 Iryna Bohovych
Iryna Bohovych Oleh Khalimonchuk
Oleh Khalimonchuk
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 13 October 2016
Sec. Signaling
Volume 4 - 2016 | https://doi.org/10.3389/fcell.2016.00109
Normal cellular physiology is critically dependent on numerous mitochondrial activities including energy conversion, cofactor and precursor metabolite synthesis, and regulation of ion and redox homeostasis. Advances in mitochondrial research during the last two decades provide solid evidence that these organelles are deeply integrated with the rest of the cell and multiple mechanisms are in place to monitor and communicate functional states of mitochondria. In many cases, however, the exact molecular nature of various mitochondria-to-cell communication pathways is only beginning to emerge. Here, we review various signals emitted by distressed or dysfunctional mitochondria and the stress-responsive pathways activated in response to these signals in order to restore mitochondrial function and promote cellular survival.
Mitochondria are semi-autonomous dynamic organelles of endosymbiotic origin involved in a plethora of vital functions including energy conversion and synthesis of key precursor metabolites, reducing equivalents, and cofactors. For more than half a century, mitochondria have been known to the general audience as the “powerhouse of the cell” (Siekvitz, 1957). This scientific meme refers to the organelle's most commonly known bioenergetic function—generation of adenosine triphosphate (ATP). This process commences by an uptake of substrates from the cytosol and is followed by their catabolic conversion via fatty acid oxidation and/or the citric acid (TCA) cycle, which yields reducing equivalents, nicotinamide adenine dinucleotide (NADH), and flavoadenine dinucleotide (FADH2) as well as multiple biosynthetic precursors. The reducing equivalents produced through the TCA cycle fuel the electron transport chain component of the mitochondrial oxidative phosphorylation system (OXPHOS), wherein flow of electrons through respiratory complexes is linked to generation of a proton gradient across the inner mitochondrial membrane (IM) that is required for ATP production by F1F0 ATP synthase. Although these functions are clearly crucial for cellular physiology and human health, the concept of mitochondria as isolated biosynthetic and bioenergetic units is insufficient to explain certain phenotypic outcomes or clinical manifestations associated with known mitochondrial dysfunctions.
The past two decades gave rise to tremendous research progress in the field of mitochondrial biology. They provided a gamut of evidence that mitochondria are deeply integrated into cellular physiology and metabolism. Mitochondria retain their own genome and transcription/translation machineries; however, because they co-evolved with their host cell, a number of originally mitochondrial genes have been transferred to the nucleus, thus contributing to the dual genetic origin of the mitochondrial proteome. That is, the vast majority of proteins comprising the mitochondrial proteome is encoded by nuclear DNA, synthesized in the cytosol, and is subsequently imported into the organelle. This bi-genomic nature of the mitochondrial proteome necessitates tightly coordinated expression of both mitochondrial and nuclear genes to produce stoichiometric amounts of its components to maintain proper organelle function. The signaling mechanisms that assure such communication are historically classified into anterograde and retrograde signaling. The former mechanisms mediate coordination of mitochondrial gene expression and—more broadly—mitochondrial function in response to endogenous and environmental homeostatic alterations sensed in the cytosol or by other organelles (usually the nucleus). In turn, retrograde signaling mechanisms monitor a variety of signals emitted by the mitochondria that allow for communicating the functional state (e.g., levels of energy production or the organelle's biosynthetic capacity) of mitochondria with other cellular compartments.
While much of the previous research focused on anterograde signaling, recent advances highlight and greatly expand the original paradigm postulated in the 1990's (Butow and Avadhani, 2004), whereby mitochondria are viewed as critical signaling hubs that take part in multiple cellular decisions. In this review, we will focus on molecular signals produced by the mitochondria to communicate homeostatic alterations and coordinate retrograde responses. We will survey several diverse groups of these signals including: (1) nucleotides; (2) precursor metabolites; (3) free radicals; (4) peptides and polypeptides; and (5) other molecules such as ions and lipids. Consequently, we will discuss their significance and impact on mitochondrial and cellular physiology.
While the generation of ATP is undoubtedly one of the major mitochondrial functions, mitochondria also provide some key precursors to other nucleotides or nucleotide-based reducing equivalents. It is therefore not surprising that cells have developed sensitive mechanisms to monitor the levels of these molecules and adjust cellular metabolic demands accordingly. In this section, we will discuss how alterations in mitochondrial or cellular levels of these mitochondria-borne molecules due to mitochondrial dysfunction or distress are recognized as an alarming signal to initiate a chain of events necessary to restore cellular energy and metabolic homeostasis.
Generation of ATP through mitochondrial oxidative phosphorylation is central to the maintenance of the optimal ATP/adenosine diphosphate (ADP) ratios within the cell (Figure 1). Under conditions of mitochondrial damage, decreased ATP production results in depletion of intracellular ATP, which leads to increased intracellular concentrations of adenosine monophosphate (AMP) or its subsequent derivative, adenosine. The latter nucleotide directly binds to the γ subunit of the energy-sensing adenosine monophosphate-activated protein kinase (AMPK) complex (Hardie et al., 2016). This regulatory binding acts in concert with an upstream regulatory protein kinase LKB1 to promote activation of AMPK (Pearce et al., 2013; Hardie et al., 2016). AMPK is a key metabolic sensor in the cell and its activation initiates multiple signaling events leading to a series of interconnected processes: (1) inhibition of ATP-dependent biosynthetic pathways to prevent wasteful use of cellular ATP; (2) stimulation of ATP production via catabolic reactions (Hall et al., 2013; Pearce et al., 2013); (3) activation of autophagic removal of damaged mitochondria (mitophagy) (Egan et al., 2011; Kim et al., 2011); and (4) metabolic re-tuning of mitochondria via promotion of mitochondrial fission (Toyama et al., 2016). These molecular events enable restoration of cellular energy homeostasis and allow cells to cope with metabolic distress.
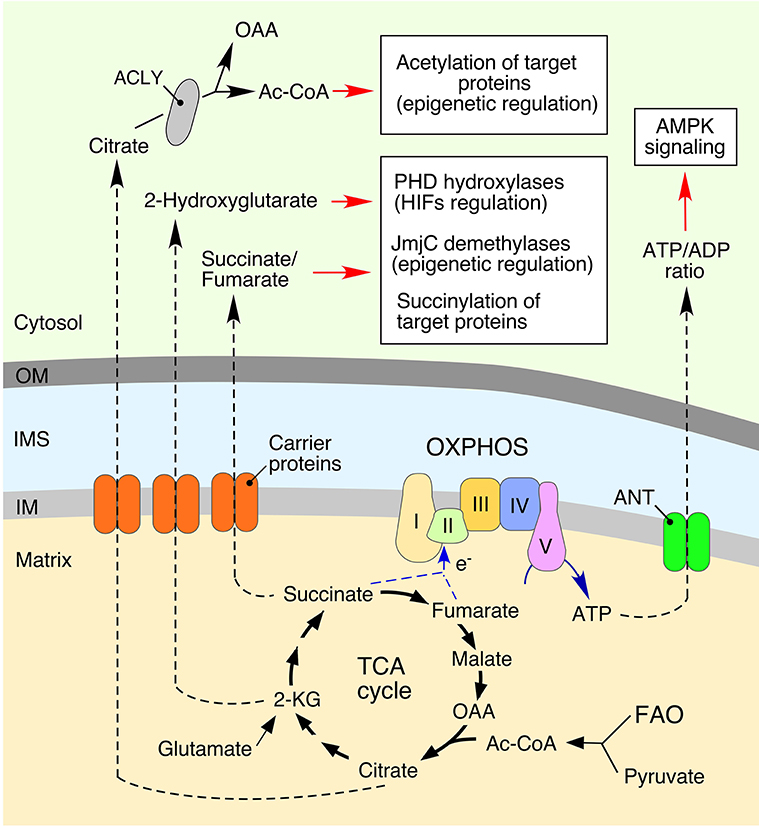
Figure 1. Mitochondria-derived metabolites regulate various cellular processes. Bioenergetic function of mitochondria is associated with generation of ATP (adenosine triphosphate) via the oxidative phosphorylation (OXPHOS) system. This process involves conversion of various substrates imported from the cytosol via the tricarboxylic acid (TCA) cycle or fatty acid oxidation (FAO). Metabolic intermediates can be exported into the cytosol to impact various signaling cascades. Mitochondria-derived citrate is converted to oxaloacetate (OAA) and acetyl coenzyme A (Ac-CoA) by ATP citrate lyase (ACLY). Subsequently, a fraction of the cytosolic Ac-CoA can be used in protein acetylation, thereby impacting multiple cellular processes (Choudhary et al., 2009; Spange et al., 2009; Wellen et al., 2009; Eisenberg et al., 2014). Another TCA cycle biosynthetic intermediate, 2-ketoglutarate (α-KG), can be converted to 2-Hydroxyglutarate. This metabolite inhibits: (1) Jumonji C domain-containing demethylases (JmjC), affecting epigenetic reprogramming (Pearce et al., 2013; Zong et al., 2016); and (2) hypoxia-inducible prolyl hydroxylases (PHDs), thus stabilizing the hypoxia-inducible factors (HIFs) and inducing a hypoxic response (Xu et al., 2011; Pearce et al., 2013; Zong et al., 2016). The TCA metabolite, fumarate, can also act as a signal-modulating factor via its ability to bind lysine residues of various proteins, resulting in a post-translational modification called succinylation. The adenine nucleotide translocator (ANT) mediates an exchange of ATP and cytosol-derived ADP (adenosine diphosphate) across the inner mitochondrial membrane (IM). Mitochondrial dysfunction derived ATP depletion perturbs ATP/ADP ratios by subsequent activation of adenosine monophosphate-activated protein kinase (AMPK), a master regulator of cellular energy homeostasis (Hall et al., 2013; Pearce et al., 2013).
