
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 27 December 2016
Sec. Cell Adhesion and Migration
Volume 4 - 2016 | https://doi.org/10.3389/fcell.2016.00149
This article is part of the Research Topic Cell adhesion and migration in the development of multicellular organisms View all 11 articles
 Yoshiyuki Matsubara1†Mikiharu Nakano2†Kazuki Kawamura1†Masaoki Tsudzuki3Jun-Ichi Funahashi4Kiyokazu Agata5Yoichi Matsuda2,6Atsushi Kuroiwa1
Yoshiyuki Matsubara1†Mikiharu Nakano2†Kazuki Kawamura1†Masaoki Tsudzuki3Jun-Ichi Funahashi4Kiyokazu Agata5Yoichi Matsuda2,6Atsushi Kuroiwa1 Takayuki Suzuki1*
Takayuki Suzuki1*Hereditary Multiple Malformation (HMM) is a naturally occurring, autosomal recessive, homozygous lethal mutation found in Japanese quail. Homozygote embryos (hmm−/−) show polydactyly similar to talpid2 and talpid3 mutants. Here we characterize the molecular profile of the hmm−/− limb bud and identify the cellular mechanisms that cause its polydactyly. The hmm−/− limb bud shows a severe lack of sonic hedgehog (SHH) signaling, and the autopod has 4 to 11 unidentifiable digits with syn-, poly-, and brachydactyly. The Zone of Polarizing Activity (ZPA) of the hmm−/− limb bud does not show polarizing activity regardless of the presence of SHH protein, indicating that either the secretion pathway of SHH is defective or the SHH protein is dysfunctional. Furthermore, mesenchymal cells in the hmm−/− limb bud do not respond to ZPA transplanted from the normal limb bud, suggesting that signal transduction downstream of SHH is also defective. Since primary cilia are present in the hmm−/− limb bud, the causal gene must be different from talpid2 and talpid3. In the hmm−/− limb bud, a high amount of GLI3A protein is expressed and GLI3 protein is localized to the nucleus. Our results suggest that the regulatory mechanism of GLI3 is disorganized in the hmm−/− limb bud.
Avian mutants have often been used to study developmental mechanisms, especially embryonic pattern formation. Some of the most well studied mutant strains in chickens are the talpids (talpid, talpid2, and talpid3) (Cole, 1942). These three talpid mutants are naturally occurring and were independently discovered. The original talpid mutation has since been lost, but talpid2 and talpid3 are still maintained in the UK and the USA. Intriguingly, these mutants share a unique phenotype characterized by polydactyly, craniofacial abnormality, autosomal recessive inheritance, and embryonic lethality. The gene responsible for talpid2 was identified as C2CD3 (Chang et al., 2014), whereas the gene responsible for talpid3 is KIAA0586 (Davey et al., 2006). These causal genes are both involved in the formation of primary cilia (Yin et al., 2009; Chang et al., 2014).
The primary cilium is thought to be necessary for intermediate sonic hedgehog (SHH) signaling because it provides a location for the processing of the transcriptional factor GLI3 (Besse et al., 2011). SHH is secreted from the Zone of Polarizing Activity (ZPA), which is located at the posterior edge of the limb bud, and determines the limb's anterior-posterior (AP) axis (Riddle et al., 1993). In the absence of SHH, GLI3 is located in the primary cilium and is phosphorylated by protein kinase A (Wang et al., 2000; Hsu et al., 2011). Phosphorylated GLI3 is ubiquitinated, resulting in partial degradation (Bhatia et al., 2006). This short form of GLI3, called GLI3R, inhibits the transcription of target genes (Wang et al., 2000). In the presence of SHH, GLI3 is maintained in a long activator form called GLI3A (Litingtung et al., 2002). GLI3A induces expression of target genes such as Patched1 (Ptch1).
Interestingly, although both C2CD3 and KIAA0586 proteins are necessary for the ciliogenesis pathway to proceed, the talpid2 and talpid3 mutants indicate they have different impacts on SHH signaling. In the talpid2 limb bud, SHH signaling is constitutively activated by the upregulation of GLI3A, which causes anterior expansion of Ptch1, Bmp4, Fgf4, and Hoxd13 expression (Rodriguez et al., 1996; Caruccio et al., 1999). In contrast, SHH signaling is abolished in the talpid3 limb bud leading to downregulation of Ptch1 and Gli1 expression, but GLI3A is still upregulated (Davey et al., 2006) as in the talpid2 mutant. It is known that in Shh deficient conditions only GLI3R is present, resulting in the formation of only digit 1 in the hindlimb and undetectable expression of Ptch1 and Gli1 (Chiang et al., 2001). The talpid3 mutant is thought to be similar, but it is still unclear why the SHH signaling pathway is defective in the talpid3 mutant despite up-regulation of GLI3A.
The HMM mutant was reported as a similar mutant phenotype to talpid in 1998 (Tsudzuki et al., 1998). It is a naturally occurring, autosomal recessive, homozygous lethal Japanese quail mutant. The gene responsible for hmm is still unknown. Homozygote embryos show polydactyly and shortened lower and upper beaks, which is slightly different from the talpid2 mutant phenotype of an extended lower beak compared to the upper beak (Chang et al., 2014). The HMM mutant also does not display the subcutaneous edema and hemorrhage over the thigh and neck regions found in the talpid2 and talpid3 mutants (Tsudzuki et al., 1998). Based on these observations, the developmental causes of the HMM mutant are likely different from the talpid2 and talpid3 mutants.
Here we characterize the molecular profile of the HMM mutant and perform a functional analysis of the cellular mechanisms that cause the mutant phenotype. Gene expression patterns indicate that SHH signaling is defective in the homozygous HMM mutant (hmm−/−), similar to the talpid3 mutant. However, the limb bud in the hmm−/− embryo still has anterior-posterior polarity with restricted anterior marker gene expression. This is different from the limb bud patterning in talpid2 and talpid3. Furthermore, we found that the ZPA in the hmm−/− limb bud does not show polarizing activity regardless of the presence of SHH protein expression. The primary cilium was present however, and we observed a high amount of GLI3A protein in the hmm−/− limb bud. These results indicate that different molecular pathways than talpid2 and talpid3 are defective in the hmm−/− limb bud.
The fertilized HMM mutant quail eggs were provided by Avian Bioscience Research center (ABRC) at Nagoya University. Embryos were staged according to Ainsworth et al. (2010). The HMM mutant shows autosomal recessive inheritance, although the causal gene, hmm, is still unknown. Heterozygous embryos showed no phenotype and were indistinguishable from wild-type embryos. Therefore we used a mixture of wild-type and heterozygous embryos as a control. Experimental procedures for isolating embryos were performed in accordance with guidelines set forth by the Regulations on Animal Experiments at Nagoya University. The embryo research was approved by Nagoya University Animal Experiment Committee (approval number 17).
Limb buds were fixed with 4% PFA overnight and then embedded in 1% low-melting agarose (Lonza). Excess agarose around the limb was removed with a razor blade. After that, agarose containing the limb buds was attached to the swivel base with Loctite for optical projection tomography (OPT) scanning (Henkel). Limb buds were treated with 100% MeOH for 3 h, and samples cleared in a 1:2 solution of benzyl alcohol (Wako): benzyl benzoate (Wako) overnight. A 3D image was taken with the OPT scanner 3001 (Bioptonics) and visualized by Avizo software (Maxnet).
Victoria blue staining was performed as described previously (Suzuki et al., 2008). Embryos were dissected in PBS and fixed in 10% Formalin overnight at room temperature. Embryos were stained overnight with 1% Victoria blue (Sigma) solution containing 1% HCl, and 70% EtOH. Embryos were washed overnight with 1% HCl in 70% EtOH solution following overnight treatment with 100% methylsalicylate to render them transparent.
In situ hybridization was performed as described previously (Suzuki et al., 2008). The following probes were used for in situ hybridizations: Hoxa13 (Yokouchi et al., 1991), Hoxd13 (Nelson et al., 1996), Fgf8, Gli3, Alx4, Lhx9, Ptch2, and Bmp2 (kindly gifted by Dr. John. F. Fallon, University of Wisconsin-Madison), Hand2 (kindly gifted by Dr. Kazuko Koshiba-Takeuchi, University of Tokyo), Shh, Gli1, and Ptch1 (kindly gifted by Dr. Yuki Sato, Kyushu university), Pax6 (kindly gifted by Dr. Yoshio Wakamatsu, Tohoku university), Pax3, Pax7, and Dbx2 (kindly gifted by Dr. Harukazu Nakamura, Tohoku university), Islet1 (589–1551 bp, GenBank NM_205414), and MyoD (155–1051 bp, GenBank NM_204214).
