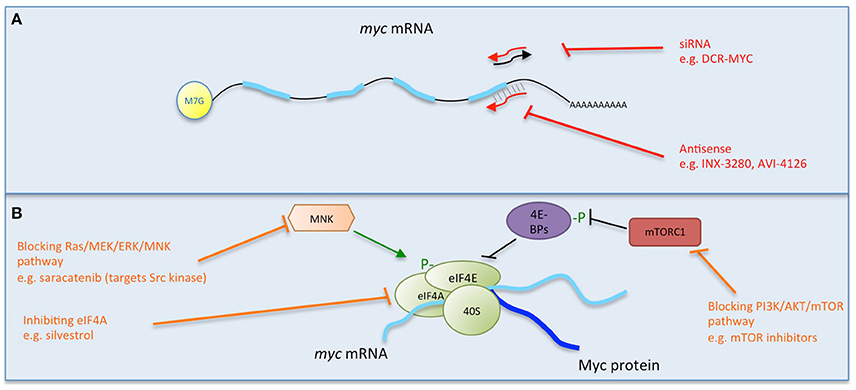
Figure 2. Multiple strategies to target Myc: interfering with myc mRNA. Direct (red) and indirect (orange) inhibitors are shown related to how they interfere with myc mRNA. (A) Causing the degradation of myc mRNA. (B) Preventing myc translation. Some examples of each inhibitor strategy are listed. Figure adapted from Koh et al. (2016).
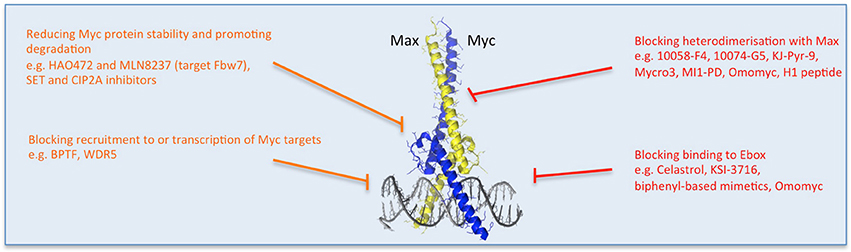
Figure 3. Multiple strategies to target Myc: reducing Myc stability and function. Direct (red) and indirect (orange) inhibitors are shown related to how they affect Myc's stability or binding to its partners or DNA. Other approaches impede Myc-dependent transcription of target genes. Some examples of each inhibitor strategy are listed. Myc/Max crystal structure is from Nair and Burley (2003) and drawn using the PyMOL Molecular Graphics System (Version 1.8 Schrodinger, LLC.).
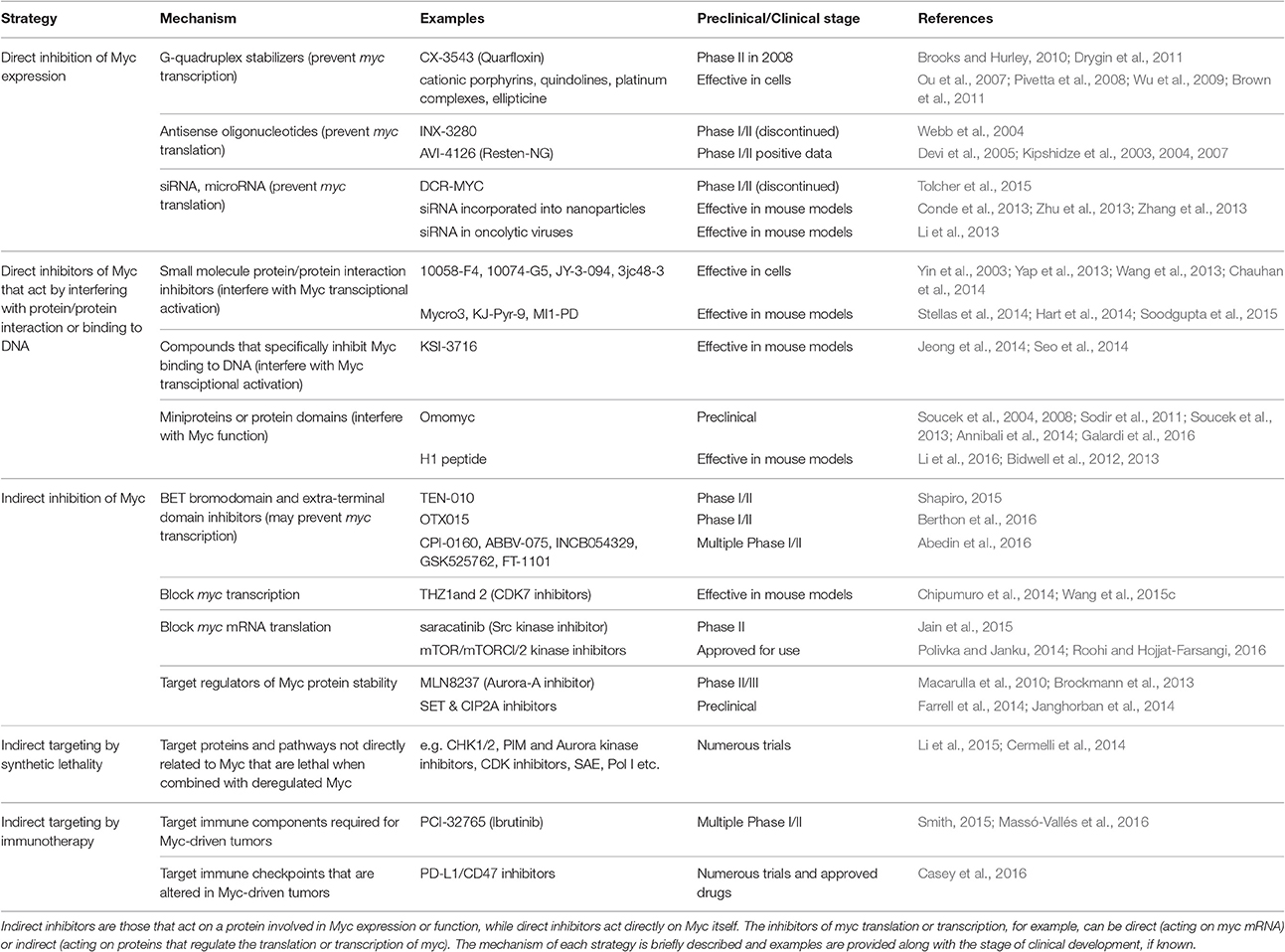
Table 1. Multiple strategies to target Myc in cancer.
G-quadruplexes (also known as G4-DNA) are tertiary structures formed in nucleic acids by sequences that are rich in guanine. The purine-rich strand in the NHE III(1) region of the Myc promoter forms G-quadruplexes (Simonsson et al., 1998, Figure 1). A number of small molecule ligands, including cationic porphyrins (Ou et al., 2007), quindolines (Pivetta et al., 2008), platinum complexes (Wu et al., 2009), and ellipticine (Brown et al., 2011) were shown to stabilize such G-quadruplexes in the myc gene, thus repressing its transcription. Notably, CX-3543 (Quarfloxin) was initally selected as a binder of the myc G-quadruplex (Chen et al., 2014) and is the only such G-quadruplex stabilizer to have reached clinical trials (entering Phase II trials for neuro-endocrine carcinomas in 2008). However, it was also shown to function by disrupting nucleolin bound to the G-quadruplexes in ribosomal DNA (Brooks and Hurley, 2009, 2010; Neidle, 2016). Since nucleolin binds to myc G-quadruplexes, Quarfloxin thus may also repress myc expression by a more indirect route (Brooks and Hurley, 2009). Development was discontinued by Cylene, although Quarfloxin has been licensed to TetraGene so development may continue.
Whether target specificity can be achieved using this strategy is not clear yet, although attempts are being made to selectively target myc (Felsenstein et al., 2016). According to many, total selectivity may not be necessary as long as the major target is a driver or provides a required function for the cancer cell, as for Myc (Neidle, 2016). Of course, as for some of the other types of inhibitors described here, the extent and severity of off-target side effects are key.
With the initial excitement over the promise of antisense as a tool to promote degradation of target mRNA, myc was first successfully attacked in vitro in multiple cell lines (Prochownik et al., 1988; Sklar et al., 1991, Figure 2A). Following these first successes, INX-3280, a 15-mer phosphorothioate oligonucleotide against the c-myc oncogene, was in Phase I and II clinical trials for the treatment of lymphoma and solid tumors more than a decade ago, but was discontinued in 2002 by Inex. A modified form incorporating a “transmembrane carrier system,” INXC-6295, was abandoned due to resource constraints.
With a slightly different strategy, AVI BioPharma (now Sarepta Therapeutics) developed AVI-4126 (Resten-RG), a phosphorodiamidate morpholino antisense oligomer (PMO) that inhibits Myc expression by preventing ribosomal assembly, thereby preventing mRNA translation (Arora et al., 2000). PMO is a modification shown to improve in vivo stability and bioavailabilty of the compound. A Phase I trial of Resten-NG carried out by AVI BioPharma enabled the determination of its bioavailability in solid tumor patients and established the feasibility of using PMOs in human cancer (Devi et al., 2005). Moreover, local delivery was feasible and safe in a related target disease, cardiovascular restenosis that involves neointimal hyperplasia (Kipshidze et al., 2003, 2004) and a Phase II study reported positive results following local delivery (Kipshidze et al., 2007; Philipp et al., 2012).
However, none of these drugs was further developed to reach the market and in fact very few antisense oligonucleotides have done so (Moreno and Pego, 2014). It is not clear though why the promising myc antisense approaches were not followed up, particularly in cancer studies and with newer nanocarrier delivery systems.
