 Klaudia Lepka
Klaudia Lepka Carsten Berndt*
Carsten Berndt*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Cell Dev. Biol. , 23 June 2016
Sec. Molecular and Cellular Pathology
Volume 4 - 2016 | https://doi.org/10.3389/fcell.2016.00063
This article is part of the Research Topic Revisiting neuroinflammation in myelin damage and repair: new molecular and cellular players View all 8 articles
Neuroinflammation in the central nervous system (CNS) is characterized by increased production of chemokines and cytokines, altered integrity of the blood-brain-barrier, influx of leukocytes as well as the activation of microglia and astroglia. Although not all characteristics are present under the following conditions, stimuli eliciting a neuroinflammatory response can be toxins, infections, autoimmune reactions, traumatic injury, psychological stress, and epileptic seizures (Vezzani et al., 2011; Barnum et al., 2012; Xanthos and Sandkühler, 2014). Further, neuroinflammation has been linked to mechanisms of disease and clinical outcomes in neurodegenerative disorders like Alzheimer's and Parkinson's disease (Amor et al., 2010). In the following, we will mainly concentrate on Multiple Sclerosis (MS) as the prototype for an autoimmune inflammatory and degenerative disorder of the CNS. According to our current understanding, the immunopathogenesis of MS is as heterogeneous as its clinical manifestations and course and may be mediated by myelin-reactive T lymphocytes, leading to oligodendroglial cell death and demyelination, as well as to bystander axonal degeneration, neuronal loss and, finally, gliosis (Hartung et al., 2014). B cells may have a fundamental role in presenting antigens to T cells and as a consequence trigger an aberrant T cell response. Moreover, upon differentiation into plasmablasts and plasma cells that manufacture antibodies (Yuseff et al., 2013; Nutt et al., 2015), they may induce demyelination through antibody-mediated complement activation (Holers, 2014). Of note, while remyelination may occur in early stages of disease, regeneration is severely compromised as the disease progresses (Kremer et al., 2016). However, the etiology and cause for disease progression and failure of recovery remain largely elusive. Regarding possible factors, reactive oxygen (ROS), and nitrogen species (RNS) have attracted increasing interest in the last two decades. Focusing on MS we will discuss the role of ROS and RNS in disease onset and progression of this disabling disease and further emphasize the role of specific redox signaling modulating protein activity and its underestimated role in the development of new therapeutic agents.
The onset of MS is characterized by inflammation-mediated demyelination due to lymphocyte infiltration from the peripheral blood and microglial activation in situ. Subtle signs of neurodegeneration are identifiable from the beginning, characterized by axonal transection within white matter lesions (Trapp et al., 1998; Kuhlmann et al., 2002). This is of clinical importance particularly in chronic stages of disease, when extended cortical demyelination occurs which in aggregate represent the pathological substrate of permanent neurological disability (Zipp and Aktas, 2006). In both disease stages accumulation of ROS and RNS has been observed (Carvalho et al., 2014). In this context, one has to consider that the terms “ROS” and “RNS” summarize a variety of molecular species which substantially differ in chemical nature, cellular localization, and biological function (Figure 1). Unfortunately, these recent advances in our understanding of redox biology still go unrecognized by many researchers.