The ZPA was isolated from the posterior side of the donor limb bud with a sharpened tungsten needle. Isolated ZPA was placed in ice-cold Tyrode solution (137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 0.2 mM Na2HPO4, 12 mM NaHCO3, 5.5 mM D-glucose) and divided into several pieces. A piece of the ZPA was stained by squirting it with DiI solution (1% DiI dissolved with 70% EtOH) in Tyrode solution, and it was implanted at the anterior side of the host limb bud with a tungsten needle.
Primary embryonic fibroblasts were isolated from the back region between the forelimb and the hindlimb at St. 35. Back tissues were dissected in PBS. After tissues were minced with a razor blade, they were incubated in 0.25% trypsin-EDTA solution (Wako) for 15 min at 37°C. An equal volume of 100% fetal calf serum was added. The supernatant of the cell suspension was plated on 3.5-cm glass-base tissue culture dish (IWAKI). The next day, cells were fixed with 4% PFA for 10 min at room temperature.
Limb buds were dissected in ice-cold PBS and fixed with 4% PFA for 15 min on ice. The PFA solution was immediately removed and fresh ice-cold PBS was added. The limb buds were treated with 30% sucrose in PBS overnight at 4°C and then embedded in compound for frozen sections (Leica). Samples were then sectioned by cryostat for immunohistochemistry.
Cells in frozen sections or fixed primary fibroblasts were permeabilized by treating with 0.2% TritonX-100 (Wako) for 20 min at room temperature, and then blocked with 3% BSA in PBS for 30 min. Anti-SHH antibody 5E1 (1:100) (DSHB), Anti-GLI3 antibody (1:100) (Santa Cruz sc-20688), and anti-acetylated Tubulin clone 6-11B-1 (1:1000) (Sigma) were diluted in 3% BSA/PBS solution. Samples were incubated overnight at 4°C with the primary antibody. The next day, samples were incubated with the secondary antibody (Alexa Fluor 488) (Thermo Fisher Scientific) diluted at 1:500 in 3% BSA/PBS solution for 2 h at room temperature. After 1 μg/ml of DAPI in PBS was added to the samples for 15 min, the samples were mounted with fluorescein mounting medium (Dako) and fluorescent images were taken with an Olympus FV1000 confocal microscope.
Fertilized eggs were incubated for 3.5 days. The limb buds of St. 23 embryos were isolated in ice-cold PBS with a tungsten needle and then collected in 1.5 mL collection tubes. After solubilizing the cells with 200 μl of lysis buffer (50 mM HEPES (pH 7.4), 150 mM NaCl, 10 mM EDTA, 1% Triton X-100, 200 mM sodium fluoride, 10% glycerol, 20 mM sodium pyrophosphate, 2 mM phenylmethane sulfonyl fluoride, 4 mM Na3VO4, 0.1 mg/ml leupeptin, and 15 mM benzamidine), the cell lysate was centrifuged at 14,000 g for 10 min. The supernatant was assayed for protein content using a Bio-Rad protein assay kit. The proteins were then resolved with SDS-polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene difluoride membranes. After blocking with 5% (w/v) non-fat dry milk in Tris-buffered saline-Tween buffer (20 mM Tris (pH 7.6), 0.14 M NaCl, and 0.1% (w/v) Tween 20), the membranes were treated with primary antibodies, Anti-GLI3 antibody (1:1000) (Santa Cruz sc-20688), and Anti-αTubulin clone B-5-1-2 (1:2500) (Sigma). The proteins were visualized using an ECL Western blotting detection system.
We previously reported that the hmm−/− embryo shows syndactylous polydactyly (Tsudzuki et al., 1998). It is still unclear how these phenotypes are induced in hmm−/− limb bud. In both talpid2 and talpid3 mutants, the limb bud is wider in size along the AP axis before cartilage condensation starts (Francis-West et al., 1995; Caruccio et al., 1999). This wider limb bud leads to expansion of the autopod, resulting in polydactyly (Litingtung et al., 2002). Therefore, we first examined the shape of the limb bud in the hmm−/− embryo. To compare the shape of the entire limb bud between the mutant and wild-type, we took OPT images (Sharpe et al., 2002) of the hind limb at St. 23, after the limb bud emerged from the body wall (Figures 1A,B). The hmm−/− limb bud was slightly wider than the hmm+/+;+/− (simplified hereafter as hmm+/−) limb bud along the AP axis (Figure 1A). The hmm−/− limb bud was also expanded along the dorso-ventral (DV) axis compared to the hmm+/− limb bud (Figure 1B). To elucidate the mechanisms underlying the expansion of the limb bud along the AP axis, we compared the limb field's AP width between hmm+/− and hmm−/− embryos. We found that the anterior boundary of the hmm−/− forelimb bud (FLB) was expanded cranially compared to that of the hmm+/− FLB (Figures 1C,E). The AP width of the hindlimb bud (HLB) appeared to be the same in both hmm+/− and hmm−/− embryos (Figures 1D,F), implying that a different mechanism is involved in the expansion of the hindlimb along the AP axis. In the talpid2 mutant, the expansion of Fgf8 expression at the apical ectodermal ridge (AER) along the AP axis is observed along with formation of a wider limb bud (Caruccio et al., 1999). Therefore, we checked the expression of Fgf8 at St. 23 and St. 26 in the hmm−/− limb bud, and found that it was expanded into both the anterior and posterior ends close to the body wall (Figures 1G–J). Expansion of Fgf8 expression was continued into St. 26, along with wider autopod formation in the hmm−/− embryo (Figures 1K–N). Taken together, these results suggest that mesenchymal cells of the hmm−/− limb bud propagate more than in the wild-type and give rise to a wider limb bud with an extended AER along the AP axis. In addition, the anterior boundary of the forelimb field is expanded in the hmm−/− embryo when the FLB is initiated.
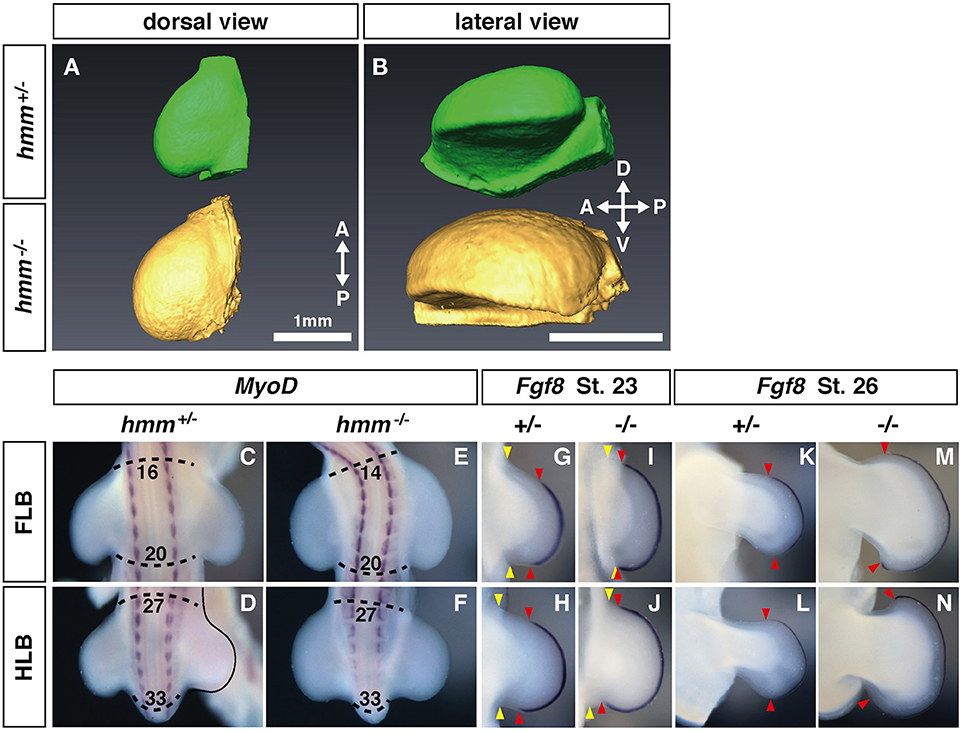
Figure 1. Expression of Fgf8 at the apical ectodermal ridge (AER) is expanded along the anterior-posterior (AP) axis in the hmm−/− limb bud. (A,B) Morphology of the limb at St. 22 as scanned by OPT. Dorsal view (A) and lateral view (B) of the hmm+/− (green) and hmm−/− (yellow) hindlimb buds. The scale bar indicates 1 mm. (C–F) In situ hybridization of MyoD. The dotted line shows the anterior and posterior end of the limb buds. Numbers indicate the position of the somite starting with the first somite. (G–N) In situ hybridization of Fgf8. Yellow arrowheads indicates the boundary between the limb bud and the body wall. Red arrowheads indicate the anterior and posterior boundaries of Fgf8 expression at the AER. A, anterior; P, posterior; D, dorsal; V, ventral; FLB, forelimb bud; HLB, hindlimb bud.