Nicotinamide adenine dinucleotide is a vital redox molecule present in the cell in both oxidized (NAD+) and reduced (NADH) forms. One distinctive feature of NAD+ is that cells can utilize it as both a cofactor and a co-substrate (Canto et al., 2015; Verdin, 2015). Cells maintain several NAD+ pools in different cellular compartments; the mitochondrial NAD+ pool is segregated from the rest of the cell due to impermeability of the mitochondrial inner membrane. Just like in the case of adenine nucleotides, the NAD+/NADH equilibrium is central to normal cellular function. In mitochondria, the optimal NAD+/NADH and FADH/FADH2 ratios are primarily maintained through OXPHOS function and the TCA cycle. In addition, the cytosolic and mitochondrial NAD+ pools are connected via malate/aspartate and glyceraldehyde-3-phosphate shuttles that promote a mitochondria-cytosol exchange of reducing equivalents (Birsoy et al., 2015; Verdin, 2015). Perturbations of OXPHOS function decrease the NAD+/NADH ratio thereby creating a deficit in mitochondria, and, ultimately, leading to cytosolic NAD+/NADH imbalance (Birsoy et al., 2015; Sullivan et al., 2015). Because glycolytic enzymes are NAD+-dependent, respiratory compromised cells divert mitochondria-destined pyruvate toward lactate production—the reaction that also yields NAD+ for use in glycolysis. These actions can subsequently activate AMPK- and MAP kinase-mediated downstream signal transduction pathways (reviewed in Canto et al., 2015). Of note, elevated cellular levels of lactate usually reflect mitochondrial dysfunction and serve as a common diagnostic marker in patients with mitochondrial disease (DiMauro and Schon, 2003; Vafai and Mootha, 2012).
As mentioned above, NAD+ is also utilized as a co-substrate by several different classes of enzymes including sirtuin protein diacetylases—a group of versatile enzymes distributed across subcellular compartments and involved in regulation of various cellular activities such as histone modification and modulation of master transcriptional regulators like forkhead box 03 (FOXO3), nuclear factor kappa B (NF-κB), and peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1α, Canto et al., 2015; Verdin, 2015). Alterations in cellular NAD+ levels may, therefore, lead to substantial changes in the activity of sirtuins and promote global transcriptional changes.
Yet another nucleotide-based signaling mechanism is thought to include the mitochondrial inner membrane-anchored enzyme dihydroorotate dehydrogenase (DHODH), which oxidizes dehydroorotate to produce orotate—the key precursor in biosynthesis of pyrimidine nucleotides, uridine triphosphate, and cytidine triphosphate (Evans and Guy, 2004). DHODH activity is critically dependent on OXPHOS activity and the presence of reduced ubiquinone as a direct electron acceptor. As such, a dysfunctional OXPHOS stalls de novo synthesis of pyrimidines, which in turn leads to stabilization and nuclear accumulation of the tumor suppressor protein p53 and subsequent transcriptional responses (Khutorenko et al., 2010).
The mitochondrion is a source of several key metabolic precursors utilized by cellular biosynthetic pathways. There is growing evidence that mitochondria-produced intermediary metabolites are not just mere biosynthetic building blocks, but also potent regulators of various cellular signaling cascades. In general, most, if not all, of the examples of mitochondrial nucleotide-mediated signaling surveyed above can be viewed from a metabolic sensing perspective. However, for the reader's convenience, we chose to discuss them separately. Here, we will focus on TCA cycle-borne biosynthetic intermediates that may exert signaling functions.
Acetyl coenzyme A (Ac-CoA) is a central donor of two-carbon units utilized in multiple biosynthetic reactions in the mitochondrion. While Ac-CoA, per se, is unable to cross mitochondrial membranes, its condensation with the four-carbon TCA cycle intermediate oxaloacetate by citrate synthase produces citrate that can be either utilized in subsequent rounds of the TCA cycle or be readily exported out of the mitochondria (Figure 1). In the cytosol, citrate is converted back to oxaloacetate and Ac-CoA, which can be used in lipid biosynthesis and protein acetylation. The latter post-translational modification is mediated by lysine acetyltransferases and can widely impact multiple cellular processes including signaling (Choudhary et al., 2009; Spange et al., 2009; Wellen et al., 2009; Eisenberg et al., 2014).
Interestingly, Ac-CoA and citrate metabolism-related signaling have been in the spotlight for quite a long time. The first retrograde response mechanism identified in respiratory deficient yeast cells in 1990s invokes metabolic remodeling to replenish mitochondrial glutamate, citrate, and Ac-CoA pools (Liao and Butow, 1993; Liu and Butow, 1999). This apparently yeast-specific pathway, known as the retrograde response gene, or RTG pathway, includes a sensor protein, Rtg2, and two transcriptional factors, Rtg1 and Rtg3 that are kept inactive in the cytoplasm through phosphorylation. Mitochondrial dysfunction appears to be sensed by Rtg2, which promotes dephosphorylation and subsequent nuclear accumulation of the Rtg1/Rtg3 tandem, thereby triggering transcriptional responses. More details on the RTG pathway can be found in the following reviews (Butow and Avadhani, 2004; Haynes et al., 2013).
Succinate, fumarate and 2-ketoglutarate are the four-carbon and five-carbon metabolites of the TCA cycle, respectively. These molecules can be exported to the cytosol through dicarboxylate carrier proteins and utilized as the donors of carbon units in a variety of biosynthetic reactions (Figure 1). However, in certain mitochondrial dysfunctions—e.g., mutations affecting the function of the TCA cycle enzymes isocytrate dehydrogenase, succinate dehydrogenase, or fumarate hydratase—cells accumulate these metabolites in both the mitochondria and cytosol (Gaude and Frezza, 2014; Parker and Metallo, 2015; Zong et al., 2016). Accumulation of 2-ketoglutarate (2-KG) is associated with its conversion to 2-hydroxyglutarate (2-HG), a potent mimetic, and inhibitor of 2-KG-dependent dioxygenases in the cell—most notably hypoxia-inducible factor prolyl hydroxylases (PHDs) (Xu et al., 2011; Pearce et al., 2013; Zong et al., 2016) and the Jumonji-domain family histone lysine demethylases (Jmj-KDMs) (Pearce et al., 2013; Zong et al., 2016). Inhibition of these enzymes leads to significant epigenetic and transcriptional changes (Pearce et al., 2013; Zong et al., 2016). Additionally, 2-HG-mediated inhibition of PHDs stabilizes hypoxia-inducible factors (HIFs), thereby activating hypoxic signaling in the cell. It is therefore not surprising that 2-HG is a recognized oncometabolite and its accumulation has been reported in many cancers (Losman and Kaelin, 2013; Zong et al., 2016).
Accumulation of succinate appears to have a similar impact on function of PHDs and has been linked to tumorigenesis (Zong et al., 2016). Accumulation of fumarate results in the metabolite's binding to lysine residues of various proteins—so called succinylation (Zhang et al., 2011). Such modifications negatively impact Kelch-like ECH associated protein 1 (KEAP1)-nuclear factor erythroid-derived 2-like (NRF2) signaling axis, wherein succinylation of KEAP1 leads to stabilization of the transcriptional factor NRF2 that drives antioxidant transcriptional responses (Adam et al., 2011, 2014). The antioxidant peptide glutathione is yet another target for succinylation—the modification depletes cellular GSH/GSSG pools, which also results in activation of NRF2-mediated antioxidant responses (Sullivan et al., 2013). Although seemingly beneficial, sustained NRF2 activation may be detrimental to the cell and has been linked to tumorigenesis (Sullivan et al., 2013).
Mitochondria-produced free radicals and their impact on biological molecules have been extensively studied since the 1950's. For decades, reactive oxygen species (ROS) and reactive nitrogen species (RNS) have been recognized as key contributors to cellular oxidative stress and are detrimental factors in many pathologies and aging (Harman, 1972; Balaban et al., 2005; Wallace, 2005). However, more recent studies established that physiological amounts of free radicals are required to mediate a number of normal cellular processes including several signaling pathways such as hypoxic signaling (Finkel, 2011; Collins et al., 2012; Sena and Chandel, 2012; Chandel, 2014; Shadel and Horvath, 2015). Because a number of outstanding reviews are available on this topic, in this section we will only highlight several key aspects of ROS and RNS-mediated mitochondrial signaling.
It is commonly accepted that the majority of cellular ROS originates from mitochondrial energy metabolism (Murphy, 2009; Figueira et al., 2013). The flow of electrons through ETC complexes is inevitably linked to a “leakage” of these reducing equivalents, exposing them to molecular oxygen. Partial reduction of O2 yields the superoxide anion (); it is estimated that ~0.2–2% of O2 consumed by mitochondria is converted to superoxide (Figueira et al., 2013). Additional factors, such as full reduction of electron carriers (e.g., under the condition of low ATP production) or elevated NAD+/NADH in the matrix (e.g., due to OXPHOS damage), can further stimulate superoxide production.
The two major sites for electron leakage are OXPHOS complexes I and III (Figure 2). Electrons in Complex I leak from the flavin mononucleotide site of the enzyme, thus producing on the matrix side of the mitochondrial inner membrane; in the case of Complex III, superoxide is produced via the reactive semiquinone intermediate on both matrix and intermembrane space (IMS)-exposed sides of the enzyme (Murphy, 2009; Figueira et al., 2013). The superoxide anion is a short-lived free radical that is readily converted to hydrogen peroxide (H2O2) by matrix-localized manganese (Mn-SOD, SOD2) or copper-zinc (Cu-Zn SOD, SOD1) superoxide dismutase in the IMS (Figure 2). Nonetheless, elevated levels of may impair the activity of Fe-S cluster-containing metabolic enzymes, particularly aconitase, whose active site contains a surface-exposed Fe-S cluster that is extremely vulnerable to oxidation (Armstrong et al., 2004). Aconitase inactivation can lead to a malfunctioning TCA cycle and accumulation of intermediary metabolites such as citrate, which will in turn impinge on cellular signaling as described in the previous section.
