 Meghan L. Marré
Meghan L. Marré Eddie A. James
Eddie A. James Jon D. Piganelli
Jon D. Piganelli
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 27 October 2015
Sec. Cell Growth and Division
Volume 3 - 2015 | https://doi.org/10.3389/fcell.2015.00067
This article is part of the Research Topic Organogenesis: from development to disease View all 10 articles
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by hyperglycemia due to progressive immune-mediated destruction of insulin-producing pancreatic islet β cells. Although many elegant studies have identified β cell autoantigens that are targeted by the autoimmune response, the mechanisms by which these autoantigens are generated remain poorly understood. Normal β cell physiology includes a high demand for insulin production and secretion in response to dynamic glucose sensing. This secretory function predisposes β cells to significantly higher levels of endoplasmic reticulum (ER) stress compared to nonsecretory cells. In addition, many environmental triggers associated with T1D onset further augment this inherent ER stress in β cells. ER stress may increase abnormal post-translational modification (PTM) of endogenous β cell proteins. Indeed, in other autoimmune disorders such as celiac disease, systemic lupus erythematosus, multiple sclerosis, and rheumatoid arthritis, abnormally modified neo-antigens are presented by antigen presenting cells (APCs) in draining lymph nodes. In the context of genetic susceptibility to autoimmunity, presentation of neo-antigens activates auto-reactive T cells and pathology ensues. Therefore, the ER stress induced by normal β cell secretory physiology and environmental triggers may be sufficient to generate neo-antigens for the autoimmune response in T1D. This review summarizes what is currently known about ER stress and protein PTM in target organs of other autoimmune disease models, as well as the data supporting a role for ER stress-induced neo-antigen formation in β cells in T1D.
Type 1 diabetes (T1D) is a chronic autoimmune disease in which insulin-producing pancreatic islet β cells are targeted and destroyed by autoreactive immune cells. Autoimmune recognition of β cells initiates processes that result in loss of β cell mass and the decline of insulin-mediated control of blood glucose levels. Eventually, the remaining β cells become insufficient to maintain normal blood glucose levels, due to reduced β cell numbers and/or to reduced insulin secretion, and chronic hyperglycemia and T1D ensue.
Given the autoimmune mechanisms of β cell destruction, a major underlying risk factor for T1D is a genetic predisposition to autoimmunity. T1D is a polygenic disease, with many genetic loci associated with disease onset. For example, polymorphisms and variants in many genes related to innate and adaptive immune cell function increase susceptibility to autoimmunity, likely by causing failure of central and peripheral immune tolerance mechanisms. With respect to central tolerance, human leukocyte antigen (HLA), which is the genetic variable with the greatest association to T1D onset (Todd et al., 1987; Dorman et al., 1990; Luca et al., 2008), shapes the adaptive immune repertoire by determining which T cells survive thymic maturation and selection. Under normal circumstances, T cells that respond too strongly to self-peptides presented by HLA are deleted or inactivated (Hogquist and Jameson, 2014). In individuals expressing autoimmune-prone polymorphisms within the HLA gene locus, these central tolerance mechanisms fail, permitting autoreactive T cells to mature, and exit the thymus (Fan et al., 2009; Geenen, 2012). With respect to peripheral tolerance, gene variants at other loci such as protein tyrosine phosphatase, non-receptor type 22 (PTPN22) may accelerate T1D onset through mechanisms that have not been fully elucidated (Pociot and McDermott, 2002; Bottini et al., 2004; Luca et al., 2008; Wallis et al., 2009). For example, some studies suggest that, in the context of genetic predisposition to autoimmunity, incomplete antigen presenting cell (APC) maturation may contribute to T1D progression. These immature APCs do not respond normally to growth factors (Serreze et al., 1993) or to inflammatory stimuli (Serreze et al., 1993; Piganelli et al., 1998). As a result, these APCs exhibit defective antigen processing and presentation that activate autoreactive T cells, but do not trigger tolerogenic mechanisms. Such failure in peripheral immune tolerance may exacerbate T1D pathology. However, as stated above, the precise mechanisms by which many of these genetic variants contribute to T1D remain unknown.
Although genetic predisposition is strongly associated with T1D progression, many epidemiological factors suggest that genetic predisposition is not sufficient to drive pathology. First, only a small portion of individuals with HLA predisposition actually progress to T1D (Knip et al., 2005). Second, monozygotic twins demonstrate relatively low concordance for T1D onset (Barnett et al., 1981; Verge et al., 1995). Third, the incidence of T1D is increasing at a rate that cannot be supported by genetic predisposition alone (Onkamo et al., 1999; Gale, 2002; DIAMOND Project Group, 2006). Finally, the age of onset and rate of progression of T1D vary greatly among patients. Together, these data support a role for environmental factors in triggering T1D onset and affecting progression. Among the many environmental triggers associated with T1D onset are viral infection (Atkinson et al., 1994; Horwitz et al., 1998, 2004; Hiemstra et al., 2001; Härkönen et al., 2002; Schulte et al., 2010), β cell exposure to chemicals (Like and Rossini, 1976; Rossini et al., 1977; Takasu et al., 1991a) or reactive oxygen species (ROS) (Piganelli et al., 2002; Tse et al., 2010; Delmastro and Piganelli, 2011; Delmastro-Greenwood et al., 2014), dysglycemia (Sosenko et al., 2009), and inflammation (Mandrup-Poulsen et al., 1987; Held et al., 1990; Jiang and Woda, 1991). Each of the environmental triggers listed here can cause β cell endoplasmic reticulum (ER) stress, suggesting that ER stress may be a common factor in disease onset. However, whether these environmental factors share common pathways to T1D remains unknown.
To understand how these factors lead to the progression of T1D, scientists have studied the non-obese diabetic (NOD) mouse. Mice of this strain develop spontaneous autoimmune diabetes with many similarities to human T1D. These similarities include genetic susceptibility at the HLA locus and other loci, and intra-islet infiltration of immune cells resulting in β cell destruction (Tochino, 1987; Leiter, 1989; Driver et al., 2012). Seminal studies with this mouse model have identified many β cell antigens targeted by the autoimmune response. These murine autoantigens include preproinsulin (Wegmann et al., 1994), glutamic acid decarboxylase (GAD65) (Tisch et al., 1993), islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) (Lieberman et al., 2003), chromogranin A (CHgA) (Stadinski et al., 2010), islet amyloid polypeptide (IAPP) (Delong et al., 2011), zinc transporter 8 (ZnT8) (Nayak et al., 2014), and 78 kDa glucose-regulated protein (GRP78) (Rondas et al., 2015). With the exception of GRP78, these proteins are also confirmed autoantigens in human T1D (Baekkeskov et al., 1990; Keller, 1990; Gorus et al., 1992; Yang et al., 2006; Wenzlau et al., 2007; Gottlieb et al., 2014) along with additional autoantigens found in humans but not yet identified in NOD mice such as tyrosine phosphatase-like insulinoma antigen 2 (IA-2) and IA-2β [also known as phosphatase homolog of granules from rat insulinomas (phogrin)] (Bonifacio et al., 1995; Lan et al., 1996), and islet cell autoantigen 69 (ICA69) (Pietropaolo et al., 1993). However, the precise mechanisms by which these β cell proteins come to be recognized and targeted by the autoimmune response in T1D remain unknown. Recent evidence suggests that some of these proteins undergo post-translational modification (PTM), generating “neo-antigens” with increased immunogenicity (Dunne et al., 2012). But whether such PTMs occur in the β cell, and what cellular processes might give rise to these PTMs in the β cell, remain unknown.
Here, we discuss cellular conditions (both physiological and pathological) that lead to protein PTM. We also review what is currently known about PTM and neo-antigen generation in target organs of other autoimmune disease models. Finally, we review the evidence supporting a role for ER stress-induced PTM in neo-antigen formation in β cells in T1D.
The ER is the organelle primarily responsible for folding and PTM of membrane-bound and secreted proteins. To accomplish these tasks, the ER lumen contains the necessary factors to support proper protein folding including molecular chaperones, ATP, an oxidizing environment to support disulfide bond formation, and millimolar concentrations of calcium (Ca2+) (Gething and Sambrook, 1992). Proteins that are properly folded exit the ER and continue toward their intended intra- or extra-cellular locations. However, improperly folded proteins remain in the ER and, when too many misfolded proteins accumulate, ER homeostasis is disrupted and ER stress ensues. ER stress activates the cytoprotective unfolded protein response (UPR), which acts to relieve ER stress and restore homeostasis by two mechanisms (Hetz, 2012). First, UPR signaling temporarily inhibits the synthesis of new non-chaperone proteins to prevent further burdening the ER machinery. Second, UPR signaling increases the expression of protein chaperones to aid in the folding of the accumulated misfolded proteins in the ER lumen.
During normal ER homeostasis, the chaperone GRP78 [also known as binding immunoglobulin protein (BiP)] binds three protein sensors of ER stress that reside in the ER membrane: protein kinase RNA (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring protein 1 (IRE1) (Bertolotti et al., 2000; Shen et al., 2002). Interaction with GRP78 keeps these proteins inactive and thereby inhibits the UPR (Figure 1A). However, when misfolded proteins accumulate in the ER, GRP78 releases these protein sensors to bind exposed hydrophobic residues in unfolded proteins. Once free from GRP78, each protein sensor initiates a signaling cascade of the UPR. PERK oligomerizes and becomes activated through autophosphorylation in trans. Activated PERK then phosphorylates the α subunit of translation initiation factor 2 (eIF2α) to attenuate mRNA translation and reduce the protein burden in the ER (Harding et al., 2000a,b). ATF6 translocates to the Golgi apparatus where it is cleaved to yield a transcription factor that initiates new chaperone synthesis to aid with folding of accumulated misfolded proteins (Haze et al., 1999). IRE1 oligomerizes and autophosphorylates in trans, enabling its endonuclease capability. IRE1 then splices X-box binding protein 1 (XBP-1) mRNA (Yoshida et al., 2001), which encodes a transcription factor that regulates proteins involved in relieving ER stress such as chaperones (Lee et al., 2003) and proteins involved in lipid synthesis to increase ER volume (Sriburi et al., 2004). Through these three signaling cascades, the UPR attempts to reduce ER stress and prevent stress-induced apoptosis (Figure 1B).