Another approach to directly inhibit Myc expression has been used successfully in vitro by lentiviral delivery of shRNA (Wang et al., 2008, Figure 2A) and also attempted in clinical trials using a lipid nanoparticle formulation to deliver myc RNAi (DCR-MYC). While preliminary trials provided evidence of destruction of myc RNA in patients and a clinical response (Tolcher et al., 2015), later trial data did not meet the company's expectations for level of knockdown or efficacy, and Dicerna has halted its development.
As a relatively poor pharmacokinetic profile seems to be a limiting development factor, many attempts are being made to overcome the rapid degradation of siRNA by its incorporation into nanoparticles. For example, gold particles modified with branched polyethyleneimine have been used as efficient and non-toxic intracellular delivery agents for c-myc siRNA both in vitro (Shaat et al., 2016) and in vivo, where they reduced lung tumor growth after intratracheal instillation (Conde et al., 2013). Similarly, folate nanoliposomes carrying siRNA targeting N-myc induce tumor cell apoptosis in a neuroblastoma model in vivo (Zhu et al., 2013), and lipid/calcium/phosphate nanoparticles combining c-myc siRNA and gemcitabine into a single nanovesicle were shown to inhibit lung tumor growth with little toxicity after systemic administration in xenograft models (Zhang et al., 2013).
A related approach making use of oncolytic viruses has also been used to successfully deliver N-myc siRNA in vivo to inhibit xenograft neuroblastoma tumor growth (Li et al., 2013). Oncolytic viruses are starting to show great promise in cancer treatment (Patel and Kratzke, 2013; Andtbacka et al., 2015).
One reason for Myc's undruggability is its intrinsically disordered nature and the fact that protein interactions occur on large, flat, structurally indistinct interfaces (Prochownik and Vogt, 2010; McKeown and Bradner, 2014). However, binding to its obligate partner Max and specific DNA recognition were shown to stabilize folded conformations of Myc's bHLH-Zip domain which, despite lacking a genuine binding pocket, could constitute a relevant target for specific inhibitors. In searching for such inhibitors, the initial yeast 2-hybrid screens and subsequent FRET and fluorescence polarization assays enabled the identification of some useful small molecule compounds that established the feasibility of inhibiting Myc/Max dimers (Yin et al., 2003; Prochownik and Vogt, 2010). Since then, though, the poor bioavailability and lack of selectivity of these for Myc has limited their use in vivo (Prochownik and Vogt, 2010; Fletcher and Prochownik, 2015).
More recently, a surface plasmon resonance (SPR) based technique to quantitate Myc/Max interaction with a DNA probe revealed that the small molecule inhibitors described below comprise 2 distinct classes that either inhibit DNA binding by disrupting Myc/Max interaction, or by distorting the pre-formed heterodimer (Wang et al., 2015a, Figure 3).
The peptide mimetic compound IIA6B17 was the first identified small molecule inhibitor of Myc/Max dimerization (Berg et al., 2002). Unfortunately, the activity of IIA6B17 also extended to c-Jun (Berg et al., 2002), as did the inhibitory effect of NY2267 (Xu et al., 2006), likely due to their similar structural features in their leucine zipper.
Another compound identified early on to affect Myc/Max interaction was 10058-F4 (Yin et al., 2003). Chemical modifications of 10058-F4 resulted in improvements in efficacy in vitro, generally correlating with their ability to disrupt Myc/Max association and DNA binding. 10058-F4 and its active analogs bind specifically to monomeric Myc, interacting with the H2/leucine zipper domain with a KD of 42 μM (Wang et al., 2007). Another small molecule arising from the same screen, 10074-G5, has a KD of 20 μM in vitro and binds to a distinct region of Myc, the basic region/H1 domain (Yin et al., 2003). Binding of both these drugs to intrinsically disordered Myc limits its ability to adopt a more defined conformation and prevents interaction with Max (Follis et al., 2008). However, both 10058-F4 and 10074-G5 are rapidly metabolized and showed poor tumor distribution, limiting their applicability in vivo (Guo et al., 2009; Clausen et al., 2010; Fletcher and Prochownik, 2015).
In fact, the therapeutic utility of potent small molecule inhibitors of Myc/Max dimerization has so far been limited by poor bioavailability, rapid metabolism, and inadequate target site penetration. Nevertheless, development of these small molecules continues in the hope of improving their in vivo characteristics (for a summary, see Fletcher and Prochownik, 2015). For example, structure-activity relationship studies of 10074-G5 led to the generation of an analog, JY-3-094, showing higher ability to disrupt the association between recombinant Myc and Max protein (Wang et al., 2013; Yap et al., 2013), but did not solve the issue of poor cell penetration. Esterification of a critical para-carboxylic acid enhanced cellular uptake, although unfortunately it impaired the ability to disrupt Myc/Max association in vitro (Wang et al., 2013).
Also related to 10074-G5 is the small molecule 3jc48-3, a congener that shows increased potency and stability in cell-based assays (Chauhan et al., 2014). More exciting still is Mycro3, for which daily treatment by oral gavage increased survival in a mouse model of pancreatic ductal carcinoma and orthotopic xenografts of human pancreatic cancer cells (Stellas et al., 2014). Finally, KJ-Pyr-9 is a new inhibitor found in a Kröhnke pyridine library, with a notably lower KD of 6.5 nM (Hart et al., 2014). In vivo, KJ-Pyr-9 effectively blocks the growth of Myc-amplified human cancer cell xenografts (Hart et al., 2014). Furthermore, it penetrates the blood-brain barrier and is not acutely toxic at doses as high as 10 mg/kg (Hart et al., 2014).
New small molecules that target Myc are also being generated thanks to novel computational techniques able to virtually screen binding to different intrinsically disordered protein conformations, maximizing the chances of structure-based drug discovery. Four compounds (with the prefix PKUMDL) show micromolar affinity for Myc and activity in cell-based assays (Yu et al., 2016).
An additional “reverse” approach involves stabilizing instead the Max/Max homodimers, thus preventing Myc/Max transactivating dimers from forming. A virtual ligand screen identified a potent stabilizer of the homodimer (NSC13728) that interferes with Myc-induced transformation and transcriptional activation (Jiang et al., 2009).
In addition, attempts are being made to incorporate the small molecules into nanoparticles for increased stability and targeted delivery. For example, an Sn2 lipase-labile pro-drug inhibitor (MI1-PD) conjugated to integrin-targeted nanoparticles extended survival in a mouse model of multiple myeloma (Soodgupta et al., 2015).
A more extensive review of these small molecule inhibitors is provided elsewhere (Chen et al., 2014). Such inhibitors have been an intense focus in Myc inhibition research for many years, and we hope that further preclinical development of promising leads is ongoing.
Other small molecule inhibitors such as MYRA-A and NSC308848 have achieved high selectivity in targeting the DNA-binding domain of Myc/Max, and preventing specific interaction with DNA (Mo and Henriksson, 2006; Mo et al., 2006, Figure 3).
Some naturally-occurring molecules have also been shown to directly interact with Myc/Max heterodimers. Celastrol and celastrol-inspired triterpenoids bind to and alter the quaternary structure of the pre-formed dimer and abrogate its DNA binding (Wang et al., 2015b).
KSI-3716 also blocks Myc/Max binding to DNA, and inhibited orthotopic tumor formation after local instillation to the bladder (Jeong et al., 2014)—the typical treatment route for this cancer type—even in gemcitabine-resistant tumors (Seo et al., 2014).
As mentioned above, though, small molecules, despite good affinity for their target in vitro, often display lack of selectivity in cells or in vivo. In order to try to overcome this issue, synthetic α-Helix “mimetics” based on biphenyl were developed (Jung et al., 2015). These have an increased interaction surface and recognize Myc/Max dimers (not free Myc) and disrupt DNA binding. However, their KD was not increased (13 μM) and specificity was not enhanced: similar activity was observed in non-cancer cells lacking Myc overexpression (Jung et al., 2015).
Miniproteins or protein domains comprise a group of structurally-related molecules based on domains from Myc family members (Figure 3). The best characterized so far and especially notable for its in vivo use, is Omomyc. Omomyc has been well validated as a gene therapy, providing the proof of concept for the feasibility of systemic Myc inhibition. It comprises the bHLH-Zip domain of Myc carrying four aminoacidic substitutions that alter its dimerization specificity, such that in addition to binding Myc's natural partner Max, it can also heterodimerize with Myc as well as homodimerize (Soucek et al., 1998, 2002; Savino et al., 2011). As a result, Omomyc acts as a dominant negative of Myc transcriptional function, being able to disrupt Myc/Max interaction, sequester Myc away from DNA and occupy the E-box with transcriptionally inactive dimers (Omomyc/Omomyc and/or Omomyc/Max). Notably, in doing so, it antagonizes all Myc family members (Soucek et al., 2008; Savino et al., 2011; Fiorentino et al., 2016). Multiple studies in mouse models of cancer demonstrated Omomyc's therapeutic impact in different types of cancer, independently of their driving mutation or tissue of origin, pointing to the key role of Myc in tumorigenesis downstream of the diverse oncogenic lesions (Soucek et al., 2004, 2008, 2013; Sodir et al., 2011; Annibali et al., 2014; Galardi et al., 2016). Importantly, in each model Omomyc showed only minimal side effects, suggesting its safety and potential applicability in patients. Work to translate its use from gene therapy to pharmacological application is currently ongoing (Peptomyc S.L.).