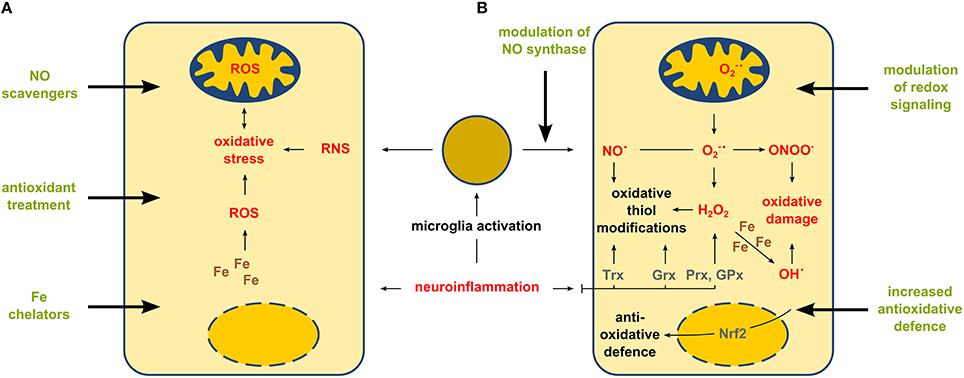
Figure 1. Gross (unspecific) treatment of oxidative stress vs. targeted modulation of enzyme-based thiol redox modifications. Neuroinflammation affects CNS cells by increasing the amounts of ROS and RNS. Panel (A) shows a simplified view on intracellular effects and therapeutic strategies aiming in the overall change of the amount of ROS and RNS. Panel (B) shows a more detailed explanation of cellular redox responses during neuroinflammation and potential therapeutic strategies aiming in the control of specific enzyme-based redox events mediated by nitric oxide synthase, transcription factors (such as Nrf2) or oxidoreductases thioredoxin (Trx), glutaredoxin (Grx) and peroxiredoxin (Prx) as well as other antioxidant enzymes, e.g., glutathione peroxidase (GPx).
The majority of these molecular species are non-radicals: All oxygen radicals are ROS, but not all ROS are oxygen radicals. Moreover, depending on the origin, cellular functions of the respective oxygen radicals could be even oppositional (Prozorovski et al., 2015). Nevertheless, increased ROS levels are a prerequisite for and a consequence of oxidative stress. Per definition, oxidative stress is an imbalance between oxidants and antioxidants in favor of the oxidants, leading to a disruption of redox signaling and control and/or molecular damage (Sies and Jones, 2007). Clearly, such an imbalance does not imply a change in the overall cellular redox state. There is no general redox state of a given cell, although some researchers still use the Nernst equation and the glutathione: glutathione disulfide redox couple to determine a cellular redox state. However, this concept ignores all other redox couples, the compartmentalization of redox potentials, and the issue that glutathione requires enzymes to exert its biological functions (Flohé, 2013; Berndt et al., 2014). In the CNS, activated immune cells like microglia are a major source of reactive species. The neural parenchyma in the CNS is highly sensitive to oxidative damage, DNA double strand breaks, membrane disruption and protein degradation, due to its high cellular metabolic activity and enrichment in polyunsaturated fatty acids (Bazinet and Layé, 2014). Further, amounts of antioxidant molecules like α-tocopherol and antioxidant enzymes like superoxide dismutases (SOD), catalase, or glutathione peroxidases (GPx) are decreased in the brain compared to other tissues (Dringen, 2000; Chiurchiù et al., 2016). These CNS-specific characteristics might reinforce mitochondrial DNA damage based on pathological accumulation of reactive species which has been invoked as a possible reason for chronic neurodegeneration as well as for failure of remyelination (Li et al., 2005; Campbell et al., 2014; Witte et al., 2014). Extensively secreted nitric oxide (NO) reacts rapidly with forming ONOO− and by this induces protein nitration in lesion areas. Nitrotyrosine is considered a hallmark of oxidative damage in neurodegenerative diseases (Pacher et al., 2007). Furthermore, iron accumulation in lesion areas promote oxidative damage of proteins, lipids, and nucleotides (Hametner et al., 2013). In summary, oxidative stress is considered as a major contributor to neuroinflammatory diseases including MS (Haider, 2015; Mahad et al., 2015). However, the direct and specific contribution of ROS and RNS to disease progression still remains elusive.