In the autopod of the hmm−/− embryo many of the digits are shortened during development (Tsudzuki et al., 1998). We therefore analyzed detailed patterns of polydactylous digits along the AP axis. In the hmm+/− HLB, digital rays 1, 2, 3, and 4 are formed correctly to their unique full lengths along the AP axis (Figure 2A). In contrast, we observed indistinguishable shortened digital rays in the hmm−/− HLB. We also found webbing along the AP axis of the digital ray sequence in the hmm−/− HLB. These results imply that anterior-posterior polarity is disrupted in the hmm−/− limb bud.
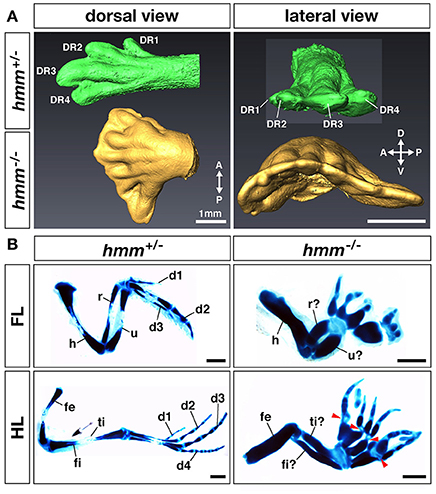
Figure 2. Anterior-posterior polarity is disrupted in the hmm−/− limb bud. (A) Morphology of the limb at St. 34 as scanned by OPT. Dorsal view and lateral view of the hmm+/− (green) and hmm−/− (yellow) hindlimb buds are shown. The scale bar indicates 1 mm. (B) The skeletal pattern of the limb at St. 36. All images are oriented with the anterior side up and the posterior side down. Red arrowheads indicate immature metatarsal-phalangeal joints. The scale bar indicates 1 mm. A, anterior; P, posterior; D, dorsal; V, ventral; DR, digital ray; FL, forelimb; HL, hindlimb; d1-d4, digit 1-digit 4; h, humerus; r, radius; u, ulna; fe, femur; ti, tibia; fi, fibula.
In order to identify the specific region where anterior-posterior polarity is disrupted, we performed victoria blue staining to visualize condensing cartilage at St. 35 (Figure 2B). In the forelimb, the hmm−/− embryo has four to eight digits with syn-, poly-, and brachdactyly, whereas the hmm+/− embryo has three digits (Table 1). The forelimb of hmm−/− has one shortened, thick humerus in the stylopod and an unidentifiable ulna/radius in the zeugopod. The hindlimb of the hmm−/− embryo also has one shortened, thick femur in the stylopod and an unidentifiable fibula/tibia in the zeugopod. The autopod of the hmm−/− hindlimb has seven to eleven digits (Table 1). Based on the morphological criteria of digit identity (number, size, and shape of the phalanges Suzuki, 2013), we assumed that the hmm−/− autopod has lost digit identity. We also found that the metacarpal/metatarsal bones were fused and the metacarpal/metatarsal-phalangeal joints were missing in the hmm−/− autopod (Figure 2B arrowhead). We observed the phalangeal joint in both the forelimb and the hindlimb, but its formation was incomplete. These results indicate that anterior-posterior polarity of both the forelimb and the hindlimb is disrupted in the hmm−/− embryo.
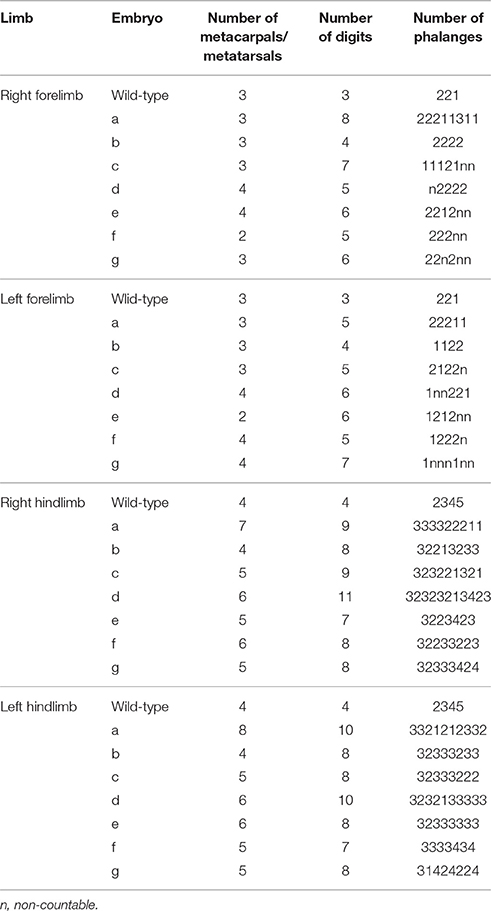
Table 1. Skeletal pattern of the autopod in HMM mutant.
To understand the molecular mechanisms of anterior-posterior pattering deficiency in hmm−/− limbs, we next examined gene expression patterns of marker genes specifically expressed at the anterior/posterior sides. Genetic antagonism between Hand2 and Gli3 is necessary to establish AP polarity at the early limb bud stage (te Welscher et al., 2002a). We found that expression of Hand2 was restricted at the posterior side in the hmm+/− limb bud. In contrast, in the hmm−/− limb bud we observed strong expression of Hand2 at the posterior side and weak expression at the anterior side (Figures 3A–D). In the hmm+/− limb bud, Gli3 was not expressed at the posterior side where Hand2 was expressed (Figures 3E,F). In contrast, Gli3 was expressed throughout the limb bud at the early stages in the hmm−/− limb bud (Figures 3G,H). Alx4 was expressed at the anterior mesoderm at St. 23 in the hmm+/− limb bud (Figures 3I,J), but its expression was downregulated though still detectable in the hmm−/− limb bud (Figures 3K,L). At St. 25, Alx4 was continuously expressed at the anterior side of the stylopod and zeugopod in the hmm+/− limb bud (Figures 3I′,J′). In the hmm−/− limb bud, its expression was still observed in the FLB (Figure 3K′) but not detectable in the HLB (Figure 3L′). Lhx9 was expressed at the anterior side at St. 22 (Figures 3M,N) and at the anterior autopod at St. 25 (Figures 3M′,N′). In contrast to the expression of Alx4, expression of Lhx9 was expanded to the posterior side in the hmm−/− limb bud (Figures 3O–P′). In particular, Lhx9 expression was observed at the posterior end of the hmm−/− hindlimb autopod (Figure 3P′). Expression of Hoxd13 was restricted to the posterior side in the hmm+/− limb bud at St. 22 (Figures 3Q,R), and at St. 25 its expression was observed at the posterior mesoderm in the autopod (Figures 3Q′,R′). Expression of Hoxd13 was similarly restricted to the posterior side in the hmm−/− limb bud (Figures 3S–T′). We checked the autopod area for Hoxa13 expression in the contra-lateral side (Figures 3U–X), and we saw that the autopod expanded along the AP axis in the hmm−/− limb bud at St. 25 (Figures 3W,X). However, expression of Hoxd13 was still restricted to the posterior side (Figures 3S′,T′), the same as in the hmm+/− autopod (Figures 3Q′,R′). At St. 29 Hoxd13 was expressed throughout the hmm+/− autopod except on the anterior side of digit 1 (Figures 3Y,Z). However, it was expressed all the way to the anterior end of the hmm−/− autopod (Figures 3a,b) along with Hoxa13 (Figures 3c–f). From these results, we conclude that the hmm−/− limb bud partially maintains AP polarity at the limb bud stage (St. 20–25), but loses AP polarity at the late autopod stage (St. 29).