Another modified Myc peptide, this time a 14 amino acid sequence from the helix 1 (H1) carboxylic region of Myc harboring 2 changes, was shown to be active against breast cancer cells in culture when fused to a fragment from Antennapedia to enable cellular uptake (Giorello et al., 1998). This has not performed well in vivo, at least partly because it does not efficiently cross the nuclear envelope. However, recently a staggered, “dual-strike” strategy was employed, whereby a first treatment with docetaxel arrested the cells in G2/M, prolonging the period of nuclear envelope disassembly, followed by a second treatment, this time with the H1 peptide (Li et al., 2016). In vivo, this procedure reduced the growth of subcutaneously-inoculated HeLa cells and prolonged animal survival (Li et al., 2016). The peptide was delivered in the macromolecular carrier HPMA.
The H1 peptide was also used for an in vivo study by fusing it to both a cell-penetrating peptide sequence and an elastin-like polypeptide (ELP) that is thermally stable and allows targeted delivery to particular tissues by local hyperthermia (Bidwell et al., 2013). This multi-functional peptide reduced tumor growth in rodent orthotopic models of glioma and breast cancer (Bidwell et al., 2012, 2013).
Another protein domain that could be used to inhibit Myc is the bHLH-Zip of Max (Montagne et al., 2012). This truncated protein spontaneously transduces into cells and inhibits Myc transcription. The idea behind this strategy would be to provide excess homodimeric Max to out-compete Myc/Max heterodimers binding to DNA.
Targeting Myc itself has often proven very challenging. Because of that, many researchers have instead opted for an indirect approach, focusing on Myc transcriptional regulation or modulation of stability and activity, by inhibiting more tractable targets and not directly hitting Myc itself. Here is an overview of these alternative approaches, once again encompassing the transcription, translation and stability of Myc, as well as controlling its activity as a transcription factor.
The BET bromodomain and extra-terminal domain inhibitors are a significant area of focus at the moment and were originally described to target Myc expression. A selective small molecule inhibitor (JQ1) of BET bromodomains was unexpectedly found to downregulate Myc (Delmore et al., 2011). JQ1 displaces the bromodomain chromatin regulators from the large super-enhancers of genes connected with multiple myeloma, notably myc (Delmore et al., 2011; Loven et al., 2013, Figure 1). JQ1 preferentially impacts on myc transcription, causing cell-cycle arrest, and cellular senescence, as well as resulting in significant anti-tumor activity in mouse models of multiple myeloma, and xenograft models of Burkitt's lymphoma and acute myeloid leukemia, diseases in which the myc gene is amplified (Delmore et al., 2011; Mertz et al., 2011).
After the initial excitement about their potential for Myc inhibition, it is now becoming clear that JQ1—and other BET inhibitors—may function by inhibiting additional oncogenic factors besides Myc (Andrieu et al., 2016), whose levels remain unaffected in some cellular contexts (Ambrosini et al., 2015; Yao et al., 2015; Bid et al., 2016; Garcia et al., 2016; Hogg et al., 2016; Donato et al., 2017). Moreover, compensatory mutations have been identified that can prevent Myc downregulation by BET inhibitors and/or their therapeutic efficacy (Shimamura et al., 2013; Fong et al., 2015; Rathert et al., 2015; Shi et al., 2016). Nevertheless, several BET inhibitors are currently in early-phase clinical trials in various hematologic malignancies (Abedin et al., 2016). Some patients in dose-escalation Phase I studies have shown complete or partial remission of acute leukemia after treatment with OTX015 (Berthon et al., 2016), and data is currently available for 3 patients treated with TEN-010 in a study of NUT-midline carcinoma showing clinical responses in both patients receiving the higher dose (Shapiro, 2015). It remains to be seen how much of the effect in patients is actually due to Myc inhibition.
An unrelated approach to blocking myc transcription targets CDK7, a catalytic subunit involved in the transcriptional initiation complex that phosphorylates serine-5 of RNA pol II (Nilson et al., 2015). A covalent inhibitor (THZ1) was developed that showed selectivity for CDK7, and specifically downregulated Myc in neuroblastoma (Chipumuro et al., 2014; Kwiatkowski et al., 2014). While THZ1 selectivity for Myc is unclear and it is possible that its therapeutic impact is due to other targets as well, it is effective at treating lung cancer (Christensen et al., 2014) and triple-negative breast cancer cell lines (Wang et al., 2015c) as well as N-Myc driven neuroblastoma in mice (Chipumuro et al., 2014). Furthermore, the analog THZ2 was developed that has improved pharmacokinetics (Wang et al., 2015c).
Another approach to interfere with Myc protein expression is to block its translation (Figure 2). myc mRNA can be translated both by 5′ cap- and internal ribosome entry site (IRES)- dependent mechanisms (Nanbru et al., 1997). The central role of mTOR in mediating the translation of mRNAs such as myc via these mechanisms suggests that targeting mTOR or upstream controllers of its activity (PI3K/PTEN/AKT and Ras/Raf/MEK/ERK) could be a fruitful strategy, and there are multiple inhibitors of these pathways.
While inhibition of mTOR alone in a mouse model of colorectal tumorigenesis failed to inhibit translation of myc, a small molecule inhibitor of eIF4A, silvestrol, effectively reduced myc translation, inhibiting tumor growth (Wiegering et al., 2015). Multiple mTOR and mTORC1/2 kinase inhibitors are currently approved for clinical use, and there is a significant focus on targeting many members of these signaling pathways (Polivka and Janku, 2014; Roohi and Hojjat-Farsangi, 2016).
Again though, inhibitors of such targets will have effects not limited to myc alone.
Recent data also indicate that concomitant targeting of HDAC and PI3K would be beneficial to the treatment of Myc-driven tumors. In particular the use of CUDC-907, a small molecule inhibitor of both HDACs and class I PI3Ks, was shown to be effective in reducing the growth and survival of Myc-transformed cancer cells and demonstrated therapeutic impact in multiple mouse models of Myc-dependent tumors (Sun et al., 2016). CUDC-907 is currently in Phase II clinical trials to study and evaluate its efficacy and safety (alone or in combination with Rituximab) in patients with Relapsed/Refractory (RR) Myc-altered Diffuse Large B-Cell Lymphoma (DLBCL), including patients with Myc alterations.
Another recent translation inhibition approach made use of Src kinase blockade with the small molecule inhibitor saracatinib in preclinical studies in premalignant breast cells and tissue. Among other effects, saracatinib inhibited the ERK1/2-MNK1-eIF4E-mediated cap-dependent translation of myc (Jain et al., 2015).
The regulation of Myc stability is complex. Numerous studies propose the targeting of ubiquitinases or phosphatases for the degradation of Myc in cancer cells. Clearly, whether these can be specific for Myc and/or have sufficiently minimal side effects is still unclear.
Myc is ubiquitinated by a number of E3 ligases, such as SCF (FBW7) and SCF (Skp2). One approach is therefore to inhibit the deubiquitinases that help stabilize Myc, such as USP28, USP38, and USP36 (Sun et al., 2015). On the other hand, proteasomal degradation could be triggered, for example by oridonin, a natural plant diterpenoid that induces FBW7-mediated Myc breakdown (Huang et al., 2012). Oridonin derivatives are in clinical trials (e.g., HAO472 for leukemia treatment), although such anticancer drugs (and ubiquitin/deubiquitinases) have numerous other potential mechanisms of action and targets, not solely limited to Myc inhibition.
It has been shown that N-Myc complexes with the Aurora-A kinase and is thus protected from proteasomal degradation (Otto et al., 2009). Aurora-A inhibitors (MLN8054 and MLN8237) disrupt the complex and promote N-Myc degradation by FBW7 ubiquitin ligase (Brockmann et al., 2013). While clinical development of MLN8054 was terminated by Millenium in 2008 due to side effects (Macarulla et al., 2010), the more potent, selective, second generation inhibitor MLN8237 (Alisertib) is currently being evaluated in multiple Phase II and III studies (based on the obligate role of Aurora kinases in mitosis). Since N-Myc is a critical target of this class of Aurora-A inhibitors, perhaps it is possible to design allosteric inhibitors that more strongly distort Aurora-A and thereby more effectively disrupt the Aurora-A/N-Myc complex, while retaining the ability of Aurora-A to promote mitotic entry, thus finding a more specific inhibitor (Brockmann et al., 2013). Notably, the Aurora kinases have turned up in multiple screens for Myc synthetic lethal targets (see later section).
Another ubiquitin ligase—HUWE1 (HECTH9, ARF-BP1, MULE)—has also been targeted by small molecule inhibitors. These reduce Myc-dependent transactivation in colorectal cancer cells, but not in stem and normal colon epithelial cells, by influencing Myc and Miz1 degradation (Peter et al., 2014).
Alternatively, the tumor suppressor protein phosphatase 2A (PP2A) targets serine 62 of Myc and causes its inactivation and destabilization (Sears, 2004). Cellular inhibitors of PP2A (the SET oncoprotein and CIP2A, the cancerous inhibitor of PP2A) are increased in human cancers and lead to overexpression of Myc (Westermarck and Hahn, 2008). Thus, inhibitors of SET and CIP2A were used to reduce Myc expression and activity, decreasing the tumorigenic potential of cancer cells (Farrell et al., 2014; Janghorban et al., 2014). Inhibitors are in preclinical development; the effects of PP2A activation though would likely not be limited to Myc degradation alone, since other pathways would be targeted as well.
