 Xavier Roucou
Xavier Roucou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 09 October 2014
Sec. Cell Death and Survival
Volume 2 - 2014 | https://doi.org/10.3389/fcell.2014.00057
This article is part of the Research TopicPromiscuous functions of the prion protein gene familyView all 13 articles
The cellular prion protein (PrPC) is a glycosylphosphatidylinositol (GPI)-anchored protein present at the cell surface. PrPC N-terminal moiety is intrinsically disordered and is able to interact with a variety of ligands. Physiological ligands have neurotrophic activity, whilst others, including protein toxic oligomers, have neurotoxic functions. These two opposite activities involve different interacting partners and result from different PrPC-activated signaling pathways. Remarkably, PrPC may be inactivated either by physiological endoproteolysis and release of the N-terminal domain, or by ectodomain shedding. Ligand-induced PrPC dimerization or enforced dimerization of PrPC indicate that PrPC dimerization represents an important molecular switch for both intracellular signaling and inactivation by the release of PrPC N-terminal domain or shedding. In this review, we summarize evidence that cell surface receptor activity of PrPC is finely regulated by dimerization.
PrPC is a cell surface protein with a bipartite structure: the N-terminal domain is disordered and the C-terminal domain is structured and contains three α-helices and two short β-strands (Wuthrich and Riek, 2001). The physiological function of PrPC is unclear, but a large body of evidence indicates that PrPC is a neuroprotective and neurotrophic protein (Linden et al., 2008). The neuroprotective function of PrPC against different insults was demonstrated in vitro in primary neurons and in cell lines, and in vivo (Roucou and LeBlanc, 2005; Lo et al., 2007). In these studies, PrPC expression was able to slow or halt cell death whilst PrPC absence did not prevent cell death. In a pioneer investigation, PrPC expression prevented cell death triggered by serum deprivation of immortalized hippocampal neurons (Kuwahara et al., 1999). Subsequent studies provided significant evidence for the implication of PrPC in cell survival. In addition to these neuroprotective effects, PrPC regulates cell proliferation, differenciation, growth, and PrPC is also important for the expansion of stem cells in culture (Martins et al., 2010; Miranda et al., 2013). Some of these trophic mechanisms have been addressed and involve the assembly of protein complexes at the cell surface.
Most of neuroprotective and neurotrophic activities result from PrPC-mediated signaling (Martins et al., 2010; Schneider et al., 2011). Thus, a large body of data indicate that GPI-anchored PrPC is a cell surface receptor or co-receptor and that its engagement with one of its numerous ligands or with antibodies activates different intracellular pathways. Cell surface receptors are generally activated by dimerization (Heldin, 1995), and this may also be valid for PrPC which forms dimers in native conditions and can be experimentally engaged with cross-linking antibodies (Mouillet-Richard et al., 2000; Rambold et al., 2008).
In prion diseases, PrPC changes conformation into a pathological conformer termed PrPSc (Prusiner, 1998). The exact mechanism of this conformational change or prion conversion is unclear but may involve the initial formation of dimers. During the process of prion conversion, PrPSc oligomerizes and form toxic oligomers that interact with PrPC and switch its neuroprotective/neurotrophic signaling to a neurotoxic signaling (Rambold et al., 2008; Resenberger et al., 2011).
In this review, I will summarize some of the most important studies on the role of dimerization on the physiological and pathological function of PrPC and PrPSc, respectively.
PrPC dimers were detected in solution in a partially purified fraction from normal bovine brain thalamus (Meyer et al., 2000), and in murine neuroblastoma N2a cells expressing Syrian hamster PrPC (Priola et al., 1995). Syrian hamster, human and bovine PrPC expressed in baculovirus and purified under native conditions spontaneously form dimers (Hundt et al., 2003). Dimerization of human PrPC was confirmed in BHK cells overexpressing PrPC and in yeast two-hybrid assays (Hundt et al., 2003). More recently, endogenous PrPC dimers were also detected by blue native PAGE in N2a cells and the dimerization domain mapped to a hydrophobic domain of the protein (amino acids 112-MAGAAAAGAVVGGLGGYMLGSA-133) (Rambold et al., 2008). Finally, PrPC dimers were detected after chemical crosslinking in crude membranes from human neuroblastoma SH-SY5Y cells and mouse brains (Rambold et al., 2008). These results convincingly demonstrate that PrPC has an intrinsic tendency to dimerize in native conditions and suggest that dimerization is important for the physiological function of PrPC.