The onset of MS is most commonly characterized by a relapsing remitting disease form (RRMS) which later progresses into a secondary progressive form (SPMS). Disease-modifying treatments (DMTs) for RRMS are known to prevent or reduce the frequency of harmful immune responses targeted to CNS antigens and thereby slow or halt progression of disease pathology and accrual of neurologic disability. The implementation of easy-to-use magnetic resonance imaging (MRI)-guided proof of concept studies has paved the way for regulatory approval of 12 DMTs for RRMS including first-line medications like INFβ and glatiramer acetate, dimethyl fumarate (DMF) as well as teriflunomide and second-line options like natalizumab and alemtuzumab (humanized monoclonal antibodies), fingolimod and the immunosuppressant mitoxantrone (Fox, 2006; Ingwersen et al., 2016). Thereby, currently available therapeutics act mainly by modulating disease-relevant early immune activation steps, but fail to address repair of already damaged brain and spinal cord areas. Moreover, molecular mechanisms of the mode of action are still not entirely known for some of these drugs. However, more recently the mechanisms of function were investigated in greater detail underlining additional neurobiological effects of several DMTs, for instance fingolimod (Foster et al., 2007), as first-in-class spingosine-1-phosphate receptor modulator (Ingwersen et al., 2012) and DMF (Dubey et al., 2015). The molecular mechanism of DMF links this drug to prevention against oxidative stress (Albrecht et al., 2012).
The identification of oxidants and antioxidants involved in disease processes raised the hope that treatment with antioxidants could combat diseases connected to oxidative stress. In the last two decades a variety of clinical studies were initiated to test the impact of antioxidant donation itself and as adjunct medication in RRMS. Surprisingly, the majority of those studies failed (Gilgun-Sherki et al., 2004; Carvalho et al., 2016) although the respective compounds such as lipid peroxyl scavengers (Hall, 1992), low molecular weight antioxidants (Hansen et al., 1995), and others showed to some extent an influence on the progression of inflammation in cell culture or animal models (Chiurchiù et al., 2016). The failure of such clinical studies might be explained by the hitherto neglected roles of specific ROS, especially H2O2 and NO, as important second messengers in cellular signaling. Thus, excess of antioxidants does not just attenuate oxidative stress, but could also interfere with anti-inflammatory response (Ohl et al., 2016) and with physiological redox signaling and thus harmfully impact recovery processes.
During recent years, redox signaling, and redox regulation emerged as one of the major physiological control mechanisms in all yet investigated cell types. Redox signaling is even a regulator of other well-established and accepted signaling pathways, e.g., phosphorylation (Corcoran and Cotter, 2013), and can affect signaling by regulation of transcription factors or enzymatic activities via thiol modifications. Thiols can undergo several reversible oxidative posttranslational modifications, e.g., nitrosylation, glutathionylation, formation of disulfides, and sulfenic acid. Key enzymes in thiol redox regulation are oxidoreductases of the thioredoxin family, namely thioredoxins (Trx), glutaredoxins (Grx), and peroxiredoxins (Prx) (Hanschmann et al., 2013; Lillig and Berndt, 2013), which display cell type specific expression in the rat CNS (Aon-Bertolino et al., 2011) and catalyze the reduction and oxidation of specific cysteinyl residues and the intracellular level of the second messenger H2O2. Another protein regulating the amount of H2O2 is GPx (Deponte, 2013).
Redox regulation of transcription is well established (Brigelius-Flohé and Flohé, 2011). Very important in defense against oxidative damage is Nuclear Factor-E2-related factor 2 (Nrf2), a transcription factor controlling the transcription of several antioxidant enzymes. Activity of Nrf2 itself is regulated by the thiol redox state of Kelch-like ECH associated protein 1 (Keap1). In its reduced state, Keap1 promotes ubiquitination and subsequent degradation of Nrf2. Oxidized Keap1 allows the accumulation of Nrf2 in the nucleus and the expression of its target genes. Keeping this in mind, important redox events induced by the formation of reactive species during disease onset and progression might be simplified as oxidative or nitrosative stress. To date, it is not clarified whether redox changes may have different roles according to disease stage. Obviously, during CNS inflammatory attacks, invading lymphocytes, and activated macrophages/microglia initiate an acute and massive ROS/RNS challenge of the tissue characterized by damage of proteins, lipids, and nucleotides and thereby leading to immediate structural demise. In contrast, mild but persistent exposure to inflammation—as found in post-acute/chronic progressive stage—may result in alteration of specific redox regulation accompanied by targeted modification of redox-sensitive signaling pathways.