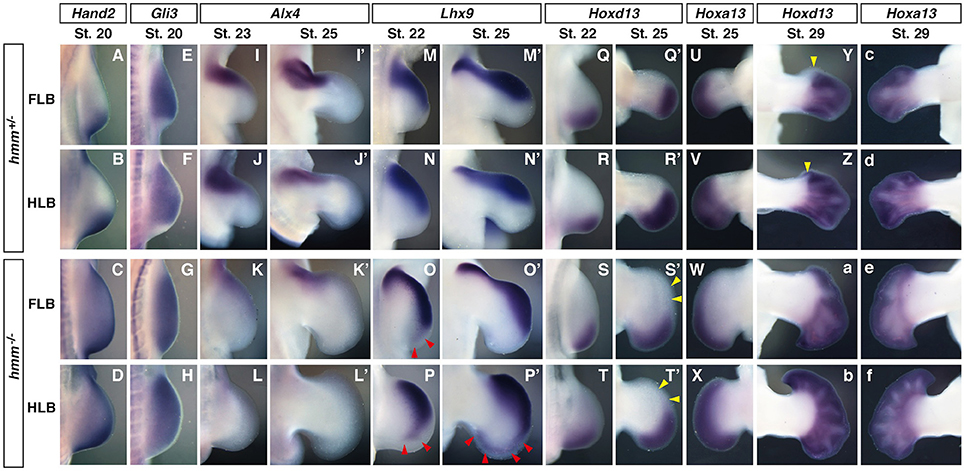
Figure 3. Anterior-posterior polarity is partially maintained at the limb bud stage in the hmm−/− embryo. In situ hybridization of Hand2 (A–D), Gli3 (E–H), Alx4 (I–L′), Lhx9 (M–P′), Hoxd13 (Q–T′,Y–b) and Hoxa13 (U–X,c–f). All images are oriented with the anterior side up and the posterior side down. (O,P,P′) Red arrowheads indicate the expanded expression domain of Lhx9 on the posterior side. (S′,T′) Yellow arrowheads indicate the region where Hoxd13 is not expressed in the autopod. (Y,Z) Yellow arrowheads indicate the region where Hoxd13 is not expressed on the anterior side of the digit 1 primordium. FLB, forelimb bud; HLB, hindlimb bud.
As described above, AP polarity in the limb bud is disrupted in the hmm−/− embryo. It is known that expression of hand2, Gli3, and Alx4 is altered by SHH signaling, which establishes anterior-posterior polarity in the limb (Takahashi et al., 1998; te Welscher et al., 2002a). We therefore examined the expression patterns of target genes downstream of SHH in the limb bud. Expression of Shh was restricted to the posterior edge of the hmm+/− limb bud (Figures 4A,B). In the hmm−/− limb bud, Shh was expressed at the posterior mesoderm; however, expression was restricted to a more proximal region of the FLB than in the hmm+/− limb bud (Figure 4C). On the other hand, the expression domain of Shh was expanded proximally in the HLB (Figure 4D).
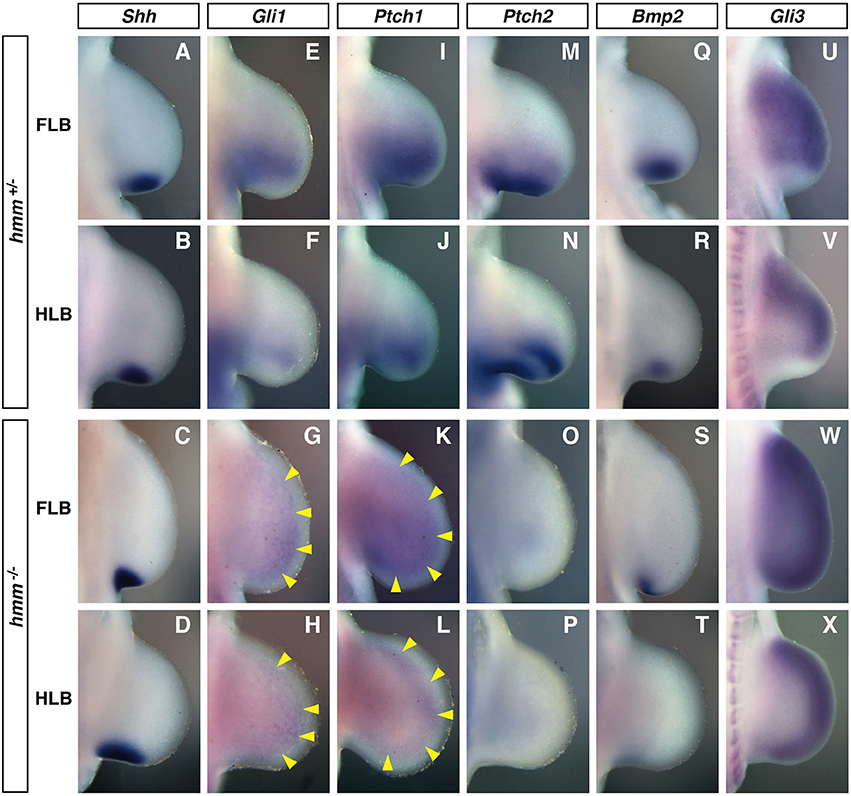
Figure 4. SHH signaling is reduced in the hmm−/− limb bud. In situ hybridization of Shh (A–D), Gli1 (E–H), Ptch1 (I–L), Ptch2 (M–P), Bmp2 (Q–T), and Gli3 (U–X) at St. 22/23. All images are oriented with the anterior side up and the posterior side down. (G–L) Yellow arrowheads indicate the weak expression of Gli1 and Ptch1. FLB, forelimb bud; HLB, hindlimb bud.
Gli1, Ptch1, Ptch2, and Bmp2 are known to be downstream target genes of SHH signaling that are expressed in response to SHH signaling (Chiang et al., 2001). In the hmm+/− limb bud, the expression domain of Gli1 was expanded to the middle part of the limb bud along the anterior-posterior axis (Figures 4E,F). In contrast, Gli1 was expressed uniformly at very low levels throughout the hmm−/− limb bud (Figures 4G,H). Similarly, Ptch1 was expressed at the posterior half of the hmm+/− limb bud (Figures 4I, J), but in the hmm−/− limb bud was expressed uniformly at very low levels along the anterior-posterior axis (Figures 4K,L). In the hmm+/− limb bud Ptch2 was expressed at the posterior mesoderm similarly to Ptch1 (Figures 4M,N), but expression was not detected in the hmm−/− limb bud (Figures 4O,P). In the hmm−/− limb bud, expression of Bmp2 was observed at the posterior edge in the FLB only, but in hmm+/− limb buds expression was observed at the posterior mesoderm in both the FLB and the HLB (Figures 4Q–T). The expression of Gli1, Ptch1, Ptch2, and Bmp2 is reduced or lost in the limb bud of the Shh−/− mouse embryo (Litingtung et al., 2002), therefore SHH signaling must be substantially reduced despite the detection of Shh expression in the hmm−/− limb bud. Reduction of SHH signaling was also observed in the neural tube of the hmm−/− embryo (Figure S1A).
We next examined limb bud expression of Gli3, a transcriptional factor that mediates SHH signaling. In the hmm+/− limb bud Gli3 was expressed in the mesenchyme in a complementary pattern to Shh expression (Figures 4A,B,U,V). In contrast, in the hmm−/− limb bud Gli3 was strongly expressed throughout the limb bud, including in the Shh-expressing region (Figures 4C,D,W,X). We therefore concluded that the syndactylous polydactyly phenotype in the hmm−/− limb bud does not result simply from a loss of SHH signaling due to a defect in Gli3 expression.