Described as “using a back door to target Myc” (Evan, 2012), in this indirect approach, the targets are the cellular changes that arise as a consequence of oncogene activation of proteins and pathways required for survival of the oncogene-addicted cells (Wang et al., 2004). Myc-mediated synthetic lethality was first described to be induced by TRAIL and DR5-agonists, taking advantage of Myc's intrinsic ability to prime cells to apoptotic stimuli (Wang et al., 2004).
More recently, SAE1/2 was identified in a genome-wide RNA interference screen to search for Myc synthetic lethal genes. This SUMOylation enzyme is required for proper mitotic spindle function, and proved necessary for Myc-driven tumorigenesis as its inhibition caused mitotic catastrophe in Myc hyperactive cells (Kessler et al., 2012). Identification of SUMO inhibitors is ongoing (Kumar et al., 2016).
A number of studies demonstrate that inhibition of CDKs is also synthetic lethal with Myc. Pharmacological inhibition of CDK2 forces embryonic fibroblasts with deregulated Myc into senescence (Hydbring et al., 2010), while CDK2 ablation induces senescence in B-cells after Myc activation, delaying lymphomagenesis (Campaner et al., 2010). Since the function of CDK2 is compensated by other CDKs in normal cells (Ortega et al., 2003), this suggests that its selective targeting could be used therapeutically, at least in Myc-driven tumors.
CDK1 inhibition is also beneficial for treating Myc-overexpressing tumors: the CDK1 inhibitor purvalanol A induces substantial apoptosis in cells overexpressing Myc, but not in cells expressing other oncogenes (Goga et al., 2007). It prolongs survival in Eμ-myc transgenic mice and lymphoma allograft models (Goga et al., 2007).
Finally, synthetic lethality was observed with CDK9. Its pharmacological inhibition or knockdown by shRNA is anti-tumorigenic both in cells and mouse models of hepatocellular carcinoma, the extent of its effect correlating with Myc levels (Huang et al., 2014).
Since several CDK inhibitors are currently in clinical trials (Lapenna and Giordano, 2009), there is clear merit in analyzing the results taking into account this potential Myc synthetic lethality.
Myc has been shown to control multiple aspects of transcription and co-transcription, as well as RNA metabolism, including splicing, mRNA stability, and translation efficiency (Koh et al., 2016). In the context of the translation machinery, Myc is able to modulate Ribosomal Biogenesis (RiBi) through the coordinated regulation of all three RNA polymerases: Pol I, Pol II, and Pol III. Selective inhibition of Pol I transcription has been proposed as a promising therapeutic approach in Myc-driven cancers (Poortinga et al., 2015), as Myc is supposed to prime cells to nucleolar stress. Remarkably, despite the risk associated to interfering with a central engine of a “housekeeping” process such as RiBi, an inhibitor of Pol I (CX-5461; Drygin et al., 2011) has recently demonstrated sufficient safety in Phase I clinical trials in patients with lymphoma and leukemia, and is now in a Phase I/II study in solid malignancies (although not confined to Myc-driven cancers only).
Myc synthetic lethality has been observed with various other targets in mouse tumor models, for example with ARK5 (Liu et al., 2012), PIM kinase (Horiuchi et al., 2016), microRNA-206 that acts by inhibiting MAP3K13 (Han et al., 2016), Aurora kinases (den Hollander et al., 2010; Yang et al., 2010), CHK1 (Murga et al., 2011; Ferrao et al., 2012), and MondoA (Carroll et al., 2015). MondoA had been previously linked to the regulation of glucose metabolism while overexpression of Myc in mammalian cells renders them addicted to certain metabolic pathways (Shim et al., 1998; Yuneva et al., 2007) thus prompting the extension of these synthetic lethal screen to genes involved in metabolism. This led to the identification of Myc synthetic lethal metabolic genes involved in glycolysis (ALDOA and PDK1) and nucleotide biosynthesis (CTPS) (Toyoshima et al., 2012); purine biosynthesis (PFAS and CAD), trans-sulphuration (CBS), mitochondrial transcription (TFAM), glycolysis (ENO3), and lipogenesis (FASN and SCD) (Carroll et al., 2015); and glutamine/glutamate (SLC1A4 and SLC25A6) (Toyoshima et al., 2012; Carroll et al., 2015).
It is well documented that Myc contributes to metabolic adaptations in cancer cells (Dang, 2012). As a proof of concept, inhibition of various metabolic targets, such as LDHA and glutaminase, reduced tumor growth, and extended survival in Myc-dependent and Myc-inducible cancer models, although the synthetic lethality with Myc was not specifically demonstrated (Hsieh and Dang, 2016).
This indirect strategy has therefore already provided a variety of additional therapeutic targets. Some clinical trials are ongoing (e.g., inhibitors of CHK1/2, PIM, and Aurora kinases). Whether these would be relevant for tumors in which Myc is not a driving oncogene has to be seen, but the fact that Myc is causally linked to most human cancers suggests that this approach warrants further clinical investigation.
This rather different approach to cancer treatment is currently receiving a significant amount of interest worldwide. One strategy would be to prime the immune system to target Myc-overexpressing tumors. As a proof of concept, immunization of mice with human Myc generated T-cells that helped protect some mice from a lethal lymphoma (Helm et al., 2013).
A more direct immunotherapy could target specific immune system components required for Myc-induced tumorigenesis, such as mast cells (Soucek et al., 2007). PCI-32765 (Ibrutinib) is an inhibitor of Bruton's tyrosine kinase (BTK) that is clinically approved for the treatment of multiple cancers (Smith, 2015; Massó-Vallés et al., 2016) and was shown to inhibit Myc-driven pancreatic islet tumor formation (Soucek et al., 2011).
Alternatively, Myc-driven tumors may downregulate the anti-tumor immune response by producing PD-L1 and CD47, two key immune checkpoints, making them eligible for treatment with immune-based therapies that target such checkpoints (Casey et al., 2016).
An impressive array of strategies has been developed for drugging Myc. These take advantage of multiple mechanisms, acting both directly and indirectly, and impacting on Myc in contrasting ways (see Table 1 for a summary). Many approaches have yielded molecules that entered clinical trials.
Early clinical studies with antisense inhibitors of myc did not progress, nor did a recent trial with myc RNAi (DCR-MYC), although the approach might still be valid in some applications. In fact, incorporation of myc siRNA into nanoparticles is an active field of research yielding novel carrier formulations with in vivo efficacy. Interference with Myc translation is an approach that has clearly advanced in the past few years and various groups and companies are pursuing it.
Similarly, the initial small molecule inhibitors showed poor bioavailability, but development of these compounds and further screens have identified compounds with improved in vivo activity, pharmacokinetics and bioavailability, even with systemic administration (e.g., Mycro3, KJ-Pyr-9). In addition, efforts are being directed toward incorporating small molecules into nanocarriers and some are starting to show in vivo efficacy (MI1-PD). Published in vivo or clinical trial data is lacking for a number of other small molecule inhibitors and natural compounds shown to target Myc in cell culture; it seems sensible to at least determine their in vivo efficacy and bioavailability and one hopes that no promising leads have fallen by the wayside.
Additional strategies such as BET inhibitors and G-quadruplex stabilizers (Quarflaxin) have progressed to clinical trials, and yet more strategies are in preclinical development, such as peptides and protein domains (Omomyc).
It is important to mention that new therapeutic opportunities constantly appear, thanks to the ever-growing knowledge regarding Myc biology and function. For example, novel strategies could soon be based on the potential inhibition of co-factors that determine Myc specific recruitment to chromatin and recognition of target genes. Among these factors, WDR5 and BPTF are probably the best characterized. WDR5 is a WD40-repeat protein that functions within the context of several chromatin-regulatory complexes (Thomas et al., 2015). WDR5 interaction with Myc facilitates its recruitment to chromatin and recognition of target genes. Similarly, in the context of chromatin access, BPTF, a core subunit of the NURF chromatin-remodeling complex, has been described as a key Myc interactor that allows its recruitment to DNA targets (Richart et al., 2016a,b). Inhibition of interaction of such proteins with Myc by small molecules could offer a novel therapeutic opportunity. Once again, it is likely that such small molecules would also affect other cellular functions that require these two important epigenetic regulatory proteins, but it is worth trying to develop molecules specific for the interface with Myc and validate them in experimental models, before discarding them a priori.
Other future strategies could include development of viral-mediated delivery of inhibitors such as shRNA, Omomyc or even Crispr to delete myc in tumor cells. It is likely, however, that the most efficient Myc inhibition strategy will not be limited to a single approach, but rather a combination of targeting methods. These may include low-dose combinations of drugs that each act to reduce Myc levels in different ways (Brockmann et al., 2013) or that act on different aspects of Myc biology.
In this context, no attempt should be left aside to finally overcome the challenge of targeting the “undruggable,” because its impact in the clinic would clearly be dramatic.
Interestingly, the search for a clinically-viable Myc inhibitor has recently been likened to the hunt for the Higgs boson (Lazo and Sharlow, 2016), which was finally discovered in 2012. After the success of that 40-year search, we can be hopeful that it will not be too long before that other elusive particle—a Myc inhibitor—emerges from clinical trials.
JW wrote the review with help from MB and LS.
We gratefully receive support from Fundación BBVA, Worldwide Cancer Research (WCR/AICR Grant #13-1182), the European Research Council (CoG Grant #617473), Instituto de Salud Carlos III (FIS Grants #PI13/01705, #PI16/01224) and the FERO Foundation. MB is supported by the Fonds de Recherche du Québec en Santé.
LS and MB are co-founders and shareholders of Peptomyc S.L., a spin-off company located at VHIO.
The other author declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all members of the Soucek lab for providing a stimulating and productive work environment and for comments on this manuscript.