Increased knowledge of enzyme-based redox events involved in disease onset and progression as well as potential redox-related modes of action of already existing drugs might pave the way for new therapeutic strategies, even approaches targeting regeneration in MS. For instance, preclinical studies revealed antioxidant properties of DMF acting via the translocation of Nrf2 into the nucleus and thereby promoting defense mechanisms against oxidative damage (Albrecht et al., 2012). Thereby, DMF treatment attenuates neuroinflammation and affects progression of MS and other neurodegenerative diseases (Johnson and Johnson, 2015; Buendia et al., 2016). It has been proposed that this mechanism is also the reason for the recently discovered neuroprotective and myelin-protective functions of DMF (Dubey et al., 2015). Of note, Nrf2 is upregulated in active MS lesions (Licht-Mayer et al., 2015). So far, the number of studies investigating the role of oxidoreductases or other antioxidant enzymes during MS is very limited, although these proteins are important during inflammatory processes, e.g., activation of macrophages (Salzano et al., 2014). Activity of GPx is dramatically decreased in cerebrospinal fluid and in serum of MS patients (Calabrese et al., 1994; Socha et al., 2014). In contrast, Prx5 as well as the mitochondrial oxidoreductases Trx2 and Prx3 are upregulated within MS lesions (Holley et al., 2007; Nijland et al., 2014) and Prx6 is increased in the spinal cord of mice that underwent experimental autoimmune encephalomyelitis, a common animal model of MS (Yun et al., 2015). Transgenic mice overexpressing Prx6 displayed attenuated blood-brain barrier leakage and neuroinflammation after induction of this model. Besides novel potent anti-inflammatory therapies, regeneration might be achieved by enzyme-based thiol redox modulation: Collapsin response mediator protein 2 (CRMP2) was proposed as a potential novel drug target for axonal regeneration after neuroinflammation (Petratos et al., 2010). Interestingly, CRMP2 mediated axonal outgrowth depends on redox regulation via Grx2 and Trx1 (Bräutigam et al., 2011; Morinaka et al., 2011).
Thus, instead of unspecific application of ROS scavengers or other broadly active antioxidants, therapies aiming at the specific modulation of enzyme-based redox regulation and signaling might be the promising future of what is called “redox medicine” (Figure 1) (Sies, 2015).
In summary, recent insights have fundamentally changed our understanding of disease-related redox processes. Obviously, the unspecific use of the term “oxidative stress” has competed with latest insights indicating that subtle changes of the redox status of single molecules have a profound effect on both, endogenous signaling pathways relevant for inflammation as well as neuroregeneration. Basic as well as translational and clinical research in this area should consider these recent shifts in paradigms regarding oxidative stress, cellular redox potentials, ROS and RNS, and redox signaling.
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Our research is funded by the Walter und Ilse Rose-Stiftung, the James and Elisabeth Cloppenburg Stiftung, the Ilselore Luckow Stiftung, the German Research foundation (priority program 1710), and the Heinrich-Heine-University graduate school iBrain.
Albrecht, P., Bouchachia, I., Goebels, N., Henke, N., Hofstetter, H. H., Issberner, A., et al. (2012). Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflammation 9:163. doi: 10.1186/1742-2094-9-163
Amor, S., Puentes, F., Baker, D., and van der Valk, P. (2010). Inflammation in neurodegenerative diseases. Immunology 129, 154–169. doi: 10.1111/j.1365-2567.2009.03225.x
Aon-Bertolino, M. L., Romero, J. I., Galeano, P., Holubiec, M., Badorrey, M. S., Saraceno, G. E., et al. (2011). Thioredoxin and glutaredoxin system proteins-immunolocalization in the rat central nervous system. Biochim. Biophys. Acta 1810, 93–110. doi: 10.1016/j.bbagen.2010.06.011
Barnum, C. J., Pace, T. W., Hu, F., Neigh, G. N., and Tansey, M. G. (2012). Psychological stress in adolescent and adult mice increases neuroinflammation and attenuates the response to LPS challenge. J. Neuroinflammation 9, 9. doi: 10.1186/1742-2094-9-9
Bazinet, R. P., and Layé, S. (2014). Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 15, 771–785. doi: 10.1038/nrn3820
Berndt, C., Lillig, C. H., and Flohé, L. (2014). Redox regulation by glutathione needs enzymes. Front. Pharmacol. 5:168. doi: 10.3389/fphar.2014.00168
Bräutigam, L., Schütte, L. D., Godoy, J. R., Prozorovski, T., Gellert, M., Hauptmann, G., et al. (2011). Vertebrate-specific glutaredoxin is essential for brain development. Proc. Natl. Acad. Sci. U.S.A. 108, 20532–20537. doi: 10.1073/pnas.1110085108
Brigelius-Flohé, R., and Flohé, L. (2011). Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 15, 2335–2381. doi: 10.1089/ars.2010.3534
Buendia, I., Michalska, P., Navarro, E., Gameiro, I., Egea, J., and León, R. (2016). Nrf2-ARE pathway: an emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 157, 84–104. doi: 10.1016/j.pharmthera.2015.11.003
Calabrese, V., Raffaele, R., Cosentino, E., and Rizza, V. (1994). Changes in cerebrospinal fluid levels of malondialdehyde and glutathione reductase activity in multiple sclerosis. Int. J. Clin. Pharmacol. Res. 14, 119–123.