In hmm−/− embryos, cells in the limb bud showed a reduction in SHH signaling despite expression of Shh (Figure 4). We therefore hypothesized that cells in the hmm−/− embryo might have lost SHH protein activity or the ability to respond to SHH protein. To test these hypotheses, we analyzed the polarizing activity of the hmm−/− limb bud. When we grafted ZPA from the hmm+/− FLB to the anterior side of the hmm+/− FLB at St. 20, strong ectopic Ptch2 expression was observed (83%, n = 6) (Figure 5A). In contrast, implantation of ZPA from the hmm+/− FLB to the hmm−/− FLB did not induce ectopic Ptch2 expression (0%, n = 4) (Figures 5B,C). We next grafted ZPA from the hmm−/− FLB to the hmm+/− FLB. Unexpectedly, ectopic Ptch2 expression was not induced (0%, n = 8) (Figures 5D,E). These results show that mesenchymal cells in the hmm−/− limb bud do not respond to SHH protein. Furthermore, despite the presence of Shh expression (Figures 4C,D), ZPA derived from the hmm−/− limb bud cannot induce expression of SHH signaling downstream targets.
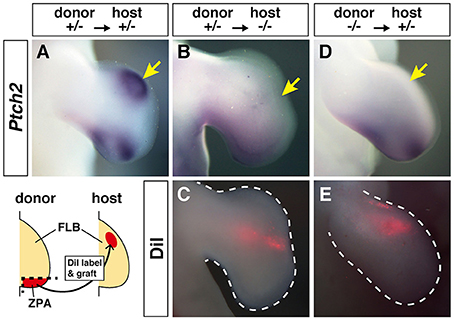
Figure 5. Zone of Polarizing Activity (ZPA) derived from the hmm−/− limb bud does not have polarizing activity. (A,B,D) In situ hybridization of Ptch2 after ZPA was implanted into the anterior limb bud. (C,E) Fluorescence of the implanted ZPA labeled with DiI is shown. The schematic drawing shows the grafting protocol of ZPA from the donor limb bud (hmm+/− or hmm−/−) to the host limb bud (hmm+/− or hmm−/−). The yellow arrows indicate the position of the implanted ZPA. FLB, forelimb bud.
We next examined the presence of SHH protein using immunohistochemistry. We found that the protein was detectable by anti-SHH antibody in the limb bud and the notochord in both hmm+/− (Figures 6A,B, Figure S1B) and hmm−/− (Figures 6C,D, Figure S1B) embryos. These observations raise the following possibilities in the hmm−/− embryo: first, the secretion pathway of the SHH protein is defective; second, the SHH protein is dysfunctional; and third, the SHH signaling pathway in the target tissues is disrupted.
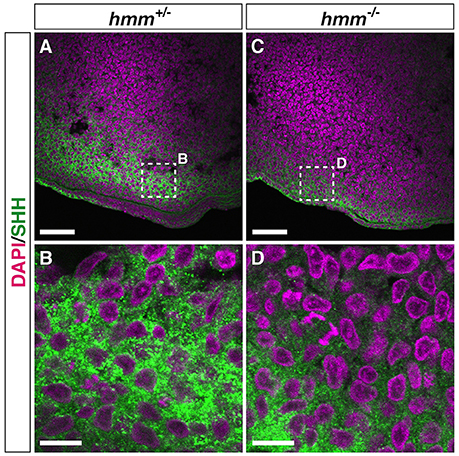
Figure 6. SHH protein is detectable by immunohistochemistry in the hmm−/− limb bud. Immunohistochemistry of SHH protein at the posterior limb bud of the St. 23 embryo is shown with fluorescent green. The nucleus is stained with DAPI. Frontal sections are oriented with the anterior side up and the posterior side down. (B,D) Higher magnification of the area enclosed by the dotted square in A and C. Scale bars, 50 μm (A,C), 10 μm (B,D).
It was recently reported that loss of primary cilia induces constitutive activation of SHH signaling through high expression of GLI3A in both talpid2 and talpid3 mutants (Davey et al., 2006; Chang et al., 2014). Therefore, we visualized the primary cilia using immunohistochemistry targeting acetylated tubulin. However, we found that primary cilia were still present in both the hmm+/− and hmm−/− limb buds (Figure 7), suggesting that loss of primary cilia is not a cause of the hmm−/− phenotype. We next performed western blotting for the GLI3 protein (Figure 8A), and found that the active form of GLI3, GLI3A, was highly expressed in both the anterior and posterior halves of the hmm−/− limb bud compared to the hmm+/− limb bud. In particular, high expression of GLI3A was observed on the anterior side of the hmm−/− limb bud where Shh is not expressed. Expression of GLI3R was not detected in our experiments. This result raises the possibility that GLI3A protein does not function normally in the hmm−/− limb bud.
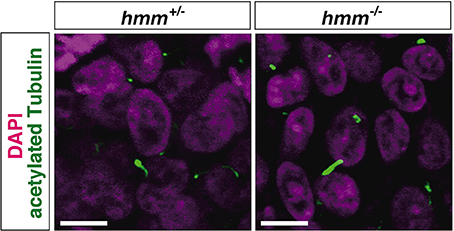
Figure 7. The primary cilium is present in hmm−/− limb bud. Immunohistochemistry of acetylated tubulin in the limb bud cells at St. 23 is shown with fluorescent green. The nucleus is stained with DAPI. Scale bars, 10 μm.
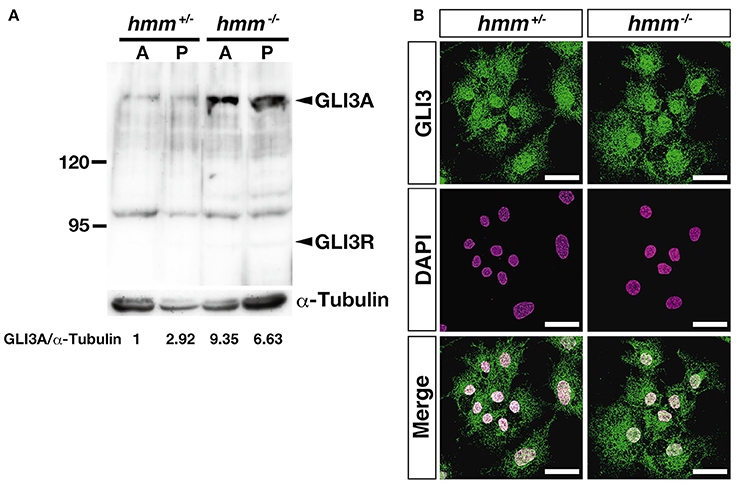
Figure 8. High amounts of GLI3A protein are expressed in the hmm−/− limb bud. (A) Western blotting of GLI3 protein and α-tubulin in the limb is shown. A and P indicate samples extracted from anterior and posterior limb buds. Relative ratio of GLI3A/α-Tubulin is shown at the bottom. (B) Immunohistochemistry of GLI3 protein in primary fibroblasts is shown with fluorescent green. The nucleus is stained with DAPI. Scale bars, 30 μm.
Finally, we examined whether transport of the GLI3 protein from the cytoplasm to the nucleus is disrupted in the hmm−/− embryo using immunohistochemistry. We observed that GLI3 protein is localized to the nucleus in both hmm+/− and hmm−/− primary fibroblast cells (Figure 8B). Taken together, our results imply that despite the presence of GLI3 protein in the nucleus, SHH signaling is abolished in the hmm−/− embryo due to a loss of function of the GLI3 protein.