Abedin, S. M., Boddy, C. S., and Munshi, H. G. (2016). BET inhibitors in the treatment of hematologic malignancies: current insights and future prospects. Onco. Targets Ther. 9, 5943–5953. doi: 10.2147/OTT.S100515
Ambrosini, G., Sawle, A. D., Musi, E., and Schwartz, G. K. (2015). BRD4-targeted therapy induces Myc-independent cytotoxicity in Gnaq/11-mutatant uveal melanoma cells. Oncotarget 6, 33397–33409. doi: 10.18632/oncotarget.5179
Andrieu, G., Belkina, A. C., and Denis, G. V. (2016). Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 19, 45–50. doi: 10.1016/j.ddtec.2016.06.004
Andtbacka, R. H., Kaufman, H. L., Collichio, F., Amatruda, T., Senzer, N., Chesney, J., et al. (2015). Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 33, 2780–2788. doi: 10.1200/JCO.2014.58.3377
Annibali, D., Whitfield, J. R., Favuzzi, E., Jauset, T., Serrano, E., Cuartas, I., et al. (2014). Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 5:4632. doi: 10.1038/ncomms5632
Arora, V., Knapp, D. C., Smith, B. L., Statdfield, M. L., Stein, D. A., Reddy, M. T., et al. (2000). c-Myc antisense limits rat liver regeneration and indicates role for c-Myc in regulating cytochrome P-450 3A activity. J. Pharmacol. Exp. Ther. 292, 921–928.
Berg, T., Cohen, S. B., Desharnais, J., Sonderegger, C., Maslyar, D. J., Goldberg, J., et al. (2002). Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 99, 3830–3835. doi: 10.1073/pnas.062036999
Berthon, C., Raffoux, E., Thomas, X., Vey, N., Gomez-Roca, C., Yee, K., et al. (2016). Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 3, e186–e195. doi: 10.1016/S2352-3026(15)00247-1
Bid, H. K., Phelps, D. A., Xaio, L., Guttridge, D. C., Lin, J., London, C., et al. (2016). The bromodomain BET inhibitor JQ1 suppresses tumor angiogenesis in models of childhood sarcoma. Mol. Cancer Ther. 15, 1018–1028. doi: 10.1158/1535-7163.MCT-15-0567
Bidwell, G. L. III, Perkins, E., Hughes, J., Khan, M., James, J. R., and Raucher, D. (2013). Thermally targeted delivery of a c-Myc inhibitory polypeptide inhibits tumor progression and extends survival in a rat glioma model. PLoS ONE 8:e55104. doi: 10.1371/journal.pone.0055104
Bidwell, G. L. III, Perkins, E., and Raucher, D. (2012). A thermally targeted c-Myc inhibitory polypeptide inhibits breast tumor growth. Cancer Lett. 319, 136–143. doi: 10.1016/j.canlet.2011.12.042
Brockmann, M., Poon, E., Berry, T., Carstensen, A., Deubzer, H. E., Rycak, L., et al. (2013). Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 24, 75–89. doi: 10.1016/j.ccr.2013.05.005
Brooks, T. A., and Hurley, L. H. (2009). The role of supercoiling in transcriptional control of MYC and its importance in molecular therapeutics. Nat. Rev. Cancer 9, 849–861. doi: 10.1038/nrc2733
Brooks, T. A., and Hurley, L. H. (2010). Targeting MYC expression through G-quadruplexes. Genes Cancer 1, 641–649. doi: 10.1177/1947601910377493
Brown, R. V., Danford, F. L., Gokhale, V., Hurley, L. H., and Brooks, T. A. (2011). Demonstration that drug-targeted down-regulation of MYC in non-hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J. Biol. Chem. 286, 41018–41027. doi: 10.1074/jbc.M111.274720
Campaner, S., Doni, M., Hydbring, P., Verrecchia, A., Bianchi, L., Sardella, D., et al. (2010). Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 12, 54–59. doi: 10.1038/ncb2004
Carroll, P. A., Diolaiti, D., McFerrin, L., Gu, H., Djukovic, D., Du, J., et al. (2015). Deregulated Myc requires MondoA/Mlx for metabolic reprogramming and tumorigenesis. Cancer Cell 27, 271–285. doi: 10.1016/j.ccell.2014.11.024
Casey, S. C., Tong, L., Li, Y., Do, R., Walz, S., Fitzgerald, K. N., et al. (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. doi: 10.1126/science.aac9935
Cermelli, S., Jang, I. S., Bernard, B., and Grandori, C. (2014). Synthetic lethal screens as a means to understand and treat MYC-driven cancers. Cold Spring Harb. Perspect. Med. 4:a014209. doi: 10.1101/cshperspect.a014209
Chauhan, J., Wang, H., Yap, J. L., Sabato, P. E., Hu, A., Prochownik, E. V., et al. (2014). Discovery of methyl 4′-methyl-5-(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)-[1,1′-biphenyl]-3-carboxylate, an improved small-molecule inhibitor of c-Myc-max dimerization. ChemMedChem 9, 2274–2285. doi: 10.1002/cmdc.201402189
Chen, B. J., Wu, Y. L., Tanaka, Y., and Zhang, W. (2014). Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int. J. Biol. Sci. 10, 1084–1096. doi: 10.7150/ijbs.10190
Chipumuro, E., Marco, E., Christensen, C. L., Kwiatkowski, N., Zhang, T., Hatheway, C. M., et al. (2014). CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139. doi: 10.1016/j.cell.2014.10.024
Christensen, C. L., Kwiatkowski, N., Abraham, B. J., Carretero, J., Al-Shahrour, F., Zhang, T., et al. (2014). Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26, 909–922. doi: 10.1016/j.ccell.2014.10.019
Clausen, D. M., Guo, J., Parise, R. A., Beumer, J. H., Egorin, M. J., Lazo, J. S., et al. (2010). In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J. Pharmacol. Exp. Ther. 335, 715–727. doi: 10.1124/jpet.110.170555
Conacci-Sorrell, M., McFerrin, L., and Eisenman, R. N. (2014). An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 4:a014357. doi: 10.1101/cshperspect.a014357
Conde, J., Tian, F., Hernández, Y., Bao, C., Cui, D., Janssen, K. P., et al. (2013). In vivo tumor targeting via nanoparticle-mediated therapeutic siRNA coupled to inflammatory response in lung cancer mouse models. Biomaterials 34, 7744–7753. doi: 10.1016/j.biomaterials.2013.06.041
Dang, C. V. (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 3:a014217. doi: 10.1101/cshperspect.a014217
Delmore, J. E., Issa, G. C., Lemieux, M. E., Rahl, P. B., Shi, J., Jacobs, H. M., et al. (2011). BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. doi: 10.1016/j.cell.2011.08.017
den Hollander, J., Rimpi, S., Doherty, J. R., Rudelius, M., Buck, A., Hoellein, A., et al. (2010). Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood 116, 1498–1505. doi: 10.1182/blood-2009-11-251074
Devi, G. R., Beer, T. M., Corless, C. L., Arora, V., Weller, D. L., and Iversen, P. L. (2005). In vivo bioavailability and pharmacokinetics of a c-MYC antisense phosphorodiamidate morpholino oligomer, AVI-4126, in solid tumors. Clin. Cancer Res. 11, 3930–3938. doi: 10.1158/1078-0432.CCR-04-2091
Donato, E., Croci, O., Sabò, A., Muller, H., Morelli, M. J., Pelizzola, M., et al. (2017). Compensatory RNA polymerase 2 loading determines the efficacy and transcriptional selectivity of JQ1 in Myc-driven tumors. Leukemia 31, 479–490. doi: 10.1038/leu.2016.182
Drygin, D., Lin, A., Bliesath, J., Ho, C. B., O'Brien, S. E., Proffitt, C., et al. (2011). Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 71, 1418–1430. doi: 10.1158/0008-5472.CAN-10-1728
Evan, G. (2012). Cancer. Taking a back door to target Myc. Science 335, 293–294. doi: 10.1126/science.1217819
Farrell, A. S., Allen-Petersen, B., Daniel, C. J., Wang, X., Wang, Z., Rodriguez, S., et al. (2014). Targeting inhibitors of the tumor suppressor PP2A for the treatment of pancreatic cancer. Mol. Cancer Res. 12, 924–939. doi: 10.1158/1541-7786.MCR-13-0542
Felsenstein, K. M., Saunders, L. B., Simmons, J. K., Leon, E., Calabrese, D. R., Zhang, S., et al. (2016). Small molecule microarrays enable the identification of a selective, quadruplex-binding inhibitor of MYC expression. ACS Chem. Biol. 11, 139–148. doi: 10.1021/acschembio.5b00577
Ferrao, P. T., Bukczynska, E. P., Johnstone, R. W., and McArthur, G. A. (2012). Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 31, 1661–1672. doi: 10.1038/onc.2011.358
Fiorentino, F. P., Tokgüun, E., Solé-Sánchez, S., Giampaolo, S., Tokgun, O., Jauset, T., et al. (2016). Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget 7, 31014–31028. doi: 10.18632/oncotarget.8826
Fletcher, S., and Prochownik, E. V. (2015). Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 1849, 525–543. doi: 10.1016/j.bbagrm.2014.03.005
Follis, A. V., Hammoudeh, D. I., Wang, H., Prochownik, E. V., and Metallo, S. J. (2008). Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem. Biol. 15, 1149–1155. doi: 10.1016/j.chembiol.2008.09.011
Fong, C. Y., Gilan, O., Lam, E. Y., Rubin, A. F., Ftouni, S., Tyler, D., et al. (2015). BET inhibitor resistance emerges from leukaemia stem cells. Nature 525, 538–542. doi: 10.1038/nature14888
Galardi, S., Savino, M., Scagnoli, F., Pellegatta, S., Pisati, F., Zambelli, F., et al. (2016). Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 17, 1872–1889. doi: 10.15252/embr.201541489
Garcia, P. L., Miller, A. L., Kreitzburg, K. M., Council, L. N., Gamblin, T. L., Christein, J. D., et al. (2016). The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene 35, 833–845. doi: 10.1038/onc.2015.126
Giorello, L., Clerico, L., Pescarolo, M. P., Vikhanskaya, F., Salmona, M., Colella, G., et al. (1998). Inhibition of cancer cell growth and c-Myc transcriptional activity by a c-Myc helix 1-type peptide fused to an internalization sequence. Cancer Res. 58, 3654–3659.