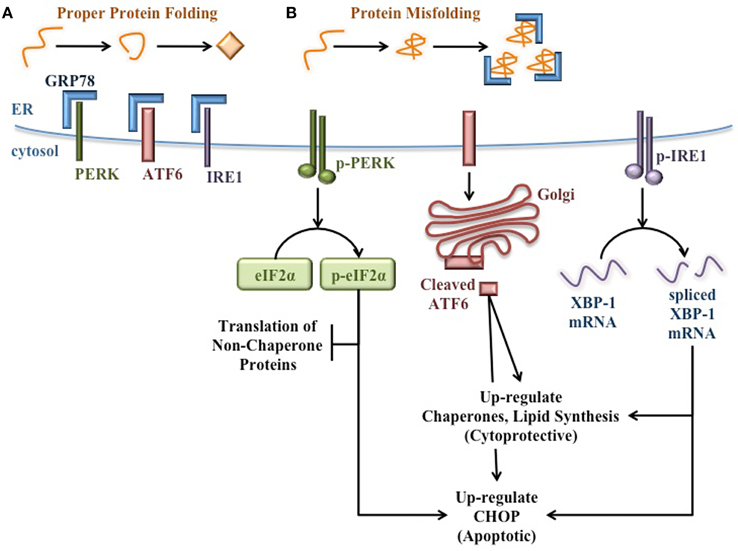
Figure 1. Signaling pathways of the unfolded protein response. (A) When protein folding proceeds normally, the protein sensors of ER stress (PERK, ATF6, and IRE1) are bound and held in their inactive state by GRP78. (B) When misfolded proteins accumulate in the ER lumen, GRP78 binds misfolded proteins, thereby releasing the protein sensors of ER stress and allowing for the activation of the cytoprotective UPR. PERK autophosphorylates in trans, then activates eIF2α by phosphorylation to attenuate translation of additional non-chaperone proteins. ATF6 translocates to the Golgi apparatus and is cleaved to yield a transcription factor that up-regulates the expression of molecular chaperones to aid in the folding of accumulated proteins in the ER. IRE1 autophosphorylates in trans and splices XBP-1 mRNA. The spliced mRNA encodes a transcription factor that up-regulates the expression of additional molecular chaperones and UPR proteins to relieve ER stress. If ER stress is too great or prolonged, the UPR induces expression of pro-apoptotic proteins such as CHOP.
However, if the burden of unfolded proteins and the subsequent ER dysfunction are too great or too prolonged, these cytoprotective functions of the UPR fail. Under these conditions, pro-apoptotic signaling pathways become activated and ultimately lead to death of the affected cell (Figure 1B). For example, the UPR induces C/EBP Homologous Protein (CHOP) expression (Wang et al., 1996), which increases ROS-mediated mitochondrial apoptosis signaling pathways (Zinszner et al., 1998; McCullough et al., 2001).
Thus, ER stress and the UPR have significant effects on cellular function and viability. Even in cells that return to homeostasis following UPR activation, ER stress and dysfunction still have consequences. For example, ER stress often results in the release of Ca2+ from the ER lumen to the cytosol. Since high Ca2+ concentrations are necessary for protein folding, this efflux of Ca2+ negatively affects ER function. Second, ER stress and dysfunction lead to abnormal protein folding and PTM, affecting protein function. Therefore, ER stress, even when temporary, may have important effects on cellular function and physiology.
All cells undergo some degree of ER stress and activate the UPR in response to improper protein folding or during times of increased protein translation. However, professional secretory cells are uniquely susceptible to ER stress as a result of their normal physiology. In addition to proteins for cellular maintenance, secretory cells are burdened with synthesizing the proteins to be secreted and the proteins required for functional secretory pathways. Thus, the demands of protein translation and folding in the ER of secretory cells are significantly greater than in nonsecretory cells. Although secretory cells contain a more fully developed ER with additional chaperones to compensate for this demand (Shimizu and Hendershot, 2009), the increased ER burden leads to increased occurrence of ER stress.
β cells, like all professional secretory cells, naturally undergo high levels of ER stress as a result of their normal secretory physiology (Araki et al., 2003a; Lipson et al., 2006a,b; Wu and Kaufman, 2006; Fonseca et al., 2007; Ortsäter and Sjöholm, 2007; Eizirik et al., 2008; Volchuk and Ron, 2010; Kim et al., 2012; Teodoro et al., 2012). Indeed, β cells undergo significant ER stress during postprandial glucose-stimulated insulin synthesis (Lipson et al., 2006a,b). β cells increase translation of preproinsulin by 50-fold in response to heightened blood glucose concentrations, reaching a production rate of 1 million molecules of preproinsulin per minute (Scheuner and Kaufman, 2008). These 1 million molecules flood the ER lumen for folding and disulfide bond formation, causing tremendous ER stress. Such cellular processes of dynamic insulin production and heightened ER stress occur from an early age. In XBP-1 splicing reporter mice, the pancreas was the first tissue to exhibit high levels of ER stress and did so as early as 16 days old post birth (Iwawaki et al., 2004). Therefore, normal insulin-secreting physiology alone significantly increases ER stress in β cells.
In addition to the high levels of inherent ER stress, many of the putative environmental triggers associated with T1D may further enhance β cell ER stress. First, Coxsackie viral infection disrupts the ER membrane (van Kuppeveld et al., 1997, 2002, 2005) releasing Ca2+ from the ER into the cytosol. Second, β cell exposure to chemicals such as streptozotocin and alloxan cause protein ADP-ribosylation (Sandler and Swenne, 1983) and ROS generation (Heikkila et al., 1976; Takasu et al., 1991b; Bedoya et al., 1996), both of which lead to protein misfolding, and also decrease ER lumen Ca2+ concentrations (Kim et al., 1994; Park et al., 1995). Third, β cell exposure to ROS from either extracellular or intracellular sources releases Ca2+ from the ER lumen into the cytosol (Favero et al., 1995; Xu et al., 1998; Görlach et al., 2006). Also, dysglycemia leads to increased glucose sensing that, as discussed above, significantly increases insulin production and secretion (Scheuner and Kaufman, 2008). Finally, pancreatic inflammation and cytokine exposure activates c-jun N-terminal (JNK) mitogen-activated protein (MAP) kinase signaling pathways (Wang et al., 2009; Lee et al., 2011). The cellular effects of each environmental trigger exacerbate β cell ER stress. Therefore, although the precise mechanisms by which these environmental triggers accelerate T1D may vary, all the factors listed here can increase β cell ER stress above the normal physiological levels. Therefore, heightened ER stress may be a common factor in early T1D pathogenesis (Figure 2).
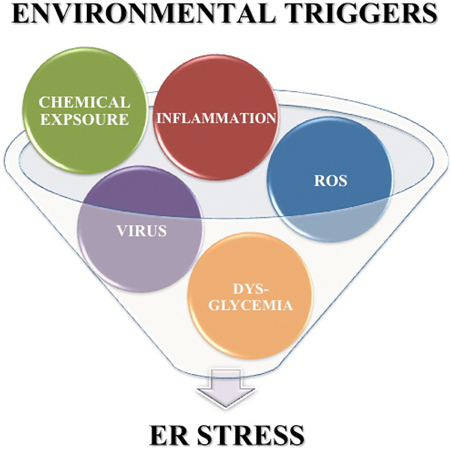
Figure 2. Environmental triggers associated with T1D exacerbate β cell ER stress. Environmental factors such as viral infection, chemicals, ROS, dysglycemia, and pancreatic inflammation are associated with onset of T1D. Each of these environmental triggers of T1D also increases β cell ER stress above the inherently high levels induced by normal β cell physiology.
ER stress and diabetes have been linked in both human and mouse studies. In studies of human islets, ER stress markers were increased in islets of T1D patients compared to islets of nondiabetic controls (Marhfour et al., 2012). In the Akita mouse model, the Ins2C96Y mutation prevents the formation of a crucial disulfide bond leading to misfolded insulin (Ron, 2002) and high ER stress in these β cells (Ron, 2002; Araki et al., 2003b; Nozaki et al., 2004). This ER stress leads to β cell apoptosis through the activation of CHOP signaling pathways (Oyadomari et al., 2002; Ron, 2002). However, inhibition of CHOP-mediated apoptosis merely delays, but does not halt, β cell loss and disease onset (Oyadomari et al., 2002). These data suggest that apoptosis may not be the only mechanism by which ER stress causes β cell death and diabetes.
In addition to folding and PTM of proteins, the ER is an important organelle for the maintenance of intracellular Ca2+ homeostasis. The ER contains the largest intracellular store of Ca2+ and is an important source of Ca2+ necessary for regulating a variety of cellular functions both in the ER lumen and in the cytosol (Meldolesi and Pozzan, 1998).
Within the ER lumen, high concentrations of Ca2+ are important for proper protein folding. Many molecular chaperones, including GRP78, are Ca2+-dependent (Ma and Hendershot, 2004). In addition, the proteins that facilitate the formation of disulfide bonds [protein disulfide isomerases (PDI)] also require Ca2+ (Nigam et al., 1994). To maintain the high concentration Ca2+ necessary for ER function, sarco/endoplasmic reticulum Ca2+ ATPases (SERCA) pumps in the ER membrane actively transport Ca2+ from the cytosol into the ER lumen (Figure 3). These pumps are regulated by existing concentrations of Ca2+ in the lumen to prevent ER Ca2+ stores from rising too high. Inhibition of these SERCA pumps prevents the movement of Ca2+ into the ER, decreasing the function of molecular chaperones and PDI, and increasing the burden of misfolded protein in the ER (Mekahli et al., 2011).
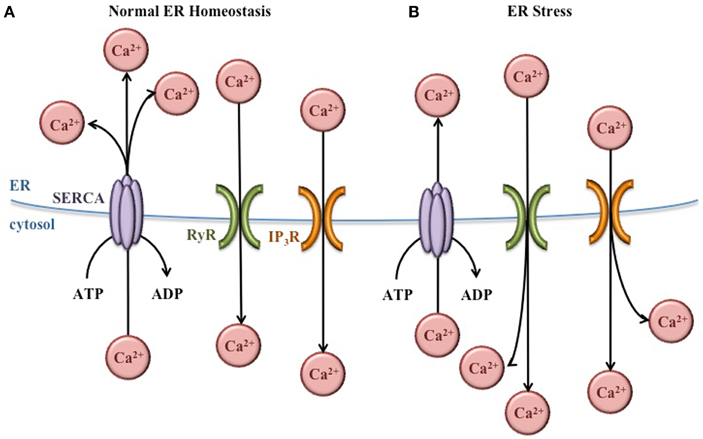
Figure 3. Regulation of ER Ca2+ concentrations. (A) Under normal conditions, Ca2+ concentrations are higher in the ER lumen than in the cytosol. This balance is maintained by SERCA pumps that bring Ca2+ into the ER lumen, and Ca2+ channels (RyR and IP3R) that release Ca2+ into the cytoplasm as needed for normal cellular signaling. (B) During ER stress, the Ca2+ gradient across the ER membrane is disturbed, leading to Ca2+ release from the ER and increased Ca2+ concentrations in the cytoplasm.