In this study, we examined the developmental properties of the hmm−/− limb bud. We found that the abnormalities of the hmm−/− limb bud develop through a different mechanism than those of the talpid2 and the talpid3 limb buds (Table 2). The hmm−/− embryo showed disruption of SHH signaling in both the limb bud and the neural tube as in talpid3 (Davey et al., 2006), whereas in the talpid2 limb bud constitutive activation of SHH signaling is observed (Caruccio et al., 1999). While the hmm−/− limb bud shows a similar phenotype to the talpid3 limb bud in terms of the SHH signaling pathway in the cells, several phenotypes are different between them. Expression of ectopic Hoxd13 was observed uniformly from the posterior to anterior mesenchyme in the talpid3 limb bud (Francis-West et al., 1995), whereas its expression was restricted posteriorly in the hmm−/− limb bud as it is in the wild-type (Figures 3S,S′,T,T′). This phenotype is unique to the hmm−/− limb bud compared to the talpid2 (Rodriguez et al., 1996) and talpid3 limb buds, indicating that the HMM mutant is a novel type of talpid mutant. It has been reported that the expression level of GLI3R regulates the Hoxd13 expression pattern along the AP axis (te Welscher et al., 2002b). The expression of Hoxd13 in the limb is downregulated in the absence of Shh expression (Ros et al., 2003) because only GLI3R is present. When GLI3R is expressed at half the wild-type level, expression of Hoxd13 can be faintly observed at the posterior side of the limb (te Welscher et al., 2002b). In the Gli3−/− limb bud, expression of Hoxd13 is expanded to the anterior side due to a lack of GLI3R protein, the same as in talpid3 mutants (Davey et al., 2006). Based on these observations, one possible reason for the different expression domains of Hoxd13 between the talpid3 and the hmm−/− limb bud is that a lower amount of GLI3R is expressed in the talpid3 limb bud than the hmm−/− limb bud due to the lack of primary cilia in the taplid3 mutant. This could result in a high expression level of GLI3A (Bangs et al., 2011) and thus expanded expression of Hoxd13 to the anterior side. In contrast, the expression level of GLI3R in the hmm−/− limb bud would be slightly higher than the talpid3 limb bud because the primary cilium is still present. These results imply that the expression level of GLI3R might be dependent on the presence or absence of the primary cilia among talpid family mutants. In our experiments, we could not detect GLI3R expression by western blotting (Figure 8A), suggesting the possibility that the antibody we used does not cross-react with quail GLI3R. Further study is needed to examine the expression level of GLI3R among talpid family mutants using different antibodies in the future.
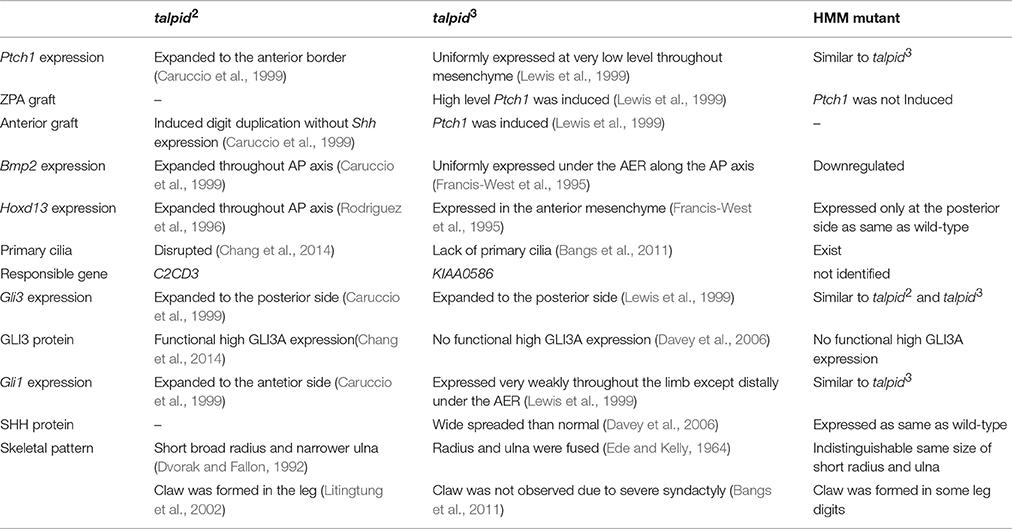
Table 2. Typical phenotypes of three talpid mutants in the limb bud.
We showed that the hmm−/− limb bud partially retained AP polarity at the limb bud stage in the absence of SHH signaling (Figure 3). Previous reports in the mouse embryo suggested that the antagonization of Hand2 by Gli3 specifies anterior-posterior polarity of the limb bud at early stages before Shh expression starts (te Welscher et al., 2002a). After Hand2/Gli3 specifies the anterior-posterior polarity in the limb bud, SHH expression in the ZPA establishes the anterior-posterior axis. We infer that immature AP polarity could be maintained downstream of the Hand2/Gli3 system in the hmm−/− limb bud.
In the hmm−/− limb bud, target genes downstream of SHH signaling (Hoxd13, and Bmp2) were expressed at much lower levels than in the talpid2 and talpid3 limb buds (Figures 3, 4, Table 2). The hmm−/− limb bud showed severe lack of SHH signaling activity despite high expression levels of the activator form of GLI3, GLI3A. We therefore assume that the function of GLI3 as a transcriptional factor is affected in the hmm−/− limb bud. Previous work has shown mice with a conditional knockout of Sufu in the limbs have polydactyly with severe hypoplasia of the humerus, distal phalanges, and a short radius and ulna. In addition, a high level of GLI3A expression was observed in the Sufu−/− limb bud (Zhulyn and Hui, 2015), but Gli1 expression was downregulated (Zhulyn et al., 2014). These phenotypes are reminiscent of the hmm−/− limb bud. Given that high levels of GLI3A expression were observed in the hmm−/− limb bud, the regulatory system of GLI3A as mediated by proteins such as SUFU might be disorganized in the hmm−/− limb bud. However, we examined expression of Sufu by in situ hybridization, and the expression appeared to be the same in both the hmm+/− and hmm−/− limb buds (data not shown). The binding of SUFU to the GLI3A/KIF7 complex leads to GLI3R formation, but this is inhibited by the SMO/KIF3A/β-ARRESTIN complex in the presence of SHH, resulting in GLI3A formation (Kovacs et al., 2008). These results suggest that the function of SUFU protein might be disorganized in the hmm−/− limb bud. When we examined the coding sequence of Gli3 derived from reverse-transcribed mRNA from the hmm−/− limb bud, we saw several abnormal splicing variants at the N-terminus and full-length ORF sequence of Gli3 (data not shown). Prior work reports that the Polydactyly Nagoya (Pdn) mouse mutant has several abnormal splicing variants of Gli3 at the N-terminus due to integration of a retrotransposon (Thien and Rüther, 1999). Homozygous Pdn mice show severe polydactyly. It is possible that the abnormal splicing variants of Gli3 we observed in the hmm−/− limb bud interfere with GLI3A activity as a dominant negative variant of GLI3.
On the other hand, our results indicate that SHH protein is dysfunctional in the hmm−/− limb bud (Figure 5) even though it was detectable by immunohistochemistry (Figure 6). In contrast, SHH protein is functional in the talpid3 limb bud (Lewis et al., 1999) (Table 2). These results also suggest that the gene responsible for hmm is different from the cause of the talpid3 mutant. We suggest that post-translational modification of SHH protein might be disrupted in the hmm−/− limb bud. After the full length of SHH protein is synthesized, autoproteolytic cleavage is induced concomitantly with cholesterol modification of the N-terminal region of SHH protein (SHH-N). After that, SHH-N is palmitoylated by skinny hedgehog (SKI) (Briscoe and Thérond, 2013) and secreted from the cells. When we examined the coding sequence of Shh expressed in the hmm−/− limb bud, it was normal. Further, ZPA derived from the hmm−/− limb bud did not induce expression of Ptch2 near the implanted region (Figure 5D). These results imply that the secretion of SHH-N protein might be disrupted by a defect in palmitoylation. Future work needs to determine if SHH-N is palmitoylated in the hmm−/− limb bud.
Altogether, our results suggest that both SHH secretion and GLI3 function are disrupted in the HMM mutant. Interestingly, mice lacking both Shh and Gli3 show similar phenotypes in both the limb bud and neural tube. The Gli3−/− limb bud still has normal expression levels of Ptch1, whereas the Shh−/−; Gli3−/− limb bud shows no expression (Litingtung et al., 2002). The hmm−/− limb bud showed very low expression of Ptch1 (Figure 4). This phenotype is more similar to the Shh−/−; Gli3−/− limb bud rather than the Gli3−/− limb bud, suggesting that the hmm−/− limb bud is not just caused by Gli3 deficient conditions. Furthermore, expression of Hoxd13 is not observed in the Shh−/− limb bud, whereas the Shh−/−; Gli3−/− limb bud has high expression of Hoxd13. The hmm−/− limb bud also showed Hoxd13 expression, suggesting that the hmm−/− limb bud is not caused by just Shh deficient conditions. In terms of neural tube development, Shh−/−; Gli3−/− mice show a milder dorsalization phenotype (Litingtung and Chiang, 2000; Persson et al., 2002) than Shh−/− mice (Chiang et al., 1996; Pierani et al., 1999). This phenotype is like the one observed in the hmm−/− neural tube (Figure S1). These observations support the idea that both SHH and GLI3 activity would be disrupted in the HMM mutant as in Shh−/−; Gli3−/− mice. In the autopod, the function of GLI3 downstream of Indian hedgehog (IHH) is necessary for cartilage growth in the digits (St-Jacques et al., 1999). Therefore, the unidentifiable, malformed digits in the hmm−/− limb bud likely result from the disruption of GLI3 function during both digit patterning and digital cartilage formation.