Goga, A., Yang, D., Tward, A. D., Morgan, D. O., and Bishop, J. M. (2007). Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 13, 820–827. doi: 10.1038/nm1606
Guo, J., Parise, R. A., Joseph, E., Egorin, M. J., Lazo, J. S., Prochownik, E. V., et al. (2009). Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother. Pharmacol. 63, 615–625. doi: 10.1007/s00280-008-0774-y
Han, H., Chen, Y., Cheng, L., Prochownik, E. V., and Li, Y. (2016). microRNA-206 impairs c-Myc-driven cancer in a synthetic lethal manner by directly inhibiting MAP3K13. Oncotarget 7, 16409–16419. doi: 10.18632/oncotarget.7653
Hart, J. R., Garner, A. L., Yu, J., Ito, Y., Sun, M., Ueno, L., et al. (2014). Inhibitor of MYC identified in a Krohnke pyridine library. Proc. Natl. Acad. Sci. U.S.A. 111, 12556–12561. doi: 10.1073/pnas.1319488111
Hartl, M. (2016). The quest for targets executing MYC-dependent cell transformation. Front. Oncol. 6:132. doi: 10.3389/fonc.2016.00132
Helm, F., Kammertoens, T., Lehmann, F. M., Wilke, A., Bruns, H., Mautner, J., et al. (2013). Targeting c-MYC with T-cells. PLoS ONE 8:e77375. doi: 10.1371/journal.pone.0077375
Hogg, S. J., Newbold, A., Vervoort, S. J., Cluse, L. A., Martin, B. P., Gregory, G. P., et al. (2016). BET inhibition induces apoptosis in aggressive B-cell lymphoma via epigenetic regulation of BCL-2 family members. Mol. Cancer Ther. 15, 2030–2041. doi: 10.1158/1535-7163.MCT-15-0924
Horiuchi, D., Camarda, R., Zhou, A. Y., Yau, C., Momcilovic, O., Balakrishnan, S., et al. (2016). PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat. Med. 22, 1321–1329. doi: 10.1038/nm.4213
Hsieh, A. L., and Dang, C. V. (2016). MYC, metabolic synthetic lethality, and cancer. Recent Results Cancer Res. 207, 73–91. doi: 10.1007/978-3-319-42118-6_4
Huang, C. H., Lujambio, A., Zuber, J., Tschaharganeh, D. F., Doran, M. G., Evans, M. J., et al. (2014). CDK9-mediated transcription elongation is required for MYC addiction in hepatocellular carcinoma. Genes Dev. 28, 1800–1814. doi: 10.1101/gad.244368.114
Huang, H. L., Weng, H. Y., Wang, L. Q., Yu, C. H., Huang, Q. J., Zhao, P. P., et al. (2012). Triggering Fbw7-mediated proteasomal degradation of c-Myc by oridonin induces cell growth inhibition and apoptosis. Mol. Cancer Ther. 11, 1155–1165. doi: 10.1158/1535-7163.MCT-12-0066
Hydbring, P., Bahram, F., Su, Y., Tronnersjo, S., Hogstrand, K., von der Lehr, N., et al. (2010). Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl. Acad. Sci. U.S.A. 107, 58–63. doi: 10.1073/pnas.0900121106
Jain, S., Wang, X., Chang, C. C., Ibarra-Drendall, C., Wang, H., Zhang, Q., et al. (2015). Src inhibition blocks c-Myc translation and glucose metabolism to prevent the development of breast cancer. Cancer Res. 75, 4863–4875. doi: 10.1158/0008-5472.CAN-14-2345
Janghorban, M., Farrell, A. S., Allen-Petersen, B. L., Pelz, C., Daniel, C. J., Oddo, J., et al. (2014). Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 111, 9157–9162. doi: 10.1073/pnas.1317630111
Jeong, K. C., Kim, K. T., Seo, H. H., Shin, S. P., Ahn, K. O., Ji, M. J., et al. (2014). Intravesical instillation of c-MYC inhibitor KSI-3716 suppresses orthotopic bladder tumor growth. J. Urol. 191, 510–518. doi: 10.1016/j.juro.2013.07.019
Jiang, H., Bower, K. E., Beuscher, A. E., Zhou, B., Bobkov, A. A., Olson, A. J., et al. (2009). Stabilizers of the Max homodimer identified in virtual ligand screening inhibit Myc function. Mol. Pharmacol. 76, 491–502. doi: 10.1124/mol.109.054858
Jung, K. Y., Wang, H., Teriete, P., Yap, J. L., Chen, L., Lanning, M. E., et al. (2015). Perturbation of the c-Myc-Max protein-protein interaction via synthetic α-helix mimetics. J. Med. Chem. 58, 3002–3024. doi: 10.1021/jm501440q
Kessler, J. D., Kahle, K. T., Sun, T., Meerbrey, K. L., Schlabach, M. R., Schmitt, E. M., et al. (2012). A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353. doi: 10.1126/science.1212728
Kipshidze, N., Iversen, P., Overlie, P., Dunlap, T., Titus, B., Lee, D., et al. (2007). First human experience with local delivery of novel antisense AVI-4126 with Infiltrator catheter in de novo native and restenotic coronary arteries: 6-month clinical and angiographic follow-up from AVAIL study. Cardiovasc. Revasc. Med. 8, 230–235. doi: 10.1016/j.carrev.2007.04.002
Kipshidze, N. N., Iversen, P., Kim, H. S., Yiazdi, H., Dangas, G., Seaborn, R., et al. (2004). Advanced c-myc antisense (AVI-4126)-eluting phosphorylcholine-coated stent implantation is associated with complete vascular healing and reduced neointimal formation in the porcine coronary restenosis model. Catheter. Cardiovasc. Interv. 61, 518–527. doi: 10.1002/ccd.20007
Kipshidze, N. N., Porter, T. R., Dangas, G., Yazdi, H., Tio, F., Xie, F., et al. (2003). Systemic targeted delivery of antisense with perflourobutane gas microbubble carrier reduced neointimal formation in the porcine coronary restenosis model. Cardiovasc. Radiat. Med. 4, 152–159. doi: 10.1016/S1522-1865(03)00184-7
Koh, C. M., Sabo, A., and Guccione, E. (2016). Targeting MYC in cancer therapy: RNA processing offers new opportunities. Bioessays 38, 266–275. doi: 10.1002/bies.201500134
Kumar, A., Ito, A., Hirohama, M., Yoshida, M., and Zhang, K. Y. (2016). Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg. Med. Chem. Lett. 26, 1218–1223. doi: 10.1016/j.bmcl.2016.01.030
Kwiatkowski, N., Zhang, T., Rahl, P. B., Abraham, B. J., Reddy, J., Ficarro, S. B., et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. doi: 10.1038/nature13393
Lapenna, S., and Giordano, A. (2009). Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 8, 547–566. doi: 10.1038/nrd2907
Lazo, J. S., and Sharlow, E. R. (2016). Drugging undruggable molecular cancer targets. Annu. Rev. Pharmacol. Toxicol. 56, 23–40. doi: 10.1146/annurev-pharmtox-010715-103440
Li, L., Sun, W., Zhang, Z., and Huang, Y. (2016). Time-staggered delivery of docetaxel and H1-S6A,F8A peptide for sequential dual-strike chemotherapy through tumor priming and nuclear targeting. J. Control. Release 232, 62–74. doi: 10.1016/j.jconrel.2016.04.021
Li, X., Zhang, X. A., Li, X., Xie, W., and Huang, S. (2015). MYC-mediated synthetic lethality for treating tumors. Curr. Cancer Drug Targets 15, 99–115. doi: 10.2174/1568009615666150121162921
Li, Y., Zhang, B., Zhang, H., Zhu, X., Feng, D., Zhang, D., et al. (2013). Oncolytic adenovirus armed with shRNA targeting MYCN gene inhibits neuroblastoma cell proliferation and in vivo xenograft tumor growth. J. Cancer Res. Clin. Oncol. 139, 933–941. doi: 10.1007/s00432-013-1406-4
Liu, L., Ulbrich, J., Muller, J., Wustefeld, T., Aeberhard, L., Kress, T. R., et al. (2012). Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature 483, 608–612. doi: 10.1038/nature10927
Loven, J., Hoke, H. A., Lin, C. Y., Lau, A., Orlando, D. A., Vakoc, C. R., et al. (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. doi: 10.1016/j.cell.2013.03.036
Macarulla, T., Cervantes, A., Elez, E., Rodriguez-Braun, E., Baselga, J., Rosello, S., et al. (2010). Phase I study of the selective Aurora A kinase inhibitor MLN8054 in patients with advanced solid tumors: safety, pharmacokinetics, and pharmacodynamics. Mol. Cancer Ther. 9, 2844–2852. doi: 10.1158/1535-7163.MCT-10-0299
Massó-Vallés, D., Jauset, T., and Soucek, L. (2016). Ibrutinib repurposing: from B-cell malignancies to solid tumors. Oncoscience 3, 147–148. doi: 10.18632/oncoscience.310
McKeown, M. R., and Bradner, J. E. (2014). Therapeutic strategies to inhibit MYC. Cold Spring Harb. Perspect. Med. 4:a014266. doi: 10.1101/cshperspect.a014266
Mertz, J. A., Conery, A. R., Bryant, B. M., Sandy, P., Balasubramanian, S., Mele, D. A., et al. (2011). Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U.S.A. 108, 16669–16674. doi: 10.1073/pnas.1108190108
Meyer, N., and Penn, L. Z. (2008). Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990. doi: 10.1038/nrc2231
Mo, H., and Henriksson, M. (2006). Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proc. Natl. Acad. Sci. U.S.A. 103, 6344–6349. doi: 10.1073/pnas.0601418103
Mo, H., Vita, M., Crespin, M., and Henriksson, M. A. (2006). Myc overexpression enhances apoptosis induced by small molecules. Cell Cycle 5, 2191–2194. doi: 10.4161/cc.5.19.3320
Montagne, M., Beaudoin, N., Fortin, D., Lavoie, C. L., Klinck, R., and Lavigne, P. (2012). The max b-HLH-LZ can transduce into cells and inhibit c-Myc transcriptional activities. PLoS ONE 7:e32172. doi: 10.1371/journal.pone.0032172
Moreno, P. M., and Pego, A. P. (2014). Therapeutic antisense oligonucleotides against cancer: hurdling to the clinic. Front. Chem. 2:87. doi: 10.3389/fchem.2014.00087
Murga, M., Campaner, S., Lopez-Contreras, A. J., Toledo, L. I., Soria, R., Montana, M. F., et al. (2011). Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 18, 1331–1335. doi: 10.1038/nsmb.2189
Nair, S. K., and Burley, S. K. (2003). X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 112, 193–205. doi: 10.1016/S0092-8674(02)01284-9
Nanbru, C., Lafon, I., Audigier, S., Gensac, M. C., Vagner, S., Huez, G., et al. (1997). Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J. Biol. Chem. 272, 32061–32066.