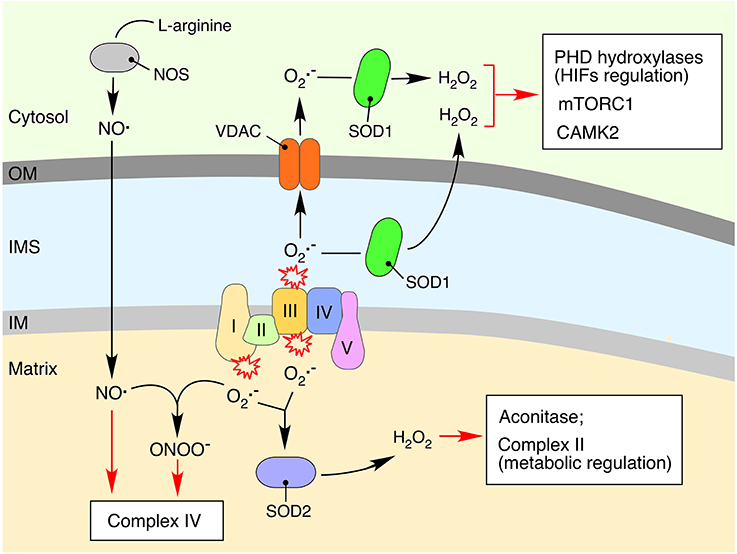
Figure 2. Impact of mitochondria-derived free radicals on cellular signaling processes. Reduction of molecular oxygen to the superoxide anion () is an unavoidable consequence of electron leakage associated with the bioenergetic function of respiratory complexes (Figueira et al., 2013). The produced is quickly converted to hydrogen peroxide (H2O2) by the mitochondrial matrix-associated manganese (SOD2) or inner membrane space (IMS)-localized copper-zinc (SOD1) superoxide dismutases (Murphy, 2009; Figueira et al., 2013). Accumulation of H2O2 can affect functioning of mitochondrial proteins, in particular those containing iron-sulfur clusters. Aconitase, a key component of the TCA cycle, is one of the mitochondrial proteins most susceptible to oxidative damage due to the surface exposed iron-sulfur cluster-containing active sites (Armstrong et al., 2004). Mitochondria-derived and H2O2 can also impact cytosolic signaling cascades via oxidation of various redox-sensitive proteins including calcium/calmodulin-dependent protein kinase 2 (CaMK2) (Erickson et al., 2008), the mammalian target of rapamycin complex 1 protein kinase (mTORC1), and PHD hydroxylases (Sena and Chandel, 2012). The cytosol-derived nitric oxide radical (NO.), a product of arginine conversion by nitric oxide synthases (NOSs), can permeate mitochondrial membranes and inhibit electron transport chain functioning through competitive inhibition of respiratory complex IV or via condensation with and formation of a potent oxidant, peroxynitrate (ONOO−) (Nisoli et al., 2003; Antunes et al., 2004; Figueira et al., 2013).
Unlike superoxide, H2O2 is a more stable molecule, which can diffuse across mitochondrial membranes. It is thought to be the primary ROS-related signal produced by the mitochondria (Collins et al., 2012). Changes in cellular H2O2 may affect several signaling-related processes. For instance, reversible oxidation of specific redox-sensitive methionine residues can modulate the activity of calcium/calmodulin-dependent protein kinase 2 (CaMK2), which controls global responses in excitable cells such as cardiomyocytes (Erickson et al., 2008). Similarly, recent data from the yeast genetic model indicate that elevated levels of endogenous mitochondria-borne H2O2 can modulate the activity of the target of rapamycin (TOR) complex 1 protein kinase (TORC1) (Pan et al., 2011; Bohovych et al., 2016). These data corroborate the results of the earlier in vitro analyses, which suggested direct redox-modulation of TORC1 (Sabrassov and Sabatini, 2005). Moreover, H2O2-triggered alterations in the redox state of metal cofactor containing enzymes can also initiate signaling responses. A well-established example of such a mechanism is the modulation of hypoxic signaling by mitochondria-derived H2O2 (Guzy et al., 2005; Hamanaka and Chandel, 2010; Sena and Chandel, 2012).
The stability of H2O2, its ability to traverse cellular membranes, and its propensity to oxidize thiol groups of surface-exposed, low-pKa cysteine residues under physiological conditions makes hydrogen peroxide-mediated post-translational modifications an important molecular event underpinning a wide range of signaling processes in the cell. The selective reactivity of H2O2 with key cysteine residues of redox-active GTPases, transcription factors, proteases, receptors, kinases, phosphatases, and protein disulfide isomerases can alter the target protein's activity, conformation, or subcellular localization, thereby modulating cellular signal cascades and outputs (reviewed in García-Santamarina et al., 2014; Russell and Cotter, 2015). The reversibility of thiol oxidation (sulfenylation) reactions is assured through the function of electron donor proteins such as thioredoxins, glutaredoxins, and sulfiredoxins, which can all efficiently reduce oxidized cysteines, thus reversing the modulatory effect of thiol oxidation (Brandes and Jakob, 2009). For instance, protein tyrosine phosphatases (PTPs) are among the best-studied components of H2O2-regulated signal transduction in mammalian cells. Initial evidence of sulfenic acid intermediate formation in response to low micromolar concentrations of H2O2 in three different PTPs (PTP1, leukocyte antigen-related phosphatase LAR, and vaccinia H1-related phosphatase VHR) was originally reported by Denu and Tanner (1998). Inactivation of the PTPs caused by sulfenylation of the catalytic cysteine can then be followed by secondary modifications like formation of disulfide bonds or sulfenyl amide linkages for further protection against overoxidation (Tanner et al., 2011). An interesting example of redox regulation involving non-catalytic cysteines was described of the human lymphoid tyrosine phosphatase (LYP). Upon oxidizing conditions, a disulfide bond is formed between the catalytic cysteine residue (Cys-227) and the cysteine residues outside of the signature motif (Cys-129) (Tsai et al., 2009). To prevent malproductive reactivation in the reducing environment, Cys-227 forms a disulfide bond with yet another non-catalytic Cys-231, suggesting the possibility of an autoregulatory circuit (Tsai et al., 2009). Multiple outstanding reviews are available on sulfenylation-based activity modulation in various protein kinases (Corcoran and Cotter, 2013; Truong and Carroll, 2013; Russell and Cotter, 2015). The examples include, but are not limited to, the signaling cascades of receptor tyrosine kinases (RTKs), the serine-threonine protein kinase, Akt, mitogen-activated kinases (MAP kinases), cytoplasmic Src (c-Src), inhibitory κB kinases (IKK), and the c-AMP-dependent protein kinase. It should be noted, however, that in many of the aforementioned cases it remains to be determined whether these H2O2-signaling events involve mitochondria-derived ROS. To date, mitochondria-produced H2O2 has been implicated in Akt/phosphoinositide-3-kinase (PI3K) signaling via sulfenylation-mediated inactivation of the phosphatase PTEN (Connor et al., 2005). Another example includes the recently described activation of two mitochondria-localized kinases, the Src family tyrosine kinase, Lyn, and the spleen tyrosine kinase, Syk, by mitochondrial ROS (Patterson et al., 2015). Activation of these kinases is important for the proper functioning of several signaling cascades including that of the mitogen-activated protein kinases JNK and Akt (Patterson et al., 2015).
The nitric oxide radical (NO.) is probably the best-characterized type of RNS. Although mitochondrial origin of NO. remains debated, it is clear that this stable free radical is able to cross mitochondrial membranes and exert several modulatory effects pertinent to mitochondrial signaling (Figure 2). Elevated levels of mitochondrial NO. may alter the mitochondrial bioenergetics status via inhibition of OXPHOS by competing with molecular oxygen for respiratory Complex IV, thereby affecting the cellular ATP/ADP ratio or driving excessive production of superoxide by the ETC (Nisoli et al., 2003; Antunes et al., 2004; Figueira et al., 2013). NO may also drive reversible modification (S-nitrosylation) of specific protein thiols to S-nitrosothiols. Such posttranslational modifications are shown to modulate properties of multiple signaling pathways including those of HIFα and NF-κB (Hess et al., 2005; Nakamura et al., 2013).
Finally, NO. can interact with mitochondrial superoxide and form yet another potent RNS, peroxynitrate (ONOO−), which also inhibits the ETC (Antunes et al., 2004) and is likely to trigger signaling responses.
The vast majority of polypeptides comprising the mitochondrial proteome are produced by cytosolic translation; newly synthesized polypeptides are imported into the organelle in an unfolded state (Neupert and Herrmann, 2007; Chacinska et al., 2009). However, several core hydrophobic subunits of OXPHOS complexes are produced by mitochondrial translation machinery and need to be stoichiometrically paired with imported polypeptides. This complex nature of the mitochondrial proteome creates a challenging protein folding environment; therefore, it is not surprising that synthesis and assembly of mitochondrial proteome components is a well-orchestrated and highly regulated process (Couvillion et al., 2016). In addition, a large number of metabolic enzymes in the matrix are prone to facile aggregation (Bender et al., 2011). Conditions of mitochondrial stress may perturb stoichiometric equilibrium and cause an accumulation of unassembled and/or misfolded proteins in various mitochondrial sub-compartments and reduce mitochondrial import. Such alterations in mitochondrial protein homeostasis (proteostasis) can be communicated via several mechanisms highlighted below. Furthermore, we will discuss how several mitochondrial polypeptides signal the initiation of an apoptotic program when mitochondrial damage exceeds the repair capacities of a cell.