In the cytosol, Ca2+ plays important roles in a variety of cellular functions including metabolism, vesicular trafficking, secretion, transcription, and apoptosis (Berridge et al., 2000). Ca2+ channels in the ER membrane such as ryanodine-receptor (RyR) and inositol 1,4,5-trisphosphate receptor (IP3R) release Ca2+ from the ER lumen into the cytosol according to its chemical gradient (Figure 3). Like the SERCA pumps, the function of these channels is regulated to prevent depletion of the ER Ca2+ concentrations (Mekahli et al., 2011).
In spite of the regulation of SERCA pumps and Ca2+ channels, the normal Ca2+ gradient across the ER membrane is altered during ER stress, leading to decreased Ca2+ in the ER and increased Ca2+ in the cytosol. These changes in Ca2+ concentrations have important effects for the cell. The ER chaperones and PDI necessary for proper protein folding depend on Ca2+, so this imbalance exacerbates ER stress and further activates the UPR. In addition, increased cytosolic Ca2+ can cause apoptosis. For instance, Ca2+ release from the ER activates the ER-associated procaspase 12 (murine) or procaspase 4 (human), which initiate the caspase cell death pathway (Nakagawa et al., 2000; Hitomi et al., 2004). Also, the Ca2+-dependent ER chaperone calreticulin (Michalak et al., 2009) activates caspase 3- and cytochrome c-dependent apoptosis pathways when ER Ca2+ concentrations decrease (Nakamura et al., 2000). Furthermore, increased cytosolic Ca2+ activates enzymes such as calpain and calciuneurin which activate mitochondria-dependent signaling cascades that ultimately lead to cellular apoptosis (Nakagawa and Yuan, 2000; Gil-Parrado et al., 2002; Kim et al., 2002; Hajnóczky et al., 2003).
Therefore, the maintenance of Ca2+ homeostasis is crucial for cellular health and function. Disruption of this Ca2+ gradient across the ER membrane has major consequences for ER function and cellular viability.
While the of activation apoptotic signaling pathways usually requires prolonged ER stress and disrupted Ca2+ gradients, other cytosolic Ca2+-depenent enzymes are activated in response to more transient ER stress and heightened cytosolic Ca2+ concentrations. In particular, two families of Ca2+-dependent PTM enzymes are activated during ER stress. The activation of these enzymes has important implications for the proteins being folded in the ER.
Tissue transglutaminase 2 (Tgase2) is a ubiquitously expressed Ca2+-dependent PTM enzyme that resides in the cytosol (Lesort et al., 1998). Tgase2 becomes activated when Ca2+ concentrations in the cytosol rise above normal physiological levels. Indeed, Tgase2 activity requires Ca2+ concentrations above what is necessary for normal cellular signaling. As such, Tgase2 usually becomes activated only when cellular homeostasis is disrupted, such as when Ca2+ is released from the ER during ER stress (Ientile et al., 2007; Kojima et al., 2010; Wilhelmus et al., 2011; Kuo et al., 2012; Verhaar et al., 2012). Once active, Tgase2 translocates to several intra- and extra-cellular compartments (Park et al., 2010) including the ER (Orru et al., 2003; Wilhelmus et al., 2011; Verhaar et al., 2012) and secretory granules (Russo et al., 2013), to modify proteins by two mechanisms (Facchiano et al., 2006): first, Tgase2 forms ε (γ-glutamyl) isopeptide bonds between gluatmine and lysine residues that crosslink proteins, and second, Tgase2 facilitates the deamidation of glutamine. PTM of proteins by Tgase2 is important for a variety of normal cellular processes (Fesus and Piacentini, 2002; Gundemir et al., 2012). For example, Tgase2 modifies caspase 3 (Yamaguchi and Wang, 2006) and mitochondrial proteins (Fok and Mehta, 2007) to regulate apoptosis, nuclear proteins to regulate gene expression (Ballestar et al., 1996; Lesort et al., 1998; Han and Park, 2000), and extracellular matrix protein to promote cell adhesion (Gaudry et al., 1999; Akimov et al., 2000) and wound healing (Haroon et al., 1999; Stephens et al., 2004; Verderio et al., 2004).
Peptidylarginine deiminases (PAD) are another family of Ca2+-dependent PTM enzymes that reside in the cytosol (Vossenaar et al., 2003b). Of the five mammalian isoforms, PAD2 is the most widely expressed, and is the isoform expressed in the pancreas (Takahara et al., 1989). PAD become activated when cytosolic Ca2+ concentrations increase to levels 100-fold above normal physiological levels (Takahara et al., 1986; Vossenaar et al., 2003b). When activated, PAD are recruited to various subcellular compartments to modify proteins (Jang et al., 2011). PAD convert arginine to citrulline, which causes a loss of a positive charge in the amino acid sequence (Rogers et al., 1977). This change in charge has significant implications for protein folding, interaction, and function (Tarcsa et al., 1996). PAD play several roles in the context of normal cellular physiology. For example, PAD target IκB kinase gamma (IKKγ) to inhibit nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation (Lee et al., 2010), target vimentin to regulate cytoskeletal disassembly (Inagaki et al., 1989), and are important in the formation of neutrophil extracellular traps (NET) (Li et al., 2010).
Although PTMs are important in normal cellular signaling and physiology, PTM of proteins may contribute to autoimmune disorders. If proteins are modified differently in peripheral tissues than in the thymus, the modified peripheral proteins may act as neo-antigens for which there is no immune tolerance (Doyle and Mamula, 2012). Indeed, a variety of PTMs are implicated in the pathology of several autoimmune diseases (Table 1). Importantly, many neo-antigens are formed through PTM by the Ca2+-dependent enzymes Tgase2 and PAD. For example, Tgase2 activity is significantly elevated in celiac disease patients (Bruce et al., 1985). Tgase2 forms intermolecular ε (γ-glutamyl) isopeptide bonds, generating dimers of itself and gliadin as well as oligomers of gliadin (Molberg et al., 1998; Fleckenstein et al., 2004). These complexes are recognized by the immune system as neo-antigens, giving rise to increased T cell responses (Molberg et al., 1998) and anti-Tgase2 antibody production (Dieterich et al., 1997). These immune responses exacerbate the inflammatory conditions in the gut (Halttunen and Mäki, 1999; Barone et al., 2007). Also, in multiple sclerosis, citrullination of myelin basic protein forms a neo-antigen to which T cells respond (Martin et al., 1994). This neo-antigen causes disease in experimental autoimmune encephalomyelitis (the mouse model of multiple sclerosis) (Zhou et al., 1995). Finally, in rheumatoid arthritis, patients develop autoantibodies to the citrullinated forms of many proteins (Schellekens et al., 1998; Masson-Bessière et al., 2001; Vossenaar et al., 2003a, 2004; Burkhardt et al., 2005; Kinloch et al., 2005). These autoantibodies are detected in the synovial fluid of rheumatoid arthritis patients at early stages of disease (van Boekel et al., 2002; Vasishta, 2002), suggesting the importance of these PAD-generated neo-antigens for disease progression.
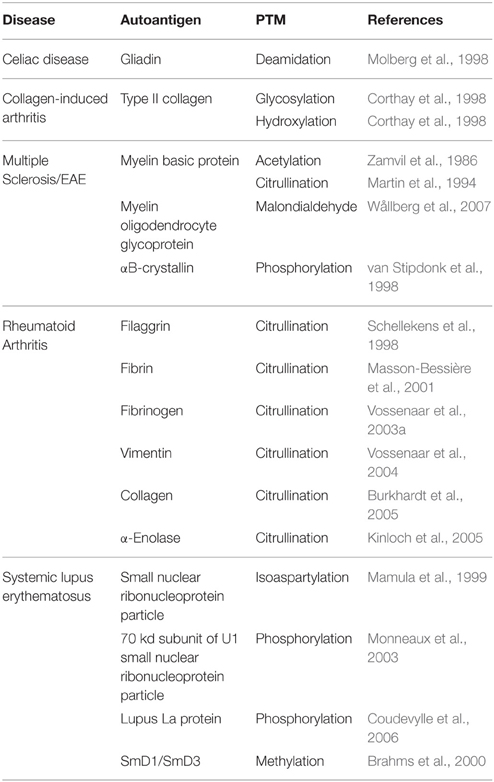
Table 1. Neo-antigens formed by PTM in autoimmune diseases.
Although Tgase2- and PAD-mediated PTMs are known to generate neo-antigens, little research has been conducted regarding the precise mechanisms by which these pathological PTMs arise in the particular cells and tissues targeted in these autoimmune disease models. However, Tgase2 and PAD, as described above, become activated under conditions that cause significantly elevated cytosolic Ca2+. The main cause of significantly elevated Ca2+ is cellular stress, especially ER stress. Therefore, ER stress may give rise to neo-antigen formation through abnormal Ca2+-dependent PTM of endogenous proteins.
Although it is well established that PTM of endogenous proteins forms neo-antigens that initiate and exacerbate the autoimmune response in many autoimmune diseases (Table 1), the role of PTM in β cell autoantigen formation long remained unexplored. However, in the last 10 years, many seminal studies have demonstrated that known murine and human β cell autoantigens exhibit greater immunogenicity after PTM (Table 2). For example, T cells from a human T1D patient recognized an oxidized epitope of proinsulin (Mannering et al., 2005). These T cell responses depended on the formation of a vicinal disulfide bond, as replacement of either cysteine with a serine residue abolished T cell responses against this peptide. In addition, PTM by the Ca2+-dependent enzymes Tgase2 and PAD increases the immunogenicity of several β cell proteins.
Table 2. Neo-antigens formed by PTM in T1D.
The first β cell autoantigen shown to elicit greater immune responses after Ca2+-dependent modification was the WE14 peptide of chromogranin A (CHgA) (Delong et al., 2012). The authors had previously demonstrated that the BDC2.5 diabetogenic CD4+ T cell clone recognizes WE14 (Stadinski et al., 2010). However, exceptionally high peptide concentrations were required for full T cell activation. In this study, Delong et al. demonstrated that treatment of WE14 with Tgase2 generated a covalently cross-linked peptide that is preferentially presented to BDC2.5 T cells, thereby increasing proliferation and IFNγ production. In addition, splenocytes isolated from pre-diabetic NOD mice responded more strongly to Tgase2-modified WE14 than to the native peptide. A subsequent study showed that WE14 was recognized by T cells from human T1D patients, and that treatment of WE14 with Tgase2 increased the response elicited from these T cells (Gottlieb et al., 2014). This confirmed the relevance of this modified β cell antigen to human T1D. Together, these studies demonstrated that Tgase2-modification of CHgA contributes to the strong activation of autoreactive immune cells in T1D.