Neidle, S. (2016). Quadruplex nucleic acids as novel therapeutic targets. J. Med. Chem. 59, 5987–6011. doi: 10.1021/acs.jmedchem.5b01835
Nilson, K. A., Guo, J., Turek, M. E., Brogie, J. E., Delaney, E., Luse, D. S., et al. (2015). THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol. Cell 59, 576–587. doi: 10.1016/j.molcel.2015.06.032
Ortega, S., Prieto, I., Odajima, J., Martin, A., Dubus, P., Sotillo, R., et al. (2003). Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 35, 25–31. doi: 10.1038/ng1232
Otto, T., Horn, S., Brockmann, M., Eilers, U., Schuttrumpf, L., Popov, N., et al. (2009). Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 15, 67–78. doi: 10.1016/j.ccr.2008.12.005
Ou, T. M., Lu, Y. J., Zhang, C., Huang, Z. S., Wang, X. D., Tan, J. H., et al. (2007). Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 50, 1465–1474. doi: 10.1021/jm0610088
Patel, M. R., and Kratzke, R. A. (2013). Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl. Res. 161, 355–364. doi: 10.1016/j.trsl.2012.12.010
Peter, S., Bultinck, J., Myant, K., Jaenicke, L. A., Walz, S., Muller, J., et al. (2014). Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE1 ubiquitin ligase. EMBO Mol. Med. 6, 1525–1541. doi: 10.15252/emmm.201403927
Philipp, S., Sack, S., Kordish, I., Brachmann, J., Hardt, S., Horn, J., et al. (2012). The appraisal-trial: evaluating RESTEN-MPTM in patients with bare metal stent de novo native coronary artery lesions. J. Clin. Exp. Cardiolog. 3:218. doi: 10.4172/2155-9880.1000218
Pivetta, C., Lucatello, L., Krapcho, A. P., Gatto, B., Palumbo, M., and Sissi, C. (2008). Perylene side chains modulate G-quadruplex conformation in biologically relevant DNA sequences. Bioorg. Med. Chem. 16, 9331–9339. doi: 10.1016/j.bmc.2008.08.068
Polivka, J. Jr., and Janku, F. (2014). Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 142, 164–175. doi: 10.1016/j.pharmthera.2013.12.004
Poortinga, G., Quinn, L. M., and Hannan, R. D. (2015). Targeting RNA polymerase I to treat MYC-driven cancer. Oncogene 34, 403–412. doi: 10.1038/onc.2014.13
Posternak, V., and Cole, M. D. (2016). Strategically targeting MYC in cancer. F1000Res. 5:F1000 Faculty Rev-408. doi: 10.12688/f1000research.7879.1
Prochownik, E. V., Kukowska, J., and Rodgers, C. (1988). c-myc antisense transcripts accelerate differentiation and inhibit G1 progression in murine erythroleukemia cells. Mol. Cell. Biol. 8, 3683–3695. doi: 10.1128/MCB.8.9.3683
Prochownik, E. V., and Vogt, P. K. (2010). Therapeutic targeting of Myc. Genes Cancer 1, 650–659. doi: 10.1177/1947601910377494
Rathert, P., Roth, M., Neumann, T., Muerdter, F., Roe, J. S., Muhar, M., et al. (2015). Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547. doi: 10.1038/nature14898
Richart, L., Carrillo-de Santa Pau, E., Rio-Machin, A., de Andres, M. P., Cigudosa, J. C., Lobo, V. J., et al. (2016a). BPTF is required for c-MYC transcriptional activity and in vivo tumorigenesis. Nat. Commun. 7:10153. doi: 10.1038/ncomms10153
Richart, L., Real, F. X., and Sanchez-Arevalo Lobo, V. J. (2016b). c-MYC partners with BPTF in human cancer. Mol. Cell Oncol. 3:e1152346. doi: 10.1080/23723556.2016.1152346
Roohi, A., and Hojjat-Farsangi, M. (2016). Recent advances in targeting mTOR signaling pathway using small molecule inhibitors. J. Drug Target. doi: 10.1080/1061186X.2016.1236112. [Epub ahead of print].
Savino, M., Annibali, D., Carucci, N., Favuzzi, E., Cole, M. D., Evan, G. I., et al. (2011). The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 6:e22284. doi: 10.1371/journal.pone.0022284
Sears, R. C. (2004). The life cycle of C-myc: from synthesis to degradation. Cell Cycle 3, 1133–1137. doi: 10.4161/cc.3.9.1145
Seo, H. K., Ahn, K. O., Jung, N. R., Shin, J. S., Park, W. S., Lee, K. H., et al. (2014). Antitumor activity of the c-Myc inhibitor KSI-3716 in gemcitabine-resistant bladder cancer. Oncotarget 5, 326–337. doi: 10.18632/oncotarget.1545
Shaat, H., Mostafa, A., Moustafa, M., Gamal-Eldeen, A., Emam, A., El-Hussieny, E., et al. (2016). Modified gold nanoparticles for intracellular delivery of anti-liver cancer siRNA. Int. J. Pharm. 504, 125–133. doi: 10.1016/j.ijpharm.2016.03.051
Shalaby, T., and Grotzer, M. A. (2016). MYC as therapeutic target for embryonal tumors: potential and challenges. Curr. Cancer Drug Targets 16, 2–21. doi: 10.2174/1568009615666150916092745
Shapiro, G. I., Dowlati, A., LoRusso, P. M., Eder, J. P., Anderson, A., Do, K. T., et al. (2015). Abstract A49: Clinically efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma (NMC). Mol. Cancer Ther. 14(12Suppl.2):A49. doi: 10.1158/1535-7163.TARG-15-A49
Shi, X., Mihaylova, V. T., Kuruvilla, L., Chen, F., Viviano, S., Baldassarre, M., et al. (2016). Loss of TRIM33 causes resistance to BET bromodomain inhibitors through MYC- and TGF-beta-dependent mechanisms. Proc. Natl. Acad. Sci. U.S.A. 113, E4558–E4566. doi: 10.1073/pnas.1608319113
Shim, H., Chun, Y. S., Lewis, B. C., and Dang, C. V. (1998). A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl. Acad. Sci. U.S.A. 95, 1511–1516.
Shimamura, T., Chen, Z., Soucheray, M., Carretero, J., Kikuchi, E., Tchaicha, J. H., et al. (2013). Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin. Cancer Res. 19, 6183–6192. doi: 10.1158/1078-0432.CCR-12-3904
Simonsson, T., Pecinka, P., and Kubista, M. (1998). DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 26, 1167–1172.