The assembly of natural PrPC dimers at the plasma membrane is associated with protective activity against the excitotoxin kainate and altering dimer formation results in cell death (Rambold et al., 2008). Based on their own data and previous data showing PrPC-mediated signaling using anti-PrPC antibodies (see below), the authors proposed that cell surface PrPC dimers induce protective signaling through an unknown transmembrane receptor. This study did not elucidate whether the formation of dimers is constitutive or depends on an unknown ligand. Also, the proportion of PrPC dimers is unknown.
Antibody-induced dimerization (also termed antibody-induced ligation or -cross-linking) is used to mimic an extracellular signal on cell surface receptors and trigger signal transduction. Although it is unclear if such strategy mimics the interaction with a partner or dimerization of the receptor, antibody-induced dimerization is largely used to engage a receptor in the absence of its ligand and relays intracellular signals. GPI-anchored proteins associate with raft domains in the plasma membrane and activate signal transduction pathways upon engagement with ligands or via antibody-induced dimerization (Robinson, 1991; Suzuki et al., 2012). For GPI-anchored proteins, signal transduction occurs through activation of intracellular tyrosine kinases including the Src-family kinases (Stefanova et al., 1991; Chen et al., 2006). Mouillet-Richard et al were the first to show that engagement of PrPC using an antibody-induced dimerization approach activates a Fyn-dependent signaling pathway in serotonergic and noradrenergic mouse cells differenciated from the murine neuroectodermal progenitor 1C11 clone (Mouillet-Richard et al., 2000). Similar results were obtained with two different antibodies, 1A8 and SAF61 targeting C-terminal epitopes. Using 4 different antibodies to induce PrPC dimerization, SAF61, Bar221, and 1A8 that target C-terminal epitopes, and SAF32 which targets epitope 79-92, NADPH oxidase was subsequently identified as the main primary target of PrPC-mediated signaling. NADPH oxidase-dependent reactive oxygen species production stimulated the phosphorylation of extracellular regulated kinases 1/2 (Erk1/2) in the 1C11 neuroectodermal precursor and its neuronal differentiated progenies, the hypothalamic GT1-7 cells, and the T lymphoid BW5147 cells (Schneider et al., 2003). PrPC signaling was dependent on Fyn in neuronal cells only, indicating specificity in the control of PrPC function. PrPC-mediated phosphorylation of Erk1/2 was independently confirmed in GT1-7 neuronal cells (Monnet et al., 2004) and in human neuroblastoma SH-SY5Y cells (Rambold et al., 2008). PrPC-induced ROS production and Erk1/2 phosphorylation was confirmed using an inducible dimerization strategy (Beland et al., 2012).
These studies lend support for a role of PrPC in signal transduction and further investigations provided more insight into the physiological consequence of PrPC signaling in neuronal 1C11 cells. In 1C11 serotonergic cells expressing 5-HT2B, 5-HT1B/D, and 5-HT2A receptor subtypes. PrPC dimerization interfered with the signaling activity of these three serotonergic receptors belonging to the GPCR family likely by modulating the recruitment of G-proteins (Mouillet-Richard et al., 2005). PrPC dimerization promoted the recruitment of the cAMP responsive element binding protein (CREB) transcription factor and the transcription of several genes with important function in cellular protection and neuronal plasticity (Pradines et al., 2008). In addition, PrPC dimerization inactivated the Glycogen Synthase Kinase 3β and activated serotonergic signaling through inhibition of the serotonin 1B receptor (Hernandez-Rapp et al., 2014). CREB recruitment and GSK3β are generally associated with cytoprotection, suggesting an important function of PrPC in cell survival and homeostasis.
For several years, these data were in contradiction with previous results indicating that antibody-induced PrPC dimerization is neurotoxic in vivo (Solforosi et al., 2004). However, these results were later invalidated with similar and other antibodies (Klohn et al., 2012). This debate is still ongoing since a recent study demonstrated that anti-PrPC antibodies induce rapid neurotoxicity in mice and cerebellar organotypic cultured slices (Sonati et al., 2012). Importantly, PrPC dimerization is unlikely to be involved in neuronal toxicity since single-chain antibodies were also toxic.
At the cell surface, PrPC interacts directly or indirectly with a variety of ligands as diverse as metals, lipids, nucleic acids, glycosaminoglycans, and other proteins (Linden et al., 2008; Beland and Roucou, 2012). In physiological conditions, it was proposed that PrPC is a scaffolding protein providing essential molecular interactions and signaling neurotrophic activities (Martins et al., 2010). PrPC ligands promoting neurotrophic activity include laminin, the 37-kDa/67-kDa laminin receptor precursor/laminin receptor, vitronectin, the neural cell adhesion molecule, and the Stress Inducible Protein 1 (Martins et al., 2010).