One of the best-understood responses to mitochondrial proteostatic stress is the mitochondrial unfolded protein response (UPRmt). Activation of UPRmt in higher eukaryotes induces the transcription of genes involved in protein folding and quality control (Pellegrino et al., 2013; Bohovych et al., 2015; Topf et al., 2016), metabolism (Lin and Haynes, 2016), and mtDNA maintenance (Lin et al., 2016). Reciprocally, UPRmt induction is linked to an inhibition of the genes related to both the TCA cycle and OXPHOS components (Nargund et al., 2015). Two key molecular facets of UPRmt were gleaned from studies in the roundworm Caenorhabditis elegans genetic model (Figure 3). One aspect involves generation of signaling peptides produced through proteolytic processing of unassembled or misfolded polypeptides by the matrix peptidase ClpXP (Haynes et al., 2010). The peptides are subsequently extruded from mitochondria into the cytosol via the HAF-1 peptide exporter and subsequently activate UPRmt by an unknown mechanism (Haynes et al., 2010, 2013). Another, likely more prominent, component of UPRmt in nematodes is represented by the versatile bZip activating transcription factor associated with stress 1 (ATFS-1) equipped with both a mitochondrial targeting sequence and a nuclear localization signal (Haynes et al., 2010; Nargund et al., 2012). Under normal conditions, ATFS-1 is targeted to mitochondria and inactivated through degradation by the LON protease. However, under conditions of mitochondrial stress that attenuate protein import into the organelle, a portion of AFTS-1 relocates to the nucleus. It was recently reported that UPRmt-associated chromatin remodeling allows nuclear ATFS-1 to bind targeted sequences (Tian et al., 2016) and promote expression of more than 400 genes encoding for proteins involved in mitochondrial proteostasis, metabolism, and innate immunity (Lin and Haynes, 2016).
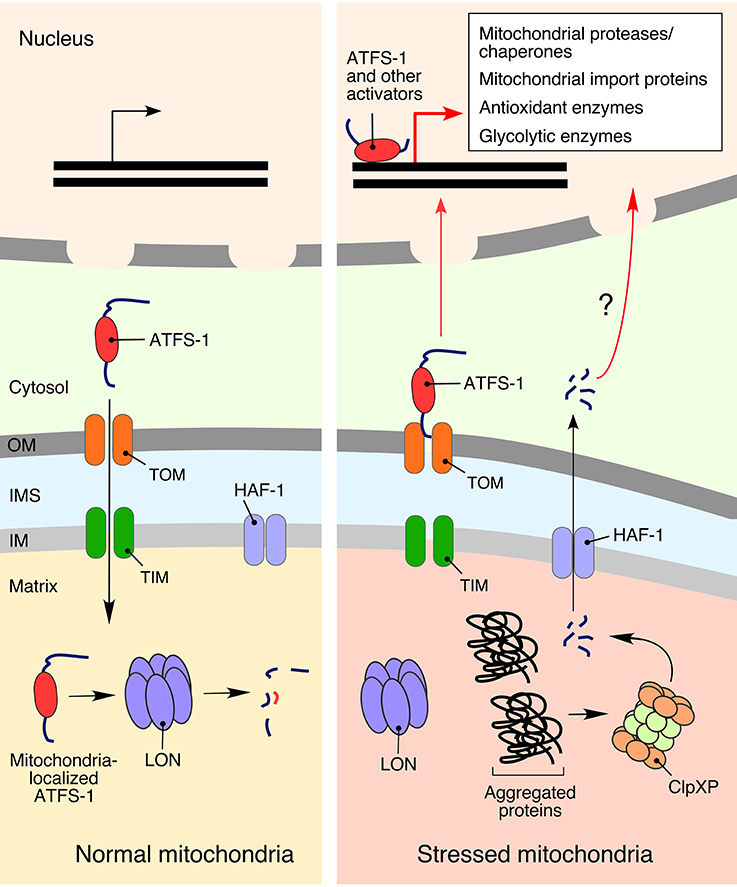
Figure 3. The mitochondrial unfolded protein response (UPRmt). Activation of UPRmt in response to mitochondrial damage is best characterized in the roundworm Caenorhabditis elegans, where it is regulated by the activating transcription factor associated with stress 1 (ATFS-1). In the absence of stress stimuli, ATFS1 is transported to the mitochondria and degraded by LON protease (Nargund et al., 2012). Conditions of mitochondrial stress stall mitochondrial import of ATFS-1, allowing a fraction of the protein to translocate into the nucleus and promote expression of more than 400 mitochondrial homeostasis-related genes (Lin and Haynes, 2016). Another, not-so-well defined, activation of UPRmt in C. elegans is associated with peptide exporter HAF-1, which translocates signaling peptides generated by the matrix peptidase ClpXP (Haynes et al., 2010).
UPRmt activation in mammalian cells is less understood. It appears to be more complex and stochastic and is achieved via a complex signaling cascade requiring sequential activation of the c-Jun N-terminal kinase, a component of AP-1 transcription factor c-Jun, and finally transcription factors C/EBPβ and C/EBP homologous protein (CHOP) (Pellegrino et al., 2013; Topf et al., 2016), which enhance the expression of so-called canonical UPRmt genes, including those that encode mitochondrial chaperones and proteases (Aldridge et al., 2007; Horibe and Hoogenraad, 2007). Interestingly, a recent study by Seiferling et al. (2016) demonstrated that CLPP appears to play no role in mammalian UPRmt activation.
The other, likely related to the UPRmt, responses reported in mammals include HTRA2 protease-mediated response in the brain (Moisoi et al., 2009) and the StAR accumulation response in steroidogenic cells (Bahat et al., 2014, 2015). The former is a brain-specific transcriptional response caused by the loss of HTRA2 in neuronal cells and a subsequent respiratory defect and accumulation of unfolded proteins in the mitochondria of these cells (Moisoi et al., 2009). The StAR overload response is triggered by elevated levels of the steroidogneic acute regulatory protein (StAR) in the mitochondrial matrix and promotes expression of several mitochondrial proteases to counter the accumulation of this protein within the mitochondria (Bahat et al., 2014, 2015). The molecular aspects of these responses are yet to be clarified.
Abnormal accumulation of mitochondrial precursor polypeptides was recently linked to activation of stress responsive pathways in the cytosol (Topf et al., 2016). Two recent publications reported a novel type of mitochondrial proteostatic response termed mPOS (mitochondrial precursor over-accumulation stress) (Wang and Chen, 2015; Wrobel et al., 2015). The mPOS is associated with defects in mitochondrial import machinery, and inner membrane integrity and function. The response appears to modulate two distinct cellular activities: (1) cytosolic protein synthesis; and (2) an unfolded protein response activated by protein mistargeting (so-called UPRam). The latter significantly differs from the canonical mitochondrial unfolded protein response (UPRmt), a pathway triggered by impaired mitochondrial proteostasis (Wrobel et al., 2015). Protein synthesis regulation by mPOS involves a global increase in cap-independent translation (and reciprocal reduction of cap-dependent translation), as well as modulation of ribosomal biogenesis (Wang and Chen, 2015; Wrobel et al., 2015). The signaling mechanisms underpinning these events remain to be determined.
A similar modulation of protein biogenesis is also related to antibiotic-induced defects in mitochondrial translation. Stalling of mitochondrial ribosomes in mouse embryonic fibroblasts by treatment with the mitochondrial translation inhibitor, actinonin, activates cellular proliferation signaling pathways such as that of p53 and MAP kinase (Richter et al., 2013).
Higher eukaryotes possess yet another elegant mechanism to signal mitochondrial distress and promote removal of dysfunctional mitochondria via selective autophagy (mitophagy). This mechanism, known as the PINK1-Parkin relay (Figure 4), composed of the phosphatase and tensin homolog (PTEN)-induced ubiquitin kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin, mediates poly-ubiquitylation of damaged mitochondria thereby priming mitophagy (Sarraf et al., 2013; Lazarou et al., 2015). Similar to ATFS-1, PINK1 is imported into mitochondria under normal conditions, wherein the protein is promptly degraded by the IM resident presenilin associated rhomboid-like protease (PARL) (Jin et al., 2010). Conditions of mitochondrial stress that perturb protein import into the organelle cause the stabilization and accumulation of PINK1 on the mitochondrial outer membrane (OM), where this kinase drives phosphorylation of Parkin as well as ubiquitin (Kondapalli et al., 2012; Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Ordureau et al., 2014). These actions promote: (1) mitochondrial retention of Parkin (Ordureau et al., 2014); (2) subsequent assembly of poly-ubiquitin chains on various OM proteins (Sarraf et al., 2013); and (3) activation of autophagosome-stimulating protein TANK-binding kinase 1, TBK1 (Heo et al., 2015). Of note, genetic mutations in the components of this pathway have been linked to familial cases of neurodegenerative diseases such as Parkinson's disease (Pickrell and Youle, 2015) and amyotrophic lateral sclerosis (Maruyama et al., 2010; Wong and Holzbaur, 2014; Cirulli et al., 2015).
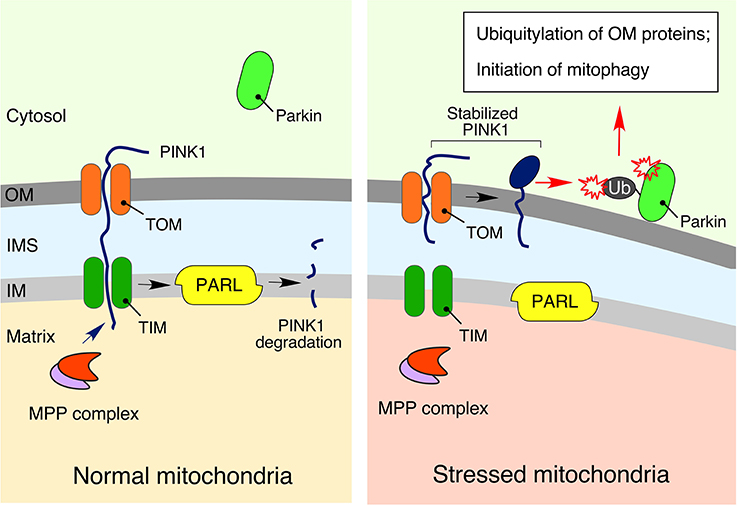
Figure 4. The PINK1-Parkin signaling relay. Under physiological conditions, phosphatase and tensin homolog (PTEN)-induced ubiquitin kinase 1 (PINK1), a key regulator of mitophagy and mitochondrial turnover, is imported into the mitochondria via the translocases of outer (TOM) and inner (TIM) membranes. It is subsequently degraded by presenilin associated rhomboid-like protease (PARL) in the IM (Jin et al., 2010). However, in response to mitochondrial damage, stabilized PINK1 accumulates on the outer membrane (OM), promoting recruitment, and activation of the E3 ubiquitin ligase parkin, followed by ubiquitylation of OM-associated proteins, and initiation of mitophagy (Kondapalli et al., 2012; Kane et al., 2014; Kazlauskaite et al., 2014; Koyano et al., 2014; Ordureau et al., 2014). More details are available in the text.