Sklar, M. D., Thompson, E., Welsh, M. J., Liebert, M., Harney, J., Grossman, H. B., et al. (1991). Depletion of c-myc with specific antisense sequences reverses the transformed phenotype in ras oncogene-transformed NIH 3T3 cells. Mol. Cell. Biol. 11, 3699–3710. doi: 10.1128/MCB.11.7.3699
Smith, M. R. (2015). Ibrutinib in B lymphoid malignancies. Expert Opin. Pharmacother. 16, 1879–1887. doi: 10.1517/14656566.2015.1067302
Sodir, N. M., Swigart, L. B., Karnezis, A. N., Hanahan, D., Evan, G. I., and Soucek, L. (2011). Endogenous Myc maintains the tumor microenvironment. Genes Dev. 25, 907–916. doi: 10.1101/gad.2038411
Soodgupta, D., Pan, D., Cui, G., Senpan, A., Yang, X., Lu, L., et al. (2015). Small molecule MYC inhibitor conjugated to integrin-targeted nanoparticles extends survival in a mouse model of disseminated multiple myeloma. Mol. Cancer Ther. 14, 1286–1294. doi: 10.1158/1535-7163.MCT-14-0774-T
Soucek, L., Buggy, J. J., Kortlever, R., Adimoolam, S., Monclus, H. A., Allende, M. T., et al. (2011). Modeling pharmacological inhibition of mast cell degranulation as a therapy for insulinoma. Neoplasia 13, 1093–1100. doi: 10.1593/neo.11980
Soucek, L., and Evan, G. (2002). Myc-Is this the oncogene from Hell? Cancer Cell 1, 406–408. doi: 10.1016/S1535-6108(02)00077-6
Soucek, L., Helmer-Citterich, M., Sacco, A., Jucker, R., Cesareni, G., and Nasi, S. (1998). Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 17, 2463–2472. doi: 10.1038/sj.onc.1202199
Soucek, L., Jucker, R., Panacchia, L., Ricordy, R., Tato, F., and Nasi, S. (2002). Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 62, 3507–3510.
Soucek, L., Lawlor, E. R., Soto, D., Shchors, K., Swigart, L. B., and Evan, G. I. (2007). Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 13, 1211–1218. doi: 10.1038/nm1649
Soucek, L., Nasi, S., and Evan, G. I. (2004). Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ. 11, 1038–1045. doi: 10.1038/sj.cdd.4401443
Soucek, L., Whitfield, J., Martins, C. P., Finch, A. J., Murphy, D. J., Sodir, N. M., et al. (2008). Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. doi: 10.1038/nature07260
Soucek, L., Whitfield, J. R., Sodir, N. M., Massó-Vallés, D., Serrano, E., Karnezis, A. N., et al. (2013). Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 27, 504–513. doi: 10.1101/gad.205542.112
Stellas, D., Szabolcs, M., Koul, S., Li, Z., Polyzos, A., Anagnostopoulos, C., et al. (2014). Therapeutic effects of an anti-Myc drug on mouse pancreatic cancer. J. Natl. Cancer Inst. 106:dju320. doi: 10.1093/jnci/dju320
Sun, K., Atoyan, R., Borek, M. A., DellaRocca, S., Samson, M. E., Ma, A. W., et al. (2016). Dual HDAC and PI3K inhibitor CUDC-907 downregulates MYC and suppresses growth of MYC-dependent cancers. Mol. Cancer Ther. 16, 285–299. doi: 10.1158/1535-7163.MCT-16-0390
Sun, X. X., Sears, R. C., and Dai, M. S. (2015). Deubiquitinating c-Myc: USP36 steps up in the nucleolus. Cell Cycle 14, 3786–3793. doi: 10.1080/15384101.2015.1093713
Thomas, L. R., Foshage, A. M., Weissmiller, A. M., and Tansey, W. P. (2015). The MYC-WDR5 nexus and cancer. Cancer Res. 75, 4012–4015. doi: 10.1158/0008-5472.CAN-15-1216
Tolcher, A. W., Papadopoulos, K. P., Patnaik, A., Rasco, D. W., Martinez, D., Wood, D. L., et al. (2015). Safety and activity of DCR-MYC, a first-in-class Dicer-substrate small interfering RNA (DsiRNA) targeting MYC, in a phase I study in patients with advanced solid tumors. J. Clin. Oncol. 33(Suppl; abstr 11006).
Toyoshima, M., Howie, H. L., Imakura, M., Walsh, R. M., Annis, J. E., Chang, A. N., et al. (2012). Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. U.S.A. 109, 9545–9550. doi: 10.1073/pnas.1121119109
Wang, H., Chauhan, J., Hu, A., Pendleton, K., Yap, J. L., Sabato, P. E., et al. (2013). Disruption of Myc-Max heterodimerization with improved cell-penetrating analogs of the small molecule 10074-G5. Oncotarget 4, 936–947. doi: 10.18632/oncotarget.1108
Wang, H., Hammoudeh, D. I., Follis, A. V., Reese, B. E., Lazo, J. S., Metallo, S. J., et al. (2007). Improved low molecular weight Myc-Max inhibitors. Mol. Cancer Ther. 6, 2399–2408. doi: 10.1158/1535-7163.MCT-07-0005
Wang, H., Mannava, S., Grachtchouk, V., Zhuang, D., Soengas, M. S., Gudkov, A. V., et al. (2008). c-Myc depletion inhibits proliferation of human tumor cells at various stages of the cell cycle. Oncogene 27, 1905–1915. doi: 10.1038/sj.onc.1210823
Wang, H., Ramakrishnan, A., Fletcher, S., and Prochownik, E. V. (2015a). A quantitative, surface plasmon resonance-based approach to evaluating DNA binding by the c-Myc oncoprotein and its disruption by small molecule inhibitors. J. Biol. Methods 2:e18. doi: 10.14440/jbm.2015.54
Wang, H., Teriete, P., Hu, A., Raveendra-Panickar, D., Pendelton, K., Lazo, J. S., et al. (2015b). Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 6, 32380–32395. doi: 10.18632/oncotarget.6116
Wang, Y., Engels, I. H., Knee, D. A., Nasoff, M., Deveraux, Q. L., and Quon, K. C. (2004). Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell 5, 501–512. doi: 10.1016/S1535-6108(04)00113-8
Wang, Y., Zhang, T., Kwiatkowski, N., Abraham, B. J., Lee, T. I., Xie, S., et al. (2015c). CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163, 174–186. doi: 10.1016/j.cell.2015.08.063
Webb, M. S., Tortora, N., Cremese, M., Kozlowska, H., Blaquiere, M., Devine, D. V., et al. (2004). Toxicity and toxicokinetics of a phosphorothioate oligonucleotide against the c-myc oncogene in cynomolgus monkeys. Antisense Nucleic Acid Drug Dev. 11, 155–163. doi: 10.1089/108729001300338681
Westermarck, J., and Hahn, W. C. (2008). Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 14, 152–160. doi: 10.1016/j.molmed.2008.02.001
Whitfield, J. R., and Soucek, L. (2012). Tumor microenvironment: becoming sick of Myc. Cell. Mol. Life Sci. 69, 931–934. doi: 10.1007/s00018-011-0860-x
Wiegering, A., Uthe, F. W., Jamieson, T., Ruoss, Y., Huttenrauch, M., Kuspert, M., et al. (2015). Targeting translation initiation bypasses signaling crosstalk mechanisms that maintain high MYC levels in colorectal cancer. Cancer Discov. 5, 768–781. doi: 10.1158/2159-8290.CD-14-1040
Wu, P., Ma, D. L., Leung, C. H., Yan, S. C., Zhu, N., Abagyan, R., et al. (2009). Stabilization of G-quadruplex DNA with platinum(II) Schiff base complexes: luminescent probe and down-regulation of c-myc oncogene expression. Chemistry 15, 13008–13021. doi: 10.1002/chem.200901943
Xu, Y., Shi, J., Yamamoto, N., Moss, J. A., Vogt, P. K., and Janda, K. D. (2006). A credit-card library approach for disrupting protein-protein interactions. Bioorg. Med. Chem. 14, 2660–2673. doi: 10.1016/j.bmc.2005.11.052
Yang, D., Liu, H., Goga, A., Kim, S., Yuneva, M., and Bishop, J. M. (2010). Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc. Natl. Acad. Sci. U.S.A. 107, 13836–13841. doi: 10.1073/pnas.1008366107
Yao, W., Yue, P., Khuri, F. R., and Sun, S. Y. (2015). The BET bromodomain inhibitor, JQ1, facilitates c-FLIP degradation and enhances TRAIL-induced apoptosis independent of BRD4 and c-Myc inhibition. Oncotarget 6, 34669–34679. doi: 10.18632/oncotarget.5785
Yap, J. L., Wang, H., Hu, A., Chauhan, J., Jung, K. Y., Gharavi, R. B., et al. (2013). Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorg. Med. Chem. Lett. 23, 370–374. doi: 10.1016/j.bmcl.2012.10.013
Yin, X., Giap, C., Lazo, J. S., and Prochownik, E. V. (2003). Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 22, 6151–6159. doi: 10.1038/sj.onc.1206641
Yu, C., Niu, X., Jin, F., Liu, Z., Jin, C., and Lai, L. (2016). Structure-based inhibitor design for the intrinsically disordered protein c-myc. Sci. Rep. 6:22298. doi: 10.1038/srep22298
Yuneva, M., Zamboni, N., Oefner, P., Sachidanandam, R., and Lazebnik, Y. (2007). Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 178, 93–105. doi: 10.1083/jcb.200703099
Zhang, Y., Peng, L., Mumper, R. J., and Huang, L. (2013). Combinational delivery of c-myc siRNA and nucleoside analogs in a single, synthetic nanocarrier for targeted cancer therapy. Biomaterials 34, 8459–8468. doi: 10.1016/j.biomaterials.2013.07.050
Keywords: Myc, oncogene, inhibitor, therapy, Omomyc, clinical application
Citation: Whitfield JR, Beaulieu ME and Soucek L (2017) Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 5:10. doi: 10.3389/fcell.2017.00010
Received: 16 December 2016; Accepted: 03 February 2017;
Published: 23 February 2017.
Edited by:
Ignacio Moreno De Alborán, Spanish National Research Council (CSIC), SpainReviewed by:
Francisco X. Real, Spanish National Cancer Research Centre, SpainCopyright © 2017 Whitfield, Beaulieu and Soucek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura Soucek, bHNvdWNla0B2aGlvLm5ldA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.