Campbell, G. R., Worrall, J. T., and Mahad, D. J. (2014). The central role of mitochondria in axonal degeneration in multiple sclerosis. Mult. Scler. 20, 1806–1813. doi: 10.1177/1352458514544537
Carvalho, A. N., Firuzi, O., Gama, M. J., van Horssen, J., and Saso, L. (2016). Oxidative stress and antioxidants in neurological diseases: is there still hope? Curr. Drug Targets. doi: 10.2174/1389450117666160401120514. [Epub ahead of print].
Carvalho, A. N., Lim, J. L., Nijland, P. G., Witte, M. E., and van Horssen, J. (2014). Glutathione in multiple sclerosis: more than just an antioxidant? Mult. Scler. 20, 1425–1431. doi: 10.1177/1352458514533400
Chiurchiù, V., Orlacchio, A., and Maccarrone, M. (2016). Is Modulation of oxidative stress an answer? The state of the art of redox therapeutic actions in neurodegenerative diseases. Oxid. Med. Cell. Longev. 2016:7909380. doi: 10.1155/2016/7909380
Corcoran, A., and Cotter, T. G. (2013). Redox regulation of protein kinases. FEBS J. 280, 1944–1965. doi: 10.1111/febs.12224
Deponte, M. (2013). Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 1830, 3217–3266. doi: 10.1016/j.bbagen.2012.09.018
Dringen, R. (2000). Metabolism and functions of glutathione in brain. Prog. Neurobiol. 62, 649–671. doi: 10.1016/S0301-0082(99)00060-X
Dubey, D., Kieseier, B. C., Hartung, H. P., Hemmer, B., Warnke, C., Menge, T., et al. (2015). Dimethyl fumarate in relapsing-remitting multiple sclerosis: rationale, mechanisms of action, pharmacokinetics, efficacy and safety. Expert Rev. Neurother. 15, 339–346. doi: 10.1586/14737175.2015.1025755
Flohé, L. (2013). The fairytale of the GSSG/GSH redox potential. Biochim. Biophys. Acta 1830, 3139–3142. doi: 10.1016/j.bbagen.2012.10.020
Foster, C. A., Howard, L. M., Schweitzer, A., Persohn, E., Hiestand, P. C., Balatoni, B., et al. (2007). Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J. Pharmacol. Exp. Ther. 323, 469–475. doi: 10.1124/jpet.107.127183
Fox, E. J. (2006). Management of worsening multiple sclerosis with mitoxantrone: a review. Clin. Ther. 28, 461–474. doi: 10.1016/j.clinthera.2006.04.013
Gilgun-Sherki, Y., Melamed, E., and Offen, D. (2004). The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. J. Neurol. 251, 261–268. doi: 10.1007/s00415-004-0348-9
Haider, L. (2015). Inflammation, iron, energy failure, and oxidative stress in the pathogenesis of multiple sclerosis. Oxid. Med. Cell. Longev. 2015:725370. doi: 10.1155/2015/725370
Hall, E. D. (1992). Novel inhibitors of iron-dependent lipid peroxidation for neurodegenerative disorders. Ann. Neurol. 32 (Suppl.), S137–S142. doi: 10.1002/ana.410320724
Hametner, S., Wimmer, I., Haider, L., Pfeifenbring, S., Brück, W., and Lassmann, H. (2013). Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 74, 848–861. doi: 10.1002/ana.23974
Hanschmann, E.-M., Godoy, J. R., Berndt, C., Hudemann, C., and Lillig, C. H. (2013). Thioredoxins, glutaredoxins, and peroxiredoxins–molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 19, 1539–1605. doi: 10.1089/ars.2012.4599