The deamidation of glutamine by Tgase2 also modulates the recognition of β cell antigens. In a recent study, deamidated peptides from many islet proteins were eluted from T1D-associated HLA-DQ proteins (van Lummel et al., 2014). These Tgase2-modified peptides bound more strongly than their unmodified counterparts to HLA-DQ molecules. Among these, a Tgase2-modified peptide from preproinsulin elicited responses from CD4+ T cells from a new-onset T1D patient. This study therefore identified novel islet peptides that become neo-antigens through PTM. This study also demonstrated stronger binding to disease-associated HLA molecules as one mechanism by which β cell neo-antigens elicit stronger autoimmune responses.
Another β cell protein shown to elicit greater immune responses after PTM is GAD65. Modification of multiple GAD65 peptides by either Tgase2 (deamidation) or PAD (deimidation) increased immunogenicity (McGinty et al., 2014). These peptides bind MHC class II molecules more strongly than the native peptides. Furthermore, T cells that recognize the modified peptides were present at higher frequencies in human T1D patients than in HLA-matched control subjects. These T cells responded to Tgase2-modified peptides of GAD65 more strongly than to the unmodified peptides and displayed a disease-relevant memory phenotype. These data demonstrated a role for Ca2+-dependent PTM in increasing immunogenicity of GAD65 peptides, and further supported a role for PTM-mediated neo-antigen generation in human T1D.
Most recently, citrullinated GRP78 was identified as an autoantigen in diabetic NOD mice (Rondas et al., 2015). CD4+ T cells from diabetic NOD mice secreted significantly higher IFNγ in response to citrullinated GRP78 compared to T cells from non-diabetic mice. In addition, new-onset diabetic NOD mice exhibited higher titers of autoantibodies that recognize modified GRP78 compared to age-matched non-diabetic mice. Importantly, these T cell responses and α-GRP78 autoantibodies specifically recognized the ctirullinated peptide, not the native peptide, demonstrating the relevance of PTM to the generation of this neo-antigen. This study, therefore, identified modified GRP78 as a novel autoantigen in the NOD mouse model of T1D.
Together, these studies demonstrate that, as in other autoimmune disorders, PTM enhances the immunogenicity of several known autoantigens in T1D. However, these studies were conducted with synthetic peptides that were modified in vitro or designed to mimic modified sequences. Whether the β cell proteins from which these peptides are derived undergo PTM within the β cell remains unknown. In addition, the mechanisms by which Tgase2 and PAD might be activated in the β cell remain undefined. However, as we have discussed here, Tgase2 and PAD are both Ca2+-dependent and known to be activated during ER stress. β cells inherently undergo particularly high levels of ER stress, which may be further increased upon exposure to environmental triggers of T1D. This high ER stress may activate Tgase2 and PAD to modify endogenous β cell proteins, generating neo-antigens. Therefore, β cell autoantigens may become immunogenic due to ER stress-induced PTM.
Many elegant and seminal studies have demonstrated that peptides derived from β cell autoantigens become more immunogenic after PTM (Mannering et al., 2005; Delong et al., 2012; Dunne et al., 2012; Gottlieb et al., 2014; McGinty et al., 2014; van Lummel et al., 2014; Rondas et al., 2015). However, the mechanisms by which these neo-antigens are modified in the β cell have not yet been elucidated. Here, we propose that the normal physiology of the β cell, together with the exposure of β cells to a variety of environmental factors, significantly raises ER stress, leading to the release of Ca2+ from the ER lumen into the cytosol. This Ca2+ flux may activate cytosolic PTM enzymes, which could modify β cell proteins, generating neo-antigens (Figure 4). Because islet β cells are inherently susceptible to high ER stress, these PTMs may occur in all β cells in all individuals. Therefore, T1D onset may not be determined by whether these neo-antigens are generated, but perhaps by genetic predisposition to autoimmunity. Individuals without a genetic predisposition to autoimmunity do not experience a failure of immune tolerance due to central and peripheral mechanisms that maintain immunological tolerance. Thus, the presentation of ER stress-induced modified neo-antigens by APCs may not activate peripheral T cells and T1D may not occur. In contrast, individuals that do harbor genetic predispositions to autoimmunity experience defects in mechanisms of immune tolerance. In these individuals, presentation of modified neo-antigens by APCs could activate autoreactive T cells and cause autoimmune destruction of β cells.
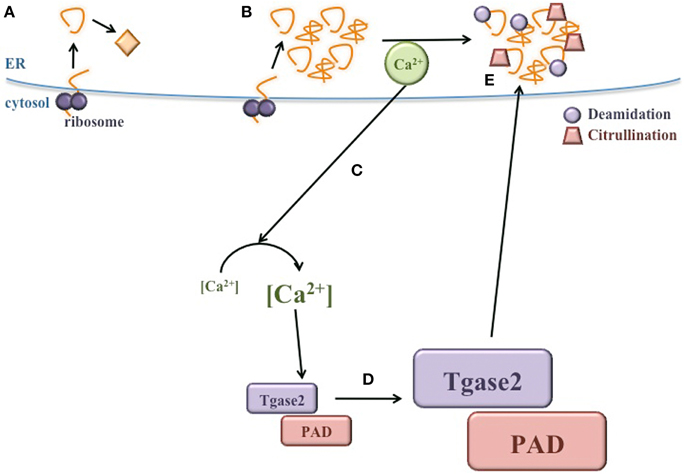
Figure 4. ER stress induces neo-antigen formation in β cells. (A) During normal conditions, proteins are translated and properly folded in the ER lumen. (B) During ER stress, proper protein folding is inhibited and misfolded proteins accumulate. (C) Ca2+ is released from the ER, significantly increasing the concentration of cytosolic Ca2+. (D) Heightened Ca2+ in the cytosol increases the activity of PTM enzymes, such as Tgase2 and PAD. (E) Activated Tgase2 and PAD modify β cell proteins, generating neo-antigens for the autoimmune response in T1D.
Once the autoimmune response is initiated, the effects of β cell ER stress are magnified. ER stress progressively increases with immune infiltration into the islet (Tersey et al., 2012). Heightened ER stress could lead to increased cytosolic Ca2+ and increased activity of Tgase2 and PAD. Recent studies have shown that Tgase2- and PAD-mediated PTMs increase the immunogenicity of peptides derived from known β cell autoantigens (Table 2). Therefore, as β cell ER stress progressively increases, these ever-more active enzymes may modify proteins beyond their physiological targets, including known β cell autoantigens. These neo-antigens could be processed and presented by APCs to T cells in draining lymph nodes. Activated immune cells returning to the islet may further increase β cell ER tress by two mechanisms. First, activated immune cells secrete cytokines that directly increase ER stress. Second, immune-mediated destruction reduces β cell mass, requiring the remaining β cells to produce more insulin per cell and augmenting the ER stress in each β cell. Increased ER stress likely leads to the generation of additional neo-antigens, further fueling the autoimmune response. Therefore, once pathology is initiated in T1D, the cycle of ER stress and neo-antigen generation likely hastens the onset of T1D and continues until the β cell mass is fully destroyed.
The recent studies that have identified modified β cell peptides as neo-antigens have opened important new areas of research in the field of T1D. Additional studies to confirm the cause of increased ER stress in the β cell, and to establish the role of ER stress in the generation of these neo-antigens, will further advance the field. In particular, understanding how these neo-antigens arise in β cells will identify opportunities for therapeutic intervention before the β cell mass is destroyed. For example, therapies that aid in proper protein folding or otherwise reduce ER stress may prevent the formation of neo-antigens. Alternatively, therapeutic agents that promote the degradation of abnormal proteins may remove neo-antigens from β cells. Either therapeutic mechanism may prevent immune-mediated recognition of β cells. Indeed, therapeutic agents that reduce ER stress or degrade misfolded proteins are effective in other disease models (Boyce et al., 2005; Ozcan et al., 2006; Harris and Rubinsztein, 2011; Zode et al., 2011; Bachar-Wikstrom et al., 2012; Hasnain et al., 2012; Engin et al., 2013; Jiang et al., 2015). Similar treatments in T1D models may reduce ER stress-induced neo-antigen formation in β cells, preventing immune destruction of β cells and preventing onset of T1D.
MM, EJ, and JP contributed to the composition of this manuscript.
This work was supported in part by the Juvenile Diabetes Research Foundation (3-PDF-2014-213-A-N to MM, 2-SRA-2014-297-Q-R to EJ, and 2-SRA-2014-296-Q-R to JP), the American Diabetes Association (1-12-BS-161 to JP), and Children's Hospital of Pittsburgh of the UPMC Health System (Cochrane-Weber Endowed Fund to JP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Gina M. Coudriet, Ph.D., and Dana M. Previte (both of University of Pittsburgh) for review of this manuscript.
APC, antigen presenting cell; ATF6, activating transcription factor 6; BiP, binding immunoglobulin protein; Ca2+, calcium; CHgA, chromogranin A; CHOP, C/EBP Homologous Protein; EAE, experimental autoimmune encephalomyelitis; eIF2α, α subunit of translation initiation factor 2; ER, endoplasmic reticulum; GAD, glutamic acid decarboxylase; GRP78, 78 kDa glucose-regulated protein; HLA, human leukocyte antigen; IA-2, tyrosine phosphatase-like insulinoma antigen 2; IAPP, islet amyloid polypeptide; ICA69, islet cell autoantigen 69; IGRP, islet-specific glucose-6-phosphatase catalytic subunit-related protein; IKKγ, IκB kinase gamma; IP3R, inositol 1, 4, 5-trisphosphate receptor; IRE1, inositol-requiring protein 1; JNK, c-jun N-terminal; MAP kinase, mitogen-activated protein kinase; NET, neutrophil extracellular traps; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOD mouse, non-obese diabetic mouse; PAD, peptidylarginine deiminases; PDI, protein disulfide isomerases; PERK, protein kinase RNA (PKR)-like ER kinase; Phogrin, phosphatase homolog of granules from rat insulinomas; PTM, post-translational modification; PTPN22, protein tyrosine phosphatase, non-receptor type 22; ROS, reactive oxygen species; RyR, ryanodine-receptor; SERCA, sacro/endoplasmic reticulum Ca2+ ATPase; Tgase2, tissue transglutaminase 2; T1D, type 1 diabetes; UPR, unfolded protein response; XBP-1, X-box binding protein 1; ZnT8, zinc transporter 8.