Interestingly, the recently described mitochondria-derived vesicles (MDV) quality control pathway that appears to mediate crosstalk between mitochondria and either peroxisomes or lysosomes (Neuspiel et al., 2008; Sugiura et al., 2012) also seems to rely on the mitophagy-independent function of the PINK1-Parkin relay (Sugiura et al., 2012).
Mitochondria are intimately involved in the control of apoptotic signaling cascades. In particular, initiation of apoptosis is associated with release of the IM and IMS proteins such as cytochrome c, apoptosis inducing factor AIF, second mitochondria-derived activator of caspase protein (SMAC), serine protease high temperature requirement protein A2 (HTRA2), and endonuclease G. Upon their release from irreversibly damaged mitochondria through the β-cell CLL/lymphoma 2 (Bcl2) family proteins-mediated mitochondrial outer membrane permeabilization, these proteins serve as signaling molecules to regulate and propel cell death processes (Tait and Green, 2010).
The small nuclear-encoded electron carrier cytochrome c plays one of the key roles in programmed cell death—its release is a critical event necessary to initiate an apoptotic program (Liu et al., 1996; Hüttemann et al., 2011). This rapid feed-forward process commences with disassociation of cytochrome c from its complexes with the mitochondria-specific phospholipid cardiolipin following the protein's detachment from the IM (Goldstein et al., 2000; Ott et al., 2002; Garrido et al., 2006). While cytochrome c-independent forms of cell death have been reported (e.g., see the review by Sevrioukova, 2011), here we will focus on mitochondrial signaling via this molecule.
Accumulation of released cytochrome c in the cytosol triggers the formation of the apoptosome—a wheel-like structure containing seven molecules of apoptotic protease-activating factor 1 (APAF-1) equaled by cytochrome c. The apoptosome complex activates the key apoptotic factor, caspase 9, which subsequently promotes the execution of the apoptotic program (Reubold and Eschenburg, 2012). Of note, cytochrome c-induced caspase activation is not always associated with cell death. Examples of the vital processes requiring cytochrome c release include platelet formation, β-cell proliferation, and monocyte-macrophage differentiation (Garrido et al., 2006). Similarly, in Drosophila melanogaster, cytochrome c serves as a crucial signaling molecule in caspase-mediated sperm cell development (Nagata, 2016).
Just like in bacteria, the initiation of mitochondrial translation requires N-formylmethionine-tRNA (Ott et al., 2016). Mitochondria-derived N-formylated peptides have long been known as potent chemoattractants for neutrophils (Carp, 1982). Further studies demonstrated binding of the polymorphonuclear neutrophils (PMN)-expressed formyl peptide receptors (FPRs) by native and synthetic peptides originated from mitochondrial Complex I subunits 4 and 6, Complex IV subunit 1 (Rabiet et al., 2005), and cytochromes b (Mukai et al., 2009; Seki et al., 2011) and c (Hokari et al., 2012). Subsequent activation of FPRs involves multiple signaling cascades controlling calcium flux, p38 kinase activation, and cytokine production (Rabiet et al., 2007). It is noteworthy that the release of formylated mitochondrial peptides following trauma is associated with inflammation, thus potentially linking injury to non-infective systemic inflammatory response syndrome and sepsis (Zhang et al., 2010).
The mammalian mitochondrial genome is known to encode only 11 proteins (Ott et al., 2016). However, this number might be extended due to the recent discovery of short open reading frames (sORFs), which encode so-called mitochondrial-derived peptides (MDPs) (Lee et al., 2016). Perhaps the best-characterized MDP is a 24-amino acid long peptide named humanin—which was originally shown to bind to the pro-apoptotic factor Bax in a manner similar to that of its nuclear-borne homolog; therefore suggesting a potential regulatory role in cell death (Guo et al., 2003). More detailed reviews of various humanin-associated cytoprotective effects can be found elsewhere (Lee et al., 2013).
Another MDP called MOTS-c (mitochondrial open reading frame of the 12S rRNA-c) was discovered more recently. The activity of this 16-amino acid long peptide has been linked to the regulation of muscle energy expenditure and insulin sensitivity. MOTS-c activates a metabolic/signaling axis comprised of the one-carbon folate cycle and AMPK, thereby providing an obesity-protective effect (Lee et al., 2015). Interestingly, the most recent study by Cobb et al. (2016) reported identification of six sORFs originating from the same genetic locus as humanin which have been named small humanin-like peptides (SHLPs). The signaling roles of SHLPs remain to be elucidated.
In this section, we will survey several additional molecules that cannot be placed into any of the above groups of mitochondria-borne signals. This does not imply that the following molecules are not linked to the pathways described above. Likewise, these signals do not appear to function on their own and are well intertwined with the signals described above and with each other. For instance, signaling via changes in mitochondrial and intracellular calcium levels is linked to ROS-mediated communication, as well as signals arising from defects in the mitochondrial genome.
The mitochondria can be viewed as an efficient Ca2+ sink that assists the endoplasmic reticulum (ER) and lysosomes in control of cellular metabolism and death via apoptosis, necrosis, and autophagy (Rizzuto et al., 2012). Active flux of Ca2+ between the ER and mitochondria is driven via the mammalian mitochondria-associated membranes (MAMs) or yeast's ER-mitochondria encounter structures (ERMES). A decrease of Ca2+ levels in the mitochondria leads to a rapid drop in ATP production, followed by activation of the AMPK signaling pathway, resulting in subsequent initiation of autophagy (La Rovere et al., 2016). Conversely, mitochondrial Ca2+ overload promotes opening of the mitochondrial permeability transition pore (mPTP), a non-specific transporting channel in the IM resulting in mitochondrial membrane depolarization, swelling, OM rupture, and release of IM and IMS residing pro-apoptotic factors (Bhosale et al., 2015; Takeuchi et al., 2015). Increased levels of Ca2+ in the cytosol can activate various soluble Ca2+-binding proteins such as calmodulin, which in turn can bind to various targets including calmodulin-dependent CaMK protein kinases, myosin light chain kinase MLCK, and calcinuerin—the key regulators of a plethora of cellular processes (Tidow and Nisse, 2013). Of note, excessive accumulation of Ca2+ and failure to clear cytoplasmic Ca2+ have been linked to necrosis initiation (Rizzuto et al., 2012).
The first insights into the impact of mitochondrial DNA (mtDNA) depletion or lesions on nuclear gene expression in mammalian cells were reported by Avadhani's lab in the late 1990's and linked these defects to decreased Ca2+ buffering capacity of the mitochondria and ultimately cytosolic calcium accumulation (Biswas et al., 1999; Amuthan et al., 2001). As outlined in the previous section, elevated levels of cytosolic trigger the activation of calcineurin and downstream transcriptional factors such as NF-κB, CHOP, and extracellular signal-regulated kinase 1 (ERK1) (Butow and Avadhani, 2004).
mtDNA, with its circular loop and numerous regions of non-methylated DNA (CpG islands), partially resembles bacterial genomes (Fang et al., 2016). Similar to bacterial pathogen-associated molecular patterns, mtDNA along with formylated peptides (see above) has been described as mitochondria-derived damage-associated molecular patterns (DAMPs) that are capable of PMNs stimulation via Toll-like receptor 9 (TLR9) and, subsequently, the p38 MAP kinase signaling cascade (Zhang et al., 2010). In addition, accumulation of mtDNA in the cytosol can activate the DNA-specific sensor, cGAS, which promotes signal transduction via the STING-TBK1-interferon regulatory factor 3 (IRF3)-dependent pathway, thereby increasing interferon-stimulated gene expression. This chain of events ultimately results in activation of an antiviral immune response (West et al., 2015). Under apoptotic conditions, oxidized mtDNA also appear to activate nucleotide-binding domain leucine-rich repeat family, pyrin domain containing 3 (NLRP3) inflammasome, and IL-1β production (Shimada et al., 2012). It is noteworthy that the NLRP3 inflammasome itself is associated with mitochondria through its interactions with mitochondrial antiviral signaling protein MAVS, mitochondrial OM GTPases mitofusins, caspase-like apoptosis regulatory protein c-FLIP, and the phospholipid cardiolipin (Elliot and Sutterwala, 2015).
mtDNA maintenance and replication requires a complex network of factors encoded by both the nuclear and the organelle's genomes (Ott et al., 2016). Mutations leading to the depletion of mtDNA have a profound effect not only on mitochondrial health, but also on the overall course of various diseases in human patients (Young and Copeland, 2016). For instance, mutated forms of the mtDNA helicase, TWINKLE, are associated with mitochondrial myopathy (MM) and infantile onset spinocerebellar ataxia (IOSCA). Mouse models for MM and IOSCA display a significant number of mtDNA replication defects upon modulation of purine and serine/glutathione biosynthesis (Nikkanen et al., 2016). Previous studies in the late-onset MM mouse model also demonstrated a fasting-like transcriptional response via the AKT1-PI3K signaling cascade upon disease progression. Particularly, increased production of the starvation-associated fibroblast growth factor 21 (Fgf21) was detected in the mutation-carrying animals, which, in turn, caused profound changes to lipid metabolism, including resistance to high-fat diet (Tyynismaa et al., 2010). CHOP10 is another transcription factor activated upon defective mtDNA expression. However, this response does not appear to be associated with the UPRmt and is likely a manifestation of a general stress response (Michel et al., 2015).