Shh−/−; Gli3−/− mice show polydactyly with unidentifiable digits similar to the hmm−/− limb bud. Recently, it was reported that Gli3, Hoxa13, and Hoxd13 triple mutant mice show more severe polydactyly than Gli3−/− mice (Sheth et al., 2012). Previous reports indicated that HoxA13 regulates cell adhesion in the chick limb bud (Yokouchi et al., 1991). It is possible that the cell adhesion molecule downstream of GLI3/HoxA13/HoxD13 is involved in the determination of digit number. In the hmm−/− limb bud, expression of Hoxd13 is expanded in the autopod (Figure 3) with a disruption of GLI3 function, resulting in irregularly spaced digital ray sequence (Figure 2). We propose that we can elucidate the mechanisms of regulating the distance between digital rays under the control of cell adhesion molecule using the HMM mutant. Furthermore, it was reported that the expression of the cell adhesion molecule N-CAM is altered in the talpid2 limb bud (Chuong et al., 1993), which implies that it contributes to the aberrant condensation and bunching of digits in this mutant (McGlinn et al., 2005). Thus, adhesion molecules are thought to be important for the pattern formation and cartilage differentiation of the digits in the autopod. Studying the HMM mutant could be especially useful for determining how cell adhesion plays a role in the development of digit number and morphology.
In conclusion, we revealed several molecular characteristics of the hmm−/− limb bud that distinguish it from the talpid2 and talpid3 limb buds. These mutants all show the common phenotype of high amounts of GLI3A expressed in the limb, but the expression of SHH downstream target genes is unique in each mutant. We need further study to understand why SHH signaling is abolished in the hmm−/− limb bud despite the presence of a high amount of GLI3A, and why the SHH protein is dysfunctional in the hmm−/− limb bud. Several new SHH signaling components and their functions have been recently reported, including IFT proteins, the Cos2-Fu system, an enzyme for Hh processing, and regulators of GLI activity like DYRK2 and MAP3K10 (Ramsbottom and Pownall, 2016). These reports indicate that the SHH signaling pathway is more complex than previously thought. We propose that further analysis of the HMM mutant will provide new insight into the SHH signaling pathway. It can also serve as a useful model system for studying pattern formation like the talpid2 and talpid3 mutants did for vertebrate morphogenesis.
YosM, MN, KK, AK, and TS conceived the project and designed the experiments. YosM performed gene-expression studies and the ZPA grafting; MN maintained HMM mutant; KK performed the immunohistochemistry. JF, KK, and TS. performed the OPT scanning. TS performed the western blot analysis. KA contributed the isolation of quail cDNA. YosM, MN, KK, MT, YoiM, AK, and TS wrote the paper.
This work was supported by KAKENHI grant no. 25111710 and 25291050.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Yo-ichi Yamamoto Shiraishi for discussions; Dr. Kaori Tsujino for collecting the quail embryos; and researchers in the Avian Bioscience Research Center for kind assistance.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcell.2016.00149/full#supplementary-material
Shh is also expressed at the notochord and floor plate, and determines the dorsal-ventral polarity of the neural tube. To check whether SHH signaling is disrupted in other tissues, we examined the expression pattern of genes downstream of SHH signaling in the neural tube (Figure S1A). In the hmm−/− embryo, Shh was expressed in the notochord but not in the floor plate. Ptch2 was expressed around the notochord in hmm+/− embryo and in the ventricular zone located in the ventral neural tube. However, this expression was not observed in the hmm−/− embryo, indicating that SHH signaling is disrupted in the neural tube as it is in the limb bud. We further checked the expression patterns of transcription factors that determine neuronal identity along the DV axis under SHH signaling (Davey et al., 2006). The expression patterns of dorsal markers Pax3 and Pax7 were not distinguishable between the hmm+/− and hmm−/− embryos. In contrast, expression of the intermediate marker Pax6 was expanded ventrally in the hmm−/− embryo. Dbx2 is also expressed at an intermediate level in the hmm+/− embryo, but its expression was reduced in the hmm−/− embryo. Islet1 expression was normally observed in the ventral spinal cord but not in the floor plate. It is notable that Islet1 expression was expanded to the most ventral region of the spinal cord in the hmm−/− embryo. These observations of dorsalization and downregulation of genes downstream of SHH indicate that SHH signaling is also disrupted in the spinal cord and neighboring tissue in hmm−/− embryos.
Figure S1. SHH signaling is reduced in the hmm−/− spinal cord. (A) In situ hybridization of Shh, Ptch2, Pax3, Pax7, Pax6, Dbx2, and Islet1 on the transverse sections of the spinal cord at St. 25. All images are oriented with the dorsal side up and the ventral side down. The red arrowhead in the Pax6 figure indicates expanded Pax6 expression at the ventral side. The yellow arrowhead in the Dbx2 figure indicates the region where expression of Dbx2 is downregulated. The red arrowhead in the Islet1 figure indicates expanded Islet1 expression at the floor plate. (B) Immunohistochemistry of SHH protein at the notochord and neural tube is shown with fluorescent green. The nucleus is stained with DAPI. Transverse sections are oriented with the dorsal side up and the ventral side down.
Ainsworth, S. J., Stanley, R. L., and Evans, D. J. R. (2010). Developmental stages of the Japanese quail. J. Anat. 216, 3–15. doi: 10.1111/j.1469-7580.2009.01173.x
Bangs, F., Antonio, N., Thongnuek, P., Welten, M., Davey, M. G., Briscoe, J., et al. (2011). Generation of mice with functional inactivation of talpid3, a gene first identified in chicken. Development 138, 3261–3272. doi: 10.1242/dev.063602
Besse, L., Neti, M., Anselme, I., Gerhardt, C., Rüther, U., Laclef, C., et al. (2011). Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 138, 2079–2088. doi: 10.1242/dev.059808
Bhatia, N., Thiyagarajan, S., Elcheva, I., Saleem, M., Dlugosz, A., Mukhtar, H., et al. (2006). Gli2 is targeted for ubiquitination and degradation by beta-TrCP ubiquitin ligase. J. Biol. Chem. 281, 19320–19326. doi: 10.1074/jbc.M513203200
Briscoe, J., and Thérond, P. P. (2013). The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 14, 416–429. doi: 10.1038/nrm3598
Caruccio, N. C., Martinez-Lopez, A., Harris, M., Dvorak, L., Bitgood, J., Simandl, B. K., et al. (1999). Constitutive activation of sonic hedgehog signaling in the chicken mutant talpid(2): Shh-independent outgrowth and polarizing activity. Dev. Biol. 212, 137–149. doi: 10.1006/dbio.1999.9321
Chang, C.-F., Schock, E. N., O'Hare, E. A., Dodgson, J., Cheng, H. H., Muir, W. M., et al. (2014). The cellular and molecular etiology of the craniofacial defects in the avian ciliopathic mutant talpid2. Development 141, 3003–3012. doi: 10.1242/dev.105924
Chiang, C., Litingtung, Y., Harris, M. P., Simandl, B. K., Li, Y., Beachy, P. A., et al. (2001). Manifestation of the limb prepattern: limb development in the absence of sonic hedgehog function. Dev. Biol. 236, 421–435. doi: 10.1006/dbio.2001.0346
Chiang, C., Litingtung, Y., Lee, E., Young, K. E., Corden, J. L., Westphal, H., et al. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413. doi: 10.1038/383407a0
Chuong, C. M., Widelitz, R. B., Jiang, T. X., Abbott, U. K., Lee, Y. S., and Chen, H. M. (1993). Roles of adhesion molecules NCAM and tenascin in limb skeletogenesis: analysis with antibody perturbation, exogenous gene expression, talpid mutants and activin stimulation. Prog. Clin. Biol. Res. 383B, 465–474.