In pathological conditions, binding of PrPSc to cell surface PrPC corrupts PrPC signaling and results in cellular toxicity (Rambold et al., 2008; Resenberger et al., 2011). This finding is particularly important as it provides a simple explanation for the observation that PrPC on the cell surface is critical for the neurotoxicity of PrPSc in prion diseases (Brandner et al., 1996; Chesebro et al., 2005). PrPC dimerization is essential for the toxicity of PrPSc (Rambold et al., 2008). PrPC is also a receptor for other toxic β-sheet oligomers, including Aβ in Alzheimer's disease (Lauren et al., 2009; Gunther and Strittmatter, 2010; Resenberger et al., 2011).
Following translocation into the endoplasmic reticulum, signal peptidase removes a N-terminal signal peptide, and a C-terminal peptide is removed prior to the attachment of the GPI anchor. Thus, human PrPC is translated as an immature 253 amino acids protein and mature PrPC is a 208 residues protein. After trafficking through the secretory pathway, a fraction of PrPC may undergo three proteolytic cleavages (Altmeppen et al., 2012). An α-cleavage between residues 110–111 and 112 in a late compartment of the secretory pathway produces PrPC1, a 17 kDa GPI-anchored C-terminal polypeptide, and a 11 kDa N-terminal polypeptide released in the extracellular space. The identity of the protease responsible for α-cleavage, termed the α-PrPase (Oliveira-Martins et al., 2010), is still unclear. A β-cleavage at amino acids 89/90 generates PrPC2, a 20 kDa GPI-anchored polypeptide, and the corresponding 8 kDa PrPN2 fragment. β-cleavage occurs at the cell surface mainly in pathological conditions; calpains execute β-cleavage in prion diseases whilst reactive oxygen species perform β-cleavage under conditions of oxidative stress. In addition, a fraction of PrPC is constitutively shed from the cell surface after proteolytic cleavage close to the GPI anchor. In vivo, the main protease responsible for PrPC shedding is the zinc metalloproteinase ADAM10 (Altmeppen et al., 2012).
In recent years, α-cleavage attracted a lot of attention because it results in the production of PrPN1, a natural PrPC metabolite with a clear neuroprotective activity against different insults in vivo, in primary neuronal cultures and in cell lines (Guillot-Sestier et al., 2009, 2012; Resenberger et al., 2011; Beland et al., 2012; Fluharty et al., 2013; Beland and Roucou, 2013a). In particular, the neuroprotection against soluble Aβ oligomers that may be the culprit species in Alzheimer's disease may pave the way for the discovery of a new class of therapeutic molecules (Beland et al., 2012; Fluharty et al., 2013). There is also some evidence that α-cleavage is increased in post-mortem brain tissues of Alzheimer's disease patients, and that PrPN1 traps Aβ into amorphous aggregates unable to transform into soluble and toxic Aβ oligomers, and that α-cleavage decrease promotes neurotoxicity in prion and Alzheimer's diseases (Pietri et al., 2013; Beland et al., 2014). PrPN1 also binds to and antagonizes the toxicity of other β-sheet rich oligomers, including PrPSc oligomers, and PrPN1-derived therapeutic molecules may help treat different neurodegenerative disorders (Resenberger et al., 2011).
The GPI-anchored PrPC1 fragment after α-cleavage of PrPC protects against prion infection of neuronal and non-neuronal cell lines and acts as a dominant-negative inhibitor of prion conversion in vivo (Lewis et al., 2009; Westergard et al., 2011). The mechanism of action of PrPC1 is unclear, but since PrPC1 is resistant to prion conversion, the authors proposed that PrPC1 competes with PrPC for binding to infectious PrPSc (Westergard et al., 2011). Thus, PrPC1-derived peptides may have therapeutic benefits in prion diseases.
As many experimental data converge to support the proposition that PrPN1 and PrPC1 are neuroprotective metabolites, two therapeutic avenues could be proposed in prion diseases: to provide exogenous PrPN1- or PrPC1-derived molecules, or to increase the natural production of PrPN1 and PrPC1 by stimulating the α-cleavage. The α-cleavage mechanism is nebulous and only two elements are known: it occurs in the late secretory pathway but the enzyme is still unknown, and the hydrophobic domain is essential for this cleavage (Bremer et al., 2010; Oliveira-Martins et al., 2010). This domain is also essential for the physiological dimerization of PrPC (Rambold et al., 2008), supporting the hypothesis of a possible connection between dimerization and α-cleavage. Using an inducible dimerization strategy with a permeable dimerizer, we were able to show that PrPC dimerization in cell lines and primary neurons increase PrPC trafficking to the plasma membrane and largely increase the production of PrPN1, PrPC1, and shed PrPC (Beland et al., 2012). After dimerization, conditioned medium containing these three metabolites strongly protected cells against toxic Aβ oligomers.