Akimov, S. S., Krylov, D., Fleischman, L. F., and Belkin, A. M. (2000). Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 148, 825–838. doi: 10.1083/jcb.148.4.825
Araki, E., Oyadomari, S., and Mori, M. (2003a). Endoplasmic reticulum stress and diabetes mellitus. Intern. Med. 42, 7–14. doi: 10.2169/internalmedicine.42.7
Araki, E., Oyadomari, S., and Mori, M. (2003b). Impact of endoplasmic reticulum stress pathway on pancreatic beta-cells and diabetes mellitus. Exp. Biol. Med. (Maywood) 228, 1213–1217.
Atkinson, M. A., Bowman, M. A., Campbell, L., Darrow, B. L., Kaufman, D. L., and Maclaren, N. K. (1994). Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J. Clin. Invest. 94, 2125–2129. doi: 10.1172/JCI117567
Bachar-Wikstrom, E., Wikstrom, J. D., Ariav, Y., Tirosh, B., Kaiser, N., Cerasi, E., et al. (2012). Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes. 62, 1227–1237. doi: 10.2337/db12-1474
Baekkeskov, S., Aanstoot, H. J., Christgau, S., Reetz, A., Solimena, M., Cascalho, M., et al. (1990). Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 347, 151–156. doi: 10.1038/347151a0
Ballestar, E., Abad, C., and Franco, L. (1996). Core histones are glutaminyl substrates for tissue transglutaminase. J. Biol. Chem. 271, 18817–18824. doi: 10.1074/jbc.271.31.18817
Barnett, A. H., Eff, C., Leslie, R. D., and Pyke, D. A. (1981). Diabetes in identical twins. A study of 200 Pairs. Diabetologia 20, 87–93. doi: 10.1007/bf00262007
Barone, M. V., Caputo, I., Ribecco, M. T., Maglio, M., Marzari, R., Sblattero, D., et al. (2007). Humoral immune response to tissue transglutaminase is related to epithelial cell proliferation in celiac disease. Gastroenterology 132, 1245–1253. doi: 10.1053/j.gastro.2007.01.030
Bedoya, F. J., Solano, F., and Lucas, M. (1996). N-monomethyl-arginine and nicotinamide prevent streptozotocin-induced double strand DNA break formation in pancreatic rat islets. Experientia 52, 344–347. doi: 10.1007/BF01919538
Berridge, M. J., Lipp, P., and Bootman, M. D. (2000). The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21. doi: 10.1038/35036035
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Bonifacio, E., Lampasona, V., Genovese, S., Ferrari, M., and Bosi, E. (1995). Identification of protein tyrosine phosphatase-like IA2 (islet cell antigen 512) as the insulin-dependent diabetes-related 37/40K autoantigen and a target of islet-cell antibodies. J. Immunol. 155, 5419–5426.
Bottini, N., Musumeci, L., Alonso, A., Rahmouni, S., Nika, K., Rostamkhani, M., et al. (2004). A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 36, 337–338. doi: 10.1038/ng1323
Boyce, M., Bryant, K. F., Jousse, C., Long, K., Harding, H. P., Scheuner, D., et al. (2005). A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 307, 935–939. doi: 10.1126/science.1101902
Brahms, H., Raymackers, J., Union, A., de Keyser, F., Meheus, L., and Lührmann, R. (2000). The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for anti-Sm autoantibodies. J. Biol. Chem. 275, 17122–17129. doi: 10.1074/jbc.M000300200
Bruce, S. E., Bjarnason, I., and Peters, T. J. (1985). Human jejunal transglutaminase: demonstration of activity, enzyme kinetics and substrate specificity with special relation to gliadin and coeliac disease. Clin. Sci. (Lond.) 68, 573–579. doi: 10.1042/cs0680573
Burkhardt, H., Sehnert, B., Bockermann, R., Engström, A., Kalden, J. R., and Holmdahl, R. (2005). Humoral immune response to citrullinated collagen Type II determinants in early rheumatoid arthritis. Eur. J. Immunol. 35, 1643–1652. doi: 10.1002/eji.200526000
Corthay, A., Bäcklund, J., Broddefalk, J., Michaëlsson, E., Goldschmidt, T. J., Kihlberg, J., et al. (1998). Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur. J. Immunol. 28, 2580–2590.
Coudevylle, N., Rokas, D., Sakarellos-Daitsiotis, M., Krikorian, D., Panou-Pomonis, E., Sakarellos, C., et al. (2006). Phosphorylated and nonphosphorylated epitopes of the La/SSB autoantigen: comparison of their antigenic and conformational characteristics. Biopolymers 84, 368–382. doi: 10.1002/bip.20458
DIAMOND Project Group (2006). Incidence and trends of childhood type 1 diabetes worldwide 1990–1999. Diabet. Med. 23, 857–866. doi: 10.1111/j.1464-5491.2006.01925.x
Delmastro, M. M., and Piganelli, J. D. (2011). Oxidative stress and redox modulation potential in type 1 diabetes. Clin. Dev. Immunol. 2011:593863. doi: 10.1155/2011/593863
Delmastro-Greenwood, M. M., Tse, H. M., and Piganelli, J. D. (2014). Effects of metalloporphyrins on reducing inflammation and autoimmunity. Antioxid. Redox Signal. 20, 2465–2477. doi: 10.1089/ars.2013.5257
Delong, T., Baker, R. L., He, J., Barbour, G., Bradley, B., and Haskins, K. (2012). Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes 61, 3239–3246. doi: 10.2337/db12-0112
Delong, T., Baker, R. L., Reisdorph, N., Reisdorph, R., Powell, R. L., Armstrong, M., et al. (2011). Islet amyloid polypeptide is a target antigen for diabetogenic CD4+ T cells. Diabetes 60, 2325–2330. doi: 10.2337/db,11-0288
Dieterich, W., Ehnis, T., Bauer, M., Donner, P., Volta, U., Riecken, E. O., et al. (1997). Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 3, 797–801. doi: 10.1038/nm0797-797
Dorman, J. S., LaPorte, R. E., Stone, R. A., and Trucco, M. (1990). Worldwide differences in the incidence of type I diabetes are associated with amino acid variation at position 57 of the HLA-DQ beta chain. Proc. Natl. Acad. Sci. U.S.A. 87, 7370–7374. doi: 10.1073/pnas.87.19.7370
Doyle, H. A., and Mamula, M. J. (2012). Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr. Opin. Immunol. 24, 112–118. doi: 10.1016/j.coi.2011.12.003
Driver, J. P., Chen, Y. G., and Mathews, C. E. (2012). Comparative genetics: synergizing human and NOD mouse studies for identifying genetic causation of type 1 diabetes. Rev. Diabet. Stud. 9, 169–187. doi: 10.1900/RDS.2012.9.169
Dunne, J. L., Overbergh, L., Purcell, A. W., and Mathieu, C. (2012). Posttranslational modifications of proteins in type 1 diabetes: the next step in finding the cure? Diabetes 61, 1907–1914. doi: 10.2337/db11-1675
Eizirik, D. L., Cardozo, A. K., and Cnop, M. (2008). The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 29, 42–61. doi: 10.1210/er.2007-0015
Engin, F., Yermalovich, A., Ngyuen, T., Hummasti, S., Fu, W., Eizirik, D. L., et al. (2013). Restoration of the unfolded protein response in pancreatic beta cells protects mice against type 1 diabetes. Sci. Transl. Med. 5, 211ra156. doi: 10.1126/scitranslmed.3006534
Facchiano, F., Facchiano, A., and Facchiano, A. M. (2006). The role of transglutaminase-2 and its substrates in human diseases. Front. Biosci. 11, 1758–1773. doi: 10.2741/1921
Fan, Y., Rudert, W. A., Grupillo, M., He, J., Sisino, G., and Trucco, M. (2009). Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J. 28, 2812–2824. doi: 10.1038/emboj.2009.212
Favero, T. G., Zable, A. C., and Abramson, J. J. (1995). Hydrogen peroxide stimulates the Ca2+ release channel from skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 270, 25557–25563. doi: 10.1074/jbc.270.43.25557
Fesus, L., and Piacentini, M. (2002). Transglutaminase 2: an enigmatic enzyme with diverse functions. Trends Biochem. Sci. 27, 534–539. doi: 10.1016/S0968-0004(02)02182-5
Fleckenstein, B., Qiao, S. W., Larsen, M. R., Jung, G., Roepstorff, P., and Sollid, L. M. (2004). Molecular characterization of covalent complexes between tissue transglutaminase and gliadin peptides. J. Biol. Chem. 279, 17607–17616. doi: 10.1074/jbc.M310198200
Fok, J. Y., and Mehta, K. (2007). Tissue transglutaminase induces the release of apoptosis inducing factor and results in apoptotic death of pancreatic cancer cells. Apoptosis 12, 1455–1463. doi: 10.1007/s10495-007-0079-3
Fonseca, S. G., Lipson, K. L., and Urano, F. (2007). Endoplasmic reticulum stress signaling in pancreatic beta-cells. Antioxid. Redox Signal. 9, 2335–2344. doi: 10.1089/ars.2007.1790
Gale, E. A. (2002). The rise of childhood type 1 diabetes in the 20th century. Diabetes 51, 3353–3361. doi: 10.2337/diabetes.51.12.3353
Gaudry, C. A., Verderio, E., Aeschlimann, D., Cox, A., Smith, C., and Griffin, M. (1999). Cell surface localization of tissue transglutaminase is dependent on a fibronectin-binding site in its N-terminal beta-sandwich domain. J. Biol. Chem. 274, 30707–30714. doi: 10.1074/jbc.274.43.30707
Geenen, V. (2012). Thymus and type 1 diabetes: an update. Diabetes Res. Clin. Pract. 98, 26–32. doi: 10.1016/j.diabres.2012.05.023
Gething, M. J., and Sambrook, J. (1992). Protein folding in the cell. Nature 355, 33–45. doi: 10.1038/355033a0
Gil-Parrado, S., Fernández-Montalván, A., Assfalg-Machleidt, I., Popp, O., Bestvater, F., Holloschi, A., et al. (2002). Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J. Biol. Chem. 277, 27217–27226. doi: 10.1074/jbc.M202945200
Görlach, A., Klappa, P., and Kietzmann, T. (2006). The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 8, 1391–1418. doi: 10.1089/ars.2006.8.1391
Gorus, F. K., Sodoyez, J. C., Pipeleers, D. G., Keymeulen, B., Foriers, A., and Van Schravendijk, C. F. (1992). Detection of autoantibodies against islet amyloid polypeptide in human serum. Lack of association with type 1 (insulin-dependent) diabetes mellitus, or with conditions favouring amyloid deposition in islets. The Belgian Diabetes Registry. Diabetologia 35, 1080–1086. doi: 10.1007/BF02221685
Gottlieb, P. A., Delong, T., Baker, R. L., Fitzgerald-Miller, L., Wagner, R., Cook, G., et al. (2014). Chromogranin A is a T cell antigen in human type 1 diabetes. J. Autoimmun. 50, 38–41. doi: 10.1016/j.jaut.2013.10.003
Gundemir, S., Colak, G., Tucholski, J., and Johnson, G. V. (2012). Transglutaminase 2: a molecular swiss army knife. Biochim. Biophys. Acta 1823, 406–419. doi: 10.1016/j.bbamcr.2011.09.012
Hajnóczky, G., Davies, E., and Madesh, M. (2003). Calcium signaling and apoptosis. Biochem. Biophys. Res. Commun. 304, 445–454. doi: 10.1016/S0006-291X(03)00616-8
Halttunen, T., and Mäki, M. (1999). Serum immunoglobulin a from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterology 116, 566–572. doi: 10.1016/S0016-5085(99)70178-2
Han, J. A., and Park, S. C. (2000). Transglutaminase-dependent modulation of transcription factor Sp1 activity. Mol. Cell 10, 612–618. doi: 10.1007/s100590000020
Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M., et al. (2000a). Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108. doi: 10.1016/S1097-2765(00)00108-8
Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H., and Ron, D. (2000b). Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 5, 897–904. doi: 10.1016/S1097-2765(00)80330-5
Härkönen, T., Lankinen, H., Davydova, B., Hovi, T., and Roivainen, M. (2002). Enterovirus infection can induce immune responses that cross-react with beta-cell autoantigen tyrosine phosphatase IA-2/IAR. J. Med. Virol. 66, 340–350. doi: 10.1002/jmv.2151
Haroon, Z. A., Hettasch, J. M., Lai, T. S., Dewhirst, M. W., and Greenberg, C. S. (1999). Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 13, 1787–1795.