Cardiolipin (CL) is a unique non-bilayer phospholipid specific to mitochondrial membranes. Mounting evidence indicates that CL contributes to a plethora of mitochondrial processes including signaling events required to initiate mitophagy and apoptotic cell death (Lu and Claypool, 2015). Under normal physiological conditions, CL is almost exclusively localized to the IM. However, mitochondrial stress stimuli trigger random CL translocation to the OM, and this event serves as a signal to activate the autophagy machinery via CL binding to its central component, microtubule-associated protein 1 light chain 3 (LC3) (Chu et al., 2013; Kagan et al., 2015). Mitophagy-specific induction by CL requires the phospholipid's remodeling by tafazzin, an evolutionary conserved monolyso-CL transacylase (Hsu et al., 2015). Mutations in tafazzin are linked to the human multisystemic disorder, Barth syndrome (Lu and Claypool, 2015). Studies in lymphoblastoid cells from Barth syndrome patients reveal an important role for CL in the translocation of the pro-apoptotic protease, caspase 8 (Gonzalvez et al., 2008).
Release of mitochondria-associated pro-apoptotic factors is believed to require selective CL oxidation by cytochrome c (Kagan et al., 2005; Choi et al., 2007); in vitro experiments suggest that cytochrome c might acquire its peroxidation activity upon interaction with the phospholipid (Basova et al., 2007). The CL-mediated decision (mitophagy vs. apoptosis) likely depends on the extent of CL externalization (Lu and Claypool, 2015).
Finally, CL is implicated as a potent immunogenic factor. First, it is capable of activating the NLRP3 inflammasome via direct binding to the NLRP3 component. As such, this event leads to caspase 1 activation and further production of the proinflammatory cytokines, IL-1β, and IL-18 (Iyer et al., 2013). Another exciting aspect is the ability of liposomes and mitochondria with CL-containing surfaces to be efficiently recognized and engulfed by macrophages in a CD36-dependent manner in vitro (Balasubramanian et al., 2015). It will be interesting to ascertain if a similar process occurs in vivo.
In this review, we have surveyed several mitochondrial signals that regulate the organelle's biogenesis and stress-response pathways. One cannot help but notice that a number of these signals and responses appear to be triggered by similar, if not the same, homeostatic insults. How mitochondria-borne signals are interrelated, and if there is any signaling hierarchy, remains to be clarified. It will be important to gain further mechanistic understanding into how mitochondrial stress signals are propagated and transferred. Because a number of the insights described above were gleaned from studies in model organisms or in vitro cell cultures, it will be equally important to determine the extent and magnitude of such signaling responses in organs and tissues. These findings may help to better understand a tissue-specific nature of certain mitochondria-related pathologies.
Another exciting question is whether these signals can be manipulated to increase mitochondrial stress tolerance and achieve therapeutic benefits in mitigating disease-related states or extending lifespan. In that respect, recent findings such as NAD+ repletion-mediated improvement of mitochondrial function and lifespan in mice (Zhang et al., 2016) or potential therapeutic effects of hypoxic preconditioning in the rodent model of mitochondrial disease (Jain et al., 2016), as well as ones of mitochondria-targeted antioxidants in the murine model of Huntington's disease (Xun et al., 2012) are encouraging and create a solid premise for future translational studies. Likewise, targeting mitochondrial signals or signaling pathways emerges as a prospective theranostic paradigm in treatment of various cancers (Zong et al., 2016).
IB analyzed the literature and co-wrote the manuscript. OK conceived the idea, analyzed the literature, co-wrote the manuscript, and designed the figures.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to those authors whose work we were unable to cite due to space limitations. We thank members of the Khalimonchuk laboratory for useful discussions and edits. OK is supported by the NIH grants R01 GM108975 and P30 GM103335 (to Nebraska Redox Biology Center).
Adam, J., Hatipoglu, E., O'Flaherty, L., Ternette, N., Sahgal, N., Lockstone, H., et al. (2011). Renal cyst formation ih Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537. doi: 10.1016/j.ccr.2011.09.006
Adam, J., Yang, M., Soga, T., and Pollard, P. J. (2014). Rare insights into cancer biology. Oncogene 33, 2547–2556. doi: 10.1038/onc.2013.222
Aldridge, J. E., Horibe, T., and Hoogenraad, N. J. (2007). Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2:e874. doi: 10.1371/journal.pone.0000874
Amuthan, G., Biswas, G., Zhang, S. Y., Klein-Szanto, A., Vijayasarathy, C., and Avadhano, N. G. (2001). Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J. 20, 1910–1920. doi: 10.1093/emboj/20.8.1910
Antunes, F., Boveris, A., and Cadenas, E. (2004). On the mechanism and biology of cytochrome oxidase inhibition by nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 101, 16774–16779. doi: 10.1073/pnas.0405368101
Armstrong, J. S., Whiteman, M., Yang, H., and Jones, D. P. (2004). The redox regulation of intermediary metabolism by a superoxide-aconitase rheostat. Bioessays 26, 894–900. doi: 10.1002/bies.20071
Bahat, A., Perlberg, S., Melamed-Book, N., Isaac, S., Eden, A., Lauria, I., et al. (2015). Transcriptional activation of LON gene by a new form of mitochondrial stress: a role for the nuclear respiratory factor 2 in StAR overload response (SOR). Mol. Cell. Endocrinol. 408, 62–72. doi: 10.1016/j.mce.2015.02.022
Bahat, A., Perlberg, S., Melamed-Book, N., Lauria, I., Langer, T., and Orly, J. (2014). StAR enhances transcription of genes encoding the mitochondrial proteases involved in its own degradation. Mol. Endocrinol. 28, 208–224. doi: 10.1210/me.2013-1275
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Balasubramanian, K., Maeda, A., Lee, J. S., Mohammadyani, D., Dar, H. H., Jiang, J. F., et al. (2015). Dichotomous roles for externalized cardiolipin in extracellular signaling: Promotion of phagocytosis and attenuation of innate immunity. Sci. Signal. 8:ra95. doi: 10.1126/scisignal.aaa6179
Basova, L. V., Kurnikov, I. V., Wang, L., Ritov, V. B., Belikova, N. A., Vlasova, I. I., et al. (2007). Cardiolipin switch in mitochondria: shutting off the reduction of cytochrome c and turning on the peroxidase activity. Biochemistry 46, 3423–3434. doi: 10.1021/bi061854k
Bender, T., Lewrenz, I., Franken, S., Baitzel, C., and Voos, W. (2011). Mitochodnrial enzymes are protected from stress-induced aggregation by mitochondrial chaperones and the Pim1/LON protease. Mol. Biol. Cell 22, 541–554. doi: 10.1091/mbc.E10-08-0718
Bhosale, G., Sharpe, J. A., Sundier, S. Y., and Duchen, M. R. (2015). Calcium signaling as a mediator of cell energy demand a trigger to cell death. Ann. N.Y. Acad. Sci. 1350, 107–116. doi: 10.1111/nyas.12885
Birsoy, K., Wang, T., Chen, W. W., Freinkman, E., Abu-Remaileh, M., and Sabatini, D. M. (2015). An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551. doi: 10.1016/j.cell.2015.07.016
Biswas, G., Adebanjo, O. A., Freedman, B. D., Anandatheerthavarada, H. K., Vijayasarathy, C., Zaidi, M., et al. (1999). Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J. 18, 522–533. doi: 10.1093/emboj/18.3.522
Bohovych, I., Chan, S. S., and Khalimonchuk, O. (2015). Mitochondrial protein quality control: the mechanisms guarding mitochondrial health. Antioxid. Redox Signal. 22, 977–994. doi: 10.1089/ars.2014.6199
Bohovych, I., Kastora, S., Christianson, S., Topil, D., Kim, H., Fangman, T., et al. (2016). Oma1 links mitochondrial protein quality control and TOR signaling to modulate physiological plasticity and cellular stress responses. Mol. Cell Biol. 36, 2300–2312. doi: 10.1128/MCB.00156-16
Brandes, S. N., and Jakob, U. (2009). Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 11, 997–1014. doi: 10.1089/ars.2008.2285
Butow, R. A., and Avadhani, N. G. (2004). Mitochondrial signaling: the retrograde response. Mol. Cell 14, 1–15. doi: 10.1016/S1097-2765(04)00179-0
Canto, C., Menzies, K. J., and Auwerx, J. (2015). NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 22, 31–53. doi: 10.1016/j.cmet.2015.05.023
Carp, H. (1982). Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med. 155, 264–275. doi: 10.1084/jem.155.1.264
Chacinska, A., Koehler, C. M., Milencovic, D., Lithgow, T., and Pfanner, N. (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644. doi: 10.1016/j.cell.2009.08.005
Chandel, N. S. (2014). Mitochondria as signaling organelles. BMC Biol. 12:34. doi: 10.1186/1741-7007-12-34
Choi, S. Y., Gonsalvez, F., Jenkins, G. M., Stomianny, C., Chretein, D., Arnoult, D., et al. (2007). Cardiolipin deficiency releases cytochrome c from the inner mitochondrial membrane and accelerates stimuli-elicited apoptosis. Cell Death Differ. 14, 597–606. doi: 10.1038/sj.cdd.4402020
Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. (2009). Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. doi: 10.1126/science.1175371
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Cirulli, E. T., Lasseigne, B. N., Petrovski, S., Sapp, P. C., Dion, P. A., Leblond, C. S., et al. (2015). Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441. doi: 10.1126/science.aaa3650
Cobb, L. J., Lee, C., Xiao, J., Yen, K., Wong, R. G., Nakamura, H. K., et al. (2016). Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 8, 796–809. doi: 10.18632/aging.100943
Collins, Y., Chouchani, E. T., James, A. M., Menger, K. E., Cocheme, H. M., and Murphy, M. P. (2012). Mitochondrial redox signaling at a glance. J. Cell Sci. 125, 801–806. doi: 10.1242/jcs.098475