Since levels of the products of both α-cleavage and shedding rose after dimerization, we concluded that the large increase of PrPC trafficking to the plasma membrane was sufficient to explain the high levels of its metabolites. This effect was fast and occurred 4 h post-dimerization. Deletion of the hydrophobic domain, the natural dimerization domain, does not prevent PrPC trafficking to the plasma membrane (Winklhofer et al., 2003). Thus, dimerization is not essential for PrPC trafficking. We proposed a model with a constitutive and dimerization-independent pathway for PrPC secretion, and a pathway regulated by dimerization. This regulated pathway would allow the cells to quickly respond to toxic insults by increasing the levels of protective PrPC metabolites (Beland and Roucou, 2013a,b).
PrPC→PrPSc conversion or prion conversion is central to prion diseases; this process is neurotoxic and PrPSc molecules assemble into infectious particles responsible for the transmission of the disease (Prusiner, 1998; Mallucci et al., 2003). Not surprisingly, numerous mechanistic studies have addressed this process using recombinant PrP (recPrP) and several experimental data have indicated an intrinsic tendency of the protein to form dimers during the initial steps of prion conversion. A fraction of Syrian hamster recPrP(90-231) forms alpha-helical dimers in solution in the presence of submicellar concentrations of SDS (Jansen et al., 2001). These dimers believed to be intermediates in prion conversion were observed by size exclusion chromatography, chemical crosslinking and analytical ultracentrifugation (Kaimann et al., 2008; Stöhr et al., 2008; Jansen et al., 2001). In these studies, non-denaturing concentrations of SDS were used to mimic membrane-like features. Using a reduction-oxidation protocol to induce the fibrillar assembly of Syrian hamster PrP(90–231), Lee and Eisenberg also observed the presence of dimeric intermediates in polyacrylamide native gels (Lee and Eisenberg, 2003). A similar conclusion was obtained with murine recPrP(23–231). During the conversion of murine PrP(23–231), an intermediate water-soluble β-sheet isoform termed PrPβ was identified (Luhrs et al., 2006). The kinetics of PrPC→PrPβ conversion suggest that dimerization is the rate-limiting step for the transition. The dimerization of murine recPrP(23–231) as a key molecular step during the conversion was confirmed in a subsequent study (Hafner-Bratkovic et al., 2011). Additionally, 3D reconstruction of murine recPrP(91–230) amyloid fibrils led to the proposition that dimers represent building units of such fibrils (Tattum et al., 2006).
These experiments were performed with non-posttranslationally modified PrPC. Yet in vivo, PrPC carries two N-glycosylations and a GPI anchor and these complex posttranslational modifications may play an important role in prion conversion. To address this issue, posttranslationally modified PrPC was purified from Chinese hamster ovary cells overexpressing Syrian hamster PrPC. This native PrPC spontaneously formed dimers stabilized by intermolecular β-sheets after insertion into artificial membranes (Elfrink et al., 2008).
Altogether, these studies support the hypothesis that dimerization is an important step for prion conversion but they did not directly test this hypothesis. To address this issue, two strategies have been used. First, two monomeric mouse PrP (23–231) were covalently linked with a linker and recombinantly purified. This tandem protein oligomerized after purification, and thioflavine T staining indicated that such oligomers were likely on the pathway of amyloid formation, although this was not demonstrated (Simoneau et al., 2007). In a second strategy, we used a conditional dimerization approach to induce chemical dimerization of mouse recPrP(23–231) and human recPrP (23-231). α-helical PrP dimers spontaneously converted into β-sheet oligomers and amyloid fibrils were detected by electron microscopy and thioflavinT staining (Roostaee et al., 2009). Importantly, these experiments were performed in physiological-like conditions in the absence of any detergent or chaotropic agents.
Models of PrP dimers and the role of dimers in prion conversion are available (Warwicker, 2000; Gauczynski et al., 2001; Tompa et al., 2002). However, in all the above studies, prion conversion was assessed by the formation of PrP amyloid fibrils and/or partial resistance to proteinase K rather than by the formation of infectious PrPSc in animal bioassays. Hence, the biological significance of PrPC dimerization for the formation of infectious PrPSc remains speculative.