Harris, H., and Rubinsztein, D. C. (2011). Control of autophagy as a therapy for neurodegenerative disease. Nat. Rev. Neurol. 8, 108–117. doi: 10.1038/nrneurol.2011.200
Hasnain, S. Z., Lourie, R., Das, I., Chen, A. C., and McGuckin, M. A. (2012). The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell Biol. 90, 260–270. doi: 10.1038/icb.2011.112
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
Heikkila, R. E., Winston, B., and Cohen, G. (1976). Alloxan-induced diabetes-evidence for hydroxyl radical as a cytotoxic intermediate. Biochem. Pharmacol. 25, 1085–1092. doi: 10.1016/0006-2952(76)90502-5
Held, W., MacDonald, H. R., Weissman, I. L., Hess, M. W., and Mueller, C. (1990). Genes encoding tumor necrosis factor alpha and granzyme a are expressed during development of autoimmune diabetes. Proc. Natl. Acad. Sci. U.S.A. 87, 2239–2243. doi: 10.1073/pnas.87.6.2239
Hetz, C. (2012). The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102. doi: 10.1038/nrm3270
Hiemstra, H. S., Schloot, N. C., van Veelen, P. A., Willemen, S. J., Franken, K. L., van Rood, J. J., et al. (2001). Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 98, 3988–3991. doi: 10.1073/pnas.071050898
Hitomi, J., Katayama, T., Eguchi, Y., Kudo, T., Taniguchi, M., Koyama, Y., et al. (2004). Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and abeta-induced cell death. J. Cell Biol. 165, 347–356. doi: 10.1083/jcb.200310015
Hogquist, K. A., and Jameson, S. C. (2014). The self-obsession of T cells: how TCR signaling thresholds affect fate “decisions” and effector function. Nat. Immunol. 15, 815–823. doi: 10.1038/ni.2938
Horwitz, M. S., Bradley, L. M., Harbertson, J., Krahl, T., Lee, J., and Sarvetnick, N. (1998). Diabetes induced by coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat. Med. 4, 781–785. doi: 10.1038/nm0798-781
Horwitz, M. S., Ilic, A., Fine, C., Balasa, B., and Sarvetnick, N. (2004). Coxsackieviral-mediated diabetes: induction requires antigen-presenting cells and is accompanied by phagocytosis of beta cells. Clin. Immunol. 110, 134–144. doi: 10.1016/j.clim.2003.09.014
Ientile, R., Caccamo, D., and Griffin, M. (2007). Tissue transglutaminase and the stress response. Amino Acids 33, 385–394. doi: 10.1007/s00726-007-0517-0
Inagaki, M., Takahara, H., Nishi, Y., Sugawara, K., and Sato, C. (1989). Ca2+-dependent deimination-induced disassembly of intermediate filaments involves specific modification of the amino-terminal head domain. J. Biol. Chem. 264, 18119–18127.
Iwawaki, T., Akai, R., Kohno, K., and Miura, M. (2004). A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 10, 98–102. doi: 10.1038/nm970
Jang, B., Shin, H. Y., Choi, J. K., Nguyen du, P. T., Jeong, B. H., Ishigami, A., et al. (2011). Subcellular localization of peptidylarginine deiminase 2 and citrullinated proteins in brains of scrapie-infected mice: nuclear localization of PAD2 and membrane fraction-enriched citrullinated proteins. J. Neuropathol. Exp. Neurol. 70, 116–124. doi: 10.1097/NEN.0b013e318207559e
Jiang, D., Niwa, M., and Koong, A. C. (2015). Targeting the IRE1alpha-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 33, 48–56. doi: 10.1016/j.semcancer.2015.04.010
Jiang, Z., and Woda, B. A. (1991). Cytokine gene expression in the islets of the diabetic biobreeding/worcester rat. J. Immunol. 146, 2990–2994.
Keller, R. J. (1990). Cellular immunity to human insulin in individuals at high risk for the development of type I diabetes mellitus. J. Autoimmun. 3, 321–327. doi: 10.1016/0896-8411(90)90150-Q
Kim, H. R., Rho, H. W., Park, B. H., Park, J. W., Kim, J. S., Kim, U. H., et al. (1994). Role of Ca2+ in alloxan-induced pancreatic beta-cell damage. Biochim. Biophys. Acta 1227, 87–91. doi: 10.1016/0925-4439(94)90111-2
Kim, M. J., Jo, D. G., Hong, G. S., Kim, B. J., Lai, M., Cho, D. H., et al. (2002). Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc. Natl. Acad. Sci. U.S.A. 99, 9870–9875. doi: 10.1073/pnas.152336999
Kim, M. K., Kim, H. S., Lee, I. K., and Park, K. G. (2012). Endoplasmic reticulum stress and insulin biosynthesis: a review. Exp. Diabetes Res. 2012:509437. doi: 10.1155/2012/509437
Kinloch, A., Tatzer, V., Wait, R., Peston, D., Lundberg, K., Donatien, P., et al. (2005). Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res. Ther. 7, R1421–R1429. doi: 10.1186/ar1845
Knip, M., Veijola, R., Virtanen, S. M., Hyoty, H., Vaarala, O., and Akerblom, H. K. (2005). Environmental triggers and determinants of type 1 diabetes. Diabetes 54(Suppl. 2), S125–S136. doi: 10.2337/diabetes.54.suppl_2.S125
Kojima, S., Kuo, T. F., Tatsukawa, H., and Hirose, S. (2010). Induction of cross-linking and silencing of Sp1 by transglutaminase during liver injury in ASH and NASH via different ER stress pathways. Dig. Dis. 28, 715–721. doi: 10.1159/000324278
Kuo, T. F., Tatsukawa, H., Matsuura, T., Nagatsuma, K., Hirose, S., and Kojima, S. (2012). Free fatty acids induce transglutaminase 2-dependent apoptosis in hepatocytes via ER stress-stimulated PERK pathways. J. Cell. Physiol. 227, 1130–1137. doi: 10.1002/jcp.22833
Lan, M. S., Wasserfall, C., Maclaren, N. K., and Notkins, A. L. (1996). IA-2, a transmembrane protein of the protein tyrosine phosphatase family, is a major autoantigen in insulin-dependent diabetes mellitus. Proc. Natl. Acad. Sci. U.S.A. 93, 6367–6370. doi: 10.1073/pnas.93.13.6367
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003
Lee, H., Park, M. T., Choi, B. H., Oh, E. T., Song, M. J., Lee, J., et al. (2011). Endoplasmic reticulum stress-induced JNK activation is a critical event leading to mitochondria-mediated cell death caused by beta-lapachone treatment. PLoS ONE 6:e21533. doi: 10.1371/journal.pone.0021533
Lee, H. J., Joo, M., Abdolrasulnia, R., Young, D. G., Choi, I., Ware, L. B., et al. (2010). Peptidylarginine deiminase 2 suppresses inhibitory {Kappa}B kinase activity in lipopolysaccharide-stimulated RAW 264.7 macrophages. J. Biol. Chem. 285, 39655–39662. doi: 10.1074/jbc.M110.170290
Lesort, M., Attanavanich, K., Zhang, J., and Johnson, G. V. (1998). Distinct nuclear localization and activity of tissue transglutaminase. J. Biol. Chem. 273, 11991–11994. doi: 10.1074/jbc.273.20.11991
Li, P., Li, M., Lindberg, M. R., Kennett, M. J., Xiong, N., and Wang, Y. (2010). PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862. doi: 10.1084/jem.20100239
Lieberman, S. M., Evans, A. M., Han, B., Takaki, T., Vinnitskaya, Y., Caldwell, J. A., et al. (2003). Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc. Natl. Acad. Sci. U.S.A. 100, 8384–8388. doi: 10.1073/pnas.0932778100
Like, A. A., and Rossini, A. A. (1976). Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 193, 415–417. doi: 10.1126/science.180605
Lipson, K. L., Fonseca, S. G., and Urano, F. (2006b). Endoplasmic reticulum stress-induced apoptosis and auto-immunity in diabetes. Curr. Mol. Med. 6, 71–77. doi: 10.2174/156652406775574613
Lipson, K. L., Fonseca, S. G., Ishigaki, S., Nguyen, L. X., Foss, E., Bortell, R., et al. (2006a). Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell. Metab. 4, 245–254. doi: 10.1016/j.cmet.2006.07.007
Luca, D., Ringquist, S., Klei, L., Lee, A. B., Gieger, C., Wichmann, H. E., et al. (2008). On the use of general control samples for genome-wide association studies: genetic matching highlights causal variants. Am. J. Hum. Genet. 82, 453–463. doi: 10.1016/j.ajhg.2007.11.003
Ma, Y., and Hendershot, L. M. (2004). ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 28, 51–65. doi: 10.1016/j.jchemneu.2003.08.007
Mamula, M. J., Gee, R. J., Elliott, J. I., Sette, A., Southwood, S., Jones, P. J., et al. (1999). Isoaspartyl post-translational modification triggers autoimmune responses to self-proteins. J. Biol. Chem. 274, 22321–22327. doi: 10.1074/jbc.274.32.22321
Mandrup-Poulsen, T., Spinas, G. A., Prowse, S. J., Hansen, B. S., Jorgensen, D. W., Bendtzen, K., et al. (1987). Islet cytotoxicity of interleukin 1. Influence of culture conditions and islet donor characteristics. Diabetes 36, 641–647. doi: 10.2337/diab.36.5.641
Mannering, S. I., Harrison, L. C., Williamson, N. A., Morris, J. S., Thearle, D. J., Jensen, K. P., et al. (2005). The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J. Exp. Med. 202, 1191–1197. doi: 10.1084/jem.20051251
Marhfour, I., Lopez, X. M., Lefkaditis, D., Salmon, I., Allagnat, F., Richardson, S. J., et al. (2012). Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 55, 2417–2420. doi: 10.1007/s00125-012-2604-3
Martin, R., Whitaker, J. N., Rhame, L., Goodin, R. R., and McFarland, H. F. (1994). Citrulline-containing myelin basic protein is recognized by T-cell lines derived from multiple sclerosis patients and healthy individuals. Neurology 44, 123–129. doi: 10.1212/WNL.44.1.123