Connor, K. M., Subbaram, S., Regan, K. J., Nelson, K. K., Mazurkiewicz, J. E., Bartholomew, P. J., et al. (2005). Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation. J. Biol. Chem. 280, 16916–16924. doi: 10.1074/jbc.M410690200
Corcoran, A., and Cotter, T. G. (2013). Redox regulation of protein kinases. FEBS J. 280, 1944–1965. doi: 10.1111/febs.12224
Couvillion, M. T., Soto, I. C., Shipkovenska, G., and Churchman, L. S. (2016). Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503. doi: 10.1038/nature18015
Denu, J., and Tanner, K. G. (1998). Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 37, 5633–5642. doi: 10.1021/bi973035t
DiMauro, S., and Schon, E. A. (2003). Mitochondrial respiratory chain diseases. N. Engl. J. Med. 348, 2656–2668. doi: 10.1056/NEJMra022567
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Eisenberg, T., Schroeder, S., Andryushkova, A., Pendl, T., Küttner, V., Bhukel, A., et al. (2014). Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme A stimulates autophagy and prolongs lifespan. Cell Metab. 19, 431–444. doi: 10.1016/j.cmet.2014.02.010
Elliot, E., and Sutterwala, F. (2015). Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 265, 35–52. doi: 10.1111/imr.12286
Erickson, J. R., Joiner, M. A., Guan, X., Kutschke, W., Yang, J., Oddis, C. V., et al. (2008). A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133, 462–474. doi: 10.1016/j.cell.2008.02.048
Evans, D. R., and Guy, H. I. (2004). Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J. Biol. Chem. 279, 33035–33038. doi: 10.1074/jbc.R400007200
Fang, C., Wei, X., and Wei, Y. (2016). Mitochondrial DNA in the regulation of innate immune responses. Protein Cell 7, 11–16. doi: 10.1007/s13238-015-0222-9
Figueira, T. R., Barros, M. H., Camargo, A. A., Castilho, R. F., Ferreira, J. C., Kowaltowski, A. J., et al. (2013). Mitochodnria asa source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid. Redox Signal. 18, 2029–2074. doi: 10.1089/ars.2012.4729
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
García-Santamarina, S., Boronat, S., and Hidalgo, E. (2014). Reversible cysteine oxidation in hydrogen peroxide sensing and signal transduction. Biochemistry 53, 2560–2580. doi: 10.1021/bi401700f
Garrido, C., Galluzzi, L., Brunet, M., Puig, P. E., Didelot, C., and Kroemer, G. (2006). Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 13, 1423–1433. doi: 10.1038/sj.cdd.4401950
Gaude, E., and Frezza, C. (2014). Defects in mitochondrial metabolism and cancer. Cancer Metab. 2:10. doi: 10.1186/2049-3002-2-10
Goldstein, J. C., Waterhouse, N. J., Juin, P., Evan, G. I., and Green, D. R. (2000). The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2, 156–162. doi: 10.1038/35004029
Gonzalvez, F., Schug, Z. T., Houtkooper, R. H., MacKenzie, E. D., Brooks, D. G., Wanders, R. J., et al. (2008). Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J. Cell Biol. 183, 681–696. doi: 10.1083/jcb.200803129
Guo, B., Zhai, D., Cabezas, E., Welsh, K., Nouraini, S., Satterthwait, A. C., et al. (2003). Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423, 456–461. doi: 10.1038/nature01627
Guzy, R. D., Hoyos, B., Robin, E., Chen, H., Liu, L., Mansfield, K. D., et al. (2005). Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408. doi: 10.1016/j.cmet.2005.05.001
Hall, J. A., Dominy, J. E., Lee, Y., and Puigserver, P. (2013). AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. J. Clin. Invest. 123, 973–979. doi: 10.1172/JCI64094
Hamanaka, R. B., and Chandel, N. S. (2010). Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biol. Sci. 35, 505–513. doi: 10.1016/j.tibs.2010.04.002
Hardie, D. G., Schaffer, B. E., and Brunet, A. (2016). AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201. doi: 10.1016/j.tcb.2015.10.013
Harman, D. (1972). The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 20, 145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x
Haynes, C. M., Fiorese, C. J., and Lin, Y. F. (2013). Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded protein response and beyond. Trends Cell Biol. 23, 311–318. doi: 10.1016/j.tcb.2013.02.002
Haynes, C. M., Yang, Y., Blais, S. P., Neubert, T. A., and Ron, D. (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529–540. doi: 10.1016/j.molcel.2010.01.015
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005). Protein s-nitorsylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166. doi: 10.1038/nrm1569
Hokari, Y., Seki, T., Nakano, H., Matsuo, Y., Fukamizu, A., Munekata, E., et al. (2012). Isolation and identification of novel neutrophil-activating cryptides hidden in mitochondrial cytochrome C. Protein Pept. Lett. 19, 680–687. doi: 10.2174/092986612800494048
Horibe, T., and Hoogenraad, N. J. (2007). The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2:e835. doi: 10.1371/journal.pone.0000835
Hsu, O., Liu, X., Zhang, J., Wang, H. G., Ye, J. M., and Shi, Y. (2015). Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy. Autophagy 11, 643–652. doi: 10.1080/15548627.2015.1023984
Hüttemann, M., Pecina, P., Rainbolt, M., Sanderson, T. H., Kagan, V. E., Samavati, L., et al. (2011). The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: from respiration to apoptosis. Mitochondrion 11, 369–381. doi: 10.1016/j.mito.2011.01.010
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Jain, I. H., Zazzeron, L., Goli, R., Alexa, K., Schatzman-Bone, S., Dhillon, H., et al. (2016). Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61. doi: 10.1126/science.aad9642
Jin, S. M., Lazarou, M., Wang, C., Kane, L. A., Narendra, D. P., and Youle, R. J. (2010). Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 191, 933–942. doi: 10.1083/jcb.201008084
Kagan, V. E., Tyurin, V. A., Jiang, J., Tyurina, Y. Y., Ritov, V. B., Amoscato, A. A., et al. (2005). Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1, 223–232. doi: 10.1038/nchembio727
Kagan, V. E., Tyurina, Y. Y., Tyurin, V. A., Mohammadyani, D., Angeli, J. P., Baranov, S. V., et al. (2015). Cardiolipin signaling mechanisms: collapse of asymmetry and oxidation. Antioxid. Redox Signal. 22, 1667–1680. doi: 10.1089/ars.2014.6219
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/BJ20140334
Khutorenko, A. A., Roudko, V. V., Chernyak, B. V., Vartapetian, A. B., Chumakov, P. M., and Evstafieva, A. G. (2010). Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 12828–12833. doi: 10.1073/pnas.0910885107
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 520, 162–166. doi: 10.1038/nature13392
La Rovere, R. M., Roest, G., Bultynck, G., and Parys, J. B. (2016). Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium. 60, 74–87. doi: 10.1016/j.ceca.2016.04.005
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, C., Kim, K. H., and Cohen, P. (2016). MOTS-c: a novel mitochondrial-derived peptide regulating muscle and fat metabolism. Free Radic. Biol. Med. [Epub ahead of print]. doi: 10.1016/j.freeradbiomed.2016.05.015
Lee, C., Yen, K., and Cohen, P. (2013). Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol. Metab. 24, 222–228. doi: 10.1016/j.tem.2013.01.005
Lee, C., Zeng, J., Drew, B. G., Sallam, T., Martin-Montalvo, A., Wan, J., et al. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 21, 443–454. doi: 10.1016/j.cmet.2015.02.009
Liao, X., and Butow, R. A. (1993). RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell 72, 61–71. doi: 10.1016/0092-8674(93)90050-Z
Lin, Y. F., and Haynes, C. M. (2016). Metabolism and UPR(mt). Mol. Cell 61, 677–682. doi: 10.1016/j.molcel.2016.02.004
Lin, Y. F., Schulz, A. M., Pellegrino, M. W., Lu, Y., Shaham, S., and Haynes, C. M. (2016). Maintenance and propagation of a deleterious mitochondrial genome be the mitochondrial unfolded protein response. Nature 533, 416–419. doi: 10.1038/nature17989
Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. doi: 10.1016/S0092-8674(00)80085-9
Liu, Z., and Butow, R. A. (1999). A transcriptional switch in the expression of yeast tricarboxylic acid cycle genes in response to a reduction or loss of respiratory function. Mol. Cell. Biol. 19, 6720–6728. doi: 10.1128/MCB.19.10.6720
Losman, J. A., and Kaelin, W. G. (2013). What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852. doi: 10.1101/gad.217406.113
Lu, Y. W., and Claypool, S. M. (2015). Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front. Genet. 6:3. doi: 10.3389/fgene.2015.00003
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurinin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.1038/nature08971
Michel, S., Canonne, M., Arnould, T., and Renard, P. (2015). Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion 21, 58–68. doi: 10.1016/j.mito.2015.01.005
Moisoi, N., Klupsch, K., Fedele, V., East, P., Sharma, S., Renton, A., et al. (2009). Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 16, 449–464. doi: 10.1038/cdd.2008.166
Mukai, H., Seki, T., Nakano, H., Hokari, Y., Takao, T., Kawanami, M., et al. (2009). Mitocryptide-2: purification, identification, and characterization of a novel cryptide that activates neutrophils. J. Immunol. 182, 5072–5080. doi: 10.4049/jimmunol.0802965
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem J. 417, 1–13. doi: 10.1042/BJ20081386
Nagata, S. (2016). Cell biology: killer enzymes tethered. Nature 533, 474–476. doi: 10.1038/nature18439
Nakamura, T., Tu, S., Akhtar, M. W., Sunico, C. R., Okamoto, S., and Lipton, S. A. (2013). Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78, 596–614. doi: 10.1016/j.neuron.2013.05.005
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P., and Haynes, C. M. (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 58, 123–133. doi: 10.1016/j.molcel.2015.02.008
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. doi: 10.1126/science.1223560
Neupert, W., and Herrmann, J. M. (2007). Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76, 723–749. doi: 10.1146/annurev.biochem.76.052705.163409
Neuspiel, M., Schauss, A. C., Braschi, E., Zunino, R., Rippstein, P., Rachubinski, R. A., et al. (2008). Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr. Biol. 18, 102–108. doi: 10.1016/j.cub.2007.12.038