PrPC forms dimers in native conditions and cell surface dimerization clearly regulates PrPC-mediated signaling and the resulting physiological neuroprotective/neurotrophic activities (Rambold et al., 2008). Intracellular dimerization also drastically increases its trafficking to the plasma membrane and the production of its natural metabolites PrPN1 and PrPC1 (Beland et al., 2012). The combination of these two effects of PrPC dimerization likely provides PrPC with a powerful neuroprotective/neurotrophic function (Figure 1). However, the flip side of the coin is that unwanted dimerization may initiate prion conversion and result in neuronal toxicity (Tompa et al., 2002). Regulating PrPC dimerization may help translate these findings into novel therapeutic interventions in neurodegenerative diseases (Beland and Roucou, 2013b).
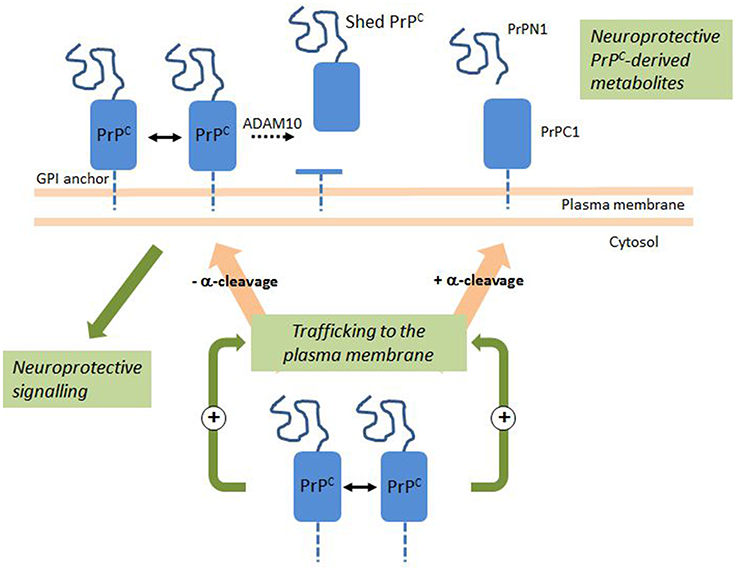
Figure 1. PrPC Dimerization activates a neuroprotective signaling pathway and the production of neuroprotective PrPC-derived metabolites. En route to the plasma membrane, a fraction of PrPC undergoes a physiological cleavage termed α-cleavage. Intracellular PrPC dimerization stimulates trafficking to the plasma membrane and the release of PrPN1 and PrPC1 at the cell surface. PrPC dimerization at the cell surface activates an intracellular signaling pathway with a neuroprotective outcome. At the cell surface, a fraction of PrPC undergoes ADAM10-mediated shedding. Double arrows indicate dimerization; dotted arrow indicates shedding. See text for details.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Altmeppen, H. C., Puig, B., Dohler, F., Thurm, D. K., Falker, C., Krasemann, S., et al. (2012). Proteolytic processing of the prion protein in health and disease. Am. J. Neurodegener. Dis. 1, 15–31.
Beland, M., Bedard, M., Tremblay, G., Lavigne, P., and Roucou, X. (2014). Abeta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol. Aging 35, 1537–1548. doi: 10.1016/j.neurobiolaging.2014.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beland, M., Motard, J., Barbarin, A., and Roucou, X. (2012). PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 32, 13255–13263. doi: 10.1523/JNEUROSCI.2236-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beland, M., and Roucou, X. (2012). The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J. Neurochem. 120, 853–868. doi: 10.1111/j.1471-4159.2011.07613.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beland, M., and Roucou, X. (2013a). Homodimerization as a molecular switch between low and high efficiency PrP C cell surface delivery and neuroprotective activity. Prion 7, 170–174. doi: 10.4161/pri.23583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beland, M., and Roucou, X. (2013b). Taking advantage of physiological proteolytic processing of the prion protein for a therapeutic perspective in prion and Alzheimer diseases. Prion 8, 106–110. doi: 10.4161/pri.27438
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brandner, S., Isenmann, S., Raeber, A., Fischer, M., Sailer, A., Kobayashi, Y., et al. (1996). Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379, 339–343. doi: 10.1038/379339a0
Bremer, J., Baumann, F., Tiberi, C., Wessig, C., Fischer, H., Schwarz, P., et al. (2010). Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 13, 310–318. doi: 10.1038/nn.2483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, Y., Thelin, W. R., Yang, B., Milgram, S. L., and Jacobson, K. (2006). Transient anchorage of cross-linked glycosyl-phosphatidylinositol-anchored proteins depends on cholesterol, Src family kinases, caveolin, and phosphoinositides. J. Cell Biol. 175, 169–178. doi: 10.1083/jcb.200512116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chesebro, B., Trifilo, M., Race, R., Meade-White, K., Teng, C., LaCasse, R., et al. (2005). Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308, 1435–1439. doi: 10.1126/science.1110837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elfrink, K., Ollesch, J., Stohr, J., Willbold, D., Riesner, D., and Gerwert, K. (2008). Structural changes of membrane-anchored native PrP(C). Proc. Natl. Acad. Sci. U.S.A. 105, 10815–10819. doi: 10.1073/pnas.0804721105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fluharty, B. R., Biasini, E., Stravalaci, M., Sclip, A., Diomede, L., Balducci, C., et al. (2013). An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 288, 7857–7866. doi: 10.1074/jbc.M112.423954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gauczynski, S., Hundt, C., Leucht, C., and Weiss, S. (2001). Interaction of prion proteins with cell surface receptors, molecular chaperones, and other molecules. Adv. Protein Chem. 57, 229–272. doi: 10.1016/S0065-3233(01)57024-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guillot-Sestier, M. V., Sunyach, C., Druon, C., Scarzello, S., and Checler, F. (2009). The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 