Masson-Bessière, C., Sebbag, M., Girbal-Neuhauser, E., Nogueira, L., Vincent, C., Senshu, T., et al. (2001). The major synovial targets of the rheumatoid arthritis-specific antifilaggrin autoantibodies are deiminated forms of the alpha- and beta-chains of fibrin. J. Immunol. 166, 4177–4184. doi: 10.4049/jimmunol.166.6.4177
McCullough, K. D., Martindale, J. L., Klotz, L. O., Aw, T. Y., and Holbrook, N. J. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001
McGinty, J. W., Chow, I. T., Greenbaum, C., Odegard, J., Kwok, W. W., and James, E. A. (2014). Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes 63, 3033–3040. doi: 10.2337/db13-1952
Mekahli, D., Bultynck, G., Parys, J. B., De Smedt, H., and Missiaen, L. (2011). Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 3:a004317. doi: 10.1101/cshperspect.a004317
Meldolesi, J., and Pozzan, T. (1998). The endoplasmic reticulum Ca2+ store: a view from the lumen. Trends Biochem. Sci. 23, 10–14. doi: 10.1016/S0968-0004(97)01143-2
Michalak, M., Groenendyk, J., Szabo, E., Gold, L. I., and Opas, M. (2009). Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 417, 651–666. doi: 10.1042/BJ20081847
Molberg, O., Mcadam, S. N., Körner, R., Quarsten, H., Kristiansen, C., Madsen, L., et al. (1998). Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 4, 713–717. doi: 10.1038/nm0698-713
Monneaux, F., Lozano, J. M., Patarroyo, M. E., Briand, J. P., and Muller, S. (2003). T cell recognition and therapeutic effect of a phosphorylated synthetic peptide of the 70K snRNP protein administered in MR/Lpr mice. Eur. J. Immunol. 33, 287–296. doi: 10.1002/immu.200310002
Nakagawa, T., and Yuan, J. (2000). Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 150, 887–894. doi: 10.1083/jcb.150.4.887
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A., et al. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403, 98–103. doi: 10.1038/47513
Nakamura, K., Bossy-Wetzel, E., Burns, K., Fadel, M. P., Lozyk, M., Goping, I. S., et al. (2000). Changes in endoplasmic reticulum luminal environment affect cell sensitivity to apoptosis. J. Cell Biol. 150, 731–740. doi: 10.1083/jcb.150.4.731
Nayak, D. K., Calderon, B., Vomund, A. N., and Unanue, E. R. (2014). ZnT8-reactive T cells are weakly pathogenic in NOD mice but can participate in diabetes under inflammatory conditions. Diabetes 63, 3438–3448. doi: 10.2337/db13-1882
Nigam, S. K., Goldberg, A. L., Ho, S., Rohde, M. F., Bush, K. T., and Sherman, M. Y. (1994). A set of endoplasmic reticulum proteins possessing properties of molecular chaperones includes Ca(2+)-binding proteins and members of the thioredoxin superfamily. J. Biol. Chem. 269, 1744–1749.
Nozaki, J. I., Kubota, H., Yoshida, H., Naitoh, M., Goji, J., Yoshinaga, T., et al. (2004). The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/akita pancreatic beta cells. Genes Cells. 9, 261–270. doi: 10.1111/j.1356-9597.2004.00721.x
Onkamo, P., Väänänen, S., Karvonen, M., and Tuomilehto, J. (1999). Worldwide increase in incidence of type I diabetes–the analysis of the data on published incidence trends. Diabetologia 42, 1395–1403. doi: 10.1007/s001250051309
Orru, S., Caputo, I., D'Amato, A., Ruoppolo, M., and Esposito, C. (2003). Proteomics identification of acyl-acceptor and acyl-donor substrates for transglutaminase in a human intestinal epithelial cell line. Implications for celiac disease. J. Biol. Chem. 278, 31766–31773. doi: 10.1074/jbc.M305080200
Ortsäter, H., and Sjöholm, Å. (2007). A busy cell—endoplasmic reticulum stress in the pancreatic beta-cell. Mol. Cell. Endocrinol. 277, 1–5. doi: 10.1016/j.mce.2007.06.006
Oyadomari, S., Koizumi, A., Takeda, K., Gotoh, T., Akira, S., Araki, E., et al. (2002). Targeted disruption of the chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109, 525–532. doi: 10.1172/JCI0214550
Ozcan, U., Yilmaz, E., Ozcan, L., Furuhashi, M., Vaillancourt, E., Smith, R. O., et al. (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140. doi: 10.1126/science.1128294
Park, B. H., Rho, H. W., Park, J. W., Cho, C. G., Kim, J. S., Chung, H. T., et al. (1995). Protective mechanism of glucose against alloxan-induced pancreatic beta-cell damage. Biochem. Biophys. Res. Commun. 210, 1–6. doi: 10.1006/bbrc.1995.1619
Park, D., Choi, S. S., and Ha, K. S. (2010). Transglutaminase 2: a multi-functional protein in multiple subcellular compartments. Amino Acids 39, 619–631. doi: 10.1007/s00726-010-0500-z
Pietropaolo, M., Castano, L., Babu, S., Buelow, R., Kuo, Y. L., Martin, S., et al. (1993). Islet cell autoantigen 69 kD (ICA69). Molecular cloning and characterization of a novel diabetes-associated autoantigen. J. Clin. Invest. 92, 359–371. doi: 10.1172/JCI116574
Piganelli, J. D., Flores, S. C., Cruz, C., Koepp, J., Batinic-Haberle, I., Crapo, J., et al. (2002). A metalloporphyrin-based superoxide dismutase mimic inhibits adoptive transfer of autoimmune diabetes by a diabetogenic T-cell clone. Diabetes 51, 347–355. doi: 10.2337/diabetes.51.2.347
Piganelli, J. D., Martin, T., and Haskins, K. (1998). Splenic macrophages from the NOD mouse are defective in the ability to present antigen. Diabetes 47, 1212–1218. doi: 10.2337/diab.47.8.1212
Pociot, F., and McDermott, M. F. (2002). Genetics of type 1 diabetes mellitus. Genes Immun. 3, 235–249. doi: 10.1038/sj.gene.6363875
Rogers, G. E., Harding, H. W., and Llewellyn-Smith, I. J. (1977). The origin of citrulline-containing proteins in the hair follicle and the chemical nature of trichohyalin, an intracellular precursor. Biochim. Biophys. Acta 495, 159–175. doi: 10.1016/0005-2795(77)90250-1
Ron, D. (2002). Proteotoxicity in the endoplasmic reticulum: lessons from the akita diabetic mouse. J. Clin. Invest. 109, 443–445. doi: 10.1172/JCI0215020
Rondas, D., Crevecoeur, I., D'Hertog, W., Ferreira, G. B., Staes, A., Garg, A. D., et al. (2015). Citrullinated glucose-regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes 64, 573–586. doi: 10.2337/db14-0621
Rossini, A. A., Appel, M. C., Williams, R. M., and Like, A. A. (1977). Genetic influence of the streptozotocin-induced insulitis and hyperglycemia. Diabetes 26, 916–920. doi: 10.2337/diab.26.10.916
Russo, L., Marsella, C., Nardo, G., Massignan, T., Alessio, M., Piermarini, E., et al. (2013). Transglutaminase 2 transamidation activity during first-phase insulin secretion: natural substrates in INS-1E. Acta Diabetol. 50, 61–72. doi: 10.1007/s00592-012-0381-6
Sandler, S., and Swenne, I. (1983). Streptozotocin, but not alloxan, induces DNA repair synthesis in mouse pancreatic islets in vitro. Diabetologia 25, 444–447. doi: 10.1007/BF00282526
Schellekens, G. A., de Jong, B. A., van den Hoogen, F. H., van de Putte, L. B., and van Venrooij, W. J. (1998). Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Invest. 101, 273–281. doi: 10.1172/JCI1316
Scheuner, D., and Kaufman, R. J. (2008). The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr. Rev. 29, 317–333. doi: 10.1210/er.2007-0039
Schulte, B. M., Kramer, M., Ansems, M., Lanke, K. H., van Doremalen, N., Piganelli, J. D., et al. (2010). Phagocytosis of enterovirus-infected pancreatic beta-cells triggers innate immune responses in human dendritic cells. Diabetes 59, 1182–1191. doi: 10.2337/db09-1071
Serreze, D. V., Gaskins, H. R., and Leiter, E. H. (1993). Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J. Immunol. 150, 2534–2543.
Shen, J., Chen, X., Hendershot, L., and Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 3, 99–111. doi: 10.1016/S1534-5807(02)00203-4
Shimizu, Y., and Hendershot, L. M. (2009). Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal. 11, 2317–2331. doi: 10.1089/ars.2009.2501
Sosenko, J. M., Palmer, J. P., Rafkin-Mervis, L., Krischer, J. P., Cuthbertson, D., Mahon, J., et al. (2009). Incident dysglycemia and progression to type 1 diabetes among participants in the diabetes prevention trial-type 1. Diabetes Care 32, 1603–1607. doi: 10.2337/dc08-2140
Sriburi, R., Jackowski, S., Mori, K., and Brewer, J. W. (2004). XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 167, 35–41. doi: 10.1083/jcb.200406136
Stadinski, B. D., Delong, T., Reisdorph, N., Reisdorph, R., Powell, R. L., Armstrong, M., et al. (2010). Chromogranin A is an autoantigen in type 1 diabetes. Nat. Immunol. 11, 225–231. doi: 10.1038/ni.1844
Stephens, P., Grenard, P., Aeschlimann, P., Langley, M., Blain, E., Errington, R., et al. (2004). Crosslinking and G-Protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 117, 3389–3403. doi: 10.1242/jcs.01188
Takahara, H., Okamoto, H., and Sugawara, K. (1986). Calcium-dependent properties of peptidylarginine deiminase from rabbit skeletal muscle. Agric. Biol. Chem. 50, 2899–2904. doi: 10.1271/bbb1961.50.2899
Takahara, H., Tsuchida, M., Kusubata, M., Akutsu, K., Tagami, S., and Sugawara, K. (1989). Peptidylarginine deiminase of the mouse. Distribution, properties, and immunocytochemical localization. J. Biol. Chem. 264, 13361–13368.