Davey, M. G., Paton, I. R., Yin, Y., Schmidt, M., Bangs, F. K., Morrice, D. R., et al. (2006). The chicken talpid3 gene encodes a novel protein essential for Hedgehog signaling. Genes Dev. 20, 1365–1377. doi: 10.1101/gad.369106
Dvorak, L., and Fallon, J. F. (1992). The talpid2 chick limb has weak polarizing activity and can respond to retinoic acid and polarizing zone signal. Dev. Dyn. 193, 40–48. doi: 10.1002/aja.1001930107
Ede, D. A., and Kelly, W. A. (1964). Developmental abnormalities in the trunk and limbs of the talpid3 mutant of the fowl. J. Embryol. Exp. Morphol. 12, 339–356.
Francis-West, P. H., Robertson, K. E., Ede, D. A., Rodriguez, C., Izpisúa-Belmonte, J. C., Houston, B., et al. (1995). Expression of genes encoding bone morphogenetic proteins and sonic hedgehog in talpid (ta3) limb buds: their relationships in the signalling cascade involved in limb patterning. Dev. Dyn. 203, 187–197. doi: 10.1002/aja.1002030207
Hsu, S.-H. C., Zhang, X., Yu, C., Li, Z. J., Wunder, J. S., Hui, C.-C., et al. (2011). Kif7 promotes hedgehog signaling in growth plate chondrocytes by restricting the inhibitory function of Sufu. Development 138, 3791–3801. doi: 10.1242/dev.069492
Kovacs, J. J., Whalen, E. J., Liu, R., Xiao, K., Kim, J., Chen, M., et al. (2008). Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 320, 1777–1781. doi: 10.1126/science.1157983
Lewis, K. E., Drossopoulou, G., Paton, I. R., Morrice, D. R., Robertson, K. E., Burt, D. W., et al. (1999). Expression of ptc and gli genes in talpid3 suggests bifurcation in Shh pathway. Development 126, 2397–2407.
Litingtung, Y., and Chiang, C. (2000). Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat. Neurosci. 3, 979–985. doi: 10.1038/79916
Litingtung, Y., Dahn, R. D., Li, Y., Fallon, J. F., and Chiang, C. (2002). Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature 418, 979–983. doi: 10.1038/nature01033
McGlinn, E., van Bueren, K. L., Fiorenza, S., Mo, R., Poh, A. M., Forrest, A., et al. (2005). Pax9 and Jagged1 act downstream of Gli3 in vertebrate limb development. Mech. Dev. 122, 1218–1233. doi: 10.1016/j.mod.2005.06.012
Nelson, C. E., Morgan, B. A., Burke, A. C., Laufer, E., DiMambro, E., Murtaugh, L. C., et al. (1996). Analysis of Hox gene expression in the chick limb bud. Development 122, 1449–1466.
Persson, M., Stamataki, D., te Welscher, P., Andersson, E., Böse, J., Rüther, U., et al. (2002). Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 16, 2865–2878. doi: 10.1101/gad.243402
Pierani, A., Brenner-Morton, S., Chiang, C., and Jessell, T. M. (1999). A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell 97, 903–915. doi: 10.1016/S0092-8674(00)80802-8
Ramsbottom, S. A., and Pownall, M. E. (2016). Regulation of Hedgehog Signalling Inside and Outside the Cell. J. Dev. Biol. 4, 23. doi: 10.3390/jdb4030023
Riddle, R. D., Johnson, R. L., Laufer, E., and Tabin, C. (1993). Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401–1416. doi: 10.1016/0092-8674(93)90626-2
Rodriguez, C., Kos, R., Macias, D., Abbott, U. K., and Izpisúa Belmonte, J. C. (1996). Shh, HoxD, Bmp-2, and Fgf-4 gene expression during development of the polydactylous talpid2, diplopodia1, and diplopodia4 mutant chick limb buds. Dev. Genet. 19, 26–32. doi: 10.1002/(SICI)1520-6408(1996)19:1<26::AID-DVG3>3.0.CO;2-2
Ros, M. A., Dahn, R. D., Fernandez-Teran, M., Rashka, K., Caruccio, N. C., Hasso, S. M., et al. (2003). The chick oligozeugodactyly (ozd) mutant lacks sonic hedgehog function in the limb. Development 130, 527–537. doi: 10.1242/dev.00245
Sharpe, J., Ahlgren, U., Perry, P., Hill, B., Ross, A., Hecksher-Sørensen, J., et al. (2002). Optical projection tomography as a tool for 3D microscopy and gene expression studies. Science 296, 541–545. doi: 10.1126/science.1068206
Sheth, R., Marcon, L., Bastida, M. F., Junco, M., Quintana, L., Dahn, R., et al. (2012). Hox genes regulate digit patterning by controlling the wavelength of a Turing-type mechanism. Science 338, 1476–1480. doi: 10.1126/science.1226804
St-Jacques, B., Hammerschmidt, M., and McMahon, A. P. (1999). Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086. doi: 10.1101/gad.13.16.2072
Suzuki, T. (2013). How is digit identity determined during limb development? Dev. Growth Differ. 55, 130–138. doi: 10.1111/dgd.12022
Suzuki, T., Hasso, S. M., and Fallon, J. F. (2008). Unique SMAD1/5/8 activity at the phalanx-forming region determines digit identity. Proc. Natl. Acad. Sci. U.S.A. 105, 4185–4190. doi: 10.1073/pnas.0707899105
Takahashi, M., Tamura, K., Büscher, D., Masuya, H., Yonei-Tamura, S., Matsumoto, K., et al. (1998). The role of Alx-4 in the establishment of anteroposterior polarity during vertebrate limb development. Development 125, 4417–4425.
te Welscher, P., Fernandez-Teran, M., Ros, M. A., and Zeller, R. (2002a). Mutual genetic antagonism involving GLI3 and dHAND prepatterns the vertebrate limb bud mesenchyme prior to SHH signaling. Genes Dev. 16, 421–426. doi: 10.1101/gad.219202
te Welscher, P., Zuniga, A., Kuijper, S., Drenth, T., Goedemans, H. J., Meijlink, F., et al. (2002b). Progression of vertebrate limb development through SHH-mediated counteraction of GLI3. Science 298, 827–830. doi: 10.1126/science.1075620
Thien, H., and Rüther, U. (1999). The mouse mutation Pdn (Polydactyly Nagoya) is caused by the integration of a retrotransposon into the Gli3 gene. Mamm. Genome 10, 205–209. doi: 10.1007/s003359900973
Tsudzuki, M., Nakane, Y., and Wada, A. (1998). Hereditary multiple malformation in Japanese quail: a possible powerful animal model for morphogenetic studies. J. Hered. 89, 24–31. doi: 10.1093/jhered/89.1.24
Wang, B., Fallon, J. F., and Beachy, P. A. (2000). Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423–434. doi: 10.1016/S0092-8674(00)80678-9
Yin, Y., Bangs, F., Paton, I. R., Prescott, A., James, J., Davey, M. G., et al. (2009). The Talpid3 gene (KIAA0586) encodes a centrosomal protein that is essential for primary cilia formation. Development 136, 655–664. doi: 10.1242/dev.028464
Yokouchi, Y., Sasaki, H., and Kuroiwa, A. (1991). Homeobox gene expression correlated with the bifurcation process of limb cartilage development. Nature 353, 443–445. doi: 10.1038/353443a0
Zhulyn, O., and Hui, C.-C. (2015). Sufu and Kif7 in limb patterning and development. Dev. Dyn. 244, 468–478. doi: 10.1002/dvdy.24249
Keywords: sonic hedgehog, polydactyly, quail, Hereditary Multiple Malformation
Citation: Matsubara Y, Nakano M, Kawamura K, Tsudzuki M, Funahashi J-I, Agata K, Matsuda Y, Kuroiwa A and Suzuki T (2016) Inactivation of Sonic Hedgehog Signaling and Polydactyly in Limbs of Hereditary Multiple Malformation, a Novel Type of Talpid Mutant. Front. Cell Dev. Biol. 4:149. doi: 10.3389/fcell.2016.00149
Received: 08 November 2016; Accepted: 13 December 2016;
Published: 27 December 2016.
Edited by:
Takaaki Matsui, Nara Institute of Science and Technology, JapanReviewed by:
Minoru Omi, Fujita Health University, JapanCopyright © 2016 Matsubara, Nakano, Kawamura, Tsudzuki, Funahashi, Agata, Matsuda, Kuroiwa and Suzuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takayuki Suzuki, c3V6dWtpLnRha2F5dWtpQGoubWJveC5uYWdveWEtdS5hYy5qcA==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.