284, 35973–35986. doi: 10.1074/jbc.M109.051086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guillot-Sestier, M. V., Sunyach, C., Ferreira, S. T., Marzolo, M. P., Bauer, C., Thevenet, A., et al. (2012). alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J. Biol. Chem. 287, 5021–5032. doi: 10.1074/jbc.M111.323626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gunther, E. C., and Strittmatter, S. M. (2010). Beta-amyloid oligomers and cellular prion protein in Alzheimer's disease. J. Mol. Med. 88, 331–338. doi: 10.1007/s00109-009-0568-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hafner-Bratkovic, I., Bester, R., Pristovsek, P., Gaedtke, L., Veranic, P., Gaspersic, J., et al. (2011). Globular domain of the prion protein needs to be unlocked by domain swapping to support prion protein conversion. J. Biol. Chem. 286, 12149–12156. doi: 10.1074/jbc.M110.213926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heldin, C. H. (1995). Dimerization of cell surface receptors in signal transduction. Cell 80, 213–223. doi: 10.1016/0092-8674(95)90404-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hernandez-Rapp, J., Martin-Lanneree, S., Hirsch, T. Z., Pradines, E., Alleaume-Butaux, A., Schneider, B., et al. (2014). A PrP(C)-caveolin-Lyn complex negatively controls neuronal GSK3beta and serotonin 1B receptor. Sci. Rep. 4:4881. doi: 10.1038/srep04881
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hundt, C., Gauczynski, S., Leucht, C., Riley, M. L., and Weiss, S. (2003). Intra- and interspecies interactions between prion proteins and effects of mutations and polymorphisms. Biol. Chem. 384, 791–803. doi: 10.1515/BC.2003.088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jansen, K., Schafer, O., Birkmann, E., Post, K., Serban, H., Prusiner, S. B., et al. (2001). Structural intermediates in the putative pathway from the cellular prion protein to the pathogenic form. Biol. Chem. 382, 683–691. doi: 10.1515/BC.2001.081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaimann, T., Metzger, S., Kuhlmann, K., Brandt, B., Birkmann, E., Höltje, H. D., et al. (2008). Molecular model of an alpha-helical prion protein dimer and its monomeric subunits as derived from chemical cross-linking and molecular modeling calculations. J. Mol. Biol. 376, 582–596. doi: 10.1016/j.jmb.2007.11.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Klohn, P. C., Farmer, M., Linehan, J. M., O'Malley, C., Fernandez de Marco, M., Taylor, W., et al. (2012). PrP antibodies do not trigger mouse hippocampal neuron apoptosis. Science 335, 52. doi: 10.1126/science.1215579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuwahara, C., Takeuchi, A. M., Nishimura, T., Haraguchi, K., Kubosaki, A., Matsumoto, Y., et al. (1999). Prions prevent neuronal cell-line death. Nature 400, 225–226. doi: 10.1038/22241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lauren, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W., and Strittmatter, S. M. (2009). Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132. doi: 10.1038/nature07761
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, S., and Eisenberg, D. (2003). Seeded conversion of recombinant prion protein to a disulfide-bonded oligomer by a reduction-oxidation process. Nat. Struct. Biol. 10, 725–730. doi: 10.1038/nsb961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lewis, V., Hill, A. F., Haigh, C. L., Klug, G. M., Masters, C. L., Lawson, V. A., et al. (2009). Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J. Neuropathol. Exp. Neurol. 68, 1125–1135. doi: 10.1097/NEN.0b013e3181b96981
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linden, R., Martins, V. R., Prado, M. A., Cammarota, M., Izquierdo, I., and Brentani, R. R. (2008). Physiology of the prion protein. Physiol. Rev. 88, 673–728. doi: 10.1152/physrev.00007.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lo, R. Y., Shyu, W. C., Lin, S. Z., Wang, H. J., Chen, S. S., and Li, H. (2007). New molecular insights into cellular survival and stress responses: neuroprotective role of cellular prion protein (PrPC). Mol. Neurobiol. 35, 236–244. doi: 10.1007/s12035-007-8003-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luhrs, T., Zahn, R., and Wuthrich, K. (2006). Amyloid formation by recombinant full-length prion proteins in phospholipid bicelle solutions. J. Mol. Biol. 357, 833–841. doi: 10.1016/j.jmb.2006.01.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mallucci, G., Dickinson, A., Linehan, J., Klöhn, P. C., Brandner, S., and Collinge, J. (2003). Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871–874. doi: 10.1126/science.1090187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martins, V. R., Beraldo, F. H., Hajj, G. N., Lopes, M. H., Lee, K. S., Prado, M. A., et al. (2010). Prion protein: orchestrating neurotrophic activities. Curr. Issues Mol. Biol. 12, 63–86.