Takasu, N., Asawa, T., Komiya, I., Nagasawa, Y., and Yamada, T. (1991a). Alloxan-induced DNA strand breaks in pancreatic islets. Evidence for H2O2 as an intermediate. J. Biol. Chem. 266, 2112–2114.
Takasu, N., Komiya, I., Asawa, T., Nagasawa, Y., and Yamada, T. (1991b). Streptozocin- and alloxan-induced H2O2 generation and DNA fragmentation in pancreatic islets. H2O2 as mediator for DNA fragmentation. Diabetes 40, 1141–1145. doi: 10.2337/diab.40.9.1141
Tarcsa, E., Marekov, L. N., Mei, G., Melino, G., Lee, S. C., and Steinert, P. M. (1996). Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J. Biol. Chem. 271, 30709–30716. doi: 10.1074/jbc.271.48.30709
Teodoro, T., Odisho, T., Sidorova, E., and Volchuk, A. (2012). Pancreatic beta-cells depend on basal expression of active ATF6alpha-p50 for cell survival even under nonstress conditions. Am. J. Physiol. Cell. Physiol. 302, C992–C1003. doi: 10.1152/ajpcell.00160.2011
Tersey, S. A., Nishiki, Y., Templin, A. T., Cabrera, S. M., Stull, N. D., Colvin, S. C., et al. (2012). Islet beta-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61, 818–827. doi: 10.2337/db11-1293
Tisch, R., Yang, X. D., Singer, S. M., Liblau, R. S., Fugger, L., and McDevitt, H. O. (1993). Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature 366, 72–75. doi: 10.1038/366072a0
Todd, J. A., Bell, J. I., and McDevitt, H. O. (1987). HLA-DQ beta gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 329, 599–604. doi: 10.1038/329599a0
Tse, H. M., Thayer, T. C., Steele, C., Cuda, C. M., Morel, L., Piganelli, J. D., et al. (2010). NADPH oxidase deficiency regulates th lineage commitment and modulates autoimmunity. J. Immunol. 185, 5247–5258. doi: 10.4049/jimmunol.1001472
van Boekel, M. A., Vossenaar, E. R., van den Hoogen, F. H., and van Venrooij, W. J. (2002). Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. 4, 87–93. doi: 10.1186/ar395
van Kuppeveld, F. J., de Jong, A. S., Melchers, W. J., and Willems, P. H. (2005). Enterovirus protein 2B Po(U)Res out the calcium: a viral strategy to survive? Trends Microbiol. 13, 41–44. doi: 10.1016/j.tim.2004.12.005
van Kuppeveld, F. J., Hoenderop, J. G., Smeets, R. L., Willems, P. H., Dijkman, H. B., Galama, J. M., et al. (1997). Coxsackievirus protein 2B modifies endoplasmic reticulum membrane and plasma membrane permeability and facilitates virus release. EMBO J. 16, 3519–3532. doi: 10.1093/emboj/16.12.3519
van Kuppeveld, F. J., Melchers, W. J., Willems, P. H., and Gadella, T. W. Jr. (2002). Homomultimerization of the coxsackievirus 2B protein in living cells visualized by fluorescence resonance energy transfer microscopy. J. Virol. 76, 9446–9456. doi: 10.1128/JVI.76.18.9446-9456.2002
van Lummel, M., Duinkerken, G., van Veelen, P. A., de Ru, A., Cordfunke, R., Zaldumbide, A., et al. (2014). Posttranslational modification of HLA-DQ binding islet autoantigens in type 1 diabetes. Diabetes 63, 237–247. doi: 10.2337/db12-1214
van Stipdonk, M. J., Willems, A. A., Amor, S., Persoon-Deen, C., Travers, P. J., Boog, C. J., et al. (1998). T cells discriminate between differentially phosphorylated forms of alphaB-crystallin, a major central nervous system myelin antigen. Int. Immunol. 10, 943–950. doi: 10.1093/intimm/10.7.943
Vasishta, A. (2002). Diagnosing early-onset rheumatoid arthritis: the role of anti-CCP antibodies. Am. Clin. Lab. 21, 34–36.
Verderio, E. A., Johnson, T., and Griffin, M. (2004). Tissue transglutaminase in normal and abnormal wound healing: review article. Amino Acids 26, 387–404. doi: 10.1007/s00726-004-0094-4
Verge, C. F., Gianani, R., Yu, L., Pietropaolo, M., Smith, T., Jackson, R. A., et al. (1995). Late progression to diabetes and evidence for chronic beta-cell autoimmunity in identical twins of patients with type I diabetes. Diabetes 44, 1176–1179. doi: 10.2337/diab.44.10.1176
Verhaar, R., Drukarch, B., Bol, J. G., Jongenelen, C. A., Musters, R. J., and Wilhelmus, M. M. (2012). Increase in endoplasmic reticulum-associated tissue transglutaminase and enzymatic activation in a cellular model of parkinson's disease. Neurobiol. Dis. 45, 839–850. doi: 10.1016/j.nbd.2011.10.012
Volchuk, A., and Ron, D. (2010). The endoplasmic reticulum stress response in the pancreatic beta-cell. Diabetes Obes. Metab. 12(Suppl. 2), 48–57. doi: 10.1111/j.1463-1326.2010.01271.x
Vossenaar, E. R., Després, N., Lapointe, E., van der Heijden, A., Lora, M., Senshu, T., et al. (2004). Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Ther. 6, R142–R150. doi: 10.1186/ar1149
Vossenaar, E. R., Nijenhuis, S., Helsen, M. M., van der Heijden, A., Senshu, T., van den Berg, W. B., et al. (2003a). Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum. 48, 2489–2500. doi: 10.1002/art.11229
Vossenaar, E. R., Zendman, A. J., van Venrooij, W. J., and Pruijn, G. J. (2003b). PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays 25, 1106–1118. doi: 10.1002/bies.10357
Wållberg, M., Bergquist, J., Achour, A., Breij, E., and Harris, R. A. (2007). Malondialdehyde modification of myelin oligodendrocyte glycoprotein leads to increased immunogenicity and encephalitogenicity. Eur. J. Immunol. 37, 1986–1995. doi: 10.1002/eji.200636912
Wallis, R. H., Wang, K., Marandi, L., Hsieh, E., Ning, T., Chao, G. Y., et al. (2009). Type 1 diabetes in the BB rat: a polygenic disease. Diabetes 58, 1007–1017. doi: 10.2337/db08-1215
Wang, Q., Zhang, H., Zhao, B., and Fei, H. (2009). IL-1beta caused pancreatic beta-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Mol. Cell. Biochem. 324, 183–190. doi: 10.1007/s11010-008-9997-9
Wang, X. Z., Lawson, B., Brewer, J. W., Zinszner, H., Sanjay, A., Mi, L. J., et al. (1996). Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 16, 4273–4280. doi: 10.1128/MCB.16.8.4273
Wegmann, D. R., Norbury-Glaser, M., and Daniel, D. (1994). Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur. J. Immunol. 24, 1853–1857. doi: 10.1002/eji.1830240820
Wenzlau, J. M., Juhl, K., Yu, L., Moua, O., Sarkar, S. A., Gottlieb, P., et al. (2007). The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc. Natl. Acad. Sci. U.S.A. 104, 17040–17045. doi: 10.1073/pnas.0705894104
Wilhelmus, M. M., Verhaar, R., Andringa, G., Bol, J. G., Cras, P., Shan, L., et al. (2011). Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in parkinson's disease brain. Brain Pathol. 21, 130–139. doi: 10.1111/j.1750-3639.2010.00429.x
Wu, J., and Kaufman, R. J. (2006). From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 13, 374–384. doi: 10.1038/sj.cdd.4401840
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237. doi: 10.1126/science.279.5348.234
Yamaguchi, H., and Wang, H. G. (2006). Tissue transglutaminase serves as an inhibitor of apoptosis by cross-linking caspase 3 in thapsigargin-treated cells. Mol. Cell. Biol. 26, 569–579. doi: 10.1128/MCB.26.2.569-579.2006
Yang, J., Danke, N. A., Berger, D., Reichstetter, S., Reijonen, H., Greenbaum, C., et al. (2006). Islet-specific glucose-6-phosphatase catalytic subunit-related protein-reactive CD4+ T cells in human subjects. J. Immunol. 176, 2781–2789. doi: 10.4049/jimmunol.176.5.2781
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Zamvil, S. S., Mitchell, D. J., Moore, A. C., Kitamura, K., Steinman, L., and Rothbard, J. B. (1986). T-cell epitope of the autoantigen myelin basic protein that induces encephalomyelitis. Nature 324, 258–260. doi: 10.1038/324258a0
Zhou, S. R., Moscarello, M. A., and Whitaker, J. N. (1995). The effects of citrullination or variable amino-terminus acylation on the encephalitogenicity of human myelin basic protein in the PL/J mouse. J. Neuroimmunol. 62, 147–152. doi: 10.1016/0165-5728(95)00112-3
Zinszner, H., Kuroda, M., Wang, X., Batchvarova, N., Lightfoot, R. T., Remotti, H., et al. (1998). CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995. doi: 10.1101/gad.12.7.982
Keywords: type 1 diabetes, β cell, ER stress, post-translational modification, neo-antigen, autoimmunity
Citation: Marré ML, James EA and Piganelli JD (2015) β cell ER stress and the implications for immunogenicity in type 1 diabetes. Front. Cell Dev. Biol. 3:67. doi: 10.3389/fcell.2015.00067
Received: 09 July 2015; Accepted: 08 October 2015;
Published: 27 October 2015.
Edited by:
Seppo J. Vainio, Oulu University, FinlandReviewed by:
Janice Blum, Indiana University School of Medicine, USACopyright © 2015 Marré, James and Piganelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jon D. Piganelli, amRwNTFAcGl0dC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.