Hansen, L. A., Willenborg, D. O., and Cowden, W. B. (1995). Suppression of hyperacute and passively transferred experimental autoimmune encephalomyelitis by the anti-oxidant, butylated hydroxyanisole. J. Neuroimmunol. 62, 69–77. doi: 10.1016/0165-5728(95)00104-A
Hartung, H.-P., Aktas, O., Menge, T., and Kieseier, B. C. (2014). Immune regulation of multiple sclerosis. Handb. Clin. Neurol. 122, 3–14. doi: 10.1016/B978-0-444-52001-2.00001-7
Holers, V. M. (2014). Complement and its receptors: new insights into human disease. Annu. Rev. Immunol. 32, 433–459. doi: 10.1146/annurev-immunol-032713-120154
Holley, J. E., Newcombe, J., Winyard, P. G., and Gutowski, N. J. (2007). Peroxiredoxin V in multiple sclerosis lesions: predominant expression by astrocytes. Mult. Scler. 13, 955–961. doi: 10.1177/1352458507078064
Ingwersen, J., Aktas, O., and Hartung, H.-P. (2016). Advances in and algorithms for the treatment of relapsing-remitting multiple sclerosis. Neurotherapeutics 13, 47–57. doi: 10.1007/s13311-015-0412-4
Ingwersen, J., Aktas, O., Kuery, P., Kieseier, B., Boyko, A., and Hartung, H.-P. (2012). Fingolimod in multiple sclerosis: mechanisms of action and clinical efficacy. Clin. Immunol. 142, 15–24. doi: 10.1016/j.clim.2011.05.005
Johnson, D. A., and Johnson, J. A. (2015). Nrf2–a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 88, 253–267. doi: 10.1016/j.freeradbiomed.2015.07.147
Kremer, D., Göttle, P., Hartung, H.-P., and Küry, P. (2016). Pushing forward: remyelination as the New Frontier in CNS diseases. Trends Neurosci. 39, 246–263. doi: 10.1016/j.tins.2016.02.004
Kuhlmann, T., Lingfeld, G., Bitsch, A., Schuchardt, J., and Brück, W. (2002). Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain J. Neurol. 125, 2202–2212. doi: 10.1093/brain/awf235
Li, J., Baud, O., Vartanian, T., Volpe, J. J., and Rosenberg, P. A. (2005). Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc. Natl. Acad. Sci. U.S.A. 102, 9936–9941. doi: 10.1073/pnas.0502552102
Licht-Mayer, S., Wimmer, I., Traffehn, S., Metz, I., Brück, W., Bauer, J., et al. (2015). Cell type-specific Nrf2 expression in multiple sclerosis lesions. Acta Neuropathol. 130, 263–277. doi: 10.1007/s00401-015-1452-x
Lillig, C. H., and Berndt, C. (2013). Glutaredoxins in thiol/disulfide exchange. Antioxid. Redox Signal. 18, 1654–1665. doi: 10.1089/ars.2012.5007
Mahad, D. H., Trapp, B. D., and Lassmann, H. (2015). Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 14, 183–193. doi: 10.1016/S1474-4422(14)70256-X
Morinaka, A., Yamada, M., Itofusa, R., Funato, Y., Yoshimura, Y., Nakamura, F., et al. (2011). Thioredoxin mediates oxidation-dependent phosphorylation of CRMP2 and growth cone collapse. Sci. Signal. 4, ra26. doi: 10.1126/scisignal.2001127
Nijland, P. G., Witte, M. E., van het Hof, B., van der Pol, S., Bauer, J., Lassmann, H., et al. (2014). Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: implications for multiple sclerosis. Acta Neuropathol. Commun. 2, 170. doi: 10.1186/s40478-014-0170-2
Nutt, S. L., Hodgkin, P. D., Tarlinton, D. M., and Corcoran, L. M. (2015). The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 15, 160–171. doi: 10.1038/nri3795
Ohl, K., Tenbrock, K., and Kipp, M. (2016). Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 277, 58–67. doi: 10.1016/j.expneurol.2015.11.010