Meyer, R. K., Lustig, A., Oesch, B., Fatzer, R., Zurbriggen, A., and Vandevelde, M. (2000). A monomer-dimer equilibrium of a cellular prion protein (PrPC) not observed with recombinant PrP. J. Biol. Chem. 275, 38081–38087. doi: 10.1074/jbc.M007114200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miranda, A., Ramos-Ibeas, P., Pericuesta, E., Ramirez, M. A., and Gutierrez-Adan, A. (2013). The role of prion protein in stem cell regulation. Reproduction 146, R91–R99. doi: 10.1530/REP-13-0100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monnet, C., Gavard, J., Mege, R. M., and Sobel, A. (2004). Clustering of cellular prion protein induces ERK1/2 and stathmin phosphorylation in GT1-7 neuronal cells. FEBS Lett. 576, 114–118. doi: 10.1016/j.febslet.2004.08.076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J. L., Lehmann, S., Launay, J. M., et al. (2000). Signal transduction through prion protein. Science 289, 1925–1928. doi: 10.1126/science.289.5486.1925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mouillet-Richard, S., Pietri, M., Schneider, B., Vidal, C., Mutel, V., Launay, J. M., et al. (2005). Modulation of serotonergic receptor signaling and cross-talk by prion protein. J. Biol. Chem. 280, 4592–4601. doi: 10.1074/jbc.M406199200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oliveira-Martins, J. B., Yusa, S., Calella, A. M., Bridel, C., Baumann, F., Dametto, P., et al. (2010). Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS ONE 5:e9107. doi: 10.1371/journal.pone.0009107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pietri, M., Dakowski, C., Hannaoui, S., Alleaume-Butaux, A., Hernandez-Rapp, J., Ragagnin, A., et al. (2013). PDK1 decreases TACE-mediated α-secretase activity and promotes disease progression in prion and Alzheimer's diseases. Nat. Med. 19, 1124–1131. doi: 10.1038/nm.3302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pradines, E., Loubet, D., Schneider, B., Launay, J. M., Kellermann, O., and Mouillet-Richard, S. (2008). CREB-dependent gene regulation by prion protein: impact on MMP-9 and beta-dystroglycan. Cell. Signal. 20, 2050–2058. doi: 10.1016/j.cellsig.2008.07.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Priola, S. A., Caughey, B., Wehrly, K., and Chesebro, B. (1995). A 60-kDa prion protein (PrP) with properties of both the normal and scrapie-associated forms of PrP. J. Biol. Chem. 270, 3299–3305. doi: 10.1074/jbc.270.7.3299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Prusiner, S. B. (1998). Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383. doi: 10.1073/pnas.95.23.13363
Rambold, A. S., Muller, V., Ron, U., Ben-Tal, N., Winklhofer, K. F., and Tatzelt, J. (2008). Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 27, 1974–1984. doi: 10.1038/emboj.2008.122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Resenberger, U. K., Harmeier, A., Woerner, A. C., Goodman, J. L., Muller, V., Krishnan, R., et al. (2011). The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 30, 2057–2070. doi: 10.1038/emboj.2011.86
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robinson, P. J. (1991). Phosphatidylinositol membrane anchors and T-cell activation. Immunol. Today 12, 35–41. doi: 10.1016/0167-5699(91)90110-F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roostaee, A., Cote, S., and Roucou, X. (2009). Aggregation and amyloid fibril formation induced by chemical dimerization of recombinant prion protein in physiological-like conditions. J. Biol. Chem. 284, 30907–30916. doi: 10.1074/jbc.M109.057950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roucou, X., and LeBlanc, A. C. (2005). Cellular prion protein neuroprotective function: implications in prion diseases. J. Mol. Med. 83, 3–11. doi: 10.1007/s00109-004-0605-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, B., Mutel, V., Pietri, M., Ermonval, M., Mouillet-Richard, S., and Kellermann, O. (2003). NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. U.S.A. 100, 13326–13331. doi: 10.1073/pnas.2235648100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schneider, B., Pietri, M., Pradines, E., Loubet, D., Launay, J. M., Kellermann, O., et al. (2011). Understanding the neurospecificity of Prion protein signaling. Front. Biosci. (Landmark Ed.) 16, 169–186. doi: 10.2741/3682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simoneau, S., Rezaei, H., Sales, N., Kaiser-Schulz, G., Lefebvre-Roque, M., Vidal, C., et al. (2007). In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 3:e125. doi: 10.1371/journal.ppat.0030125