Nikkanen, J., Forsström, S., Euro, L., Paetau, I., Kohnz, R. A., Wang, L., et al. (2016). Mitochondrial DNA replication defects disturb cellular dNTP pools and remodel one-carbon metabolism. Cell Metab. 23, 635–648. doi: 10.1016/j.cmet.2016.01.019
Nisoli, E., Clementi, E., Paolucci, C., Cozzi, V., Tonello, C., Sciorati, C., et al. (2003). Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899. doi: 10.1126/science.1079368
Ordureau, A., Sarraf, S. A., Duda, D. M., Heo, J. M., Jedrychowski, M. P., Sviderskiy, V. O., et al. (2014). Quantitative proteomics of reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56, 360–375. doi: 10.1016/j.molcel.2014.09.007
Ott, M., Amunts, A., and Brown, A. (2016). Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 85, 1.1–1.25. doi: 10.1146/annurev-biochem-060815-014334
Ott, M., Robertson, J. D., Gogvadze, V., Zhitovsky, B., and Orrenius, S. (2002). Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. U.S.A. 99, 1259–1263. doi: 10.1073/pnas.241655498
Pan, Y., Schroeder, E. A., Ocampo, A., Barrientos, A., and Shadel, G. S. (2011). Regulation of yeast chronological life span by TORC1 via adaptivemitochondrial ROS signaling. Cell Metab. 13, 668–678. doi: 10.1016/j.cmet.2011.03.018
Parker, S. J., and Metallo, C. M. (2015). Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 152, 54–62. doi: 10.1016/j.pharmthera.2015.05.003
Patterson, H. C., Gerbeth, C., Thiru, P., Vogtle, N. F., Knoll, M., Shahsafaei, A., et al. (2015). A respiratory chain controlled signal transduction cascade in the mitochondrial intermembrane space mediates hydrogen peroxide signaling. Proc. Natl. Acad. Sci. U.S.A. 112, E5679–E5688. doi: 10.1073/pnas.1517932112
Pearce, E. L., Poffenberger, M. C., Chang, C. H., and Jones, R. G. (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454. doi: 10.1126/science.1242454
Pellegrino, M., Nargund, A., and Haynes, C. M. (2013). Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410–416. doi: 10.1016/j.bbamcr.2012.02.019
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin and mitochondrial fidelity in Parkinson's disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Rabiet, M. J., Huet, E., and Boulay, F. (2005). Human mitochondria-derived N-formylated peptides are novel agonists equally active on FPR and FPRL1, while Listeria monocytogenes-derived peptides preferentially activate FPR. Eur. J. Immunol. 35, 2486–2495. doi: 10.1002/eji.200526338
Rabiet, M. J., Huet, E., and Boulay, F. (2007). The N-formyl peptide receptors and the anaphylatoxin C5a receptors: an overview. Biochimie 89, 1089–1106. doi: 10.1016/j.biochi.2007.02.015
Reubold, T. F., and Eschenburg, S. (2012). A molecular view on signal transduction by the apoptosome. Cell Signal. 24, 1420–1425. doi: 10.1016/j.cellsig.2012.03.007
Richter, U., Lahtinen, T., Marttinen, P., Myohanen, M., Greco, D., Cannini, G., et al. (2013). A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr. Biol. 23, 535–541. doi: 10.1016/j.cub.2013.02.019
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi: 10.1038/nrm3412
Russell, E. G., and Cotter, T. G. (2015). New insight into the role of Reactive Oxygen Species (ROS) in cellular signal-transduction processes. Int. Rev. Cell Mol. Biol. 319, 221–254. doi: 10.1016/bs.ircmb.2015.07.004
Sabrassov, D. D., and Sabatini, D. M. (2005). Redox regulation of the nutrient-sensitive Raptor-mTOR pathway and complex. J. Biol. Chem. 280, 39505–39509. doi: 10.1074/jbc.M506096200
Sarraf, S. A., Raman, M., Guarani-Pereira, V., Sowa, M. E., Huttlin, E. L., Gygi, S. P., et al. (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. doi: 10.1038/nature12043
Seiferling, D., Szczepanowska, K., Becker, C., Senft, K., Hermans, S., Maiti, P., et al. (2016). Loss of CLPP alleviates mitochondrial cardiomyopathy without affecting the mammalian UPRmt. EMBO Rep. 17, 953–964. doi: 10.15252/embr.201642077
Seki, T., Fukamizu, A., Kiso, Y., and Mukai, H. (2011). Mitocryptide-2, a neutrophil-activating cryptide, is a specific endogenous agonist for formyl-peptide receptor-like 1. Biochem. Biophys. Res. Commun. 404, 482–487. doi: 10.1016/j.bbrc.2010.12.007
Sena, L. A., and Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. doi: 10.1016/j.molcel.2012.09.025
Sevrioukova, I. F. (2011). Apoptosis-inducing factor: structure, function, and redox regulation. Antioxid. Redox Signal. 14, 2545–2579. doi: 10.1089/ars.2010.3445
Shadel, G. S., and Horvath, T. L. (2015). Mitochondrial ROS signaling in organismal homeostasis. Cell 163, 560–569. doi: 10.1016/j.cell.2015.10.001
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Siekvitz, P. (1957). Powerhouse of the cell. Sci. Am. 197, 131–144. doi: 10.1038/scientificamerican0757-131
Spange, S., Wagner, T., Heinzel, T., and Kramer, O. H. (2009). Acetylation of non-histone proteins modulates cellular signaling at multiple levels. Int. J. Biochem. Cell Biol. 41, 185–198. doi: 10.1016/j.biocel.2008.08.027
Sugiura, A., McLelland, G. L., Fon, E. A., and McBride, H. M. (2012). A new pathway for mitochondrial quality control: mitochondria-derived vesicles. EMBO J. 33, 2142–2156. doi: 10.15252/embj.201488104
Sullivan, L. B., Gui, D. Y., Hosiso, A. M., Bush, L. N., Freinkman, E., and Vander Heiden, M. G. (2015). Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563. doi: 10.1016/j.cell.2015.07.017
Sullivan, L. B., Martinez-Garcia, E., Nguyen, H., Mullen, A. R., Dufour, E., Sudarshan, S., et al. (2013). The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236–248. doi: 10.1016/j.molcel.2013.05.003
Tait, S. W. G., and Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. doi: 10.1038/nrm2952
Takeuchi, A., Kim, B., and Matsuoka, S. (2015). The destiny of Ca2+ released by mitochondria. J. Physiol. Sci. 65, 11–24. doi: 10.1007/s12576-014-0326-7
Tanner, J. J., Parsons, Z. D., Cummings, A. H., Zhou, H., and Gates, K. S. (2011). Redox regulation of protein tyrosine phosphatases: structural and chemical aspects. Antioxid. Redox Signal. 15, 77–97. doi: 10.1089/ars.2010.3611
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell 165, 1197–1208. doi: 10.1016/j.cell.2016.04.011
Tidow, H., and Nisse, P. (2013). Structural diversity of calmodulin binding to its target sites. FEBS J. 280, 5551–5565. doi: 10.1111/febs.12296
Topf, U., Wrobel, L., and Chacinska, A. (2016). Chatty mitochondria: keeping balance in cellular protein homeostasis. Trends Cell. Biol. 26, 577–586. doi: 10.1016/j.tcb.2016.03.002
Toyama, E. Q., Herzig, S., Courchet, J., Lewis, T. L. Jr. Loson, O. C., Hellberg, K., et al. (2016). AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. doi: 10.1126/science.aab4138
Truong, T. H., and Carroll, K. S. (2013). Redox regulation of protein kinases. Crit. Rev. Biochem. Mol. Biol. 48, 332–356. doi: 10.3109/10409238.2013.790873
Tsai, S. J., Sen, U., Zhao, L., Greenleaf, W., Dasgupta, J., Fiorillo, E., et al. (2009). Crystal structure of the human lymphoid tyrosine phosphatase catalytic domain: insights into redox regulation. Biochemistry 48, 4838–4845. doi: 10.1021/bi900166y
Tyynismaa, H., Carroll, C. J., Raimundo, N., Ahola-Erkkila, S., Wenz, T., Ruhanen, H., et al. (2010). Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 19, 3948–3958. doi: 10.1093/hmg/ddq310
Vafai, S. B., and Mootha, V. K. (2012). Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383. doi: 10.1038/nature11707
Verdin, E. (2015). NAD+ in aging, metabolism and neurodegeneration. Science 350, 1208–1213. doi: 10.1126/science.aac4854
Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Wang, X., and Chen, X. J. (2015). A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 524, 481–484. doi: 10.1038/nature14859
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., and Thompson, C. B. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. doi: 10.1126/science.1164097
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
Wong, Y. C., and Holzbaur, E. L. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U.S.A. 111, E4439–E4448. doi: 10.1073/pnas.1405752111
Wrobel, L., Topf, U., Bragoszewski, P., Wiese, S., Sztolsztener, M. E., Oelieklaus, S., et al. (2015). Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524, 485–488. doi: 10.1038/nature14951
Xun, Z., Rivera-Sanchez, S., Ayala-Pena, S., Lim, J., Budworth, H., Skoda, E. M., et al. (2012). Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington's disease. Cell Rep. 2, 1137–1142. doi: 10.1016/j.celrep.2012.10.001
Xu, W., Yang, H., Liu, Y., Yang, Y., Wang, P., Kim, S. H., et al. (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of a-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. doi: 10.1016/j.ccr.2010.12.014
Young, M. J., and Copeland, W. C. (2016). Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 38, 52–62. doi: 10.1016/j.gde.2016.03.005
Zhang, H., Ryu, D., Wu, Y., Gariani, K., Wang, X., Luan, P., et al. (2016). NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443. doi: 10.1126/science.aaf2693
Zhang, Q., Raoof, M., Chen, Y., Sumi, Y., Sursal, T., Junger, W., et al. (2010). Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107. doi: 10.1038/nature08780
Zhang, Z., Tan, M., Xie, Z., Dai, L., Chen, Y., and Zhao, Y. (2011). Identofocation of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63. doi: 10.1038/nchembio.495
Keywords: mitochondria, retrograde signaling, metabolites, reactive oxygen species, mitochondrial stress
Citation: Bohovych I and Khalimonchuk O (2016) Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 4:109. doi: 10.3389/fcell.2016.00109
Received: 25 July 2016; Accepted: 16 September 2016;
Published: 13 October 2016.
Edited by:
Simon Rousseau, McGill University, CanadaCopyright © 2016 Bohovych and Khalimonchuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oleh Khalimonchuk, b2toYWxpbW9uY2h1azJAdW5sLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.