Pacher, P., Beckman, J. S., and Liaudet, L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424. doi: 10.1152/physrev.00029.2006
Petratos, S., Azari, M. F., Ozturk, E., Papadopoulos, R., and Bernard, C. C. (2010). Novel therapeutic targets for axonal degeneration in multiple sclerosis. J. Neuropathol. Exp. Neurol. 69, 323–334. doi: 10.1097/NEN.0b013e3181d60ddb
Prozorovski, T., Schneider, R., Berndt, C., Hartung, H.-P., and Aktas, O. (2015). Redox-regulated fate of neural stem progenitor cells. Biochim. Biophys. Acta 1850, 1543–1554. doi: 10.1016/j.bbagen.2015.01.022
Salzano, S., Checconi, P., Hanschmann, E.-M., Lillig, C. H., Bowler, L. D., Chan, P., et al. (2014). Linkage of inflammation and oxidative stress via release of glutathionylated peroxiredoxin-2, which acts as a danger signal. Proc. Natl. Acad. Sci. U.S.A. 111, 12157–12162. doi: 10.1073/pnas.1401712111
Sies, H. (2015). Oxidative stress: a concept in redox biology and medicine. Redox Biol. 4, 180–183. doi: 10.1016/j.redox.2015.01.002
Sies, H., and Jones, D. P. (2007). “Oxidative stress,” in Encyclopedia of Stress, 2nd Edn., ed G. Fink (Amsterdam: Elsevier), 47–48.
Socha, K., Kochanowicz, J., Karpińska, E., Soroczyńska, J., Jakoniuk, M., Mariak, Z., et al. (2014). Dietary habits and selenium, glutathione peroxidase and total antioxidant status in the serum of patients with relapsing-remitting multiple sclerosis. Nutr. J. 13:62. doi: 10.1186/1475-2891-13-62
Trapp, B. D., Peterson, J., Ransohoff, R. M., Rudick, R., Mörk, S., and Bö, L. (1998). Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 338, 278–285. doi: 10.1056/NEJM199801293380502
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Revi. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Witte, M. E., Mahad, D. J., Lassmann, H., and van Horssen, J. (2014). Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 20, 179–187. doi: 10.1016/j.molmed.2013.11.007
Xanthos, D. N., and Sandkühler, J. (2014). Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 15, 43–53. doi: 10.1038/nrn3617
Yun, H.-M., Park, K.-R., Kim, E.-C., and Hong, J. T. (2015). PRDX6 controls multiple sclerosis by suppressing inflammation and blood brain barrier disruption. Oncotarget 6, 20875–20884. doi: 10.18632/oncotarget.5205
Yuseff, M.-I., Pierobon, P., Reversat, A., and Lennon-Duménil, A.-M. (2013). How B cells capture, process and present antigens: a crucial role for cell polarity. Nature Rev. Immunol. 13, 475–486. doi: 10.1038/nri3469
Keywords: antioxidants, clinical trials, multiple sclerosis, redox signaling, oxidative stress
Citation: Lepka K, Berndt C, Hartung H-P and Aktas O (2016) Redox Events As Modulators of Pathology and Therapy of Neuroinflammatory Diseases. Front. Cell Dev. Biol. 4:63. doi: 10.3389/fcell.2016.00063
Received: 12 May 2016; Accepted: 08 June 2016;
Published: 23 June 2016.
Edited by:
Veronique E. Miron, The University of Edinburgh, UKCopyright © 2016 Lepka, Berndt, Hartung and Aktas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carsten Berndt, Y2Fyc3Rlbi5iZXJuZHRAaGh1LmRl;
Orhan Aktas, b3JoYW4uYWt0YXNAaGh1LmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.