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Solforosi, L., Criado, J. R., McGavern, D. B., Wirz, S., Sanchez-Alavez, M., Sugama, S., et al. (2004). Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303, 1514–1516. doi: 10.1126/science.1094273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sonati, T., Reimann, R. R., Falsig, J., Baral, P. K., O'Connor, T., Hornemann, S., et al. (2012). The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 501, 102–106. doi: 10.1038/nature12402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stefanova, I., Horejsi, V., Ansotegui, I. J., Knapp, W., and Stockinger, H. (1991). GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science 254, 1016–1019. doi: 10.1126/science.1719635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stöhr, J., Weinmann, N., Wille, H., Kaimann, T., Nagel-Steger, L., Birkmann, E., et al. (2008). Mechanisms of prion protein assembly into amyloid. Proc. Natl. Acad. Sci. U.S.A. 105, 2409–2414. doi: 10.1073/pnas.0712036105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, K. G., Kasai, R. S., Hirosawa, K. M., Nemoto, Y. L., Ishibashi, M., Miwa, Y., et al. (2012). Transient GPI-anchored protein homodimers are units for raft organization and function. Nat. Chem. Biol. 8, 774–783. doi: 10.1038/nchembio.1028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tattum, M. H., Cohen-Krausz, S., Thumanu, K., Wharton, C. W., Khalili-Shirazi, A., Jackson, G. S., et al. (2006). Elongated oligomers assemble into mammalian PrP amyloid fibrils. J. Mol. Biol. 357, 975–985. doi: 10.1016/j.jmb.2006.01.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tompa, P., Tusnady, G. E., Friedrich, P., and Simon, I. (2002). The role of dimerization in prion replication. Biophys. J. 82, 1711–1718. doi: 10.1016/S0006-3495(02)75523-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Warwicker, J. (2000). Modeling a prion protein dimer: predictions for fibril formation. Biochem. Biophys. Res. Commun. 278, 646–652. doi: 10.1006/bbrc.2000.3829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Westergard, L., Turnbaugh, J. A., and Harris, D. A. (2011). A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J. Biol. Chem. 286, 44234–44242. doi: 10.1074/jbc.M111.286195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Winklhofer, K. F., Heske, J., Heller, U., Reintjes, A., Muranyi, W., Moarefi, I., et al. (2003). Determinants of the in vivo folding of the prion protein. A bipartite function of helix 1 in folding and aggregation. J. Biol. Chem. 278, 14961–14970. doi: 10.1074/jbc.M209942200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: prion protein trafficking, dimerization, signaling, neuroprotection, neurodegeneration
Citation: Roucou X (2014) Regulation of PrPC signaling and processing by dimerization. Front. Cell Dev. Biol. 2:57. doi: 10.3389/fcell.2014.00057
Received: 04 August 2014; Paper pending published: 25 August 2014;
Accepted: 19 September 2014; Published online: 09 October 2014.
Edited by:
Sophie Mouillet-Richard, Institut National de la Santé et de la Recherche Médicale, FranceReviewed by:
Markus Glatzel, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2014 Roucou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xavier Roucou, Département de Biochimie (Z8-2001), Faculté de Médecine, Université de Sherbrooke, 3201 Jean Mignault, Sherbrooke, QC J1E4K8, Canada e-mail:eGF2aWVyLnJvdWNvdUB1c2hlcmJyb29rZS5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.